

Abstract
Bacterial secretory preproteins are translocated across the inner membrane post‐translationally by the SecYEG‐SecA translocase. Mature domain features and signal peptides maintain preproteins in kinetically trapped, largely soluble, folding intermediates. Some aggregation‐prone preproteins require chaperones, like trigger factor (TF) and SecB, for solubility and/or targeting. TF antagonizes the contribution of SecB to secretion by an unknown molecular mechanism. We reconstituted this interaction in vitro and studied targeting and secretion of the model preprotein pro‐OmpA. TF and SecB display distinct, unsuspected roles in secretion. Tightly associating TF:pro‐OmpA targets the translocase at SecA, but TF prevents pro‐OmpA secretion. In solution, SecB binds TF:pro‐OmpA with high affinity. At the membrane, when bound to the SecA C‐tail, SecB increases TF and TF:pro‐OmpA affinities for the translocase and allows pro‐OmpA to resume translocation. Our data reveal that TF, a main cytoplasmic folding pathway chaperone, is also a bona fide post‐translational secretory chaperone that directly interacts with both SecB and the translocase to mediate regulated protein secretion. Thus, TF links the cytoplasmic folding and secretion chaperone networks.
Keywords: outer membrane protein A, protein targeting, SecB, Sec system, trigger factor
Subject Categories: Membrane & Intracellular Transport, Protein Biosynthesis & Quality Control
The cytoplasmic chaperone Trigger Factor (TF) interacts with SecB and is targeted with the secretory preprotein proOmpA to the translocase.
Introduction
In post‐translational, Sec‐dependent secretion ribosome‐released nascent polypeptide chains delay their folding for the duration of their cytoplasmic transit 1 and resume it in distinct cellular locations on the trans side of the membrane 2. A total of 505 different proteins, carrying signal peptides (SPs) fused N‐terminally to their mature domains, follow this process in Escherichia coli (E. coli) K‐12 1, 3, 4, 5, 6. Secretion occurs through an inner membrane channel (~2.5 nm) formed by the SecYEG proteins 7. Translocation starts once SecA, an ATPase motor that shuttles between cytoplasm and membrane, peripherally associates with SecYEG 8, and the preprotein binds bivalently onto SecA, via both its SP and its mature domain 1, 2, 9, 10.
Secretory proteins constitute a novel protein class with enhanced disorder, reduced hydrophobicity and aggregation propensity 6, 11, 12, 13. Their unusual structural features directly impact the membrane targeting process. Most of them form slow‐folding intermediates that can remain soluble for long, in vitro 11. This may render them largely independent of chaperones or SPs, both previously thought essential for maintaining them unfolded thus allowing targeting and translocation competence 14, 15, 16, 17, 18, 19, even in vivo 9, 11. Slow folding, with or without chaperones, exposes mature domain targeting signals (MTSs) 9 that mediate targeting to the translocase with nanomolar affinity and allow export 9, 20, 21.
Chaperones are often abundant in cells, some of them particularly under stress 17 and can occupy strategic locations, e.g. bound to the ribosome or in the periplasm. A balanced cooperation of chaperone networks is essential for cell viability 22. Two chaperones proposed to have specific roles in protein secretion in E. coli K‐12 are as follows: (i) the tetrameric SecB. Found only in some proteobacteria 23, it is proposed to be the main secretory pathway chaperone since it binds unfolded preproteins and interacts with SecA 24, via complexing the C‐terminal tail of the latter with low micromolar affinity 25. Yet, SecB clients in E. coli may be ~20 1. (ii) Trigger factor (TF), a ubiquitous cytoplasmic dimer and ribosome‐bound monomer, commonly facilitates folding of cytoplasmic folder clients or hands them over to downstream foldases such as DnaK and GroEL 26, 27. TF has been circumstantially implicated in the secretory pathway due to its interaction with ~20% of nascent secretory polypeptides at the ribosomal exit tunnel 6. In vitro, TF maintains the translocation competence of secretory polypeptides like pro‐OmpA 28, 29 and solubilizes 19 aggregation‐prone secretory proteins 6, 30.
Due to its high intracellular concentration {~40 μM TF; (Appendix Table S1 at 1}, TF is commonly bound to ribosomes (concentration ~20 μM 1, 31) because of its ~1 μM K d for them 27, 32. It patrols the ribosome exit tunnel 33, 34 and contributes, together with other ribosome‐bound factors, to the folding and sorting of cytoplasmic proteins 27 and the sorting of co‐ and post‐translationally targeted secretory proteins 32, 35, 36, 37 through an interplay with the signal recognition particle 32, 36 and with ribosome‐bound SecA 38. TF protects hydrophobic regions of secretory nascent chains 28, 33, 39 using four hydrophobic patches per monomer 16. However, TF is not essential for in vivo secretion 40 and its precise role in preprotein sorting, targeting and solubility remains elusive.
SecB, a dimer of dimers, interacts with secretory chains 41 via a long hydrophobic groove along a surface formed by all its protomers and undergoes slight conformational changes upon binding to secretory preproteins 42. SecB prevents or delays preprotein folding and may relay preproteins to cytoplasmic or SecYEG‐bound SecA 43, 44. Initiation of ATP hydrolysis by SecA causes SecB release 1, 43.
K‐12 cells devoid of SecB 22, 45, TF 22 or both 22 are viable although SecB or TF deletion causes some increase in intracellular protein aggregation 46, compromised cell division 40 and transcriptome alterations 47. This cellular adaptation to their absence has been attributed to the built‐in redundancy of chaperone networks 48. In the absence of TF and SecB, more ribosomes become membrane‐associated and translocation may become more co‐translational 22, 49. At low temperatures, deletion of secB causes lethality; this phenotype is restored if additionally, the TF‐encoding tig gene is deleted 22. Apparently, high intracellular TF concentration is not tolerated for certain clients depending on their affinities; relief by SecB suggests that some of them might be secretory. TF has been previously shown to slow down protein secretion 50. These observations suggested that TF and SecB may somehow cooperate in post‐translational protein export 22, 50.
Non‐ATPase chaperones like TF and SecB are thought to act primarily as holdases 42, 51, 52, 53, 54; i.e., they would bind and remain bound onto a secretory chain with a low k off until the latter is delivered to the translocase 16, 42. Both TF and SecB can also bind preproteins 16, 42 that do not need chaperones for solubility, targeting and secretion 9, 11.
We investigated the independent roles and functions of TF and SecB and their potential cooperation in preprotein targeting and translocation. We studied the type and affinity of the interactions between TF, SecB and the model secretory protein pro‐OmpA (outer membrane protein A). The conformation of non‐folded pro‐OmpA was previously analysed using biophysical tools including hydrogen–deuterium exchange‐mass spectrometry (HDX‐MS) 11. It contains substantial secondary but no tertiary structure and is soluble under reducing conditions 11, 55. The OmpA mature domain will only fold after insertion in the outer membrane 56.
Our data reveal that both TF and SecB interact with pro‐OmpA but differently. TF acting as a tight and SecB as a weak pro‐OmpA holdase can sequester it away, thereby avoiding the formation of off‐pathway, translocation‐incompetent states. Tightly associating TF:pro‐OmpA complexes do arrive at the Sec translocase, but pro‐OmpA is not released for secretion. Three new SecB roles were revealed as follows: (i) it complexes TF:pro‐OmpA in solution; (ii) it increases the individual TF and TF:pro‐OmpA affinities for the translocase, and (iii) it acts as a release factor at the translocase for TF‐bound pro‐OmpA by binding to the C‐tail of SecA. These multi‐level roles may explain why balanced TF:SecB ratios are essential for viability. These previously unknown TF interactions with SecB and the translocase place TF on the post‐translational secretory pathway ushering a subset of secretory proteins.
Results
An optimal SecB/TF ratio is essential in vivo
The ratio of SecB to TF is maintained optimal in vivo 22, 50. This requirement is so important that even in wild‐type MC4100 cells, elevated synthesis of TF in trans in the presence of the transcriptional inducer of the tetracycline‐activated promoter anhydrotetracycline (AHT) leads to either lethality (Fig 1A, 16°C; compare row 2 to row 1) or to a severe growth defect (30°C). Correspondingly, strain MC4100ΔsecB that produces TF in the absence of SecB displays lethality at the non‐permissive temperature of 16°C (Fig 1A, row 3), but this is corrected by either expressing secB in trans from a plasmid (row 4) or by additionally deleting the tig gene that encodes TF (row 5) 22. If in this otherwise completely viable strain MC4100ΔtigΔsecB, tig is expressed in trans from a plasmid, lethality is re‐capitulated in the presence of AHT but also, to a lesser extent, in its absence (row 6). The effect is so severe that in trans, TF synthesis becomes lethal even at the otherwise permissive temperature of 30°C (row 6, right).
Figure 1. An optimal TF/SecB ratio is essential for cell viability and in vitro translocation of pro‐OmpA.
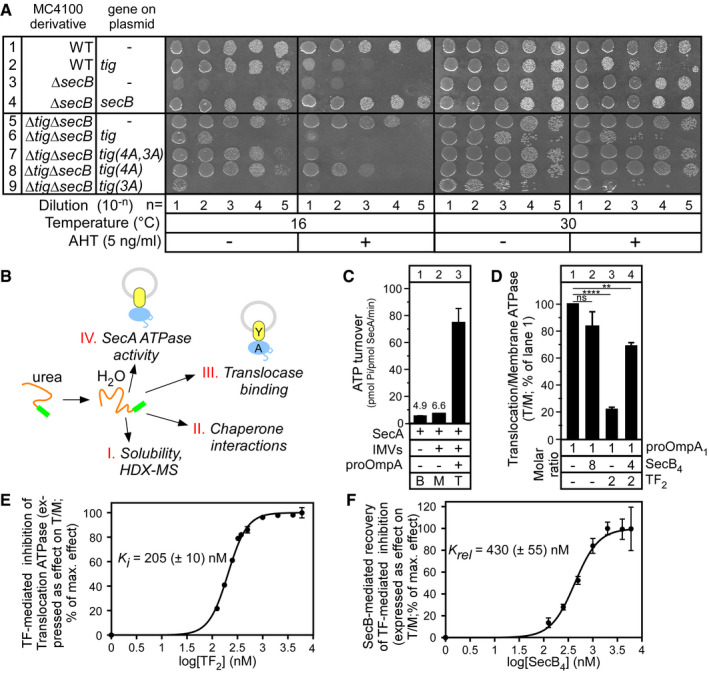
- In vivo genetic complementation of the E. coli MC4100 wild‐type (WT) and secB or/and tig knock‐out derivatives by either an empty vector or one carrying the secB or tig (encoding TF) genes or derivatives (see “Plasmid” table in Appendix), as indicated. Serial dilutions of a culture (OD600 = 0.5) were spotted (12 μl) on LB‐Ampicillin plates containing or not anhydrotetracycline (AHT; 5 ng/ml) and grown at 16°C or 30°C (as indicated). n = 3–7 biological replicates.
- Schematic summary of the assays used in this study. Preproteins (purified commonly in 6 M urea; except in Appendix Fig S6B) were diluted in an aqueous buffer (final urea < 0.2 M) and their physicochemical properties and functional interactions with Sec system components analysed. Y: SecYEG; A: SecA.
- SecA ATPase activity determination in solution (basal; B), or plus SecYEG IMVs; (membrane; M) or plus pro‐OmpA (pre‐treated with 10 mM DTT; 5 mM EDTA; 20 min; 4°C; translocation; T). n = 21 biological replicates. Mean values (± SEM) are shown.
- pro‐OmpA translocation ATPase activity stimulation of SecA (as in C) in the absence or presence of TF2 or/and SecB4, added at the indicated molar excess over pro‐OmpA1. The T/M ratios were calculated; the one in the absence of chaperones was considered as 100%, and all other values were expressed as % of this. n = 2–4 biological replicates. Mean values (± SEM) are shown. Unpaired parametric t‐test, 95% confidence interval: ns: not significant (P = 0.1032); ****P < 0.0001; **P = 0.0037.
- Titrated TF‐mediated inhibition (0–6 μM range) of pro‐OmpA‐stimulated translocation ATPase activity (as in C). Values were normalized (effectmax = 100%; effectmin = 0%) and plotted (n = 1–4 biological replicates; mean values ± SEM) versus the log10[TF2]. The K i was determined using a variable slope fit (log[inhibitor] versus normalized response; GraphPad Prism).
- SecB‐mediated relief of TF‐mediated inhibition of pro‐OmpA translocation ATPase activity (as in C). TF2 (1 μM) and SecB4 (0–6 μM) were used. The T/M ratios were calculated, normalized (as in D) and plotted (n = 2–3 biological replicates; mean values ± SEM) versus the log10[SecB4]. An apparent relief constant (K rel) was determined (as in E).
Source data are available online for this figure.
To identify TF regions important for inhibition, we used the composite derivative tig (4A,3A) in which two main functional regions of TF are compromised by alanyl mutagenesis: its client binding site region 2 (4A: M374A, Y378A, V384A and F387A) 51 and its ribosome binding domain (3A: F44A, R45A and K46A) 57 and tig(4A,3A) no longer inhibit viability (Fig 1A, lane 7). Using the individually mutated derivatives 4A and 3A that are compromised for client 51 and ribosome 57 binding affinities, respectively, we determined that TF‐mediated growth inhibition requires primarily optimal client (lane 8; tig(4A)) and much less so ribosome (lane 9; tig(3A)) association. All TF derivatives remained stable and detectable in similar amounts (Fig EV1A).
Figure EV1. TF intracellular levels, pro‐OmpA‐induced translocation ATPase assay and SecB affinity of SecA‐YEG IMVs (related to Figs 1, 2 and 4).
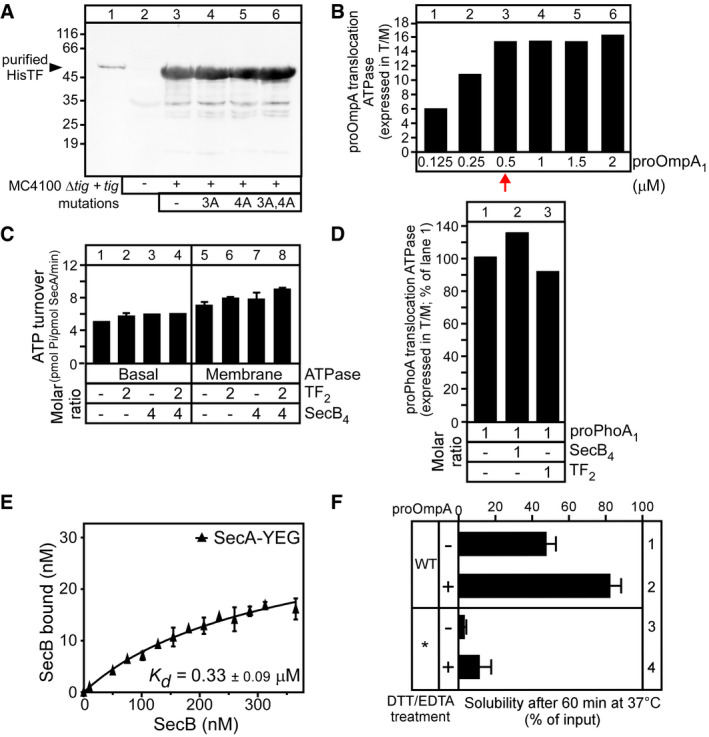
- Intracellular levels of TF or derivatives in MC4100ΔsecBΔtigcells carrying pASK IBA 7 plus vector with tig or derivatives. Cells were grown at 37°C, and gene expression was induced by addition of AHT (OD600 = 0.3; 37°C; 5 ng/ml AHT; 3 h). Equal number of cells (normalized by OD600) was analysed on 12% SDS–PAGE and immune‐stained using α‐TF. Purified His‐TF (20 ng; lane 1) served as a molecular weight/protein amount marker. n = 3; a representative experiment is shown.
- Titration of pro‐OmpA to define a linear range for ATPase stimulation. pro‐OmpA was titrated (0–2 μM; direct dilution from 6 M to 0.12 M urea) into reactions (50 μl; Buffer E) containing SecA (0.4 μM) and SecYEG‐IMVs (0.4 μM SecY). The translocation ATPase was measured, and the ratio of translocation over membrane (T/M) ATPase is shown. n = 1. Unless otherwise indicated, 0.5 μM pro‐OmpA was used for all ATPase experiments (red arrow).
- SecA basal or membrane ATPase activity is not affected neither by SecB nor by TF. The basal (0.4 μM SecA) and membrane (plus SecYEG‐IMVs; 0.4 μM SecY) ATPase was measured (50 μl; Buffer E) without or with TF2, SecB4 or both (as indicated; added at the molar excess over pro‐OmpA1 that was used in Fig 1D). n = 1–2 biological replicates; mean values ± SEM are shown.
- TF does not affect the translocation of pro‐PhoA. pro‐PhoA1 (3 μM) was added to reactions [50 μl; Buffer E; SecA (0.4 μM); SecYEG‐IMVs (0.4 μMSecY)] that contained, or not (as indicated), TF2 or SecB4 (at the indicated molar excess over pro‐PhoA); as in Panel (B). n = 1.
- Equilibrium dissociation constant (Kd) of SecB for SecA bound to SecYEG‐IMVs. A concentration range of SecB (0–400 nM) was mixed with SecA (0.4 μM) bound on SecYEG (0.8 μM in 50 μl reactions). Data, analysed by non‐linear regression, represent average values; error bars: standard mean error (SEM) is shown; n = 6 biological replicates.
- The aggregation propensity of pro‐OmpA* under different conditions. pro‐OmpA (wild‐type; WT) and pro‐OmpA* (mutant) pre‐treated (10 mM DTT; 5 mM EDTA; 20 min; 4°C) or not were diluted out of chaotrope (Buffer C; < 0.2 M urea) and incubated (60 min; 37°C). Aggregates were removed by centrifugation (20,000 g; 10 min; 4°C), and the amount of protein remaining soluble was determined via spectroscopic measurements (280 nm; NanoDrop 2000; Thermo). n = 6 biological replicates; mean values ± SEM are shown.
Source data are available online for this figure.
Based on the above, we hypothesized that excess of TF might sequester away some critical secretory proteins, downstream of its ribosome‐associated state, and thus, becomes deleterious for the secretory pathway. We presumed that SecB might antagonise, directly or indirectly, this tight TF association with secretory clients. To probe this hypothesis, we reconstituted the reaction in vitro.
SecB relieves TF‐inhibited translocation of pro‐OmpA in vitro
To investigate the deleterious effect of TF, we used an in vitro preprotein translocation assay. pro‐OmpA, retained non‐folded in chaotrope, was diluted into an aqueous buffer in the absence (Fig 1B, I and II) or presence (III and IV) of Sec pathway components. We previously established that under these assay conditions, preproteins commonly retain their non‐folded, soluble structures for long 11. Hence, we were able to study under one regime their structural dynamics, using hydrogen–deuterium exchange‐mass spectrometry (HDX‐MS) (Fig 1B and I) 10, 11, as well as their functional interaction with chaperones like TF and SecB (Fig 1B, II), or/and with the SecYEG‐SecA translocase (Fig 1B, III). Addition of the latter, in inverted inner membrane vesicles (IMVs) that had been urea‐stripped of any peripheral components, allowed us to study the effect of soluble factors added in trans on either docking onto the translocase (Fig 1B, III) or on the ability of SecA to hydrolyse ATP (Fig 1B, IV) 10, 58.
We first monitored ATP turnover by SecA (Fig 1B, IV) as it catalyses protein translocation (Fig 1C) 58. The low‐level basal ATPase activity of SecA[basal (B) ATPase, lane 1] is stimulated ~1.4‐fold by the addition of SecYEG‐containing IMVs (membrane (M) ATPase, lane 2) and a further ~11‐fold when pro‐OmpA is diluted from 6 M to 0.12 M urea into the reaction (translocation (T) ATPase, lane 3). Equimolar SecYEG‐SecA ratios were used (0.4 μM), and following titration experiments (Fig EV1B, lane 3), pro‐OmpA was added at 0.5 μM, the maximal concentration that still yields a linear range of ATPase stimulation.
Similar experiments were performed in the presence of TF and SecB, added alone or together. Since TF or SecB has no effect on the basal or membrane ATPase of SecA (Fig EV1C), we focused on the translocation ATPase. To compare across the different conditions, the ratio of translocation to membrane ATPase (T/M) in the absence of TF or SecB (Fig 1C, lanes 2–3) was considered as 100% and all other ratios were expressed as a percent of this value (Fig 1D, lanes 1–4). Addition of SecB alone had no adverse effect on translocation ATPase activity (lane 2). In contrast, the presence of TF led to extensive inhibition of pro‐OmpA‐stimulated translocation ATPase activity (lane3) that was significantly relieved when SecB was also present (lane 4). The TF‐mediated repression of translocation ATPase was specific to pro‐OmpA, a known preferred TF client 28, 39, 51, and was not observed with other preproteins to which TF also binds in vitro (e.g. pro‐PhoA; Fig EV1D).
We titrated the effect of TF, using 0–6 μM TF dimer (TF2, the native quaternary state), on the translocation ATPase of SecA, yielding a K i of 205 (± 10) nM (Fig 1E), suggesting high‐affinity binding of TF for pro‐OmpA.
Next, we titrated the effect of SecB, using 0–6 μM SecB tetramer (SecB4, the native quaternary state), on the inhibition of the translocation ATPase caused by 1 μM of TF. The effect of SecB on the T/M ratio was determined, normalized (effectmax = 100%) and plotted against the log‐transformed SecB4 concentration. Using the same fit as before, we determined a relief constant (K rel) of 425 (± 10) nM (Fig 1F) that suggested high‐affinity binding of SecB4 with either TF:pro‐OmpA or the SecYEG‐SecA translocase (see below). SecB4 binds to the latter with an affinity of 200–330 nM (Fig EV1E) 44.
Presumably, TF binding either prevents pro‐OmpA from being targeted to the translocase or, post‐targeting, it prevents pro‐OmpA from being translocated. SecB might act at either one of these steps.
Tight binding of TF to pro‐OmpA
We further probed TF complex formation with pro‐OmpA, first using native‐PAGE (Fig 2A). Unlike TF (Fig 2A, lane 2; apparent mass ~100 kDa), non‐folded pro‐OmpA is not resolved by native‐PAGE as a sharp band (lane 9). However, addition of increasing molar excess of TF2 to pro‐OmpA resulted in increasing formation of a species migrating slower than TF, indicative of complex formation (lanes 3–8).
Figure 2. TF is a tight holdase for translocation‐competent pro‐OmpA.
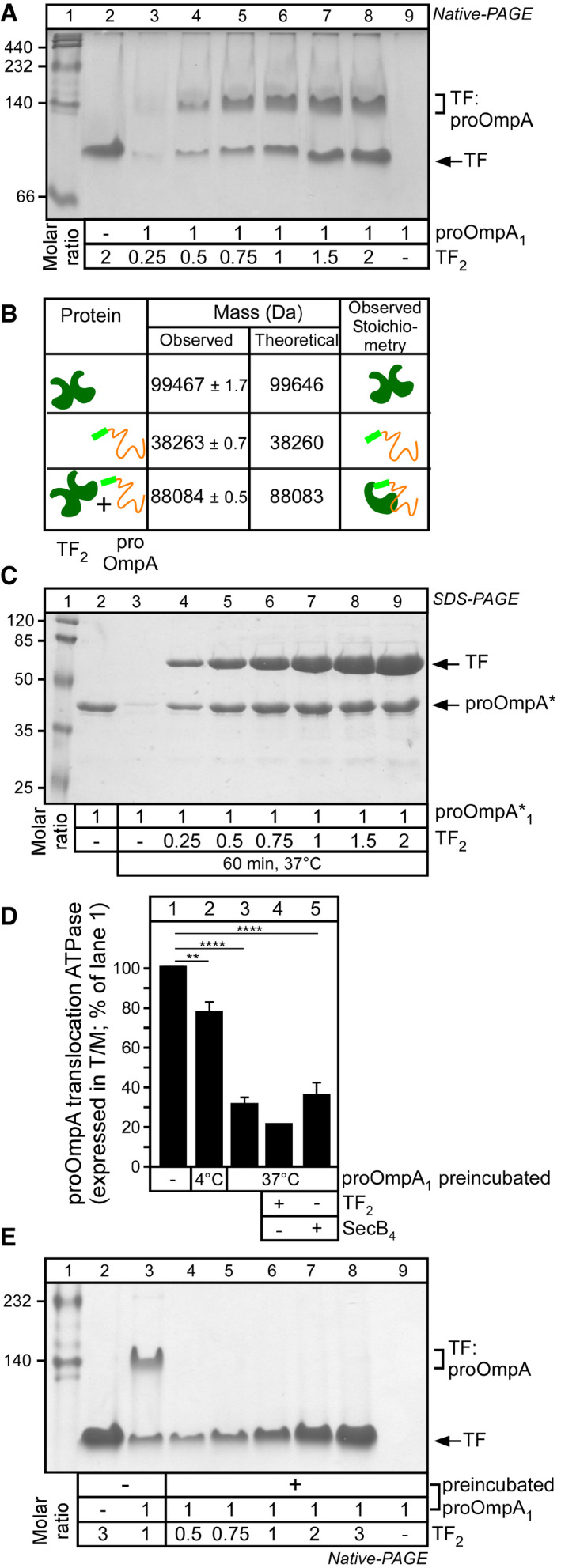
- TF–pro‐OmpA physical interaction in solution. pro‐OmpA1 (5 μM) was incubated (50 μl; Buffer C; < 0.2 M urea; 37°C; 60 min) with the indicated molar excess of TF2. Soluble proteins were analysed on 10% native‐PAGE (4 mA; 16 h; 4°C) and Coomassie Blue stained. A representative experiment is shown; n = 4 biological replicates.
- TF–pro‐OmpA intact native‐MS analysis. Mass spectra of TF2 (20 μM; Buffer G), pro‐OmpA (20 μM; pre‐treated with 10 mM DTT; Buffer G) or their mixture were acquired in near‐native conditions using a Synapt G2 mass spectrometer and analysed using MassLynX 4.1, and masses were deconvoluted using MaxEnt 1 and shown together with the theoretical masses and deduced stoichiometries. n = 2 biological replicates.
- TF solubilizes pro‐OmpA*. pro‐OmpA*1(5 μM) was incubated (50 μl; Buffer C; < 0.2 M urea;37°C; 60 min) alone or in the presence of TF. Proteins in the soluble fraction were analysed on 15% SDS–PAGE and Coomassie Blue stained. Lane 2: 2.84 μg pro‐OmpA*. Representative experiment is shown; n = 5 biological replicates.
- pro‐OmpA pre‐incubated at 37°C fails to stimulate SecA ATPase activity. pro‐OmpA (0.5 μM) was either diluted directly (lane 1) or after preincubation alone (10 min; Buffer D; < 0.2 M urea; either at 4°C or 37°C) into reactions containing SecYEG‐SecA alone or together with TF2 or SecB4(2 and 4× molar excess over pro‐OmpA, respectively). T/M ratios (as in Fig 1D) are shown (n = 8 biological replicates; mean values ± SEM; unpaired parametric t‐test, 95% confidence interval: **P = 0.0017; ****P < 0.0001).
- pro‐OmpA pre‐incubated at 37°C does not interact with TF. pro‐OmpA1 (5 μM) was pre‐incubated (Buffer D; < 0.2 M urea; 37°C; 10 min) or not (lane 3), prior to addition of the indicated molar excess of TF2 (analysed as in A). Representative experiment is shown; n = 4 biological replicates.
Source data are available online for this figure.
TF self‐dimerizes with an affinity of ~1.5–2 μM 59 (Appendix Fig S1) yielding a mass determined by native‐mass spectrometry (native‐MS) of 99,467 ± 1.7 Da (Fig 2B). Binding to pro‐OmpA (mass of 38,263 ± 0.7 Da) yields a TF:pro‐OmpA complex of 88,084 ± 0.5 Da, consistent with a 1:1 stoichiometry (Fig 2B; Appendix Fig S2). 1:1 stoichiometry was also reported for TF:pro‐PhoA 16.
TF binding prevents pro‐OmpA from aggregating
Next, we probed the ability of TF to prevent pro‐OmpA aggregation (Fig 1B, II). For this specific assay, we resorted to using pro‐OmpA*, an aggregation‐prone derivative (Appendix Fig S1B) 11, incubated under conditions that maximally promote its aggregation (5 μM; 60 min at 37°C; without DTT/EDTA pre‐treatment; Fig EV1F) in the absence (Fig 2C, lane 3) or presence of TF (lanes 4–9). Insoluble material was removed by centrifugation before soluble supernatants were analysed by SDS–PAGE and native‐PAGE, Coomassie Blue staining and densitometry. While pro‐OmpA* alone aggregated extensively (Fig 2C, compare lanes 2 and 3; Appendix Fig S3), its solubility increased in a TF concentration‐dependent manner (lanes 4–9).
We concluded that TF association can prevent pro‐OmpA aggregation by forming a stable complex corroborating previous data 28, 29.
TF does not associate with off‐pathway pro‐OmpA folding intermediates
pro‐OmpA that is diluted into aqueous buffer and incubated for 10 min at 4°C before being added to SecYEG‐SecA (Fig 2D, lane 2) is slightly less efficient in stimulating SecA translocation ATPase from the same protein that is diluted in the presence of SecYEG‐SecA (compare lane 2 to 1). Moreover, if pro‐OmpA is incubated for 10 min at 37°C prior to its addition to SecYEG‐SecA, a 70–80% loss of stimulation was detected (lane3). Aggregation, that could justify this loss of activity, was not observed (Appendix Fig S1C). We therefore presumed that acquisition of off‐pathway folding intermediates, similar to the ones we previously detected 11, might be responsible for reduced translocation ATPase stimulation.
To test this directly, we monitored pro‐OmpA folding kinetics (at 4°C and 37°C), by global HDX‐MS analysis 11, 60, and in the same course of time, we followed its ability to stimulate the translocation ATPase of SecA (Fig EV2A and B, respectively). At 4°C, within 1–10 min, pro‐OmpA acquires two intermediates that take up less deuterium (D) than the fully deuterated (FD) control, i.e. are more folded (I1 and I2; Fig EV2A, middle; compare to FD on top) and which remain on‐pathway for translocation for 60 min as judged by the T/M ratio (Fig EV2B, middle). In contrast, at 37°C pro‐OmpA rapidly shifts to a single intermediate, detectable for at least 1 h, that takes up less D than I1 yet more than I2 (Fig EV2A, bottom) and which rapidly loses the ability to stimulate translocation ATPase (Fig EV2B, bottom). Once the I3 intermediate was preformed (10 min; aqueous buffer; 37°C; Fig EV2A, bottom), mixing it with TF did not lead to complex formation detectable by native‐PAGE analysis (Fig 2E; compare to Fig 2A).
Figure EV2. Time kinetics of pro‐OmpA folding at different temperatures, monitored by global HDX‐MS analysis and translocation ATPase (related to Figs 2 and 3).
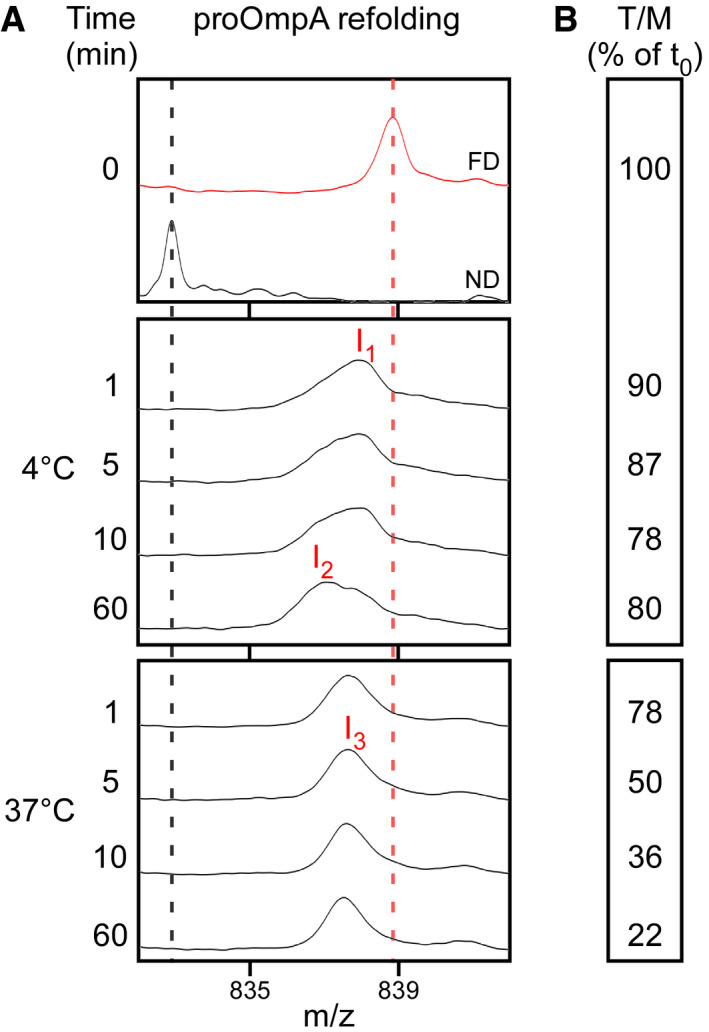
-
A, BPre‐treated pro‐OmpA was diluted from chaotrope into aqueous Buffer D and incubated at the indicated temperature. (A) At the indicated time points, samples were deuterated (100‐sec pulse; 4°C or 37°C) quenched on ice (formic acid; pD: 2.1) and analysed by global HDX‐MS. Samples were compared to the fully deuterated (FD) and non‐deuterated (ND) controls. The (+44) charge state is shown. n = 3. m/z: mass/charge ratio. (B) At the indicated time points, pro‐OmpA samples [0.5 μM; for time points 10 and 60 min, a centrifugation step (20,000 g; 10 min; 4°C) has been added] were added to translocation ATPase assays. ATPase activity was measured (as in Fig 1C) and transformed to T/M ratio as in Fig 1D. n = 2. T/M: ratio of translocation and membrane ATPase.
Source data are available online for this figure.
We concluded that pro‐OmpA can acquire mis‐folded intermediates that take it off‐pathway for translocation, and these are no longer recognizable by TF.
TF:pro‐OmpA complexes, but not TF, bind to the SecYEG‐SecA translocase
We next sought to determine which stage of the pro‐OmpA translocation reaction TF inhibits. To define whether inhibition lies prior to or post‐translocase targeting, we preformed TF:pro‐OmpA complexes in solution and quantified the subsequent delivery of pro‐OmpA onto the SecYEG‐SecA translocase. In this assay system, the binding of titrated concentrations of a [35S]‐labelled protein (as indicated) to SecA bound to SecYEG in IMVs is measured 9, 10, 61.
Both pro‐OmpA and OmpA bind to SecYEG‐bound SecA with high affinities when diluted from chaotrope straight into the SecYEG‐SecA reaction [0.47 (± 0.07) and 0.65 (± 0.13) μM, respectively; Fig 3A, lane 2, top and middle]. In contrast, the off‐pathway I3 intermediate has no affinity for the translocase (Fig 3A, lane 2, bottom), in agreement with its lack of stimulation of the translocation ATPase of SecA (Figs 2D, lane 3; and EV4B).
Figure 3. Equilibrium dissociation constants (K d s) of proteins and complexes for the Sec translocase.
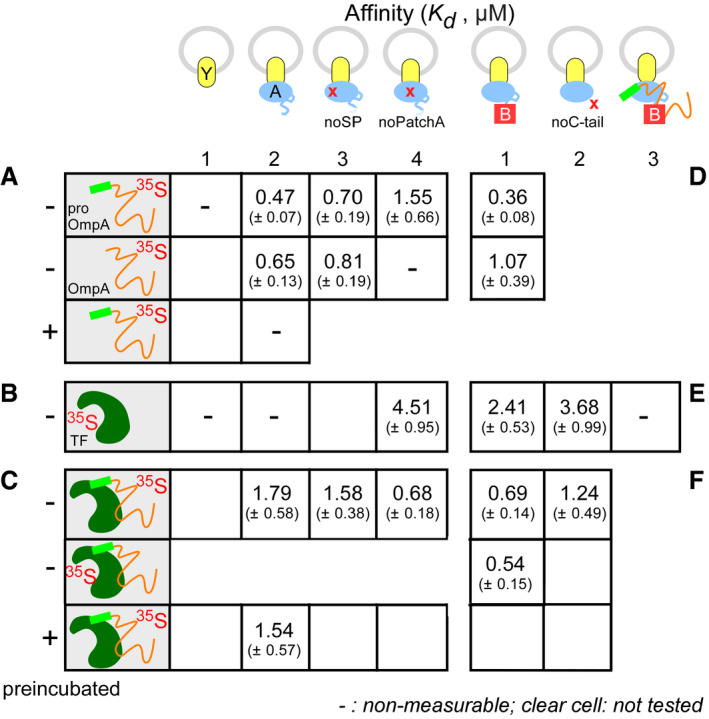
-
A–FEquilibrium dissociation constants (Kd) of protein ligands (as indicated) for SecYEG‐IMVs with or without SecA or its derivatives or SecB. Data analysed by non‐linear regression and average values with standard mean error (SEM) are shown; n = 6–9 biological replicates.
Source data are available online for this figure.
Figure EV4. The TF:pro‐OmpA:SecB super‐complex analysed by ITC and native‐PAGE (related to Fig 5).
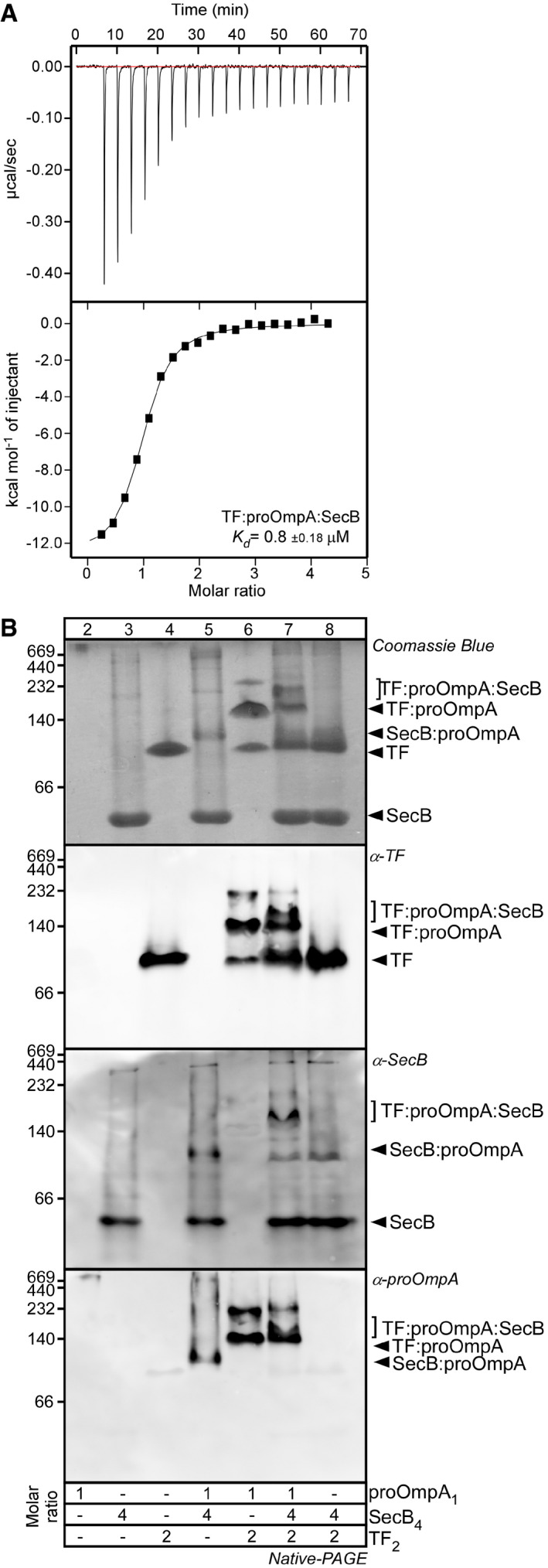
- Kd determination of TF:pro‐OmpA:SecB complex in solution by ITC. TF2:pro‐OmpA1 (15 μM; cell) was titrated with SecB4 (128 μM; syringe) in Buffer H at 25°C using an iTC200 (Malvern). The injection profile (raw data; top panel) and the calorimetric binding isotherm (bottom panel) are shown. A representative experiment is shown; n = 3.
- Clear native‐PAGE analysis of TF:pro‐OmpA:SecB complex. The indicated proteins and their complexes were analysed as in Fig 2A. Gels were stained with Coomassie Blue (top) or with the indicated polyclonal antisera. Representative experiments are shown; n = 3 biological repeats.
As seen for other preproteins 61, neither pro‐OmpA nor OmpA binds to SecYEG when SecA is absent (Fig 3A, lane 1). Like pro‐PhoA 9, 61, pro‐OmpA docks to SecYEG‐bound SecA bivalently. SecA(noSP), an alanyl substituted derivative that cannot bind SPs 62, still binds pro‐OmpA and OmpA with the K d of the mature domain (lane 3). Similarly, SecA(noPatchA), an alanyl substituted derivative of the mature domain binding site on SecA 9, cannot bind OmpA but still binds pro‐OmpA (lane 4).
Trigger factor alone displayed no measurable binding to SecYEG‐bound (Fig 3B, lane 2) or to soluble (Appendix Fig S4) SecA. In contrast, TF:pro‐OmpA bound to SecYEG‐SecA with a K d of 1.79(± 0.58) μM (Fig 3C). We obtained identical results with either TF:[35S]‐pro‐OmpA (row 1) or [35S]‐TF:pro‐OmpA (row 2), suggesting that TF remains complexed with pro‐OmpA. This binding remains unaffected by a 10‐min incubation of the TF:pro‐OmpA complex at 37°C prior to its addition to SecYEG‐SecA (Fig 3C). Thus, TF binding prevents formation of the translocation incompetent I3 intermediate (compare to Fig 3A, bottom; preincubation of pro‐OmpA alone). However, the affinities of pro‐OmpA and of TF:pro‐OmpA for the Sec translocase differ [K d = 0.47 (± 0.07) vs. 1.79 (± 0.58) μM, respectively; Fig 3A and C]. We presume TF binding biases a different pro‐OmpA “state” or shields binding sites, hence leading to a drop in the affinity for the SecA receptor. Mutations in the SP binding site of SecA did not alter the K d of TF:pro‐OmpA [1.79 (± 0.58) vs. 1.58 (± 0.38) μM; Fig 3C] although they did so for free pro‐OmpA (Fig 3A), corroborating our hypothesis. TF‐bound pro‐OmpA may dock onto SecA using mainly PatchA, the mature domain binding site 9. TF:pro‐OmpA binds better to the SecYEG‐bound SecA (noPatchA) than that to the wild‐type translocase [K d = 0.68 (± 0.18) vs. 1.79 (± 0.58) μM; Fig 3C] and better than the free pro‐OmpA to the same mutant receptor [K d = 0.68 (± 0.18) μM (Fig 3C) vs. 1.55 (± 0.66) μM (Fig 3A)]. These observations support the possibility that pro‐OmpA adopts an altered conformation upon TF binding or/and even that TF has its own independent contribution to SecA binding (see below).
We concluded that while TF does not bind to the SecA‐SecYEG translocase with a measurable K d, it accompanies the pro‐OmpA client as it binds onto its SecA receptor and remains bound to it. Clearly, TF becomes deleterious for translocation at a post‐targeting step.
SecB prevents pro‐OmpA from acquiring off‐pathway folding intermediates
SecB relieves the inhibitory effect of TF on pro‐OmpA translocation (Fig 1D, lane 4). Given the data above, we entertained three hypotheses: (i) SecB may bind to pro‐OmpA molecules released from TF, that we consider unlike, taking into account the determined K i (Fig 1E); (ii) SecB binding to soluble TF:pro‐OmpA complexes releases pro‐OmpA in solution allowing its independent self‐targeting to the translocase; c. SecB binding at or near to translocase‐docked TF:pro‐OmpA complexes releases pro‐OmpA for subsequent translocation.
We first examined whether SecB exerts an effect directly on pro‐OmpA. For this, the ability of SecB to prevent formation of the I3 intermediate was monitored by incubating 0.5 μM pro‐OmpA with a range of SecB concentrations (0–2 μM SecB4), at 37°C, for 10 min, prior to the SecYEG‐SecA translocase addition and measuring the translocation ATPase of SecA. The effect of SecB on the T/M ratio was determined, normalized, plotted and analysed as in Fig 1F. SecB interacts with pro‐OmpA directly, with a solubilization parameter K sol of 190 (± 45) nM, prevents formation of the I3 intermediate and leads to high translocation ATPase (Fig 4A). Moreover, in another functional assay similar to that in Fig 2C, SecB promotes the solubility of the aggregation‐prone pro‐OmpA* (Fig 4B).
Figure 4. Physical and functional interaction of SecB with pro‐OmpA and TF .
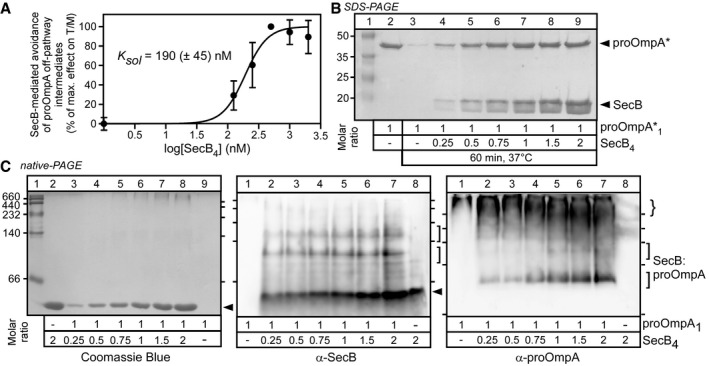
- SecB maintains soluble translocation‐competent states of pro‐OmpA at 37°C. pro‐OmpA1 (0.5 μM) was incubated with a range of SecB4 concentrations (0–2 μM; Buffer D; < 0.2 M urea; 10 min, 37°C). Soluble samples (20,000 g; 10 min; 4°C) were added to translocation ATPase reactions (as in Fig 1C). Normalized T/M ratios were plotted (n = 2–8 biological replicates; mean values ± SEM) and an apparent solubilization constant (K sol) determined (as in Fig 1F).
- SecB protects pro‐OmpA* from aggregation. pro‐OmpA*1 (5 μM) was incubated (50 μl; Buffer C; 37°C; 60 min) alone or in the presence of the indicated molar excess of SecB4, and pro‐OmpA* in the soluble fraction (after centrifugation) was analysed by SDS–PAGE (as in Fig 2C). Lane 2: 2.84 μg pro‐OmpA*. Representative experiment is shown; n = 4 biological replicates.
- SecB physically interacts with pro‐OmpA. pro‐OmpA1 (5 μM) was incubated (50 μl; Buffer C; < 0.2 M urea; 4°C; 60 min), with the indicated molar excess of SecB4 (filled arrow), analysed by native‐PAGE as in Fig 2A and stained as indicated. Brace: higher order pro‐OmpA species. Representative experiment is shown; n = 5 biological replicates.
Source data are available online for this figure.
However, in contrast to what was observed with TF:pro‐OmpA (Fig 2A), SecB:pro‐OmpA (Fig 4C) or SecB:pro‐OmpA* (Appendix Fig S3) complexes could not be detected on Coomassie Blue‐stained clear native‐PAGE gels (Fig 4C, left). Immunostaining with α‐SecB (middle) and α‐pro‐OmpA (right) antisera revealed two distinct but minor populations of SecB:pro‐OmpA complexes (brackets), while most of pro‐OmpA entered the gel and was detectable in high apparent masses (brace). A Kd,app of 3 μM was calculated (Appendix Fig S3), suggesting a high Koff and rapid dissociation. Native‐MS detected SecB as a homotetrameric complex of 68764 ± 0.7 Da (Appendix Fig S5) that forms a 1:1 heterocomplex of 107,108 ± 1.4 Da with pro‐OmpA.
Given the weaker Kd, ~15‐fold less than that of TF for pro‐OmpA (Fig 1E), it seems unlikely that the first hypothesis can adequately explain how SecB relieves TF‐mediated inhibition with such fast kinetics.
Soluble TF:pro‐OmpA:SecB quaternary super‐complexes
We next examined whether SecB can associate with TF:pro‐OmpA complexes in solution using four methods.
Firstly, we used native‐MS and determined the association of the TF:pro‐OmpA complex with the SecB apoprotein. TF:pro‐OmpA associates with SecB to form a super‐complex of 157,311 consistent only with a TF1:pro‐OmpA1:SecB4 stoichiometry (Figs 5A and B, and EV3).
Figure 5. TF:pro‐OmpA:SecB super‐complex analysis.
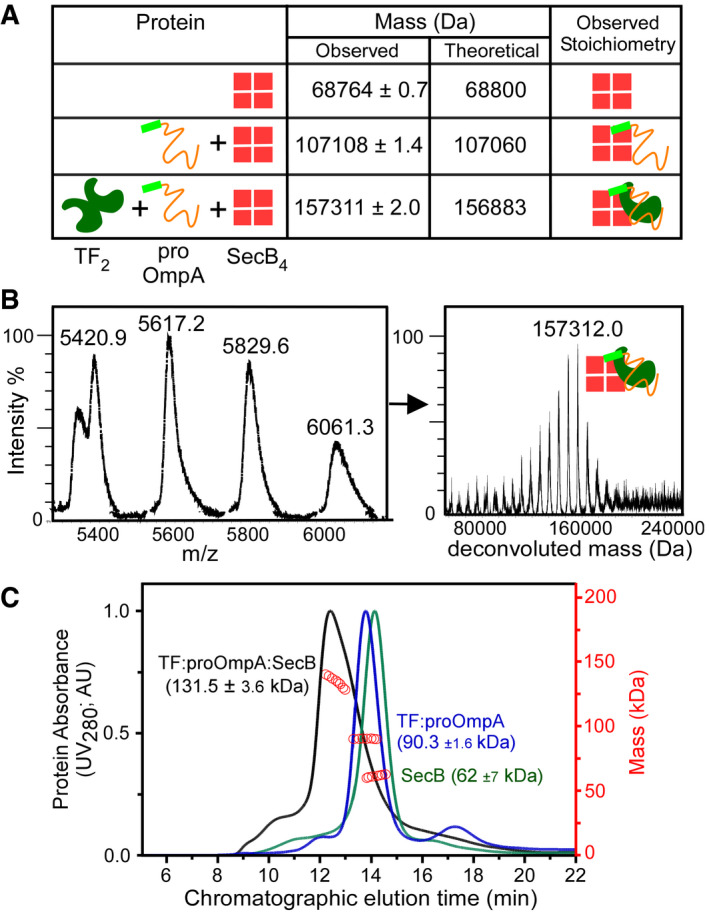
- Analysis of pro‐OmpA:SecB and TF:pro‐OmpA:SecB complexes by native‐MS. SecB4 (30 μM; Buffer G), SecB4 (40 μM) and pro‐OmpA (20 μM; pre‐treated with 10 mM DTT; buffer G) and of SecB4 (40 μM), pro‐OmpA (20 μM) and TF2 (20 μM) in Buffer G were analysed as in Fig 2B (raw data in Fig EV3). n = 2 biological replicates.
- Blow up of the ESI‐mass spectrum of the TF:pro‐OmpA:SecB4 super‐complex from A (complete dataset in Fig EV3). Left: charge state distributions, at the indicated m/z range. Right: masses deconvoluted from the mass spectra (as in Fig 2B). Inset cartoon depicts stoichiometry. Representative spectrum is shown; n = 2 biological replicates.
- TF:pro‐OmpA:SecB super‐complex analysis using GPC‐MALS in Buffer A. TF1:pro‐OmpA1 (10 μM each; green), SecB4 (40 μM; blue) and TF1:pro‐OmpA1:SecB4 (10, 10 and 40 μM, respectively; black). n = 5 biological replicates; mean mass: 137 kDa (± 6 kDa) representative experiment is shown; red circles: determined mass (kDa); AU: arbitrary units.
Source data are available online for this figure.
Figure EV3. Native‐MS of TF:pro‐OmpA:SecB (related to Fig 5).
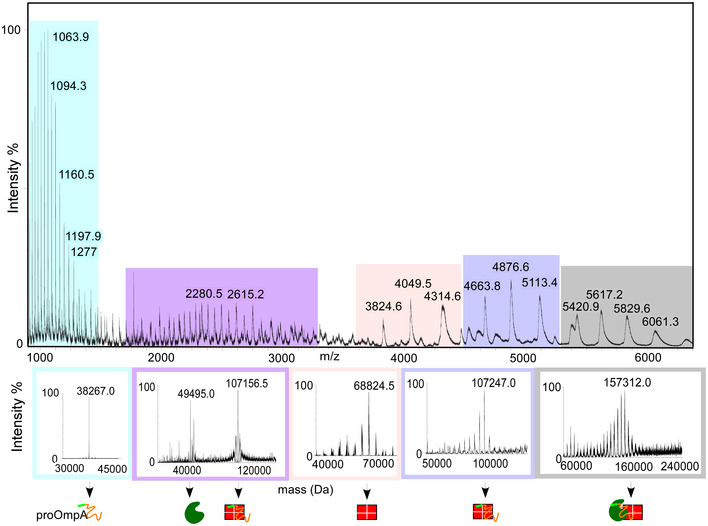
ESI‐mass spectrum and charge state distributions of different populations detected during native‐MS analysis of TF:pro‐OmpA:SecB complex. pro‐OmpA, [TF1 and (SecB4:pro‐OmpA)1], SecB4, (SecB4:pro‐OmpA) and the super‐complex (TF:pro‐OmpA:SecB4)1 are coloured in cyan, violet, light pink, light purple and grey, respectively. The lower panel shows the different populations in mass spectra of the complex deconvoluted to derive the indicated molecular masses.
Secondly, we used isothermal titration calorimetry (ITC). While SecB has no measurable affinity for TF (Appendix Fig S4B), titrating SecB4 in preformed equimolar TF:pro‐OmpA complexes revealed an affinity of 0.8 (± 0.18) μM and a stoichiometry of one SecB4 per TF:pro‐OmpA (Fig EV4A).
Thirdly, soluble TF:pro‐OmpA:SecB complexes were observed by gel permeation chromatography coupled online to multiangle laser light scattering (GPC‐MALS; Fig 5C). Upon mixing TF:pro‐OmpA with SecB4, a quantitative shift was observed to a higher mass peak, suggesting formation of a holo‐complex that displayed some polydispersity. Ensemble measurements yielded a mean mass of ~137 (± 6) kDa; consistent with TF1:pro‐OmpA1 bound to 2–4 SecB.
Fourthly, using native‐PAGE (Fig EV4B) SecB4 migrates with an apparent mass of ~50 kDa in this gel system, seen after Coomassie Blue staining (Fig EV4B, top, lane 3). When SecB is mixed together with TF:pro‐OmpA (lane7) a new, slower migrating species appears with higher apparent mass than that of TF:pro‐OmpA (lane 6) and the intensity of the TF:pro‐OmpA complex band is reduced. This presumed ternary TF:pro‐OmpA:SecB super‐complex is not detectable when SecB4 is added to pro‐OmpA (lane 5) or to TF2 (lane 8) alone. The TF:pro‐OmpA:SecB super‐complex is immuno‐stained using anti‐sera to each of the three partner proteins (Fig EV4B, bottom panels).
Taken together, these data demonstrate that SecB forms a previously unknown high‐affinity quaternary complex with TF and pro‐OmpA in solution in vitro, and therefore, likely in the cytoplasm. Formation of this stable complex suggests that SecB binding to TF:pro‐OmpA is unlikely to promote significant pro‐OmpA release.
SecYEG/SecA‐bound SecB optimizes TF and TF:pro‐OmpA binding to SecA
SecB does not affect pro‐OmpA or OmpA affinity for the translocase significantly [0.47 (± 0.07) or 0.65 (± 0.13) μM (Fig 3A) vs. 0.36 (± 0.08) or 1.07 (± 0.39) μM (Fig 3D), respectively]. However, although TF shows no measurable affinity for the wild‐type translocase (Fig 3B, lane 2), it acquired a substantial one in the presence of SecB [2.41 (± 0.53) μM; Fig 3E, lane 1]. Since the direct binding of TF to SecB in solution is beyond detection (Fig EV4B, lanes 8; Appendix Fig S4), this suggested that somehow, SecB binding to the translocase might provide additional receptor sites or/and allosterically alters SecA, leading to TF binding. In support of this hypothesis SecB improved TF:pro‐OmpA binding onto the wild‐type translocase; 0.69 (± 0.14) (Fig 3F, lane 1) vs. 1.79 (± 0.58) μM (Fig 3C, top, lane 2). However, despite the presence of SecB, if pro‐OmpA is already docked on SecA, TF no longer recognizes the wild‐type translocase (Fig 3E, lane 3). We entertain two possibilities: the translocase binding sites for TF that SecB had exposed are hidden by pro‐OmpA or a pro‐OmpA‐induced conformational change in the translocase makes these sites unavailable to TF binding 61.
SecB interacts with the SecA C‐tail region, known to co‐ordinate metal ions like Zn2+ 25, 43, 63. Moreover, the C‐tail residues 849–854 form a β‐strand 9, 62, 64 that binds to PatchA, the mature domain binding site of SecA 9, thereby acting as a substrate mimic 62. A SecA(noC‐tail) derivative, missing the 70 C‐terminal residues, is fully functional in vivo 65 and was shown to be inhibited by TF in a genetic complementation assay (Appendix Fig S6A). We quantified the binding of TF to the SecA(noC‐tail) (Fig 3E, lane 2). In the absence of SecB, TF exhibited an affinity for the SecYEG‐bound SecA(noC‐tail) [3.68 (± 0.99) μM] similar to the one seen in the presence of SecB for the wild‐type translocase [2.41 (± 0.53) μM; lane 1]. Our inability to detect TF interaction with SecA(noC‐tail) in solution (by ITC; Appendix Fig S4C) suggested that SecA(noC‐tail) becomes primed for its interaction with TF by its prior binding to SecYEG.
Our results reveal a previously unsuspected TF:Sec translocase interaction that is conditional on a specific SecA conformation, induced by SecB binding. Attesting to this, SecA(noC‐tail) undergoes measurable conformational changes, detectable by local HDX‐MS (Fig EV5), that may underlie its conversion to a higher affinity TF receptor. This new unexpected role of SecB renders it an important determinant for the targeting of either TF or TF:pro‐OmpA complexes to the translocase.
Figure EV5. Local HDX‐MS analysis of the SecA(noC‐tail) variant (related to Fig 6).
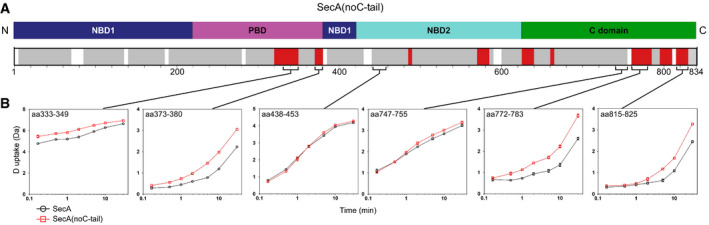
- Linear map of SecA(noC‐tail) with the four structural domains indicated the following: nucleotide binding domain 1 (NBD1, dark blue), preprotein binding domain (PBD, magenta), nucleotide binding domain 2 (NBD2, cyan) and C‐terminal domain (C domain, green). Local HDX‐MS experiments on SecA and SecA(noC‐tail) were carried out in identical conditions and directly compared. Removal of the C‐tail results in increased dynamics across SecA. Regions with increased dynamics are indicated in red; those that show no differences between SecA and SecA(noC‐tail) in grey, regions for which no data are available are in white. n = 3.
- Representative D uptake plots for selected peptides from different regions of SecA. D uptake values from SecA (black) and SecA(noC‐tail) (red) are plotted with respect to D labelling time. Peptides located mainly in PBD, and the C domain showed increased dynamics in SecA(noC‐tail), while most other regions of SecA showed no major differences. Representative experiment is shown; n = 3; aa: amino acid.
SecB requires the SecA C‐tail to overcome the TF‐mediated secretion inhibition
SecB exhibits high affinity for soluble 25 or membrane‐bound (0.2–0.33 μM; Fig EV1E; 44) SecA and is released from it upon ATP hydrolysis 41. Although SecB binding to SecA occurs in a C‐tail‐dependent manner 25, 43, 44, 63, the C‐tail of SecA per se is not essential in vivo or in vitro 65. Corroborating this, pro‐OmpA stimulated the translocation ATPase of SecA(noC‐tail) (Fig 6A, lane 1) similarly to that of wild‐type SecA (Fig 1C and D) 65. SecB addition had no effect (Fig 6A, lane 2). TF suppressed this ATPase by 80% (lane 3), similarly to the effect seen on wild‐type SecA (Fig 1D, lane 3). However, in contrast to what was seen for the wild‐type SecA (Fig 1D, compare lanes 3 and 4), further addition of SecB, even up to 12 molar excess (Fig 6A, lane 5), did not relieve the TF‐mediated inhibition (compare lanes 4 and 5 to 3).
Figure 6. SecB‐mediated, SecA C‐tail‐dependent release of TF‐associated pro‐OmpA for translocation.
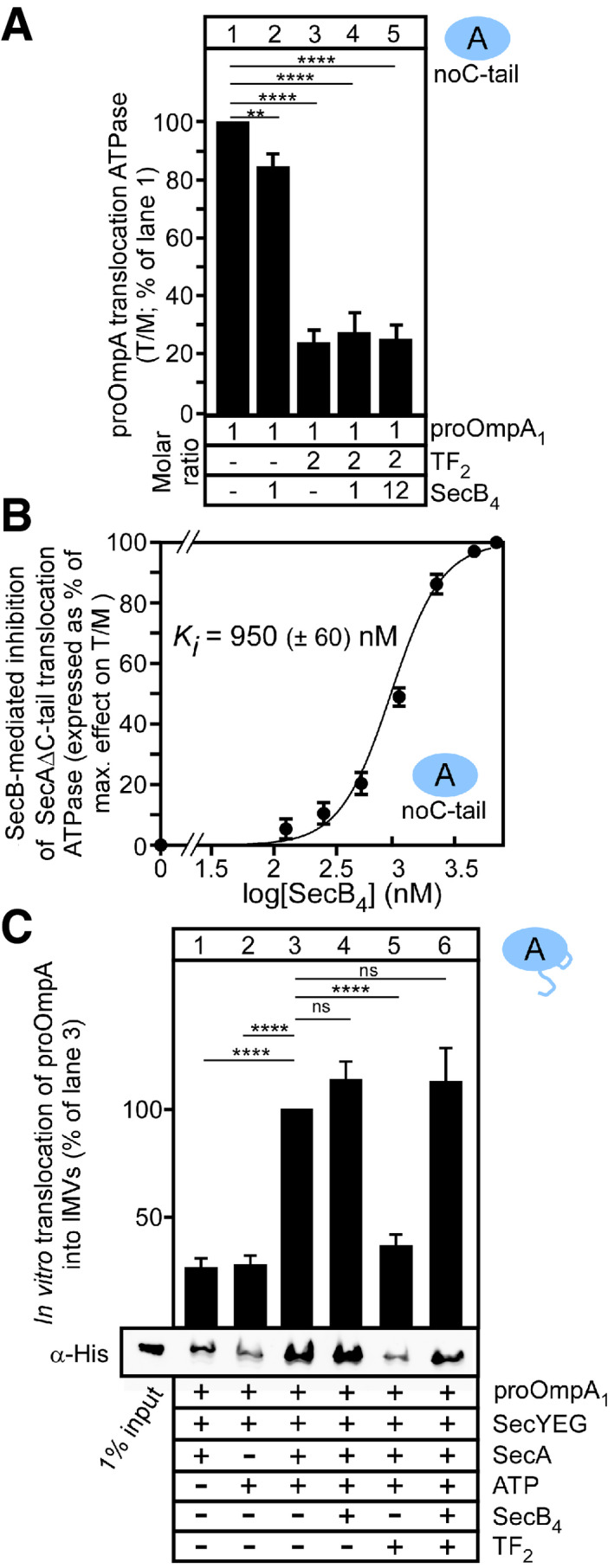
- SecB fails to restore pro‐OmpA translocation ATPase activity stimulation of SecA(noC‐tail) in the presence of TF. pro‐OmpA translocation ATPase activity stimulation (as in Fig 1C; SecB molar excess over pro‐OmpA indicated). T/M ratio of SecA(noC‐tail) in the absence of TF or SecB = 100%; lane 1; all other values were expressed as a % of this value. n = 4 biological replicates; mean values ± SEM. Unpaired parametric t‐test, 95% confidence interval: **P = 0.0048; ****P < 0.0001.
- Excess of SecB inhibits the pro‐OmpA‐stimulated translocation ATPase activity of SecA(noC‐tail). ATPase measured as in Fig 1C; SecB concentration range: 0–6 μM. Values were normalized (effectmax = 100%; effectmin = 0%) and plotted (n = 3 biological replicates; mean values ± SEM) versus the log10[SecB4]. The K i was determined as in Fig 1E.
- TF and SecB‐regulated, SecA‐dependent in vitro translocation of pro‐OmpA into SecYEG/A‐IMVs, analysed after proteolytic treatment. Translocated, protease‐resistant pro‐OmpA was visualized using immunostaining (α‐His) in the absence or presence of TF2 or/and SecB4 (as indicated). Translocation in the absence of TF or SecB was considered 100% (lane 3); all other values were expressed as a percentage of this. A summary of all repeats is presented (mean ± SEM; n = 6 biological replicates; ns: not significant (P = 0.1568 for lane 3 vs. 4; P = 0.4835 for lane 3 vs. 6); Unpaired parametric t‐test, 95% confidence interval: ****P < 0.0001) with a representative Western blot (bottom).
Source data are available online for this figure.
Notably, even SecB when added to translocation ATPase reactions with SecA(noC‐tail) becomes deleterious at high concentrations and inhibits pro‐OmpA‐stimulated translocation ATPase (Fig 5B). This presumably results from SecB exerting its holdase function on pro‐OmpA but being unable to productively interact with the translocase in the absence of the SecA C‐tail.
We concluded that productive SecB binding to the SecA C‐tail is necessary to relieve the TF‐ or SecB‐mediated inhibition of pro‐OmpA translocation.
SecYEG/SecA‐bound SecB acts as a preprotein‐ and TF‐exchange factor
Our data demonstrated that the TF:pro‐OmpA complexes arrive at and bind to the translocase (Fig 3C). Yet, pro‐OmpA is probably not liberated from TF as indicated by the difference in the affinity of TF:pro‐OmpA [1.79 (± 0.58) μM; Fig 3C] to that of free pro‐OmpA [0.47 (± 0.07) μM; Fig 3A]. This affinity is restored when SecB is present [0.69 (± 0.14) μM; Fig 3F]. We hypothesized that SecB, that recognizes the TF:pro‐OmpA complex even in solution [0.8 (± 0.18) μM; Fig EV4), can only somehow dissociate TF from pro‐OmpA onto the translocase, thus allowing pro‐OmpA translocation.
We directly tested this hypothesis by using an in vitro pro‐OmpA translocation assay 9. pro‐OmpA is translocated into the lumen of IMVs and thus becomes protease‐resistant, in a SecA and ATP‐dependent manner (Fig 6C, lanes 1–3). SecB seems to slightly increase pro‐OmpA translocation (lane 4). Addition of TF alone inhibits (lane 5), but further addition of SecB restores (lane 6) pro‐OmpA translocation. Similar results were obtained with [35S]‐pro‐OmpA synthesized in the absence of urea, thus excluding a chaotrope‐related artefact (Appendix Fig S6B).
We concluded that SecB bound at the SecYEG‐SecA translocase acts as an exchange factor, causing pro‐OmpA release from TF to permit its translocation.
Discussion
Trigger factor is one of the ~200 universally conserved proteins in the bacteria 27, 66, 67 and has a well‐understood role in cytoplasmic protein folding 27 (Fig 7A, left), but its precise role in post‐translational secretion (Fig 7A, right) has remained unclear. A perplexing observation has been that TF retards protein export in vivo, since tig mutations increased the Sec‐dependent export of some clients 22, 50, 68. When overexpressed, TF actually compromises translocation in vivo 50 and viability 22. As SecB relieves this deleterious effect 22, it was anticipated that overexpressed TF may specifically block a secretion pathway step possibly through its ribosome‐associating capacity: ribosome‐bound TF sterically hinders the association of ribosomes with the SecYEG translocase 22.
Figure 7. Models of TF involvement in the post‐translational secretory pathway.
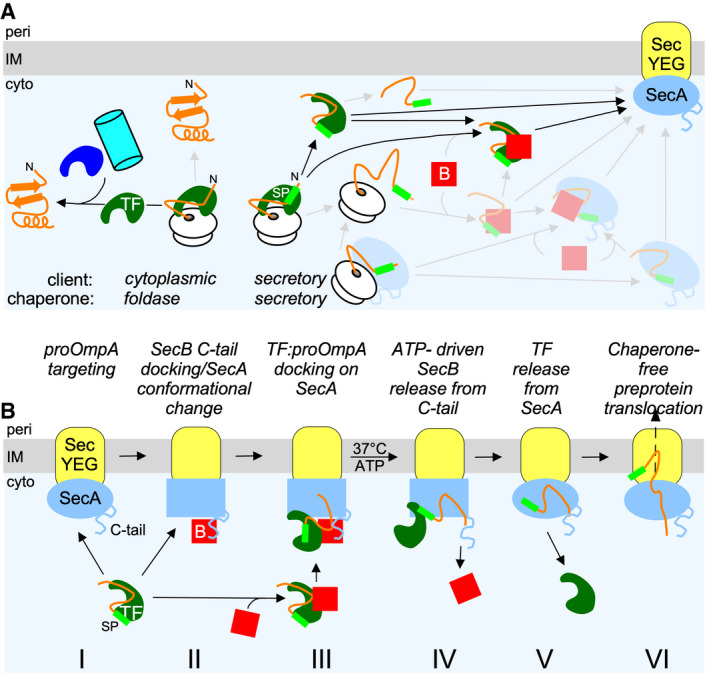
- Cartoon of the contribution of TF in cytoplasmic protein folding (left) and secretory protein targeting (right, black arrows). The secretory targeting factors SecB and SecA that act independently (grey arrows) or interface with TF are also shown. Blue, cyan: other folding chaperones.
- Schematic model of TF:pro‐OmpA targeted to the translocase with or without contributions from SecB and subsequent SecA C‐tail/SecB‐mediated control of pro‐OmpA translocation. Oval to rectangle transition in SecA signifies conformational changes. B: SecB; cyto: cytoplasm; IM: inner membrane; peri: periplasm.
We now show that TF is a bona fide secretory pathway chaperone as follows: (i) it forms tight complexes with a subset of secretory preproteins such as pro‐OmpA, co‐ 69 and post‐translationally (Figs 1E and 2A; 29) and can prevent their aggregation (Fig 2C; 28, 39) or acquisition of off‐pathway folding intermediates (Figs 2D and 3C, and EV2); (ii) when complexed to pro‐OmpA it binds to SecB in solution (Figs 5B and C, EV3 and EV4A and B) and to SecYEG‐bound SecA with high affinity (Fig 3C); (iii) in the presence of SecB, but not in its absence, TF binds to SecYEG‐bound SecA with high affinity even in the absence of pro‐OmpA (Fig 3E); and d. TF does not allow pro‐OmpA, that is co‐targeted to the translocase, to proceed to translocation (Figs 1D and 6A and B) unless it is first recycled by SecB acting via the SecA C‐tail (Figs 1D and 6A and B). Reconstitution of the TF‐mediated inhibition reaction in vitro and use of the non‐ribosome binding 3A mutant (Fig 1A, lane 8) revealed that the role of TF is largely ribosome‐independent and hence post‐translational. This intricate and remarkable network of interactions in which TF physically participates was previously unsuspected and extends the functional interactome of TF to the Sec translocase. The association of TF with the membrane may explain, at least partly, why in proteomics experiments TF is systematically identified in the inner membrane peripherome 70.
That TF specializes on only a limited set of secretory clients might explain its non‐essentiality for protein secretion. Of the 108 secretory chains that TF recognizes on ribosomes 6, 69, only 10 are found to be TF‐associated after synthesis 6. A second reason is that targeting in the secretion pathway has in‐built redundancy (Fig 7A, right), as does the folding pathway (Fig 7A, left) 48. For example, as an alternative to using TF, pro‐OmpA might be briefly complexed with SecB (Fig 4A–C; 44, 71) or SecA 72 or a complex of both 44, 72. The intracellular concentrations of TF, SecB, SecA and their affinities for each other and for pro‐OmpA (Figs 3 and EV1, and EV4A) will direct/orchestrate these interactions “on demand” in vivo. Since all three chaperones can make use of either the SP or the multiple mature domain stretches of a preprotein 9, 16, 42, even multiple different chaperones may assemble on the same preprotein, as proposed for TF and DnaK 73, or for multiple TFs interacting with the same unfolded secretory polypeptide 16.
Trigger factor does not display a generic preference for slightly hydrophobic outer membrane porin β‐barrels although it interacts strongly with some of them. It associates with ~25% of all nascent outer membrane proteins that constitute < 3% of all its nascent interactors 6, 69, co‐ or post‐translationally (e.g. OmpX; Appendix Fig S3) 6, 74. Therefore, it is unclear why TF would specialize on pro‐OmpA as a client. It may recognize particular structural features on pro‐OmpA. Alternatively, such specialization may have derived from pro‐OmpA's disposition to acquire “off‐translocation pathway” intermediates (Figs 2E and EV2) and/or the fact that pro‐OmpA is one of the most abundant cellular proteins (~100,000 to ~150,000 copies/cell 1). Any disruption or slow down of secretion flow will result in the accumulation of large amounts of cytoplasmic pro‐OmpA. TF may sequester these molecules queuing for translocation to prevent their mis‐folding or aggregation and ensure their delivery to the translocase, once sufficient amounts of SecB/SecA become available. This hypothesis is in line with the fact that non‐stressed cells do not need TF to accomplish pro‐OmpA secretion since there is no perturbation of the secretion flow and hence no accumulation of pro‐OmpA molecules 40.
Secretory protein recognition and targeting by TF must be governed by some specificity to avoid futile interactions: e.g. neither TF:pro‐OmpA should be passed on to cytoplasmic foldases, nor TF:cytoplasmic clients should reach and stochastically interfere with the operation of the translocase. Kinetic “bottlenecks” may render such “illicit” interactions less likely, such as short TF:client lifetimes or, downstream chaperones at high concentrations and/or elevated affinities for TF:clients. Correct vectorial transfer to the right pathway, cytoplasmic or secretory, may be additionally secured by SPs for a fraction of exported proteins. SPs may act as high affinity, secretion pathway‐only handles, for tighter interactions with TF and subsequently with SecB and/or SecA (Fig 7A, right). For example, SecB binding to TF:pro‐OmpA, with an increased affinity (Fig EV4A), will bias TF:pro‐OmpA to stay on the secretory pathway and reach the translocase. In contrast, SecA may chaperone cytoplasmic proteins for folding, thus additionally diverting them from the secretion pathway 75.
Non‐ATPase chaperones, such as TF and SecB, are assumed to behave like typical holdases 16, i.e. will bind tightly, with long lifetimes, to a secretory polypeptide chain and prevent it from acquiring a folded state or aggregating. As most secretory proteins, including pro‐OmpA, can maintain soluble non‐folded or partially folded states in solution for long and are frequently disordered 11, this is not often/frequently necessary. However, if the need arises, TF residing at the ribosomal exit tunnel, its cytoplasmic concentration and high affinity for clients can secure a rapid interaction with secretory clients, preventing not only aggregation but also off‐pathway folding intermediates (Figs 1E and 2A and C, and EV4B).
Of note, complexes of pro‐OmpA with SecB, considered as the typical pro‐OmpA chaperone 54, can be stabilized under certain conditions (Figs 4 and 6B; Appendix Fig S5) 76, 77, were weaker than those of TF (Figs 1F and 4C; Appendix Fig S3D). Nevertheless, SecB rescued pro‐OmpA from both mis‐folding (Fig 4A) and aggregation (Fig 4B) 55. The apparent high K off of this interaction goes against the typical holdase concept. Perhaps even fleeting interactions with SecB return pro‐OmpA to soluble states, reminiscent of binding/release cycles that clients undergo on foldase chaperones 78, 79, while at elevated concentrations, the SecB holdase is stabilized (Fig 6B).
TF:secretory client complexes, with or without SecB, are specifically targeted to SecYEG‐bound SecA (Fig 3). In this previously unsuspected reaction, TF:secretory client complexes bind to the translocase through the independent affinities of the secretory chain, TF and SecB for SecA and for each other. One remarkable aspect of this process extends the role of SecB beyond that of a typical chaperone. SecB bound via the SecA C‐tail acts as a recycling regulator for TF:pro‐OmpA complexes, overcoming the TF‐mediated inhibition of pro‐OmpA translocation (Fig 6A and C). The precise mechanistic, structural and dynamic basis of this process requires future dissection. Nevertheless, a key event in the process seems to be the conformational dynamics of the SecA motor, the only ATP hydrolysing enzyme in the dynamic holo‐complex and, evidently, anticipated to be the core ATP‐driven partner recycling component. Local HDX‐MS (Fig EV5) and single molecule‐FRET analysis 80, 81 (S. Krishnamurthy, N. Eleftheriadis, K. Karathanou, S. Smit, A.G. Portaliou, K.E. Chatzi, M.F. Sardis, S. Karamanou, A. Bondar, G. Gouridis & A. Economou, in preparation) revealed such conformational events in various parts of the SecA structure including its preprotein binding domain. The enhanced affinity for TF of the two SecA derivatives, SecA(noPatchA) and SecA(noC‐tail) (Fig 3B and E), raises the possibility that these two elements that are juxtaposed in the SecA structure, one binding mature domains (PatchA) and the other SecB (C‐tail), may be central switches that modulate SecA conformation (Fig EV5) and, consequently, the affinity for TF.
Taken together, these and past data led to a simple working hypothesis for the translocation of secretory proteins co‐targeted to the translocase with TF (Fig 7B). TF:pro‐OmpA complexes are targeted to the translocase and bind to SecA with (II, III) or without (I) SecB. In the former case, binding is ~3 times tighter and could be attributed to SecB providing binding surfaces directly to TF and/or preprotein and to SecB‐driven conformational changes onto SecA mediated through SecB occupancy of the SecA C‐tail (III). Initiation of the translocation ATPase cycle expels SecB (IV) 43, and SecA conformation is altered to a state that promotes dissociation of the TF:pro‐OmpA complex. pro‐OmpA is released from TF and binds with high affinity to SecA in a way that is no longer available for TF binding (Fig 3E). As a result of a reduced affinity for the SecA receptor, TF is expelled to the cytoplasmic pool (Fig 7B, V). SecA‐bound pro‐OmpA proceeds with translocation (VI).
Materials and Methods
For a complete list of strains (Appendix Table S1), plasmids (Appendix Table S2), primers (Appendix Table S3), buffers (Appendix Table S4), see Appendix.
In vivo genetic complementation assay
Wild‐type MC4100, MC4100ΔsecB, MC4100ΔtigΔsecB and BL21‐19 cells transformed with plasmids carrying, or not, secA or secAΔCtail, tig or secB were grown overnight at 30°C in LB supplemented with ampicillin (100 μg/ml) and gentamicin (10 μgr/ml) if needed. Cultures were diluted (1/50) into fresh LB and grown at 30°C until OD600 = 0.5. Serial dilutions were spotted on freshly prepared LB plates containing antibiotics without or with anhydrotetracycline (AHT; 5 ng/ml) and without or with L‐arabinose (0.1%w/v).
Chaperone‐mediated solubilization assay using SDS–PAGE
Chaperones (in Buffer A) and pro‐OmpA*(in Buffer B; without DTT/EDTA treatment) were mixed together at different molar ratios in Buffer C, incubated (37°C; 60 min) and centrifuged (20,000 g; 4°C; 10 min; Sigma 1‐16K). Soluble proteins were analysed on a 15% acrylamide SDS–PAGE gel and Coomassie Blue stained. pro‐OmpA signals were quantified by scanning densitometry (Image J; https://imagej.net/Welcome).
Chaperone‐pro‐OmpA binding assay using native‐PAGE
Chaperones (Buffer A) and pro‐OmpA (Buffer B; DTT‐ and EDTA‐treated) were mixed together at different molar ratios in aqueous Buffer C, or Das indicated, incubated (37°C; 60 min) and centrifuged (20,000 g; 4°C; 10 min; Sigma 1‐16K). Soluble proteins were separated on a 10% acrylamide native‐PAGE gel (4 mA; 4°C; 16 h) and Coomassie Blue stained.
Cell growth, gene overexpression and cell lysis
Escherichia coli Lemo21, BL21.19 or Tuner cells transformed with plasmids expressing preproteins, mature domains or chaperones (see “Plasmids” table S2 in Appendix), or BL31(DE3) cells, transformed with pET610 (carrying the his‐secYEG operon), were grown in LB (37°C; 100 mg/ml Ampicillin; till OD600 = 0.6), and gene expression was induced (0.1 mM IPTG; 3 h; 30°C) 11. For preproteins, 4 mM sodium azide was added in the cell culture, 10 min prior to IPTG induction to prevent SecA‐dependent secretion and thus SP cleavage 82. Cells were harvested (5,000 g; 4°C; 15 min; Avanti J‐26S XPI, JLA 8.1000 rotor; Beckman) and resuspended in Buffer I (preproteins, mature domains) or Buffer J (His‐TF) or Buffer K (SecB) or Buffer L (SecA and derivatives) or Buffer M (SecYEG‐IMVs). Cells were lysed using a French press (8,000 psi; 3–5 passes; pre‐cooled cylinder at 4°C; Thermo). Unbroken cells were removed (3,000 g; 10 min; Sigma 3‐16KL; rotor 11180), and supernatants were centrifuged further (35,000 g; 30 min; 4°C; Optima XPN‐80, Beckman) to separate soluble from membrane proteins or/and inclusion bodies.
Protein purification
Denaturing protein purification of His‐tagged proteins
The pellet of lysed cells was solubilized in Buffer N (Dounce homogenizing) and centrifuged (35,000 g; 30 min; Optima XPN‐80; 45 Ti fixed‐angle rotor; Beckman) 9, 11. The urea‐solubilized supernatant was diluted with Buffer O to 6 M urea before applying it on a column packed with Ni+2‐NTA agarose resin (1 ml/5 μgr protein; Qiagen) pre‐equilibrated with Buffer P (gravity flow; 1 ml/min). The column was washed sequentially with Buffer P and Buffer Q (10 column volumes). Proteins were eluted with Buffer R, incubated with EDTA (10 mM; 30 min; 4°C), dialysed (12–14,000 Da molecular weight cut‐off, Medicell Membranes Ltd.; Buffer S; 12 h; 4°C), aliquoted and stored at −20°C. All purification and dialysis buffers were supplemented with 5 mM and 2.5 mM MgCl2, respectively, to prevent SP cleavage 83.
Non‐denaturing purification of His‐tagged proteins
The supernatant of lysed cells was loaded on a column packed with Ni+2‐NTA Agarose resin (1/ml resin/10 μgr of protein; Qiagen), pre‐equilibrated with Buffer T (gravity flow; 1 ml/min) and washed with Buffer U 9, 11. Proteins were eluted with Buffer V, incubated with EDTA (10 mM; 30 min, 4°C) and dialysed first in Buffer W (3 h; 4°C) and then in Buffer X (12 h; 4°C). Proteins were aliquoted and stored at −20°C.
Purification of untagged SecB
The soluble supernatant was loaded on a column packed with a Q resin (5 ml; flow rate 1 ml/min; GE Healthcare) washed sequentially with 10 column volumes of Buffer Y, Buffer Z and Buffer AA 9, 10, 11. SecB was eluted with Buffer AB and further purified using preparative gel permeation chromatography (HiLoad Superdex 200; Buffer AC; ÄKTA Pure System; GE Healthcare). Peak fractions (5 ml) were collected, pooled and dialysed: Buffer W (12 h; 4°C) then in Buffer X (12 h; 4°C). Protein aliquots were stored at −20°C.
Purification of untagged SecA and derivatives
Following cell lysis, the soluble supernatant was loaded (2 ml/min) on a pre‐equilibrated home‐prepared Cibacron Blue resin (Sigma) 9, 10, 11. The column was washed (Buffer AD; 10 column volumes; 2 ml/min; ÄKTA Pure System; GE Healthcare), and SecA was eluted using a linear gradient (Buffer AD to Buffer AE; 4 column volumes; 2 ml/min), in 5 ml fractions, and concentrated, and NaCl was adjusted to 1 M final concentration. Following DTT treatment (20 mM; 15 min; 4°C), SecA was loaded on a preparative HiLoad Superdex 200 (GE Healthcare) gel permeation chromatography column (Buffer AC). Peak fractions (5 ml) were collected, concentrated, treated with 10 mM DTT and re‐loaded on a second HiLoad Superdex 200 (GE Healthcare) pre‐equilibrated with Buffer A. Fractions containing SecA were pooled and dialysed in Buffer X (12 h; 4°C). Protein aliquots were stored at −20°C.
Determination of protein concentration
Protein concentrations were determined spectroscopically (280 nm; NanoDrop 2000; Thermo) 9, 11. The molecular weight and extinction coefficient of proteins were determined using the Expasy server (http://web.expasy.org/protparam/). Spectroscopic measurements were also employed to determine the soluble/aggregation character of proteins alone (Fig EV3B). The initial protein concentration (the input = 100%, at 0 min) was compared to protein concentration in the supernatants after centrifugation (20,000 g; 4°C; 10 min; Sigma 1‐16K) after an incubation (time and temperature as indicated). Wherever indicated, pre‐treatment of protein stock solutions was with 10 mM DTT, 5 mM EDTA (20 min; 4°C).
Preparation of inverted inner membrane vesicles (IMVs)
Following lysis by French press and removal of unbroken cells (4,000 g; 10 min; Sigma 3‐16KL; rotor 11180), samples were ultra‐centrifuged (95,000 g; 90 min; 4°C; 45 Ti fixed‐angle rotor; Optima XPN‐80, Beckman) 9, 11. The membrane pellet was resuspended (2 ml; Buffer AF; Dounce‐homogenizer), loaded on top of a 5‐step sucrose gradient (1.9; 1.7; 1.5; 1.3; 1.1 M sucrose in Buffer AF; 6 ml each layer) and centrifuged to equilibrium (84,000 g; 16 h, 4°C; SW 32 Ti swinging bucket rotor, Optima XPN‐80, Beckman). IMVs, collected from gradient fraction 2, were resuspended (Buffer AG) and re‐centrifuged (95,000 g; 90 min; 4°C; fixed‐angle T647.5 rotor, Sorvall). The membrane pellet was resuspended and Dounce‐homogenized in Buffer AH (20 min; 4°C), loaded on top of an equal volume of Buffer AI and centrifuged (95,000 g; 90 min; 4°C;T647.5 rotor; Beckman). IMVs were collected 84, Dounce‐homogenized in 2 ml of Buffer D and extruded through a 100‐nm pore size filter (15–21 passes; Avestin LiposoFast‐Basic system), to obtain unilamellar vesicles of similar diameter, and stored in aliquots at −80°C.
Measuring SecA ATPase activity
ATP hydrolysis was measured in Buffer E (50 μl) 85, 86, 87. To determine basal ATPase activity, 0.4 μM SecA was added; for membrane ATPase, SecYEG‐IMVs were added (0.4 μM SecY; for membrane/basal ratio 1.4; 85); and for translocation ATPase, preprotein (as indicated) was added. SecB and TF were added (as indicated). In the case preincubation is indicated, the preprotein (−/+ chaperone) was incubated (Buffer D; 40 μl; 10 min; indicated temperature) and centrifuged (20,000 g; 10 min; 4°C) and the supernatant was added to SecA‐SecYEG (as for translocation ATPase setup). After incubation at 37°C (20 min for basal and membrane; 10 min for translocation), the released inorganic phosphate (Pi) was measured using malachite green reagent and the K cat (pmol Pi/pmol SecA protomer/min) was determined.
[35S]‐labelling of proteins in vitro
Proteins were labelled with [35S]‐methionine (1,000 Ci/mMole; Perkin‐Elmer) during in vitro synthesis, using a TNT Quick coupled Transcription/Translation system (Promega) according to the manufacturer's instructions 9, 11. [35S]‐labelled proteins were separated from the free unincorporated radiolabelled amino acids by centrifugal gel filtration (1‐ml homemade insulin syringe columns; packed with G‐50 resin; pre‐equilibrated with Buffer F for TF and SecB or Buffer AJ for pro‐OmpA; centrifuged for 5 min (4°C; 3,000 g; Sigma 3‐16KL; rotor 11180). Flow‐through was further centrifuged to remove ribosomes (436,000 g; 20 min; 4°C; rotor TL‐100; Optima Max‐XP, Beckman‐Coulter) and stored at −20°C. For in vitro translation/translocation reactions, following synthesis of [35S]‐pro‐OmpA and removal of ribosomes, protein in Buffer F was immediately used for translocation reactions (see below).
Determination of equilibrium dissociation constants (Kd) for SecYEG‐bound SecA
SecA (0.4 μM) mixed with SecYEG‐IMVs (0.4 μM SecY) and SecB (1 μM; whenever indicated) was pre‐incubated (4°C; 10 min; in 10 μl of Buffer F) 9, 11, 61. Pre‐treated pro‐OmpA or OmpA stocks (10 mM DTT; 20 min; 4°C; then centrifuged at 20,000 g; 20 min; 4°C) were diluted in Buffer F in order to achieve a range of 0–2.4 μM in 200 μl. For TF, a range of 0–24 μM was used and for SecB 0–1 μM. Where indicated, pro‐OmpA or TF:pro‐OmpA dilutions were pre‐incubated (10 min; 37°C) prior to addition to the binding reactions. Proteins were added to SecA‐SecYEG(‐SecB) binding reactions (final 0–0.6 μM range for SecB, OmpA, pro‐OmpA; 0–6 μM for TF). 2 μl of [35S]‐protein was added to all samples as a tracer. Samples were incubated (20 min; 4°C), overlaid on an equal volume of BSA/Sucrose cushion (Buffer AK) and centrifuged (436,000 g; 20 min; 4°C; rotor TL‐100; Optima Max‐XP, Beckman‐Coulter). The pellet (containing IMVs and IMV‐bound proteins) was resuspended (300 μl Buffer F), and samples were immobilized on a nitrocellulose membrane (Protran, 0.4 mm) using a vacuum manifold (Bio‐Dot apparatus; Bio‐Rad). Bound [35S]‐labelled proteins on IMVs were visualized using a high‐resolution phosphor storage screen (GE Healthcare) on a Typhoon FLA 9500 system (GE Healthcare; default system settings) and quantified by Image Quant software (GE Healthcare). Data were analysed by non‐linear regression (one binding site fit; GraphPad Prism 6). For the determination of each K d, n = 6; 20 concentration points were used. Binding of proteins to the SecYEG‐IMVs in the absence of SecA showed only non‐saturable binding assumed to be on lipids.
pro‐OmpA in vitro translocation
pro‐OmpA (Buffer AJ or F) was pre‐treated (10 mM DTT; 5 mM EDTA; 20 min; 4°C) and centrifuged (20,000 g; 10 min; 4°C) 9. Translocation reactions (100 μl; Buffer F) with SecA (0.8 μM), SecYEG‐IMVs (0.8 μM SecY), pro‐OmpA (3 μM), TF2 (3 μM), SecB4 (3 μM) and ATP (2 mM) were incubated (30 min; 37°C) and transferred on ice. Non‐translocated molecules were digested by addition of 1 mg/ml proteinase K (Roche; 20 min; 4°C). Proteins were precipitated with 25% w/v TCA (60 min; 4°C), analysed by SDS–PAGE (12% gels) and visualized by immunostaining with α‐His antisera (Serotec) using α‐mouse horseradish peroxidase‐coupled secondary antibody (Jackson ImmunoResearch Laboratories, Inc.). Signals were visualized using the Supersignal West Pico kit (Thermo Fisher Scientific) in a CCD‐camera system (LAS‐4000; GE Healthcare) and quantified using Image J (https://imagej.net).
Native‐mass spectrometry (Native‐MS)
Trigger factor was exchanged in Buffer G (overnight; 4°C). pro‐OmpA was diluted in the dialysed TF in a 1:1 molar ratio and incubated (10 min) before it was subjected to mass spectrometric analysis. 30 μM of the sample was injected for native‐mass analysis using electrospray ionization mass spectrometer with a Q‐TOF mass analyser (Synapt G2 HDMS, Waters); sodium iodide (2 mg/ml) was used as a calibrant. The operating parameters for the spectral acquisition were as follows: capillary voltage, 1.8 kV; sample cone voltage, 60 V; extraction cone voltage, 2 V; source temperature, 37°C; desolvation temperature, 150°C; backing pressure, 5.9 mbar; source pressure, 2.08 e‐3 mbar; and Trap, 1.44 e‐3 mbar. Spectra were acquired in the range of 900–13,000 m/z in positive ion V‐mode. The molecular mass of the recorded spectra was calculated using MassLynx software (MassLynx version 4.1) by deconvoluting the mass spectra using MaxEnt 1.
Global and local hydrogen–deuterium exchange‐mass spectrometry (HDX‐MS)
Protein folding experiments
pro‐OmpA (Buffer AL) was pre‐treated (10 mM DTT; 5 mM EDTA; 20 min; 4°C) and centrifuged (20,000 g; 10 min; 4°C) 11. Folding was initiated by diluting pro‐OmpA to 0.2 M urea with pre‐warmed/chilled Buffer AM, at the indicated temperature and monitored up to 1 h (tfold = 1 min, 5 min, 10 min, 15 min and 1 h).
Global pulsed HDX‐MS
Lyophilized Buffer AM was freshly reconstituted in D2O (99.9% atom D, Sigma‐Aldrich 151882). At the indicated tfold, samples containing 0.85 μM pro‐OmpA were isotope labelled in 95.5% (v/v) D2O (pD:8.0; 100 s; indicated temperature). Non‐deuterated (ND) samples were prepared similarly, using protiated Buffer AM. Fully deuterated (FD) controls were labelled in Buffer AN (pD:8.0; 4°C; 1 h). All samples were quenched with pre‐chilled formic acid (ice; final pD = 2.5), snap‐frozen in liquid nitrogen and stored at −80°C until MS analysis (max 2 days; nanoACQUITY UPLC System with HDX technology—Synapt G2 ESI‐Q‐TOF; Waters). The UPLC sample chamber temperature was set at 0.2°C. pro‐OmpA (42.6 pmol) was desalted [250 μl/min, 2 min; MassPREP Micro Desalting Column (Waters); 0.23% (v/v) formic acid] and eluted [40 μl/min; 5%–90% linear gradient of 0.23% (v/v) formic acid in acetonitrile]. Spectra were acquired in the 400–2,000 m/z range (capillary voltage, 3.0 kV; sampling cone, 40 V; extraction cone, 3.6 V; source temperature, 80°C; and desolvation gas flow, 500 L/h at 150°C). Continuous calibration was achieved by co‐infusing Leucine Enkephalin (2 ng/μl in 50% acetonitrile–0.1% formic acid; 5 μl/min; Waters).
Local HDX‐MS
SecA and SecA (noC‐tail) were prepared in 100 μM stock concentrations. The deuterium exchange reaction was initiated by diluting 200 pmol of protein into D2O Buffer AO at a 1:10 ratio (final D2O concentration 90%). Continuous deuterium labelling was carried out for various timepoints (10 s, 30 s, 1 min, 2 min, 5 min, 10 min, 30 min and 2 days) prior to quenching with pre‐chilled quench solution (formic acid, 4 mM TCEP, 1 mg/ml fungal protease XII). The exchange reaction was mixed with quench solution at a 1:1 ratio to obtain a final pH of 2.5, and the reaction was incubated (2 min; 4°C). 100 pmol of SecA was injected into a nanoACQUITY UPLC System with HDX technology (Waters, UK) coupled to a SYNAPT G2 ESI‐Q‐TOF mass spectrometer. For enhanced peptide coverage, SecA was digested in 2 steps, first with fungal protease XIII 88 at the quench step, and subsequently digested online on a home packed immobilized pepsin (Sigma) cartridge (2 mm × 2 cm, Idex), at 16°C. HPLC and MS parameters were set as previously described 10. Peptide identification was carried out using the ProteinLynx Global server (Waters, UK). Deuterium exchange data were analysed using DynamX (Waters, UK) 10, 60.
Gel permeation coupled online to multiangle laser light scattering (GPC‐MALLS)
Multiangle light scattering experiments were performed online after gel‐permeation chromatography on a Superdex HR200 10/300GL mounted on an HPLC system (Optilab T‐rex; Wyatt) coupled to a photodiode array detector (SPD‐M10AVP; Shimadzu), a multiangle light scattering detector (DAWN‐EOS; Wyatt) and a refractive index detector (RID10A; Shimadzu) 9, 85. TF1:pro‐OmpA1 (10 μM each) and/or SecB4 (40 μM) were loaded using a 100 μl injection loop, in Buffer A, and chromatographed at 22°C at 0.8 ml/min. Data collection, analysis and plotting were performed using Astra v.5.0 software (Wyatt).
Isothermal titration calorimetry (ITC)
TF:pro‐OmpA and SecB samples were extensively dialysed (Buffer H) 61. All solutions were filtered (0.45 μm) and thoroughly degassed (20 min; gentle stirring under vacuum). The cell was filled with TF2:pro‐OmpA1 (15 μM; 300 μl) and the syringe with protein ligands as indicated (128 μM; 100 μl). For the titration, 2 μl injections, at 4‐min intervals (cell temperature 25°C, constant stirring at 300 rpm), were used. The experiments were performed using a MicroCal iTC200 System (GE Healthcare). Data were analysed with MicroCal Origin software version 7.0 (GE Healthcare).
Author contributions
JDG, AGP, BS, SKa and SKr purified proteins. JDG performed solubility, native‐PAGE, ATPase and in vivo experiments. AGP performed molecular cloning, native‐PAGE, membrane affinity, in vitro translocation and ATPase, TF expression, in vivo growth and GPC‐MALS experiments. BS performed ITC, native‐MS experiments and in vivo experiments. SKr performed local HDX‐MS. SKa supervised and helped with biochemical and biophysical assays and data analysis and performed global HDX‐MS experiments. AE, JDG, AGP and SKa wrote and revised the paper with contributions from BS. SKa and AE conceived and managed the project. All authors reviewed the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Acknowledgements
This paper is dedicated to the memory of Prof. Olaf Schneewind. We are grateful to P. Genevaux for strains; K. Chatzi, N. Famelis and L. Emmanouilidis for preliminary observations; and A. Tsirigotaki and N. Eleftheriadis for suggestions and biochemicals. Research in our lab was funded by grants (to AE): RiMembR (Vlaanderen Onderzoeksprojecten; #G0C6814N; FWO); MeNaGe (RUN #RUN/16/001; KU Leuven); CARBS (#G093519N; FWO); ProFlow (FWO/F.R.S.‐ FNRS “Excellence of Science—EOS” programme grant #30550343) and (to AE and SK): FOscil (ZKD4582—C16/18/008; KU Leuven). JDG was an FWO doctoral fellow (#1S53916N; FWO); SKr was an FWO [PEGASUS]² Marie Skłodowska‐Curie Fellow (#12Q1417N; FWO).
EMBO Reports (2020) 21: e49054
Contributor Information
Anastassios Economou, Email: tassos.economou@kuleuven.be.
Spyridoula Karamanou, Email: lily.karamanou@kuleuven.be.
Data availability
All raw data used for figures and expanded view figures are provided as source data files.
References
- 1. Tsirigotaki A, De Geyter J, Sostaric N, Economou A, Karamanou S (2017) Protein export through the bacterial Sec pathway. Nat Rev Microbiol 15: 21–36 [DOI] [PubMed] [Google Scholar]
- 2. De Geyter J, Tsirigotaki A, Orfanoudaki G, Zorzini V, Economou A, Karamanou S (2016) Protein folding in the cell envelope of Escherichia coli . Nat Microbiol 1: 16107 [DOI] [PubMed] [Google Scholar]
- 3. Rapoport TA, Jungnickel B, Kutay U (1996) Protein transport across the eukaryotic endoplasmic reticulum and bacterial inner membranes. Annu Rev Biochem 65: 271–303 [DOI] [PubMed] [Google Scholar]
- 4. Cross BC, Sinning I, Luirink J, High S (2009) Delivering proteins for export from the cytosol. Nat Rev Mol Cell Biol 10: 255–264 [DOI] [PubMed] [Google Scholar]
- 5. Smets D, Loos MS, Karamanou S, Economou A (2019) Protein transport across the bacterial plasma membrane by the sec pathway. Protein J38: 262–273 [DOI] [PubMed] [Google Scholar]
- 6. Loos MS, Ramakrishnan R, Vranken W, Tsirigotaki A, Tsare EP, Zorzini V, Geyter J, Yuan B, Tsamardinos I, Klappa M et al (2019) Structural basis of the subcellular topology landscape of Escherichia coli . Front Microbiol 10: 1670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Veenendaal AK, van der Does C, Driessen AJ (2004) The protein‐conducting channel SecYEG. Biochim Biophys Acta 1694: 81–95 [DOI] [PubMed] [Google Scholar]
- 8. Vrontou E, Economou A (2004) Structure and function of SecA, the preprotein translocase nanomotor. Biochim Biophys Acta 1694: 67–80 [DOI] [PubMed] [Google Scholar]
- 9. Chatzi KE, Sardis MF, Tsirigotaki A, Koukaki M, Sostaric N, Konijnenberg A, Sobott F, Kalodimos CG, Karamanou S, Economou A (2017) Preprotein mature domains contain translocase targeting signals that are essential for secretion. J Cell Biol 216: 1357–1369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sardis MF, Tsirigotaki A, Chatzi KE, Portaliou AG, Gouridis G, Karamanou S, Economou A (2017) Preprotein conformational dynamics drive bivalent translocase docking and secretion. Structure 25: 1056–1067.e1056 [DOI] [PubMed] [Google Scholar]
- 11. Tsirigotaki A, Chatzi KE, Koukaki M, De Geyter J, Portaliou AG, Orfanoudaki G, Sardis MF, Trelle MB, Jorgensen TJD, Karamanou S et al (2018) Long‐lived folding intermediates predominate the targeting‐competent secretome. Structure 26: 695–707.e695 [DOI] [PubMed] [Google Scholar]
- 12. Zhou J, Dunker AK (2018) Regulating protein function by delayed folding. Structure 26: 679–681 [DOI] [PubMed] [Google Scholar]
- 13. Orfanoudaki G, Markaki M, Chatzi K, Tsamardinos I, Economou A (2017) MatureP: prediction of secreted proteins with exclusive information from their mature regions. Sci Rep 7: 3263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Park S, Liu G, Topping TB, Cover WH, Randall LL (1988) Modulation of folding pathways of exported proteins by the leader sequence. Science 239: 1033–1035 [DOI] [PubMed] [Google Scholar]
- 15. Beena K, Udgaonkar JB, Varadarajan R (2004) Effect of signal peptide on the stability and folding kinetics of maltose binding protein. Biochemistry 43: 3608–3619 [DOI] [PubMed] [Google Scholar]
- 16. Saio T, Guan X, Rossi P, Economou A, Kalodimos CG (2014) Structural basis for protein antiaggregation activity of the trigger factor chaperone. Science 344: 1250494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hartl FU, Bracher A, Hayer‐Hartl M (2011) Molecular chaperones in protein folding and proteostasis. Nature 475: 324–332 [DOI] [PubMed] [Google Scholar]
- 18. Hegde RS, Bernstein HD (2006) The surprising complexity of signal sequences. Trends Biochem Sci 31: 563–571 [DOI] [PubMed] [Google Scholar]
- 19. Bechtluft P, van Leeuwen RG, Tyreman M, Tomkiewicz D, Nouwen N, Tepper HL, Driessen AJ, Tans SJ (2007) Direct observation of chaperone‐induced changes in a protein folding pathway. Science 318: 1458–1461 [DOI] [PubMed] [Google Scholar]
- 20. Derman AI, Puziss JW, Bassford PJ Jr, Beckwith J (1993) A signal sequence is not required for protein export in prlA mutants of Escherichia coli . EMBO J 12: 879–888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Prinz WA, Spiess C, Ehrmann M, Schierle C, Beckwith J (1996) Targeting of signal sequenceless proteins for export in Escherichia coli with altered protein translocase. EMBO J 15: 5209–5217 [PMC free article] [PubMed] [Google Scholar]
- 22. Ullers RS, Ang D, Schwager F, Georgopoulos C, Genevaux P (2007) Trigger Factor can antagonize both SecB and DnaK/DnaJ chaperone functions in Escherichia coli . Proc Natl Acad Sci USA 104: 3101–3106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chatzi KE, Sardis MF, Karamanou S, Economou A (2013) Breaking on through to the other side: protein export through the bacterial Sec system. Biochem J 449: 25–37 [DOI] [PubMed] [Google Scholar]
- 24. Sala A, Bordes P, Genevaux P (2014) Multitasking SecB chaperones in bacteria. Front Microbiol 5: 666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Randall LL, Crane JM, Liu G, Hardy SJ (2004) Sites of interaction between SecA and the chaperone SecB, two proteins involved in export. Protein Sci 13: 1124–1133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Castanie‐Cornet MP, Bruel N, Genevaux P (2014) Chaperone networking facilitates protein targeting to the bacterial cytoplasmic membrane. Biochim Biophys Acta 1843: 1442–1456 [DOI] [PubMed] [Google Scholar]
- 27. Hoffmann A, Bukau B, Kramer G (2010) Structure and function of the molecular chaperone Trigger Factor. Biochim Biophys Acta 1803: 650–661 [DOI] [PubMed] [Google Scholar]
- 28. Crooke E, Guthrie B, Lecker S, Lill R, Wickner W (1988) ProOmpA is stabilized for membrane translocation by either purified E. coli trigger factor or canine signal recognition particle. Cell 54: 1003–1011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lecker S, Lill R, Ziegelhoffer T, Georgopoulos C, Bassford PJ Jr, Kumamoto CA, Wickner W (1989) Three pure chaperone proteins of Escherichia coli–SecB, trigger factor and GroEL–form soluble complexes with precursor proteins in vitro . EMBO J 8: 2703–2709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Niwa T, Kanamori T, Ueda T, Taguchi H (2012) Global analysis of chaperone effects using a reconstituted cell‐free translation system. Proc Natl Acad Sci USA 109: 8937–8942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bakshi S, Siryaporn A, Goulian M, Weisshaar JC (2012) Superresolution imaging of ribosomes and RNA polymerase in live Escherichia coli cells. Mol Microbiol 85: 21–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bornemann T, Holtkamp W, Wintermeyer W (2014) Interplay between trigger factor and other protein biogenesis factors on the ribosome. Nat Commun 5: 4180 [DOI] [PubMed] [Google Scholar]
- 33. Kaiser CM, Chang HC, Agashe VR, Lakshmipathy SK, Etchells SA, Hayer‐Hartl M, Hartl FU, Barral JM (2006) Real‐time observation of trigger factor function on translating ribosomes. Nature 444: 455–460 [DOI] [PubMed] [Google Scholar]
- 34. Merz F, Boehringer D, Schaffitzel C, Preissler S, Hoffmann A, Maier T, Rutkowska A, Lozza J, Ban N, Bukau B et al (2008) Molecular mechanism and structure of Trigger Factor bound to the translating ribosome. EMBO J 27: 1622–1632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Patzelt H, Rudiger S, Brehmer D, Kramer G, Vorderwulbecke S, Schaffitzel E, Waitz A, Hesterkamp T, Dong L, Schneider‐Mergener J et al (2001) Binding specificity of Escherichia coli trigger factor. Proc Natl Acad Sci USA 98: 14244–14249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ariosa A, Lee JH, Wang S, Saraogi I, Shan SO (2015) Regulation by a chaperone improves substrate selectivity during cotranslational protein targeting. Proc Natl Acad Sci USA 112: E3169–E3178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Singh R, Kraft C, Jaiswal R, Sejwal K, Kasaragod VB, Kuper J, Burger J, Mielke T, Luirink J, Bhushan S (2014) Cryo‐electron microscopic structure of SecA protein bound to the 70S ribosome. J Biol Chem 289: 7190–7199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Huber D, Jamshad M, Hanmer R, Schibich D, Doring K, Marcomini I, Kramer G, Bukau B (2017) SecA cotranslationally interacts with nascent substrate proteins in vivo . J Bacteriol 199: e00622‐16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Crooke E, Wickner W (1987) Trigger factor: a soluble protein that folds pro‐OmpA into a membrane‐assembly‐competent form. Proc Natl Acad Sci USA 84: 5216–5220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Guthrie B, Wickner W (1990) Trigger factor depletion or overproduction causes defective cell division but does not block protein export. J Bacteriol 172: 5555–5562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bechtluft P, Nouwen N, Tans SJ, Driessen AJ (2010) SecB–a chaperone dedicated to protein translocation. Mol BioSyst 6: 620–627 [DOI] [PubMed] [Google Scholar]
- 42. Huang C, Rossi P, Saio T, Kalodimos CG (2016) Structural basis for the antifolding activity of a molecular chaperone. Nature 537: 202–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Fekkes P, van der Does C, Driessen AJ (1997) The molecular chaperone SecB is released from the carboxy‐terminus of SecA during initiation of precursor protein translocation. EMBO J 16: 6105–6113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hartl FU, Lecker S, Schiebel E, Hendrick JP, Wickner W (1990) The binding cascade of SecB to SecA to SecY/E mediates preprotein targeting to the E. coli plasma membrane. Cell 63: 269–279 [DOI] [PubMed] [Google Scholar]
- 45. Baars L, Ytterberg AJ, Drew D, Wagner S, Thilo C, van Wijk KJ, de Gier JW (2006) Defining the role of the Escherichia coli chaperone SecB using comparative proteomics. J Biol Chem 281: 10024–10034 [DOI] [PubMed] [Google Scholar]
- 46. Ullers RS, Luirink J, Harms N, Schwager F, Georgopoulos C, Genevaux P (2004) SecB is a bona fide generalized chaperone in Escherichia coli . Proc Natl Acad Sci USA 101: 7583–7588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Fan D, Liu L, Zhu L, Peng F, Zhou Q, Liu C (2017) Global analysis of the impact of deleting trigger factor on the transcriptome profile of Escherichia coli . J Cell Biochem 118: 141–153 [DOI] [PubMed] [Google Scholar]
- 48. Calloni G, Chen T, Schermann SM, Chang HC, Genevaux P, Agostini F, Tartaglia GG, Hayer‐Hartl M, Hartl FU (2012) DnaK functions as a central hub in the E. coli chaperone network. Cell Rep 1: 251–264 [DOI] [PubMed] [Google Scholar]
- 49. Wang S, Yang CI, Shan SO (2017) SecA mediates cotranslational targeting and translocation of an inner membrane protein. J Cell Biol 216: 3639–3653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lee HC, Bernstein HD (2002) Trigger factor retards protein export in Escherichia coli . J Biol Chem 277: 43527–43535 [DOI] [PubMed] [Google Scholar]
- 51. Saio T, Kawagoe S, Ishimori K, Kalodimos CG (2018) Oligomerization of a molecular chaperone modulates its activity. Elife 7: e35731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hoffmann A, Becker AH, Zachmann‐Brand B, Deuerling E, Bukau B, Kramer G (2012) Concerted action of the ribosome and the associated chaperone trigger factor confines nascent polypeptide folding. Mol Cell 48: 63–74 [DOI] [PubMed] [Google Scholar]
- 53. Liu CP, Perrett S, Zhou JM (2005) Dimeric trigger factor stably binds folding‐competent intermediates and cooperates with the DnaK‐DnaJ‐GrpE chaperone system to allow refolding. J Biol Chem 280: 13315–13320 [DOI] [PubMed] [Google Scholar]
- 54. Bechtluft P, Kedrov A, Slotboom DJ, Nouwen N, Tans SJ, Driessen AJ (2010) Tight hydrophobic contacts with the SecB chaperone prevent folding of substrate proteins. Biochemistry 49: 2380–2388 [DOI] [PubMed] [Google Scholar]
- 55. Lecker SH, Driessen AJ, Wickner W (1990) ProOmpA contains secondary and tertiary structure prior to translocation and is shielded from aggregation by association with SecB protein. EMBO J 9: 2309–2314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kleinschmidt JH (2003) Membrane protein folding on the example of outer membrane protein A of Escherichia coli . Cell Mol Life Sci 60: 1547–1558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Bukau B, Deuerling E, Pfund C, Craig EA (2000) Getting newly synthesized proteins into shape. Cell 101: 119–122 [DOI] [PubMed] [Google Scholar]
- 58. Gouridis G, Karamanou S, Koukaki M, Economou A (2010) In vitro assays to analyze translocation of the model secretory preprotein alkaline phosphatase. Methods Mol Biol 619: 157–172 [DOI] [PubMed] [Google Scholar]
- 59. Maier R, Eckert B, Scholz C, Lilie H, Schmid FX (2003) Interaction of trigger factor with the ribosome. J Mol Biol 326: 585–592 [DOI] [PubMed] [Google Scholar]
- 60. Tsirigotaki A, Papanastasiou M, Trelle MB, Jorgensen TJ, Economou A (2017) Analysis of translocation‐competent secretory proteins by HDX‐MS. Methods Enzymol 586: 57–83 [DOI] [PubMed] [Google Scholar]
- 61. Gouridis G, Karamanou S, Gelis I, Kalodimos CG, Economou A (2009) Signal peptides are allosteric activators of the protein translocase. Nature 462: 363–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Gelis I, Bonvin AM, Keramisanou D, Koukaki M, Gouridis G, Karamanou S, Economou A, Kalodimos CG (2007) Structural basis for signal‐sequence recognition by the translocase motor SecA as determined by NMR. Cell 131: 756–769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Breukink E, Nouwen N, van Raalte A, Mizushima S, Tommassen J, de Kruijff B (1995) The C terminus of SecA is involved in both lipid binding and SecB binding. J Biol Chem 270: 7902–7907 [DOI] [PubMed] [Google Scholar]
- 64. Hunt JF, Weinkauf S, Henry L, Fak JJ, McNicholas P, Oliver DB, Deisenhofer J (2002) Nucleotide control of interdomain interactions in the conformational reaction cycle of SecA. Science 97: 2018–2026 [DOI] [PubMed] [Google Scholar]
- 65. Karamanou S, Sianidis G, Gouridis G, Pozidis C, Papanikolau Y, Papanikou E, Economou A (2005) Escherichia coli SecA truncated at its termini is functional and dimeric. FEBS Lett 579: 1267–1271 [DOI] [PubMed] [Google Scholar]
- 66. Wegrzyn RD, Deuerling E (2005) Molecular guardians for newborn proteins: ribosome‐associated chaperones and their role in protein folding. Cell Mol Life Sci 62: 2727–2738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Ries F, Carius Y, Rohr M, Gries K, Keller S, Lancaster CRD, Willmund F (2017) Structural and molecular comparison of bacterial and eukaryotic trigger factors. Sci Rep 7: 10680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Bowers CW, Lau F, Silhavy TJ (2003) Secretion of LamB‐LacZ by the signal recognition particle pathway of Escherichia coli . J Bacteriol 185: 5697–5705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Oh E, Becker AH, Sandikci A, Huber D, Chaba R, Gloge F, Nichols RJ, Typas A, Gross CA, Kramer G et al (2011) Selective ribosome profiling reveals the cotranslational chaperone action of trigger factor in vivo . Cell 147: 1295–1308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Papanastasiou M, Orfanoudaki G, Koukaki M, Kountourakis N, Sardis MF, Aivaliotis M, Karamanou S, Economou A (2013) The Escherichia coli peripheral inner membrane proteome. Mol Cell Proteomics 12: 599–610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Reusch RN (2012) Insights into the structure and assembly of Escherichia coli outer membrane protein A. FEBS J 279: 894–909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. de Keyzer J, van der Does C, Kloosterman TG, Driessen AJ (2003) Direct demonstration of ATP‐dependent release of SecA from a translocating preprotein by surface plasmon resonance. J Biol Chem 278: 29581–29586 [DOI] [PubMed] [Google Scholar]
- 73. Teter SA, Houry WA, Ang D, Tradler T, Rockabrand D, Fischer G, Blum P, Georgopoulos C, Hartl FU (1999) Polypeptide flux through bacterial Hsp70: DnaK cooperates with trigger factor in chaperoning nascent chains. Cell 97: 755–765 [DOI] [PubMed] [Google Scholar]
- 74. Arifuzzaman M, Maeda M, Itoh A, Nishikata K, Takita C, Saito R, Ara T, Nakahigashi K, Huang HC, Hirai A et al (2006) Large‐scale identification of protein‐protein interaction of Escherichia coli K‐12. Genome Res 16: 686–691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Eser M, Ehrmann M (2003) SecA‐dependent quality control of intracellular protein localization. Proc Natl Acad Sci USA 100: 13231–13234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Nishiyama K, Tokuda H (2010) Preparation of a highly translocation‐competent proOmpA/SecB complex. Protein Sci 19: 2402–2408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Zhou Q, Sun S, Tai P, Sui SF (2012) Structural characterization of the complex of SecB and metallothionein‐labeled proOmpA by cryo‐electron microscopy. PLoS One 7: e47015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Tang YC, Chang HC, Roeben A, Wischnewski D, Wischnewski N, Kerner MJ, Hartl FU, Hayer‐Hartl M (2006) Structural features of the GroEL‐GroES nano‐cage required for rapid folding of encapsulated protein. Cell 125: 903–914 [DOI] [PubMed] [Google Scholar]
- 79. Clerico EM, Tilitsky JM, Meng W, Gierasch LM (2015) How hsp70 molecular machines interact with their substrates to mediate diverse physiological functions. J Mol Biol 427: 1575–1588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Vandenberk N, Karamanou S, Portaliou AG, Zorzini V, Hofkens J, Hendrix J, Economou A (2019) The preprotein binding domain of SecA displays intrinsic rotational dynamics. Structure 27: 90–101.e106 [DOI] [PubMed] [Google Scholar]
- 81. Ernst I, Haase M, Ernst S, Yuan S, Kuhn A, Leptihn S (2018) Large conformational changes of a highly dynamic pre‐protein binding domain in SecA. Commun Biol 1: 130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Oliver DB, Cabelli RJ, Dolan KM, Jarosik GP (1990) Azide‐resistant mutants of Escherichia coli alter the SecA protein, an azide‐sensitive component of the protein export machinery. Proc Natl Acad Sci USA 87: 8227–8231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Paetzel M (2014) Structure and mechanism of Escherichia coli type I signal peptidase. Biochim Biophys Acta 1843: 1497–1508 [DOI] [PubMed] [Google Scholar]
- 84. Chang CN, Blobel G, Model P (1978) Detection of prokaryotic signal peptidase in an Escherichia coli membrane fraction: endoproteolytic cleavage of nascent f1 pre‐coat protein. Proc Natl Acad Sci USA 75: 361–365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Gouridis G, Karamanou S, Sardis MF, Scharer MA, Capitani G, Economou A (2013) Quaternary dynamics of the SecA motor drive translocase catalysis. Mol Cell 52: 655–666 [DOI] [PubMed] [Google Scholar]
- 86. Karamanou S, Vrontou E, Sianidis G, Baud C, Roos T, Kuhn A, Politou AS, Economou A (1999) A molecular switch in SecA protein couples ATP hydrolysis to protein translocation. Mol Microbiol 34: 1133–1145 [DOI] [PubMed] [Google Scholar]
- 87. Sianidis G, Karamanou S, Vrontou E, Boulias K, Repanas K, Kyrpides N, Politou AS, Economou A (2001) Cross‐talk between catalytic and regulatory elements in a DEAD motor domain is essential for SecA function. EMBO J 20: 961–970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Wowor AJ, Yan Y, Auclair SM, Yu D, Zhang J, May ER, Gross ML, Kendall DA, Cole JL (2014) Analysis of SecA dimerization in solution. Biochemistry 53: 3248–3260 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Data Availability Statement
All raw data used for figures and expanded view figures are provided as source data files.