

Abstract
The intestinal epithelium is a paradigm of adult tissue in constant regeneration that is supported by intestinal stem cells (ISCs). The mechanisms regulating ISC homeostasis after injury are poorly understood. We previously demonstrated that IκBα, the main regulator of NF‐κB, exerts alternative nuclear functions as cytokine sensor in a subset of PRC2‐regulated genes. Here, we show that nuclear IκBα is present in the ISC compartment. Mice deficient for IκBα show altered intestinal cell differentiation with persistence of a fetal‐like ISC phenotype, associated with aberrant PRC2 activity at specific loci. Moreover, IκBα‐deficient intestinal cells produce morphologically aberrant organoids carrying a PRC2‐dependent fetal‐like transcriptional signature. DSS treatment, which induces acute damage in the colonic epithelium of mice, results in a temporary loss of nuclear P‐IκBα and its subsequent accumulation in early CD44‐positive regenerating areas. Importantly, IκBα‐deficient mice show higher resistance to damage, likely due to the persistent fetal‐like ISC phenotype. These results highlight intestinal IκBα as a chromatin sensor of inflammation in the ISC compartment.
Keywords: fetal‐like phenotype, intestinal stem cells, IκBα, polycomb, regeneration
Subject Categories: Chromatin, Epigenetics, Genomics & Functional Genomics; Regenerative Medicine; Signal Transduction
IκBα resides in the nucleus of intestinal stem cells and its deficiency leads to aberrant PRC2 activity, a fetal‐like ISC phenotype and defective differentiation. IκBα‐deficient mice show resistance to intestinal damage, likely due to their fetal‐like ISCs.
Introduction
NF‐κB is an essential pathway regulating immune responses and inflammation along evolution 1. In general, it comprises a complex combination of elements that varies depending on the triggering signal and the cellular–organismal system. The pathway also includes several negative feedback loops that prevent excessive and sustained activation under both basal and activated conditions. The main inhibitors of NF‐κB factors are the cytoplasmic IκB proteins that are phosphorylated and degraded in response to signaling leading to NF‐κB release, nuclear translocation, and gene transcription. Importantly, various IκBs including IκBα are rapidly resynthesized following NF‐κB activation thus imposing a robust post‐activation repression of the pathway 2, 3, 4, 5.
IκBα‐deficient mice provide strong evidence that IκBα is an inhibitor that attenuates excessive NF‐κB activation. Although they do not show overt developmental defects, IκBα‐deficient mice die around postnatal day 7 due to massive inflammation of the skin, which is NF‐κB‐dependent 6, 7, 8. Histological analysis of mice at days 4–5 after birth demonstrated that mutant skin displayed defective differentiation with increased proliferation of basal keratinocytes 8, 9. To study whether the observed phenotype was cell‐autonomous, keratinocyte‐specific knockout mice (Ikba K5Δ/K5Δ) were generated 8. These mutant mice display altered growth and dermatitis with skin abnormalities including acanthosis and hyperkeratosis, without the lethality observed in the total KO. However, double Iκbα K5Δ/K5Δ and T‐cell‐specific Iκbα lckΔ/lckΔ reproduced the phenotype observed in total KO mice, demonstrating that alterations of keratinocyte and T‐cell functions are key to the phenotype of IκBα‐deficient mice.
In addition to the critical involvement of NF‐κB activity in the skin phenotype of IκBα‐deficient mice, other studies have also documented that basal keratinocytes contain a SUMOylated (at K21) form of P‐IκBα that binds chromatin and regulate transcription of a subset of differentiation‐related genes through modulation of the polycomb repression complex 2 (PRC2) 9. Activity of the PRC2 complex, which executes the trimethylation of lysine 27 of histone H3 (H3K27me3), is essential for proper homeostasis of the skin 10, 11, 12 and maintenance of intestinal stem cell (ISC) function 13, 14, 15. Interestingly, IκBαNES mice carrying a mutation in the nuclear export signal (NES) of IκBα, which renders the protein primarily nuclear and unable to drive NF‐κB activation, exhibited an expansion of the interfollicular epidermal stem cell compartment 9 that was not accompanied by the lethal phenotype of total IκBα KO mice 16.
The skin and the intestinal epithelium are the prototype of adult tissues with constant renewal. ISCs are essential to sustain the intestinal integrity during lifespan and comprise a variety of populations including the rapidly cycling Lgr5‐positive (Lgr5+) cells located at the bottom of intestinal crypts 17 and more quiescent populations located around the +4 position of the crypt, initially characterized by the expression of high Bmi1 levels 18, 19. Interestingly, ISCs when cultured under defined conditions are capable of generating 3D structures called organoids 20, 21, which structurally and functionally resemble the intestinal epithelium including the capacity of self‐renewal and the differentiation toward the various intestinal lineages. In contrast, ISCs isolated from the fetal intestine do not produce organoids but structures that remain in a spherical shape with an absence of differentiated lineages 22, 23. It was recently shown that intestinal damage leads to re‐initiation of the fetal program characterized by increased proliferation, loss of the Lgr5+ population, and activation of a whole transcriptional signature downstream of IFNγ signaling, leading to enhanced regeneration capacity 24, 25.
The possibility that IκBα, in an NF‐κB‐dependent or NF‐κB‐independent manner, participates in ISC regulation under physiologic or pathologic situations has not been specifically addressed. We here show that P‐IκBα is pervasively localized in the nucleus of intestinal epithelial crypt cells, with IκBα deficiency imposing a fetal‐like phenotype to ISCs that is linked to defective lineage differentiation and altered PRC2 activity. Moreover, we present data suggesting a role for IκBα in intestinal tissue regeneration after damage.
Results
Nuclear localization and chromatin binding of phosphorylated IκBα in intestinal crypt cells, including the Lgr5+ ISCs
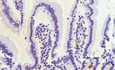
By immunofluorescence analysis in 2‐month‐old mice, we detected total IκBα predominantly distributed in the cytoplasm of differentiated intestinal cells but restricted to the nucleus of crypt cells (Fig 1A), including the vast majority of the Lgr5+ ISCs (as determined in the Lgr5GFP reporter mouse) (Figs 1B and EV1A). Using two different specific antibodies against phosphorylated IκBα, which is degraded in the cytoplasm but persists in the nucleus when SUMOylated 9, 26, we detected a consistent nuclear signal restricted to the intestinal crypt compartment of the small intestine (Figs 1C and EV1B) comprising 40–50% of the Lgr5+ population (Figs 1D and EV1C). However, nuclear P‐IκBα was not exclusive of Lgr5+ cells but it was present in a variable proportion of cells from positions +1 to +10 (from the crypt base) reaching a maximum at positions +5 to +9 (Fig 1E). Double IF analysis demonstrated that P‐IκBα was restricted to the EpHB2‐high cell population (including progenitors and stem cells), although excluded from the terminally differentiated LYZ1+ Paneth cells that also localizes at the crypt base (Figs 1F and G, and EV1D). By subcellular fractionation of intestinal cells followed by WB analysis, we found that nuclear IκBα did not correspond to the canonical 37‐kDa protein (the main form detected in the cytoplasmic fraction) but displayed an apparent molecular weight of 70 kDa, compatible with SUMOylated IκBα 9, 26 (Fig 1H and I). We next determined the expression pattern of P‐IκBα in the developing mouse intestine. Nuclear P‐IκBα was already present in most intestinal cells at developmental stage E12.5 and E14.5 and subsequently restricted to the inter‐villus regions at later development stages, ending in a crypt‐restricted distribution in the adult. This expression pattern runs in parallel with that observed for the ISC marker Bmi1 (Fig 1J).
Figure 1. Nuclear localization and chromatin binding of P‐IκBα in intestinal crypts.
-
A, BImmunofluorescence (IF) analysis of IκBα in sections from murine small intestine of 2‐month‐old WT (A) and Lgr5‐GFP reporter mice (B). Scale bars, 50 μm.
-
C, DIF analysis of P‐IκBα in the intestine of 2‐month‐old WT (C) and Lgr5‐GFP reporter mice (D), and quantification of P‐IκBα positivity inside the Lgr5‐GFP population. A minimum of 30 crypts was counted in 3 Lgr5‐GFP mice. Scale bars in C, 25 μm and in D, 50 μm.
-
EDistribution of P‐IκBα‐positive cells according to their position in the intestinal crypt from 200 crypts counted.
-
FDouble IF analysis of 2‐month‐old murine small intestine sections with the indicated antibodies and quantification of P‐IκBα positivity inside the EphB2+ population. Seventy crypts were counted. Bars represent mean values ± standard error of the mean (s.e.m.). Scale bars, 50 μm.
-
GDouble IF analysis of murine small intestine sections with the indicated antibodies. Scale bars, 50 μm.
-
HWestern blot analysis of cytoplasmic (CYT) and nuclear (NUC) extracts from isolated intestinal crypt cells.
-
IWestern blot analysis of crypt nuclear extracts immunoprecipitated with anti‐SUMO2/3 antibody.
-
JIF analysis of developing intestines from E12.5 to E17.5 with the indicated antibodies. Scale bars, 25 μm.
-
KRepresentation of IκBα distribution in the indicated genomic regions obtained from ChIP‐sequencing analysis (n = 4).
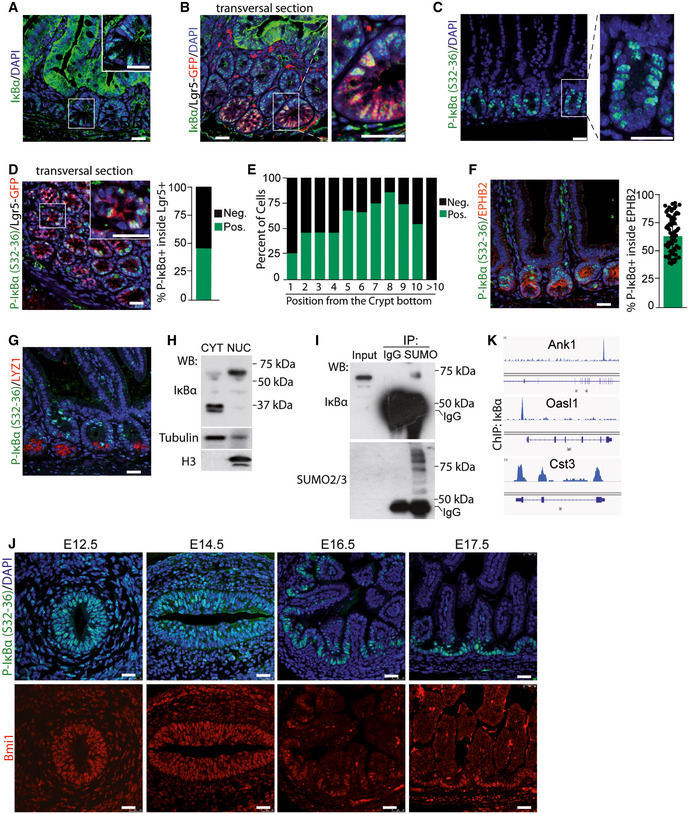
Figure EV1. P‐IκBα localizes in the nucleus of intestinal crypt cells and binds chromatin at specific genes.
-
AIHC analysis of P‐IκBα (S36) in intestinal mouse sections.
-
B–DIF analysis in sections from murine small intestine of 2‐month‐old Lgr5‐GFP reporter mice (B and C) and WT mice (D).
-
ETable indicating the genes identified in IκBα ChIP‐sequencing of P6 WT intestinal cells with P.adj < 0.05. n = 4 2‐month‐old WT mice were analyzed.
Finally, chromatin immunoprecipitation (ChIP)‐sequencing analysis of intestinal crypt cells demonstrated direct binding of IκBα to the promoter region of a limited number of genes (Figs 1K and EV1E).
These results indicate that SUMOylated IκBα specifically localizes in the nucleus of progenitor and ISCs during development and in the adult mice where it interacts with chromatin.
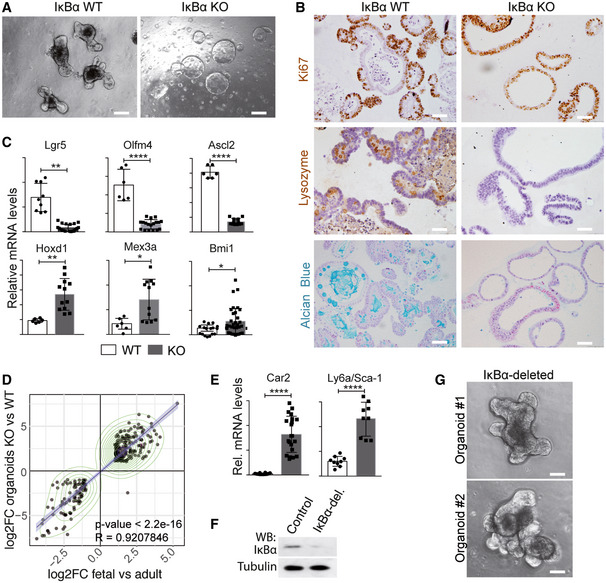
Altered intestinal homeostasis in IκBα‐deficient mice
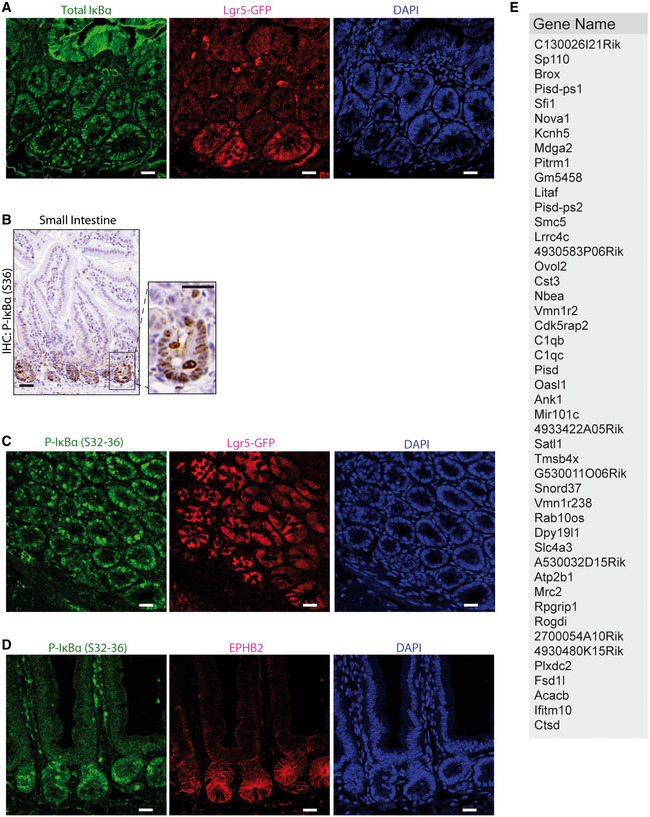
To study the contribution of IκBα to intestinal homeostasis, we analyzed the phenotype of conventional IκBα knockout (KO) mice 6, 7. As previously shown, IκBα KO mice are indistinguishable from wild‐type (WT) or heterozygous (HET) littermates at birth, although they die around postnatal day 7 (P7) due to severe skin inflammation 6, 7, 8. We noticed a significant growth delay in the KO animals starting at day P4 (Fig 2A and B). Further examination of P6 IκBα KO intestines revealed that they were significantly shorter than WT or HET intestines (Fig 2C). By immunohistochemical staining (IHC) analysis of P6 intestinal sections, we found that IκBα KO imposed a significant expansion of the proliferative (Ki67+) compartment, with the presence of Ki67+ cells distributed all along the crypt‐villus axis. In addition, IκBα KO mice showed a severe defect in the differentiation of mature intestinal lineages as illustrated by the virtual absence of lysozyme (LYZ1)‐expressing Paneth cells and a significant reduction in the number of Alcian Blue‐positive mucus‐secreting goblet cells (Fig 2D). We also analyzed compound IκBε;IκBβ KO and IκBαNES mice, which are viable and phenotypically comparable to WT littermates. We found that IκBβ;IκBε KO mice do not show any defect in the intestinal architecture, proliferation, or differentiation as determined by IHC analysis of 2‐month‐old intestines (Fig 2E). Similarly, 2‐month‐old IκBαNES intestines, which showed nuclear accumulation of P‐IκBα restricted to the crypt compartment (Fig 2F), displayed normal morphology with a number of Ki67+ cells and lysozyme‐expressing Paneth cells comparable to the WT, and a slight increase in the amount of Alcian Blue‐positive goblet cells (Fig 2G).
Figure 2. Altered intestinal homeostasis in IκBα‐deficient mice.
-
ARepresentative images of P4 and P6 mice of the indicated IκBα genotypes.
-
B, CQuantification of the animal weight (B) and intestinal length (C) from 50 WT/HET and 15 KO mice analyzed.
-
D–GImmunohistochemical (IHC), IF, and Alcian Blue staining with quantification of P6 IκBα WT and KO intestines (D), 2‐month‐old IκBβ;IκBε KO (E), and 2‐month‐old IκBαNES mice (F and G). A minimum of 50 crypts/villus was counted per genotype (3 mice).
These results indicate that total IκBα deficiency leads to defective intestinal homeostasis, which is ortholog‐specific, and rescued by the predominantly nuclear IκBαNES mutant.
IκBα deficiency imposes a fetal‐like phenotype to intestinal stem cells
To further investigate the putative impact of IκBα on ISC activity, we obtained P6 WT (n = 2) and IκBα KO (n = 3) intestinal cells and FACS purified them based on EphB2 expression (Fig 3A) for subsequent RNA‐sequencing analysis. As a control, we confirmed that cells displaying high EphB2 levels were confined to the crypt bottom in both IκBα genotypes (Fig 3B). Moreover, we verified that sorted WT EphB2‐high cells contained nuclear P‐IκBα (Fig 3C) and specifically expressed the ISC markers Lgr5 and Ascl2 27, whereas the differentiation marker Mucin2 (Muc2) was restricted to EphB2‐negative (Fig EV2A). Analysis of RNA‐sequencing data showed clustering of the samples depending on EphB2 high/low and the IκBα genotype (Fig 3D). Importantly, we observed that canonical ISC genes were down‐regulated in EphB2‐high KO cells, including Ascl2, Olfm4, and Lgr5 (Fig EV2B). A decrease in the expression of canonical ISC markers together with reduced differentiation has recently been described as the intestinal fetal signature 24, 25. We observed a significant direct correlation between genes miss‐regulated in KO cells and genes differentially expressed in the fetal compared with adult ISCs (Fig 3E), with 11 out of 15 most up‐regulated genes in the IκBα KO EphB2‐high cells belonging to the fetal signature including Ly6a/Sca‐1 or Lrig1 (Fig 3F) 24, 25. Gene Set Enrichment Analysis (GSEA) of the IκBα KO EphB2‐high signature showed a significant enrichment in TNFα‐driven NF‐κB and interferon‐gamma (IFNγ) signaling, which was previously associated also with the fetal‐like ISC reversion induced by intestinal damage 24. In contrast, signaling pathways known to support adult ISC activity such as Wnt and Notch 20 were markedly altered in IκBα KO cells (Fig 3G).
Figure 3. IκBα‐deficient intestinal stem cells display a fetal‐like phenotype.
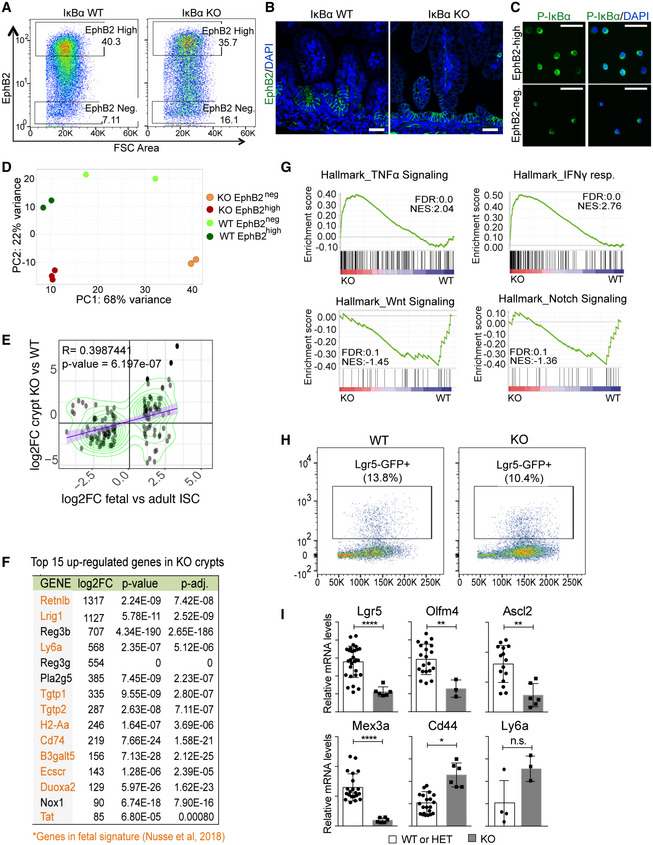
- Representative image of the intestinal populations purified in the cell sorting experiments.
- IF analysis of EphB2 in P6 WT and IκBα KO intestine.
- P‐IκBα IF of EphB2‐high or EphB2‐negative sorted cells.
- Clustering analysis of the indicated cell populations based on their transcriptional profiles obtained in RNA‐seq. Three mice per genotype were initially processed although 1 EphB2neg KO replicate was excluded from the analysis due to insufficient number of reads in the RNA‐seq.
- Correlation plot of the indicated differentially expressed gene (DEG) sets. Values for DEG in fetal and adult ISC were obtained from Nusse et al (2018).
- Table indicating the top 15 up‐regulated genes in the IκBα KO EphB2‐high signature including 11 genes in the fetal ISC signature (in orange).
- GSEA of EphB2‐high cells indicating the enrichment of relevant ISC‐related pathways.
- Representative image of the Lgr5+ population purified in the cell sorting experiments.
- qPCR analysis of the indicated genes in purified Lgr5+ cells from the different IκBα backgrounds. Four WT and 1 KO mice were analyzed with at least three technical replicates. Bars represent mean values ± standard error of the mean (s.e.m.). P values were derived from an unpaired t‐test, two‐tailed, ****P‐value < 0.0001, **P‐value < 0.005, *P‐value < 0.05; n.s.: no significant.
Figure EV2. IκBα KO intestines showed alterations in ISC population.
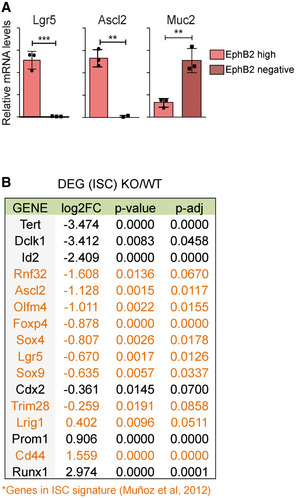
- qPCR analysis of EphB2‐high and EphB2‐negative sorted cells from P6 WT intestine. Three technical replicates were analyzed. Bars represent mean values ± standard error of the mean (s.e.m.); P values were derived from an unpaired t‐test, two‐tailed, ***P‐value < 0.0005, **P‐value < 0.005.
- Table indicating the ISC genes differentially expressed between IκBα WT and KO EphB2‐high sorted cells (RNA‐seq, n = 3 WT and n = 3 KO), including genes of ISC signature 43 (in orange).
Then, we used the Lgr5GFP reporter mouse to FACS‐purify single ISC cells based on GFP expression. We detected a slight decrease in the percent of LGR5GFP+ cells in the IκBα KO intestine (Fig 3H). Intriguingly, inside this particular ISC population we observed a transcriptional decline of the canonical ISC genes Lgr5, Olfm4, Ascl2, and Mex3a, in contrast with the ISC marker Cd44 and the fetal gene Ly6a that were slightly increased (Fig 3I). These results indicated that IκBα deficiency impairs maturation of fetal ISCs toward the adult identity.
IκBα‐deficient intestines show altered PRC2 distribution and function
As IκBα is the main regulator of the NF‐κB pathway, we anticipated that aberrant NF‐κB activity might be at the base of the observed intestinal defects of IκBα KO mice. By IHC analysis of P6 WT and IκBα KO intestines, we did not observe any nuclear RelA/p65 or c‐Rel in the epithelial cells of either genotype (Fig 4A). By ChIP analysis of P6 intestines, we failed to detect any significant alteration in the number and distribution of RelA/p65 peaks in the different IκBα genotypes (Figs 4B and EV3A), and we did not detect reliable peaks in the c‐Rel precipitation. Similarly, we did not find any alteration in the levels of active Notch1 (ICN1) or β‐catenin, the downstream effectors of Notch and Wnt pathways, which were found deregulated in the RNA‐seq analysis (Fig 4C and D).
Figure 4. Normal NF‐κB but altered PRC2 function in IκBα‐deficient intestine.
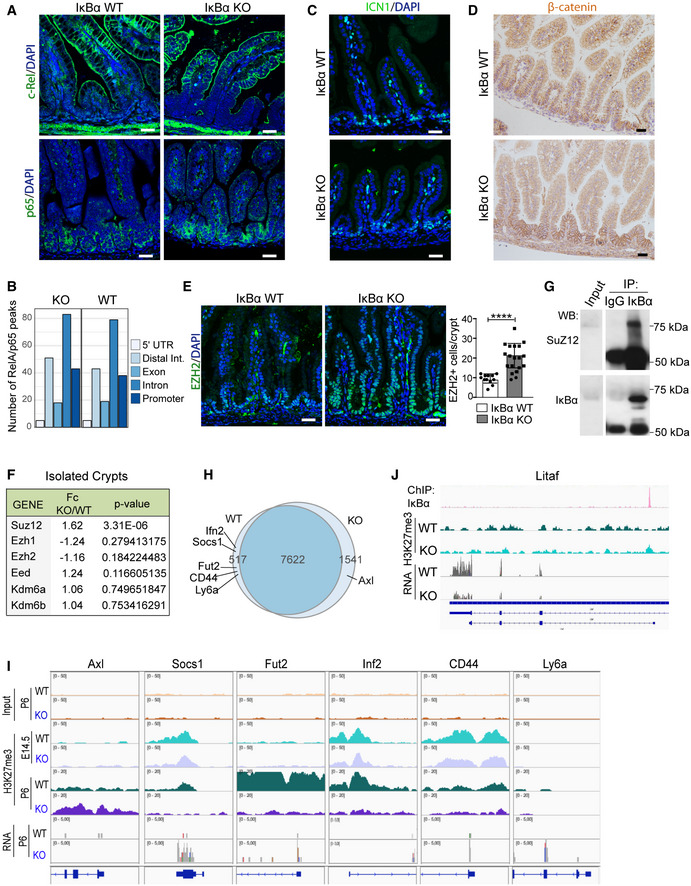
-
AIF analysis of c‐Rel and RelA/p65 in P6 WT and IκBα KO intestines.
-
BNumber of peaks from p65 ChIP‐sequencing analysis associated with the different genomic localizations was obtained merging two biological replicates per condition (n = 2 P6 WT and n = 2 P6 IκBα KO intestinal crypt cells). Data from individual experiments have been deposited at GSE131187.
-
C, DIF analysis of active Notch1 (ICN1) (C) and β‐catenin (D) in the indicated IκBα backgrounds at P6 intestines.
-
EIF analysis of the PRC2 protein EZH2 and quantification of the EZH2‐positive cells per crypt in n = 3 P6 animals per genotype were analyzed (a minimum of 15 crypts was counted per genotype). Bars represent mean values ± standard error of the mean (s.e.m.); P values were derived from an unpaired t‐test, two‐tailed, ****P‐value < 0.0001.
-
FTable indicating the methylation‐related genes (PRC2 members and KDM6 demethylase) differentially expressed between IκBα WT and KO EphB2‐high sorted cells (RNA‐seq).
-
GWestern blot analysis of IκBα IP from crypt nuclear extracts.
-
HVenn diagram representing the distribution of genes detected as differentially methylated in the H3K27me3 ChIP‐sequencing analysis across P6 IκBα genotypes (n = 2 WT and n = 2 KO mice).
-
IRepresentation of H3K27me3 distribution at embryonic E14.5 (n = 2 WT and n = 2 KO mice) and postnatal P6 (n = 2 WT and n = 2 KO mice) (from ChIP‐sequencing) and expression levels (from RNA‐sequencing) in the genomic region of the indicated fetal ISC genes.
-
JRepresentation of IκBα (n = 4) and H3K27me3 (n = 2) distribution (from ChIP‐sequencing) and expression levels (from RNA‐sequencing) in the genomic region of Litaf.
Figure EV3. p65 and H3K27me3 ChIP‐sequencing analysis showed no differences along genomic regions.
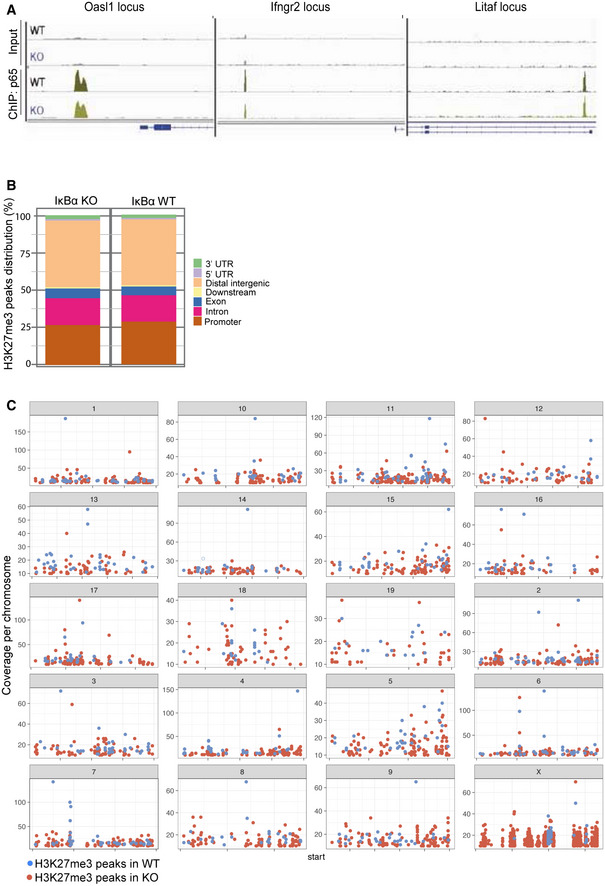
- Representation of p65 distribution (from ChIP‐sequencing, n = 2 WT and n = 2 KO) in the indicated genomic regions.
- Graph showing the distribution of H3K27me3 peaks in the indicated genomic locations in IκBα WT and IκBα KO.
- Graph showing the chromosomal distribution of H3K27me3 peaks in IκBα WT and IκBα KO.
As we previously described the interaction of IκBα and PRC2 at the chromatin level, we analyzed the distribution of the PRC2 catalytic subunit EZH2 in the intestine of WT and IκBα KO mice. We detected nuclear EZH2 localized in the intestinal crypts of both WT and KO mice, with IκBα KO displaying a significantly higher number of EZH+ cells per crypt‐villus unit (Fig 4E), although we did not observe any significant alteration at the transcriptional level, except for slight increase of Suz12 in KO cells (Fig 4F). Moreover, by Co‐IP experiments we demonstrated the interaction of the PRC2 subunit SUZ12 with SUMOylated IκBα in the nucleus of crypt cells (Fig 4G). By ChIP‐sequencing analysis of H3K27me3 from P6 WT (n = 2) and IκBα KO (n = 2) intestines, we found a comparable occurrence of this mark in both genotypes (7,622 common regions) showing a distribution at the genomic regions (intergenic, promoter, or gene body) that was comparable between genotypes (Fig EV3B) and randomly scattered across chromosomes (Fig EV3C). However, we identified a limited number of genes (2,058) that were associated with peaks differentially represented in the KO (1,541) and WT (517) mice including genes related to the fetal ISC identity such as Axl, Socs1, or Ly6a (Fig 4H and I). Notice that these fetal ISC genes were similarly methylated in the embryo at E14.5 but they display a differential methylation pattern at P6 (postnatal) that inversely correlated with their expression levels (Fig 4I). In addition, and despite the limited number of genes identified in the initial IκBα ChIP assay (see Fig EV1E), several IκBα targets such as Sfi1, Pisd, and Litaf were also differentially expressed in IκBα KO EphB2‐high cells (23 differentially expressed out of 49 IκBα targets) associated with local changes in H3K27me3 levels at the region bound by IκBα (Fig 4J).
These results indicate that IκBα is required for establishing the proper H3K27 methylation pattern in a subset of genes including those that define ISC identity. Moreover, our results suggest that the ISC phenotype of IκBα KO intestine is NF‐κB‐independent but PRC2‐dependent.
IκBα‐deficient‐derived organoids display a fetal ISC phenotype and defective differentiation
To study IκBα function in a better‐defined ISC model, we used the 3D organoid system. Organoids derived from postnatal day 6 (P6) intestinal cells were morphologically indistinguishable from that produced of adult cells. We detected nuclear P‐IκBα in these organoids that was restricted to the crypt‐like pockets (Fig EV4A) where ISCs are localized 21. Importantly, intestinal cells from P6 IκBα KO mice repeatedly failed to generate well‐differentiated organoids but produced spherical structures (spheroids) resembling tumor‐ or fetal‐derived organoids (Fig 5A). By IHC analysis of P6 organoids, we confirmed that IκBα KO spheroids mostly consisted of proliferating Ki67+ cells, which displayed a total absence of LYZ1+ Paneth cells and a severe decrease in the number of goblet cells as determined by Alcian Blue staining (Fig 5B), similar to that observed in the IκBα KO intestine.
Figure EV4. P‐IκBα is expressed in organoid pockets, and its deficiency leads to transcriptional alterations in differentiation markers and pathways, which are NF‐κB‐ and Wnt/β‐catenin‐independent.
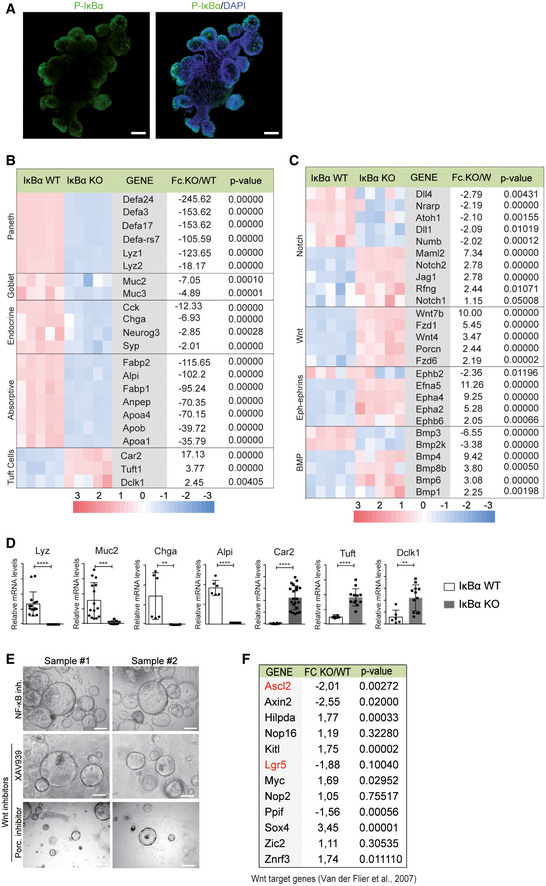
-
AIF analysis of P‐IκBα in WT organoids.
-
B, CSelection of differentially expressed genes associated with intestinal lineages (B) and molecular pathways (C) identified by microarray assay from WT and IκBα KO‐derived organoids. Five different organoids per genotype were analyzed, which were generated from at least 2 different mice per genotype.
-
DValidation of relevant genes by qPCR. Three technical replicates of a minimum of two organoids per condition were analyzed. Bars represent mean values ± standard error of the mean (s.e.m.); P values were derived from an unpaired t‐test, two‐tailed, ****P‐value < 0.0001, ***P‐value < 0.0005, **P‐value < 0.005.
-
ERepresentative stereoscopic images of IκBα KO organoids treated as indicated.
-
FTable indicating the Wnt/β‐catenin target genes 44 differentially expressed between IκBα WT and KO organoids (expression microarray, n = 5 WT and n = 5 KO). In red, Wnt target ISC genes.
Figure 5. IκBα‐deficient‐derived organoids display a fetal ISC phenotype.
- Representative stereoscopic images of P6 WT and IκBα KO‐derived intestinal organoids.
- IHC analysis with the indicated antibodies and Alcian Blue staining of WT and IκBα KO‐derived organoids.
- qPCR analysis of the indicated ISC genes in WT and IκBα KO‐derived organoids.
- Correlation plots of the indicated differentially expressed gene (DEG) sets obtained in expression microarray (5 technical replicates of a minimum of two organoids per genotype were analyzed). Values for DEG in fetal and adult organoids were obtained from 23.
- qPCR analysis of the indicated fetal genes in WT and IκBα KO‐derived organoids.
- Western blot analysis of control and IκBα‐deleted organoids using CRISPR‐Cas9 technology.
- Representative stereoscopic images of WT and IκBα‐deleted organoids.
We performed microarray expression analysis of WT (n = 5) and IκBα KO (n = 5) organoids and found that 5,832 genes were differentially expressed among genotypes with P‐value < 0.001. Genes down‐regulated in the IκBα KO comprise those related to intestinal differentiation of all major lineages including Paneth (Defa and Lyz), goblet (Muc2 and Muc3), entero‐endocrine cells (Chga or Neurog3), and enterocytes (Alpi, Fabp, and Anpep), in contrast with genes related to tuft cell differentiation such as Dclk1 and Tuft1 that were significantly up‐regulated (Fig EV4B and D). Consistent with defective intestinal differentiation, we also found altered expression of genes of the Wnt, BMP, Notch, and Ephrin/Eph pathways (Fig EV4C).
Surprisingly, but similar to that observed in the analysis of intestinal crypt cells, several ISC genes were highly deregulated in the IκBα KO organoids including Lgr5, Olfm4, and Ascl2 (Fig 5C). Moreover, differential expression of IκBα deficient organoids significantly correlated (R = 0.92; P < 0.0001) with the differential expression observed in organoids derived from fetal intestine 23 (Fig 5D). The overexpression of selected fetal genes in IκBα KO organoids was demonstrated by qPCR (Fig 5E). We next knocked out IκBα by CRISPR‐Cas9 in already formed WT organoids (Fig 5F). CRISPR‐Cas9 KO of IκBα in WT intestinal organoids did not significantly affect their differentiated morphology (Fig 5G), suggesting that regulation of ISC maturation by IκBα is exerted during development and is not continuously required to maintain adult ISCs under standard growing conditions.
These results indicated that organoids derived from IκBα‐deficient intestinal cells mimic the fetal bias that is found in the IκBα KO intestinal crypt cells in vivo.
Fetal‐like phenotype of IκBα‐deficient cells is PRC2‐, Notch‐, and interferon‐dependent
Then, we used specific inhibitors and genetic strategies to evaluate the functional involvement of candidate pathways in the fetal‐like phenotype of IκBα KO organoids. We first speculated that increased NF‐κB signaling due to IκBα deficiency might contribute to the observed phenotype. However, blockage of the NF‐κB upstream kinase IKKβ with BAY65‐5811 did not affect the spheroidal morphology of IκBα KO organoids after one month of treatment (Fig EV4E). Then, we transduced IκBα KO organoids with shRNA against RelA/p65 and c‐Rel. Reduction of p65 and c‐Rel levels alone or in combination did not affect spheroidal organoid morphology (Fig 6A). Accordingly, p65 and c‐Rel knockdown did not revert ISC gene expression or differentiation blockage to the WT conditions (Fig 6B). In contrast, we observed a strong up‐regulation of the NF‐κB target gene Cxcl10 after viral transduction of control shRNA that was totally abrogated by RelA and or c‐Rel depletion (Fig 6B), demonstrating that this gene is NF‐κB‐dependent. Importantly, incubation with the PRC2/EZH2 inhibitor EPZ‐6438 led to the sporadic restoration of crypt‐like pockets arising from spherical structures. The number and complexity of budding organoids progressively increased with time of treatment (Fig 6C). qPCR analysis of IκBα KO organoids untreated or treated for 1 month with EPZ‐6438 indicated a transcriptional conversion toward the WT phenotype with up‐regulation of the canonical stem cell markers Lgr5 and Cd44, and the differentiation markers Lyz and Muc2 (Fig 6D). Importantly, expression levels of Lrig1 and Ly6a, which were up‐regulated in IκBα KO organoids, remained primarily unaffected or reduced following treatment, likely reflecting the heterogeneity of the culture in which only a low percentage of KO organoids revert toward WT phenotype (Fig 6D).
Figure 6. Fetal ISC phenotype of IκBα‐deficient organoids is PRC2‐, Notch‐, and IFN‐dependent.
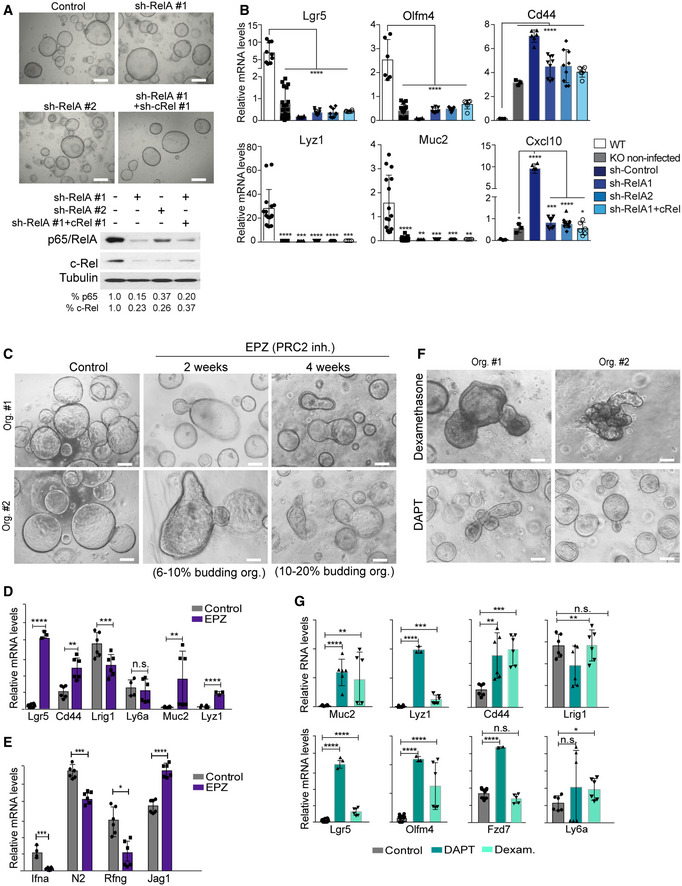
-
ARepresentative stereoscopic images and Western blot analysis of IκBα KO organoids infected with the indicated shRNA for NF‐κB elements.
-
BqPCR analysis of the indicated ISC and differentiation genes in the different shRNA‐treated organoids. The canonical NF‐κB target gene Cxcl10 is shown as positive control of the experiment.
-
CRepresentative stereoscopic images of IκBα KO organoids untreated or treated with the PRC2 inhibitor EPZ‐6438.
-
D, EqPCR analysis of the indicated adult and fetal ISC genes (D) and elements of the Notch and IFN pathways (E) (differentially expressed in IκBα KO organoids) in IκBα KO organoids untreated or treated with the PRC2 inhibitor EPZ‐6438.
-
FRepresentative stereoscopic images of IκBα KO organoids treated with dexamethasone as IFN inhibitor and DAPT as Notch inhibitor.
-
GqPCR analysis of IκBα KO organoids untreated or treated with dexamethasone and DAPT.
Next, we determine the putative effects of PRC2 inhibition on the expression of Notch and IFN pathway elements that were found miss‐regulated in the IκBα KO organoids. Inhibition of PRC2 led to altered expression of IFNα (Ifna), Notch2 (N2), radical Fringe (Rfng), and Jag1 (Fig 6E) suggesting that Notch and IFN pathways could be downstream effectors (at least partially) of the PRC2‐dependent defects of IκBα KO organoids. Consistently, treatment of IκBα KO organoids with the Notch inhibitor DAPT or the IFN inhibitor dexamethasone produces morphologic (Fig 6F) and transcriptional changes (Fig 6G) that were comparable to EPZ‐6438 (Fig 6D). In contrast, we did not observe any evidence of differentiation but a general decline in organoid growth following Wnt/β‐catenin pathway inhibition with XAV939 or the Porcupine inhibitors (Fig EV4E). These results are in agreement with the absence of Wnt target genes over‐activation in IκBα KO organoids as determined by analysis of transcriptomic data (Fig EV4F).
Functional involvement of IκBα in the regenerating intestinal epithelium
Intestinal repair after damage has been linked to the acquisition of fetal‐like traits in the ISC compartment, which is triggered by the immune response and changes in the extracellular matrix 24, 25. Thus, we used the DSS‐induced colitis model to investigate the possibility that IκBα participates in the process of intestinal regeneration by regulating ISC function. We first determine P‐IκBα distribution in the colon of control mice, and mice treated with DSS for 5 days and sacrificed at day 6. Analysis of colonic tissue showed the presence of nuclear P‐IκBα at the bottom of the crypts under homeostasis (untreated) conditions. In contrast, DSS‐treated animals showed a virtual loss of nuclear P‐IκBα‐positive cells (Figs 7A and EV5A), with P‐IκBα detection restricted to sporadic cells (see asterisks) in areas of acute damage, which were identified by the presence of epithelial strings that express the ISC markers Olfm4, which is absent from the normal murine colonic mucosa, and CD44, and displayed moderate levels of proliferation (as determined by Ki67 staining) (Fig 7A and B). By double IF, we confirmed the presence of P‐IκBα‐positive cells in the Olfm4‐positive area (Fig 7C). In contrast, adjacent areas where regeneration had already started contained massive amounts of nuclear P‐IκBα in highly proliferative crypt‐like structures that were Olfm4‐negative but CD44‐positive (Figs 7A and B, and EV5A). Examination of p65 and c‐Rel distribution in the regenerating epithelium demonstrated that nuclear (active) p65/NF‐κB staining was also restricted to areas of acute damage (Fig EV5B), suggesting that loss of nuclear P‐IκBα in this area might be linked to IKK activation. Interestingly, we demonstrated that Olfm4 and Cd44 transcription was induced by the pro‐inflammatory cytokine TNFα in intestinal organoids, which was IκBα‐dependent (they are not induced in IκBα KO organoids at any time point of TNFα treatment) but NF‐κB‐independent (since canonical NF‐κB targets such as Cxcl10 and A20 are consistently activated by TNFα in KO organoids) (Fig 7D).
Figure 7. Nuclear accumulation and functional involvement of IκBα in the regenerating intestinal epithelium.
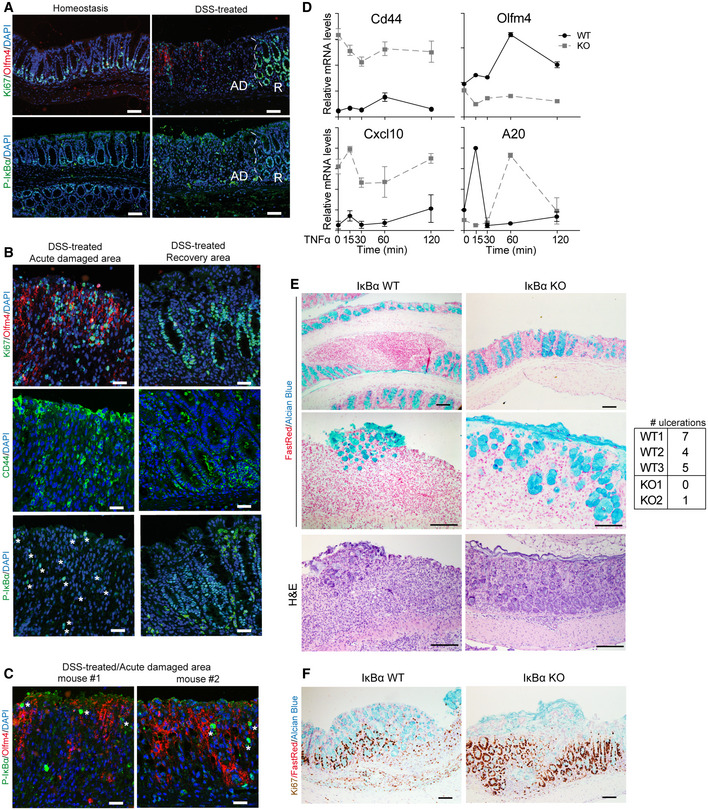
-
AIF analysis of colonic tissue from control and DSS‐treated animals including acute damaged (AD) areas and recovery (R) areas.
-
B, CIF analysis of P‐IκBα and the indicated markers in the areas of acute damage and recovery (B) and colocalization of P‐IκBα with the ISC marker Olfm4 in acute damaged areas (C). Asterisks indicate the presence of rare cells with nuclear P‐IκBα.
-
DqPCR analysis of the indicated genes in WT and IκBα KO organoids treated with TNFα and collected at the indicated time‐points. Three technical replicates of organoids from each genotype were analyzed.
-
E, FRepresentative images of the colonic tissue from 2‐month‐old DSS‐treated WT and IκBα KO mice are shown. Alcian Blue staining was used to identify the mucus‐secreting goblet cells in the colonic glands and Ki67 as proliferation marker. Nuclear counterstain is Fast Red. The table shows number of ulcerations present in the intestines of mice analyzed (3 WT, 2 IκBα KO).
Figure EV5. P‐IκBα is lost in damaged areas, where NF‐κB pathway is active, and accumulated in regenerating crypts.
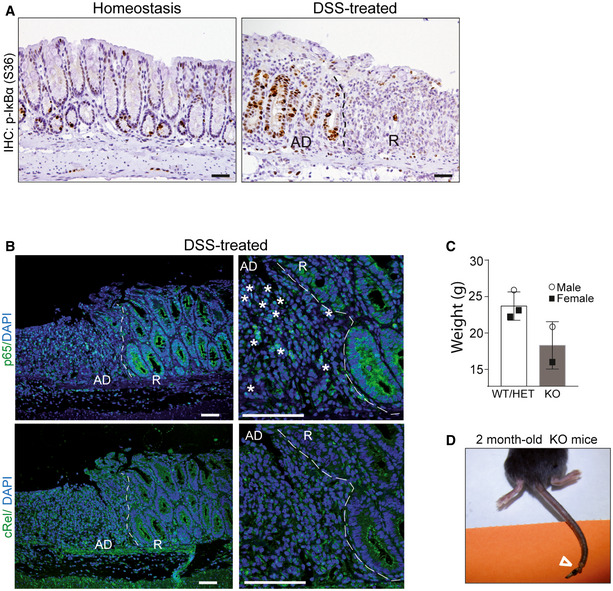
-
AIHC analysis of P‐IκBα (S36) in the colonic tissue of control and DSS‐treated mice including acute damaged (AD) areas and recovery (R) areas.
-
BIF analysis of the NF‐κB subunits p65/RelA and c‐Rel in areas of acute damage and recovery. Asterisks indicate the presence of nuclear p65 in acute damage areas adjacent to the recovery region. The pictures show acute damaged (AD) areas and recovery (R) areas.
-
C, DGraph indicating the reduced weight (A) and representative image of the skin defects (see arrowhead) (D) observed in the 2‐month‐old IκBα KO mice. Bars represent mean values ± standard error of the mean (s.e.m.).
Interestingly, in our specific‐pathogen‐free (SPF) animal facility, few IκBα KO animals evaded the postnatal critical period and survived for, at least, 2 months after birth (hereafter denoted as IκBα KO survivors). We noticed lower weight in these KO animals (Fig EV5C), associated with the presence of skin lesions (Fig EV5D). To study the role of nuclear IκBα during the regeneration processes, we treated 2‐month‐old WT and IκBα KO survivor mice with DSS as described above. At the time of sacrifice, the colonic mucosa of WT animals displayed extensive areas of ulceration with glandular depletion (as indicated by the absence of Alcian Blue‐positive goblet cells), fibrosis of lamina propria, and inflammation mostly composed of lymphocytes with occasional neutrophils, as expected. In contrast, DSS‐treated IκBα KO mice showed little evidence of ulceration in the colonic tissue (Fig 7E). IHC analysis of the proliferation marker Ki67 indicated the presence of highly proliferative areas in the DSS‐treated IκBα KO mucosa (Fig 7F), suggestive of active regeneration.
Together, our data indicate that IκBα is required for the proper regulation of ISC function under homeostasis and might play a role in tissue regeneration after injury (see model in Fig 8).
Figure 8. Model for IκBα contribution to ISC development and regeneration.
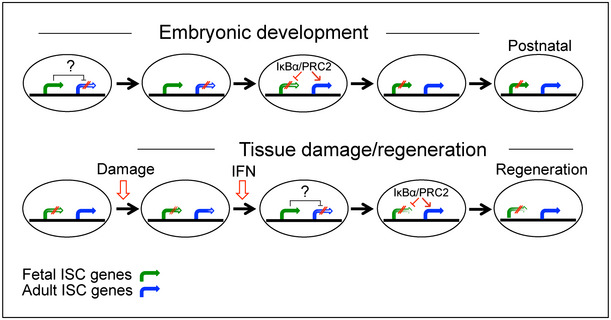
In brief, nuclear IκBα is required for the PRC2‐mediated timely repression of fetal ISC genes during development and activation of adult ISC genes. In the absence of IκBα, expression of fetal ISC genes is sustained after birth resulting in defective ISC maturation (upper panel). During regeneration (lower panel), specific signals such as IFN impose a temporary fetal ISC phenotype that is required for subsequent tissue regeneration 24, 25. Next, nuclear IκBα massively accumulates at regeneration areas to repress fetal ISC gene expression thus facilitating ISC maturation and tissue repair. Paradoxically, IκBα KO mice do not show evidence of colonic ulceration after DSS treatment suggesting that fetal‐like ISC (imposed by IκBα deficiency) is intrinsically resistant to injury.
Discussion
We here describe a previously unappreciated phenotype of IκBα KO mice in intestinal homeostasis and ISC identity. Specifically, we found that IκBα‐deficient ISCs transcriptionally and functionally resemble the fetal‐like ISCs previously observed in mice exposed to helminth infection or DSS‐induced epithelial damage 24, 25, but also in the organoids obtained from fetal intestinal cells 23. The severity of this ISC phenotype imposed by IκBα deficiency is surprising since we observed that only 50% of the Lgr5+ crypt cells (that constitute a very homogeneous population) are P‐IκBα‐positive. However, we do think that nuclear IκBα is cycling between the non‐phosphorylated and phosphorylated forms (in response to specific stimuli not yet defined) leading to waves of gene activation/repression.
Interestingly, we found a significant association between the transcriptional IκBα KO ISC signature and genes of the IFN pathway, which was demonstrated as a driver of the fetal ISC identity 24. Whether IκBα acts as chromatin suppressor of one or several IFN elements, or IκBα deficiency imposes some increased NF‐κB activity, which in turn facilitates IFN transcription as previously shown 28, 29, remains to be elucidated in this context. However, the fetal‐like phenotype of IκBα KO organoids was not rescued by IKKβ inhibition or knocking down c‐Rel or p65, suggesting that canonical NF‐κB pathway is not required to maintain this phenotype, in agreement with the apparently normal intestinal differentiation of mice carrying constitutively active IKKβ 30. However, it is formally possible that IκBα deficiency imposes some degree of basal NF‐κB activity that impacts on ISC function. Analysis of specific compound IκBα;Rel (RelA and c‐Rel)‐deficient mice but most importantly of tissue‐specific IκBα KO mice would help to elucidate the relative contribution of NF‐κB activity to the fetal ISC phenotype. In contrast, altered PRC2 activity was consistently detected in IκBα KO intestinal cells as indicated by aberrant H3K27me3 distribution in specific regulatory regions including several IFN‐related gene promoters, which is expected to impact in ISC homeostasis. Moreover, we were able to directly demonstrate the physical association between nuclear SUMOylated IκBα and the PRC2 subunit SUZ12 in IP experiments from intestinal crypt cell lysates, further indicating that intestinal IκBα modulates PRC2 function. Importantly, and different from what was found following helminth infection, the fetal ISC phenotype imposed by IκBα deficiency is intrinsic to epithelial cells, as it is observed in the organoid system independent of an inflammatory environment. It is also possible that IκBα deficiency perturbs ISC maturation during development through both NF‐κB‐dependent and NF‐κB‐independent mechanisms, and this phenotype is subsequently maintained in a cell‐autonomous NF‐κB‐independent manner. Since H3K27me3 ChIP from WT and KO embryos at early developmental stage (E14.5) did not show significant differences, we speculated that repression of the fetal ISC phenotype and maturation toward the adult state mediated by IκBα through PRC2 may occur at later stages of intestinal development once villi are formed and differentiated cells start to appear. Thus, PRC2 activity is likely to show two different waves of activity in the intestine, a first general wave that is IκBα‐independent and a second one that is IκBα‐dependent and affects specific subsets of genes including those related to ISC maturation from a previous fetal state. This requirement seems to be different in non‐homeostatic in vivo conditions, since IκBα loss in the damage tissue is associated with a partial conversion of cells into a transient fetal‐like phenotype (revealed by increased expression of the ISC genes Olfm4 and Cd44).
A similar mechanism may regulate fetal‐like‐to‐adult ISC conversion in response to injury following activation of IκBα target genes in response to inflammatory cytokines such as TNFα. Our results support a role for nuclear P‐IκBα as a crucial modulator of the switch between adult and fetal ISCs in the process of regeneration after damage. Moreover, our result suggests that IκBα regulates the transition from fetal to adult ISC phenotype by directly affecting H3 methylation at the promoter regions of specific fetal ISC genes such as Axl, Fut2, and Ly6a that might be driven by inflammatory cytokines. In this sense, we found that TNFα imposes a rapid activation of several ISC genes including Cd44 and Olfm4, which are not canonical NF‐κB target genes, and are not activated in the IκBα KO cells (whereas canonical NF‐κB such as Cxcl10 and A20 is effectively activated). Further investigation focused on the regulatory regions of these ISC genes is required to better understand how they individually or collectively respond to specific insults, environmental fluctuations, or inflammatory responses involving specific cytokines such as TNFα. It would be also important to test whether IκBα regulates the capacity of enterocyte progenitors 31 and post‐mitotic Paneth cells 32, 33 to produce adult or fetal ISCs after damage, as several authors have proposed.
In summary, we have uncovered a novel function for IκBα that may represent a general link between ISC homeostasis, inflammation, and response to damage and pathogens. Our hypothesis, awaiting further substantiation, is that IκBα acts as a molecular switch for stimulus‐mediated activation of a subset of ISC genes, likely through modulation of PRC2 activity.
Materials and Methods
Animal studies
C57BL/6J WT mice were purchased from The Jackson Laboratories. IκBα knockout (KO) (B6;129S4‐Nfkbiatm1Bal/J) mice and Lgr5GFP‐CreERT (B6.129P2‐Lgr5tm1(cre/ERT2)Cle/J) mice have the same genetic C57BL/6J background and were obtained from The Jackson Laboratories, and IκBαNES mice were previously described 16. Compound IκBβ;IκBε KO mice were generously donated by Dr. Alexander Hoffmann (University of California, LA). In all procedures, animals were kept under pathogen‐free conditions, and animal work was conducted according to the guidelines from the Animal Care Committee at the Generalitat de Catalunya. The Committee for Animal Experimentation at the Institute of Biomedical Research of Bellvitge (Barcelona) approved these studies.
Induction of colitis in mice
For colon inflammation studies, mice were given 2% DSS (w/v) [MP Biochemicals 216011090 MW, 36,000–50,000 Da] ad libitum in the drinking water, for 5 days, and were sacrificed at day 6. Colons were dissected, fixed in 10% formalin, and paraffin‐embedded for immunohistochemical analysis.
Organoid culture
For organoid generation, intestinal crypts were disaggregated mechanically, filtered in 70‐μm cell strainer, and seeded in Matrigel [Corning Ref. 354234] as described 20, 21. Organoids were expanded by serial passaging and kept frozen in liquid nitrogen for being used in subsequent experiments. In the inhibitor experiments, fresh medium containing the different compound was added to 3D cultures every 48 h. Reagents used are the following: 5 μM TNFα [Biological Industries Ref. 30‐T252‐B], IKKβ inhibitor 1 μM BAY 65‐5811 [Bayer Ref.], PRC2 inhibitor 5 μM EPZ‐6438 (EZH2 inhibitor) [MedChemExpress Ref. HY‐13803], Wnt inhibitor 5 μM XAV939 [Sigma Ref.X3004] and 1 μM C59 [Abcam Ref. ab142216], Notch inhibitor 5 μM DAPT (γ‐secretase inhibitor) [Calbiochem Ref. 565770], and glucocorticoid 20 μM Dexamethasone [Sigma Ref. D4902].
Organoid infection
MISSION shRNA for RelA [Sigma TRCN0000055346] and c‐Rel [Sigma TRCN0000225712] lentiviral plasmids was transfected in HEK293T cells. For CRISPR‐Cas9 experiments, lentiCRISPR v2 [Addgene #52961] was used. gRNA against the first exon of Nfkbia gene was designed using the Benchling [Biology Software] (the sequence of the 3 different guides used is as follows: 5′‐TGGACGATCGCCACGAC‐3′, 5′‐CGCCCTACCGAGTTCTTCC‐3′, 5′‐GCAGCAGCTCACGG‐3′). lentiCRISPR v2 with selected gRNA and lentiviral vectors were transfected in HEK293T cells. One day after transfection, media was changed, and viral particles were collected 24 h later and then concentrated using Ultracentrifuge Optima™ XPN‐100—IVD (Biosafe) (Beckman Coulter).
For organoid transduction, organoids from 4‐day cultures were disaggregated (15 min 37°C with trypsin for IκBα KO and mechanical disaggregation for wt organoids). The cell pellet was resuspended in concentrated virus that was diluted in complete organoid culture medium, centrifuged for 1 h at 150 g, and incubated for 4 h at 37°C. Cells were then washed in complete organoid culture medium, seeded in Matrigel [Corning Ref. 354234], and cultured as described above.
Cell fractionation and Western blot (WB)
For cytoplasm and nuclear separations, cells were lysed in 10 mM HEPES, 1.5 mM MgCl2, 10 mM KCl, and 0.05% NP‐40 (pH 7.9) for 10 min on ice and centrifuged at 850 g. Supernatants were recovered as the cytoplasmic fraction, and the pellets were lysed in Laemmli buffer (1× SDS–PAGE buffer + β‐mercaptoethanol (BME) [Sigma Ref. M‐3148]). Lysates were analyzed by Western blotting using standard SDS–polyacrylamide gel electrophoresis (SDS–PAGE) techniques. In brief, protein samples were boiled in Laemmli buffer, run in polyacrylamide gels, and transferred onto polyvinylidene difluoride (PVDF) membranes [Millipore Ref. IPVH00010]. Membranes were incubated overnight at 4°C with the appropriate primary antibodies (Appendix Table S1), extensively washed and then incubated with specific secondary horseradish peroxidase‐linked antibodies from Dako [Ref. P0260 and P0448]. Peroxidase activity was visualized using the enhanced chemiluminescence reagent [Biological Industries Ref. 20‐500‐120] and autoradiography films [GE Healthcare Ref. 28906835].
Immunohistochemical staining
Tissues and organoids were fixed in 4% formaldehyde overnight at room temperature and embedded in paraffin. 4‐μm paraffin‐embedded sections were first de‐paraffinized. IHC was performed following standard techniques with EDTA‐ or citrate‐based antigen retrieval, overnight incubation with primary antibodies (Appendix Table S2), and developed with the Envision+ System HRP Labelled Polymer anti‐Rabbit [Dako Ref. K4003] or anti‐Mouse [Dako Ref. K4001] and 3,3′‐diaminobenzidine (DAB) [Dako Ref. K3468]. Samples were mounted in DPX mountant [Sigma Ref. 06522]. Images were obtained with an Olympus BX61 microscope.
Immunofluorescence analysis
For tissue and organoid immunofluorescence, the same protocol as IHC was followed. However, the samples were developed with TSA™ Plus Cyanine 3/Fluorescein System [PerkinElmer Ref. NEL753001KT] and mounted in ProLong™ Diamond Antifade Mountant plus DAPI [Thermo Scientific Ref. P36971]. Images were taken in an SP5 upright confocal microscope (Leica). Antibodies used can be found in Appendix Table S3.
Hematoxylin and eosin staining
Previously de‐paraffinized sections were incubated with hematoxylin [Merck Ref. 1092530500] 30 s, tap water 5 min, 80% ethanol 0.15% HCl 30 s, water 30 s, 30% ammonia water (NH3(aq)) 30 s, water 30 s, 96% ethanol 5 min, eosin [Bio‐Optica Ref. 05‐10003/L] 3 s, and absolute ethanol 1 min. Samples were dehydrated and mounted in DPX mountant [Sigma Ref. 06522], and images were obtained with an Olympus BX61 microscope.
Alcian blue staining
Previously de‐paraffinized sections were incubated with 3% acetic acid in H2O for 3 min, immersed in Alcian Blue (10 mg/ml pH2.5) [Merck Ref. 101647] for 2‐10 min, and counterstained with Nuclear Fast Red solution [Sigma Ref. 6409‐77‐4] for 10 min. Samples were mounted in DPX mountant [Sigma Ref. 06522], and images were obtained with an Olympus BX61 microscope.
Immunoprecipitation
Nucleus from crypt cells were isolated with buffer pH 7.9 10 mM HEPES, 1.5 mM MgCl2, 10 mM KCl, 0.5 mM DTT, 0.05% NP40 plus ubiquitin/ubiquitin‐like peptidase inhibitor PR‐619 [Tebu‐Bio SI9619], protease [Complete Mini, Roche Ref. 11836153001], and phosphatase [PhosSTOP, Roche Ref. 4906845001] inhibitor cocktail. The nucleus was lysed with RIPA buffer (0.1% DOC, 0.1% SDS, 1% Triton X‐100, 10 mM Tris–HCl pH 8.0, 140 mM NaCl, 1 mM EDTA, 0.25 mM EGTA, 10 mM Na‐butyrate, 0.1 mM Na‐orthovanadate, in H2O). After centrifugation, the SUMOylated proteins in supernatant were precipitated with anti‐SUMO2‐3 [10 μl of Abcam ab81371] antibody or IκBα was precipitated with anti‐IκBα antibody [25 μl Santa Cruz sc‐371]. Rabbit and mouse IgG were used as negative controls (4 μg each). The antibodies were incubated with nuclear lysates overnight and the complexes precipitated with protein A‐sepharose beads [GE Healthcare, Ref. 17‐0780‐01] 2 h at 4°C.
qRT–PCR analysis
Total RNA was extracted with the RNeasy Mini Kit [Qiagen Ref. 74004], and the RT‐First Strand cDNA Synthesis Kit [GE Healthcare Life Sciences Ref. 27‐9261‐01] was used to produce cDNA. qRT–PCR was performed in LightCycler 480 system using SYBR Green I Master Kit [Roche Ref. 04887352001]. The primers used are listed in Appendix Table S4.
RNA‐sequencing experiments and data analysis
In all experiments, we extracted total RNA from three mice per genotype using RNeasy Micro Kit [Qiagen Ref. 74004]. The RNA concentration and integrity were determined using Agilent Bioanalyzer [Agilent Technologies]. Libraries were prepared at the Genomics Unit of PRBB (Barcelona, Spain) using standard protocols, and cDNA was sequenced using Illumina® HiSeq platform, obtaining ~ 25–30 million 50‐bp single‐end reads per sample. Adapter sequences were trimmed with Trim Galore. Sequences were filtered by quality (Q > 30) and length (> 20 bp). Filtered reads were mapped against the latest release of the mouse reference genome (mm10) using default parameters of TopHat (v.2.1.1) 34, and expressed transcripts were then assembled. High‐quality alignments were fed to HTSeq (v.0.9.1) 35 to estimate the normalized counts of each expressed gene. Differentially expressed genes between different conditions were explored using DESeq2 R package (v.1.20.0) 36. Plots were done in R. RNA‐sequencing data are deposited at the GEO database with accession number GSE131187.
Chromatin immunoprecipitation and ChIP‐sequencing analysis
Intestinal crypts were subjected to ChIP as previously described 9, 37. Briefly, formaldehyde crosslinked cell extracts were sonicated, and chromatin fractions were incubated for 16 h with anti‐IκBα [abcam ab32518], anti‐H3K27me3 [Millipore 07‐449], anti‐c‐Rel [Abcam ab227519], and anti‐p65 [Abcam ab16502] antibodies in RIPA buffer and then precipitated with protein A/G‐sepharose [GE Healthcare, Refs. 17‐0618‐01 and 17‐0780‐01]. Crosslinkage was reversed, and 6–10 ng of precipitated chromatin was directly sequenced in the genomics facility of Parc de Recerca Biomèdica de Barcelona (PRBB) using Illumina® HiSeq platform. Raw single‐end 50‐bp sequences were filtered by quality (Q > 30) and length (length > 20 bp) with Trim Galore 38. Filtered sequences were aligned against the reference genome (mm10 release) with Bowtie2 39. MACS2 software 40 was run first for each replicate, and then combining all replicates, using unique alignments (q‐value < 0.1). Peak annotation was performed with ChIPseeker package 41, and functional enrichment analysis with enrichR 42, using the latest version of GO annotations. Peak visualization was done with Integrative Genomics Viewer (IGV). ChIP‐sequencing data are deposited at the GEO database with accession number GSE131187.
Cell sorting
Crypt cells obtained after mechanical disaggregation and 70 μm filtration were trypsinized [Biological Industries Ref. 03‐050‐1A] 15 min at 37°C. Cells were filtered through 40‐μm pore diameter cell strainers. Incubation with APC‐EphB2 antibody [BD Pharmingen Ref. 564699] was performed for 20 min, and the cells were sorted in an Influx™ Sorter [BD Biosciences]. For IF, EphB2 high or low were cytospin onto poly‐l‐lysine‐coated slides (100 g, 3 min).
For Lgr5+ cell isolation in Lgr5GFP‐CreERT mice, the detection of endogenous GFP was used for sorting.
Expression microarray
We extracted total RNA from five replicates of a minimum of two organoids per genotype using RNeasy Micro Kit [Qiagen Ref. 74004]. Purity and integrity of the RNA were assessed by spectrophotometry and nanoelectrophoresis using the NanoDrop ND‐2000 spectrophotometer (NanoDrop Technologies, USA) and the Nano lab‐on‐a‐chip assay for total eukaryotic RNA using Bioanalyzer 2100 (Agilent Technologies, USA), respectively. RNA samples were processed according to the manuals GeneChip WT PLUS Reagent kit (P/N 703174 Rev. 2) and Expression Wash, Stain and Scan User Manual (P/N 702731 Rev. 3) (Thermo Fisher). Samples were hybridized to the mouse Clariom S Array (Thermo Fisher) in a GeneChip hybridization oven 640. For the statistical analysis, R programming (version 3.4.2) was used with different packages from Bioconductor and the Comprehensive R Archive Network (CRAN). Normalization was performed using the Robust Multi‐array Average algorithm (RMA) included in aroma.affymetrix package. Differential expression (DE) analysis was assessed with the limma package. Gene annotations were provided by Affymetrix for the Clariom S array (NetAffx na36, mm10 genome assembly). The location of each Transcript Cluster ID (TCI) (Start, Stop, Strand and Chromosome) was used to map into the UCSC database (April 2014mm10) to obtain genes matching the same coordinates. Data from microarray analysis are deposited at the GEO database with accession number GSE130937.
Quantification and statistical analysis
Statistical parameters, including number of events quantified, standard deviation, and statistical significance, are reported in the figures and in the figure legends. Statistical analysis has been performed using GraphPad Prism 6 software (GraphPad), and P < 0.05 is considered significant. Two‐sided Student's t‐test was used to compare differences between two groups. Each experiment shown in the manuscript has been repeated at least twice.
Author contributions
AB, SM, and LE conceptualized the study, designed the experiments, and wrote the manuscript. LM performed experiments and wrote the manuscript. JB, EL‐A, MG, JG, CR‐H, DL, MG‐F, and SA‐G performed biochemical assays and in vitro and in vivo experiments. YG analyzed ChIP‐sequencing and RNA‐sequencing data. RB and ARN treated mice with DSS and provided tissue samples. MI performed the histopathological analysis of intestinal samples.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Review Process File
Acknowledgements
We thank all members of Espinosa and Bigas laboratories for helpful discussions and technical support. We also thank the animal facility, FACS facility, and genomic facility of the PRBB for their technical support. We thank Alexander Hoffmann for providing the IκBε;IκBβ KO mice and for helpful discussions during the preparation of the manuscript. This work was funded by grants from Instituto de Salud Carlos III FEDER (PIE15/00008 and PI16/00437) and Generalitat de Catalunya 2017SGR135. LM is a recipient for 2015FI‐B00806 and 2016FI‐B1 00110 and JG for PERIS SLT002/16/00070 fellowship programs from Generalitat de Catalunya.
EMBO Reports (2020) 21: e49708
Contributor Information
Anna Bigas, Email: abigas@imim.es.
Lluís Espinosa, Email: lespinosa@imim.es.
Data availability
RNA‐Seq and ChIP‐seq data: GEO database GSE131187 https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE131187
Expression microarray: GEO database GSE130937 https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE130937
References
- 1. Hoffmann J, Akira S (2013) Innate immunity. Curr Opin Immunol 25: 1–3 [DOI] [PubMed] [Google Scholar]
- 2. Arenzana‐Seisdedos F, Turpin P, Rodriguez M, Thomas D, Hay RT, Virelizier JL, Dargemont C (1997) Nuclear localization of I kappa B alpha promotes active transport of NF‐kappa B from the nucleus to the cytoplasm. J Cell Sci 110(Pt 3): 369–378 [DOI] [PubMed] [Google Scholar]
- 3. Chiao PJ, Miyamoto S, Verma IM (1994) Autoregulation of I kappa B alpha activity. Proc Natl Acad Sci USA 91: 28–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Le Bail O, Schmidt‐Ullrich R, Israel A (1993) Promoter analysis of the gene encoding the I kappa B‐alpha/MAD3 inhibitor of NF‐kappa B: positive regulation by members of the rel/NF‐kappa B family. EMBO J 12: 5043–5049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sun SC, Ganchi PA, Ballard DW, Greene WC (1993) NF‐kappa B controls expression of inhibitor I kappa B alpha: evidence for an inducible autoregulatory pathway. Science 259: 1912–1915 [DOI] [PubMed] [Google Scholar]
- 6. Beg AA, Sha WC, Bronson RT, Baltimore D (1995) Constitutive NF‐kappa B activation, enhanced granulopoiesis, and neonatal lethality in I kappa B alpha‐deficient mice. Genes Dev 9: 2736–2746 [DOI] [PubMed] [Google Scholar]
- 7. Klement JF, Rice NR, Car BD, Abbondanzo SJ, Powers GD, Bhatt PH, Chen CH, Rosen CA, Stewart CL (1996) IkappaBalpha deficiency results in a sustained NF‐kappaB response and severe widespread dermatitis in mice. Mol Cell Biol 16: 2341–2349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rebholz B, Haase I, Eckelt B, Paxian S, Flaig MJ, Ghoreschi K, Nedospasov SA, Mailhammer R, Debey‐Pascher S, Schultze JL et al (2007) Crosstalk between keratinocytes and adaptive immune cells in an IkappaBalpha protein‐mediated inflammatory disease of the skin. Immunity 27: 296–307 [DOI] [PubMed] [Google Scholar]
- 9. Mulero MC, Ferres‐Marco D, Islam A, Margalef P, Pecoraro M, Toll A, Drechsel N, Charneco C, Davis S, Bellora N et al (2013) Chromatin‐bound IkappaBalpha regulates a subset of polycomb target genes in differentiation and cancer. Cancer Cell 24: 151–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ezhkova E, Pasolli HA, Parker JS, Stokes N, Su IH, Hannon G, Tarakhovsky A, Fuchs E (2009) Ezh2 orchestrates gene expression for the stepwise differentiation of tissue‐specific stem cells. Cell 136: 1122–1135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mejetta S, Morey L, Pascual G, Kuebler B, Mysliwiec MR, Lee Y, Shiekhattar R, Di Croce L, Benitah SA (2011) Jarid2 regulates mouse epidermal stem cell activation and differentiation. EMBO J 30: 3635–3646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mulder KW, Wang X, Escriu C, Ito Y, Schwarz RF, Gillis J, Sirokmany G, Donati G, Uribe‐Lewis S, Pavlidis P et al (2012) Diverse epigenetic strategies interact to control epidermal differentiation. Nat Cell Biol 14: 753–763 [DOI] [PubMed] [Google Scholar]
- 13. Chiacchiera F, Rossi A, Jammula S, Zanotti M, Pasini D (2016) PRC2 preserves intestinal progenitors and restricts secretory lineage commitment. EMBO J 35: 2301–2314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Koppens MA, Bounova G, Gargiulo G, Tanger E, Janssen H, Cornelissen‐Steijger P, Blom M, Song JY, Wessels LF, van Lohuizen M (2016) Deletion of polycomb repressive complex 2 from mouse intestine causes loss of stem cells. Gastroenterology 151: 684–697 e12 [DOI] [PubMed] [Google Scholar]
- 15. Oittinen M, Popp A, Kurppa K, Lindfors K, Maki M, Kaikkonen MU, Viiri K (2017) Polycomb repressive complex 2 enacts Wnt signaling in intestinal homeostasis and contributes to the instigation of stemness in diseases entailing epithelial hyperplasia or neoplasia. Stem Cells 35: 445–457 [DOI] [PubMed] [Google Scholar]
- 16. Wuerzberger‐Davis SM, Chen Y, Yang DT, Kearns JD, Bates PW, Lynch C, Ladell NC, Yu M, Podd A, Zeng H et al (2011) Nuclear export of the NF‐kappaB inhibitor IkappaBalpha is required for proper B cell and secondary lymphoid tissue formation. Immunity 34: 188–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Barker N, van Es JH, Kuipers J, Kujala P, van den Born M, Cozijnsen M, Haegebarth A, Korving J, Begthel H, Peters PJ et al (2007) Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 449: 1003–1007 [DOI] [PubMed] [Google Scholar]
- 18. Sangiorgi E, Capecchi MR (2008) Bmi1 is expressed in vivo in intestinal stem cells. Nat Genet 40: 915–920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yan KS, Chia LA, Li X, Ootani A, Su J, Lee JY, Su N, Luo Y, Heilshorn SC, Amieva MR et al (2012) The intestinal stem cell markers Bmi1 and Lgr5 identify two functionally distinct populations. Proc Natl Acad Sci USA 109: 466–471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sato T, van Es JH, Snippert HJ, Stange DE, Vries RG, van den Born M, Barker N, Shroyer NF, van de Wetering M, Clevers H (2011) Paneth cells constitute the niche for Lgr5 stem cells in intestinal crypts. Nature 469: 415–418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sato T, Vries RG, Snippert HJ, van de Wetering M, Barker N, Stange DE, van Es JH, Abo A, Kujala P, Peters PJ et al (2009) Single Lgr5 stem cells build crypt‐villus structures in vitro without a mesenchymal niche. Nature 459: 262–265 [DOI] [PubMed] [Google Scholar]
- 22. Fordham RP, Yui S, Hannan NR, Soendergaard C, Madgwick A, Schweiger PJ, Nielsen OH, Vallier L, Pedersen RA, Nakamura T et al (2013) Transplantation of expanded fetal intestinal progenitors contributes to colon regeneration after injury. Cell Stem Cell 13: 734–744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mustata RC, Vasile G, Fernandez‐Vallone V, Strollo S, Lefort A, Libert F, Monteyne D, Perez‐Morga D, Vassart G, Garcia MI (2013) Identification of Lgr5‐independent spheroid‐generating progenitors of the mouse fetal intestinal epithelium. Cell Rep 5: 421–432 [DOI] [PubMed] [Google Scholar]
- 24. Nusse YM, Savage AK, Marangoni P, Rosendahl‐Huber AKM, Landman TA, de Sauvage FJ, Locksley RM, Klein OD (2018) Parasitic helminths induce fetal‐like reversion in the intestinal stem cell niche. Nature 559: 109–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yui S, Azzolin L, Maimets M, Pedersen MT, Fordham RP, Hansen SL, Larsen HL, Guiu J, Alves MRP, Rundsten CF et al (2018) YAP/TAZ‐dependent reprogramming of colonic epithelium links ECM remodeling to tissue regeneration. Cell Stem Cell 22: 35–49 e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Desterro JM, Rodriguez MS, Hay RT (1998) SUMO‐1 modification of IkappaBalpha inhibits NF‐kappaB activation. Mol Cell 2: 233–239 [DOI] [PubMed] [Google Scholar]
- 27. Merlos‐Suarez A, Barriga FM, Jung P, Iglesias M, Cespedes MV, Rossell D, Sevillano M, Hernando‐Momblona X, da Silva‐Diz V, Munoz P et al (2011) The intestinal stem cell signature identifies colorectal cancer stem cells and predicts disease relapse. Cell Stem Cell 8: 511–524 [DOI] [PubMed] [Google Scholar]
- 28. Garoufalis E, Kwan I, Lin R, Mustafa A, Pepin N, Roulston A, Lacoste J, Hiscott J (1994) Viral induction of the human beta interferon promoter: modulation of transcription by NF‐kappa B/rel proteins and interferon regulatory factors. J Virol 68: 4707–4715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Thanos D, Maniatis T (1995) Identification of the rel family members required for virus induction of the human beta interferon gene. Mol Cell Biol 15: 152–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Guma M, Stepniak D, Shaked H, Spehlmann ME, Shenouda S, Cheroutre H, Vicente‐Suarez I, Eckmann L, Kagnoff MF, Karin M (2011) Constitutive intestinal NF‐kappaB does not trigger destructive inflammation unless accompanied by MAPK activation. J Exp Med 208: 1889–1900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tetteh PW, Basak O, Farin HF, Wiebrands K, Kretzschmar K, Begthel H, van den Born M, Korving J, de Sauvage F, van Es JH et al (2016) Replacement of lost Lgr5‐positive stem cells through plasticity of their enterocyte‐lineage daughters. Cell Stem Cell 18: 203–213 [DOI] [PubMed] [Google Scholar]
- 32. Schmitt M, Schewe M, Sacchetti A, Feijtel D, van de Geer WS, Teeuwssen M, Sleddens HF, Joosten R, van Royen ME, van de Werken HJG et al (2018) Paneth cells respond to inflammation and contribute to tissue regeneration by acquiring stem‐like features through SCF/c‐Kit signaling. Cell Rep 24: 2312–2328 e7 [DOI] [PubMed] [Google Scholar]
- 33. Yu S, Tong K, Zhao Y, Balasubramanian I, Yap GS, Ferraris RP, Bonder EM , Verzi MP, Gao N (2018) Paneth cell multipotency induced by notch activation following injury. Cell Stem Cell 23: 46–59 e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL (2013) TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14: R36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Anders S, Pyl PT, Huber W (2015) HTSeq–a Python framework to work with high‐throughput sequencing data. Bioinformatics 31: 166–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Love MI, Anders S, Kim V, Huber W (2015) RNA‐Seq workflow: gene‐level exploratory analysis and differential expression. F1000Res 4: 1070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Aguilera C, Hoya‐Arias R, Haegeman G, Espinosa L, Bigas A (2004) Recruitment of IkappaBalpha to the hes1 promoter is associated with transcriptional repression. Proc Natl Acad Sci USA 101: 16537–16542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Krueger B, Friedrich T, Forster F, Bernhardt J, Gross R, Dandekar T (2012) Different evolutionary modifications as a guide to rewire two‐component systems. Bioinform Biol Insights 6: 97–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Langmead B, Salzberg SL (2012) Fast gapped‐read alignment with Bowtie 2. Nat Methods 9: 357–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W et al (2008) Model‐based analysis of ChIP‐Seq (MACS). Genome Biol 9: R137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yu G, Wang LG, He QY (2015) ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 31: 2382–2383 [DOI] [PubMed] [Google Scholar]
- 42. Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A et al (2016) Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res 44: W90–W97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Muñoz J, Stange DE, Schepers AG, van de Wetering M, Koo BK, Itzkovitz S, Volckmann R, Kung KS, Koster J, Radulescu S et al (2012) The Lgr5 intestinal stem cell signature: robust expression of proposed quiescent ‘+4’ cell markers. EMBO J 31: 3079–3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Van der Flier LG, Sabates‐Bellver J, Oving I, Haegebarth A, De Palo M, Anti M, Van Gijn ME, Suijkerbuijk S, Van de Wetering M, Marra G et al (2007) The intestinal Wnt/TCF signature. Gastroenterology 132: 628–632 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Review Process File
Data Availability Statement
RNA‐Seq and ChIP‐seq data: GEO database GSE131187 https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE131187
Expression microarray: GEO database GSE130937 https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE130937