

Abstract
Isobaric peptide termini labeling (IPTL) is an attractive protein quantification method because it provides more accurate and reliable quantification information than traditional isobaric labeling methods (e.g., TMT and iTRAQ) by making use of the entire fragment-ion series instead of only a single reporter ion. The multiplexing capacity of published IPTL implementations is, however, limited to three. Here, we present a selective maleylation-directed isobaric peptide termini labeling (SMD-IPTL) approach for quantitative proteomics of LysC protein digestion. SMD-IPTL extends the multiplexing capacity to 4-plex with the potential for higher levels of multiplexing using commercially available 13C/15N labeled amino acids. SMD-IPTL is achieved in a one-pot reaction in three consecutive steps: (1) selective maleylation at the N-terminus; (2) labeling at the ε-NH2 group of the C-terminal Lys with isotopically labeled acetyl-alanine; (3) thiol Michael addition of an isotopically labeled acetyl-cysteine at the maleylated N-terminus. The isobarically labeled peptides are fragmented into sets of b- and y-ion clusters upon LC-MS/MS, which convey not only sequence information but also quantitative information for every labeling channel and avoid the issue of ratio distortion observed with reporter-ion-based approaches. We demonstrate the SMD-IPTL approach with a 4-plex labeled sample of bovine serum albumin (BSA) and yeast lysates mixed at different ratios. With the use of SMD-IPTL for labeling and a narrow precursor isolation window of 0.8 Th with an offset of −0.2 Th, accurate ratios were measured across a 10-fold mixing range of BSA in a background of yeast proteome. With the yeast proteins mixed at ratios of 1:5:1:5, BSA was detected at ratios of 0.94:2.46:4.70:9.92 when spiked at 1:2:5:10 ratios with an average standard deviation of peptide ratios of 0.34.
Proteome quantification gives information on the relative amounts of a large number of proteins between samples.1−3 The existing mass-spectrometry-based proteome-wide quantitative methods can be classified into label-free proteomics4,5 and label-based proteomics.6−8 Even though advanced data-acquisition schemes and algorithms have been developed for label-free proteomics, the limited throughput and signal variation due to, among others, variable sample loss during workup and changing ionization efficiency between injections argue in favor of multiplexed, label-based proteomics. Multiplexed quantification approaches (e.g., ICAT,6,9,10 SILAC,11−13 iTRAQ,7,14 TMT,8,15 and IPTL16−21) exploit different combinations of heavy and light isotopes to differentially label peptides, which enables simultaneous sample workup and LC-MS/MS analysis of multiple samples in a single experiment. The commonly used isotopes are 13C, 15N, 18O, and 2H, with 2H being less popular because of the potential risk of altering the peptide retention time.22−25
The existing multiplexing strategies can also be classified into two categories, MS1 quantification and MS2 quantification, on the basis of the stage at which peptides are quantified.26 For MS1 quantification, also called isotopic quantification, such as SILAC12 and ICAT,6 the same peptide from different samples will be labeled with different isotopic tags, which results in the same peptide showing multiple precursor ions at the MS1 level. Relative quantification is achieved by comparing the intensities or peak areas of the precursor ions at the MS1 level. Therefore, any isotopic quantification method will at least double the complexity of the MS1 spectrum, which further aggravates the already challenging issue of a limited sampling capacity of precursor ions for MS/MS fragmentation across a chromatographic peak. In contrast, the MS2 quantification methods use isobarically labeled peptides, so the same peptide originating from different samples will have the same mass. After fragmentation, the isobarically labeled peptide will release a unique reporter ion (TMT and iTRAQ) or peptide fragment ions (IPTL), which can be used to reveal the quantification information. The MS2 quantification methods not only allow for the straightforward quantification of multiple samples in a single MS2 spectrum but also further reduce the required instrument time. The most widely used isobaric quantification methods are those using reporter-ion tags (TMT and iTRAQ) because of the multiplex capacity and well-developed data-processing software. However, reporter-ion-based quantification methods suffer from ratio distortion,26−31 which is particularly serious in complex samples, arising from the cofragmentation of multiple peptides passing the precursor-ion selection window. These peptides release identical reporter ions that are indistinguishable in MS2. To correct for the ratio distortion, several methods have been proposed, such as additional gas-phase purification31 and MultiNotch MS3.27 An alternative isobaric method that gives rise to multiple quantification ions per peptide and is therefore less affected by the cofragmentation problem is isobaric peptide termini labeling (IPTL). IPTL was first reported in 2009 by Koehler et al.,16 but the approach is not as extensively used as TMT and iTRAQ, presumably because of the limited multiplexing capacity, especially when avoiding deuterium labeling.
The initially reported IPTL method showed relative quantification of two samples of LysC digested proteins, where the peptides were crosswise-modified at the C- and N-terminus with a pair of complementary isotopic tags, resulting in isobarically labeled peptides that were fragmented into product-ion clusters. The peptide and protein ratios can be inferred by comparing the intensities of the individual y- and b-series fragment ions. Even in the case of cofragmentation of two or more peptides, fragment ions can usually be correctly attributed. IPTL potentially permits more accurate and reliable quantification with multiple quantification data points per spectrum, for each y- and b-ion, and suffers less from cofragmentation. Consequently, a number of optimized methods and applications have been reported in the past 10 years, such as selective succinylation17 and dimethylation19,32−35 based IPTL (triplex-IPTL19 and triplex-QITL21), SILAC34,35 or proteolytic 18O labeling33,36 combined with IPTL (IVTAL,18 G-IVTL,34 QITL,33 and diDO-IPTL36), and pseudoisobaric dimethyl labeling20,37−40 (pIDL,37 PITL,38 SWATH-pseudo-IPTL,20 and MdFDIA41). Most of the methods are for duplex labeling or triplex at most, which means that the multiplexing capacity of IPTL is still far less than that of TMT, which has been extended to 16 labeling channels in a single LC-MS/MS run.42 Recently, Liu et al.40 reported the pseudoisobaric dimethyl labeling (m-pIDL) method, which increased the multiplex capacity to 6-plex. m-pIDL does not suffer from cofragmentation while relying on a wide isolation window of 10 Th. A potential limitation is the utilization of deuterium in the isotopic tags, which carries the risk of changing the retention time of labeled peptides.
To improve the IPTL multiplex capacity with nondeuterium tags, we propose the selective maleylation-directed isobaric peptide termini labeling (SMD-IPTL) method, which is based on selective maleylation at the N-termini of LysC digested peptides. The performance of SMD-IPTL was assessed at the 4-plex level by spiking different amounts of bovine serum albumin (BSA) into a yeast proteome background. SMD-IPTL can be extended to the 7-plex level using commercially available 13C- or 15N-labeled cysteine and alanine.
Experimental Section
Details of the used chemicals and materials, the synthesis of isotopically labeled acetyl-cysteine and the acetyl-alanine p-nitrophenol ester, LC purification, LysC digestion, LC/MS/MS analysis, and database searching and quantification can be found in the Supporting Information.
Optimization of Selective Maleylation at the Peptide N-Terminus
Solutions of different pH values (7.0, 6.5, 6.0, 5.5, and 5.0) were prepared with 100 mM sodium acetate and acetic acid.17 Subsequently, the peptide WLYRAK was dissolved in solutions of different pH values (7.0, 6.5, 6.0, 5.5, and 5.0) to a concentration of 10 μM. Then 4 μg/μL maleic anhydride was freshly prepared in acetonitrile and 2 μL was added to 100 μL of each WLYRAK solution. The reaction tube was shaken at room temperature for 30 min. The reaction was tracked with LC-MS. Maleylation on LysC peptides was further optimized by infusing 50 μg/μL maleic anhydride with a syringe pump at a flow rate of 0.4 μL/min into 25 μg of LysC peptides dissolved in 1 mL of sodium acetate–acetic acid solution at pH 5.5 for 1 h.
2-Plex Labeling of Maleylated WLYRAK
Maleylated peptide solution (100 μL) was dried in a vacuum concentrator, followed by the addition of 100 μL of 50 mM sodium tetraborate, and the pH was adjusted to 9 with 500 mM NaOH. Then 100 mM 13C1-acetyl-alanine p-nitrophenol ester (13C1-Ac-Ala-PNP) containing one 13C label in the acetyl group or acetyl-alanine-p-nitrophenol ester (Ac-Ala-PNP), which contains no 13C label, was prepared in dimethyl formamide. Subsequently, 2 μL of p-nitrophenol ester was added to the maleylated peptide solution and incubated for 1 h at room temperature. To ensure complete labeling, 2 μL of p-nitrophenol ester was added again and incubated for 30 min more. Afterward, the pH of acetyl-alanine labeled solutions was adjusted to 9. Subsequently, 5 μL of 400 mM 13C1-acetyl-cysteine (13C1-Ac-Cys-OH), which contains one 13C label in the acetyl group, was added to the Ac-Ala-PNP labeled solution. Conversely, 5 μL of 400 mM acetyl-cysteine (Ac-Cys-OH) was added to the 13C1-Ac-Ala-PNP labeled solution. The reaction solutions were bubbled with argon for 5 min and incubated overnight at 55 °C. Finally, potentially formed esters at the hydroxyl groups of Ser, Thr, or Tyr and excess PNP ester were hydrolyzed by treatment with 5% hydroxylamine43 for 5 min at 55 °C prior to desalting the samples by SPE using the STAGE (STop And Go Extraction) TIPS Desalting Procedure44 followed by LC-MS analysis.
4-Plex Isobaric Labeling of LysC Peptides
Maleylated LysC peptides (400 μL) of BSA or yeast protein was mixed with 50 μL of 100 mM sodium tetraborate and the pH was adjusted to 9 with 5 M NaOH. The solution was split into four tubes for labeling reactions of four channels. Two microliters of 100 mM Ac-Ala-PNP, 13C1-Ac-Ala-PNP, 13C2-Ac-Ala-PNP, and 13C3-Ac-Ala-PNP was respectively added to the four tubes of maleylated LysC peptide solution and incubated for 1 h at room temperature. To ensure complete labeling, 2 μL of p-nitrophenol ester was added again and incubated for 30 min more. Afterward, the pH of acetyl-alanine-p-nitrophenol ester labeled solutions was adjusted to 9. Ten microliters of 400 mM 13C3-Ac-Cys-OH was added to the Ac-Ala-PNP labeled solution, 10 μL of 400 mM 13C2-Ac-Cys-OH was added to the 13C1-Ac-Ala-PNP labeled solution, 10 μL of 400 mM 13C1-Ac-Cys-OH was added to the 13C2-Ac-Ala-PNP labeled solution, and 10 μL of 400 mM Ac-Cys-OH was added to the 13C3-Ac-Ala-PNP labeled solution. The reaction solutions were bubbled with argon for 5 min and incubated overnight at 55 °C. Finally, potentially formed esters at the hydroxyl groups of Ser, Thr, or Tyr and excess PNP ester were hydrolyzed by treatment with 5% hydroxylamine43 for 5 min at 55 °C prior to desalting the samples by SPE using the STAGE (STop And Go Extraction) TIPS Desalting Procedure44 followed by LC-MS analysis.
Results and Discussion
Improving the Multiplexing Capacity of IPTL
SMD-IPTL not only retains all of the merits of IPTL16,17,19 but also improves it by (I) increasing the multiplex capacity to 4-plex or more with readily available isotopically labeled amino acids and (II) utilizing non-deuterium labeled tags to ensure that isobarically labeled peptides have the same retention time.
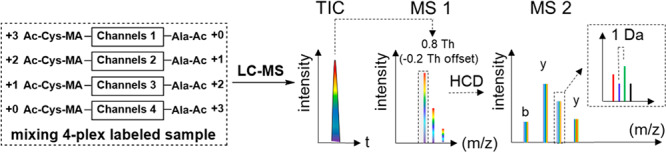
Inspired by selective succinylation17 and thiol Michael addition on maleic derivatives,45 we assumed that maleic anhydride has comparable selective reactivity for the peptide N-terminus as succinic anhydride. The introduced maleic derivatives at the N-terminus can be used for further modification. We decided to use readily available 13C and 15N labeled amino acids as tag building blocks. As shown in Figure 1A, the isobaric labeling can be achieved in a one-pot reaction with three consecutive steps: (1) selective maleylation at the N-terminus; (2) labeling with acetyl-alanine-p-nitrophenol ester at the ε-NH2 group of the C-terminal Lys; (3) thiol Michael addition at the double bond of the newly maleylated N-terminus. As a result, the LysC peptides will be isobarically and differentially labeled at the N- and C-terminus with a complementary pair of isotopically labeled acetyl-cysteine and acetyl-alanine, respectively. With commercially available isotopically labeled cysteine and alanine, seven isobaric combinations can potentially be made, without the need for deuterium labels, as shown in Figure 1B. The isobarically labeled peptides derived from different samples are mixed prior to LC-MS/MS analysis (Figure 1C) and have identical retention times and masses. The MS2 peak intensities of y- and b-ion series fragments for each labeling channel are extracted and their intensity ratios represent the difference in the amount of the peptides in each sample in the mixture, as shown in the inset in Figure 1C.
Figure 1.

Scheme of SMD-IPTL. (A) Labeling steps of SMD-IPTL. (B) Seven possible combinations of isotopically labeled acetyl-cysteine and acetyl-alanine. The atom marked with “*” denotes 13C or 15N. (C) LC-MS/MS process for a mixture of 7-plex labeled samples.
Optimization of the Selective Maleylation Reaction
Crosswise labeling of the N-terminal α-NH2 group and Lys ε-NH2 group with a pair of complementary tags is the prerequisite for IPTL. The challenge of IPTL therefore lies in the selective labeling of these two forms of the −NH2 group with readily available tags. Both selective succinylation17 and selective dimethylation32 have been used in IPTL, exploiting the pKa difference between α- and ε-amino groups to selectively label the α-NH2 group at a specific pH. Maleic anhydride has a similar structure and reactivity as succinic anhydride, but the carbon double bond in the maleic anhydride permits further derivatization after maleylation, which can be used to insert an isotopic tag at the peptide N-terminus.45 We used the peptide WLYRAK to investigate the reactivity and selectivity of maleylation at various pH values (pH 7.0, 6.5, 6.0, 5.5, and 5.0) and in water. As shown in Figure 2A, selectivity of maleylation for the α-NH2 group increases as the pH decreases. However, the reaction is not complete at pH 5.0, so pH 5.5 presents the best compromise between overall reaction yield and selectivity with an overall yield of more than 90% and a double maleylation of 5–8% (see Figure S5 for the MS2 spectrum of the N-maleylated peptide Ma-WLYRAK). The selective maleylation reaction was further optimized by infusion of 50 μg/μL maleic anhydride with a syringe pump at a flow rate of 0.4 μL/min into the peptide solution at pH 5.5. As shown in Figure S6, double maleylation was further reduced to less than 2%. The amount of double-maleylated peptide did not increase when the infusion time was prolonged. On the basis of these results, sodium acetate at pH 5.5 and slow infusion of maleic anhydride with a syringe pump at 0.4 μL/min were used for optimal selective maleylation.
Figure 2.

Optimization of maleylation and isobaric labeling of peptide WLYRAK. (A) Maleylation in various pH buffers. XICs of m/z 418.74, 467.74, and 516.74 were combined for all pH values. Peak a in the extracted ion chromatograms is the unmodified peptide; peak b is the peptide selectively maleylated on the α-NH2 group; peak c is the double-maleylated peptide on both α-NH2 and ε-NH2 groups. (B) One-pot isobaric labeling reaction steps. Extracted Ion Chromatograms (XICs) of m/z 467.74, 524.27, and 605.78 were combined for all steps. Peak b is the single-maleylated peptide (Ma-WLYRAK); peak d is the peptide after labeling with acetyl-alanine at the ε-NH2 group (Ma-WLYRAK-Ac-Ala); peak e is the peptide after thiol Michael addition of acetyl-cysteine (Ac-Cys-Ma-WLYRAK-Ac-Ala). Mass spectra are shown to the right of the chromatograms. (C) Enlarged view of the y3-ion in the MS2 spectrum for different mixing ratios of f, 13C1-Ac-Cys-WLYRAK-Ac-Ala, and g, Ac-Cys-WLYRAK-Ac-Ala-13C1. (D) MS2 spectrum of 1:1 mixed 13C1-Ac-Cys-WLYRAK-Ac-Ala and Ac-Cys-WLYRAK-Ac-Ala-13C1. The peak marked with “*” is the precursor ion having lost the Ac-Ala group.
One-Pot Isobaric Labeling of Peptide WLYRAK
After selective maleylation at the α-NH2 group, 13C1-Ac-Ala-PNP or Ac-Ala-PNP was used to react with the ε-NH2 group of the C-terminal Lys at pH 9. As shown in Figure 2B, Ma-WLYRAK can be completely converted to Ma-WLYRAK-Ac-Ala-13C1 or Ma-WLYRAK-Ac-Ala. Subsequently, after the pH was adjusted to 9, the complementary isotopic form of acetyl-cysteine was incorporated to generate 13C1-Ac-Cys-WLYRAK-Ac-Ala and Ac-Cys-WLYRAK-Ac-Ala-13C1. Although the newly inserted N-terminal maleyl group is less reactive to thiol than maleimide, Tian et al. demonstrated that the maleyl group can efficiently react with cysteine and mercaptoethanol.45 However, we found that the thiol Michael addition was slow, taking more than 30 h to complete. After screening various additives, including triethylamine,46 hexylamine, proline,47 dimethylphenylphosphine,46 and sodium tetraborate,48 we found that sodium tetraborate enables full labeling of the N-terminal maleyl group in 15 h (Figure S7).
To demonstrate the feasibility of SMD-IPTL, isobarically labeled 13C1-Ac-Cys-WLYRAK-Ac-Ala and Ac-Cys-WLYRAK-Ac-Ala-13C1 were mixed at various ratios (1:1, 1:5, 1:10, 1:20, 5:1, 10:1, and 20:1) followed by LC-MS/MS analysis. As shown in Figure 2D, the fragment ions cover the entire b-ion series and several y-ions, which means that fragmentation of the peptide backbone works well after tags were inserted on both termini. Notably, every y- and b-ion appeared as a peak pair in the spectrum, as shown in Figure 2C (enlarged view of the y3-ion for different mixing ratios). The relative intensity of the light and heavy peaks is consistent with the corresponding mixing ratio. Figure S8 shows the correlation between experimental and theoretical ratios over the tested mixing ratios, which indicates that accurate quantification can be achieved over a 20-fold dynamic range for peptide WLYRAK.
Reducing Isotope Interference of Fragment Ions by Using a Narrow Precursor Isolation Window
In the MS2 spectra of IPTL, every labeling channel has a set of unique fragment ions, which represent the main advantage of IPTL by providing more accurate and reliable quantification information because the entire fragment-ion series contains ratio information instead of only the reporter ion in the TMT and iTRAQ reporter-ion-based approaches. Figure 3A–D shows the y5-ion of isobarically labeled SEIAHRFK (a BSA-derived peptide after LysC digestion) from four labeling channels with an interval of 1 Da between the channels. However, with a precursor isolation window (IW) of 2 Th, which is the default setting in data-dependent acquisition (DDA) proteomics on Orbitrap mass spectrometers, deducing quantification ratios from intensities of fragment ions is complicated because of the interference of the natural 13C contribution to the isotopologue pattern of the fragment ions.28 As shown in Figure 3E, setting the isolation window to 2 Th in the LC-MS/MS analysis of a 1:1:1:1 mixed BSA sample resulted in an MS2 spectrum (Figure 3F) in which the ratio of the y5-ions did not correspond to the expected 1:1:1:1 ratio because there is a small additional 13C peak next to the four major peaks. The use of a narrow precursor isolation window29,49 has been reported to reduce the 13C contribution to the fragment ions. As shown in Figure 3G, when the isolation window was set at 0.8 Th with −0.2 Th offset to specifically select the monoisotopic peak, the ratio between the y5-ions perfectly matched the expected 1:1:1:1 ratio (Figure 3H). However, on the Q Exactive plus mass spectrometer, the precursor-ion selection automatically shifts the isolation window to the center on the highest intensity peak, rather than the monoisotopic peak, which means that for peptides above ∼2 kDa multiple isotopologues are cofragmented. This means that, for the labeled peptide VPQVSTPTLVEVSRSLGK (2273.14 Da), even with a narrow isolation window of 0.8 Th and an offset of −0.2 Th (Figure 3I), the 13C contribution still affected the MS2 spectrum. As shown in Figure 3J–L, the 13C contribution increases with fragment-ion mass. Therefore, for the MS2 spectra derived from peptides where the isolation window is centered on the first 13C isotopologue, only the small fragment ions y2, b1, and b2 were used for data processing. The y1-ion was never used for quantification because all peptides in LysC digestion have the same y1-ion, which may distort ratios by peptide cofragmentation similar to reporter-ion-based approaches.40 Fragment ions other than the main b- and y-ion series, such as those having H2O and NH3 loss, were also ignored because of the increased likelihood of convolution between them.
Figure 3.

Reducing interference of the endogenous 13C contribution to the fragment ions by narrowing the width of the precursor isolation window (IW). Peptides are from the 4-plex labeled LysC digested BSA. (A) y5-ion of 13C3-Ac-Cys-Ma-SEIAHRFK-Ac-Ala; (B) y5-ion of 13C2-Ac-Cys-Ma-SEIAHRFK-Ac-Ala-13C1; (C) y5-ion of 13C1-Ac-Cys-Ma-SEIAHRFK-Ac-Ala-13C2; (D) y5-ion of Ac-Cys-Ma-SEIAHRFK-Ac-Ala-13C3; (E) IW of 2 Th to select the precursor ion of isobarically labeled SEIAHRFK mixed at a ratio of 1:1:1:1; (F) y5-ion of isobarically labeled SEIAHRFK mixed at a ratio of 1:1:1:1 and fragmented with an IW of 2 Th; (G) IW of 0.8 Th with a −0.2 Th offset to select the precursor ion of isobarically labeled SEIAHRFK mixed at a ratio of 1:1:1:1; (H) y5-ion of isobarically labeled SEIAHRFK mixed at a ratio of 1:1:1:1 and fragmented with an IW of 0.8 Th with a −0.2 Th offset; (I) IW of 0.8 Th with a −0.2 Th offset to select the precursor ion of isobarically labeled VPQVSTPTLVEVSRSLGK mixed at a ratio of 1:1:1:1; (J), (K), and (L), respectively show the b1-, y6-, and y15-ions of isobarically labeled VPQVSTPTLVEVSRSLGK mixed at a ratio of 1:1:1:1 and fragmented with an IW of 0.8 Th with a −0.2 Th offset.
After preselection of the fragment ions suitable for quantification, the next step is to calculate the ratios at the spectrum, peptide, and protein levels sequentially. As shown in Figure 4A, the log2-normalized ratio has a better convergence to zero as the intensity of fragment ions increases, which means that more intense fragments are more reliable and should contribute more to the calculated ratio. Consequently, the ratio at the spectrum level was calculated as the normalized ratio of the sum of all fragment-ion intensities from the same labeling channel (Figures S3 and S4). The same trend of intensities is apparent at the peptide level (Figure 4B). Thus, the ratio at the peptide level was calculated by using the spectra with the three highest total peak intensities and the ratio at the protein level was calculated by using the peptides with the three highest total peak intensities. According to this calculation, on the basis of medians of the log2-normalized measured ratios, the 1:5:1:5 and 1:2:5:10 mixed 4-plex labeled BSA ratios were determined to be 1.10:5.28:1.05:4.50 and 1.06:2.60:4.63:9.51 at the peptide level, as shown in parts (C) and (D), respectively, of Figure 4.
Figure 4.

Normalized ratios distribution of 4-plex labeled BSA mixed at various ratios. (A) Normalized ratios of all assigned fragment ions from 1:1:1:1 mixed 4-plex labeled BSA; (B) normalized ratios of identified peptides from 1:1:1:1 mixed 4-plex labeled BSA; (C) normalized ratios of identified peptides from 1:5:1:5 mixed 4-plex labeled BSA; (D) normalized ratios of identified peptides from 1:2:5:10 mixed 4-plex labeled BSA.
4-Plex Labeling of LysC Peptides of the Yeast Proteome
To evaluate the efficiency and reproducibility of SMD-IPTL labeling reactions and the quantification accuracy on a complex sample, 4-plex labeling was performed on the LysC peptides from a yeast proteome. Three replicate maleylation reactions were performed and the maleylation yield of each peptide was determined as the percentage of intensity of the N-terminal maleylated form to the total intensity of all possible forms including unmodified, N-terminal maleylated, C-terminal maleylated, and both N- and C-terminal maleylated peptide. As shown in Table 1 and Figure S9, around 95% of the peptides had a labeling yield higher than 90% and the average coefficient of variation (CV) of three replicates was 14.0%. The efficiency and reproducibility of subsequent labeling steps of the four channels with four pairs of complementary acetyl-cysteine and acetyl-alanine-p-nitrophenol ester can be found in Figure S10. More than 98% of identified peptides had a total yield above 98% and an average CV of 1.3%.
Table 1. Efficiency Evaluation of Three Replicates of Maleylation on LysC Yeast Peptides.
| number of peptides |
||||||
|---|---|---|---|---|---|---|
| yield >95% | 90–95% | 85–90% | 80–85% | yield <80% | CV | |
| replicate 1 | 2175 | 95 | 39 | 21 | 72 | 14.0% |
| replicate 2 | 2056 | 82 | 53 | 21 | 69 | 13.1% |
| replicate 3 | 2083 | 90 | 33 | 16 | 84 | 14.8% |
To investigate the quantification accuracy of SMD-IPTL in a complex sample, the 4-plex labeled LysC yeast peptides were mixed at a ratio of 1:1:1:1 and analyzed with an isolation window of 0.8 Th and an offset of −0.2 Th. As shown in Figure 5A, the log2-normalized ratios are mainly distributed over the range from −0.4 to 0.4. The medians of log2-normalized ratios of the four labeling channels are 0.03, 0.03, −0.06, and −0.03, respectively (Figure 5A). On the protein level (337 proteins were identified), 97.0% of the proteins were quantified within a 2-fold range and 91.7% of proteins were quantified within a 1.5-fold range.
Figure 5.

Various ratios of 4-plex labeled yeast and BSA-yeast samples. (A) Normalized ratios of a yeast sample mixed at a ratio of 1:1:1:1 [150:150:150:150 (ng)]; (B) normalized ratio of a BSA-yeast proteome sample that consists of the 1:2:5:10 [1:2:5:10 (ng)] mixed 4-plex labeled BSA and the 1:5:1:5 [50:250:50:250 (ng)] mixed 4-plex labeled yeast.
Quantifying BSA in a Background of Yeast Proteins
To investigate the dynamic range of SMD-IPTL in a complex background, a BSA-yeast proteome sample was prepared. 4-Plex labeled LysC peptides of BSA at a ratio of 1:2:5:10 and 4-plex labeled LysC peptides of yeast proteins at the ratio of 1:5:1:5 were mixed separately. The yeast peptides were then mixed with peptides of BSA, according to the scheme shown in Figure 5B. The mixed BSA-yeast sample was analyzed with an isolation window of 0.8 Th and an offset of −0.2 Th. To assess the quantification of the yeast proteins, as shown in Figure 5B, the medians of log2-normalized protein ratios of the four labeling channels were determined as −0.01, 2.37, 0.15, and 2.29, respectively, close to the theoretical medians of 0, 2.32, 0, and 2.32. In the channels of Ac-Cys-Ma-peptides-Ac-Ala-13C3 and 13C2–Ac-Cys-Ma-peptides-Ac-Ala-13C1, 4 times lower amounts of yeast peptides were mixed than in the other two channels, and the quantified protein ratios therefore had a larger variation. The 1:5:1:5 mixed channels of yeast had an average standard deviation of all log2 ratios of 0.40. For 4-plex labeled BSA, the log2-normalized protein ratios were −0.09:1.30:2.23:3.31, which is close to the log2-theoretical ratios of 0.00:1.00:2.32:3.32.
Although SMD-IPTL exhibits good quantification capability across a 10-fold dynamic range, there is still room for improvement with respect to peptide identification, which is a prerequisite for calculating theoretical fragment ions as required for relative peptide quantification. In the MS2 spectra of SMD-IPTL, the number of fragment ions is multiplied by the number of labeling channels, which provides more accurate quantification information. However, this also makes identification more challenging because of the more complex MS2 spectra, which are not properly handled by existing protein identification software50,51 because unmatched fragment-ion peaks reduce the peptide score. The development of identification algorithms capable of handling IPTL data would further improve the identification of peptides in SMD-IPTL samples and hence the number of quantified proteins. In addition, data acquisition software that is capable of specifically selecting the monoisotopic peak for all peptides in combination with a narrow precursor isolation window would further improve the quantification accuracy of SMD-IPTL. We observed that peptide ionization efficiency decreased slightly after isobaric labeling, which might be caused by modification of the amino groups of the peptides. Finally, the multiplexing capacity of SMD-IPTL can be further improved by extending the range of isotopic forms of Cys and Ala and the use of mass spectrometers with sufficient resolution to distinguish 13C and 15N isotopes in fragment ions.53
Conclusions
SMD-IPTL not only retains all the advantages of IPTL approaches but also improves the multiplexing capacity to at least 4-plex labeling with available non-deuterium isotopically labeled amino acids. SMD-IPTL is readily expandable to 7-plexing following the same strategy. Isobaric labeling of peptides is achieved in a one-pot reaction comprising three steps. We applied a precursor-ion isolation window of 0.8 Th with an offset of −0.2 Th during data acquisition, which significantly helped deducing protein ratios from intensities of fragment ions. Finally, the proteome quantification capability of SMD-IPTL was demonstrated with the 4-plex labeling of a yeast proteome sample spiked with BSA over a 10-fold dynamic range. Further improvements in data acquisition and data analysis software are needed to make the SMD-IPTL approach more widely used in the future.
Acknowledgments
We gratefully acknowledge the China Scholarship Council (CSC) for a Ph.D. fellowship to X.T. We thank Prof. Bernd Thiede for providing the quantification software isobariQ. X.T. thanks Jos Hermans for helping with the LC-MS analyses. This work was partially funded by the Open Technology Programme of Toegepaste en Technische Wetenschappen (TTW) with project number 15230, which is financed by The Netherlands Organisation for Scientific Research (NWO).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.analchem.0c01059.
Chemicals and materials; synthesis of isotopically labeled acetyl-cysteine and the acetyl-alanine-p-nitrophenol ester; LC purification; LysC digestion; LC-MS/MS analysis; synthesis of isotopically labeled acetyl-cysteine; synthesis of the isotopically labeled acetyl-alanine-p-nitrophenol ester; LC-MS of isotopically labeled acetyl-cysteine; LC-MS of the acetyl-alanine-p-nitrophenol ester; data-processing workflow for a 4-plex labeled sample; examples of calculating ratios at the fragment ion, PSM, peptide, and protein level; MS/MS spectrum of Ma-WLYRAK; optimization of the selective maleylation reaction by syringe pump infusion and the use of different catalysts and additives; correlation of measured ratios and mixing ratios at the peptide level; maleylation of Lys-C yeast peptides; 4-plex labeling of Ma-Lys-C yeast peptides (PDF)
The authors declare no competing financial interest.
Notes
The raw mass spectrometry proteomics data of the yeast and BSA-yeast sample have been deposited at the ProteomeXchange Consortium via the PRIDE54 partner repository with the data set identifier PXD018589.
After this paper was published ASAP on May 12, 2020, a correction was made to Figure 3 (peaks were missing in panel I). The corrected version was reposted May 13, 2020.
Supplementary Material
References
- Zhang Y.; Fonslow B. R.; Shan B.; Baek M.-C.; Yates J. R. Chem. Rev. 2013, 113, 2343–2394. 10.1021/cr3003533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauniyar N.; Yates J. R. J. Proteome Res. 2014, 13, 5293–5309. 10.1021/pr500880b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chahrour O.; Cobice D.; Malone J. J. Pharm. Biomed. Anal. 2015, 113, 2–20. 10.1016/j.jpba.2015.04.013. [DOI] [PubMed] [Google Scholar]
- Bondarenko P. V.; Chelius D.; Shaler T. A. Anal. Chem. 2002, 74, 4741–4749. 10.1021/ac0256991. [DOI] [PubMed] [Google Scholar]
- Asara J. M.; Christofk H. R.; Freimark L. M.; Cantley L. C. Proteomics 2008, 8, 994–999. 10.1002/pmic.200700426. [DOI] [PubMed] [Google Scholar]
- Gygi S. P.; Rist B.; Gerber S. A.; Turecek F.; Gelb M. H.; Aebersold R. Nat. Biotechnol. 1999, 17, 994–999. 10.1038/13690. [DOI] [PubMed] [Google Scholar]
- Ross P. L.; Huang Y. N.; Marchese J. N.; Williamson B.; Parker K.; Hattan S.; Khainovski N.; Pillai S.; Dey S.; Daniels S.; Purkayastha S.; Juhasz P.; Martin S.; Bartlet-Jones M.; He F.; Jacobson A.; Pappin D. J. Mol. Cell. Proteomics 2004, 3, 1154–1169. 10.1074/mcp.M400129-MCP200. [DOI] [PubMed] [Google Scholar]
- Thompson A.; Schäfer J.; Kuhn K.; Kienle S.; Schwarz J.; Schmidt G.; Neumann T.; Hamon C. Anal. Chem. 2003, 75, 1895–1904. 10.1021/ac0262560. [DOI] [PubMed] [Google Scholar]
- Colangelo C. M.; Williams K. R. Methods in molecular biology (Clifton, N.J.) 2006, 328, 151–158. 10.1385/1-59745-026-X:151. [DOI] [PubMed] [Google Scholar]
- Yi E. C.; Li X. J.; Cooke K.; Lee H.; Raught B.; Page A.; Aneliunas V.; Hieter P.; Goodlett D. R.; Aebersold R. Proteomics 2005, 5, 380–387. 10.1002/pmic.200400970. [DOI] [PubMed] [Google Scholar]
- Jiang H.; English A. M. J. Proteome Res. 2002, 1, 345–350. 10.1021/pr025523f. [DOI] [PubMed] [Google Scholar]
- Ong S. E.; Blagoev B.; Kratchmarova I.; Kristensen D. B.; Steen H.; Pandey A.; Mann M. Mol. Cell. Proteomics 2002, 1, 376–386. 10.1074/mcp.M200025-MCP200. [DOI] [PubMed] [Google Scholar]
- Zhu H.; Pan S.; Gu S.; Bradbury E. M.; Chen X. Rapid Commun. Mass Spectrom. 2002, 16, 2115–2123. 10.1002/rcm.831. [DOI] [PubMed] [Google Scholar]
- Ow S. Y.; Cardona T.; Taton A.; Magnuson A.; Lindblad P.; Stensjö K.; Wright P. C. J. Proteome Res. 2008, 7, 1615–1628. 10.1021/pr700604v. [DOI] [PubMed] [Google Scholar]
- Dayon L.; Hainard A.; Licker V.; Turck N.; Kuhn K.; Hochstrasser D. F.; Burkhard P. R.; Sanchez J. C. Anal. Chem. 2008, 80, 2921–2931. 10.1021/ac702422x. [DOI] [PubMed] [Google Scholar]
- Koehler C. J.; Strozynski M.; Kozielski F.; Treumann A.; Thiede B. J. Proteome Res. 2009, 8, 4333–4341. 10.1021/pr900425n. [DOI] [PubMed] [Google Scholar]
- Koehler C. J.; Arntzen M. O.; Strozynski M.; Treumann A.; Thiede B. Anal. Chem. 2011, 83, 4775–4781. 10.1021/ac200229w. [DOI] [PubMed] [Google Scholar]
- Nie A. Y.; Zhang L.; Yan G. Q.; Yao J.; Zhang Y.; Lu H. J.; Yang P. Y.; He F. C. Anal. Chem. 2011, 83, 6026–6033. 10.1021/ac201035f. [DOI] [PubMed] [Google Scholar]
- Koehler C. J.; Arntzen M. O.; de Souza G. A.; Thiede B. Anal. Chem. 2013, 85, 2478–2485. 10.1021/ac3035508. [DOI] [PubMed] [Google Scholar]
- Zhang S.; Chen L. F.; Shan Y. C.; Sui Z. G.; Wu Q.; Zhang L. H.; Liang Z.; Zhang Y. K. Analyst 2016, 141, 4912–4918. 10.1039/C6AN00388E. [DOI] [PubMed] [Google Scholar]
- Jiang H. C.; Yin H. R.; Xie L. Q.; Zhang Y.; Zhang L.; Yang P. Y.; Lu H. J. Anal. Chim. Acta 2018, 1001, 70–77. 10.1016/j.aca.2017.11.004. [DOI] [PubMed] [Google Scholar]
- Koehler C. J.; Arntzen M. O.; Thiede B. Rapid Commun. Mass Spectrom. 2015, 29, 830–836. 10.1002/rcm.7170. [DOI] [PubMed] [Google Scholar]
- Zhang R.; Sioma C. S.; Thompson R. A.; Xiong L.; Regnier F. E. Anal. Chem. 2002, 74, 3662–3669. 10.1021/ac025614w. [DOI] [PubMed] [Google Scholar]
- Di Palma S.; Raijmakers R.; Heck A. J. R.; Mohammed S. Anal. Chem. 2011, 83, 8352–8356. 10.1021/ac2018074. [DOI] [PubMed] [Google Scholar]
- Boutilier J. M.; Warden H.; Doucette A. A.; Wentzell P. D. J. Chromatogr. B: Anal. Technol. Biomed. Life Sci. 2012, 908, 59–66. 10.1016/j.jchromb.2012.09.035. [DOI] [PubMed] [Google Scholar]
- Mertins P.; Udeshi N. D.; Clauser K. R.; Mani D.; Patel J.; Ong S. e.; Jaffe J. D.; Carr S. A. Mol. Cell. Proteomics 2012, 11, M111.014423. 10.1074/mcp.M111.014423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ting L.; Rad R.; Gygi S. P.; Haas W. Nat. Methods 2011, 8, 937–940. 10.1038/nmeth.1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuhr M.; Haas W.; McAlister G. C.; Peshkin L.; Rad R.; Kirschner M. W.; Gygi S. P. Anal. Chem. 2012, 84, 9214–9221. 10.1021/ac301962s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnett M.; Yeung E.; Wuhr M. Anal. Chem. 2018, 90, 5032–5039. 10.1021/acs.analchem.7b04713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bantscheff M.; Boesche M.; Eberhard D.; Matthieson T.; Sweetman G.; Kuster B. Mol. Cell. Proteomics 2008, 7, 1702–1713. 10.1074/mcp.M800029-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenger C. D.; Lee M. V.; Hebert A. S.; McAlister G. C.; Phanstiel D. H.; Westphall M. S.; Coon J. J. Nat. Methods 2011, 8, 933–935. 10.1038/nmeth.1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin H.; Wang F.; Zhang Y.; Hu Z.; Song C.; Wu R.; Ye M.; Zou H. Chem. Commun. 2012, 48, 6265–6267. 10.1039/c2cc31705b. [DOI] [PubMed] [Google Scholar]
- Yang S. J.; Nie A. Y.; Zhang L.; Yan G. Q.; Yao J.; Xie L. Q.; Lu H. J.; Yang P. Y. J. Proteomics 2012, 75, 5797–5806. 10.1016/j.jprot.2012.07.011. [DOI] [PubMed] [Google Scholar]
- Xie L. Q.; Nie A. Y.; Yang S. J.; Zhao C.; Zhang L.; Yang P. Y.; Lu H. J. Analyst 2014, 139, 4497–4504. 10.1039/C4AN00728J. [DOI] [PubMed] [Google Scholar]
- Cao T.; Zhang L.; Zhang Y.; Yan G. Q.; Fang C. Y.; Bao H. M.; Lu H. J. Anal. Chem. 2017, 89, 11468–11475. 10.1021/acs.analchem.7b02654. [DOI] [PubMed] [Google Scholar]
- Waldbauer J.; Zhang L.; Rizzo A.; Muratore D. Anal. Chem. 2017, 89, 11498–11504. 10.1021/acs.analchem.7b02752. [DOI] [PubMed] [Google Scholar]
- Zhou Y.; Shan Y. C.; Wu Q.; Zhang S.; Zhang L. H.; Zhang Y. K. Anal. Chem. 2013, 85, 10658–10663. 10.1021/ac402834w. [DOI] [PubMed] [Google Scholar]
- Zhang S.; Wu Q.; Shan Y. C.; Zhou Y.; Zhang L. H.; Zhang Y. K. J. Proteomics 2015, 114, 152–160. 10.1016/j.jprot.2014.11.014. [DOI] [PubMed] [Google Scholar]
- Di Y.; Zhang Y.; Zhang L.; Tao T.; Lu H. J. Anal. Chem. 2017, 89, 10248–10255. 10.1021/acs.analchem.7b01635. [DOI] [PubMed] [Google Scholar]
- Liu J. H.; Zhou Y.; Shan Y. C.; Zhao B. F.; Hu Y. C.; Sui Z. G.; Liang Z.; Zhang L. H.; Zhang Y. K. Anal. Chem. 2019, 91, 3921–3928. 10.1021/acs.analchem.8b04806. [DOI] [PubMed] [Google Scholar]
- Di Y.; Zhang Y.; Zhang L.; Tao T.; Lu H. Anal. Chem. 2017, 89, 10248–10255. 10.1021/acs.analchem.7b01635. [DOI] [PubMed] [Google Scholar]
- Thompson A.; Wölmer N.; Koncarevic S.; Selzer S.; Böhm G.; Legner H.; Schmid P.; Kienle S.; Penning P.; Höhle C.; Berfelde A.; Martinez-Pinna R.; Farztdinov V.; Jung S.; Kuhn K.; Pike I. Anal. Chem. 2019, 91, 15941–15950. 10.1021/acs.analchem.9b04474. [DOI] [PubMed] [Google Scholar]
- Abello N.; Kerstjens H. A. M.; Postma D. S.; Bischoff R. J. Proteome Res. 2007, 6, 4770–4776. 10.1021/pr070154e. [DOI] [PubMed] [Google Scholar]
- Rappsilber J.; Ishihama Y.; Mann M. Anal. Chem. 2003, 75, 663–670. 10.1021/ac026117i. [DOI] [PubMed] [Google Scholar]
- Tian S. S.; Zheng S. Z.; Han Y. P.; Guo Z. C.; Zhai G. J.; Bai X.; He X.; Fan E. G.; Zhang Y. K.; Zhang K. Anal. Chem. 2017, 89, 8259–8265. 10.1021/acs.analchem.7b01120. [DOI] [PubMed] [Google Scholar]
- Chan J. W.; Hoyle C. E.; Lowe A. B.; Bowman M. Macromolecules 2010, 43, 6381–6388. 10.1021/ma101069c. [DOI] [Google Scholar]
- Kumar A.; Akanksha Tetrahedron 2007, 63, 11086–11092. 10.1016/j.tet.2007.08.033. [DOI] [Google Scholar]
- Hussain S.; Bharadwaj S. K.; Chaudhuri M. K.; Kalita H. Eur. J. Org. Chem. 2007, 2007, 374–378. 10.1002/ejoc.200600691. [DOI] [Google Scholar]
- Virreira Winter S.; Meier F.; Wichmann C.; Cox J.; Mann M.; Meissner F. Nat. Methods 2018, 15, 527–530. 10.1038/s41592-018-0037-8. [DOI] [PubMed] [Google Scholar]
- Xie L.-Q.; Zhang L.; Nie A.-Y.; Yan G.-Q.; Yao J.; Zhang Y.; Yang P.-Y.; Lu H.-J. Proteomics 2015, 15, 3755–3764. 10.1002/pmic.201400513. [DOI] [PubMed] [Google Scholar]
- Arntzen M. Ø.; Koehler C. J.; Barsnes H.; Berven F. S.; Treumann A.; Thiede B. J. Proteome Res. 2011, 10, 913–920. 10.1021/pr1009977. [DOI] [PubMed] [Google Scholar]
- Werner T.; Sweetman G.; Savitski M. F.; Mathieson T.; Bantscheff M.; Savitski M. M. Anal. Chem. 2014, 86, 3594–3601. 10.1021/ac500140s. [DOI] [PubMed] [Google Scholar]
- Perez-Riverol Y.; Csordas A.; Bai J.; Bernal-Llinares M.; Hewapathirana S.; Kundu D. J.; Inuganti A.; Griss J.; Mayer G.; Eisenacher M.; Pérez E.; Uszkoreit J.; Pfeuffer J.; Sachsenberg T.; Yılmaz Ş.; Tiwary S.; Cox J.; Audain E.; Walzer M.; Jarnuczak A. F.; et al. Nucleic Acids Res. 2019, 47, D442–D450. 10.1093/nar/gky1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.