

Abstract
Allograft inflammatory factor-1 (AIF-1) is a cytoplasmic, scaffold signal transduction protein constitutively expressed in inflammatory cells, but inducible in vascular smooth muscle cells (VSMCs) in response to injury or inflammatory stimuli. Although several basic science and population studies have reported increased AIF-1 expression in human and experimental atherosclerosis, a direct causal effect of AIF-1 expression on development of atherosclerosis has not been reported. The purpose of this study is to establish a direct relationship between AIF-1 expression and development of atherosclerosis. AIF-1 expression is detected VSMC in atherosclerotic lesions from ApoE−/− mice, but not normal arteries from wild-type mice. AIF-1 expression can be induced in cultured VSMC by stimulation with oxidized LDL (ox-LDL). Transgenic mice in which AIF-1 expression is driven by the G/C modified SM22 alpha promoter to restrict AIF-1 expression to VSMC develop significantly increased atherosclerosis compared with wild-type control mice when fed a high-fat diet (P = 0.022). Cultured VSMC isolated from Tg mice demonstrated significantly increased migration in response to ox-LDL compared with matched controls (P < 0.001). VSMC isolated from Tg mice and cultured human VSMC which over express AIF-1 demonstrated increased expression of MMP-2 and MMP-9 mRNA and protein and increased NF-κB activation in response to ox-LDL as compared with wild-type control mice. VSMC which over express AIF-1 have significantly increased uptake of ox-LDL, and increased CD36 expression. Together, these data suggest a strong association between AIF-1 expression, NF-κB activation, and development of experimental atherosclerosis.
Keywords: Allograft inflammatory factor-1, Atherosclerosis, Vascular smooth muscle cell, Nuclear factor kappa B, Oxidized LDL
1. Introduction
Vascular smooth muscle cells (VSMC) play an important role in development and progression of atherosclerosis. In early lesions, the VSMC response to cytokines synthesized by activated immune cells is manifested many ways. VSMC can migrate into the intima and proliferate, creating a cap between the lumen of the vessel and the lipid laden core of the lesion [1–3]. VSMC can synthesize chemokines and adhesion molecules which enhance inflammatory cell transmigration into the lesion, thus propagating development of the growing plaque. VSMC can take up LDL in various forms, with subsequent autocrine activating effects [4–6]. Oxidized LDL can induce growth factor and inflammatory cytokine expression, leading to VSMC proliferation and inflammatory cell recruitment [6–8]. Collectively, these studies suggest that ox-LDL can activate VSMC directly by activation of VSMC signal transduction cascades, and indirectly, thorough autocrine and paracrine cytokine production. Molecules which mediate signal transduction cascades in response to cytokines and ox-LDL in VSMC are potentially important targets of anti-atherosclerotic modalities. Proteins which are inducibly expressed in response to injury or inflammation make even more specific targets of these therapies.
Allograft inflammatory factor-1 (AIF-1) is constitutively expressed in lymphoid tissue but not expressed in unstimulated arteries or cultured VSMC, but is rapidly induced in vivo in response to injury, and in cultured VSMC by inflammatory cytokines [9]. AIF-1 is a cytoplasmic, calcium binding protein, with molecular signatures of a scaffold-signaling protein, including several PDZ interaction domains, which are important in mediating interactions of multi-protein complexes. We have determined an important role for AIF-1 in VSMC pathophysiology in several vascular injury models. Our previous study has shown that chronic over expression of AIF-1 in transgenic mice increases intimal hyperplasia in response to carotid artery ligation injury [10]. Abrogation of AIF-1 expression by adenoviral delivery of AIF-1-specific siRNA significantly decreases intimal hyperplasia in the rat carotid artery balloon angioplasty model [11]. AIF-1 expression is induced in allografted arteries in a rat model of carotid allograft, and persistent expression of AIF-1 in allografted human hearts correlates with development of transplant restenosis [9,12]. Several basic science and human population studies have described AIF-1 expression in atherosclerotic tissue. For example, AIF-1 mRNA expression was significantly increased in human atherosclerotic lesions versus healthy sections of aorta [13]. Transgenic mice in which AIF-1 over expression was driven by the macrophage-specific CD11b promoter developed increased number and size of atherosclerotic lesions [14]. It was concluded that this was due primarily to increased phagocytotic activity of macrophage in these mice. Similarly, we previously found that knock down of AIF-1 in macrophage reduces their migration and proliferation in response to ox-LDL [15]. In a study designed to identify genomic and epigenetic DNA alterations in human atherosclerotic plaque, microsatellite loss of heterozygosity was found in chromosome 6p21.3, corresponding to the promoter region of AIF-1 gene[16]. Rearrangements in this region significantly correlated with atherosclerotic risk.
While these results suggest that AIF-1 is an inflammation-responsive signaling protein that plays a central role in regulation of VSMC activation and development of vasculopathies, a causal relationship between AIF-1 expression and development of atherosclerosis in vivo has not been established. Further, the cellular pathways and molecular mechanisms responsible for AIF-1 activity in oxidized LDL-stimulated VSMC has not been characterized. We hypothesized that AIF-1 would be involved in VSMC involvement in atherosclerosis generation in mice. Because AIF-1 is constitutively expressed in leukocytes, we utilized a smooth muscle cell specific transgenic mouse in which AIF-1 expression is driven the G/C-modified SM22α promoter [17], which we have previously utilized to determine AIF-1 role in ligation injury [10]. The specific role of AIF-1 in VSMC development and progression of atherosclerosis is currently unknown. This report shows the effects of AIF-1 over expression in VSMC and development of atherosclerosis, LDL uptake, and NF-κB activation.
2. Materials and methods
2.1. Generation of AIF-1 transgenic mouse and induction of atherosclerosis
The G/C-modified SM22α promoter construct, identification of founder mice, AIF-1 VSMC-restricted expression, and breeding has been described previously [10]. This promoter has been modified such that expression is maintained even in the context of injury in which VSMC de-differentiate, and has been successfully used in several studies [10,17]. Age and gender-matched wild-type and transgenic littermates in the C57Bl6 background were used for these studies. Mice were fed high fat pro-inflammatory chow diet (21% fat, 1.25% cholesterol, 0.5% cholate) for 16 weeks then sacrificed.
2.2. En face staining and quantification of aortic lesions
Abdominal aortas were en face stained for atherosclerotic lesions with Sudan IV as previously described [18]. Briefly, mice were anesthetized with 0.05 cc of a 1:1 mixture of ketamine and xylazine by intramuscular injection then perfusion fixed with 20 mL PBS followed by 20 mL of 10% neutral-buffered formalin. Adventitial fat was removed from the aortic arch to the iliac bifurcation, arteries were filleted open and pinned to a dissecting tray then fixed in 10% neutral-buffered formalin overnight at room temperature. Arteries were stained with Sudan IV (0.5, w/v), and mounted to slides. Photographs were taken with a Spot Diagnostic Instruments camera at 10× magnification. Fatty lesion area was quantified using Image Pro Plus software and expressed as a percent of the total aorta area for each mouse. Values were compared using a student’s t-test.
2.3. Immunohistochemistry
Cross sections from aorta from 24-week-old ApoE−/− mice fed standard-chow were incubated with biotinylated secondary antibody (1:200) followed by avidin–biotin–peroxidase complex in a Vectastain Elite kit (both from Vector Labs, Burlingame, CA). The reaction product was visualized with DAB (Vector Labs) used as the chromogenic substrate, which produces a reddish-brown stain, and counterstained with hematoxylin. For immunofluorescence, primary antibody incubation was followed by a 30-min incubation with secondary antibody conjugated to AlexaFluor 568 (red) and AlexaFluor 488 (green) (Molecular Probes Inc., Eugene, OR).
2.4. VSMC culture, proliferation, and migration
VSMCs were isolated from wild-type and transgenic mouse abdominal aortas, and human VSMC cultured as previously described [9,10]. Human VSMC were transduced with 30 MOI AdAIF-1 or AdLacZ adenovirus as we described [11]. Migration assays were performed as described [10,11]. Briefly, 6.5 mm-diameter Transwell Boyden chamber plates (Costar) with 8-μm polycarbonate membrane pore size were seeded with VSMC in medium containing 0.5% FCS. PDGF at 20 mg/ml was placed in the lower chamber and cells were incubated for 2 h at 37 °C, at which time cells were fixed and stained. VSMC that migrated to the lower surface of the membrane were quantitated by counting 4 high powered fields per membrane. Experiments were performed in triplicate from VSMC isolated from three different transgenic and wild-type mice.
2.5. RT-PCR, Western blotting, and cellular fractionation
RNA isolation and qRT-PCR, were performed as previously described [9]. Briefly, total RNA was isolated from VSMC, isolated with Tri-Reagent (MRC Inc., Cincinnati, OH), reverse transcribed using random hexamers and PCR amplified using an Eppendorf Realplex4 Mastercycler as described [19]. Multiple mRNAs (Ct values) were quantitated simultaneously by the software. Primer pairs were as follows: GAPDH: forward: CGA CAG TCA GCC GCA TCT T, reverse: CCC CAT GGT GTC TGA GCG; SRA(1) forward: GAT TGG GAA CAT TCT CAG ACC TT, reverse: CTT GTC CAA AGT GAG CTG CCT T; CD36 forward: GAG AAC TGT TAT GGG GCT AT, reverse: ACA GAC CAA CTG TGG TAG; CXCL16 forward: GAG CTC ACT CGT CCC AAT GAA, reverse, GGA GAG GGC TGA GGT GGG GG; MMP2 forward: TCC AGG GGA CAT CCT ATG ACA G, reverse; TCT CAG GGG AGA AGC CAT ACT TC; MMP9 forward: CTC CAC ACC TCA GAA CCA ATC T; reverse: CAC CCG AGT GTA ACC ATA GGG. VSMC extracts and blotting was performed as described previously [9,19]. Briefly, after SDS-PAGE and transfer, membranes were incubated with a 1:3000–5000 dilution of primary antibody, and a 1:5000 dilution of secondary antibody. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH), Iκb alpha, NF-κB subunit p65, histone 2A, and secondary antibody were from Cell Signaling Inc. AIF-1 antibody has been described [9]. MMP2, MMP9, and CD36 antibody were from AbCam Inc. Cellular fractionation for NF-κB translocation was performed as we previously described [19]. Reactive proteins were visualized using enhanced chemiluminescence (Amersham) according to manufacturer’s instructions. Relative intensity of bands was normalized to GAPDH and SMC actin, and quantitated by scanning image analysis and the Image J densitometry program.
2.6. Flow cytometry for lipid uptake
Uptake of fluorescent-labeled oxidized LDL was measured by flow cytometry using DiI-ox-LDL(Kalen Biomedical Inc.) as described [20]. Briefly, hVSMC transduced by either AdAIF-1 or AdLacZ were grown to 70% confluence. After 24 h, they were serum starved in 0.1% serum-containing media for 48 h. VSMC were then incubated in the dark with DiI-ox-LDL (5 μg/ml) for 2 h at 37 °C. After incubation VSMC were washed 2× with PBS, trypsinized, harvested with 1% BSA in PBS and subjected to flow cytometry analysis (FACS Calibur, Becton Dickinson). The mean of DiI-ox-LDL fluorescence intensity was obtained from 10,000 cells. DCF fluorescence was read on FL2(525–625) in log scale.
2.7. Statistics
Results are expressed as mean±SE. Differences between groups were evaluated with the use of ANOVA to evaluate differences between individual mean values or by t tests where appropriate. Differences were considered significant at a level of P < 0.05.
3. Results
AIF-1 is induced in atherosclerotic lesions and induced by oxidized LDL in cultured VSMC. We have previously described inducible expression of AIF-1 in angioplasty and ligation-injured arteries, and in cultured VSMC by stimulation with serum and select cytokines. To determine if AIF-1 expression was induced by atherogenic stimuli, two experiments were performed. First, immunohistochemistry using anti-AIF-1 antibody was performed using aorta from 24-week-old ApoE−/− mice fed standard-chow. Fig. 1A shows abundant positive immunoreactivity in medial and intimal VSMC, and very little in a murine artery from a normal non-atherosclerosis-susceptible mouse. Dual-color immunofluorescence was used to co-localize AIF-1 expression with cell types present in the lesion. Fig. 1B shows that AIF-1 is expressed in both medial and cap VSMC. AIF-1 also co-localizes with CD45-positive cells in the lesion (Supplemental Data Fig. 1). Most notable was AIF-1 expression in medial and cap VSMC in the lesion. To determine if atherosclerotic stimuli could induce AIF-1 expression in VSMC in a more controlled environment, we challenged cultured, primary human VSMC with 30 μg/ml oxidized LDL (ox-LDL) for various times. Fig. 1C shows that AIF-1 protein expression is induced by ox-LDL in VSMC.
Fig. 1.

AIF-1 expression is induced by atherogenic stimuli. (A) AIF-1 is not expressed in a normal mouse carotid artery, but is detected in VSMC in lesion from aortic arch of ApoE−/− mouse fed normal chow. Red-brown indicates positive staining. Image is 400×. (B) Fluorescence immunohistochemistry in above lesion. Co-localization of AIF-1antibody (red) with cap and medial VSMC (green), merged, and negative control. Arrows point to examples of co-positive VSMC. 600× magnification for all. (C) Induction ofAIF-1 protein expression in cultured human VSMC by ox-LDL. Human coronary artery VSMC were serum-starved for 48 h, then stimulated with 30 μg ox-LDL for the indicated times. Extracts were blotted with anti-AIF-1 and GAPDH (loading control) antibodies. Western Blot shown is representative.
AIF-1 transgenic mice develop increased atherosclerotic lesions. To determine a causal role for AIF-1 expression in atherosclerosis, we utilized the previously described AIF-1 transgenic mouse. In this mouse, AIF-1 expression is driven by the G/C-modified SM22α promoter to restrict AIF-1 over expression to arterial VSMC [10]. This was necessary because as AIF-1 is constitutively expressed in immune cells, and it was important to demonstrate a VSMC-specific role for AIF-1 in development of atherosclerosis. The G/C-modified promoter is modified to not silence as VSMC de-differentiate [17]. AIF-1 transgene expression compared with wild-type mouse is shown in Supplemental Data Fig. 2. Wild-type and AIF-1 Tg mice were fed an atherogenic diet for 16 weeks, at which time the aortas of these mice were excised and stained with Sudan IV to quantify total lesion area. Fig. 2 shows that AIF-1 transgenic mice develop significantly increased plaque area compared with wild-type control mice, with 6.36 ± 1.2% vs. 2.53±0.7% aortic lesion area for AIF-1 Tg and matched controls, respectively (n = 9, P = 0.021). Serum lipid profile was determined, and total plasma cholesterol and triglyceride levels were statistically similar between the two groups of mice (Fig. 2C). Serum triglyceride for wild-type mice was 60.8 ± 7.4 mg/dl compared with 56.3 ± 6.6 for AIF-1 transgenic. Serum cholesterol for wild-type mice was±109.6 ± 10.7 mg/dl compared with 106.2 ± 10.4 for AIF-1 Tg.
Fig. 2.
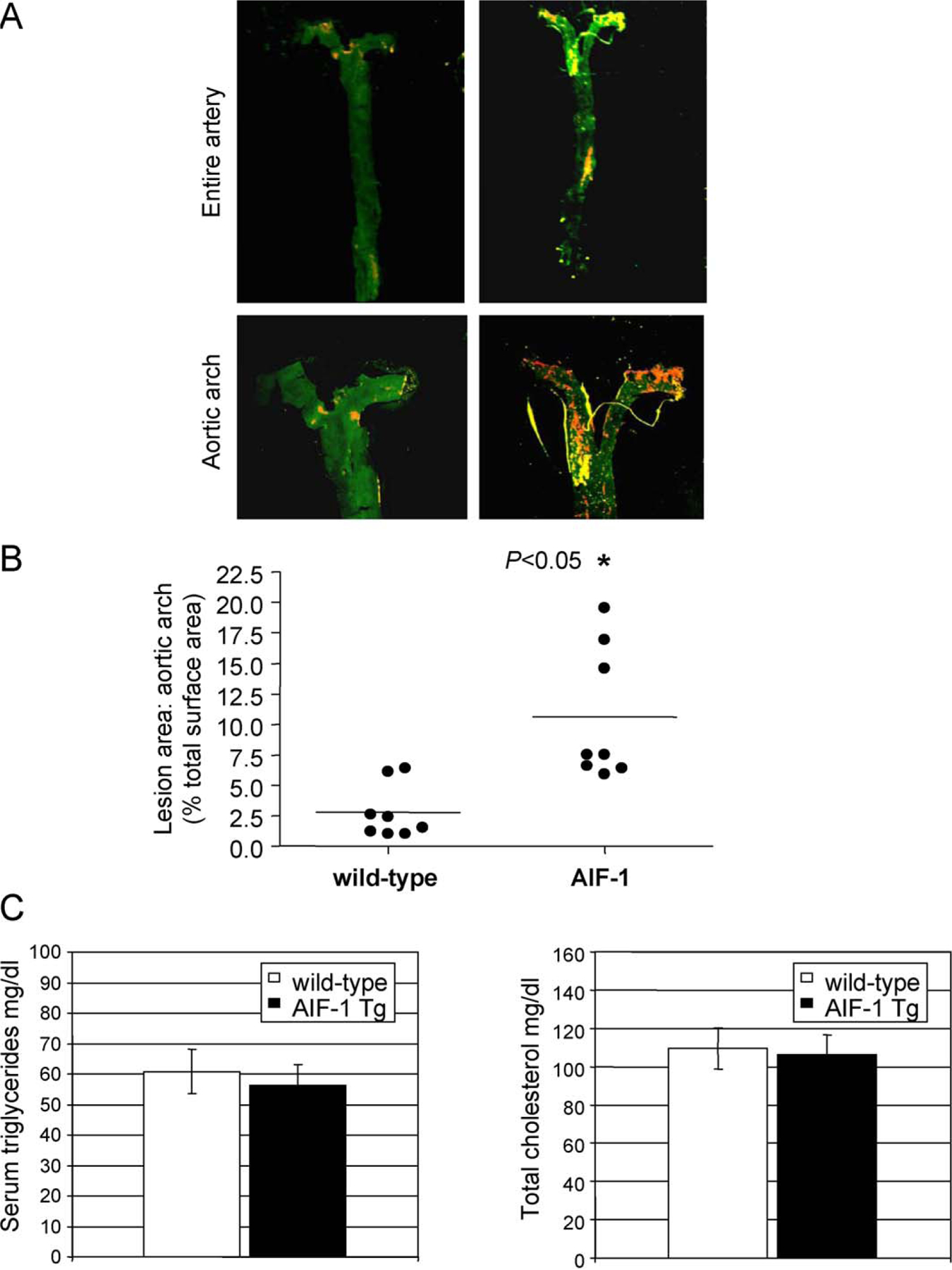
AIF-1 expression increases atherosclerosis. (A) Representative pseudo-colored photographs of Sudan IV stained abdominal aorta and higher magnification of aortic arch. Yellow and orange areas represent atherosclerotic lesions. (B) Quantification of lesion size. Age and sex-matched littermates were fed atherogenic diet for 16 weeks, and lesion size quantified by en face staining. P < 0.05 transgenic vs. wild-type littermates, n = 8. (C) Lipoprotein profiles do not differ between AIF-1 Tg and wild-type mice. Serum triglyceride and total cholesterol levels of AIF-1 Tg and WT mice. Differences between age and sex-matched groups were not significant for either lipid product. Triglyceride P = 0.3, cholesterol P = 0.8 (n = 8 per group). (For interpretation of the references to color in this figure legend, the reader is referred to the web version of the article.)
VSMC from AIF-1 transgenic mice migrate more rapidly in response to ox-LDL. Migration of VSMC in response to chemotactic factors is an important contributing factor to atherosclerotic lesion development, and oxidized lipids are chemotactic for VSMC [25]. To determine if AIF-1 expression influenced VSMC migration in response to ox-LDL, wild-type and VSMC isolated from transgenic mice were isolated, and seeded into Boyden chambers, and differences in chemotaxis quantitated by counting cells which migrated in response to 30 μg/ml ox-LDL. Fig. 3A demonstrates that cultured VSMC from both wild-type and transgenic mice migrated in response to ox-LDL. VSMC isolated from Tg mice demonstrated significantly increased migration, both in response to ox-LDL and in serum-free media compared with wild-type VSMC from matched controls (39.6 ± 2.5 vs. 93.0 ± 6.4 cells/HPF for wild-type and AIF-1 Tg, P < 0.001 for serum-free, and (58.4 ± 4 vs. 130.5 ± 5.1 cells/HPF for wild-type and AIF-1 Tg P < 0.001). AIF-1 Tg VSMC stimulated with ox-LDL migrate significantly more rapidly than those in serum free media (P = 0.02).
Fig. 3.
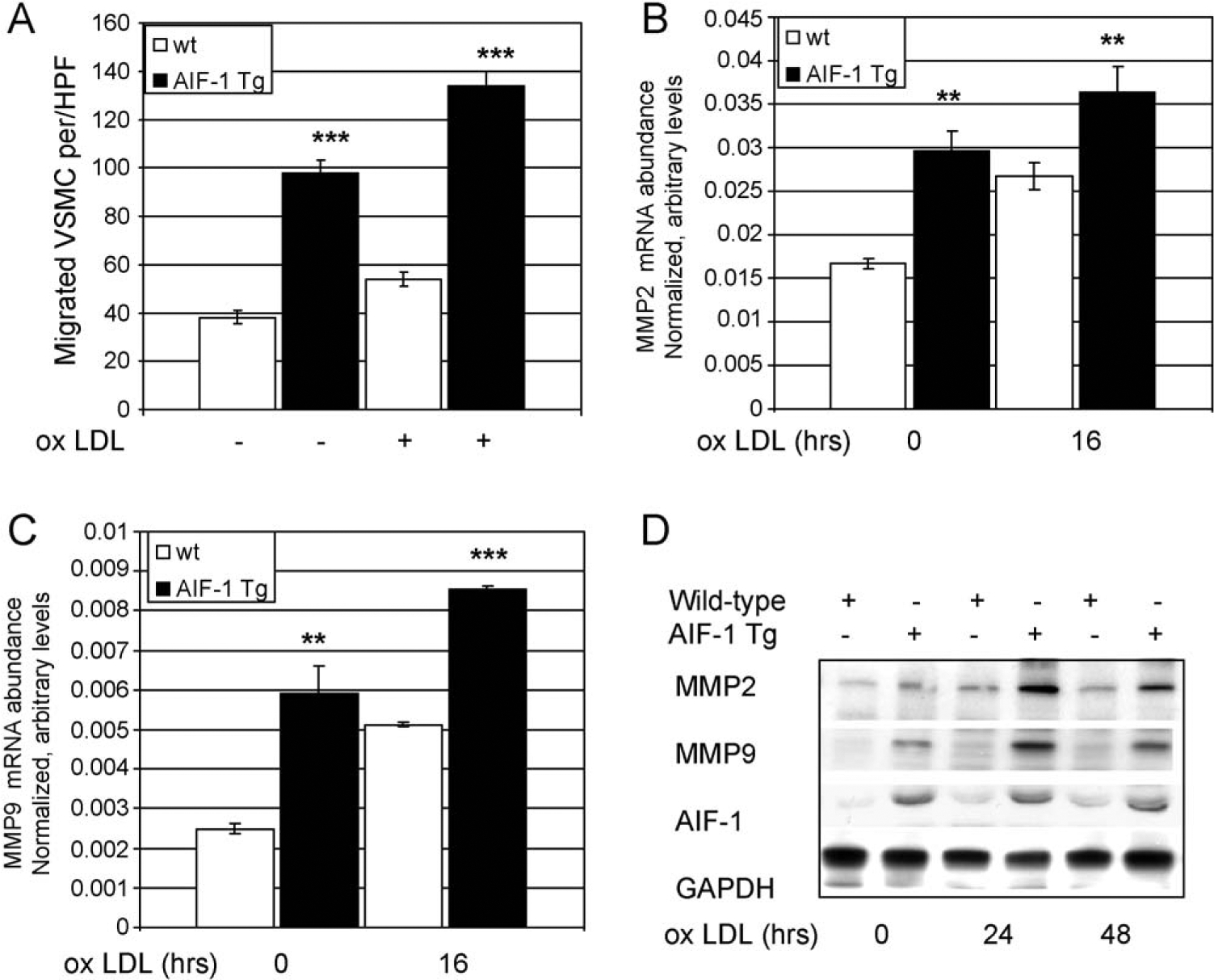
(A) VSMC from AIF-1 transgenic mice migrate more rapidly in response to ox-LDL. VSMC from transgenic and wild-type mice were seeded into Boyden chambers, and differences in chemotaxis quantitated by counting cells which migrated in response to 30 μg/ml ox-LDL and in serum-free media compared with wild-type VSMC from matched controls (P < 0.01). Data shown from at least three independently performed experiments. (B and C) VSMC from AIF-1 transgenic mice express significantly increased MMP2 and MMP9 mRNA compared with control VSMC (P < 0.01 or 0.001 for all time points) quantitated by quantitative RT-PCR. (D) MMP2 and MMP9 protein is increased in VSMC from AIF-1 transgenic mice compared with control VSMC. Lysates from untreated and ox-LDL stimulated VSMC were immunoblotted with the antibodies shown. Western Blot shown is representative of at least three.
VSMC which over express AIF-1 have increased matrix metalloproteinase (MMPs) expression. Matrix metalloproteinase (MMPs) expression is an important component of SMC migration [5,25]. To determine if AIF-1 expression influenced expression of MMP-2 and MMP-9, VSMC were stimulated with 30 μg/ml ox-LDL, and MMP-2 and MMP-9 mRNA expression quantitated by quantitative reverse transcription PCR (qRT-PCR). Fig. 3B and C shows that both MMP2 and MMP9 mRNA abundance are significantly increased in both serum-deprived and ox-LDL stimulated VSMC. This increase in mRNA expression was verified at the protein level as lysates from AIF-1 Tg VSMC have a greater abundance of MMP9 and MMP2 than do wild-type VSMC (Fig. 3D). It is interesting to note that both basal levels and ox-LDL-induced levels of mRNA and protein are increased in the AIF-1 Tg VSMC relative to wild-type.
VSMC which over express AIF-1 have increased NF-κB activation. Expression of numerous genes implicated in the response to injury and atherosclerosis are regulated by the transcription factor NF-κB, including MMP-2 and MMP-9 [3]. NF-κB can be activated by ox-LDL in VSMC [21]. We hypothesized that one mechanism whereby atherosclerotic burden was increased in AIF-1 Tg VSMC was by activation of NF-κB. For these experiments, VSMC were serum starved for 72 h, then stimulated with 30 μg/ml ox-LDL. IκB expression was determined by western blot. Fig. 4A shows that VSMC from AIF-1 Tg mice had increased basal levels of activated NF-κB, as determined by IκB degradation. Importantly, cultured human VSMC infected with AdAIF-1 also demonstrate increased NF-κB activation compared with Adenoviral transduced controls (Fig. 4B). As further verification of AIF-1-mediated NF-κB activation, VSMC from transgenic mice and AdAIF-1 transduced human VSMC were stimulated with ox-LDL, and NF-κB activation assayed by nuclear translocation of the p65 subunit of NF-κB. Fig. 4C shows that significantly more p65 localizes to the nucleus in VSMC which over express AIF-1 compared with control VSMC. Noteworthy is nuclear accumulation of p65 in unstimulated VSMC which over express AIF-1, suggesting constitutive activation of NF-κB in the absence of stimuli.
Fig. 4.
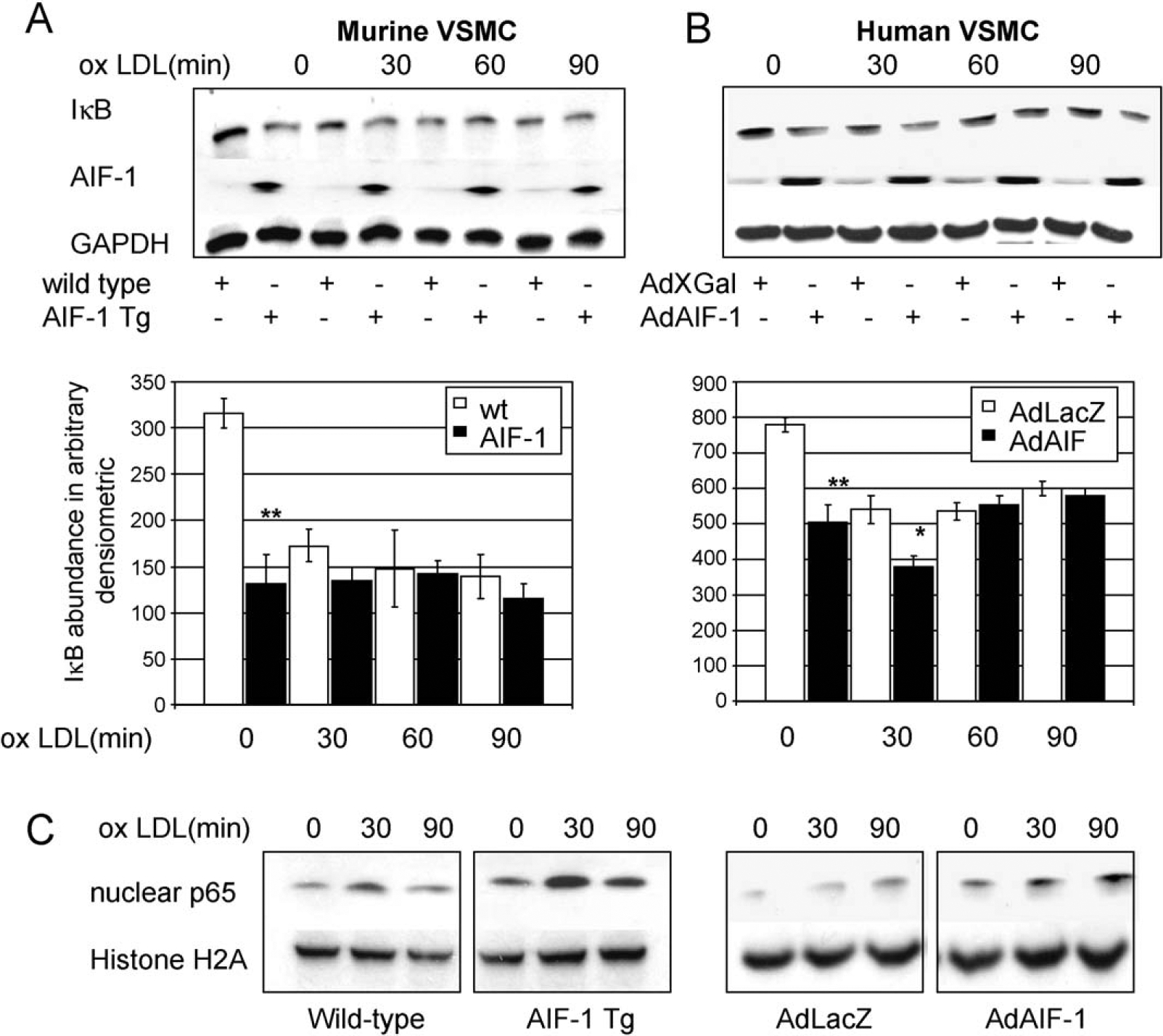
VSMC which over express AIF-1 have increased NF-κB activation. Representative western blots showing NF-κB activation in ox-LDL stimulated. (A) AIF-1 Tg and WT mouse VSMC, and (B) human VSMC transduced with AdAIF-1 or AdLacZ. Densitometry analysis from at least three independently performed experiments showed less IKBα in both AIF-1 Tg and AdAIF-1 transduced VSMC at baseline (P < 0.001) WT or adenoviral transduced controls. (C) Translocation of p65 subunit of NF-κB is increased in AIF-1 Tg VSMC and AdAIF-1 human VSMC compared with control VSMC. Lysates from unstimulated and ox-LDL stimulated VSMC were fractionated and nuclear fractions immunoblotted with antibody to p65 subunit of NF-κB, and histone H2A as a nuclear loading control. Western blot shown is representative of at least three.
VSMC which over express AIF-1 show increased lipid uptake. VSMC express receptors which internalize modified ox-LDL as ligand, accumulate large amounts of cholesterol esters, and become foam cells [1,22]. We hypothesized that AIF-1 expression would increase VSMC uptake of ox-LDL and contribute to foam cell formation. To test this hypothesis, human VSMC were transfected with 30 MOI of AIF-1 or LacZ adenovirus. After 48 h VSMC were starved in 0.1% FCS for 24 h, then incubated with 5 μg/ml of fluorescently labeled ox-LDL (Dil-ox-LDL) for 2 h. Uptake was quantitated by FACS analysis. Fig. 5A shows that VSMC which over express AIF-1 significantly increase their uptake of oxidized LDL by an average of 37% (417.7 ± 11.8 vs. 586.7 ± 15.4 mean fluorescent units for control vs. AdAIF-1, respectively (P < 0.01). This suggests that AIF-1 expression can increase oxidized lipid uptake in VSMC.
Fig. 5.

VSMC which over express AIF-1 show increased lipid uptake and increased CD36 mRNA abundance. (A) Human VSMC were transfected with 30 MOI of AIF-1 or LacZ adenovirus. After 48 h VSMC were starved in 0.1% FCS for 24 h, then incubated with 5 μg/ml of fluorescently labeled ox-LDL (Dil-ox-LDL) for 2 h. Uptake was quantitated by FACS analysis. (B) Adenoviral transduced human VSMC were treated as in “A”, at which time RNA was extracted and receptor abundance quantitated by quantitative RT-PCR. CD36 mRNA was significantly more abundant at all time points assayed, compared with control VSMC (P < 0.05 for all time points). (C) AdAIF-1 VSMC express more CD36 protein compared with AdLacZ controls. Lysates from unstimulated and ox-LDL stimulated VSMC were immunoblotted with the antibodies shown. Western Blot shown is representative of at least three.
VSMC which over express AIF-1 show increased CD36 mRNA abundance. VSMC, like macrophage, internalize ox-LDL by several receptors, including SRA(1), CD36, and CX-CL16 [22]. We hypothesized that one mechanism for increased ox-LDL uptake was by an increase in scavenger receptor abundance. Human VSMC were transfected with 30 MOI of AIF-1 or LacZ adenovirus. After 48 h VSMC were starved for 24 h, then incubated with 30 μg/ml ox-LDL for various times, at which time RNA was extracted and receptor abundance quantitated by quantitative RT-PCR. Fig. 5B shows that AIF-1 expression significantly increased mRNA abundance of CD36 at all time points assayed, compared with control cells. This increase in mRNA expression was verified at the protein level as lysates from AdAIF-1 cells demonstrate greater abundance of CD36 protein compared with AdLacZ controls (Fig. 5C). Interestingly, neither SRA(1) nor CXCL16 mRNA levels were significantly different between AdAIF-1 and AdLacZ transduced VSMC (Supplemental Data Fig. 3).
4. Discussion
The major findings of this report are that AIF-1 expression is induced in VSMC by atherogenic stimuli, and that constitutive AIF-1 expression in VSMC results in increased atherosclerotic burden in AIF-1 transgenic mice. We have previously shown that AIF-1 is expressed in macrophage in human atherosclerotic arteries, and that atherogenic stimuli induce AIF-1 expression in cultured, primary macrophages [15]. Others have shown that AIF-1 mRNA is detected in human atherosclerotic plaque [13]. Further, genomic rearrangements in the AIF-1 loci are associated with increased diagnosis of atherosclerosis [16]. A macrophage-specific AIF-1 transgenic mouse developed increased atherosclerotic lesions compared with control mice [14]. The present study extends all of those observations and indicates that transgenic mice in which AIF-1 is over expressed in VSMC develop significantly increased atherosclerotic lesions. This was not attributed to increased triglyceride concentration, as levels of triglyceride and cholesterol were the same in transgenic and control mice. This is important, and novel, as activated, synthetic VSMC are found in atherosclerotic lesions, and play an important role in the initiation and propagation of atherosclerosis [1–5]. For this study we utilized a previously developed a VSMC specific AIF-1 transgenic mouse in which AIF-1 expression is driven by the G/C modified SM22α promoter[10]. When ligated, these mice developed increased intimal hyperplasia in response to arterial injury [10]. These mice are in the C57Bl6 background, which develop plaque when fed a high-fat, pro-inflammatory diet [23,24]. It was important to utilize this mouse as AIF-1 is constitutively expressed in immune cells, and use of these mice would enable us to attribute any effects to VSMC-derived AIF-1, rather than modification of the immune response.
It was important to identify a mechanism for these in vivo effects. Oxidized LDL has different effects on different cell types. It is anti-proliferative to EC and mildly proliferative to monocytes, but is strongly proliferative to VSMC [8]. Interestingly, we did not see increased proliferation in response to ox-LDL, in spite of reports by ourselves and other groups that increased expression of AIF-1 is proliferative [10,11,26,27]. VSMC migration into the developing lesion is one of the earliest cellular events in plaque formation [12]. VSMC which over express AIF-1 migrate more rapidly in response to ox-LDL than do wild-type control VSMC. This is similar to what we have reported on increased migration in response to PDGF [28]. Interestingly, we observed no statistically significant differences in inflammatory cell infiltration between AIF-1 Tg and wild-type mice. Similarly, pro-inflammatory cytokine expression also was not increased in VSMC cultured from transgenic mice compared with wild-type mice. Together, these data implied that AIF-1 effects were not pro-inflammatory, and prompted us to investigate elsewhere for AIF-1 observed effects on VSMC activation and development of atherosclerosis.
An important aspect of the VSMC response to injury requires migration from the media to the lumen of the artery, which requires synthesis of proteases to degrade the extracellular matrix. Matrix metalloproteinase (MMPs) expression is an important component of SMC migration [5]. MMP-9 is increased in atherosclerotic plaque [3], and inhibition of MMP9 decreases VSMC migration in response to PDGF [25]. The increase in MMP2 and MMP9 mRNA expression led us to investigate if AIF-1 expression led to activation of a transcription factor or factors.
Many cellular responses to oxidized lipids and inflammatory stimuli are mediated by the transcription factor NF-κB, and NF-κB contributes to regulation of expression of MMP2 and MMP9 [21]. One report suggests that in a cancer cell line, AIF-1 expression is associated with activation of NF-κB, and we hypothesized that this transcription factor would be activated in AIF-1 transgenic VSMC[26]. Constitutive over expression of AIF-1 leads to increased activation of NF-κB in VSMC isolated from transgenic mice. Further, human VSMC transduced with AIF-1 adenovirus show a similar increase in NF-κB activation, suggesting a conservation of AIF-1 function among species. Most interesting in this experiment was the observation that NF-κB is activated in serum-deprived, unstimulated VSMC, in both transgenic mouse and human VSMC. This is reminiscent of previous studies, in which we observed that AIF-1 over expression results in VSMC in a primed, activated state. In these studies, even when VSMC are serum-starved, AIF-1 over expressing VSMC continue to proliferate, and basal levels of proliferative proteins such as cyclins are detectible [27]. Further, low levels of p38 MAPK were activated in AIF-1 transgenic VSMC [10]. While the precise mechanisms of AIF-1 activation of NF-κB remain unclear, the observation of constitutive NF-κB activation are consistent with these prior reports, and together provide a potential mechanism for increased migration and expression of MMP2 and MMP9.
VSMC express receptors for lipid and can form foam cells, thus affecting the lipid content of the atherosclerotic plaque. VSMC which over express AIF-1 have increased uptake of oxidized LDL. A potential mechanism may be increased abundance of CD36 mRNA in AIF-1 over expressing VSMC. While this is likely not the only mechanism whereby AIF-1 expression increases lipid uptake into VSMC, it may well be a contributing factor. In macrophage, CD36 is a major receptor for ox-LDL internalization and is required for formation of foam cells [29,30]. Scavenger receptor knock out mice have reduced atherosclerosis [1,29]. CD36 engagement can also induce signal transduction events, including NF-κB activation [22]. Agents which reduce NF-κB activity result in a decrease in CD36 expression[30]. This increase in CD36 mRNA abundance may be consistent with transcriptional activation by NF-κB. Future studies need to determine if the increased CD36 is responsible for increased NF-κB activity, or augmented NF-κB activation is responsible for increased CD36 mRNA abundance.
In summary, there are several novel points to this study; first, AIF-1 expression is increased in VSMC in atherosclerotic plaque; second, VSMC-restricted over expression of AIF-1 leads to increased atherosclerotic plaque in AIF-1 transgenic mice; third, constitutive expression of AIF-1 leads to increase VSMC chemotaxis to oxidized LDL, increased NF-κB activation and lipid uptake. Together, the significance of these data indicate that the intracellular signaling protein AIF-1 is an important target of anti-atherosclerotic modalities, and that VSMC activation can contribute to the development of atherosclerosis.
Supplementary Material
Funding
This work was supported by grant HL-63810 from the National Heart Lung, and Blood Institute; grant 0455562U from the American Heart Association; and grant 146643428 from the Roche Organ Transplant Research Foundation, to M.V.A.
Footnotes
Appendix A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.atherosclerosis.2011.07.095.
References
- [1].Doran AC, Meller N, McNamara CA. Role of smooth muscle cells in the initiation and early progression of atherosclerosis. Arterioscler Thromb Vasc Biol 2008;5:812–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Pei H, Wang Y, Miyoshi T, et al. Direct evidence for a crucial role of the arterial wall in control of atherosclerosis susceptibility. Circulation 2006;114:2382–9. [DOI] [PubMed] [Google Scholar]
- [3].Galis ZS, Sukhova GK, Lark MW, Libby P. Increased expression of matrix metalloproteinases and matrix degrading activity in vulnerable regions of human atherosclerotic plaques. J Clin Invest 1994;94:2493–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Libby P Inflammation in atherosclerosis. Nature 2002;420:a868–74. [DOI] [PubMed] [Google Scholar]
- [5].Ross R Atherosclerosis—an inflammatory disease. N Engl J Med 1999;340:115–26. [DOI] [PubMed] [Google Scholar]
- [6].Raines E, Ferri N. Cytokines affecting endothelial and smooth muscle cells in vascular disease. J Lipid Res 2005;46:1081–92. [DOI] [PubMed] [Google Scholar]
- [7].Singer C, Sonemany S, Baker K, Gerthoffer W. Synthesis of immune modulators by smooth muscles. BioEssays 2004;26:646–55. [DOI] [PubMed] [Google Scholar]
- [8].Chisolm GM 3rd, Chai Y. Regulation of cell growth by oxidized LDL. Free Radic Biol Med 2000;28:1697–707. [DOI] [PubMed] [Google Scholar]
- [9].Autieri MV, Carbone C, Mu A. Expression of allograft inflammatory factor-1 is a marker of activated human vascular smooth muscle cells and arterial injury. Arterioscler Thromb Vasc Biol 2000;20(7):1737–44. [DOI] [PubMed] [Google Scholar]
- [10].Sommerville LJ, Kelemen SE, Autieri MV. Increased smooth muscle cell activation and neointima formation in response to injury in AIF-1 transgenic mice. Arterioscler Thromb Vasc Biol 2008;28(1):47–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Sommerville LJ, Xing C, Kelemen SE, Eguchi S, Autieri MV. Inhibition of allograft inflammatory factor-1 expression reduces development of neointimal hyperplasia and p38 kinase activity. Cardiovasc Res 2009;81(1): 206–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Autieri MV, Kelemen S, Thomas BA, et al. Allograft inflammatory factor-1 expression correlates with cardiac rejection and development of cardiac allograft vasculopathy. Circulation 2002;106(17):2218–23. [DOI] [PubMed] [Google Scholar]
- [13].Seo D, Wang T, Dressman H, et al. Gene expression phenotypes of atherosclerosis. Arterioscler Thromb Vasc Biol 2004;24(10):1922–7. [DOI] [PubMed] [Google Scholar]
- [14].Mishima T, Iwabuchi K, Fujii S, et al. Allograft inflammatory factor-1 augments macrophage phagocytotic activity and accelerates the progression of atherosclerosis in ApoE−/− mice. Int J Mol Med 2008;21(2):181–7. [PubMed] [Google Scholar]
- [15].Tian Y, Kelemen SE, Autieri MV. Inhibition of AIF-1 expression by constitutive siRNA expression reduces macrophage migration, proliferation, and signal transduction initiated by atherogenic stimuli. Am J Physiol Cell Physiol 2006;290(4):C1083–91. [DOI] [PubMed] [Google Scholar]
- [16].Arvanitis DA, Flouris GA, Spandidos DA. Genomic rearrangements on VCAM1, SELE, APEG1 and AIF1 loci in atherosclerosis. J Cell Mol Med 2005;9(1): 153–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Wamhoff BR, Hoofnagle MH, Burns A, et al. A G/C element mediates repression of the SM22alpha promoter within phenotypically modulated smooth muscle cells in experimental atherosclerosis. Circ Res 2004;95:981–8. [DOI] [PubMed] [Google Scholar]
- [18].Zhang C, Zheng H, Yu Q, et al. A practical method for quantifying atherosclerotic lesions in rabbits. J Comp Pathol 2010;142:122–8. [DOI] [PubMed] [Google Scholar]
- [19].Cuneo AA, Herrick D, Autieri MV. Il-19 reduces VSMC activation by regulation of mRNA regulatory factor HuR and reduction of mRNA stability. J Mol Cell Cardiol 2010;49(4):647–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Osto E, Kouroedov A, Mocharla P, et al. Inhibition of protein kinase Cbeta prevents foam cell formation by reducing scavenger receptor A expression in human macrophages. Circulation 2008;118(21):2174–82. [DOI] [PubMed] [Google Scholar]
- [21].Brand K, Page S, Walli AK, Neumeier D, Baeuerle PA. Role of nuclear factor-kappa B in atherogenesis. Exp Physiol 1997;82(2):297–304. [DOI] [PubMed] [Google Scholar]
- [22].Murphy JE, Tedbury PR, Homer-Vanniasinkam S, Walker JH, Ponnambalam S. Biochemistry and cell biology of mammalian scavenger receptors. Atherosclerosis 2005;182(September (1)):1–15. [DOI] [PubMed] [Google Scholar]
- [23].Daugherty A, Rateri DL. Development of experimental designs for atherosclerosis studies in mice. Methods 2005;36(2):129–38. [DOI] [PubMed] [Google Scholar]
- [24].Tangirala RK, Rubin EM, Palinski W. Quantification of atherosclerosis in murine models: correlation between lesions in the aortic origin and in the differences in the extent of lesions between sexes in LDL receptor-deficient and apolipoprotein E-deficient mice. J Lipid Res 1995;36(11):2320–8. [PubMed] [Google Scholar]
- [25].Kenagy RD, Hart CE, Stetler-Stevenson WG, Clowes AW. Primate smooth muscle cell migration from aortic explants is mediated by endogenous platelet-derived growth factor and basic fibroblast growth factor acting through matrix metalloproteinases 2 and 9. Circulation 1997;96:3555–60. [DOI] [PubMed] [Google Scholar]
- [26].Liu S, Tan WY, Chen QR, et al. Daintain/AIF-1 promotes breast cancer proliferation via activation of the NF-kappaB/cyclin D1 pathway and facilitates tumor growth. Cancer Sci 2008;99(5):952–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Autieri MV, Carbone CM. Overexpression of allograft inflammatory factor-1 promotes proliferation of vascular smooth muscle cells by cell cycle deregulation. Arterioscler Thromb Vasc Biol 2001;21(9):1421–6. [DOI] [PubMed] [Google Scholar]
- [28].Autieri MV, Kelemen SE, Wendt KW. AIF-1 is an actin-polymerizing and Rac1-activating protein that promotes vascular smooth muscle cell migration. Circ Res 2003;92(10):1107–14. [DOI] [PubMed] [Google Scholar]
- [29].Mehta JL, Sanada N, Hu CP, et al. Deletion of LOX-1 reduces atherogenesis in LDLR knockout mice fed high cholesterol diet. Circ Res 2007;100(11):1634–42. [DOI] [PubMed] [Google Scholar]
- [30].Mandosi E, Fallarino M, Gatti A, et al. Atorvastatin downregulates monocyte CD36 expression, nuclear NFkappaB and TNFalpha levels in type 2 diabetes. J Atheroscler Thromb 2010;17(6):539–45. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.