

Abstract
In recent years, the outcome of mantle cell lymphoma (MCL) has improved, especially in younger patients, receiving cytarabine-containing chemoimmunotherapy and autologous stem cell transplantation. Nevertheless, a proportion of MCL patients still experience early failure. To identify biomarkers anticipating failure of intensive chemotherapy in MCL, we performed target resequencing and DNA profiling of purified tumor samples collected from patients enrolled in the prospective FIL-MCL0208 phase 3 trial (high-dose chemoimmunotherapy followed by autologous transplantation and randomized lenalidomide maintenance). Mutations of KMT2D and disruption of TP53 by deletion or mutation associated with an increased risk of progression and death, both in univariate and multivariate analysis. By adding KMT2D mutations and TP53 disruption to the MIPI-c backbone, we derived a new prognostic index, the “MIPI-genetic” (“MIPI- g”). The “MIPI-g” improved the model discrimination ability compared to the MIPI-c alone, defining three risk groups: i) low-risk patients (4-year progression free survival and overall survival of 72.0% and 94.5%); ii) inter-mediate-risk patients (4-year progression free survival and overall survival of 42.2% and 65.8%) and iii) high-risk patients (4-year progression free survival and overall survival of 11.5% and 44.9%). Our results: i) confirm that TP53 disruption identifies a high-risk population characterized by poor sensitivity to conventional or intensified chemotherapy; ii) provide the pivotal evidence that patients harboring KMT2D mutations share the same poor outcome as patients harboring TP53 disruption; and iii) allow to develop a tool for the identification of high-risk MCL patients for whom novel therapeutic strategies need to be investigated. (Trial registered at clinicaltrials.gov identifier: NCT02354313).
Introduction
The introduction of high dose cytarabine-containing chemoimmunotherapeutic regimens and autologous transplantation (ASCT) have considerably improved the outcome of young fit mantle cell lymphoma (MCL) patients. Nonetheless, approximately 20-25% of MCL patients demonstrate inadequate efficacy of intensified chemoimmunotherapy as they are either primary refractory or relapse within 2 years from ASCT.1–5
Clinical and pathological scores, including the MCL international prognostic index (MIPI),6 the Ki-67 proliferative index,7 and their combination in the MIPI-c score, stratify MCL patients in groups at different risk of relapse.8 However, none of these tools has sufficient positive predictive value to trigger the development of tailored schedules specifically designed for high risk patients.9
Several recurrent mutations have been described in MCL, affecting DNA repair genes and cell cycle regulators (TP53, ATM, CCND1), epigenetic regulation genes (KMT2D, WHSC1) and cell-signaling pathways genes (NOTCH1-2, BIRC3, TRAF2).10–12 The proof of principle that MCL genetics can impact on disease outcome stems from studies that have focused on the TP53 tumor suppressor gene, including both mutations and 17p deletions.13–17
We prospectively assessed the clinical impact of a panel of genomic alterations in a cohort of young MCL patients treated with high dose chemoimmunotherapy and ASCT from the Fondazione Italiana Linfomi (FIL) MCL0208 phase 3 trial.18 The results document that KMT2D mutations associate with poor outcome in MCL and, along with TP53 mutations and 17p deletions, might be integrated in a new prognostic score to segregate a subgroup of patients who obtain minimal or no benefit from intensive chemoimmunotherapy. The prognostic score was validated in an independent series of cases.
Methods
Patients series
The FIL-MCL0208 (NCT02354313) is a phase 3, multicenter, open-label, randomized, controlled study, designed to determine the efficacy of lenalidomide as maintenance versus observation in young (18-65 years old), fit, advanced stage (Ann arbor II-IV) MCL patients after first line intensified and high-dose chemo-immunotherapy followed by ASCT. Cases of non-nodal MCL were excluded.19 The clinical trial, as well as the ancillary mutational study, were approved by the Ethical Committees of all the enrolling Centers. All patients provided written informed consent for the use of their biological samples for research purposes, in accordance with Institutional Review Boards requirements and the Helsinki’s declaration. Clinical results of the fist interim analysis of the trial were already presented.18 Further information are supplied in the Online Supplementary Materials and Methods.
Biological samples
Tumor cells were sorted from the baseline bone marrow (BM) samples by immunomagnetic beads (CD19 MicroBeads,human-Miltenyi Biotec GmbH, Bergisch Gladbach, Germany) and stocked as dry pellets.
Tumor DNA was extracted according to DNAzol protocol (Life Technologies). Germline DNA was obtained from peripheral blood (PB) mononuclear cells collected under treatment and proven to be tumor free by minimal residual disease (MRD) analysis. Further information are supplied in the Online Supplementary Materials and Methods.
Next generation sequencing (NGS)
A targeted resequencing panel (target region: 37’821 bp) (Online Supplementary Table S1) including the coding exons and splice sites of seven genes (ATM, TP53, CCND1, WHSC1, KMT2D, NOTCH1 exon 34, BIRC3) that are recurrently mutated in ≥5% of MCL tumors was specifically designed.10–12 We also included in the panel TRAF220 and CXCR4.21 NGS libraries preparation was performed using TruSeq Custom Amplicon sequencing assay according to manufacturer’s protocol (Illumina, Inc., San Diego, CA, USA). Multiplexed libraries (n=48 per run) were sequenced using 300-bp paired-end runs on an Illumina MiSeq sequencer, (median depth of coverage 2,356x). A robust and previously validated bioinformatics pipeline was used for variant calling (Online Supplementary Materials and Methods). Copy number variation analysis methods22,23 are supplied in the Online Supplementary Materials and Methods.
MRD analysis
For MRD purposes, MCL diagnostic BM and PB samples were investigated for immunoglobulin heavy chain (IGH) gene rearrangements and BCL1/IGH MTC by qualitative PCR.24–26 Both BM and PB samples were analyzed for MRD at specific time points during and after treatment. Further information are supplied in the Online Supplementary Materials and Methods.
Statistical analysis
The primary outcome of the clinical study was progression-free survival (PFS) and secondary outcomes included overall survival (OS).27 The adjusted effects of mutations and exposure variables (MIPI-c and blastoid variant) on PFS and OS were estimated by Cox regression. To compare clinical baseline features between patients enrolled in the molecular study and patients not included in the analysis, we used Mann-Whitney test for continuous variables and Pearson’s χ2 test for categorical variables. Statistical analyses were performed using Stata 13.0 and R 3.4.1. Further information are supplied in the Online Supplementary Materials and Methods.
Validation set
The Nordic Lymphoma Group MCL2 and MCL3, phase 2, prospective trials17 were used for independent validation of our findings. In particular, the raw sequencing data of the study by Eskelund et al. were reanalyzed according to our bioinformatics pipeline (detailed before), to get a uniform mutation calling.
Results
Patients characteristics
Out of the 300 patients enrolled in the FIL-MCL0208 clinical trial, 186 (62%) were provided with CD19+ sorted tumor cells from the BM and were evaluable for both mutations and copy number abnormalities. Moreover, four more patients were provided with the copy number abnormalities data only. Baseline features of the cases included in the molecular study overlapped with those cases not included in the molecular analysis because of a lack of tumor material in the BM aspirates. As expected, tumor cells were obtained more frequently in cases with BM infiltration documented by morphological or flow-cytometry analysis (Table 1). Overall, this observation did not introduce a selection bias, since cases evaluable for genomic studies showed a similar outcome to cases not analyzed, both in terms of PFS and OS (Online Supplementary Figure S1).
Table 1.
Clinical and biological baseline characteristics of the patients included and not included in the molecular analysis.

Description of genomic alterations
At least one somatic non-synonymous mutation affecting genes of the target region was observed in 69.8% of patients (130 of 186) (Figure 1, Online Supplementary Figure S2 and Online Supplementary Table S2). Mutated genes were ATM (41.9%), followed by WHSC1 (15.6%), KMT2D (12.4%), CCND1 (11.8%), TP53 (8.1%), NOTCH1 (7.5%), BIRC3 (5.9%) and TRAF2 (1.1%). KMT2D deletion occurred in 1.6% of patients (3 of 190) and TP53 deletion in 13.2% patients (25 of 190). TP53 was inactivated by mutations or deletions in 31 of 186 (16.6%) cases, including 8 of 186 (4.3%) mutated/deleted cases, 16 of 186 (8.6%) deleted but not mutated cases, and 7 of 186 (3.7%) mutated but not deleted cases. KMT2D was inactivated by mutations or deletions in 25 of 186 (13.4%) cases, including 1 of 186 (<1%) mutated/deleted case, 2 of 186 (<1%) deleted but non mutated cases, and 22 of 186 (11.8%) mutated but not deleted cases.
Figure 1.

Overview on prevalence and molecular spectrum of non-synonymous somatic mutations discovered in patients’ tumor DNA. Heatmap representing the mutational profiles of 186 mantle cell lymphoma (MCL) cases, genotyped on tumor DNA (and four additional patients with copy number abnormalities data only). Each column represents one patient, each row represents one gene. The fraction of patients with mutations in each gene is plotted on the right. The number of aberrations in a given patient is plotted above the heatmap.
KMT2D mutations and TP53 disruption associate with poor outcome in MCL
By univariate analysis, mutations of KMT2D were associated with poor clinical outcome in terms of both PFS and OS. At 4 years, the PFS of KMT2D mutated patients was 33.2% versus 63.7% (P<0.001) in wild-type (WT) cases (Figure 2A). The OS of KMT2D mutated patients was 62.3% versus 86.8% (P=0.002) in WT patients (Figure 2B). Consistent with previous reports, both TP53 mutations and deletion associated with shorter PFS and OS at 4 years (Figure 2C-D and Figure 3). In detail, the negative prognostic impact for TP53 disruption was equal for all the three inactivation modalities, which were then considered as a single group for further analyses (Online Supplementary Figure S3). No further survival analysis was performed on KMT2D deletions, given the low frequency of this genetic lesion. All the other investigated mutations did not show a strong association with PFS or OS (Online Supplementary Figure S4-6 and Online Supplementary Table S3).
Figure 2.

Prognostic impact of KMT2D and TP53 mutations. Kaplan-Meier estimates of progression free survival and overall survival of KMT2D (A, B), and TP53 (C, D) mutated versus wild-type (WT) patients. Cases harboring mutations (mut) in these genes are represented by the yellow line. Cases WT for these genes are represented by the blue line. The Log-rank statistics P-values are indicated adjacent curves.
Figure 3.

Prognostic impact of TP53 deletion. Kaplan-Meier estimates of progression free survival (PFS) (A) and overall survival (OS) (B) of TP53 deleted versus wild-type patients. Cases with TP53 deletion are represented by the yellow line. Cases without TP53 (del) deletion are represented by the blue line. The Log-rank statistics P-values are indicated adjacent curves.
Patients harboring TP53 disruption were significantly enriched in known high-risk features of MCL. Indeed, 48.3% of the TP53 disrupted patients had Ki-67 ≥30%, 37.9% scored in the higher MIPI-c risk classes (i.e. “inter-mediate-high” and “high”), and 22.6% showed blastoid morphology. Conversely, 45.5% of cases harboring KMT2D mutations scored in the higher MIPI-c risk classes but did not associate with Ki-67 expression or blastoid morphology (Online Supplementary Table S4). Moreover, KMT2D mutated patients showed slightly higher beta-2 microglobulin (B2M) median values, as well as higher prevalence of B symptoms and bulky disease (>5 cm) than WT patients (all P<0.05). Interestingly, also TP53 disrupted patients showed slightly higher B2M median values (P<0.05) than WT patients (Online Supplementary Table S4) and were associated with a high rate of disease progression during treatment (9 of 31 patients, 29%). Moreover, TP53 disrupted patients reached lower levels of MRD negativity after ASCT, if compared with WT ones: 35% versus 58% in BM (P=0.06) and 58% versus 80% in PB (P=0.04), respectively. Similar trends were seen for KMT2D mutated patients (46% vs. 55% in BM and 58% vs. 79%), albeit not statistically significant (Online Supplementary Table S5). Analogous to the Nordic Lymphoma Group MCL2 and MCL3 trials,17 also in our study morphological BM involvement was significantly associated with the presence of mutations in any of the genes analysed (P<0.05). However, both TP53 disruptions and KMT2D mutations were equally distributed in patients with and without BM involvement (P=0.26 and P=0.32, respectively).
By multivariate analysis adjusted for the validated risk factors MIPI-c and blastoid variant, both KMT2D mutations and TP53 disruptions maintained an independent increased hazard of progression and death (Table 2 and Online Supplementary Table S6). Patients carrying at least one of these genetic lesions, namely KMT2D mutations, TP53 mutations or deletion (n=49/186, 26.3%), had a 4-year PFS of 32.0% versus 69.9% of WT patients (P<0.0001) and a 4-year OS of 65.1% versus 90.3% (P<0.0001), respectively (Figure 4).
Table 2.
Uniavariate and multivariate Cox-regression analysis in terms of progression free survival and overall survival.

Figure 4.

Prognostic impact of combined KMT2D mutations and TP53 disruption. Kaplan-Meier estimates of (A) progression free survival (PFS) and (B) overall survival (OS) of patients harboring KMT2D mutations and/or TP53 disruption (mutations and/or deletions). Cases harboring at least 1 of these 3 genetic lesions are represented by the yellow line. Cases without these genes are represented by the blue line. The Log-rank statistics P-values are indicated adjacent curves.
Integration of a genetic score into the MIPI-c: the “MIPI-g” model
In order to integrate the clinical impact of KMT2D mutations and TP53 disruptions into the MIPI-c prognostic index (complete data available for 172 patients), we assigned a score to each of the single variables, based on the multivariate Cox regression analysis. MIPI-c low, low-intermediate and intermediate-high risk classes scored 0 points, MIPI-c high-risk class scored 1 point, while KMT2D mutations as well as TP53 disruption scored 2 points (Table 3). Patients were then grouped into three risk classes, according to their total score, in the “MIPI-g” index, namely: i) 0 points, low risk group (LR 121 patients, 70.3%); ii) 1-2 points, intermediate risk group (IR 38 patients, 22.1%); iii) ≥3 points, high risk group (HR 13 patients, 7.6%). PFS and OS at 4-years for low-, intermediate-, and high-risk groups were 72.0%, 42.2%, 11.5% (P<0.0001) and 94.5%, 65.8%, 44.9% (P<0.0001), respectively (Figure 5). The MIPI-g index improved the model discrimination ability, with a C-statistics of 0.675 for PFS (bootstrapping corrected 0.654) and 0.776 for OS (boot-strapping corrected 0.747), as compared to MIPI-c alone (C-statistics 0.592 and 0.7, respectively).
Table 3.
The MIPI-g score.
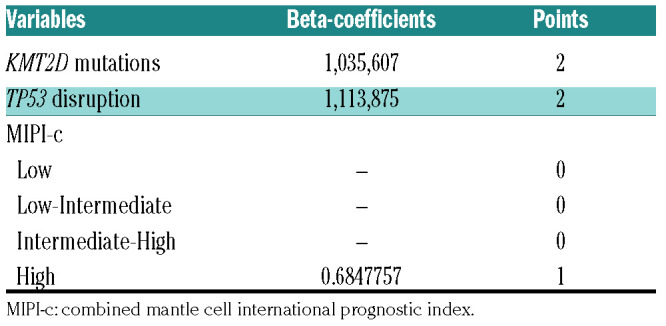
Figure 5.

The “MIPI-g” model. Kaplan-Meier estimates of (A) progression free survival (PFS) and (B) overall survival (OS) of patients harboring KMT2D mutations and/or TP53 disruption (mutations and/or deletions) integrated into the MIPI-c. Low MIPI-g risk cases are represented by the blue line, intermediate MIPI-g cases by the yellow line and high MIPI-g cases by the red line. The Log-rank statistics P-values are indicated for adjacent curves.
Validation set
Most KMT2D variants considered in the Nordic study have been removed by our mutational calling, since these were missense variants not reported in COSMIC. At the end of the re-analysis, from the original 28 mutations, 21 were excluded. Two previously unrecognized frameshift mutations have been identified by our bioinformatics pipeline, overall accounting for a total of nine KMT2D mutations (all disrupting, as expected for KMT2D) in the Nordic validation series. In the Nordic validation series, KMT2D mutated patients showed a similar increased risk for OS, with a median OS of 12.7 years (95% confidenec interval [CI] not evaluable) for WT versus 8.4 (95% CI: 0-17.6) for mutated cases. The Nordic validation series also replicated the MIPI-g score. The re-analysis of TP53 mutations confirmed the original data of Eskelund et al., with median OS of 12.7 (95% CI not evaluable) for WT cases and 2.0 years (95% CI: 1.2-2-8) for mutated cases. Consistently, also the MIPI-g validation on the Nordic series showed similar results: 4-year OS for LR (n=103), IR (n=36) and HR (n=13) MIPI-g groups were 91.3%, 72.2%, 15.4%.
Discussion
To identify new molecular predictors in MCL, we performed targeted resequencing and DNA profiling of purified tumor samples collected from young patients enrolled in the ASCT-based prospective FIL-MCL0208 phase 3 trial (NCT02354313). Our study documents that: i) KMT2D mutations are a novel, independent, adverse genetic biomarker in MCL, impacting both on PFS and OS (Figure 2A-B); ii) TP53 disruptions (both mutations and deletion) prospectively confirm their adverse prognostic value in young MCL patients receiving high-dose chemo-immunotherapy followed by ASCT, both in terms of PFS and OS (Figures 2C-D and Figure 3); iii) identification of either KMT2D mutations or TP53 disruption (or both) defines a HR group of young MCL patients whose outcome is still not satisfactory despite intensive immunochemotherapy and ASCT (Figure 4); iv) these bio-markers may be incorporated into a “MIPI-g” model, accounting for three risk classes (Figure 5), that improves the C-statistics discrimination ability on survival, if compared to MIPI-c alone.
The adverse prognostic value of TP53 mutations in MCL has been already observed in some retrospective series,13–17 and has been recently confirmed in a combined series from two, ASCT-based, phase 2 trials of the Nordic Lymphoma Group.17 TP53 deletions impacted on both PFS and OS in the randomized, phase 3 European MCL Network “Younger” trial,16 while these data were not confirmed by multivariate analysis in the Nordic study, due to the high association with TP53 mutations.17 Our prospective study performed in a similar patient population of young MCL patients demonstrates that the presence of either TP53 mutations or deletions or both associates with poor prognosis. Importantly, although TP53 aberrations associated with elevated Ki-67, higher MIPI-c classes and blastoid morphology, their impact on survival was independent of these known risk factors. Moreover, TP53 disrupted patients show higher levels of MRD positivity after ASCT, as described in the Online Supplementary Table S5. Finally, some previous studies reported also a negative impact of NOTCH1 mutations in univariate analysis,10,17 however in our cohort these mutations were not an independent predictor of survival, as most of them co-occurred with TP53 mutations.
In the FIL-MCL0208 trial, KMT2D mutations emerged as a novel biomarker heralding chemo-immunotherapy failure, with a predictive value similar to that of TP53 aberrations. KMT2D (lysine methyltransferase 2D), also known as MLL2, acts as a tumor suppressor gene mutated in several B-cell lymphoma types, including 10-15% of MCL.28–31 Even though KMT2D mutated patients of the FIL-MCL0208 trial scored in the HR MIPI-c classes, they showed neither elevated Ki-67 nor blastoid morphology, suggesting that KMT2D mutations capture high-risk patients not otherwise identifiable through conventional pathologic parameters.
To the best of our knowledge, the adverse impact on cancer survival of KMT2D mutations has not been documented to date. No impact on survival was found for KMT2D mutations in the Nordic study.17 The lack of impact on survival of KMT2D mutations in the Nordic MCL series might be related to two main reasons. First, in the Nordic series, most KMT2D mutations were missense sequence variants (15 of 28) not reported as somatic variants in the COSMIC database, and therefore not fulfilling the criteria of “true” mutations. Conversely, in our series 74% of KMT2D mutations were protein truncating events, as expected.28–31 Second, since Eskelund et al. performed mutational analysis in unsorted BM samples, the low or absent tumor content of many cases might lead to underestimate28 “true” mutations. By applying our bioinformatics pipeline to the raw sequencing data of the MCL2 and MCL3 Nordic Lymphoma Group trials, we validated the poor prognostic role of KMT2D mutations in an independent prospective cohort.
The independent adverse prognostic value of TP53 and KMT2D aberrations prompted us to integrate the molecular results into the MIPI-c,8 aiming at further improving its ability to discriminate high-risk patients. The “MIPI-g” was able to divide patients into three risk classes, on the basis of a simple score given to each variable (namely: MIPI-c class, TP53 disruption and KMT2D mutations). Patients in the HR “MIPI-g” groups may deserve new treatments, and a simple tool like the MIPI-g might be pro-posed in a future, “tailored” trial to select HR MCL patients for targeted experimental strategies.
Our study suffers from some limitations. The analyses were performed only on CD19+ sorted BM cells and no tissue control is available at the moment; this issue might represent a limit for the extrapolation of the results to lymph-node samples, as across-compartment heterogeneity of the mutational landscape is described in MCL.10 However, the CD19+ selection approach we used, increasing the purity of tumor cells and, consequently, the sensitivity of our mutational approach, ensured that all the analyzed samples are representative of MCL. Therefore, we set a variant allele frequency (VAF) threshold of 10% to call a mutation, accordingly to ERIC guidelines for the mutational analysis of the TP53 gene in chronic lymphocytic leukemia.32 Although we acknowledge that the present validation relies on a limited number of KMT2D mutated patients, we noted that the Nordic trials are currently the only prospective studies with prompt available mutational data, adequate clinical follow-up and similar characteristics (i.e. patients age and treatment schedule), to validate our original findings from the FIL-MCL0208 trial.
The impact of lenalidomide maintenance in the FIL-MCL0208 trial on the described genetic aberrations has not been addressed, as complete data on randomization are not available yet. However, it should be noted that due to the high number of progressive diseases in the aberrant TP53/KMT2D group, 27 patients have been finally randomized but only nine actually started lenalidomide maintenance. Therefore, it is unlikely that lenalidomide might play a clear role in driving the outcome of these patients and the trial will probably not be able to fully address this issue even with a longer follow-up.
In conclusion, our results show that KMT2D mutated and/or TP53 disrupted younger MCL patients are a HR population, characterized by poor sensitivity even to intensified chemo-immunotherapy. Given the negative prognostic impact of these genetic lesions, they might be used to select HR patients for novel therapeutic approaches that can circumvent these detrimental genetic lesions. As in other lymphoid disorders, novel non-chemotherapeutic strategies specifically designed for HR patients need to be investigated in MCL. Besides the approved drugs lenalidomide and ibrutinib, new molecules such as the BCL-2 inhibitor venetoclax might be very promising for these chemorefractory patients, especially for TP53 disrupted cases.33,34 Moreover, as the majority of KMT2D mutated and/or TP53 disrupted patients of our series actually achieve a response, though short-lasting after ASCT, an alternative consolidation with allogeneic transplantation deserves investigation.
Supplementary Material
Footnotes
Check the online version for the most updated information on this article, online supplements, and information on authorship & disclosures: www.haematologica.org/content/105/6/1604
Funding
This work was supported by: Progetto di Rilevante Interesse Nazionale (PRIN2009) from the Ministero Italiano dell’Università e della Ricerca (MIUR), Roma, Italy [7.07.02.60 AE01]; Progetto di Ricerca Sanitaria Finalizzata 2009 [RF-2009-1469205] and 2010 [RF-2010-2307262 to S.C.], A.O.S. Maurizio, Bolzano/Bozen, Italy; Fondi di Ricerca Locale, Università degli Studi di Torino, Italy; Fondazione Neoplasie Del Sangue (Fo.Ne.Sa), Torino, Italy; Fondazione CRT (project codes: 2016.0677 and 2018.1284), Torino, Italy; Associazione DaRosa, Torino, Italy; Molecular bases of disease dissemination in lymphoid malignancies to optimize curative therapeutic strategies, (5x1,000 No. 21198), Associazione Italiana per la Ricerca sul Cancro Foundation, Milan, Italy; Progetto Ricerca Finalizzata RF-2011-02349712, Ministero della Salute, Rome, Italy; and PRIN 2015ZMRFEA_004, MIUR, Rome, Italy; Partially funded by the AGING Project – Department of Excellence – DIMET, Università del Piemonte Orientale, Novara, Italy; Grant No. KFS-3746-08-2015, Swiss Cancer League, Bern, Switzerland; Grant No. KLS-3636-02-2015, Swiss Cancer League, Bern, Switzerland; Grant No. 320030_169670/1 Swiss National Science Foundation, Berne, Switzerland; iCARE No. 17860, Associazione Italiana per la Ricerca sul Cancro and Unione Europea, Milan, Italy; Fondazione Fidinam, Lugano, Switzerland; Nelia & Amadeo Barletta Foundation, Lausanne, Switzerland; Fondazione Ticinese per la Ricerca sul Cancro, Bellinzona, Switzerland and Novara-AIL Onlus, Novara, Italy.
References
- 1.Eskelund CW, Kolstad A, Jerkeman M, et al. 15-year follow-up of the Second Nordic Mantle Cell Lymphoma trial (MCL2): prolonged remissions without survival plateau. Br J Haematol. 2016;175(3):410–418. [DOI] [PubMed] [Google Scholar]
- 2.Hermine O, Hoster E, Walewski J, et al. Addition of high-dose cytarabine to immunochemotherapy before autologous stem-cell transplantation in patients aged 65 years or younger with mantle cell lymphoma (MCL Younger): a randomised, open-label, phase 3 trial of the European Mantle Cell Lymphoma Network. Lancet. 2016;388(10044):565–575. [DOI] [PubMed] [Google Scholar]
- 3.Delarue R, Haioun C, Ribrag V, et al. CHOP and DHAP plus rituximab followed by autologous stem cell transplantation in mantle cell lymphoma: a phase 2 study from the Groupe d’Etude des Lymphomes de l’Adulte. Blood. 2013;121(1):48–53. [DOI] [PubMed] [Google Scholar]
- 4.Romaguera JE, Fayad LE, Feng L, et al. Ten-year follow-up after intense chemoimmunotherapy with Rituximab-HyperCVAD alternating with Rituximab-high dose methotrexate/cytarabine (R-MA) and without stem cell transplantation in patients with untreated aggressive mantle cell lymphoma. Br J Haematol. 2010;150(2):200–208. [DOI] [PubMed] [Google Scholar]
- 5.Le Gouill S, Thieblemont C, Oberic L, et al. Rituximab after autologous stem-cell transplantation in mantle-cell lymphoma. N Engl J Med. 2017;377(13):1250–1260. [DOI] [PubMed] [Google Scholar]
- 6.Hoster E, Dreyling M, Klapper W, et al. A new prognostic index (MIPI) for patients with advanced-stage mantle cell lymphoma. Blood. 2008;111(2):558–565. [DOI] [PubMed] [Google Scholar]
- 7.Hoster E, Klapper W, Hermine O, et al. Confirmation of the mantle-cell lymphoma International Prognostic Index in randomized trials of the European Mantle-Cell Lymphoma Network. J Clin Oncol. 2014;32(13):1338–1346. [DOI] [PubMed] [Google Scholar]
- 8.Hoster E, Rosenwald A, Berger F, et al. Prognostic value of Ki-67 index, cytology, and growth pattern in mantle-cell lymphoma: Results from randomized trials of the european mantle cell lymphoma network. J Clin Oncol. 2016;34(12):1386–1394. [DOI] [PubMed] [Google Scholar]
- 9.Dreyling M, Ferrero SE. The role of targeted treatment in mantle cell lymphoma: Is transplant dead or alive¿ Haematologica. 2016;101(2):104–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Beà S, Valdés-Mas R, Navarro A, et al. Landscape of somatic mutations and clonal evolution in mantle cell lymphoma. Proc Natl Acad Sci U S A. 2013;110(45):18250–18255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang J, Jima D, Moffitt AB, et al. The genomic landscape of mantle cell lymphoma is related to the epigenetically determined chromatin state of normal B cells. Blood. 2014;123(19):2988–2996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rahal R, Frick M, Romero R, et al. Pharmacological and genomic profiling identifies NF-κB-targeted treatment strategies for mantle cell lymphoma. Nat Med. 2014;20(1):87–92. [DOI] [PubMed] [Google Scholar]
- 13.Kumar A, Yang W, Bantilan KS, et al. Prognostic significance of genomic alterations in mantle cell lymphoma. Blood. 2016;128(22):4115. [Google Scholar]
- 14.Nordström L, Sernbo S, Eden P, et al. SOX11 and TP53 add prognostic information to MIPI in a homogenously treated cohort of mantle cell lymphoma - a Nordic Lymphoma Group study. Br J Haematol. 2014;166(1):98–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Halldórsdóttir AM, Lundin A, Murray F, et al. Impact of TP53 mutation and 17p deletion in mantle cell lymphoma. Leukemia. 2011;25(12):1904–1908. [DOI] [PubMed] [Google Scholar]
- 16.Delfau-Larue MH, Klapper W, Berger F, et al. High-dose cytarabine does not over-come the adverse prognostic value of CDKN2A and TP53 deletions in mantle cell lymphoma. Blood. 2015;126(5):604–611. [DOI] [PubMed] [Google Scholar]
- 17.Eskelund CW, Dahl C, Hansen JW, et al. TP53 mutations identify younger mantle cell lymphoma patients who do not benefit from intensive chemoimmunotherapy. Blood. 2017;130(17):1903–1910. [DOI] [PubMed] [Google Scholar]
- 18.Cortelazzo S, Martelli M, Ladetto M, et al. High dose sequential chemotherapy with rituximab and ASCT as first line therapy in adult MCL patients: clinical and molecular response of the MCL0208 trial, a FIL study. Haematologica. 2015;100(s1):3–4.25552677 [Google Scholar]
- 19.Swerdlow SH, Campo E, Pileri SA, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016;127(20):2375–2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Meissner B, Kridel R, Lim RS, et al. The E3 ubiquitin ligase UBR5 is recurrently mutated in mantle cell lymphoma. Blood. 2013;121(16):3161–3164. [DOI] [PubMed] [Google Scholar]
- 21.Roccaro AM, Sacco A, Jimenez C, et al. C1013G/CXCR4 acts as a driver mutation of tumor progression and modulator of drug resistance in lymphoplasmacytic lymphoma. Blood. 2014;123(26):4120–4131. [DOI] [PubMed] [Google Scholar]
- 22.Rinaldi A, Kwee I, Young KH, et al. Genome-wide high resolution DNA profiling of hairy cell leukaemia. Br J Haematol. 2013;162(4):566–569. [DOI] [PubMed] [Google Scholar]
- 23.Kwee IW, Rinaldi A, de Campos CP, et al. Fast and robust segmentation of copy number profiles using multi-scale edge detection. BioRxiv. 2016. 10.1101/056705. [DOI]
- 24.Voena C, Ladetto M, Astolfi M, et al. A novel nested-PCR strategy for the detection of rearranged immunoglobulin heavy-chain genes in B cell tumors. Leukemia. 1997;11(10):1793–1798. [DOI] [PubMed] [Google Scholar]
- 25.Stamatopoulos K, Kosmas C, Belessi C, et al. Molecular analysis of bcl-1/IgH junctional sequences in mantle cell lymphoma: potential mechanism of the t(11;14) chromosomal translocation. Br J Haematol. 1999;105(1):190–197. [DOI] [PubMed] [Google Scholar]
- 26.Pott C. Minimal residual disease detection in mantle cell lymphoma: technical aspects and clinical relevance. Semin Hematol. 2011;48(3):172–184. [DOI] [PubMed] [Google Scholar]
- 27.Cheson BD, Pfistner B, Juweid ME, et al. Revised response criteria for malignant lymphoma. J Clin Oncol. 2007;25(5):579–586. [DOI] [PubMed] [Google Scholar]
- 28.Froimchuk E, Jang Y, Ge K. Histone H3 lysine 4 methyltransferase KMT2D. Gene. 2017;627:337–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Varettoni M, Zibellini S, Defrancesco I, et al. Pattern of somatic mutations in patients with Waldenström macroglobulinemia or IgM monoclonal gammopathy of undetermined significance. Haematologica. 2017;102(12):2077–2085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Spina V, Khiabanian H, Messina M, et al. The genetics of nodal marginal zone lymphoma. Blood. 2016;128(10):1362–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pasqualucci L, Trifonov V, Fabbri G, et al. Analysis of the coding genome of diffuse large B-cell lymphoma. Nat Genet. 2011;43(9):830–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Malcikova J, Tausch E, Rossi D, et al. ERIC recommendations for TP53 mutation analysis in chronic lymphocytic leukemia-update on methodological approaches and results interpretation. Leukemia. 2018;32(5):1070–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Davids MS, Roberts AW, Seymour JF, et al. Phase I first-in-human study of venetoclax in patients with relapsed or refractory non-hodgkin lymphoma. J Clin Oncol. 2017;35(8):826–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tam CS, Anderson MA, Pott C, et al. Ibrutinib plus venetoclax for the treatment of mantle-cell lymphoma. N Engl J Med. 2018;378(13):1211–1223. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.