

Abstract
BRD4, a member of the bromodomain and extraterminal domain (BET) family, has emerged as a promising epigenetic target in cancer and inflammatory disorders. All reported BET family ligands bind within the bromodomain acetyl-lysine binding sites and competitively inhibit BET protein interaction with acetylated chromatin. Alternative chemical probes that act orthogonally to the highly-conserved acetyl-lysine binding sites may exhibit selectivity within the BET family and avoid recently reported toxicity in clinical trials of BET bromodomain inhibitors. Here, we report the first identification of a ligandable site on a bromodomain outside the acetyl-lysine binding site. Inspired by our computational prediction of hotspots adjacent to nonhomologous cysteine residues within the C-terminal BRD4 bromodomain (BRD4-BD2), we performed a midthroughput mass spectrometry screen to identify cysteine-reactive fragments that covalently and selectively modify BRD4. Subsequent mass spectrometry, NMR, and computational docking analyses of electrophilic fragment hits revealed a novel ligandable site near Cys356 that is unique to BRD4 among human bromodomains. This site is orthogonal to the BRD4-BD2 acetyl-lysine binding site as Cys356 modification did not impact binding of the pan-BET bromodomain inhibitor JQ1 in fluorescence polarization assays nor an acetylated histone peptide in AlphaScreen assays. Finally, we tethered our top-performing covalent fragment to JQ1 and performed NanoBRET assays to provide proof of principle that this orthogonal site can be covalently targeted in intact human cells. Overall, we demonstrate the potential of targeting sites orthogonal to bromodomain acetyl-lysine binding sites to develop bivalent and covalent inhibitors that displace BRD4 from chromatin.
Graphical Abstract
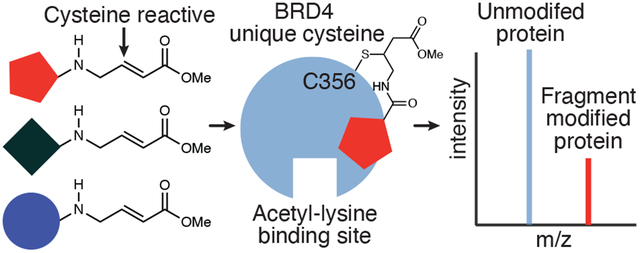
Introduction
Lysine acetylation is an abundant protein post-translational modification (PTM) (1, 2) impacting diverse cellular processes (3, 4). Histone lysine acetylation was originally identified as a transcriptionally activating PTM because acetylation neutralizes electrostatic histone-DNA interactions, decondensing chromatin and allowing access of transcription machinery (5). Beyond modulating histone-DNA contacts, lysine acetylation mediates histone-protein interactions that define the chromatin transcriptional state (6). Acetylation-dependent histone-protein interactions are primarily mediated by bromodomains; ~110 amino-acid (aa) protein modules that selectively bind acetyl-lysine residues (7). Of the 61 human bromodomains (7), eight are distributed across four proteins (BRDT, BRD2, BRD3 and BRD4) that comprise the bromodomain and extraterminal domain (BET) family. Each BET protein contains two N-terminal bromodomains in tandem followed by an extraterminal protein-protein interaction domain (8). The tandem bromodomains and extraterminal domain function together to recruit transcription factors to sites of chromatin acetylation (9).
Since their discovery in 2010, pan-BET inhibitors like JQ1 (10) that non-selectively bind the acetyl-lysine binding sites of all eight BET bromodomains with equal nanomolar potency have enhanced understanding of BET bromodomain function. Pan-BET inhibitors were quickly found to have potential therapeutic uses in cancer (11–15), fibrosis (16, 17), as well as cardiovascular (18, 19), inflammatory (20–22) and autoimmune disease (23, 24). At least 14 pan-BET inhibitors (many of which bear structural resemblance to JQ1) are currently in clinical trials, predominantly for various forms of cancer (25). However, dose-limiting toxicities remain a barrier to the progression of pan-BET inhibitors through FDA-approval (26–29) and highlight the need to develop more selective chemical tools to study BET bromodomain function and pave the way toward safer therapeutics.
The four BET family members have overlapping but non-redundant functions in how they regulate transcription and their roles in pathogenesis and disease progression (8, 30). Functional N- and C-terminal bromodomains (BD1 and BD2, respectively) are both necessary to afford full BET protein activity (8, 31, 32). As a result, small molecules targeting subsets of BET bromodomains may address certain pathogenic processes more specifically than pan-BET inhibitors. Across BET proteins, BD1 and BD2 show less homology (~44% identity) within the same BET protein compared to the greater homology of the four BD1 or the four BD2 bromodomains (>75% identity) across BET proteins (33). Consequently, the only successful attempts to selectively target subsets of the eight BET bromodomains have targeted all four BD1 (34, 35) or all four BD2 bromodomains (36), which has yielded small molecules with biological effects distinct from those of pan-BET inhibitors. For instance, treatment of oligodendrocyte progenitor cells with olinone, a compound with ~100-fold selectivity for BD1 compared to BD2 BET bromodomains, facilitates cellular differentiation while this process is inhibited by pan-BET inhibitors (35). MS402 binds BRD4-BD1 with ~10-fold selectivity relative to BRD4-BD2 (37) and selectively inhibits Th17 cell differentiation compared to pan-BET inhibitors that broadly affect Th1, Th2 and Th17 cell differentiation (37). RVX-208, a compound exhibiting 15- to 30-fold selectivity toward BD2 compared to BD1 BET bromodomains (36), induces Apolipoprotein A-I and high-density lipoprotein and has emerged as a potential therapeutic in clinical trials of cardiovascular disease (38–40).
Improving the potency of pan-BET inhibitors may lower therapeutic doses and avoid toxicities and off-target effects. For instance, inhibitors that bivalently engage both BD1 and BD2 within a BET protein (e.g. MT1) demonstrate subnanomolar affinities (41) and potencies in cellular and in vivo assays orders of magnitude greater than monovalent counterparts (42). Treatment with bivalent pan-BET inhibitors results in prolonged and transcriptionally distinct effects relative to monovalent inhibitors (41, 43). In addition to effects resulting from enhanced affinity, differences between bivalent and monovalent inhibitors may be attributed to induction of large conformational changes of the tandem bromodomains by bivalent inhibitors. Irreversible BET inhibition is another promising strategy to maximize the potency of BET-targeted molecules. Proteolysis targeting chimeras (PROTACs) that target BET proteins for degradation have been developed by linking pan-BET inhibitors to Von Hippel-Lindau or Cereblon E3 ligase ligands (44, 45). These BET degraders demonstrated longer lasting c-Myc suppression and stronger antiproliferative effects in lymphoma and leukemia cell lines compared to monovalent reversible pan-BET inhibitors (44, 45). Pre-clinical studies of BET PROTACs have shown positive effects (46, 47) and demonstrate transcriptional effects that differ from those caused by reversible pan-BET inhibitors (48). Covalent ligands represent another route to irreversible BET inhibition. Epoxide-conjugated BET inhibitors that covalently target a conserved methionine within BET acetyl-lysine binding sites exhibited prolonged transcriptional and anti-proliferative effects in leukemia cell lines compared to non-covalent compounds (49).
To date, no compounds have been reported that selectively bind an individual BET protein. The lack of selectivity of pan-BET inhibitors is a barrier to the elucidation of the biological functions of individual BET proteins and the design of effective BET-targeted therapies (50). BRD4 is the most studied BET protein and carries out crucial transcriptional functions through direct interactions with positive transcription elongation factor b (P-TEFb) (51–53) and Mediator (54, 55). Consequently, BRD4 is thought to be the relevant target in the majority of pathogenic contexts associated with positive responses to pan-BET inhibition (56). However, in many cases this hypothesis is not fully supported as functions of BRD4 are often ascribed based on the effects of pan-BET inhibitors (50). Therefore, chemical tools that selectively target BRD4 are desired.
Here, we identified a novel small molecule binding site on BRD4-BD2 and present the first reported strategy for chemically targeting a bromodomain using a site orthogonal to the acetyl-lysine binding site. Computational analyses identified potential small-molecule binding sites adjacent to nonhomologous cysteine residues that are unique to BRD4-BD2 among human bromodomains. Screening a library of 200 cysteine-reactive fragments against BRD4-BD2 using intact protein mass spectrometry (MS) demonstrated that the site near BRD4 Cys356 could be accessed by small molecules. Selective covalent labeling of BRD4-BD2 relative to other BET bromodomains was shown using MS, differential scanning fluorimetry (DSF) and nuclear magnetic resonance (NMR) spectroscopy. The most efficient covalent fragment was tethered to the pan-BET inhibitor JQ1 (10) using a polyethylene glycol (PEG) linker to design a novel class of chemical tools that target BRD4 in NanoBRET cell-based assays in a bivalent and covalent manner. We anticipate further optimization of covalent fragments that target Cys356 will lead to BRD4-selective chemical probes and starting points for BRD4-selective drug development.
Results and Discussion
BRD4-BD2 contains two nonhomologous cysteines next to predicted binding sites
BRD4-BD2 contains two nonhomologous cysteine residues (Cys356 and Cys391) not present in the remaining seven BET bromodomains (Figure 1A). More broadly, these cysteine residues are unique to BRD4-BD2 among the 61 bromodomains encoded in the human genome (7). Furthermore, the Cys356 and Cys391 sulfhydryls are both surface exposed in published crystal structures (e.g. PDB ID: 4KV4). FTMap analysis (57) identified a strong (cluster strength [CS] = 18) hot spot ~5 Å from the Cys356 sulfhydryl (Figure 1B). A weak hot spot (CS = 3) was predicted ~10 Å from the Cys391 sulfhydryl (Figure 1C). FTMap hot spots with CS > 16 are predicted to be ligandable. For reference, the highly ligandable BRD4-BD2 acetyl-lysine binding site (10, 21) was predicted to contain three separate hotspots (CS = 24, 19 and 5) (Figure 1D). This analysis suggested we may be able to discover novel small molecules that bind near and covalently modify Cys356 of BRD4.
Figure 1:
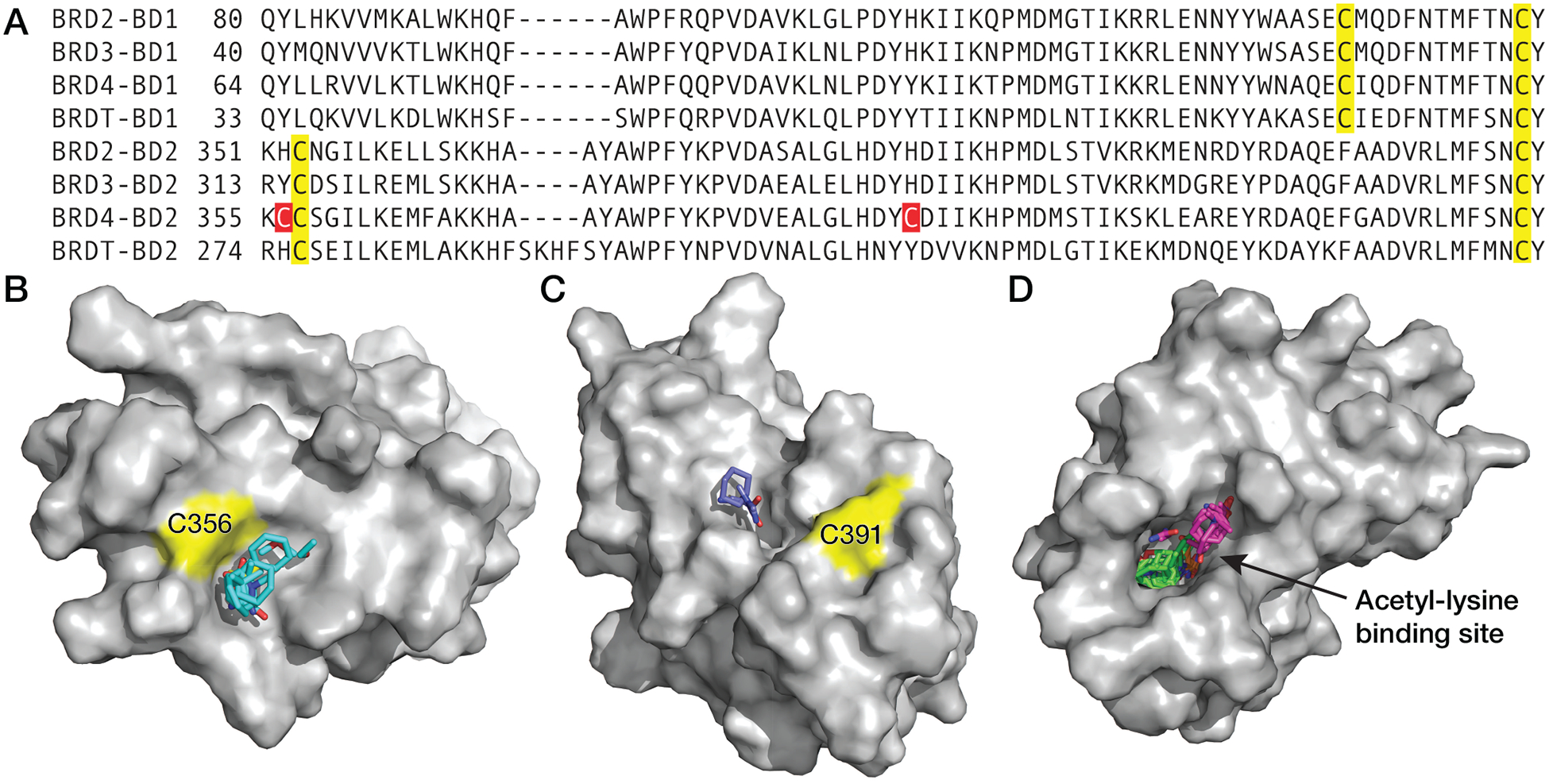
The second bromodomain of BRD4 (BRD4-BD2) contains two nonhomologous and surface exposed cysteine residues. (A) Structure-based sequence alignment of BET family bromodomains demonstrating BRD4-BD2 contains two nonhomologous cysteine residues (red) not present in other BET bromodomains and three homologous cysteine residues (yellow) that are present in the BD1, BD2, or all BET bromodomains. (B) FTMap analysis of BRD4-BD2 (PDB ID: 4KV4) identified a strong hot spot near BRD4 Cys356 (cluster strength = 18), predicting this site is highly ligandable (CS > 16) (57). (C) A weak hot spot was also identified near BRD4 Cys391 (CS = 3). (D) For comparison, three separate hot spots were predicted within the deep and hydrophobic acetyl-lysine binding site (CS = 24, 19 and 5).
Cysteine-reactive fragment screen yields selective and destabilizing BRD4-BD2 modifiers
To identify small molecules that bind at predicted sites adjacent to nonhomologous BRD4-BD2 cysteine residues, 200 fragments that all contain a cysteine-reactive Michael acceptor functional group (58) were screened against BRD4-BD2 in pools of 10 by intact protein mass spectrometry (MS) (Figure 2A & S1; Table S1). In this screen, 20 μM BRD4-BD2 was incubated with pools of 10 compounds, each at 100 μM (1 mM combined fragment concentration) for 8 hours at 25 °C. Twenty protein/fragment complexes were detected with relative abundance ≥20% of the unmodified BRD4-BD2 base peak and mass error <1 Da, resulting in an initial 10% hit rate. Of these twenty fragments, seven were selected for further investigation according to fragment availability (Figure 2B). Treatment of 10 μM BRD4-BD2 with 1 mM each of the seven fragments for 8 hours at 25 °C induced biphasic DSF curves (probability of biphasic vs. monophasic fit >3.3×1022 for 26, 66, 67, 115, 142, and 154 and >2.4×104 for 56) (Figure 2C; Table S2), consistent with mixtures of unmodified and covalently-modified BRD4-BD2. In contrast, BRD4-BD2 samples containing DMSO alone produced monophasic DSF curves (probability monophasic vs. biphasic fit >4.7) (Table S2), consistent with a uniform unmodified protein population. Of the seven fragments tested, five (26, 66, 115, 142, and 154) significantly decreased the melting temperature of BRD4-BD2 between 6.9 and 8.1 °C (Table S2). In contrast, JQ1 binding to the acetyl-lysine binding site was previously shown to increase the thermal stability of BRD4-BD2 by 7 to 9 °C (10), suggesting a unique binding site for these fragments relative to BET bromodomain acetyl-lysine binding site ligands. The four fragments (26, 66, 115, and 142) producing the greatest change in thermal stability measured by DSF were further stratified according to the rate they modify 100 μM BRD4-BD2 at 1 mM fragment concentration over the course of 8 hours (25 °C) as measured by intact protein MS (Figure 2D & S2; Table S3). Fragments 142, 26, and 66 demonstrated the fastest kinetics of covalent reaction with second order rate constants of 2.84 ± 0.16, 3.05 ± 0.11, and 2.61 ± 0.14 M−1s−1, respectively (Figure 2E; Table S4), and were selected for further analysis.
Figure 2:
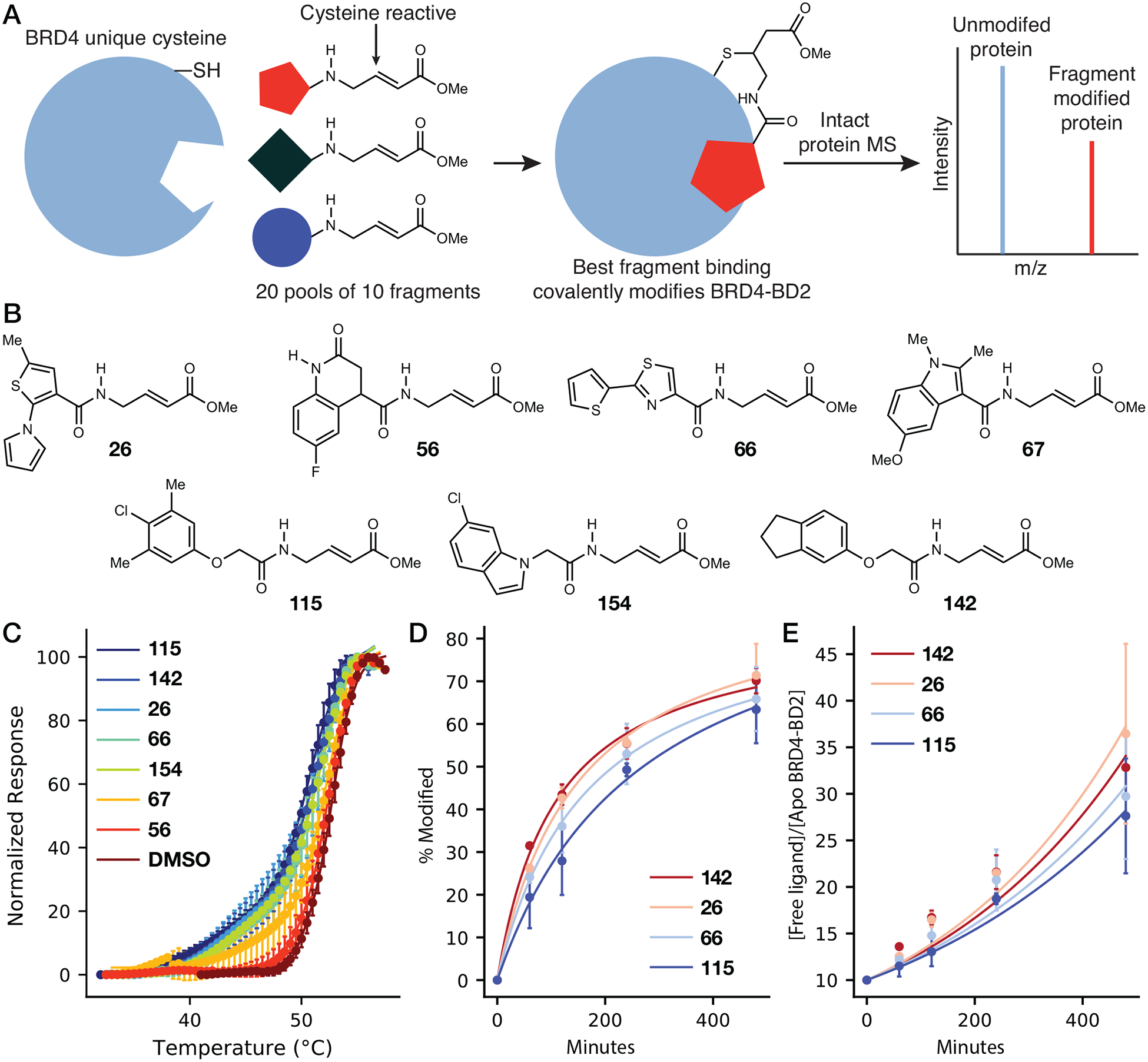
An intact protein mass spectrometry-based screen identifies cysteine reactive fragments that covalently modify BRD4-BD2, resulting in thermal destabilization. (A) Schematic depicting covalent fragment screening using intact protein mass spectrometry (MS). (B) Chemical structures of electrophilic fragment hits chosen from the primary intact protein MS screen. (C) Traces resulting from differential scanning fluorimetry (DSF) analysis of BRD4-BD2 after incubation with electrophilic fragment hits or DMSO. Each point represents an average of three replicates. Covalent fragment labeling decreases the thermal stability of BRD4-BD2 and results in biphasic curves. (D) Intact protein MS time course of BRD4-BD2 modification after incubation with covalent fragments. At each time point, the singly modified BRD4-BD2 peak was divided by the sum of the singly modified peak and the base peak (Figure S2) to yield values corresponding to fraction of BRD4-BD2 modified. (E) Pseudo-first order rate analysis of BRD4-BD2 covalent fragment labeling over the time course shown in (D).
Counterscreen against other human bromodomains demonstrates selectivity towards BRD4-BD2 cysteines
To determine if fragments 26, 66, and 142 selectively bind and react at a unique cysteineadjacent site on BRD4-BD2 among BET bromodomains, the reactivity of each fragment toward BRD4-BD1 and BRD3-BD2 was compared to that of BRD4-BD2. Together, BRD4-BD1 and BRD3-BD2 contain all three homologous cysteine residues in BET bromodomains (Figure 1A) (7). The 142 methyl ester was further tested for reactivity towards the additional BET bromodomains BRDT-BD1, BRD2-BD2, and BRD3-BD1 (Figure S3A–C; Table S5). Since BET bromodomains comprise only one of the eight phylogenetic families of human bromodomains (family II) (7), covalent labeling of 142 methyl ester was also tested toward human bromodomains from other families including PCAF (family I), CBP (III), BRD9 (IV), TAF1-BD2 (VII), and PBRM1-BD6 (VIII) (Figure S3D–H; Table S5). Each fragment-protein mixture was incubated for 8 hours (25 °C) at a 100:1 molar ratio (1 mM fragment, 10 μM bromodomain) and subsequently analyzed using intact protein MS (Figure 3 & S3; Table S5). For BRD4-BD2, 91.4, 77.1 and 64.2% of the total bromodomain peak intensity corresponded to single modification by fragments 142, 26, and 66, respectively. In contrast, <4.% of total bromodomain peak intensity corresponded to covalent modification when either BRD4-BD1 or BRD3-BD2 was incubated with any of these three fragments. Furthermore, incubation of 142 methyl ester with additional BET bromodomains or bromodomains from other phylogenetic families (see above) did not result in detectable levels of covalent modification by 142 methyl ester. TAF1-BD2 (family VII) was the exception to this trend as incubation with 142 methyl ester resulted in 16.7% of total bromodomain signal intensity corresponding to covalent modification (Figure S3G), which is modest reactivity compared to that measured for BRD4-BD2 (91.4%) under the same conditions. These analyses are consistent with all three fragments being selective for BRD4-BD2 over other BET bromodomains (family II). In addition, 142 methyl ester demonstrated selectivity toward BRD4-BD2 among other human bromodomains from phylogenetic families I, II, III, IV, VII, and VIII. We hypothesized this selectivity resulted from selective fragment reactivity toward one of the two nonhomologous cysteine residues (Cys356 or Cys391) in BRD4 (Figure 1A).
Figure 3:
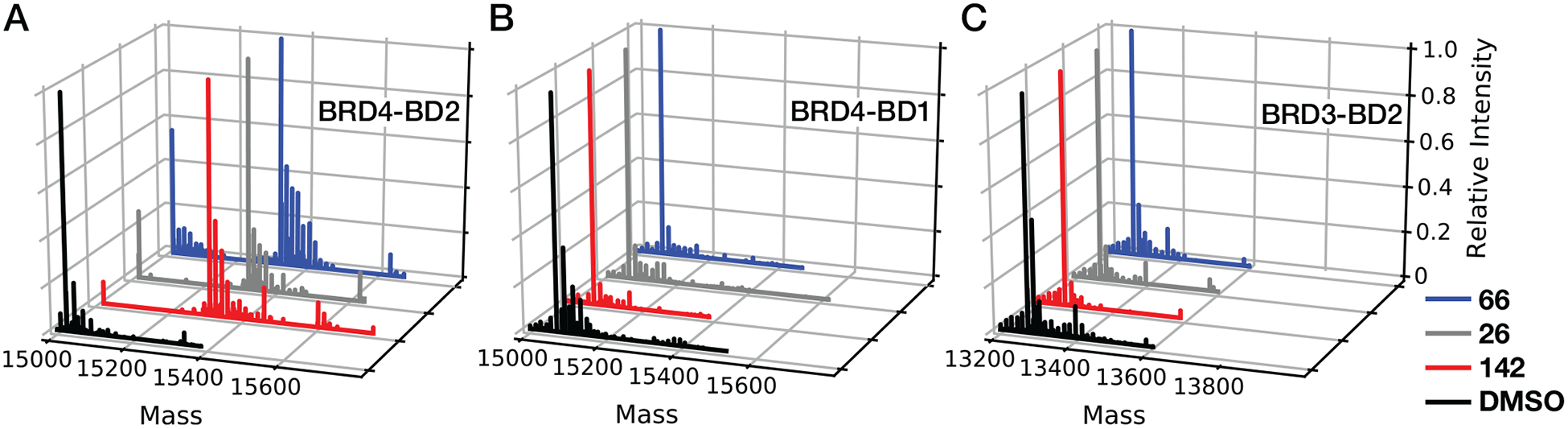
Covalent fragment hits exhibit selective reactivity toward BRD4-BD2 relative to other BET bromodomains. Deconvoluted MS spectra of mixtures containing (A) BRD4-BD2, (B) BRD4-BD1, or (C) BRD3-BD2 after incubation with DMSO (black) or fragment 142 (red), 66 (blue), or 26 (grey). Since BRD4-BD1 and BRD3-BD2 together contain all three homologous BET bromodomain cysteine residues (highlighted in yellow in Figure 1A), these results suggest the three covalent fragment hits selectively react with one of the two BRD4 nonhomologous cysteine residues, Cys356 or Cys391 (Figure 1A).
Fragment hits selectively modify BRD4 Cys356
Top-down MS fragmentation of covalently-modified BRD4-BD2 indicated either Cys356 or Cys357 (p = 7.2×10−39) is the primary site of reaction with 142 methyl ester (Figure S4; Table S6), the most potent BRD4-BD2 modifier (Figure 3A). Although top-down MS fragmentation analysis could not distinguish between Cys356 and Cys357, we hypothesized Cys356 was the predominately modified cysteine since Cys357 is invariant among all BD2 BET bromodomains (BRD2-BD2 and BRD3-BD2 contain a homologous cysteine residue to Cys357 but were not modified by the covalent fragments; Figure 1A, 3C, S3B) and Cys357 is not solvent exposed in BRD4-BD2 crystal structures (see e.g. PDB ID: 4KV4). To establish Cys356 as the site of BRD4-BD2 modification, fragments 26, 66 and 142 were incubated with BRD4-BD2 C356A for 8 hours (25 °C) at a 10:1 molar ratio and analyzed using intact protein MS (Figure 4; Table S7). To further stratify fragment hits according to selectivity toward BRD4 Cys356, BRD4-BD2 concentrations were increased to 100 μM (compared to 10 μM in previous screens; Figure 3) to favor potentially less kinetically favored reactions with additional BRD4-BD2 cysteine residues and provide a more stringent test of cysteine selectivity. Indeed, the resulting MS spectra of BRD4-BD2 WT contain a third major peak corresponding to modification by two fragments (Figure 4). Of the compounds tested, 142 methyl ester demonstrated the greatest selectivity toward a single cysteine residue on BRD4-BD2 (59.5% of total BRD4-BD2 peak intensity corresponding to single modification, compared with 43.4 and 47.3% for fragments 66 and 26, respectively). We hypothesize at higher protein concentrations, these fragments react at the surface exposed Cys391 residue (Figure 1C) or a conserved cysteine residue (Figure 1A) in addition to Cys356 in BRD4-BD2. Initial modification of Cys356 thermally destabilizes BRD4-BD2 (Figure 2C), perhaps rendering Cys391 or another cysteine residue more accessible to covalent reaction with fragments. In contrast to BRD4-BD2 WT, spectra of BRD4-BD2 C356A after incubation with each fragment only exhibit two major peaks corresponding to unlabeled bromodomain and modification by a single fragment. Furthermore, only 19.2, 16.7, and 17.7% of total BRD4-BD2 C356A peak intensity corresponded to modification by fragments 66, 26, and 142, respectively, compared to 73.3, 78.8, and 73.1% for the WT construct. These results confirm identified electrophilic fragments primarily react covalently at Cys356 within BRD4-BD2 with 142 methyl ester exhibiting the greatest selectivity for Cys356.
Figure 4:
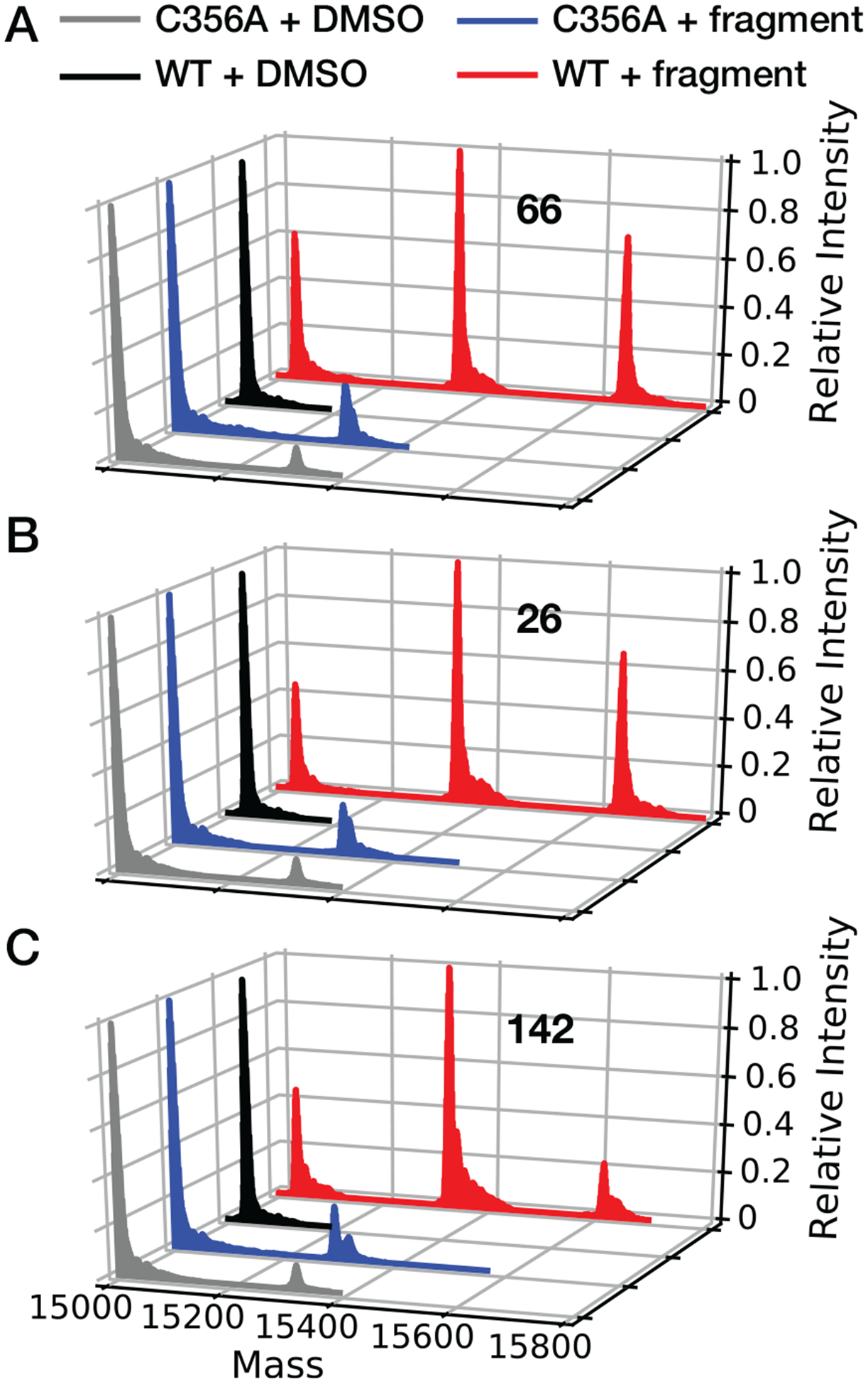
Cys356 is the primary site of BRD4-BD2 modification by covalent fragment hits. Deconvoluted MS spectra resulting from mixtures containing WT (red and black) or C356A BRD4-BD2 (blue and gray) and fragment (A) 26, (B) 66, or (C) 142. C356A mutation results in >85% loss of modification levels for all fragments examined.
Structural mapping of a ligandable site on BRD4-BD2 orthogonal to the acetyl-lysine binding site
To further elucidate the fragment binding site surrounding Cys356, NMR 1H, 15N heteronuclear single-quantum coherence (HSQC) experiments were performed with 500 μM uniformly 15N-labeled BRD4-BD2 (Figure S5) and spectra collected from samples treated with 1 mM 142 methyl ester for 8 hours at 25 °C were compared to spectra collected from a DMSO control (Figure S6). Five residues in BRD4-BD2 underwent chemical shift perturbations (CSPs) that were greater than two standard deviations above the mean CSP across the entire protein (Figure 5A). Covalent docking using the CovDock module developed by Schrödinger (59), predicted poses in which 142 methyl ester occupied the hot spot (CS = 18) identified adjacent to Cys356 in FTMap analysis (Figure 1B, 5B). In the 23 crystal and NMR structures of BRD4-BD2 deposited in the PDB that resolve Cys356, the amide nitrogen atoms of each of the five residues (S351, E352, Q353, G359 and F450) exhibiting the greatest CSPs are located on average 10, 8, 6, 7 and 12 Å, respectively, from the Cys356 sulfur atom. S351, E352, Q353, and G359 comprise the N-terminal surface of the BRD4-BD2 αZ helix (7), directly adjacent to Cys356 (Figure 5B). F450 resides within the bromodomain core on the C helix (7), consistent with a global shift in the orientation of the four-helix bundle that may be responsible for the significant decrease in thermal stability upon covalent modification of Cys356 (Figure 2C). In addition, of the nine residues undergoing CSPs between one and two standard deviations above the mean, five (K362 on the αZ helix and A415, E451, M457 and E460 on the C helix) include amide nitrogen atoms that reside between 10 and 13 Å from the Cys356 sulfur atom. The remaining 4 residues (W374, K395, E438 and A441) are located >20 Å from Cys356 and likely are affected by a global shift in the four-helix bundle tertiary structure upon Cys356 modification. However, any conformational changes induced by covalent fragment modification of Cys356 did not impact binding of JQ1 (Figure S7A) or a biotinylated histone H4 peptide diacetylated at lysines 5 and 8 (H4K5/8diacetyl) (Figure S7B) to the BRD4-BD2 acetyl-lysine binding site. This lack of detectable impact on JQ1 or acetylated histone peptide binding is consistent with the large distances (28 ± 5 Å) between the backbone nitrogens exhibiting CSP magnitudes greater than two standard deviations above the mean after modification by 142 methyl ester and the side-chain nitrogen of the Asn433 residue necessary for acetyl-lysine binding. In contrast, titration of 15N-labeled BRD4-BD2 with a H4K5/8diacetyl peptide induced the greatest magnitude CSPs (Figure 5C–D) at residues (A371, Y372, D381, Y390, and D392) that include backbone nitrogen atoms located relatively close to the Asn433 side-chain nitrogen atom (12 ± 2 Å) compared to the Cys356 side-chain sulfur atom (27 ± 6 Å). Collectively, these results indicate 142 methyl ester binds at a novel site on BRD4-BD2 adjacent to Cys356 that is completely orthogonal to the acetyl-lysine binding site.
Figure 5:
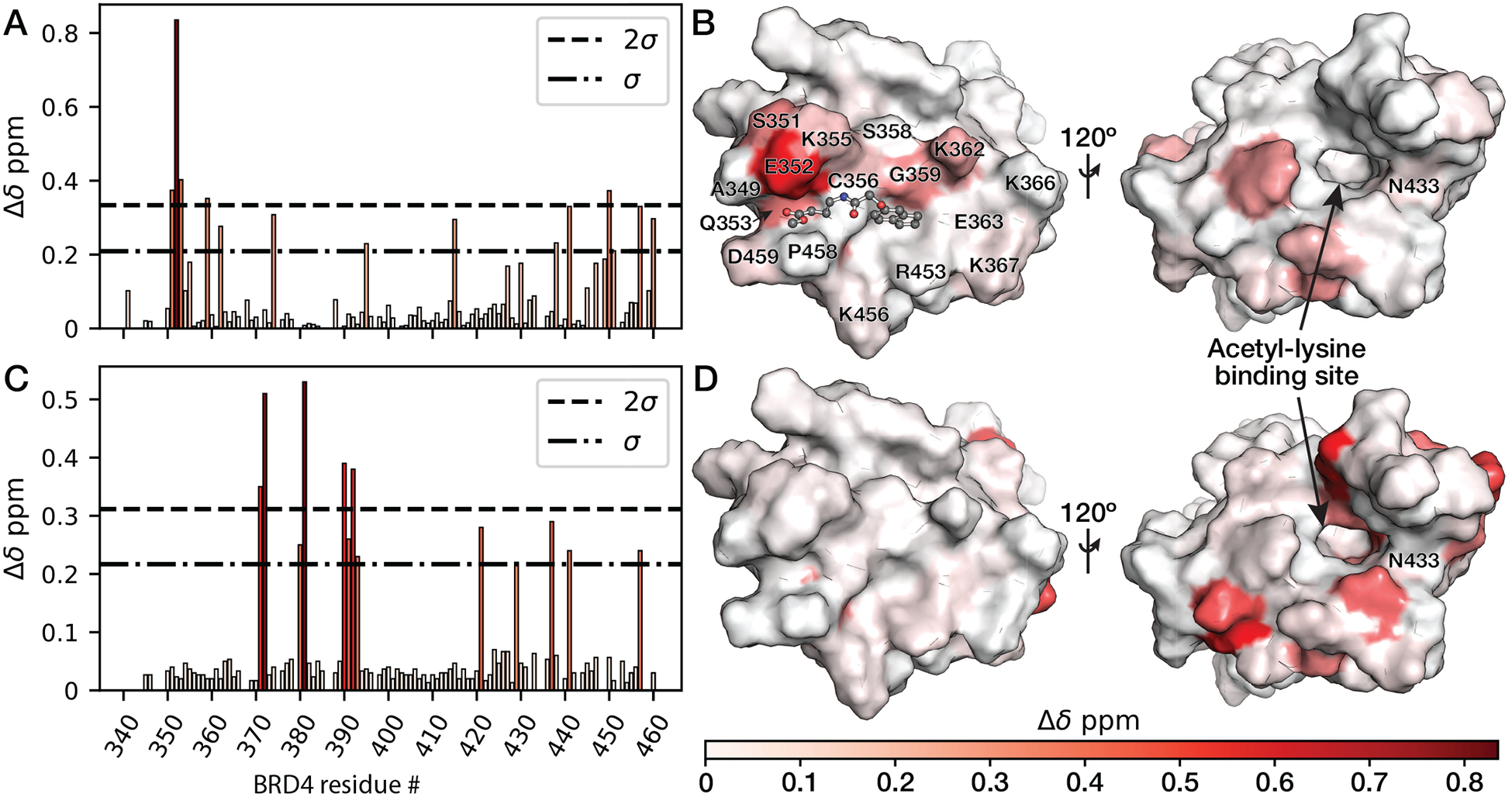
The 142 methyl ester binds to a site that is near Cys356 and orthogonal to the acetyl-lysine binding site. (A) Bar graph depicting BRD4-BD2 1H, 15N CSP magnitudes upon covalent modification with 142 methyl ester in 1H, 15N HSQC experiments. (B) Mapping of CSPs induced by 142 methyl ester in NMR HSQC experiments onto a structure of 142 methyl ester covalently docked at BRD4-BD2 (PDB ID: 4KV4) Cys356 by Michael addition using the Schrödinger small-molecule discovery suite. (C) Bar graph depicting 1H, 15N CSP magnitudes after titration with a histone H4 (residues 1–12) peptide diacetylated at lysines 5 and 8. (D) Mapping of CSPs induced by the diacetylated histone H4 peptide onto a crystal structure of BRD4-BD2 (PDB ID: 4KV4).
Fragment linking to design bivalent and covalent BRD4 inhibitors with cellular activity
To determine if covalent fragments could alkylate BRD4-BD2 Cys356 in a cellular context and begin developing a novel strategy for selective BRD4 inhibition, fragment 142 was tethered to the pan-BET bromodomain inhibitor JQ1 (10) using a polyethylene glycol (PEG) linker to create JQ1-PEG2-142 (Figure 6A). Since previous studies demonstrated various amide substitutions of the JQ1 t-butyl ester group minimally affect BET bromodomain binding (60, 61), we hypothesized the JQ1 side of JQ1-PEG2-142 would maintain targeting of all eight BET bromodomains with nanomolar affinity. In addition, we hypothesized that the covalent fragment half of JQ1-PEG2-142 would selectively and covalently react with only BRD4 Cys356 among BET cysteine residues when the JQ1 half is bound to BRD4-BD1 (Figure 6B), resulting in bivalent and covalent inhibition of BRD4. To test this hypothesis, a previously-described BRD4 bioluminescence resonance energy transfer (BRET) biosensor construct (41) was used consisting of the BRD4 tandem bromodomains flanked dually on the C-terminus by a nanoluciferase (NL) tag and on the N-terminus by a HaloTag (HT). In this construct, the NL tag and fluorophore-labeled HT produce NanoBRET signal in a manner that inversely correlates with the distance separating the two bromodomains (Figure 6C). JQ1-PEG2-142 treatment of HEK293 cells transfected with the BRD4 NanoBRET biosensor construct resulted in increased NanoBRET signal (EC50 = 247 ± 64 nM; Table S8), indicating JQ1-PEG2-142 induces a conformational change in BRD4 that brings the two bromodomains into proximity (Figure 6C). The bivalent BET bromodomain inhibitor MT1 that simultaneously binds the acetyl-lysine binding sites of both BD1 and BD2 of all four BET proteins (42) and the monovalent BET bromodomain inhibitor JQ1 (10) were used as positive and negative controls for induction of NanoBRET signal from the BRD4 biosensor construct (Figure 6C). Importantly, JQ1-PEG2-142 did not induce a detectable increase in NanoBRET signal with the BRD4 C356A mutant biosensor construct (Figure 6C), indicating Cys356 is necessary for the mechanism of bidentate engagement of BRD4 by JQ1-PEG2-142. As a control for expression and proper folding of the C356A biosensor, MT1 maintained induction of NanoBRET signal for the C356A biosensor with an EC50 value (62 ± 7 nM) within 2-fold that of MT1 with the wild-type construct (43 ± 6 nM) (Table S8). Since the NanoBRET signal decreased at higher MT1 concentrations, likely due to a high-dose hook effect (62) commonly observed at high concentrations of multivalent ligands, MT1 concentrations above 1 μM were not included in nonlinear fitting to determine EC50 values. In addition, the potency of JQ1-PEG2-142 in increasing NanoBRET signal with the wild-type biosensor construct directly correlated with the potency of JQ1-PEG2-142 in displacing N-terminal NL-tagged full-length BRD4 from C-terminal HT histone 3.3 in HEK293 cells (IC50 = 105 ± 62 nM) (Figure 6D; Table S9). In these studies, JQ1-PEG2-142 displaced NL-BRD4 from chromatin with a potency within error of that exhibited by JQ1 and MT1 (135 ± 69 nM and 80 ± 64, respectively; Figure 6D). In summary, these data suggest the covalent fragment portion of JQ1-PEG2-142 can engage a novel orthogonal binding site adjacent to Cys356 in intact human cells.
Figure 6:
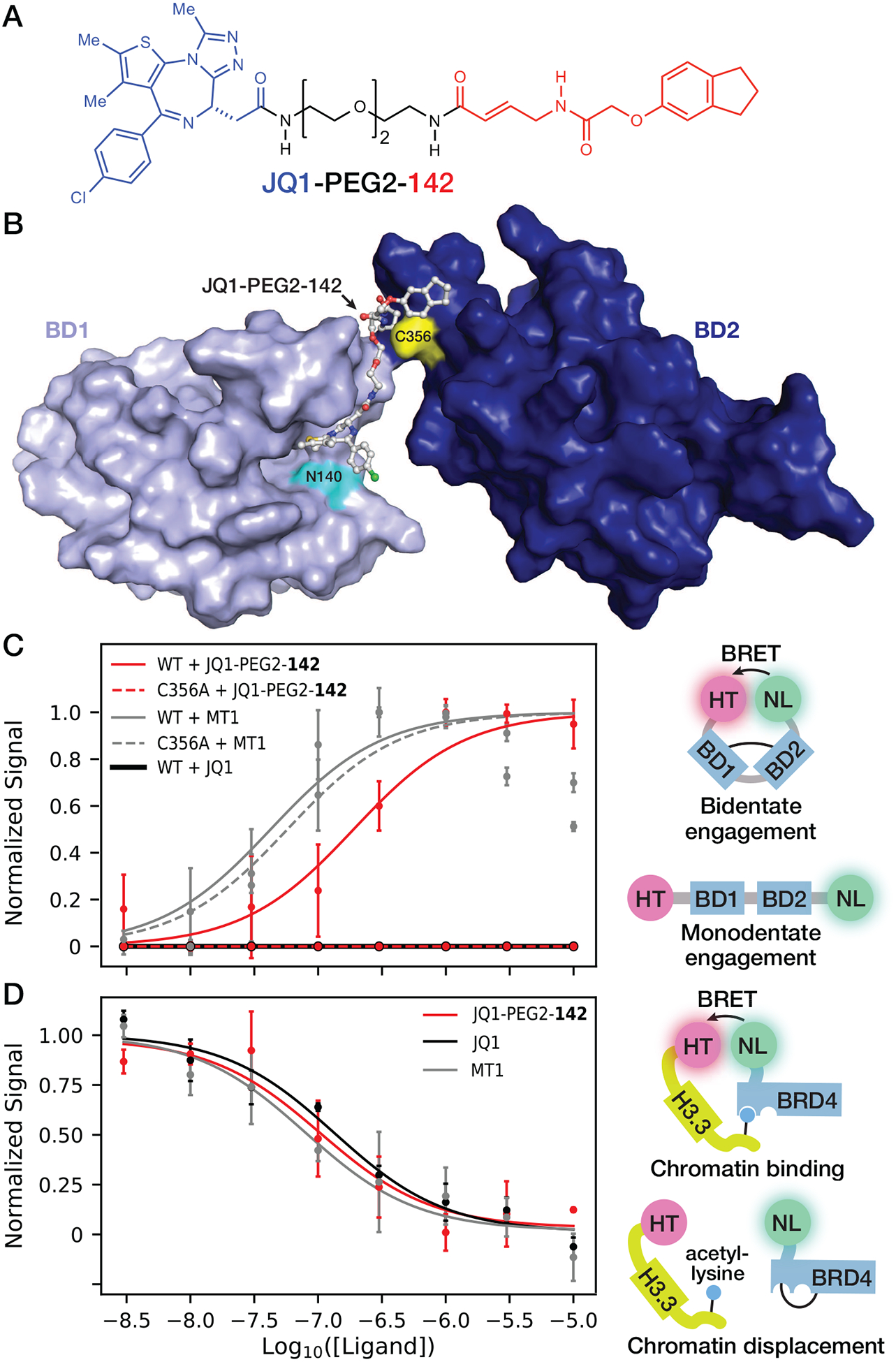
Chemically tethering fragment 142 to JQ1 yields bivalent inhibitors of BRD4 that are active in intact cells. (A) Chemical structure of JQ1 tethered to fragment 142 via a PEG linker. (B) Rosetta model of BRD4-BD1 (PDB ID: 3MXF) docked onto the JQ1 side of JQ1-PEG2-142 after covalent docking to BRD4-BD2 Cys356 (PDB ID: 4KV4) using Schrödinger CovDock (59). (C) NanoBRET assay for bivalent binding to the BRD4 tandem bromodomains. HEK293 cells transfected with either WT or C356A mutant HT-BRD4-BD1/BD2-NL biosensor were treated with 3 nM to 10 μM JQ1-PEG2-142, JQ1 or MT1 and NanoBRET signal was measured as an indicator of conformational change induced by bivalent compound binding. (D) HEK293 cells co-transfected with NL-BRD4 and histone H3.3-HT were titrated with 3 nM to 10 μM JQ1-PEG2-142, JQ1 or MT1 and NanoBRET signal was measured as an indicator of NL-BRD4 displacement from chromatin incorporated with histone H3.3-HT. For NanoBRET experiments, each replicate was normalized according to theoretical minima and maxima of the three-parameter curve fits. Each point represents the average of two or more normalized biological replicates.
Potential for covalent-fragment screening to identify novel chemical probes of bromodomain-containing epigenetic proteins
BET proteins are potential therapeutic targets in a broad range of pathological processes (6, 56, 63). However, recent reports of toxicity and lack of efficacy at tolerable doses in clinical trials (26, 28) raise concerns about the promise of pan-BET bromodomain inhibitor FDA-approval. An untested hypothesis is that inhibitors of individual BET proteins may maintain therapeutic activity while avoiding toxic side-effects under specific disease contexts (25, 50). However, the widespread use of pan-BET bromodomain inhibitors has made it difficult to assign functions to specific BET family members. Of the three somatic BET proteins (BRD2, BRD3 and BRD4), only BRD4 contains an extended C-terminus that regulates P-TEFb-mediated RNA polymerase II activation (51–53, 64, 65) and is involved in recruiting the Mediator transcription co-activation complex to promoters and enhancers (66–68). These activities uniquely implicate BRD4 in activating MYC transcription (69, 70) and facilitating NF-κB-mediated inflammatory cytokine production (71–73). As a result, BRD4-selective chemical probes are greatly desired to determine whether additional inhibition of BRD2 and BRD3 is therapeutically useful or toxic in models of cancer and inflammation driven disease. To date, there are no known compounds that selectively target BRD4 bromodomains with nanomolar cellular activity. For example, Ouyang et al. described a compound (FL-411) exhibiting >25-fold selectivity toward the BRD4 bromodomains over to other BET bromodomains and inhibited proliferation of breast cancer cell lines with IC50 values >1.6 μM (74). Another recent study reported a xanthine derivative that binds BRD4-BD1 with >10-fold affinity compared to other BET bromodomains, but the usefulness of this probe is limited by relatively weak affinity (Kd = 5 μM) (75). Otherwise, attempts to pharmacologically target individual BET bromodomains have been unsuccessful due to the high sequence and structural similarity of the acetyl-lysine binding sites. These difficulties highlight the utility of developing novel strategies to chemically target bromodomains that do not rely solely on small molecule binding within highly conserved acetyl-lysine binding sites.
Development of covalent probes has gained renewed interest in recent years, particularly following FDA-approval of covalent proteasome and kinase inhibitors (76). In addition, recent proteome-wide screens using libraries of cysteine-reactive small-molecules have identified covalently-ligandable sites on regions of proteins that have not previously been targeted by or accessible to reversible ligands (77–80). Covalent bromodomain inhibition was recently achieved through addition of epoxides (49) or dichlorotriazines (81) to known reversible bromodomain ligands. An alternative approach to develop covalent probes involves screening reactive molecules against a protein target to identify novel binding sites near reactive residues (82, 83). Here, we screened a library of electrophilic fragments with defined cysteine reactivity (58) using intact protein MS to identify a novel small molecule binding site adjacent to a nonhomologous cysteine residue on the surface of BRD4-BD2, but orthogonal to the acetyl-lysine binding site (Figure 1). Initial fragment hits were stratified based on compound availability, impact on BRD4-BD2 thermal stability and kinetics of covalent modification (Figure 2). The three fragment hits (26, 66 and 142) selected for further analysis exhibited selectivity for BRD4-BD2-specific cysteine residues over homologous cysteine residues within BRD3-BD2 and BRD4-BD1 (Figure 3). Fragments 26, 66 and 142 each were further shown to predominantly covalently modify the nonhomologous cysteine Cys356 within BRD4-BD2 (Figure 4). Although modification by all three fragments thermally destabilized BRD4-BD2, no significant effect was observed on ligand binding to the BRD4-BD2 acetyl-lysine binding site (Figure S7). These results are consistent with the discovery of a novel small-molecule binding site on BRD4-BD2 adjacent to BRD4 Cys356 that is completely orthogonal to the acetyl-lysine binding site (Figure 5).
To incorporate additional BET bromodomain targeting into covalent probe design, we tethered the pan-BET inhibitor JQ1 (10) to the most efficient BRD4-BD2-labeling covalent fragment 142 using a PEG linker (Figure 6A). In silico modeling studies (Figure 6B) indicated the PEG2 linker is not long enough for the JQ1 portion to simultaneously bind to the BRD4-BD2 acetyl-lysine binding site while the covalent fragment portion covalently reacts with Cys356. As a result, we hypothesized the JQ1 portion of JQ1-PEG2-142 would maintain binding to all BET bromodomains and only when bound to BRD4-BD1 would the electrophilic portion of each molecule react with Cys356, aided by higher effective concentrations of the covalent fragment near BRD4-BD2 on the same protein (Figure 6B). JQ1-PEG2-142 induced increased NanoBRET signal emitted by a BRD4 tandem bromodomain biosensor in HEK293 cells, consistent with closer proximity between N- and C-terminal BRD4 bromodomains in the presence of JQ1-PEG2-142 (Figure 6C). While this NanoBRET assay cannot distinguish between bivalent binding to BD1 and BD2 on the same BRD4 protein (intramolecular) versus on two separate BRD4 proteins (intermolecular), we hypothesize relatively high effective concentrations of two bromodomains on the same BRD4 protein favors intramolecular binding, as demonstrated in previous studies of bivalent BET bromodomain inhibitors (41).
JQ1-PEG2-142 displaced NL-BRD4 from chromatin containing H3.3-HT in HEK293 cells with a potency that was within error of that for JQ1 and MT1 (Figure 6D). Additionally, treatment with JQ1-PEG2-142 and JQ1 also attenuated c-Myc expression in K562 chronic myelogenous leukemia cells to a similar extent (Figure S8). These similar potencies between JQ1-PEG2-142 and JQ1 in cell-based assays of BRD4 bromodomain inhibition imply conjugation of fragment 142 neither results in significant undesired reactivity toward other cellular thiols nor impairs JQ1-PEG2-142 cell permeability and entry into the nucleus.
To determine the concentration of covalent fragment at which global reactivity toward cysteines within a cellular proteome is observed, HEK293T and K562 cell lysates were treated with alkyne versions of fragment 142 linked via an ester or amide (142 propargyl ester or 142 propargyl amide). Detectable non-specific protein thiol labeling was not observed below fragment concentrations of 100 μM (Figure S9). Similarly, 142 methyl ester and 142 methyl amide did not compete with alkyne-conjugated iodoacetamide (IA-alkyne) for cysteine labeling in HEK293T and K562 cell lysates below fragment concentrations of 100 μM (Figure S10). Importantly, the fragment 142 concentrations required to observe global cysteine labeling were >10-fold higher than those needed for JQ1-PEG2-142 to achieve maximal displacement of NL-BRD4 from H3.3-HT in HEK293T cells (Figure 6D). Furthermore, our inability to detect increased NanoBRET signal from the C356A mutant BRD4 biosensor (Figure 6C) indicates Cys356 is a relevant site of covalent modification by fragment 142 in intact human cells when the local concentration is high enough due to JQ1 binding to BRD4-BD1.
We had hoped bivalent and covalent properties associated with BRD4 binding by JQ1-PEG2-142 would afford increased potency and selectivity in displacing BRD4 from chromatin compared with JQ1 alone. Compounds such as MT1 that bivalently engage both acetyl-lysine sites within BET tandem bromodomains have exhibited >10 fold increased potency in cell-based assays compared to equivalent monovalent inhibitors (41, 43), although we observed similar potency between JQ1 and MT1 in our NL-BRD4/H3.3-HT displacement assays suggesting that we may already be at the limit of detection for this NanoBRET assay (Figure 6D; Table S9). Inhibitors that covalently react within BET bromodomain acetyl-lysine binding sites also demonstrate enhanced potency in cell-based assays compared to equivalent unreactive compounds (49). Conversely, the reversible unreactive BET bromodomain inhibitors used in this previous study inhibited BRD4 tandem bromodomain binding to histone H4 peptides with up to 30-fold weaker IC50 values compared to JQ1 (49). Therefore, tethering a weaker (micromolar) affinity reversible BET bromodomain ligand to our covalent fragments may afford a greater dynamic range when measuring additional affinity gained through covalent modification of BRD4 compared to our current strategy that used JQ1, which already binds with nanomolar affinity. In addition, increased potency may not have been exhibited by JQ1-PEG2-142 relative to JQ1 because of relatively low levels of Cys356 covalent modification in a cellular context. Relatively low levels of covalent modification may have resulted from our amide linking strategy for generating JQ1-PEG2-142, which reduced the cysteine reactivity of the Michael acceptor relative to 142 methyl ester. Indeed, replacing the ester with an amide in 142 (Figure S10) and the alkyne-conjugated version of fragment 142 (Figure S9) substantially impaired fragment labeling of cysteine residues in cell lysates. These results are consistent with NMR rate studies demonstrating the Michael acceptor in 142 methyl ester reacts more rapidly with N-acetylcysteine (6.94 × 10−4 ± 2 × 10−6 M−1s−1) compared to 142 methyl amide (5.63 × 10−4 ± 6 × 10−6 M−1s−1) (Figure S11). In future studies, we plan to explore additional JQ1-PEG2-142 analogs with higher cysteine-reactivity and compounds that contain weaker binding BD1-selective BET bromodomain inhibitors (34, 35) in favor of the nanomolar binding pan-BET inhibitor JQ1.
Conclusions
This study identifies a novel small molecule binding site within BRD4 that is accessible through covalent targeting of Cys356. This is the first report of chemical probes targeting a bromodomain surface orthogonal to the acetyl-lysine binding site. As Cys356 is unique to BRD4 and absent in all other human bromodomains (7), this study lays the groundwork for future development of chemical probes with BRD4-specific properties. In addition, bivalent binding modes accessed by JQ1-PEG2-142 imposes spatial constraints on BRD4 tandem bromodomains that may impact protein-protein interactions at surfaces orthogonal to the bromodomain acetyl-lysine binding sites. Screening approaches, like the approach described here, seeking out small molecule binding sites orthogonal to bromodomain acetyl-lysine binding sites will be important to probe acetylation-independent functions of bromodomain-containing proteins and identify unexpected means of achieving selectivity. Generation of novel BET protein chemical probes acting orthogonal to existing BET inhibitors that target the acetyl-lysine binding site will enhance investigations of BET protein biology and help address critical questions and toxicity issues regarding the potential of BET inhibitors as therapeutics in the clinic.
Methods
FTMap computational analysis
Detection of ligandable surfaces on BRD4-BD2 was carried out using FTMap (57). The structure of BRD4 (PDB ID: 4KV4) was submitted to the FTMap server using the default settings (http://ftmap.bu.edu/ (57)). The PyMOL Molecular Graphics System (Version 2.3, Schrödinger LLC) was used to analyze results and render figures.
Covalent fragment intact protein mass spectrometry screen
Recombinantly purified BRD4-BD2 (10 μM) in 50 mM HEPES, 150 mM NaCl, and 0.1 mM EDTA pH 7.5 was combined with mixtures of 10 fragments (10 mM DMSO stock solutions, final concentrations: 100 μM of each fragment and 1% v/v DMSO). The reaction mixture was incubated for 8 h at 25 °C before being quenched by passage through Zeba gel filtration columns (Thermo Fisher Scientific, 7 kDa MWCO) to remove unreacted fragments. Protein solutions were immediately applied to Peptide Protein C18 300 Å UltraMicroSpin Columns (3–30 μg), washed with 10 × 200 μL H2O (0.1% v/v formic acid) and eluted with 80% v/v acetonitrile in H2O (0.1% v/v formic acid). Protein samples were diluted to 50% v/v acetonitrile in H2O (0.1% v/v formic acid) prior to intact protein MS analysis via direct injection using a Thermo Q Exactive Hybrid Quadropole instrument in positive ion mode. Raw MS spectra were deconvoluted to neutral monoisotopic masses using the Xtract algorithm implemented by the Thermo Scientific Xcalibur software with default parameters (i.e. signal-to-noise threshold for peak detection = 2 using charge states ≤ 10). For single compound validations and bromodomain selectivity, 10 μM of individual bromodomains were incubated for 8 h with 1 mM of fragment 26, 66 or 142 (1% v/v DMSO) and prepared and analyzed by intact protein MS as described above. For BRD4-BD2 C356A labeling experiments, 100 μM of either BRD4-BD2 WT or BRD4-BD2 C356A were combined with 1 mM fragment 26, 66 or 142, prepared as described above and analyzed by intact protein LC-MS using a Thermo LTQ XL Linear Ion Trap instrument. The resulting MS spectra were deconvoluted to neutral average masses using the Zscore algorithm implemented by the MagTran software (84) with default parameters (i.e. Charge range = 0 – 100, signal-to-noise threshold for peak detection = 5, mass accuracy = 0.05 Da, and charge was determined using the charge envelope only). Spectra were plotted to include all deconvoluted masses detected that were greater than the mass for the corresponding unmodified bromodomain.
Differential scanning fluorimetry
Mixtures containing 10 μM BRD4-BD2 in 50 mM HEPES, 150 mM NaCl and 0.1 mM EDTA pH 7.5 and 1 mM fragment with 1% v/v DMSO or DMSO control were incubated at 25 °C for 8 h, at which time reactions were quenched by passage through Zeba gel filtration columns (7 kDa MWCO). Labeled protein solutions were diluted 1:2 with 10× SYPRO orange (Sigma-Aldrich) dye (final concentrations: 5 μM BRD4-BD2, 5× SYPRO orange). 30 μL of each mixture was added to a PCR plate, and melting curves were generated by monitoring SYPRO Orange fluorescence in FRET mode over a temperature gradient of 20–95 °C over 50 min using a CFX96 real-time PCR detection system (Bio-Rad). Fluorescence values before the minimum and after 2 readings past the maximum fluorescence were excluded from curve fitting. The resulting curves were normalized to RFU values between 0 and 100 and fit using the python lmfit package to either monophasic or biphasic Boltzmann Sigmoidal equations, respectively:
and monophasic versus biphasic fits were selected for each sample based on the Akaike information criterion.
Covalent labeling mass spectrometry timecourses
Mixtures containing 100 μM BRD4-BD2 in 50 mM HEPES, 150 mM NaCl and 0.1 mM EDTA pH 7.5 and 1 mM fragment with 1% v/v DMSO were incubated at 25 °C for 0, 1, 2, 4, and 8 h. Reactions were quenched by passage through Zeba gel filtration columns (7 kDa MWCO) and the protein samples were immediately applied to Peptide Protein C18 300 Å UltraMicroSpin Columns (3–30 μg), washed with 10 × 200 μL H2O (0.1% v/v formic acid) and eluted with 80% v/v acetonitrile in H2O (0.1% v/v formic acid). Protein samples were diluted to 50% v/v acetonitrile in H2O (0.1% v/v formic acid) prior to LC-MS analysis using an LTQ XL Linear Ion Trap instrument. MS spectra were deconvoluted to neutral average masses using the Zscore algorithm implemented by the MagTran software (84) with default parameters (i.e. Charge range = 0 – 100, signal-to-noise threshold for peak detection = 5, mass accuracy = 0.05 Da, and charge was determined using the charge envelope only). At each time point, the fraction of covalently modified BRD4-BD2 was determined by dividing the deconvoluted BRD4-BD2 + fragment peak area by the sum of the deconvoluted BRD4-BD2 and BRD4-BD2 + fragment peak areas.
Protein-detected NMR spectroscopy
To study the binding mode of 142 methyl ester, 300 μL of 500 μM uniformly labeled 15N-BRD4-BD2 was incubated with 1 mM 142 methyl ester (1% v/v DMSO) or DMSO control for 8 h and reactions were quenched as described above. The resulting protein samples were dialyzed into 100 mM sodium phosphate pH 6.5, 50 mM NaCl, and 5 mM DTT for NMR analysis. NMR 1H, 15N HSQC experiments were collected at 25 °C on a Bruker Avance II 500 MHz spectrometer. To compare the binding site of 142 methyl ester to an established BET bromodomain acetyl-lysine binding site ligand, uniformly labeled 15N-BRD4-BD2 (500 μM) was titrated with a histone H4 (residues 1–12) peptide diacetylated at lysines 5 and 8 (H4K5/8diacetyl) at concentrations of 3.4 and 6.8 mM in 100 mM sodium phosphate pH 6.5, 50 mM NaCl, and 5 mM DTT. 1H, 15N HSQC experiments consisted of 16 scans with 1024 and 210 complex points in the 1H and 15N dimensions, respectively. Backbone resonance assignments for BRD4-BD2 were obtained from published values (85). In addition, 15N HSQC, HNCA, HNCACB, HNCO, HN(CO)CA, and HN(CO)CACB experiments were used to confirm assignments and distinguish between overlapping peaks in the 1H, 15N HSQC spectrum. All NMR data were processed by using the NMRPipe (86) and SPARKY (87) was used for resonance assignment. Chemical shifts were measured using SPARKY (87). Total 1H, 15N chemical shift perturbations were calculated as:
Spectral overlays were generated using the python package nmrglue (88).
In silico covalent docking
The structure of BRD4-BD2 (PDB ID: 4KV4) was imported into the Schrödinger Maestro suite (v. 10.7) by using the Protein Preparation wizard and subjected to restrained minimization by using the OPLS2005 force field. Three-dimensional structures of 142 methyl ester were prepared for docking using the LigPrep application in Maestro (v. 3.9), and lig- and ionization states at pH 7.0 ± 2.0 were generated by using Epik (v. 3.7). CovDock (v. 1.2) (59) was used to covalently dock 142 methyl ester to BRD4-BD2 Cys356 using Michael addition chemistry.
Protein modeling
JQ1-PEG2-142 was docked to BRD4-BD2 Cys356 (PDB ID: 4KV4) using the Schrödinger covalent docking module as described above. The crystal structure of BRD4-BD1 bound to JQ1 (PDB ID: 3MXF) was then superimposed onto the covalently docked structure of BRD4-BD2 in PyMOL using the JQ1 substructure coordinates and the 3MXF JQ1 molecule was removed. The resulting protein complex was then relaxed and locally docked using Rosetta (89) in full-atom mode using -ex1 and -ex2aro flags and with the covalently labeled BRD4-BD2 Cys356 residue parametrized as an unnatural amino acid.
NanoBRET assays
Compound treatments and NanoBRET measurements in HEK293 cells were carried out as previously described (41). 2 mL of HEK293 human embryonic kidney cells (2 × 105 cells/mL) cultured in DMEM containing 2 mM glutamine and 1% v/v Pen/Strep and 10% v/v FBS were seeded into each well of a six-well culture plate (Corning) and allowed to attach for 4–6 h. For intramolecular biosensor experiments, cells were transfected with 1 μg of a HaloTag-BRD4-BD1/BD2-NanoLuc (aa 44–460 for BRD4) fusion (Promega) or HaloTag-BRD4-BD1/BD2(C356A)-NanoLuc variant (site-directed mutagenesis performed by Gen-script) fusion expression vectors. For BRD4 chromatin displacement experiments, cells were co-transfected with 2 μg Histone H3.3-HaloTag (NCBI reference sequence NM_002107) and 0.2 μg NanoLuc-BRD4 full-length (aa 1–1,362) (NCBI reference sequence NP_490597) expression vectors (Promega) (41).
After 24 h transfection, cells were harvested and resuspended in OptiMEM (Life Technologies) a density of 3 × 105 cells/mL in the absence (control) or presence (experimental) of 100 nM HaloTag NanoBRET 618 fluorescent ligand (Promega). Cells were then seeded into white, flat-bottomed tissue-culture-treated plates (Costar), with 90 μL of cell suspension per well, to which test compounds or vehicle (final 0.1% v/v DMSO concentration were added as 10×dilutions in OptiMEM at varying concentrations. Plates were incubated for 24 h at 37 °C in the presence of 5% CO2. NanoBRET Nano-Glo Substrate (Promega) was added to both control and experimental samples at a final concentration of 10 μM. Plates were read within 10 min using a Tecan Spark plate reader. A corrected NanoBRET ratio was calculated, defined as the ratio of the emission at >610 nm/450 nm for experimental samples minus the emission at >610 nm/450 nm for control samples. The data were fit to either the following EC50 equation:
or IC50 equation:
using the lmfit python package and normalized according to theoretical minima and maxima of the three-parameter curve fits.
Supplementary Material
Acknowledgement
We thank F. Peterson for technical assistance with NMR experiments, the Office of Research and the Research Computing Center of the Medical College of Wisconsin (MCW) for help with Schrödinger and computational server resources, and R. Mishra and the Center for Molecular Innovation and Drug Discovery at Northwestern University for assisting with the initial design of the library of electrophilic fragments. BRD4 NanoBRET constructs were a generous gift from D. Daniels at Promega. Mass spectrometry analyses were performed in the MCW Center for Biomedical Mass Spectrometry Research. Synthetic chemistry instrumentation was provided by the MCW Cancer Center. This work was supported by the National Institutes of Health grants R01DK119359 (B.C.S.), R01GM115632 (A.V.S.) and T32GM105538 (S.G.K.), the American Heart Association grant 15SDG25830057 (B.C.S.), the American Cancer Society institutional research grants 14-247-29-IRG and 86-004-26-IRG (B.C.S.) and 93-037-18-IRG (A.V.S.), the American Diabetes Association grant 1-18-IBS-068 (B.C.S.), the Michael Keelan Research Foundation (B.C.S.), the Advancing a Healthier Wisconsin endowment (B.C.S.), Northwestern University (A.V.S.), Pew Charitable Trusts (A.V.S.), an ACS Medicinal Chemistry Predoctoral Fellowship (S.G.K.), and a CLP Lambert Fellowship (Z.X.). M.D.O. is a member of the Medical Scientist Training Program at Medical College of Wisconsin, which is supported in part by National Institutes of Health Training Grant T32GM080202 from the National Institute of General Medical Sciences. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Supporting Information Available
- Supporting information: PDF file containing additional methods, synthetic procedures, Figure S1–S11, Table S1–S9, and supporting references.
- Supporting information: Excel file containing the compound ID number, mix number, molecular weight, monoisotopic mass, and smiles string for each of the 200 covalent fragments in the initial screening library.
References
- 1.Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, Olsen JV, and Mann M (2009) Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 325, 834–840. [DOI] [PubMed] [Google Scholar]
- 2.Kim SC, Sprung R, Chen Y, Xu Y, Ball H, Pei J, Cheng T, Kho Y, Xiao H, Xiao L, Grishin NV, White M, Yang XJ, and Zhao Y (2006) Substrate and functional diversity of lysine acetylation revealed by a proteomics survey. Mol Cell 23, 607–618. [DOI] [PubMed] [Google Scholar]
- 3.Choudhary C, Weinert BT, Nishida Y, Verdin E, and Mann M (2014) The growing landscape of lysine acetylation links metabolism and cell signalling. Nat Rev Mol Cell Biol 15, 536–550. [DOI] [PubMed] [Google Scholar]
- 4.Verdin E, and Ott M (2015) 50 years of protein acetylation: from gene regulation to epigenetics, metabolism and beyond. Nat Rev Mol Cell Biol 16, 258–264. [DOI] [PubMed] [Google Scholar]
- 5.Allfrey VG, Faulkner R, and Mirsky AE (1964) Acetylation and Methylation of Histones and their Possible Role in the Regulation of RNA Synthesis. Proc Natl Acad Sci U S A 51, 786–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Muller S, Filippakopoulos P, and Knapp S (2011) Bromodomains as therapeutic targets. Expert Rev Mol Med 13, e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Filippakopoulos P, Picaud S, Mangos M, Keates T, Lambert JP, Barsyte-Lovejoy D, Felletar I, Volkmer R, Müller S, Pawson T, Gingras AC, Arrow-smith CH, and Knapp S (2012) Histone recognition and large-scale structural analysis of the human bromodomain family. Cell 149, 214–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Taniguchi Y (2016) The Bromodomain and Extra-Terminal Domain (BET) Family: Functional Anatomy of BET Paralogous Proteins. Int J Mol Sci 17, E1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rahman S, Sowa ME, Ottinger M, Smith JA, Shi Y, Harper JW, and Howley PM (2011) The Brd4 extraterminal domain confers transcription activation independent of pTEFb by recruiting multiple proteins, including NSD3. Mol Cell Biol 31, 2641–2652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, Morse EM, Keates T, Hickman TT, Felletar I, Philpott M, Munro S, McKeown MR, Wang Y, Christie AL, West N, Cameron MJ, Schwartz B, Heightman TD, La Thangue N, French CA, Wiest O, Kung AL, Knapp S, and Bradner JE (2010) Selective inhibition of BET bromodomains. Nature 468, 1067–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Basheer F, and Huntly BJ (2015) BET bromodomain inhibitors in leukemia. Exp Hematol 43, 718–731. [DOI] [PubMed] [Google Scholar]
- 12.Belkina AC, and Denis GV (2012) BET domain co-regulators in obesity, inflammation and cancer. Nat Rev Cancer 12, 465–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chaidos A, Caputo V, and Karadimitris A (2015) Inhibition of bromodomain and extra-terminal proteins (BET) as a potential therapeutic approach in haematological malignancies: emerging preclinical and clinical evidence. Ther Adv Hematol 6, 128–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ghoshal A, Yugandhar D, and Srivastava AK (2016) BET inhibitors in cancer therapeutics: a patent review. Expert Opin Ther Pat 26, 505–522. [DOI] [PubMed] [Google Scholar]
- 15.Sahai V, Redig AJ, Collier KA, Eckerdt FD, and Munshi HG (2016) Targeting BET bromodomain proteins in solid tumors. Oncotarget 7, 53997–54009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tang X, Peng R, Phillips JE, Deguzman J, Ren Y, Apparsundaram S, Luo Q, Bauer CM, Fuentes ME, DeMartino JA, Tyagi G, Garrido R, Hogaboam CM, Denton CP, Holmes AM, Kitson C, Stevenson CS, and Budd DC (2013) Assessment of Brd4 inhibition in idiopathic pulmonary fibrosis lung fibroblasts and in vivo models of lung fibrosis. Am J Pathol 183, 470–479. [DOI] [PubMed] [Google Scholar]
- 17.Tang X, Peng R, Ren Y, Apparsundaram S, Deguzman J, Bauer CM, Hoffman AF, Hamilton S, Liang Z, Zeng H, Fuentes ME, Demartino JA, Kitson C, Stevenson CS, and Budd DC (2013) BET bromodomain proteins mediate downstream signaling events following growth factor stimulation in human lung fibroblasts and are involved in bleomycin-induced pulmonary fibrosis. Mol Pharmacol 83, 283–293. [DOI] [PubMed] [Google Scholar]
- 18.Anand P, Brown JD, Lin CY, Qi J, Zhang R, Artero PC, Alaiti MA, Bullard J, Alazem K, Margulies KB, Cappola TP, Lemieux M, Plutzky J, Bradner JE, and Haldar SM (2013) BET bromodomains mediate transcriptional pause release in heart failure. Cell 154, 569–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Spiltoir JI, Stratton MS, Cavasin MA, Demos-Davies K, Reid BG, Qi J, Bradner JE, and McKinsey TA (2013) BET acetyl-lysine binding proteins control pathological cardiac hypertrophy. J Mol Cell Cardiol 63, 175–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Belkina AC, Nikolajczyk BS, and Denis GV (2013) BET protein function is required for inflammation: Brd2 genetic disruption and BET inhibitor JQ1 impair mouse macrophage inflammatory responses. J Immunol 190, 3670–3678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nicodeme E, Jeffrey KL, Schaefer U, Beinke S, Dewell S, Chung CW, Chandwani R, Marazzi I, Wilson P, Coste H, White J, Kirilovsky J, Rice CM, Lora JM, Prinjha RK, Lee K, and Tarakhovsky A (2010) Suppression of inflammation by a synthetic histone mimic. Nature 468, 1119–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang G, Liu R, Zhong Y, Plotnikov AN, Zhang W, Zeng L, Rusinova E, Gerona-Nevarro G, Moshkina N, Joshua J, Chuang PY, Ohlmeyer M, He JC, and Zhou MM (2012) Down-regulation of NF-κB transcriptional activity in HIV-associated kidney disease by BRD4 inhibition. J Biol Chem 287, 28840–28851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bandukwala HS, Gagnon J, Togher S, Greenbaum JA, Lamperti ED, Parr NJ, Molesworth AM, Smithers N, Lee K, Witherington J, Tough DF, Prinjha RK, Peters B, and Rao A (2012) Selective inhibition of CD4+ T-cell cytokine production and autoimmunity by BET protein and c-Myc inhibitors. Proc Natl Acad Sci U S A 109, 14532–14537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mele DA, Salmeron A, Ghosh S, Huang HR, Bryant BM, and Lora JM (2013) BET bromodomain inhibition suppresses TH17-mediated pathology. J Exp Med 210, 2181–2190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pervaiz M, Mishra P, and Günther S (2018) Bromodomain Drug Discovery - the Past, the Present, and the Future. Chem Rec 18, 1808–1817. [DOI] [PubMed] [Google Scholar]
- 26.Lewin J, Soria JC, Stathis A, Delord JP, Peters S, Awada A, Aftimos PG, Bekradda M, Rezai K, Zeng Z, Hussain A, Perez S, Siu LL, and Massard C (2018) Phase Ib Trial With Birabresib, a Small-Molecule Inhibitor of Bromodomain and Extraterminal Proteins, in Patients With Selected Advanced Solid Tumors. J Clin Oncol 36, 3007–3014. [DOI] [PubMed] [Google Scholar]
- 27.Nakagawa A, Adams CE, Huang Y, Hamarneh SR, Liu W, Von Alt KN, Mino-Kenudson M, Hodin RA, Lillemoe KD, Fernández-Del Castillo C, Warshaw AL, and Liss AS (2016) Selective and reversible suppression of intestinal stem cell differentiation by pharmacological inhibition of BET bromodomains. Sci Rep 6, 20390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Postel-Vinay S, Herbschleb K, Massard C, Woodcock V, Soria JC, Walter AO, Ewerton F, Poelman M, Benson N, Ocker M, Wilkinson G, and Middleton M (2019) First-in-human phase I study of the bromodomain and extraterminal motif inhibitor BAY 1238097: emerging pharmacokinetic/pharmacodynamic relationship and early termination due to unexpected toxicity. Eur J Cancer 109, 103–110. [DOI] [PubMed] [Google Scholar]
- 29.Siu KT, Ramachandran J, Yee AJ, Eda H, Santo L, Panaroni C, Mertz JA, Sims Iii RJ, Cooper MR, and Raje N (2017) Preclinical activity of CPI-0610, a novel small-molecule bromodomain and extra-terminal protein inhibitor in the therapy of multiple myeloma. Leukemia 31, 1760–1769. [DOI] [PubMed] [Google Scholar]
- 30.Stonestrom AJ, Hsu SC, Werner MT, and Blobel GA (2016) Erythropoiesis provides a BRD’s eye view of BET protein function. Drug Discov Today Technol 19, 23–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chiang CM (2014) Nonequivalent response to bromodomain-targeting BET inhibitors in oligodendrocyte cell fate decision. Chem Biol 21, 804–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang R, Li Q, Helfer CM, Jiao J, and You J (2012) Bromodomain protein Brd4 associated with acetylated chromatin is important for maintenance of higher-order chromatin structure. J Biol Chem 287, 10738–10752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nakamura Y, Umehara T, Nakano K, Jang MK, Shirouzu M, Morita S, Uda-Tochio H, Hamana H, Terada T, Adachi N, Matsumoto T, Tanaka A, Horikoshi M, Ozato K, Padmanabhan B, and Yokoyama S (2007) Crystal structure of the human BRD2 bromodomain: insights into dimerization and recognition of acetylated histone H4. J Biol Chem 282, 4193–4201. [DOI] [PubMed] [Google Scholar]
- 34.Divakaran A, Talluri SK, Ayoub AM, Mishra NK, Cui H, Widen JC, Berndt N, Zhu JY, Carlson AS, Topczewski JJ, Schonbrunn EK, Harki DA, and Pomerantz WCK (2018) Molecular Basis for the N-Terminal Bromodomain- and-Extra-Terminal-Family Selectivity of a Dual Kinase-Bromodomain Inhibitor. J Med Chem 61, 9316–9334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gacias M, Gerona-Navarro G, Plotnikov AN, Zhang G, Zeng L, Kaur J, Moy G, Rusinova E, Rodriguez Y, Matikainen B, Vincek A, Joshua J, Casaccia P, and Zhou MM (2014) Selective chemical modulation of gene transcription favors oligodendrocyte lineage progression. Chem Biol 21, 841–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Picaud S, Wells C, Felletar I, Brotherton D, Martin S, Savitsky P, Diez-Dacal B, Philpott M, Bountra C, Lingard H, Fedorov O, Müller S, Brennan PE, Knapp S, and Filippakopoulos P (2013) RVX-208, an inhibitor of BET transcriptional regulators with selectivity for the second bromodomain. Proc Natl Acad Sci U S A 110, 19754–19759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cheung K, Lu G, Sharma R, Vincek A, Zhang R, Plotnikov AN, Zhang F, Zhang Q, Ju Y, Hu Y, Zhao L, Han X, Meslamani J, Xu F, Jaganathan A, Shen T, Zhu H, Rusinova E, Zeng L, Zhou J, Yang J, Peng L, Ohlmeyer M, Walsh MJ, Zhang DY, Xiong H, and Zhou MM (2017) BET N-terminal bromodomain inhibition selectively blocks Th17 cell differentiation and ameliorates colitis in mice. Proc Natl Acad Sci U S A 114, 2952–2957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nicholls SJ, Ray KK, Johansson JO, Gordon A, Sweeney M, Halliday C, Kulikowski E, Wong N, Kim SW, and Schwartz GG (2018) Selective BET Protein Inhibition with Apabetalone and Cardiovascular Events: A Pooled Analysis of Trials in Patients with Coronary Artery Disease. Am J Cardiovasc Drugs 18, 109–115. [DOI] [PubMed] [Google Scholar]
- 39.Shishikura D, Kataoka Y, Honda S, Takata K, Kim SW, Andrews J, Psaltis PJ, Sweeney M, Kulikowski E, Johansson J, Wong NCW, and Nicholls SJ (2019) The Effect of Bromodomain and Extra-Terminal Inhibitor Apabetalone on Attenuated Coronary Atherosclerotic Plaque: Insights from the ASSURE Trial. Am J Cardiovasc Drugs 19, 49–57. [DOI] [PubMed] [Google Scholar]
- 40.Wasiak S, Tsujikawa LM, Halliday C, Stotz SC, Gilham D, Jahagirdar R, Kalantar-Zadeh K, Robson R, Sweeney M, Johansson JO, Wong NC, and Kulikowski E (2018) Benefit of Apabetalone on Plasma Proteins in Renal Disease. Kidney Int Rep 3, 711–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Waring MJ, Chen H, Rabow AA, Walker G, Bobby R, Boiko S, Bradbury RH, Callis R, Clark E, Dale I, Daniels DL, Dulak A, Flavell L, Holdgate G, Jowitt TA, Kikhney A, McAlister M, Méndez J, Ogg D, Patel J, Petteruti P, Robb GR, Robers MB, Saif S, Stratton N, Svergun DI, Wang W, Whittaker D, Wilson DM, and Yao Y (2016) Potent and selective bivalent inhibitors of BET bromodomains. Nat Chem Biol 12, 1097–1104. [DOI] [PubMed] [Google Scholar]
- 42.Tanaka M, Roberts JM, Seo HS, Souza A, Paulk J, Scott TG, DeAngelo SL, Dhe-Paganon S, and Bradner JE (2016) Design and characterization of bivalent BET inhibitors. Nat Chem Biol 12, 1089–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ren C, Zhang G, Han F, Fu S, Cao Y, Zhang F, Zhang Q, Meslamani J, Xu Y, Ji D, Cao L, Zhou Q, Cheung KL, Sharma R, Babault N, Yi Z, Zhang W, Walsh MJ, Zeng L, and Zhou MM (2018) Spatially constrained tandem bromodomain inhibition bolsters sustained repression of BRD4 transcriptional activity for TNBC cell growth. Proc Natl Acad Sci U S A 115, 7949–7954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lu J, Qian Y, Altieri M, Dong H, Wang J, Raina K, Hines J, Winkler JD, Crew AP, Coleman K, and Crews CM (2015) Hijacking the E3 Ubiquitin Ligase Cereblon to Efficiently Target BRD4. Chem Biol 22, 755–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Winter GE, Buckley DL, Paulk J, Roberts JM, Souza A, Dhe-Paganon S, and Bradner JE (2015) DRUG DEVELOPMENT. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science 348, 1376–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sun B, Fiskus W, Qian Y, Rajapakshe K, Raina K, Coleman KG, Crew AP, Shen A, Saenz DT, Mill CP, Nowak AJ, Jain N, Zhang L, Wang M, Khoury JD, Coarfa C, Crews CM, and Bhalla KN (2018) BET protein proteolysis targeting chimera (PROTAC) exerts potent lethal activity against mantle cell lymphoma cells. Leukemia 32, 343–352. [DOI] [PubMed] [Google Scholar]
- 47.Zhang X, Lee HC, Shirazi F, Baladandayuthapani V, Lin H, Kuiatse I, Wang H, Jones RJ, Berkova Z, Singh RK, Lu J, Qian Y, Raina K, Coleman KG, Crews CM, Li B, Hailemichael Y, Thomas SK, Wang Z, Davis RE, and Orlowski RZ (2018) Protein targeting chimeric molecules specific for bromodomain and extra-terminal motif family proteins are active against pre-clinical models of multiple myeloma. Leukemia 32, 2224–2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Berthon C, Raffoux E, Thomas X, Vey N, Gomez-Roca C, Yee K, Taussig DC, Rezai K, Roumier C, Herait P, Kahatt C, Quesnel B, Michallet M, Recher C, Lokiec F, Preudhomme C, and Dombret H (2016) Bromodomain inhibitor OTX015 in patients with acute leukaemia: a dose-escalation, phase 1 study. Lancet Haematol 3, e186–e195. [DOI] [PubMed] [Google Scholar]
- 49.Kharenko OA, Patel RG, Brown SD, Calosing C, White A, Lakshmi-narasimhan D, Suto RK, Duffy BC, Kitchen DB, McLure KG, Hansen HC, van der Horst EH, and Young PR (2018) Design and Characterization of Novel Covalent Bromodomain and Extra-Terminal Domain (BET) Inhibitors Targeting a Methionine. J Med Chem 61, 8202–8211. [DOI] [PubMed] [Google Scholar]
- 50.Andrieu G, Belkina AC, and Denis GV (2016) Clinical trials for BET inhibitors run ahead of the science. Drug Discov Today Technol 19, 45–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bisgrove DA, Mahmoudi T, Henklein P, and Verdin E (2007) Conserved P-TEFb-interacting domain of BRD4 inhibits HIV transcription. Proc Natl Acad Sci U S A 104, 13690–13695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Itzen F, Greifenberg AK, Bösken CA, and Geyer M (2014) Brd4 activates P-TEFb for RNA polymerase II CTD phosphorylation. Nucleic Acids Res 42, 7577–7590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schröder S, Cho S, Zeng L, Zhang Q, Kaehlcke K, Mak L, Lau J, Bisgrove D, Schnölzer M, Verdin E, Zhou MM, and Ott M (2012) Two-pronged binding with bromodomain-containing protein 4 liberates positive transcription elongation factor b from inactive ribonucleoprotein complexes. J Biol Chem 287, 1090–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bhagwat AS, Roe JS, Mok BYL, Hohmann AF, Shi J, and Vakoc CR (2016) BET Bromodomain Inhibition Releases the Mediator Complex from Select cis-Regulatory Elements. Cell Rep 15, 519–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pelish HE, Liau BB, Nitulescu II, Tangpeerachaikul A, Poss ZC, Da Silva DH, Caruso BT, Arefolov A, Fadeyi O, Christie AL, Du K, Banka D, Schneider EV, Jestel A, Zou G, Si C, Ebmeier CC, Bronson RT, Krivtsov AV, Myers AG, Kohl NE, Kung AL, Armstrong SA, Lemieux ME, Taatjes DJ, and Shair MD (2015) Mediator kinase inhibition further activates super-enhancer-associated genes in AML. Nature 526, 273–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shi J, and Vakoc CR (2014) The mechanisms behind the therapeutic activity of BET bromodomain inhibition. Mol Cell 54, 728–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kozakov D, Grove LE, Hall DR, Bohnuud T, Mottarella SE, Luo L, Xia B, Beglov D, and Vajda S (2015) The FTMap family of web servers for determining and characterizing ligand-binding hot spots of proteins. Nat Protoc 10, 733–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kathman SG, Xu Z, and Statsyuk AV (2014) A fragment-based method to discover irreversible covalent inhibitors of cysteine proteases. J Med Chem 57, 4969–4974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhu K, Borrelli KW, Greenwood JR, Day T, Abel R, Farid RS, and Harder E (2014) Docking covalent inhibitors: a parameter free approach to pose prediction and scoring. J Chem Inf Model 54, 1932–1940. [DOI] [PubMed] [Google Scholar]
- 60.Jung M, Philpott M, Müller S, Schulze J, Badock V, Eberspächer U, Moos-mayer D, Bader B, Schmees N, Fernández-Montalván A, and Haendler B (2014) Affinity map of bromodomain protein 4 (BRD4) interactions with the histone H4 tail and the small molecule inhibitor JQ1. J Biol Chem 289, 9304–9319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Olp MD, Zhu N, and Smith BC (2017) Metabolically Derived Lysine Acylations and Neighboring Modifications Tune the Binding of the BET Bromodomains to Histone H4. Biochemistry 56, 5485–5495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Roy RD, Rosenmund C, and Stefan MI (2017) Cooperative binding mitigates the high-dose hook effect. BMC Syst Biol 11, 74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang CY, and Filippakopoulos P (2015) Beating the odds: BETs in disease. Trends Biochem Sci 40, 468–479. [DOI] [PubMed] [Google Scholar]
- 64.Jang MK, Mochizuki K, Zhou M, Jeong HS, Brady JN, and Ozato K (2005) The bromodomain protein Brd4 is a positive regulatory component of P-TEFb and stimulates RNA polymerase II-dependent transcription. Mol Cell 19, 523–534. [DOI] [PubMed] [Google Scholar]
- 65.Yang Z, Yik JH, Chen R, He N, Jang MK, Ozato K, and Zhou Q (2005) Recruitment of P-TEFb for stimulation of transcriptional elongation by the bromodomain protein Brd4. Mol Cell 19, 535–545. [DOI] [PubMed] [Google Scholar]
- 66.Donner AJ, Ebmeier CC, Taatjes DJ, and Espinosa JM (2010) CDK8 is a positive regulator of transcriptional elongation within the serum response network. Nat Struct Mol Biol 17, 194–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jiang YW, Veschambre P, Erdjument-Bromage H, Tempst P, Conaway JW, Conaway RC, and Kornberg RD (1998) Mammalian mediator of transcriptional regulation and its possible role as an end-point of signal transduction pathways. Proc Natl Acad Sci U S A 95, 8538–8543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wu SY, and Chiang CM (2007) The double bromodomain-containing chromatin adaptor Brd4 and transcriptional regulation. J Biol Chem 282, 13141–13145. [DOI] [PubMed] [Google Scholar]
- 69.Toyoshima M, Howie HL, Imakura M, Walsh RM, Annis JE, Chang AN, Frazier J, Chau BN, Loboda A, Linsley PS, Cleary MA, Park JR, and Grandori C (2012) Functional genomics identifies therapeutic targets for MYC-driven cancer. Proc Natl Acad Sci U S A 109, 9545–9550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zuber J, Shi J, Wang E, Rappaport AR, Herrmann H, Sison EA, Magoon D, Qi J, Blatt K, Wunderlich M, Taylor MJ, Johns C, Chicas A, Mulloy JC, Kogan SC, Brown P, Valent P, Bradner JE, Lowe SW, and Vakoc CR (2011) RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature 478, 524–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Huang B, Yang XD, Zhou MM, Ozato K, and Chen LF (2009) Brd4 coactivates transcriptional activation of NF-kappaB via specific binding to acetylated RelA. Mol Cell Biol 29, 1375–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hargreaves DC, Horng T, and Medzhitov R (2009) Control of inducible gene expression by signal-dependent transcriptional elongation. Cell 138, 129–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zou Z, Huang B, Wu X, Zhang H, Qi J, Bradner J, Nair S, and Chen LF (2014) Brd4 maintains constitutively active NF-κB in cancer cells by binding to acetylated RelA. Oncogene 33, 2395–2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ouyang L, Zhang L, Liu J, Fu L, Yao D, Zhao Y, Zhang S, Wang G, He G, and Liu B (2017) Discovery of a Small-Molecule Bromodomain-Containing Protein 4 (BRD4) Inhibitor That Induces AMP-Activated Protein Kinase-Modulated Autophagy-Associated Cell Death in Breast Cancer. J. Med. Chem 60, 9990–10012. [DOI] [PubMed] [Google Scholar]
- 75.Raux B, Voitovich Y, Derviaux C, Lugari A, Rebuffet E, Milhas S, Priet S, Roux T, Trinquet E, Guillemot JC, Knapp S, Brunel JM, Fedorov AY, Collette Y, Roche P, Betzi S, Combes S, and Morelli X (2016) Exploring Selective Inhibition of the First Bromodomain of the Human Bromodomain and Extra-terminal Domain (BET) Proteins. J Med Chem 59, 1634–1641. [DOI] [PubMed] [Google Scholar]
- 76.Singh J, Petter RC, Baillie TA, and Whitty A (2011) The resurgence of covalent drugs. Nat Rev Drug Discov 10, 307–317. [DOI] [PubMed] [Google Scholar]
- 77.Backus KM, Correia BE, Lum KM, Forli S, Horning BD, Gonzalez-Paez GE, Chatterjee S, Lanning BR, Teijaro JR, Olson AJ, Wolan DW, and Cravatt BF (2016) Proteome-wide covalent ligand discovery in native biological systems. Nature 534, 570–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bateman LA, Nguyen TB, Roberts AM, Miyamoto DK, Ku WM, Huffman TR, Petri Y, Heslin MJ, Contreras CM, Skibola CF, Olzmann JA, and Nomura DK (2017) Chemoproteomics-enabled covalent ligand screen reveals a cysteine hotspot in reticulon 4 that impairs ER morphology and cancer pathogenicity. Chem. Commun. (Camb.) 53, 7234–7237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Roberts AM, Miyamoto DK, Huffman TR, Bateman LA, Ives AN, Akopian D, Heslin MJ, Contreras CM, Rape M, Skibola CF, and Nomura DK (2017) Chemoproteomic Screening of Covalent Ligands Reveals UBA5 As a Novel Pancreatic Cancer Target. ACS Chem. Biol 12, 899–904. [DOI] [PubMed] [Google Scholar]
- 80.Maurais AJ, and Weerapana E (2019) Reactive-cysteine profiling for drug discovery. Curr Opin Chem Biol 50, 29–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.D’Ascenzio M, Pugh KM, Konietzny R, Berridge G, Tallant C, Hashem S, Monteiro O, Thomas JR, Schirle M, Knapp S, Marsden B, Fedorov O, Bountra C, Kessler BM, and Brennan PE (2019) An Activity-Based Probe Targeting Non-Catalytic, Highly Conserved Amino Acid Residues within Bromodomains. Angew. Chem. Int. Ed. Engl 58, 1007–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kathman SG, and Statsyuk AV (2016) Covalent Tethering of Fragments For Covalent Probe Discovery. Medchemcomm 7, 576–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Resnick E, Bradley A, Gan J, Douangamath A, Krojer T, Sethi R, Geurink PP, Aimon A, Amitai G, Bellini D, Bennett J, Fairhead M, Fedorov O, Gabizon R, Gan J, Guo J, Plotnikov A, Reznik N, Ruda GF, D?az-S?ez L, Straub VM, Szommer T, Velupillai S, Zaidman D, Zhang Y, Coker AR, Dowson CG, Barr HM, Wang C, Huber KVM, Brennan PE, Ovaa H, von Delft F, and London N (2019) Rapid Covalent-Probe Discovery by Electrophile-Fragment Screening. J. Am. Chem. Soc 141, 8951–8968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhang Z, and Marshall AG (1998) A universal algorithm for fast and automated charge state deconvolution of electrospray mass-to-charge ratio spectra. J Am Soc Mass Spectrom 9, 225–233. [DOI] [PubMed] [Google Scholar]
- 85.Shi J, Wang Y, Zeng L, Wu Y, Deng J, Zhang Q, Lin Y, Li J, Kang T, Tao M, Rusinova E, Zhang G, Wang C, Zhu H, Yao J, Zeng YX, Evers BM, Zhou MM, and Zhou BP (2014) Disrupting the interaction of BRD4 with diacetylated Twist suppresses tumorigenesis in basal-like breast cancer. Cancer Cell 25, 210–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, and Bax A (1995) NM-RPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR 6, 277–293. [DOI] [PubMed] [Google Scholar]
- 87.Lee W, Tonelli M, and Markley JL (2015) NMRFAM-SPARKY: enhanced software for biomolecular NMR spectroscopy. Bioinformatics 31, 1325–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Helmus JJ, and Jaroniec CP (2013) Nmrglue: an open source Python package for the analysis of multidimensional NMR data. J Biomol NMR 55, 355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sircar A, Chaudhury S, Kilambi KP, Berrondo M, and Gray JJ (2010) A generalized approach to sampling backbone conformations with RosettaDock for CAPRI rounds 13–19. Proteins 78, 3115–3123. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.