

Abstract
Peripheral T-cell lymphoma comprises a heterogeneous group of mature non-Hodgkin lymphomas. Their diagnosis is challenging, with up to 30% of cases remaining unclassifiable and referred to as “not otherwise specified”. We developed a reverse transcriptase-multiplex ligation-dependent probe amplification gene expression profiling assay to differentiate the main T-cell lymphoma entities and to study the heterogeneity of the “not specified” category. The test evaluates the expression of 20 genes, including 17 markers relevant to T-cell immunology and lymphoma biopathology, one Epstein-Barr virus-related transcript, and variants of RHOA (G17V) and IDH2 (R172K/T). By unsupervised hierarchical clustering, our assay accurately identified 21 of 21 ALK-positive anaplastic large cell lymphomas, 16 of 16 extranodal natural killer (NK)/T-cell lymphomas, 6 of 6 hepatosplenic T-cell lymphomas, and 13 of 13 adult T-cell leukemia/lymphomas. ALK-negative anaplastic lymphomas (n=34) segregated into one cytotoxic cluster (n=10) and one non-cytotoxic cluster expressing Th2 markers (n=24) and enriched in DUSP22-rearranged cases. The 63 TFH-derived lymphomas divided into two subgroups according to a predominant TFH (n=50) or an enrichment in Th2 (n=13) signatures. We next developed a support vector machine predictor which attributed a molecular class to 27 of 77 not specified T-cell lymphomas: 17 TFH, five cytotoxic ALK-negative anaplastic and five NK/T-cell lymphomas. Among the remaining cases, we identified two cell-of-origin subgroups corresponding to cytotoxic/Th1 (n=19) and Th2 (n=24) signatures. A reproducibility test on 40 cases yielded a 90% concordance between three independent laboratories. This study demonstrates the applicability of a simple gene expression assay for the classification of peripheral T-cell lymphomas. Its applicability to routinely-fixed samples makes it an attractive adjunct in diagnostic practice.
Introduction
Peripheral T-cell lymphomas (PTCL) are a diverse group of neoplasms representing 10-15% of all lymphomas worldwide, with large geographic variation. According to the 2017 revision of the World Health Organization (WHO) classification of lymphoid neoplasms, PTCL comprise up to 30 entities derived from various subsets of mature T or natural killer (NK) cells.1 The heterogeneity and rarity of these tumors, combined with their complex immunophenotypic profile and partially overlapping features across different entities, make their diagnosis particularly challenging. In addition, there is a high variability in the diagnostic workup among pathologists, which may account for relatively poor reproducibility of the diagnoses.2–4 Although most cases can be ascribed to specific disease entities, approximately one-third of PTCL not fulfilling the criteria for other entities remain unclassifiable and are categorized “by default” as PTCL-not otherwise specified (NOS).
The classification of PTCL has undergone major changes over the past years with the incorporation of much new information on their genetic background and taking into account the notion that PTCL arise from discrete subsets of normal T cells. In recent years, the description of the signature and mutational landscape of PTCL has generated novel molecular biomarkers to refine the diagnostic criteria for some entities. Notably, the expression of TFH markers and the presence of genetic lesions associated with angioimmunoblastic T-cell lymphoma (AITL) (such as RHOA, TET2, DNMT3A, and IDH2 mutations), found in a significant proportion of PTCL-NOS,5–10 led to the reclassification of these as “nodal PTCL with a TFH phenotype” (TFH-PTCL) in the revised WHO classification.1 Among anaplastic large cell lymphoma (ALCL), the identification of recurrent rearrangements of the ALK gene led to ALK-positive ALCL being referred to as a definitive separate entity (ALCL ALK+), and to reconsider ALCL without ALK rearrangement as a distinct but genetically heterogeneous group comprising subtypes characterized by alterations of the DUSP22/IRF4 or TP63 genes with distinct clinical, pathological and biological features.11 Among the remaining PTCL-NOS category, two molecular subgroups defined by the expression of the TBX21 and GATA3 transcription factors have been proposed,12,13 with a worse prognosis suggested for GATA3-positive cases.13–16 In daily diagnostic practice, however, high-throughput technologies are difficult to integrate. Moreover, the immunohistochemical surrogates are not fully validated and require an increasingly large panel of antibodies, and their evaluation may be problematic or present limitations.3,17
Here, we designed a simple targeted mRNA expression profiling assay based on reverse transcriptase-multiplex ligation-dependent probe amplification (RT-MLPA), using a panel of molecular markers relevant to the characterization of PTCL. We first assessed the accuracy of this assay in the classification of PTCL entities other than PTCL-NOS, and then used the assay to study the heterogeneity of PTCL-NOS. Our findings support this RT-MLPA assay as a robust and useful tool, suitable for the routine classification of PTCL and, therefore, promoting an optimal clinical management of PTCL patients.
Methods
Patients and tumor samples
A series of 270 lymphoma samples were selected within the framework of the multicentric T-cell lymphoma consortium (TENOMIC) of the Lymphoma Study Association (LYSA). All cases had been reviewed by at least two expert hematopathologists, according to the criteria of the recently up-dated WHO classification.1 The series was enriched in nodal TFH-PTCL (TFH-PTCL) defined by the expression of at least two TFH markers among CD10, BCL6, CXCL13, PD1, ICOS and in PTCL-NOS defined as a diagnosis of exclusion of any well-defined entity. The design of the study is summarized in Online Supplementary Figure S1. Briefly, a classification cohort (n=230) was used to train a support vector machine (SVM) classifier and a diagnostic cohort (n=40) was used to evaluate its inter-laboratory reproducibility on formalin-fixed paraffin-embedded (FFPE) samples. The study was approved by the local ethics committee (CPP Ile de France IX 08-009).
RT-MLPA assay gene expression profiling
RNA extracted from frozen and/or FFPE tumor samples was applied to RT-MLPA, as described18 (Online Supplementary Methods). Briefly, this targeted multiplex assay consists of the hybridization and ligation of specific probes on cDNA, followed by PCR amplification. We designed 41 probes (Eurofins MWG Operon, Ebersberg, Germany) targeting 20 genes, selected for their relevance to PTCL classification (Table 1). RT-MLPA results were compared to Affymetrix HG-U133-plus-2.0 gene expression data in 72 previously reported cases.18,19
Table 1.
Gene panel designed for the reverse transcriptase-multiplex ligation-dependent probe amplification assay.

Bioinformatic analysis
A web interface was developed for the complete analysis of the RT-MLPA results (https://bioinfo.calym.org/RTMLPA). An SVM was developed to classify PTCL samples: two-thirds of the 184 PTCL of the classification cohort, which clustered in defined molecular branches according to the clustering (n=230), were randomly selected to train the classifier, which was validated in the remaining one-third of cases. A bootstrap resampling process was used to build 100 independent training and validation series. A definitive SVM predictor was thus developed using the 184 cases. This supervised learning model assigns a class to every PTCL sample. Therefore, we integrated the distance to the centroid of the predicted class for each sample to avoid classifying distant samples into the same group. The analytical process is detailed in the Online Supplementary Methods.
Histopathology and molecular validation
RT-MLPA signatures were correlated to immunochemical data, including expression of GATA3 and TBX21. The cut off for positive immunohistochemical staining was 10% of presumed neoplastic cells (Online Supplementary Methods). Fluorescence in situ hybridization (FISH) for DUSP22/IRF4 rearrangement was performed in 20 ALCL. Mutations were validated using polymerase chain reaction (PCR) allele-specific and/or targeted deep sequencing.20,21 Technical details are presented in the Online Supplementary Methods.
Data analysis
Affymetrix and RT-MLPA gene expression values were correlated using Spearman’s correlation test. Correlations between immunohistochemical results and RT-MLPA gene expression values were evaluated using Wilcoxon’s rank-sum test. Unsupervised hierarchical analysis was performed using the Ward method.
Overall and progression-free survival analysis was performed using the Kaplan-Meier method and the log-rank test. The Mann-Whitney test was used to analyze continuous data and the Fisher exact test to analyze categorical data.
Results
Design and validation of the RT-MLPA assay
The study design is presented in Online Supplementary Figure S1. The gene set and sequences of the RT-MLPA probes are shown in Table 1 and Online Supplementary Table S1, respectively. The panel was designed to include several genes encoding immunohistochemical or genetic markers routinely used for the diagnosis of PTCL and genes of interest selected from previous transcriptomic and genomic studies.9,10,12,13,18 It includes genes related to the major CD4 and CD8 T-cell subsets, genes defining the main subsets of Th cells [TFH (CXCL13, CXCR5, ICOS, BCL6), Th1 (TBX21, IFNg), Th2 (GATA3, CCR4), and Treg (FOXP3)], as well as genes encoding cytotoxic molecules (PRF, GZMB). CD30 and ALK were chosen to identify ALCL and CD56 and EBER1 (Epstein-Barr virus encoding small RNA) were selected to identify hepatosplenic T-cell lymphoma (HSTL) and NKTCL. We also included the RHOAG17V and IDH2R172K/T variants, as the most prevalent hotspot mutations of TFH-derived PTCL.
We obtained RT-MLPA profiles for all 230 PTCL of the classification cohort. Representative RT-MLPA profiles for each entity are shown in Online Supplementary Figure S2. TFH-PTCL profiles were characterized by the expression of a combination of TFH genes (CXCL13, CXCR5, ICOS, and BCL6), together with frequent RHOA and/or IDH2 variants. ATLL expressed Th2 markers (GATA3 and CCR4) and ICOS, with variable levels of FOXP3. NKTCL showed high expression of EBER1 and GZMB, as well as Th1 markers (TBX21 and IFNg). HSTL expressed CD56, GATA3, TBX21, and BCL6. ALK-positive ALCL expressed ALK, CD30, PRF, and GZMB. ALK-negative ALCL comprised two distinct profiles, with or without expression of PRF and GZMB. The non-cytotoxic ALCL showed high expression of CD30 and Th2 markers (GATA3 and CCR4) but not PRF or GZMB. Unexpectedly, RT-MLPA identified ALK expression in a case of ALCL initially considered ALK-negative (based on negative immunostaining with the ALK1 clone), leading to reclassification to ALK-positive ALCL. This was further confirmed by IHC using an alternative antibody (D5F3 clone) (Online Supplementary Figure S3).
Paired RT-MLPA profiles and Affymetrix gene expression data available in 72 cases (23 AITL and 49 PTCL-NOS) were compared.18,19 There were significant correlations for each evaluable gene (TNFRSF8/CD30, PRF, GZMB, GATA3, CXCL13, ICOS, CD8, BCL6, CD4, FOXP3, CCR4, CXCR5, and TBX21) (Online Supplementary Figure S4). RT-MLPA and immunohistochemical data scores also showed significant correlations for the 12 evaluable markers (Online Supplementary Figure S5). There was also a good correlation with the EBER in situ hybridization results, showing the capacity of the assay to correctly detect EBV infection. RT-MLPA profiles performed in duplicates in 20 PTCL on RNA extracted from both frozen and FFPE samples, showed a strong correlation (rho>0.7, Spearman correlation test) (Online Supplementary Figure S6).
RT-MLPA identified 33 of 33 RHOAG17V and 9 of 10 IDH2 R172K/T mutations, detected by either AS-quantitative PCR and/or next-generation sequencing (NGS) studies. The only RT-MLPA failure corresponded to an AITL with an IDH2R172K mutant with a 2.8% allele frequency, which was only detected by NGS (Online Supplementary Table S2).
Unsupervised analysis highlights heterogeneity among ALK-negative ALCL and TFH-PTCL
Given the expected heterogeneity of PTCL-NOS, we first restricted our analyses to specified PTCL entities (not taking into account PTCL-NOS). Unsupervised hierarchical clustering performed on 153 such cases (30 AITL, 33 TFH-PTCL, 16 NKTCL, 13 ATLL, 6 HSTL, 21 ALK-positive ALCL, and 34 ALK-negative ALCL) identified two main branches separating cytotoxic and non-cytotoxic entities (Figure 1).
Figure 1.

Unsupervised hierarchical clustering of peripheral T-cell lymphoma (PTCL) entities other than PTCL-not otherwise specified (NOS) using reverse transcriptase-multiplex ligation-dependent probe amplification (RT-MLPA) (n=153). The assay was used to classify angioimmunoblastic T-cell lymphomas (AITL) (n=30), PTCL with T-follicular helper phenotype (TFH) (n=33), anaplastic large cell lymphomas (ALCL) (n=55), adult T-cell lymphomas (ATLL) (n=13), hepatosplenic T-cell lymphoma (HSTL) (n=6), and natural killer (NK)-TCL (n=16). Differential gene expression is depicted according to a red (positive) to blue (negative) color scale, and concordance with histopathological diagnosis (Pathology). Two main branches were observed: the left branch divided in 6 HTSL (C1), 50 TFH-PTCL/AITL (C2), 12 ATLL with 13 TFH-PTCL (C3), and 24 ALK-negative ALCL (C4), and the right branch contained two clusters of 16 NKTCL (C5) and 31 cytotoxic ALCL (C6).
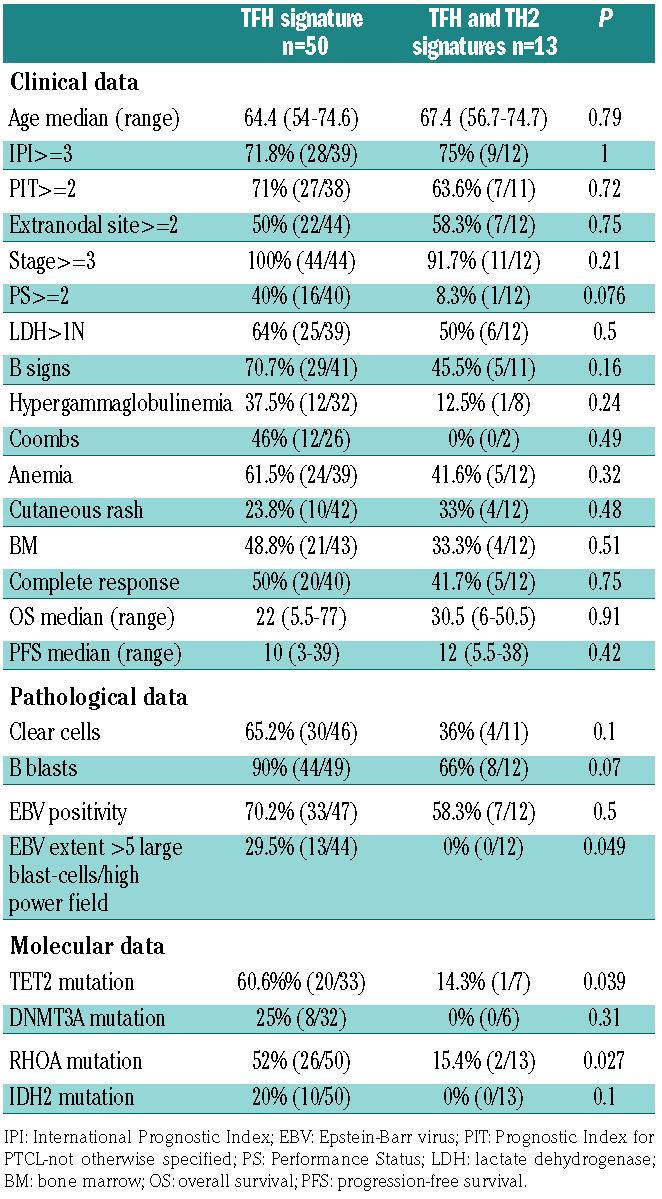
The cytotoxic branch is divided into two clusters, one very homogeneous cluster (C5) comprising the 16 NKTCL, and a second cluster (C6) composed of 31 cytotoxic ALCL (21 ALK-positive and 10 ALK-negative). The other branch is divided into four clusters (C1-4). The C1 cluster contained the six HSTL. The 63 PTCL with a TFH phenotype (AITL and TFH-PTCL) distributed along the two clusters C2 and C3. The C2 cluster comprised a major group of AITL/TFH-PTCL characterized by a TFH signature (C2, n=50). In addition to 12 ATLL, 13 TFH-PTCL and one AITL in the C3 cluster showed an enrichment in TFH and Th2 markers. Interestingly, RHOA mutations were identified in 26 of 50 (52%) and 2 of 13 (15%) of the C2 and C3 clusters, respectively (P=0.027). By immunohistochemistry, these TFH-PTCL in the C3 cluster showed expression of GATA3 (in more than 50% of tumor cells) in 9 of 12 (75%) contributive cases (Figure 2). The C4 cluster contained 24 ALK-negative non-cytotoxic ALCL with Th2 signature, with 8 of 16 contributive cases showing DUSP22 rearrangement by FISH.
Figure 2.

Nodal peripheral T-cell lymphomas (PTCL) with a double TFH/Th2 phenotype and a molecular Th2 signature. (A) Diffuse proliferation of large pleomorphic cells; this case would be classified as TFH PTCL according to the World Health Organization 2017, based on the expression of 2 TFH markers, i.e. PD1 (B) and BCL6 (C), but disclosed strong nuclear staining for GATA3 in virtually all tumor cells (D) and, although less uniform, FOXP3 (E). Few tumor cells also expressed CD30 (F). Reverse transcriptase-multiplex ligation-dependent probe amplification (RT-MLPA) profile showed a Th2 signature and classified in the Th2 class by the support vector machine (SVM).
PTCL-NOS distribute among distinct clusters using unsupervised clustering
When applied to all 230 PTCL samples (including 77 PTCL-NOS), unsupervised clustering showed that the majority of PTCL-NOS (n=48 of 77, 62.3%) clustered within four of the six previous clusters as they showed gene signatures in common with molecular TFH-PTCL (C2, n=6), TFH/Th2 PTCL (C3, n=19), NKTCL (C5, n=5), and cytotoxic ALCL (C6, n=18) (Figure 3). Despite a variable expression of CD30 by immunohistochemistry, 18 PTCL-NOS distributed within the cluster of cytotoxic ALCL based on the expression of cytotoxic markers plus TBX21 and IFN, consistent with a possible Th1 origin. Accordingly, 8 of 12 of these cases tested for TBX21 by immunohistochemistry were positive. This molecular subgroup is further referred to as “cytotoxic/Th1 PTCL” according to its signature. In addition, 19 other PTCL-NOS cases, all with negative HTLV-1 serologies, clustered with ATLL, based on an enrichment in Th2 molecules GATA3 and CCR4, and are referred to as molecular “Th2 PTCL”. Finally, 29 PTCL-NOS did not cluster within any of the defined branches, and segregated with 19 other cases (4 AITL, 12 TFH-PTCL, 1 ATLL).
Figure 3.

Unsupervised hierarchical clustering of peripheral T-cell lymphomas (PTCL) including PTCL-not otherwise specified (NOS) (n=230). PTCL-NOS distributed among six defined clusters represented by colored bars under the heat map (from left to right): C1 (red), C2 (green), C3 hepatosplenic T-cell lymphoma (HSTL) (purple), C4 angioimmunoblastic T-cell lymphomas AITL/TFH-derived PTCL (blue and light blue), C5 natural killer (NK)-TCL (yellow), and “cytotoxic anaplastic large cell lymphomas (ALCL)” (orange and red). The 77 PTCL-NOS (gray in Pathology) are distributed among the subgroups.
Support vector machine classifier accurately classifies specified PTCL and identifies subgroups within PTCL-NOS
We next built a support vector machine (SVM) model to assign each case to a class based on the RT-MLPA data (Figure 4A). One hundred and eighty-four PTCL corresponding to the molecular groups defined according to the latter clustering (Figure 3) were used for the construction of the model and to define the molecular classes: 45 TFH-PTCL/AITL, 21 NKTCL, 42 Th2 PTCL-NOS/ATLL, 50 cytotoxic-ALCL, 20 non-cytotoxic ALCL, and the six HSTL. The 46 PTCL which did not cluster within these six defined branches were not considered to develop the classifier. The SVM algorithm accurately assigned 140 of 153 specified PTCL to the correct categories: 16 of 16 NKTCL, 13 of 13 ATLL, 6 of 6 HSTL, 31 of 31 cytotoxic ALCL, 24 of 24 non-cytotoxic ALK-negative ALCL cases, and 50 of 63 AITL/TFH-PTCL. Interestingly, 11 TFH-PTCL without RHOA mutation were classified as molecular Th2 PTCL, one as molecular cytotoxic/Th1, and one AITL was distant from the barycenter of the TFH/AITL class and not classified. Of the 77 remaining PTCL-NOS, 70 (91%) were classified by the SVM as TFH/AITL (C2, n=17), cytotoxic/Th1 PTCL (C6, n=19), ALK-negative ALCL (C6, n=5), NKTCL (C5, n=5), while 24 cases were molecularly designated Th2 PTCL (C3). Finally, seven cases, which were distant to the barycenter of their predicted SVM class (2 Th2, 3 TFH/AITL, 2 cytotoxic/Th1), could not be attributed a molecular class by the SVM. Figure 4B illustrates the subgroups of PTCL-NOS as evidenced in the principal component analysis (PCA). A correlation of the SVM class with the histopathological data of the 77 PTCL-NOS is presented in Online Supplementary Figure S7.
Figure 4.

Bioinformatic model for the analysis of reverse transcriptase-multiplex ligation-dependent probe amplification (RT-MLPA) data. (A) The support vector machine (SVM) model attributes a predicted (rectangle) or provisional (oval) class for each peripheral T-cell lymphoma (PTCL) sample. Post-tests based on ALK and CD30 were designed to distinguish between the subgroups in the heterogeneous “cytotoxic/ALCL-like” category. (B) principal component analysis (PCA) plot of the SVM classification for PTCL-not otherwise specified (NOS) (n=77) showed three main molecular categories among: TFH/angioimmunoblastic T-cell lymphomas (AITL) (blue), Th2 (light blue), and cytotoxic/Th1 (green). The latter also comprised NKTCL-like (yellow) and anaplastic large cell lymphomas (ALCL)-like (orange) subgroups. Seven cases were unclassified (red).
Clinico-pathological correlations of the support vector machine classes
Survival data were available for 88.7% (204 of 230) of the patients. Median duration of follow up was 122 months [interquartile range (IQR) = 80.5-173]. Among the 132 of 175 patients with follow-up data available who received anthracyclin-based chemotherapy, the median overall survival (OS) and progression-free survival (PFS) were 15 months (IQR=6-51.5) and nine months (IQR=3-36), respectively. The outcome of PTCL patients was poor (5-year OS=27%), except for those with ALK-positive ALCL (5-year OS=70%) (Figure 5A).
Figure 5.

Kaplan-Meier survival analysis. (A) Overall survival of the 108 patients corresponding of the main reverse transcriptase-multiplex ligation-dependent probe amplification (RT-MLPA) subgroups: 11 anaplastic large cell lymphomas (ALCL) ALK+, 10 cytotoxic ALCL ALK−, 24 non-cytotoxic ALCL ALK−, and 63 TFH/angioimmunoblastic T-cell lymphomas (AITL). (B) Overall survival of the main RT-MLPA subgroups according to the DUSP22 status within the non-cytotoxic ALCL ALK-negative category. (C) Overall survival of 43 peripheral T-cell lymphomas (PTCL)-not otherwise specified (NOS) according to the molecular status by RT-MLPA (19 cytotoxic/TH1 and 24 TH2). (D) Overall survival of 30 PTCL-NOS according to the immunohistochemistry data (19 GATA3, 11 TBX21).
Considering the two clusters observed among PTCL with a TFH phenotype (Figure 1), we failed to demonstrate any significant clinical difference (Table 2). Interestingly, RHOAG17V mutations were found in 52% and 15.4% respectively among the C2 (TFH) and C3 (Th2) clusters (P=0.02). TET2 mutations were observed in 60.6% and 14.3% of the C2 and C3 clusters (P=0.039). DNMT3A and IDH2 mutations were found respectively in 25% and 20% of the C2 cluster but none within the C3 (P=not significant, n.s).
Table 2.
Clinical, pathological and molecular features of the two subgroups of TFH-peripheral T-cell lymphomas (PTCL).
Among ALCL, non-cytotoxic ALK-negative ALCL were characterized by the expression of Th2 mRNA (GATA3 and CCR4) and GATA3 expression by immunohistochemistry in 11 of 12 informative cases. Non-cytotoxic and cytotoxic ALK-negative ALCL disclosed similar PFS and OS (Figure 5A). However, despite similar main clinical characteristics (Online Supplementary Table S3), patients with a DUSP22-rearrangement (n=8) tended to have a favorable outcome (5-year OS=62.5%) close to that of ALK-positive ALCL patients (70% 5-year OS), compared to the very poor prognosis of patients without DUSP22 rearrangement (n=8) (5-year OS=12.5%; P=0.07) (Figure 5B). No TP63 rearrangement was detected in this series.
Finally, within the limitations of size of the current series, among PTCL-NOS, there was no significant difference in patient’s outcome with respect to their “Th1/cytotoxic” or “Th2” molecular signatures (Figure 5C) or immunohistochemical profiles (Figure 5D).
Reproducibility of the RT-MLPA assay among three centers in routine practice
We evaluated the reproducibility of the RT-MLPA assay in the diagnostic setting by testing 40 FFPE PTCL samples in three independent centers. A concordance in the diagnostic class proposed by the classifier was observed between the three centers for 36 (90%) samples (Table 3), with a strong correlation between the RT-MLPA values for each gene (Online Supplementary Table S4). Among the 36 concordant samples, the SVM class was in accordance with the pathological diagnosis for 32 cases (89%). The four discrepancies consisted of two tumor-cell rich AITL assigned to the Th2/ATLL-like group, as previously observed in the classification cohort, one AITL with a prominent cytotoxic T-cell environment assigned to the cytotoxic/Th1-like group, and one ATLL with a double CD4/CD8 phenotype that was not classified.
Table 3.
Reproducibility of the reverse transcriptase-multiplex ligation-dependent probe amplification (RT-MLPA) assay among three laboratories (Center 1, Center 2, and Center 3).

Discussion
The classification of PTCL is often challenging and poorly reproducible, with a recent study showing a 31.5% rate of discrepancy between the referral and expert diagnoses,4 likely due to the complexity of these rare neoplasms and the wide range of practices among pathologists and laboratories.3 Hsi et al. pointed out the limited number of immunohistochemical markers assessed in routine practice, especially the TFH markers, resulting in a poor characterization of PTCL and a high frequency of PTCL-NOS diagnosed in the US.22 The ligation-dependent RT-PCR assay has been reported to be a simple and robust assay applicable to FFPE samples that can be used to classify DLBCL into GCB or ABC subtypes.23,24 Here, we expanded on this as RT-MLPA can contribute to classify the main specified categories of non-cutaneous PTCL in routine practice. This assay, which can be performed relatively quickly (48 hours), only requires equipment already in regular use, including a thermocycler and genetic fragment size analyzer. The profiles are publicly accessible and easy to interpret through a dedicated website. Finally, the assay is cost-effective (approximately $5 per sample).23 RT-MLPA is a useful tool in combination with pathological evaluation to characterize PTCL, especially when immunohistochemistry is flawed or incomplete. In addition to evaluating the expression of Th-differentiation antigens and markers suitable for immunohistochemistry, the current RT-MLPA assay also provides genetic information, such as RHOA and IDH2 mutations, which are highly relevant for the diagnosis of PTCL of TFH origin,19 even though the RHOA G17V mutation has also been reported in a small minority of ATLL,25 as observed in one case of our series. The accurate classification of specified PTCL other than NOS entities in most cases corroborates the relevance of the designed gene panel. Altogether, although some markers in our RT-MLPA assay might not be useful in every PTCL case, this “one fits all” assay evaluates diagnostic markers covering the different PTCL entities in a systematic and cost-effective way.
In addition, the RT-MLPA assay highlighted the heterogeneity in the gene signature of ALK-negative ALCL and PTCL of TFH origin, as defined in the up-dated WHO classification. We observed that a significant proportion of ALK-negative ALCL display a distinct signature, with expression of CD30 and Th2 genes, but no cytotoxic markers. These cases showed a dense and cohesive pattern of CD30-positive anaplastic large cells but, in contrast to common ALCL, only a few hallmark cells, an absence of EMA, and a frequently preserved T-cell program with most T-cell antigens being retained. Genetically, this group appeared heterogeneous but was enriched in cases of DUSP22/IRF4 rearrangement (in 8 of 16 non-cytotoxic cases vs. in only 1 of 10 cytotoxic ALK- ALCL; P=0.09). Despite the absence of any significant morphological or immunophenotypic difference between cases with or without DUSP22 rearrangement, we further hypothesize that DUSP22 status is of clinical relevance with better survival of DUSP22-rearranged ALCL.11,26 In the absence of TP63 rearrangement (data not shown), further investigations are needed to explore the genetic abnormalities in the DUSP22-non rearranged cases.
Within the umbrella category of nodal PTCL of TFH derivation, comprising AITL and a number of nodal PTCL previously classified under PTCL-NOS but expressing two or more TFH markers, our study interrogates the biological and clinical significance of two subgroups.1 Indeed, whereas the majority (79%) were attributed to the TFH/AITL class by the molecular classifier, a minority disclosed, in addition to TFH markers, enrichment in Th2 genes. We confirmed a “mixed” TFH/Th2 immunophenotype for nine TFH-PTCL showing a Th2 signature by RT-MLPA. It has been reported that Th1 and Th2 cells can express TFH markers, and conversely that TFH cells have the capacity to express Th1 or Th2 cytokines.23,24 It has also been suggested that a subset of TFH cells may originate from Th2 lymphocytes in the presence of B cells and that TFH cells can acquire GATA3 expression.25,26 Overall, these data raise the question of Th cell plasticity and the specificity of the criteria required to assess TFH-derived PTCL. Indeed, the current definition of a TFH phenotype based on two or more TFH markers may have some limitations in certain cases,1 and the integration of genetic markers, made possible by the RT-MLPA assay, may be helpful. In this respect, when comparing the “TFH/AITL” class to the other SVM classes, it appeared that, among the four TFH genes in our RT-MLPA panel (BCL6, CXCL13, CXCR5, ICOS), ICOS and then BCL6 appeared less specific than CXCL13 and CXCR5 (data not shown). Further studies, however, are needed to determine whether cases with a “mixed” TFH-Th2 RNA profile or immunophenotype should be considered to be TFH-PTCL or Th2 PTCL-NOS with associated TFH markers. The almost complete absence of RHOA and TET2 mutation in the cases with a Th2-like profile may support the latter hypothesis.
Our targeted RT-MLPA panel confirmed two subgroups among PTCL-NOS, based on the expression of TBX21 and cytotoxic markers or GATA3 and CCR4 in 39% and 31% of cases, respectively, expanding the findings of previous studies.13,14 The prognostic relevance of these two groups is controversial.27 Within the limits of our retrospective study, we failed to demonstrate any significant correlation with outcome between the Th1 and Th2 molecular or phenotypic subgroups. In addition, all TBX21 cases in our series had a cytotoxic profile by RT-MLPA and immunohistochemistry. Among PTCL-NOS with a cytotoxic phenotype, the RT-MLPA assay highlighted a small group of cases with an EBV signature, now referred to as “nodal T/NK lymphoma EBV-positive” according to the revised WHO classification.1 Whether these cases, confirmed to be EBV-positive in almost all neoplastic cells by EBER ISH, are related to extranodal NK/T lymphoma nasal-type warrants further investigation. Of note, the Th2 signature based on the expression of GATA3 and CCR4 in our panel was characteristic, although not specific, of ATLL. The expression of GATA3 and CCR4 together with variable expression of FOXP3 in ATLL is in accordance with a recent study showing that the HBZ transcript induces CCR4 expression in CD4 T cells by enhancing GATA3 expression, whereas FOXP3 expression was inconsistent in ATLL. However, the distinction between PTCL-NOS with a Th2-like signature and ATLL requires an investigation into HTLV1 serology and/or viral integration.28,29
Finally, our SVM model proposed a class for most cases, with few discrepancies. It may be a useful tool in combination with pathological evaluation, especially when immunohistochemistry is not conclusive or not available. In this series, 23% of cases diagnosed by default as PTCL-NOS due to incomplete or failure in immunohistochemistry were classified as TFH/AITL by our assay. Unclassified or misclassified cases by RT-MLPA were limited to TFH-PTCL or AITL rich in reactive CD8-positive cytotoxic cells known to be abundant in a proportion of AITL,30 or cytotoxic PTCL with various reactive TFH cells. These cases illustrate the contribution of the microenvironment to the molecular signature, especially when tumor cell content is low or heterogeneous, a common problem encountered in all gene-expression methods. Therefore, RT-MLPA results should be interpreted in the light of clinical context, as well as biological and histopathological findings. It is worthy of note that our assay does not provide a final solution for PTCL classification, but rather a step forward that requires extensive reworking.
Overall, this study demonstrates the applicability of a robust and dedicated RT-MLPA assay which is easily transposable to the diagnostic workflow. It is simple to use, can be applied to FFPE and frozen samples, integrates genetic features, and is cost-effective; these all make it an attractive alternative to high-throughput technologies in routine practice. Implementation of RT-MLPA in large studies in the future, especially in the setting of clinical trials, may confirm how this adjunct tool can help better classify PTCL and therefore improve the management of these patients in the era of personalized medicine.
Supplementary Material
Acknowledgments
The authors would like to thank the LYSA-Pathology and the Plateforme de Ressources Biologiques from Henri Mondor Hospital for its technical assistance and the participants of the Tenomic consortium (see Online Supplementary Appendix). This work was supported in part by institutional grants from the Institut National du Cancer (INCA), the Fondation pour la Recherche Médicale (FRM, Equipe Labellisée DEQ20160334875), the Leukemia Lymphoma Society (LLS), the Fondation Force Hémato and the Institut Carnot (CALYM), MEDIC Foundation.
Appendix. Participants of the Tenomic consortium
A. Martin, Hôpital Avicenne, Bobigny, France; I. Soubeyran, P. Soubeyran, Institut Bergonié, Bordeaux, France; A. Pilon, O. Tournilhac, CHU Estaing, Clermont-Ferrand, France; P. Gaulard, C. Copie-Bergman, M.H. Delfau, J. Moroch, E. Poullot, F. Lemonnier, F. Le Bras, J. Dupuis, C. Haioun, Hôpital Henri Mondor, Créteil, France; T. Petrella, L. Martin, JN. Bastié, O. Casasnovas CHU,Dijon, France; B. Fabre, R. Gressin, CHU, Grenoble, France; L. de Leval,B. Bisig, E. Missiaglia, MP Dobay, A. Cairoli, CHUV, Lausanne, Suisse; C. Bonnet, J. Somja, CHU Sart-Tilman,Liège, Belgique; M.C. Copin, B. Bouchindhomme, F. Morschhauser, CHU, Lille, France; B. Petit, M. Delage, A. Jaccard, Hôpital Dupuytren, Limoges, France; F. Berger, B. Coiffier, A. Traverse-Glehen, L. Genestier, E. Bachy, CHU Sud, Lyon, France; T. Rousset, G. Cartron, V. Szablewski, Hôpital Gui de Chauliac-St Eloi, Montpellier, France; S. Thiebault, B. Drenou, Hôpital E. Muller, Mulhouse, France; K. Montagne, C. Bastien, S. Bologna, CHU de Brabois, Nancy, France; C. Bossard, S. Le Gouill, Hôtel-Dieu, Nantes, France; J. Brière, V. Meignin, C. Gisselbrecht, J. Soulier, Hôpital St Louis, Paris, France; B. Fabiani, A. Aline-Fardin, P. Coppo, Hôpital Saint-Antoine, Paris, France; F. Charlotte, J. Gabarre, Hôpital Pitié-Salpétrière, Paris, France; T. Molina, J. Bruneau, D. Canioni, E. Macintyre, V. Asnafi, D. Sibon, R. Delarue, JP Jaïs, Hôpital Necker, Paris, France; M. Parrens, J.P. Merlio, K. Bouabdallah, Hôpital Haut Lévêque, Bordeaux, France; S. Maugendre-Caulet, P. Tas,F. Llamas-Gutierrez T. Lamy, CHU Pontchaillou, Rennes, France; J.M. Picquenot, E.L Veresezan, F. Drieux, P. Ruminy, F. Jardin, C. Bastard, Centre H Becquerel, Rouen, France; M. Peoch, J. Cornillon, CHU, Saint Etienne, France; L. Lamant, C. Laurent, L. Ysebaert, Hôpital Purpan, Toulouse, France; J. Bosq, P. Dartigues, V. Ribrag, P. Dessen, G Meurice, Institut G Roussy, Villejuif, France; M. Patey, A. Delmer, Hôpital R. Debré, Reims, France; J.F. Emile, K. Jondeau, Hôpital Ambroise Paré, Boulogne, France; M.C. Rousselet, M. Hunault, A. Clavert, CHU, Angers, France ; C. Legendre, S. Castaigne, A.L. Taksin, CH Versailles, Le Chesnay, France; J. Vadrot, B. Joly, A. Devidas, CH Sud Francilien, Corbeil, France; G. Damaj, CHU Caen, France; F Radvanyi, E. Chapeaublanc, Institut Curie, Paris, France; S. Spicuglia, CIML, Marseille, France; C. Thibault, IGBMC, Illkirsch, France; V. Fataccioli, project coordinator, Hôpital Henri Mondor, Créteil, France.
Footnotes
Check the online version for the most updated information on this article, online supplements, and information on authorship & disclosures: www.haematologica.org/content/105/6/1582
References
- 1.Swerdlow S., Campo E., Harris NL, et al. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues (IARC WHO Classification of Tumours). Revised 4th Edition. Vol. 2 2017. [Google Scholar]
- 2.Herrera AF, Crosby-Thompson A, Friedberg JW, et al. Comparison of referring and final pathology for patients with T-cell lymphoma in the National Comprehensive Cancer Network: Second-Opinion Pathology Review of TCL. Cancer. 2014;120(13):1993–1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hsi ED, Said J, Macon WR, Rodig SJ, et al. Diagnostic Accuracy of a Defined Immunophenotypic and Molecular Genetic Approach for Peripheral T/NK-cell Lymphomas: A North American PTCL Study Group Project. Am J Surg Pathol. 2014;38(6):768–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Laurent C, Baron M, Amara N, et al. Impact of Expert Pathologic Review of Lymphoma Diagnosis: Study of Patients From the French Lymphopath Network. J Clin Oncol. 2017;35(18):2008–2017. [DOI] [PubMed] [Google Scholar]
- 5.Lemonnier F, Couronne L, Parrens M, et al. Recurrent TET2 mutations in peripheral T-cell lymphomas correlate with TFH-like features and adverse clinical parameters. Blood. 2012;120(7):1466–1469. [DOI] [PubMed] [Google Scholar]
- 6.Couronné L, Bastard C, Bernard OA. TET2 and DNMT3A mutations in human T-cell lymphoma. N Engl J Med. 2012;366(1):95–96. [DOI] [PubMed] [Google Scholar]
- 7.Cairns RA, Iqbal J, Lemonnier F, et al. IDH2 mutations are frequent in angioimmunoblastic T-cell lymphoma. Blood. 2012;119(8):1901–1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Manso R, Sánchez-Beato M, Monsalvo S, et al. The RHOA G17V gene mutation occurs frequently in peripheral T-cell lymphoma and is associated with a characteristic molecular signature. Blood. 2014;123(18):2893–2894. [DOI] [PubMed] [Google Scholar]
- 9.Palomero T, Couronné L, Khiabanian H, et al. Recurrent mutations in epigenetic regulators, RHOA and FYN kinase in peripheral T cell lymphomas. Nat Genet. 2014;46(2):166–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sakata-Yanagimoto M, Enami T, Yoshida K, et al. Somatic RHOA mutation in angioimmunoblastic T cell lymphoma. Nat Genet. 2014;46(2):171–175. [DOI] [PubMed] [Google Scholar]
- 11.Parrilla Castellar ER, Jaffe ES, Said JW, et al. ALK-negative anaplastic large cell lymphoma is a genetically heterogeneous disease with widely disparate clinical outcomes. Blood. 2014;124(9):1473–1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Iqbal J, Weisenburger DD, Greiner TC, et al. Molecular signatures to improve diagnosis in peripheral T-cell lymphoma and prognostication in angioimmunoblastic T-cell lymphoma. Blood. 2010;115(5):1026–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Iqbal J, Wright G, Wang C, et al. Gene expression signatures delineate biological and prognostic subgroups in peripheral T-cell lymphoma. Blood. 2014;123(19):2915–2923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang T, Feldman AL, Wada DA, et al. GATA-3 expression identifies a high-risk subset of PTCL, NOS with distinct molecular and clinical features. Blood. 2014;123(19):3007–3015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang W, Wang Z, Luo Y, Zhong D, Luo Y, Zhou D. GATA3 expression correlates with poor prognosis and tumor-associated macrophage infiltration in peripheral T cell lymphoma. Oncotarget. 2016;7(40):65284–65294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Heavican TB, Bouska A, Yu J, et al. Genetic drivers of oncogenic pathways in molecular subgroups of peripheral T-cell lymphoma. Blood. 2019;133(15):1664–1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.de Jong D, Rosenwald A, Chhanabhai M, et al. Immunohistochemical Prognostic Markers in Diffuse Large B-Cell Lymphoma: Validation of Tissue Microarray As a Prerequisite for Broad Clinical Applications—A Study From the Lunenburg Lymphoma Biomarker Consortium. J Clin Oncol. 2007;25(7):805–812. [DOI] [PubMed] [Google Scholar]
- 18.de Leval L, Rickman DS, Thielen C, et al. The gene expression profile of nodal peripheral T-cell lymphoma demonstrates a molecular link between angioimmunoblastic T-cell lymphoma (AITL) and follicular helper T (TFH) cells. Blood. 2007;109(11): 4952–4963. [DOI] [PubMed] [Google Scholar]
- 19.Dobay MP, Lemonnier F, Missiaglia E, et al. Integrative clinicopathological and molecular analyses of angioimmunoblastic T-cell lymphoma and other nodal lymphomas of follicular helper T-cell origin. Haematologica. 2017;102(4):e148–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alirkilicarslan AL, Dupuy A, Pujals A, et al. Expression of TFH markers and detection of RHOA p. G17V and IDH2 p. R172K/S mutations in cutaneous localizations of angioimmunoblastic T-cell lymphomas. Am J Surg Pathol. 2017;41(12):1581–1592. [DOI] [PubMed] [Google Scholar]
- 21.Dupuy A, Lemonnier F, Fataccioli V, et al. Multiple ways to detect IDH2 mutations in angioimmunoblastic T cell lymphoma: from immunohistochemistry to Next Generation Sequencing. J Mol Diagn. 2018;20(5):677–685. [DOI] [PubMed] [Google Scholar]
- 22.Hsi ED, Horwitz SM, Carson KR, et al. Analysis of Peripheral T-cell Lymphoma Diagnostic Workup in the United States. Clin Lymphoma Myeloma Leuk. 2017; 17(4):193–200. [DOI] [PubMed] [Google Scholar]
- 23.Mareschal S, Ruminy P, Bagacean C, et al. Accurate Classification of Germinal Center B-Cell–Like/Activated B-Cell–Like Diffuse Large B-Cell Lymphoma Using a Simple and Rapid Reverse Transcriptase–Multiplex Ligation-Dependent Probe Amplification Assay: A CALYM Study. J Mol Diagn. 2015;17(3):273–283. [DOI] [PubMed] [Google Scholar]
- 24.Bobée V, Ruminy P, Marchand V, et al. Determination of Molecular Subtypes of Diffuse Large B-Cell Lymphoma Using a Reverse TranscriptaseMultiplex Ligation-Dependant Probe Amplification Classifier: a CALYM Study. J Mol Diagn. 2017;19(6): 892–904. [DOI] [PubMed] [Google Scholar]
- 25.Nagata Y, Kontani K, Enami T, et al. Variegated RHOA mutations in adult T-cell leukemia/lymphoma. Blood. 2016;127(5): 596–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pedersen MB, Hamilton-Dutoit SJ, Bendix K, et al. DUSP22 and TP63 rearrangements predict outcome of ALK-negative anaplastic large cell lymphoma: a Danish cohort study. Blood. 2017;130(4):554–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Maura F, Agnelli L, Leongamornlert D, et al. Integration of transcriptional and mutational data simplifies the stratification of peripheral T cell lymphoma. Am J Hematol. 2019;94(6):628–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Matutes E. Adult T-cell leukaemia/lymphoma. J Clin Pathol. 2006;60(12):1373–1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Licata MJ, Janakiram M, Tan S, et al. Diagnostic challenges of adult T-cell leukemia/lymphoma in North America – a clinical, histological, and immunophenotypic correlation with a workflow proposal. Leuk Lymphoma. 2018;59(5):1188–1194. [DOI] [PubMed] [Google Scholar]
- 30.Gaulard P, de Leval L. The microenvironment in T-cell lymphomas: Emerging themes. Semin Cancer Biol. 2014;24:49–60. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.