

Abstract
CD1d‐restricted invariant natural killer T (iNKT) cells constitute a common glycolipid‐reactive innate‐like T‐cell subset with a broad impact on innate and adaptive immunity. While several microbial glycolipids are known to activate iNKT cells, the cellular mechanisms leading to endogenous CD1d‐dependent glycolipid responses remain largely unclear. Here, we show that endoplasmic reticulum (ER) stress in APCs is a potent inducer of CD1d‐dependent iNKT cell autoreactivity. This pathway relies on the presence of two transducers of the unfolded protein response: inositol‐requiring enzyme‐1a (IRE1α) and protein kinase R‐like ER kinase (PERK). Surprisingly, the neutral but not the polar lipids generated within APCs undergoing ER stress are capable of activating iNKT cells. These data reveal that ER stress is an important mechanism to elicit endogenous CD1d‐restricted iNKT cell responses through induction of distinct classes of neutral lipids.
Keywords: antigen‐presenting cells, ER stress, lipid antigens, neutral lipid, NKT cells
Subject Categories: Immunology, Membrane & Intracellular Transport
Upon ER stress immunogenic lipids are produced that activate NKT cells. Defining the conditions required to generate endogenous high immunogenic lipid antigens paves the way for the design of novel lipids with immunomodulatory function.

Introduction
The endoplasmic reticulum (ER) plays a crucial role in protein quality control and lipid biosynthesis within the cell. When altered cellular states perturb ER functions, a phenomenon termed “ER stress” activates signal transduction pathways collectively known as the unfolded protein response (UPR) 1, 2. The UPR is comprised of three core stress transducers, namely ER‐resident transmembrane proteins inositol‐requiring enzyme 1α (IRE1α), pancreatic ER eukaryotic translation initiation factor (eIF)‐2α kinase (PERK) and activating transcription factor (ATF)6. These transducers function to reduce ER stress by upregulating transcripts that control protein‐folding capacity within the ER, promote degradation of misfolded proteins via ER‐associated protein degradation (ERAD) and regulate biogenesis of the ER membranes 3.
Stimulation of membrane phospholipid synthesis during UPR has been shown to require the transcription factor X‐box binding protein 1 (XBP‐1) 4, a downstream target of IRE1α to regulate free fatty acid, triglyceride and cholesterol synthesis 5, 6. Sterol regulatory element‐binding proteins (SREBPs) are transcription factors that regulate the expression of genes involved in lipid synthesis and signalling networks to maintain lipid metabolism and homeostasis within the cell 7, 8, 9. SREBPs are implicated in numerous pathogenic processes such as obesity, diabetes mellitus, non‐alcoholic fatty liver disease, non‐alcoholic steatohepatitis, neurodegenerative diseases and cancer 10. During ER stress, PERK‐mediated (eIF)‐2α phosphorylation causes SREBP activation 11, 12. Similarly, the main ER stress regulator, the chaperone 78 kDa glucose‐regulated protein (GRP78, also known as BiP), suppresses ER stress‐regulated SREBP activation in the liver and improves hepatic steatosis and insulin sensitivity 13. Thus, the UPR collectively coordinates protein quality control as well as lipid biosynthesis during ER stress to re‐establish cellular homeostasis. A number of studies have established a mechanistic link between UPR and dysregulation of sphingolipid metabolism, and it is thought that these processes can contribute to the development of inflammatory diseases 14, 15, 16.
Lipids may also serve as an important source of immunological antigens. CD1 molecules, a family of MHC class I‐like glycoproteins, are specialized in presenting self‐ and non‐self‐lipids to T lymphocytes 17. The human genome encodes five CD1 genes, CD1a, CD1b, CD1c, CD1d and CD1e, whereas only CD1d is expressed in rodents. In the ER, nascent CD1 heavy chains are folded and assembled with β2‐microglobulin (β2m) in a process coordinated by the ER chaperones calnexin and calreticulin 18, 19. Proper folding and stabilization of CD1b and CD1d is thought to be facilitated by the loading of phospholipids derived from the ER membrane 20, 21 and is mediated by an ER chaperone called microsomal triglyceride transfer protein (MTP) 22. Invariant natural killer T (iNKT) cells are a specialized subset of T cells that recognize self‐ and foreign lipids presented by CD1 molecules 23, 24. A hallmark is their ability to secrete large amounts of T helper (Th)1, Th2 and Th17 cytokines as well as chemokines shortly after activation 25. Consequently, iNKT cells have been suggested to play a role in either the amelioration or exacerbation of disease models like type I diabetes, lupus, cancer, asthma, atherosclerosis, rheumatoid arthritis, experimental autoimmune encephalomyelitis (EAE) and inflammatory bowel disease (IBD) 25, 26. Most murine and human iNKT cells recognize a non‐self‐glycosphingolipid called α‐galactosylceramide (α‐GalCer) 27 which induces strong NKT cell activation. However, to date, no microbial or self‐lipid antigen has been described with similarly potent antigenic capacity. In this study, we demonstrate that the IRE1α or PERK pathway within APCs induces a lipid biosynthetic program that regulates the generation of distinct classes of endogenous neutral lipids. Moreover, neutral lipids generated under ER stress are loaded onto CD1d, leading to potent iNKT cell stimulation, highlighting a novel endogenous mechanism responsible for triggering these immunomodulatory T cells.
Results
ER‐stressed APCs promote iNKT cell activation
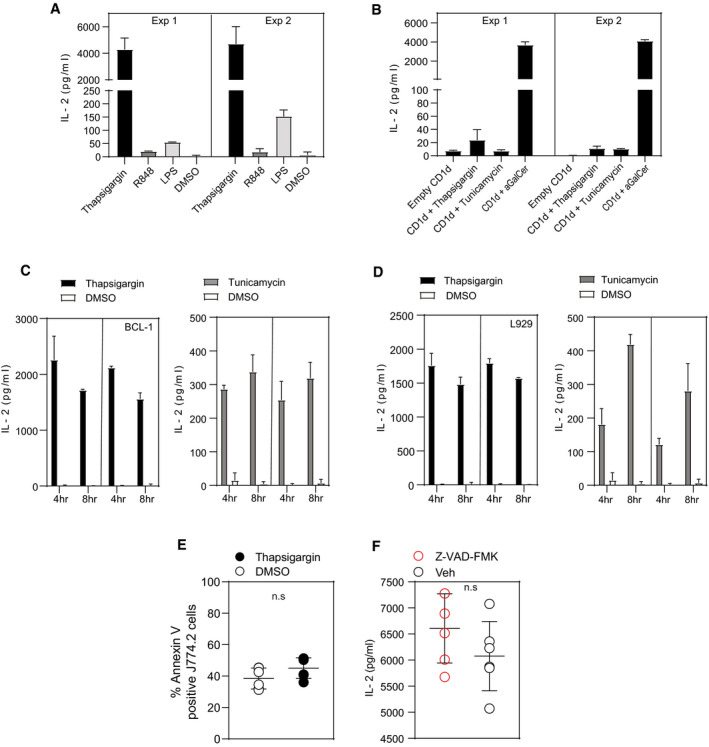
The UPR is accompanied by upregulation in lipid synthesis enzymes suggesting that lipid synthesis is needed for resolving ER stress 28, 29, 30. However, whether this process impacts generation of immunogenic lipid antigens is unknown. To test this hypothesis, we initially cultured murine and human macrophage cell lines with chemical agents known to induce UPR such as thapsigargin or tunicamycin, R848 or LPS at various time points, washed them and then co‐cultured them with iNKT cell hybridomas. As shown in Fig 1, treatment of the macrophage cell lines J774.2 and U937 results in strong activation of the NKT cell hybridoma N38‐2C12. The strongest iNKT cell autoreactivity was detected with thapsigargin‐treated macrophages (for 4 h) when compared to tunicamycin or LPS, whereas R848‐ or DMSO‐treated cells displayed negligible levels of IL‐2 cytokine release (Figs 1A and B, and EV1A). Despite the extensive washing procedure, we cannot exclude the potential presence of residual spore fractions of tunicamycin or thapsigargin in the medium of the macrophages, which could have been sufficient to trigger iNKT cells. Therefore, as a control, we directly incubated iNKT cell hybridoma cells in a CD1d protein‐bound plate loaded with tunicamycin or thapsigargin. However, here we found no induction of IL‐2 (Fig EV1B). Analysis of UPR inducers in other murine cell lines (such as the B‐lymphoma (Bcl‐1) or fibroblast (L929) cell lines) confirmed that stressed cells induce NKT cell activation, suggesting that the observation was not restricted to macrophages only, but that in general induction of UPR in diverse APCs is associated with iNKT activation (Fig EV1C and D). Moreover, our results also exclude the effect of apoptosis‐mediated iNKT activation because there was no difference in Annexin V levels, and importantly, blocking apoptosis using a pan‐caspase inhibitor had no effect on IL‐2 secretion by 2C12 cells (Fig EV1E and F).
Figure 1. iNKT cell activation by antigen‐presenting cells undergoing UPR.
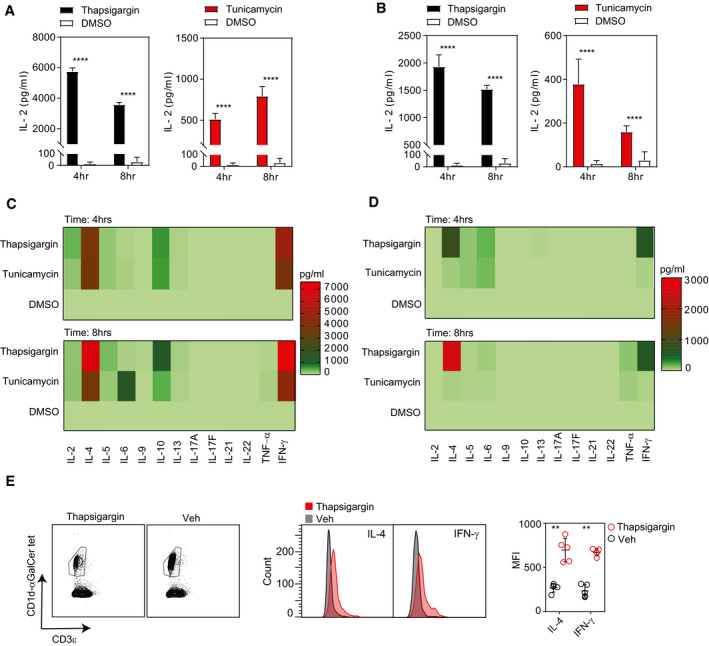
-
A, BBar graph represents IL‐2 secretion by NKT cell hybridoma (2C12) co‐cultured with (A) a murine macrophage cell line (J774.2) or with (B) a human macrophage cell line (U937) treated with either thapsigargin (black bars), tunicamycin (red bars) or DMSO (white bars). Graphs show mean ± SEM from n = 3 biological replicates. ****P < 0.0001 (unpaired t‐test).
-
C, DThe heat map represents cytokine secretion by in vitro expanded (C) murine splenic iNKT cells or (D) human peripheral blood iNKT cells co‐cultured either with thapsigargin‐ or tunicamycin‐ or DMSO‐treated murine BMDMs. The heat map shows the average result of two pooled biological replicates.
-
EWild‐type C57BL/6 mice were injected intravenously with thapsigargin‐loaded PLGA nanoparticles or vehicle, and 12 h later, hepatic iNKT cells were analysed by flow cytometry. Histograms represent expression of intracellular IL‐4 and IFN‐γ levels in hepatic iNKT cells from mice injected with thapsigargin‐loaded PLGA nanoparticles (red histogram) or vehicle‐treated (grey histogram) mice. Dot plots represent MFI for intracellular IL‐4 and IFN‐γ expression in hepatic iNKT cells from thapsigargin‐loaded PLGA nanoparticles (red dots) or vehicle‐treated (black dots) mice. Data plots show mean ± SEM from n = 5 mice per group. **P ≤ 0.001 (Mann–Whitney U‐test).
Source data are available online for this figure.
Figure EV1. Induction of UPR in diverse APCs is associated with iNKT activation.
-
ABar graph represents IL‐2 secretion by 2C12 NKT hybridoma upon co‐culture with J774.2 treated with either thapsigargin, R848, LPS or DMSO. Co‐culture with thapsigargin‐treated J774.2 cells resulted in significant increase of IL‐2 secretion by 2C12, whereas R848‐ or LPS‐treated J774.2 cells induced small increases or negligible levels of IL‐2 secretion by 2C12 when compared to DMSO controls. In total, n = 2 biological replicates were performed (two technical replicates per biological replicate). Graphs show mean ± SEM of each biological replicate.
-
BIL‐2 secretion by 2C12 is not due to the direct activation of 2C12 cells by either thapsigargin or tunicamycin. In total, n = 2 biological replicates were performed (two technical replicates per biological replicate). Graphs show mean ± SEM of each biological replicate.
-
C, DIL‐2 secretion by NKT cell hybridoma 2C12 is not restricted to macrophages undergoing UPR but also occurs in diverse APCs such as murine Bcl‐1 and L929 cell lines. In total, n = 2 biological replicates were performed (two technical replicates per biological replicate). Graphs show mean ± SEM of each biological replicate.
-
EDot plot represents little or no significant difference in the induction of apoptosis by thapsigargin (50 nM) treatment in J774.2 cells when compared to DMSO treatment. Graphs show mean ± SEM from n = 3 biological replicates (two technical replicates per biological replicate). n.s (non‐significant) (unpaired t‐test).
-
FDot plot represents that the activation of 2C12 NKT hybridoma is not due to cells undergoing apoptosis upon treating the J774.2 cells with thapsigargin. Graphs show mean ± SEM from n = 5 individual mice per condition. n.s (non‐significant) (Mann–Whitney U‐test).
Source data are available online for this figure.
We next examined whether UPR induction within primary macrophages regulates TCR‐dependent iNKT activation. For these experiments, murine bone marrow‐derived macrophages (BMDM) and human peripheral‐derived macrophages were stimulated in vitro with either thapsigargin or tunicamycin, and subsequently co‐cultured with murine or human CD1d tetramer‐sorted iNKT cells from C57Bl/6 mice or healthy human peripheral blood, respectively. As shown in Fig 1C and D, the primary murine and human APC undergoing UPR induces mixed Th1 and Th2 cell‐type iNKT cytokine responses, whereas type 17 cytokines were not induced. Interestingly, intravenous (i.v.) administration of PLGA nanoparticles loaded with thapsigargin significantly increased the levels of IL‐4 and IFN‐γ cytokine‐positive iNKT cells in the liver of CD57Bl/6 mice as compared to vehicle‐treated particles (Fig 1E). Collectively, these results suggest that UPR inducers cause primary APCs to potently activate both mouse and human iNKT cells leading to IL‐4 and IFN‐γ release.
UPR‐induced iNKT cell activation is CD1d‐dependent
iNKT cells are prototypic innate‐like T cells, and as such, their activation may occur through cytokine‐dependent activation such as IL‐12 or by glycolipid‐mediated TCR recognition through CD1d presentation 31, 32, 33. We therefore evaluated the nature of the observed iNKT cell autoreactivity upon UPR stress. First, we co‐cultured NKT cell hybridoma N38‐2C12 with J774.2 stimulated with either thapsigargin or α‐GalCer in the presence of CD1d blocking monoclonal antibody or an isotype control Ab (Fig 2A). The results indicate that thapsigargin‐induced NKT cell activation relies on CD1d, and the degree of NKT cell activation was comparable with the response elicited by the strong prototypic agonist α‐GalCer. To evaluate whether inflammatory cytokines produced by APCs undergoing UPR might be involved in the observed iNKT activation, we first performed cytokine arrays in supernatant collected from primary macrophages treated with thapsigargin or tunicamycin. This assay provided evidence that the ER‐stressed macrophages produced negligible levels of cytokine release except for IL‐6 and TNF‐α (Fig EV2A). We confirmed these findings by performing co‐cultures of NKT hybridomas in the presence of J774.2 cell line with CD1d knockdown and with primary macrophages derived from CD1d−/− mice stimulated with thapsigargin. Both resulted in reduced iNKT cell autoreactivity versus the untransfected parental cell line or primary macrophages derived from CD1d+/+ mice, respectively (Fig 2B and C). Theoretically, enhanced iNKT cell autoreactivity may occur secondary to changes in surface levels of CD1d on APC during ER stress. We therefore analysed the CD1d surface expression by APCs treated with thapsigargin (Fig EV3A and B). The data, however, indicate that CD1d surface expression by APCs remains unaltered during ER stress. Furthermore, we have been able to exclude a role for direct effect of thapsigargin or tunicamycin in binding CD1d and activating iNKT cells as drug addition into plate‐bound CD1d proteins failed to activate iNKT cells (Fig EV1B). Taken together, these findings point to a strong induction of iNKT cell autoreactivity in response to UPR activators in APCs, which is strictly CD1d‐dependent.
Figure 2. Autoreactivity of iNKT cell is CD1d‐dependent.
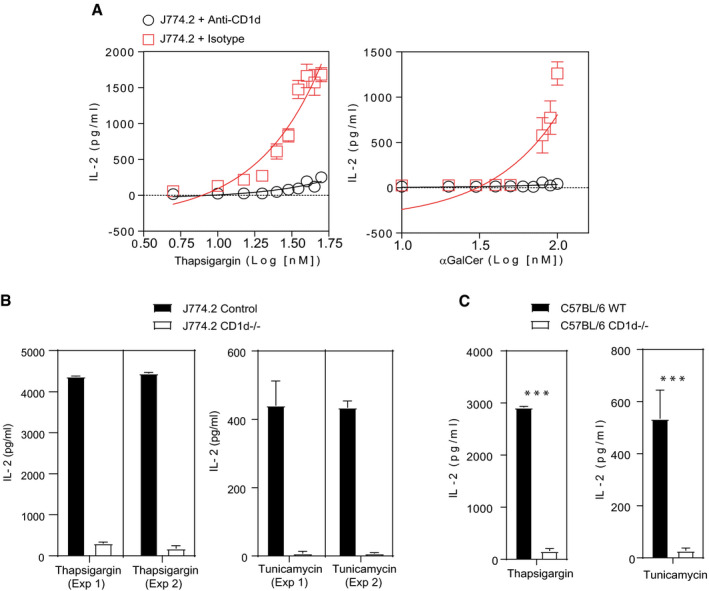
- Thapsigargin‐ or α‐GalCer‐treated J774.2 cells were co‐cultured with 2C12 in the presence or absence of the murine CD1d‐specific antibody (black circles) or isotype (red squares) for 16 h. The addition of an anti‐CD1d antibody blocked the activation of 2C12 demonstrating that the NKT cell activation was CD1d‐specific. Graphs show mean ± SEM from n = 2 biological replicates.
- Bar graph represents IL‐2 secretion by NKT cell hybridoma (2C12) co‐cultured murine macrophage cell line (J774.2) treated either with thapsigargin‐ or tunicamycin‐treated CD1d knockdown (white bars) or untransfected (black bars). Knockdown of CD1d blocked IL‐2 secretion by 2C12. In total, n = 2 biological replicates were performed (three technical replicates per biological replicate). Graphs show mean ± SEM of each biological replicate.
- Bar graph represents IL‐2 secretion by NKT cell hybridoma (2C12) co‐cultured either with thapsigargin‐ or tunicamycin‐treated primary BMDM isolated from CD1d−/− (white bars) or wild‐type (black bars) mice. 2C12 co‐cultured with CD1d−/− BMDMs resulted in reduced IL‐2 secretion. Graphs show mean ± SEM from n = 3 biological replicates. ***P < 0.001 (unpaired t‐test).
Source data are available online for this figure.
Figure EV2. Autoreactivity of iNKT cells induced by UPR is cytokine‐ and TLR‐independent.
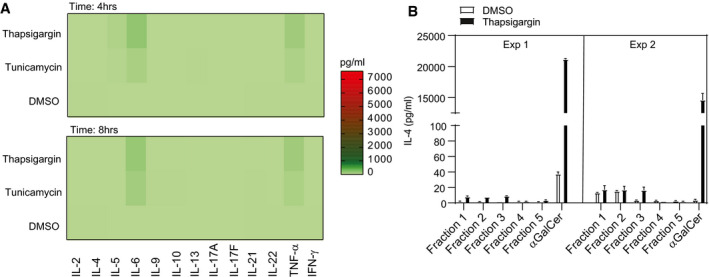
- Heat map (A) represents cytokine secretion by primary murine BMDMs treated with either thapsigargin, tunicamycin or DMSO. Except for IL‐6 and TNF‐α, BMDMs displayed negligible levels of cytokines. Heat map shows the average result of two pooled biological replicates.
- Neutral (Fractions 1–3) and polar lipids (Fractions 4–5) were isolated from murine bone marrow‐derived macrophages stimulated with thapsigargin or DMSO and fractionated using solid‐phase extraction (SPE) cartridges, co‐cultured with purified NKT2 subsets. Bar graph represents IL‐4 release by NKT2 cells by CD1d protein‐bound neutral and polar lipid fractions isolated from thapsigargin (black bars)‐ or DMSO (white bars)‐treated primary murine macrophages. In total, n = 2 biological replicates were performed (two technical replicates per biological replicate). Graphs show mean ± SEM of each biological replicate.
Source data are available online for this figure.
Figure EV3. CD1d surface expression is not altered in ER‐stressed macrophages.
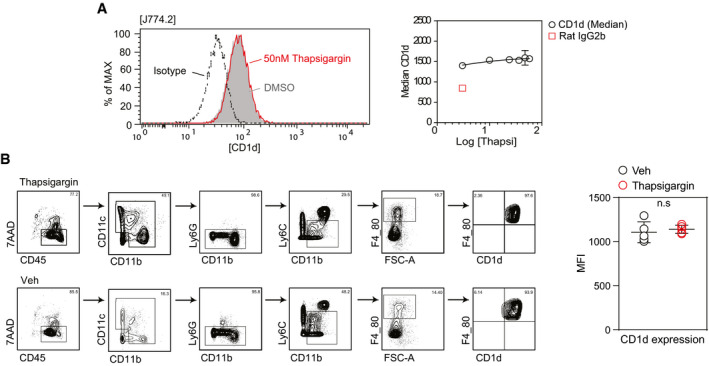
- Cell surface expression of CD1d is not modulated upon addition of thapsigargin in J774.2 macrophages. Histograms depict surface expression of CD1d of J774.2 treated with thapsigargin (red) or DMSO (grey) versus isotype control (black) by flow cytometry (left panel). One representative histogram is shown. Graph (right panel) represents mean ± SEM surface expression of CD1d by J774.2 treated with different concentration of thapsigargin (black circle) versus isotype control (red square box). Experiments were performed twice.
- Contour (left panel) represents the gating strategy for the CD1d expression on hepatic macrophage population gated on live (7AAD−) CD45+CD11b+ Ly6G− Ly6Clo/int F4/80+), and dot plot (right panel) shows that CD1d surface expression (MFI) is not changed in hepatic macrophage populations upon intravenous injection of PLGA particles loaded with thapsigargin. Data represent n = 5 individual mice. Bars and error bar represents mean ± SEM. n.s (non‐significant) (Mann–Whitney U‐test).
Source data are available online for this figure.
IRE1α and PERK control UPR‐induced iNKT cell activation
During ER stress, UPR transmits survival signals through three sensory systems, PERK, IRE1α and ATF6 cascades to restore cellular homeostasis. To assess activation of these UPR pathways in macrophages in response to thapsigargin or tunicamycin, we analysed the expression of the three sensors and a relevant downstream target gene by qPCR analyses. As expected, the three sensors as well as the XBP1/ATF6 downstream target gene Bip and the PERK downstream target gene Chop were strongly induced by thapsigargin and tunicamycin (Fig 3A and B). The UPR signalling pathways and activation of IRE1α‐XBP1 or PERK‐ATF4 or ATF6α have roles in controlling the transcriptional regulation of genes involved in lipid metabolism 34, 35, 36, 37, 38. We therefore sought to delineate the functional impact of interfering with the different UPR sensory systems through genetic and pharmacological approaches.
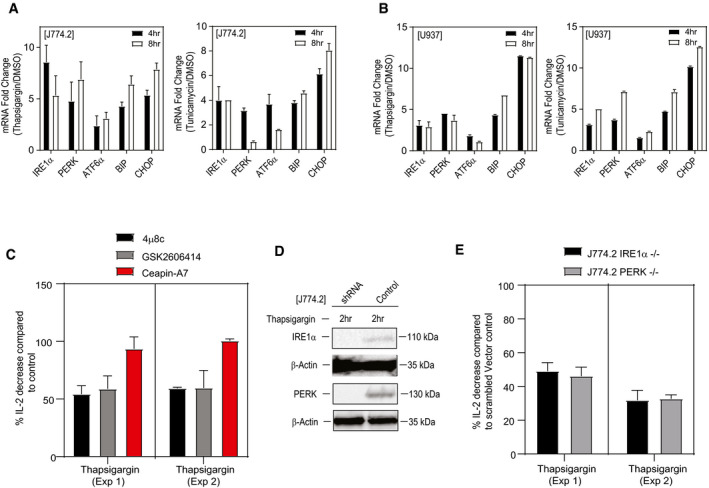
Figure 3. Autoreactivity of iNKT cell requires both the IRE1α and PERK pathway.
-
A, BqPCR analyses showing the expression (fold induction) of UPR signature genes compared to DMSO in murine and human macrophage cell lines, J774.2 (A) and U937 (B) stimulated with either thapsigargin, tunicamycin or DMSO for 4 or 8 h. Graphs show mean ± SEM from n = 2 biological replicates (two technical replicates per biological replicate).
-
CBar graph represents percentage of IL‐2 secretion by NKT cell hybridoma (2C12) co‐cultured with murine macrophage cell line (J774.2) treated with either thapsigargin or tunicamycin in the presence of IRE1α inhibitor (4μ8c) (black bar) or PERK inhibitor (GSK) (grey bar) or ATF6α inhibitor (Ceapin‐A7) (red bar), respectively. Inhibition of IRE1α or PERK resulted in significant reduction in IL‐2 secretion by iNKT hybridoma compared to controls. In total, n = 2 biological replicates were performed (two and four technical replicates per biological replicate, respectively). Graphs show mean ± SEM of each biological replicate.
-
DImmunoblots represent the knockdown efficiency for IRE1α and PERK protein in J774.2 cell line transduced with lentiviral particles carrying gRNA against the IRE1α or PERK gene.
-
EBar graph represents percentage of IL‐2 secretion by NKT cell hybridoma (2C12) co‐cultured with thapsigargin‐treated murine IRE1α or PERK knockdown macrophage cell line (J774.2). Knockdown of IRE1α (black bar) or PERK (grey bar) resulted in significant reduction in IL‐2 secretion by iNKT hybridoma. In total, n = 2 biological replicates were performed (three technical replicates per biological replicate). Graphs show mean ± SEM of each biological replicate.
Source data are available online for this figure.
To understand which UPR signalling arm promotes the highest degree of iNKT cell activation, we performed co‐cultures of NKT cell hybridomas with J774.2 cells treated with inhibitors of IRE1α (4μ8c) 39, PERK (GSK2606414) 40 or AFT6α (Ceapin‐A7) 41. As shown in Fig 3C, chemical inhibition of IRE1α or PERK resulted in a significant reduction in IL‐2 secretion by iNKT hybridoma versus controls (Fig 3C). By contrast, inhibition of the ATF6α had no effect on the activation of iNKT cells. Further, co‐culture of NKT cell hybridoma with control versus IRE1α or PERK shRNA knockdown J774.2 cells (for validation of the knockdown, see Fig 3D) resulted in a decreased cytokine production by iNKT cells (Fig 3E). Overall, whereas initial studies implicated UPR inducers in antigenicity of iNKT cells, this study now directly implicates specific arms of the UPR in APCs leading to iNKT cell activation. Thus, CD1d‐dependent activation of iNKT cells by APCs requires both the IRE1α and PERK branches of the UPR.
UPR‐induced endogenous neutral rather than polar lipids activate iNKT cells
We reasoned that (an) appropriate endogenous lipid antigen(s) should be contained within lipid extracts from J774.2 cells treated with UPR inducers, since cells undergoing ER stress can be efficiently recognized by iNKT cells. To identify lipid species generated during UPR with the highest immunogenic potential, total lipids were isolated from macrophages treated with either thapsigargin or tunicamycin using a modified Folch extraction protocol 42. The resulting organic fractions were further fractionated using solid‐phase extraction (SPE) cartridges 43 and tested for their ability to stimulate NKT hybridomas. Plate‐bound mCD1d protein was pre‐incubated with the lipid fractions and then washed to remove unbound material prior to the addition of the iNKT cells. Pre‐treatment of the mCD1d fusion protein with the lipid fractions 1–3 of the thapsigargin‐ or tunicamycin‐treated J774.2 cells resulted in a markedly augmented IL‐2 release by the NKT hybridomas compared to the mCD1d fusion protein treated with control lipid fractions (Fig 4A). In contrast, the mCD1d fusion protein pre‐incubated with the lipid fractions 4–6 induced only a small increase in IL‐2 production (Fig 4A). Further HPLC‐Q‐TOF‐MS analyses verified that the SPE fractionation resulted in a clear separation of non‐polar lipids DAG and ceramide from the polar sphingolipids including monohexosylceramide and sphingomyelin (Fig EV4). Based on these findings, we concluded that the iNKT cell activation was not caused by the polar lipids, but rather by the neutral lipids. Previous reports have suggested that CD1d protein undergoes acidification during the process of antigen loading in intracellular compartments 44, 45. We therefore sought to examine the effect of acidic pH on endogenous antigen presentation by the mCD1d protein. Murine CD1d fusion protein was incubated with fractionated lipid fractions diluted into citrate/phosphate buffer solutions having pH 7.0 or pH 4.0 at a different ratio of antigen to protein and assayed for recognition by iNKT cells (Fig 4B). Recognition of the neutral lipid fractions was enhanced approximately twofold after antigen pre‐incubation at pH 4.0 compared to a neutral pH 7.0. Maximal IL‐2 release was reproducibly observed for the neutral lipid fractions pre‐incubated at pH 4.0, while IL‐2 production was significantly lower for polar lipid fractions pre‐incubated at pH 4.0 (Fig 4B). To determine if the neutral lipid fractions can activate primary iNKT cells, we performed the CD1d plate assay using fractionated lipids isolated either from murine or human primary macrophages. The experiment was performed by loading mCD1d and huCD1d fusion protein with the neutral lipid fractions isolated from thapsigargin‐treated murine or human primary macrophages, respectively, at pH 4.0 and subsequently co‐cultured with murine or human CD1d tetramer‐sorted and polarized NKT cells from C57Bl/6 mice or healthy human peripheral blood, respectively. The results showed increased IFN‐γ release by NKT1 cells isolated from C57Bl/6 mice or healthy human peripheral blood, respectively, when compared to the CD1d fusion protein treated with DMSO‐treated lipid fractions (Fig 4C and D). In contrast, the murine and human CD1d fusion protein pre‐incubated with the polar lipid fractions induced only a small increase in IFN‐γ production (Fig 4C and D). In addition, loading of murine CD1d fusion protein either with the neutral or polar lipid fractions resulted in less or no significant IL‐4 release by NKT2 cells compared to the CD1d fusion protein treated with DMSO‐treated lipid fractions (Fig EV2B). We therefore concluded that the neutral lipid fractions isolated from thapsigargin‐treated macrophages are able to steer NKT cells towards Th1 responses.
Figure 4. Fractionated neutral lipid from stimulated macrophages activates iNKT cells.
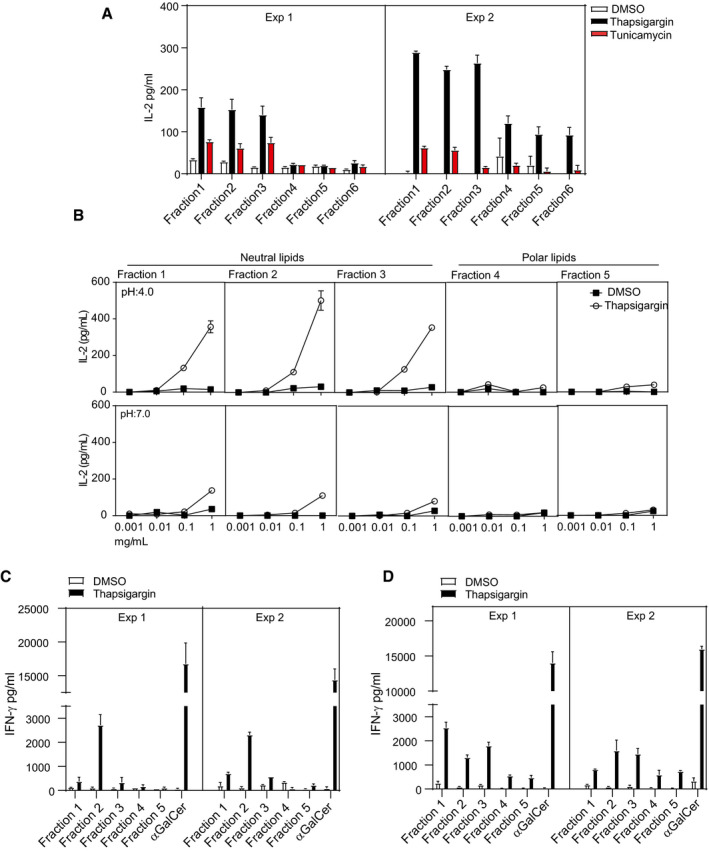
- Neutral (Fractions 1–3) and polar lipids (Fractions 4–6) were isolated from J774.2 macrophages stimulated with thapsigargin, tunicamycin or DMSO. Lipids were fractionated using solid‐phase extraction (SPE) cartridges. Bar graph represents NKT cell activation by CD1d protein‐bound neutral lipids fractions isolated from thapsigargin (black bars)‐ or tunicamycin (red bars)‐treated J774.2 cells. Polar lipid fractions induce very little IL‐2 secretion by 2C12. In total, n = 2 biological replicates were performed (two technical replicates per biological replicate). Graphs show mean ± SEM of each biological replicate.
- Graph represents enhancement of loading with neutral lipid fractions isolated from thapsigargin‐treated J774.2 at acidic pH and reduced mCD1d loading at neutral pH. Graphs show mean ± SEM from n = 2 biological replicates (two technical replicates per biological replicate).
- Neutral (Fractions 1–3) and polar lipids (Fractions 4–5), isolated from murine bone marrow‐derived macrophages stimulated with thapsigargin or DMSO and fractionated using solid‐phase extraction (SPE) cartridges, were co‐cultured with purified NKT1 subsets. Bar graphs represent IFN‐γ release by NKT1 cells induced by CD1d protein‐bound neutral lipid fractions isolated from thapsigargin (black bars)‐ or DMSO (white bars)‐treated primary murine macrophages. Polar lipid fractions induce a minimal amount of IFN‐γ secretion by the NKT1 sublineage. In total, n = 2 biological replicates were performed (two technical replicates per biological replicate). Graphs show mean ± SEM of each biological replicate.
- Neutral (Fractions 1–3) and polar lipids (Fractions 4–5) were isolated from primary macrophages derived from human peripheral blood stimulated with thapsigargin or DMSO and fractionated using solid‐phase extraction (SPE) cartridges. Bar graph represents IFN‐γ release by NKT1 cells by CD1d protein‐bound neutral lipid fractions isolated from thapsigargin (black bars)‐ or DMSO (white bars)‐treated human primary macrophages. Polar lipid fractions induce only minimal amounts of IFN‐γ secretion by the NKT1 sublineage. In total, n = 2 biological replicates were performed (two technical replicates per biological replicate). Graphs show mean ± SEM of each biological replicate.
Source data are available online for this figure.
Figure EV4. HPLC‐Q‐TOF‐MS analysis of thapsigargin‐treated monocyte fractions.
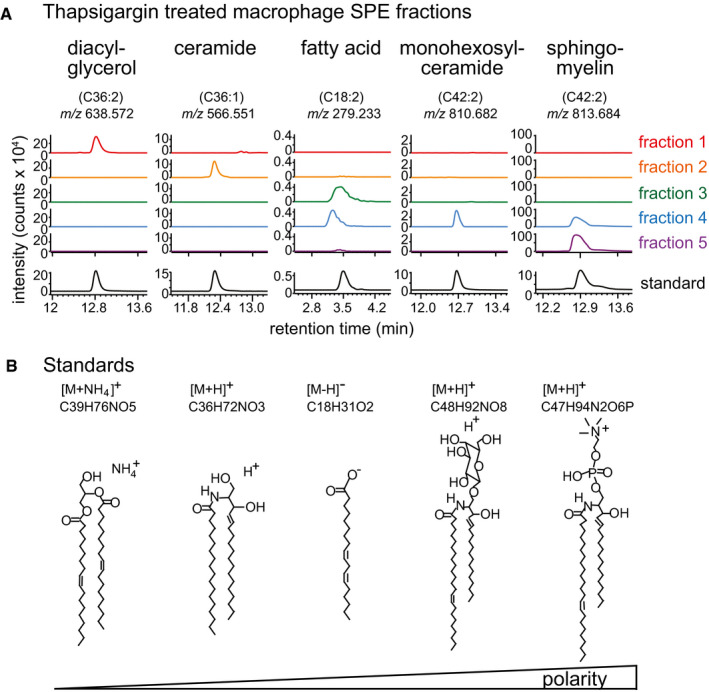
- The solid‐phase extraction (SPE) fractions of thapsigargin‐treated macrophages were normalized to 1 mg/ml. Ten micrograms from each fraction was analysed by the reverse‐phase HPLC‐QTOF‐MS in both positive and negative ion modes. Each of the five SPE fractions is complex mixtures, which were analysed for m/z values matching benchmark molecules (coloured traces), corresponding to known m/z values and measured retention times (black) for common neutral lipids (ceramides, diacylglycerols), fatty acid, glycolipid or polar sphingolipids (sphingomyelin). As expected for this SPE method, Fractions 1 and 2 contained neutral lipids and molecules with higher polarity eluted in later fractions.
- Standards from low polarity to high polarity of lipid classes were chosen based on [Ref. 43].
UPR‐mediated iNKT activation requires endosomal/lysosomal recycling of CD1d molecules
The acquisition of self‐antigens may occur at distinct intracellular sites as CD1d can traffic through sections of the endosomal vesicular system before being re‐expressed at the cell surface. Hence, recycling of CD1d between various endomembrane compartments and the cell surface for antigen presentation depends upon the tyrosine motif present in the cytoplasmic tail of CD1d 46. We therefore assessed the impact of internationalization and recycling of CD1d by comparing responses of UPR in CD1d‐transfected APCs (A20 CD1d) in the presence or absence of this cytoplasmic sorting tyrosine motif [(A20 tail deleted (TD) CD1d]. As shown in Fig 5A, ER‐stressed APCs elicited markedly lower levels of iNKT cell autoreactivity when lacking the cytoplasmic tail of CD1d. Studies have shown that during inflammation and infection, iNKT activation is controlled by rapid degradation of the two catabolic enzymes such as ASAH1 acid ceramidase and GLA/GAA α‐glycosidase, which leads to the accumulation of stimulatory endogenous glycosphingolipids 47, 48. We therefore investigated whether ER‐stressed macrophages downregulate transcripts encoding glycosphingolipids‐degrading enzymes such as acid ceramidase (ASAH1), α‐glucosidase (GAA) and α‐galactosidase (GLA). Our results suggest that the transcripts of these catabolic enzymes such as α‐glucosidase (GAA), α‐galactosidase (GLA) and acid ceramidase (ASAH1) were reduced in thapsigargin‐treated J774.2 cells when compared to DMSO controls (Fig 5B). Similarly, the inhibition of α‐/β‐glucosidases in J774.2 cells with castanospermine or deoxygalactonojirimycin resulted in enhanced IL‐2 secretion by 2C12 cells (Fig 5C). All together, these data suggest that degradation of these catabolic enzymes during UPR in macrophages leads to potential accumulation of stimulatory glycosphingolipids. To further evaluate the nature of the observed iNKT cell autoreactivity upon UPR stress linked to synthesis of α‐linked glycosylceramides 49, we co‐cultured NKT cell hybridoma 2C12 with J774.2 stimulated with either thapsigargin or α‐GalCer in the presence of L363 blocking monoclonal antibody or an isotype control Ab (Fig 5D). In contrast to the clear inhibition of IL‐2 secretion by 2C12 hybridoma when J774.2 macrophage cells incubated with α‐GalCer were treated with the L363 antibody, no such effect was observed in thapsigargin‐treated cells. The results suggest that ER stress‐mediated NKT cell activation was due to generation of novel stimulatory endogenous iNKT ligands but not due to the accumulation of α‐linked glycosylceramides.
Figure 5. UPR‐induced neutral lipids require CD1d endosomal/lysosomal recycling.
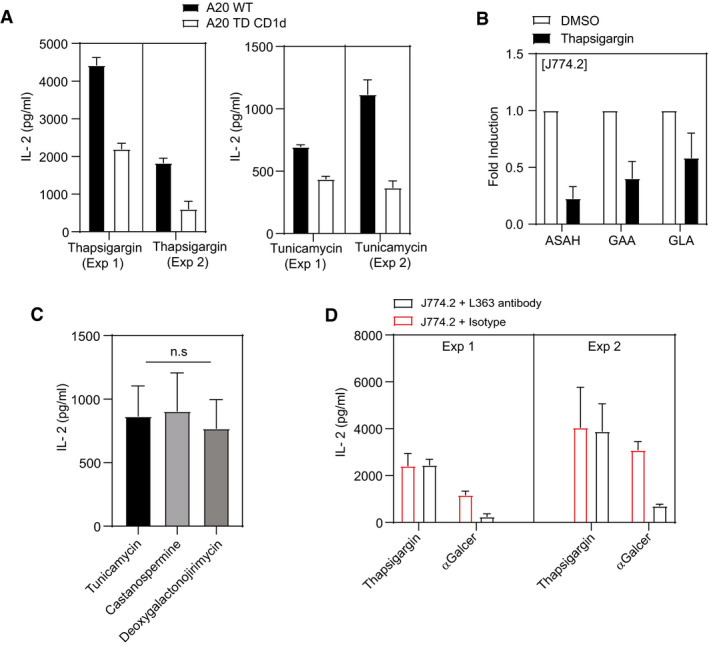
- Bar graph represents that recycling of CD1d between various endomembrane compartments and the cell surface is required for optimal IL‐2 secretion by NKT cells. In total, n = 2 biological replicates were performed (three technical replicates per biological replicate). Graphs show mean ± SEM of each biological replicate.
- qPCR analyses showing the expression (fold induction) of genes encoding for glycosphingolipid degrading enzymes compared to DMSO in murine macrophage cell lines J774.2 stimulated with either thapsigargin or DMSO for 8 h. In total, n = 2 biological replicates were performed (two technical replicates per biological replicate). Graphs show mean ± SEM of each biological replicate.
- Glycolipid processing in intracellular compartments is not required prior to presentation to NKT cells. IL‐2 secretion in UPR‐induced NKT cell activation in the presence or absence of castanospermine or deoxygalactonojirimycin. Graphs show mean ± SEM from n = 3 biological replicates. n.s (non‐significant) (one‐way ANOVA) biological replicates.
- L363 antibody does not impact UPR‐induced NKT cell activation. 2C12 NKT cells were co‐cultured with ER‐stressed macrophages J774.2 cells and treated with L363 antibody or isotype control. α‐GalCer‐induced responses served as positive control. In total, n = 2 biological replicates were performed (four technical replicates per biological replicate). Graphs show mean ± SEM of each biological replicate.
Source data are available online for this figure.
Discussion
For several years, it has been known that CD1d‐expressing APCs can induce proliferation, cytokine secretion and/or cytolysis of CD1d‐restricted T cells in the absence of foreign antigen. This autoreactivity was described in early studies for CD1a‐, CD1c‐restricted T cells and later for group 2 or CD1d‐restricted T cells 50, 51. However, while several microbially derived glycolipid antigens have been discovered, highlighting the role of iNKT cells in host defence, the mechanisms leading to generation of endogenous lipid antigens are less clear. In this study, we reveal that ER stress inducers as well as defined mediators of the UPR in APCs play a pivotal role in CD1d‐mediated iNKT cell responses by generating endogenous neutral lipid antigens in APCs able to trigger these cells. This implies two arms of the UPR sensors, IRE1α‐XBP1 and PERK‐ATF4, which promote induction or accumulation of endogenous neutral lipids, in the activation of iNKT cells in a CD1d‐dependent manner. This pathway was found to be conserved in both mice and humans suggesting an evolutionary important mechanism of iNKT cell stimulation under sterile ER stress conditions.
The UPR plays an important immune role through a direct crosstalk between ER stress‐induced signalling pathways and immune responses. The IRE1α/XBP1s axis regulates immune cell survival and antigen‐presenting abilities 52, 53, 54. IRE1α regulates expression of several members of the MHC‐I antigen presentation pathway 55 and also plays an important role in cross‐presentation which may predominantly connect to the phagosomal pathway 54. Functional studies with cDC1 and cDC2 cells show that antigen presentation via MHC‐II is unaffected in the absence of XBP1 54. However, in ovarian cancer, XBP1 ablation in DCs results in infiltration of CD4+ T cells. This suggests a negative role of XBP1 in CD4+ T‐cell activation under conditions of sustained ER stress 56. The results presented in this study indicate that the ER‐stressed APCs also play a role in activation of iNKT cells via CD1d, a prototypic non‐classical MHC molecule. During ER stress, the components of UPR are involved in the regulation of membrane lipid biosynthesis and ER biogenesis to maintain cellular homeostasis 4, 38. We now show that ER‐stressed APCs also induce generation of endogenous lipid antigens presented by CD1d molecules to iNKT cells. Thapsigargin‐mediated induction of UPR in macrophages was associated with highest level of activation of CD1d‐restricted iNKT cells when compared to tunicamycin, R848 or LPS, which may be linked to distinct mechanisms of action to perturb the ER function 57, 58.
Activated stimulated iNKT cells are well known to produce large quantities of type 1, type 2 as well as type 17 cytokines. In vivo, they may impact diverse immune reactions in response to various pathogens and tumours 23, 25, 59, 60, 61. In vitro UPR induction in APCs activates both murine and human iNKT cells which in turn secrete predominantly IL‐4, IFN‐γ, along with IL‐10 in mice. In vivo administration of thapsigargin‐loaded nanoparticles also led to iNKT cell activation as measured by intracellular cytokine production (Fig 1E). Previous studies have demonstrated that intravenous (i.v.) administered nanoparticles are sequestered largely by the Kupffer cells in liver compared to other periphery organs 62. Therefore, we injected the most efficient biodegradable PLGA nanoparticles of 1 μm size loaded with thapsigargin to understand the in vivo interaction between Kupffer and iNKT cells. In line with the in vitro results, administration of thapsigargin‐loaded nanoparticles in mice significantly increased IL‐4 and IFN‐γ in hepatic iNKT cells when compared to vehicle controls. Intriguingly, however, ER‐stressed APCs failed to induce IL‐17 which is of particular interest as it suggests that this pathway may not play a major role in the inflammatory effector roles ascribed to iNKT17 cells but rather impacts iNKT1 function.
Previously, we showed that the UPR sensor IRE1α plays a key role in regulating cytokine production in iNKT cells by affecting cytokine mRNA stability. This was restricted to certain iNKT cell subsets: iNKT1 and iNKT17, but not iNKT2 cells, which were impacted by IRE1α 63. This illustrates that UPR has pleiotropic and cell subset‐dependent roles on the immune system. Because the UPR and, particularly, IRE1α control cytokine production of iNKT cells 63, we wanted to rule out that a similar mechanism would also be in place in ER‐stressed APCs. Hence, in addition to the strong T‐cell receptor (TCR) signal induced by high‐affinity lipid antigens which can activate iNKT cells directly in CD1d‐dependent manner, iNKT cells may also be activated by a weak TCR signal by low‐affinity lipid antigens in combination with cytokines such as IL‐12 secreted by the activated APC 33, 64, 65. However, our data are most consistent with a model that ER‐stressed APCs induce iNKT cell activation through a CD1d‐dependent but cytokine‐independent mechanism. First, there was no direct effect of thapsigargin or tunicamycin in binding CD1d and activating iNKT cells. Second, the UPR‐mediated responses were clearly CD1d‐dependent as demonstrated by the CD1d blocking assays and the lack of response in CD1d KO APC. Furthermore, ER‐stressed APCs in the absence of iNKT cells induced very little, if any, secretion of cytokines. Therefore, it is more likely that exposure to ER stress inducers in APCs results in generation of CD1d‐restricted lipid antigens which trigger iNKT cell responses. This mirrors the effect of the activation of PERK as well as three other kinases of different stress pathways on the immunopeptidome. By selective translation of mRNAs harbouring upstream open reading frames in the 5′ untranslated region, tracer peptides were found to be processed and presented in MHC class I molecules to antigen‐specific T cells 66.
In addition to phospholipids, ER membrane also contains sphingolipids such as ceramide, sphingosine‐1‐phosphate (S1P) and sphingosine. After identifying α‐GalCer, a powerful marine sponge sphingolipid antigen recognized in antigen screening 67, it was found that iNKT cells can be activated by glycosphingolipids (GSL). Endogenous sphingolipids have since been examined as potential lipid antigens for iNKT cells. During ER stress, alteration in lipid metabolism is needed in order to maintain homeostasis to prevent ER stress 29. Here, IRE1α, ATF6 and PERK appear to function collectively and failure to do so results in the suppression of metabolic genes involved in lipid metabolism. Of note, the liver‐specific deletion of X‐box binding protein 1 (XBP1), a transcription factor activated by IRE1, resulted in a profound hypocholesterolaemia and hypotriglyceridaemia in adult mice, suggesting that de novo synthesis of lipids in the liver is regulated in part by XBP14. Studies have shown that PERK‐mediated eIF‐2α phosphorylation results in activation of C/EBP and peroxisome proliferator‐activated receptor‐γ (PPARγ), which contribute to the induction of lipogenic genes. Also, the PERK‐ATF4 pathway plays an important role in promoting lipogenesis both in the liver and other tissues such as adipose tissue 10, 35, 37. However, the functional significance of the interactions between altered lipid metabolism regulated by UPR and other immune cells has not been explored in detail. Our data suggest that activation of IRE1α‐XBP1 and PERK‐ATF4 pathways to restore ER homeostasis within APCs and the resulting lipid metabolic phenotype promotes an ER stress‐adapted CD1d‐restricted T‐cell response. These findings suggest that failure of the UPR to restore ER stress perturbs CD1d‐restricted NKT cell biology which may contribute to chronic inflammatory diseases. In future work, this hypothesis could be explored in relevant disease models.
The search for novel intracellular lipid antigens capable of activating CD1d‐restricted T cells has been the subject of intense research interest. Although a few candidate endogenous lipid antigens have been identified, most activate CD1d‐restricted T cells only very weakly 61. A major difficulty in identifying novel lipid antigens with high immunogenic potential is that such candidate lipids are expressed at very low concentrations in resting, or even stimulated antigen‐presenting cells. Our findings are novel in the sense that we have identified a cellular state, namely ER stress, during which immunogenic neutral lipids capable of activating CD1d‐restricted T cells are generated. Furthermore, in vitro CD1d plate‐bound assays suggest that neutral lipids generated during UPR require acidic pH for efficient loading to CD1d. Consistent with earlier studies 46, 68, it is likely that under lower pH conditions, the binding groove of mCD1d protein might be more exposed to solvent, resulting in better binding of antigens compared to neutral pH. Moreover, our A20CD1dTD assays suggest that CD1d protein translocation between intracellular compartments and the cellular membrane is required for optimal iNKT activation during UPR. Previous studies have shown that degradation of the catabolic enzymes namely ASAH1 or GAA or GLA controls the activation of iNKT cells by accumulating α‐GalCers 49. Surprisingly, our in vitro assays using the L363 antibody suggest other potential endogenous glycosphingolipids are accumulated during UPR in macrophages which resulted in activation of iNKT cells. Consistent with our findings, a recent study 69 shows that ER‐stressed macrophages require CD1d recycling in the endosome and lysosome for optimal iNKT activation. Even though our results suggest an upregulation of endogenous lipids bound to CD1d molecules during UPR that activate iNKT cells, whether ER stress implies local biosynthesis of new agonist lipids, or it rather plays an indirect role in the upregulation of molecular pathways related to the endo/lysosomal handling of lipids to CD1d warrants further investigation. Studies have shown that in addition to iGB3 or endogenous α‐GalCer other glycosphingolipids may also involve in selecting iNKT cells in thymus during its development 49, 70. However, identification of those endogenous ligands that promote positive selection of iNKT cells remains unclear. Since ER stress in thymocytes facilitates their survival 71, there is a more likely possibility that UPR‐mediated accumulation of endogenous glycosphingolipids may be involved in positive selection of iNKT cells in thymus. Therefore, identification of endogenous ligands generated during UPR may facilitate better understanding of thymic iNKT cell development. Also, given the myriad of inflammatory diseases linked to the UPR pathway, our data suggest that the UPR‐mediated induction of endogenous glycolipids provides a long‐sought‐after mechanism for how UPR may impact chronic inflammatory diseases such as inflammatory bowel diseases or hepatitis.
Materials and Methods
Mice
C57BL/6 and CD1d1/2−/− (B6 background) mice were purchased from Jackson Laboratory (USA). All mice were maintained under specific pathogen‐free conditions in the Animal Care Facility of Ghent University. Experiments were performed with age‐ and sex‐matched mice at 10–12 weeks of age. All experiments were approved by the Institutional Animal Care and Ethics Committee.
Flow cytometry and antibodies
iNKT cells were sorted on FACSAria III (BD Biosciences). Murine antibodies used were CD19‐PerCP‐Cy5.5 (eBio1D3), βTCR‐FITC (H57‐597), CD4‐APC‐eFluor780 (RM4‐5), CD8α‐e450 (53‐6.7), IL4‐FITC (11B11), IFNγ‐FITC (XMG1.2), MHC‐II‐FITC (114.15.2), F4/80‐PE (BM8), CD170‐APC (IRNM44N), CD11C‐PE‐Cy7 (N4/8) and Ly6G‐PE Texas red (all from Thermo Fisher scientific); CD11b‐APC‐Cy7 (M1/70), Ly6C‐BV510 (HK1.4), CD45‐BV605 (30‐F11), and CD1d‐e450 (1B1) (all from BD). Human antibodies used were TCRVβ11‐FITC (C21) and 7‐AAD‐PerCP‐Cy5.5 (all from BD). iNKT cells were stained at 4°C for 30 min using α‐GalCer‐loaded CD1d tetramers provided by NIH Tetramer Core Facility, USA, followed by antibody staining. iNKT cells are defined as iNKT (TCRβ+CD1d‐αGC+); for Annexin V, staining set (Thermo Scientific) was used according to the manufacturer's instructions. For intracellular proteins, the FoxP3/transcription factor staining buffer set (eBioscience) was used according to the manufacturers’ recommendations.
Cell suspension preparations
Single‐cell suspensions from murine spleen were resuspended in PBS and layered over Ficoll gradients. Human peripheral blood mononuclear cells (PBMCs) were isolated by layering the whole blood over Ficoll–Paque. iNKT cells were enriched by using mouse CD5 (Ly‐1)‐microbeads (Miltenyi Biotec) or human untouched T cells‐Dynabeads (Invitrogen), respectively, and resuspended in PBS containing 1 mM EDTA and 0.5% BSA.
Cell culture
J774.2 and U937 cell lines from ATCC were cultured in DMEM or RPMI‐1640 with 10% FBS, 2 mM glutamine, and 100 units/ml penicillin and streptomycin (all from Gibco) (complete medium). 2C12 hybridoma was obtained from Mitch Kronenberg (La Jolla Institute for Allergy and Immunology) and cultured in complete DMEM supplemented with 5.5 × 102 μM μ‐ME, (Sigma‐Aldrich). Bone marrow‐derived macrophages (BMDMs) were isolated from C57BL/6 or CD1d−/− mice by flushing the femur and tibia with RPMI‐1640 medium. The bone marrow cells were resuspended in complete RPMI‐1640 medium supplemented with 40 ng/ml murine M‐CSF. Cells were cultured in petri dishes for 8 days at 37°C and 5% CO2 with medium change every 3 days. Human peripheral‐derived macrophages were isolated by layering whole blood over Ficoll–Paque. Isolated peripheral mononuclear cells were resuspended in complete RPMI‐1640 medium supplemented with 40 ng/ml human M‐CSF. Cells were cultured in petri dishes for 6 days at 37°C and 5% CO2 with medium change every 2 days. For all co‐culture experimental setups, J774.2 or U937 or primary macrophages cells were plated at a density of 1.0 × 105 cells/well in flat‐bottom 96‐well plate and stimulated with thapsigargin (50 nM) or tunicamycin (3 μg/ml) or DMSO (all from Sigma‐Aldrich) for 4 and 8 h in complete DMEM or RPMI‐1640 medium. For blocking experiments, the J774.2 cells were incubated with thapsigargin (50 nM) in the presence or absence of 10 μg/ml murine anti‐CD1d (1B1) or α‐GalCer‐CD1d complex (L363) antibody for 8 h. For inhibitors studies, the J774.2 cells were incubated with thapsigargin in the presence of castanospermine (50 nM) or deoxynojirimycin (50 nM). After incubation, cells were washed twice with PBS and a total of 0.5 × 105 iNKT cells or 2C12/well resuspended in complete RPMI‐1640 or DMEM supplemented with 5.5 × 102 μM μ‐ME were added. Supernatants were collected after 16 h of co‐culture and used for cytokine measurements by ELISA or by using fluorescence‐encoded beads (LEGENDplex).
In vitro expansion of iNKT cells
Sorted murine splenic bulk iNKT cells were resuspended in complete RPMI‐1640 medium containing 5.5 × 102 μM μ‐ME, murine IL‐2 (10 ng/ml), IL‐12 (1 ng/ml) and soluble α‐CD28 (5 μg/ml; all from Thermo Fisher Scientific) and stimulated with plate‐bound α‐CD3ε (3 μg/ml) for 2 days. Cells were then rested for 2 days and resuspended in complete RPMI‐1640 medium containing 5.5 × 102 μM μ‐ME and murine IL‐7 (2 U/ml; Thermo Fisher Scientific) for 4 days. iNKT cells were then re‐stimulated with plate‐bound α‐CD3ε (3 μg/ml) for a further 3 days and subsequently expanded for an additional 10 days, as described before 72. Human iNKT cells were expanded by means of 6B11Ab (Thermo Fisher) and IL‐2 (Roche) as described before 73.
Administration of thapsigargin‐loaded PLGA nanoparticles
5 mg of PLGA (poly lactic‐co‐glycolic acid), 2.5 μl DOPE‐rhodamine (10 mg/ml in ethanol) and 500 μg thapsigargin were dissolved in 200 μl dichloromethane and emulsified in a 0.1% aqueous polyvinyl alcohol solution by ultrasonication. The resulting emulsion was diluted with 20 ml of a 0.05% aqueous polyvinyl alcohol solution and subsequently stirred 2 h at 40°C. The resulting solution was filtered using a 40‐μm filter to remove larger particulates. The sizes of the particles (1 μm) were confirmed by optical microscopy. C57BL/6 mice were tail vein injected with 200 μl of PLGA nanoparticles suspension or 200 μl vehicle using 29‐gauge insulin needle, and hepatic iNKT cells were analysed 12 h post‐injection by flow cytometry.
Lentiviral production and lentiviral infection
Lentivirus production was performed by co‐transfecting murine Ern1 (K4522205) or Eif2ak3 (K4376505) or scrambled (K010) pLenti‐U6‐sgRNA‐SFFV‐Cas9‐2A‐Puro plasmid with the packaging plasmid pLenti‐P2A/P2B (LV003) in HEK293T cells using Lipofectamine 2000 (Thermo Fisher Scientific). Lentiviral particles were collected by following the manufacturer's protocol for Lentiviral Expression Systems (Applied Biological Materials Inc.). For lentiviral infection, viral particles were mixed with 4 μg/ml polybrene and added to J774.2 cell lines. After 48 h of infection, stable cells were selected using puromycin at 2 μg/ml.
Quantitative real‐time RT–PCR
RNA was extracted from J774.2 or U937 stimulated with thapsigargin (50 nM) or tunicamycin (3 μg/ml) or DMSO (all from Sigma‐Aldrich) for 4 and 8 h using the miRNeasy Mini Kit (QIAGEN, Valencia, CA). cDNA was produced and amplified using the QuantiTect Whole Transcriptome Kit (QIAGEN, Valencia, CA). qPCR was performed with the FastStart SYBR Green Master Mix (Roche, Basel, Switzerland) and detected with a LightCycler 480 System (Roche, Basel, Switzerland). Samples were normalized against at least 3 of the following housekeeping genes: Rpl13a, Atp5b, Eif4b, Cyc1, B2m, Ubc, Actb, Gapdh, Sdha or Tubb4a using qBasePlus software (Biogazelle NV, Belgium). Primer sequences are listed in Table EV1.
Western blot
Cells were stimulated with thapsigargin (50 nM) (all from Sigma‐Aldrich) for 2 h. Cells were spun for 10 min at 400 g at 4°C. After centrifugation, the medium was removed and washed with ice‐cold PBS. Cells were lysed for 15 min on ice in E1A buffer (1% NP40, 20 mM HEPES, pH 7.9, 250 mM NaCl, 1 mM EDTA and protease/phosphatase inhibitors). Insoluble material was discarded by cold centrifugation at 13,500 g, and a fixed amount of lysate was mixed with sample buffer before separation by 10% SDS–PAGE. Gels were transferred onto 0.45‐μM nitrocellulose membranes (Bio‐Rad). After transferring to a membrane, proteins were incubated overnight in the presence of anti‐IRE1α or anti‐PERK antibody (1:1,000; Cell Signaling) at 4°C followed by incubation with horseradish peroxidase‐conjugated anti‐rabbit IgG antibody (1:10,000; Cell Signaling) for 1 h at room temperature. Protein bands were visualized by enhanced chemiluminescence kit (Thermo Scientific). Membranes were stripped for 15 min using stripping buffer (Cell Signaling) and reprobed with anti‐β‐Actin antibody (1:1,000; Santa Cruz Biotechnology).
Fractionation of total lipids
50 × 106 of J774.2 or U937 or primary murine or human macrophage cells were treated for 8 h with 50 nM thapsigargin or DMSO. After incubation, the cells were washed with cold PBS after which they were pelleted and snap‐frozen in dry ice. Pellets were resuspended in 2 ml PBS followed by the addition of 4 ml of chloroform and methanol (1:1). After thorough mixing, the cells were centrifuged at 3,750 g for 10 min to remove insoluble/precipitated protein. 1 ml of 50 mM citric acid, 2 ml of water and 1 ml of chloroform were added and centrifuged at 3,750 g for 20 min with brake. The lower phase, containing lipids of interest, was transferred to a new tube, dried down under a stream of nitrogen and resuspended in the desired amount of chloroform. Amino‐propyl columns (SUPELCO Superclean LC‐NH2‐100 mg, 1 ml) were pre‐equilibrated with 2–3 ml n‐hexane. Lipids in chloroform applied to the column. The samples were allowed to adsorb to the matrix by percolation through the cartridge by gravity. Lipid fractions were then eluted sequentially as follows: Fraction 1 was eluted with 1.4 ml of ethyl acetate–hexane 15:85; Fraction 2 was eluted with 1.6 ml of chloroform–methanol 23:1 (v/v); Fraction 3 with 1.8 ml of diisopropyl ether–acetic acid 98:5 (v/v); Fraction 4 with 2 ml of acetone–methanol 9:1.35 (v/v); and Fraction 5 with 2 ml chloroform–methanol 2:1 (v/v). The fractions were then dried by passing a stream of nitrogen gas and used for further analyses.
CD1d plate‐bound assay
Fractionated lipids isolated either from thapsigargin‐ or DMSO‐treated J774.2 cells were dried by passing a stream of nitrogen gas and then resuspended in 50 mM pH 4 or pH 7 citrate buffer supplemented with 0.25% CHAPS. The lipid was then mixed with biotinylated CD1d (provided by the NIH Tetramer Facility) that was also diluted into citrate‐CHAPS. Lipids were then bound to CD1d for overnight, after which the pH was adjusted to 7.4 with 1 M Tris pH 9. For binding CD1d onto 96‐well plates, 5 μg of CD1d protein was added per well for 1 h at room temperature. Plates were then extensively washed with sterile 1× PBS before the addition of 50 × 104 N38‐2C12 hybridoma cells or primary murine or human expanded NKT1 per well in complete RPMI medium. After 16‐h incubation at 37°C and 5% CO2, supernatants were then collected and analysed for IL‐2 by ELISA or by using fluorescence‐encoded beads (LEGENDplex).
Mass spectrometry
The solid‐phase extraction purified fractions of thapsigargin‐treated macrophages were weighed and normalized to 1 mg/ml in the mobile phase solvent A (see below). The samples (10 μl injection) were run on an Agilent Poroshell 120 A, EC‐C18, 3 × 50 mm, 1.9 μm reverse‐phase column equipped with an Agilent EC‐C18, 3 × 5 mm, 2.7 μm guard column and analysed by using Agilent 6530 Accurate‐Mass Q‐TOF/1260 series HPLC instrument. The mobile phases were (A) 2 mM ammonium formate in methanol/water (95/5; V/V) and (B) 3 mM ammonium formate in 1‐propanol/cyclohexane/water (90/10/0.1; v/v/v). In a 20‐min run, the solvent gradients are as follows: 0–4 min, 100% A; 4–10 min, from 100% A to 100% B; 10–15 min, 100% B; 15–16 from 100% B to 100% A; 16–20 min, 100% A. All the lipid standards diacylglycerol (DAG, D0138) and linoleic acid (18:2 FA, L1376) were purchased from Sigma‐Aldrich. C18 ceramide (d18:1/18:0) (#860518), C24:1 galactosylceramide (d18:1/24:1) (#860546) and C24:1 sphingomyelin (SM, #860593) were purchased from Avanti Polar Lipids.
Statistical analysis
Statistical testing was performed using GraphPad Prism software.
Author contributions
SG, MD, DBM and DE designed the experiments. SG performed experiments and wrote the manuscript. EV, MM, KV, DG, EC and T‐YC performed experiments. BGDG and FG synthesized nanoparticles. SJ, MD and DE supervised the work. DBM, KV, SJ, MD and DE wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Table EV1
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Acknowledgements
We thank Prof. Dale Godfrey (University of Melbourne) and Prof. Daniel Pellicci (University of Melbourne) for their valuable feedback. We thank the NIH Tetramer Core Facility for CD1d protein and CD1d tetramer production. S.G holds a post‐doctoral fellowship from the Fund for Scientific Research–Flanders (FWO). Research was supported by grants from the Group‐ID Multidisciplinary Platform (MRP) of Ghent University (to D.E and M.B.D.), Fund for Scientific Research–Flanders and an Excellence of Science (EOS) Grant (to D.E.) and Stichting tegen Kanker, Belgium (to D.E.) and the NIH (R01 AR048632 (to D.B.M.)).
EMBO Reports (2020) 21: e48927
Data availability
No primary datasets have been generated and deposited.
References
- 1. Hetz C (2012) The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol 13: 89–102 [DOI] [PubMed] [Google Scholar]
- 2. Walter P, Ron D (2011) Unfolded protein response: from stress pathway to homeostatic regulation. Science 334: 1081–1086 [DOI] [PubMed] [Google Scholar]
- 3. Ron D, Walter P (2007) Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev cell Biol 8: 519–529 [DOI] [PubMed] [Google Scholar]
- 4. Lee A, Chu GC, Neal N, Glimcher LH (2005) XBP‐1 is required for biogenesis of cellular secretory machinery of exocrine glands. EMBO J 24: 4368–4380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Calfon M, Zeng H, Urano F, Till JH, Hubbard SR, Harding HP, Clark SG, Ron D (2002) IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP‐1 mRNA. Nature 415: 92–96 [DOI] [PubMed] [Google Scholar]
- 6. Feely RA, Reay D, Hewitt N, Grace J, Smith K, Wickett ME (2008) Regulation of hepatic lipogenesis by the transcription factor XBP1. Science 320: 1492–1497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yokoyama C, Wang X, Briggs MR, Admon A, Wu J, Hua X, Goldatein JL, Brown MS (1993) SREBP‐1, a basic‐helix‐loop‐helix‐leucine zipper protein that controls transcription of the low density lipoprotein receptor gene. Cell 75: 187–197 [PubMed] [Google Scholar]
- 8. Yang F, Vought BW, Satterlee JS, Walker AK, Sun ZJ, Watts JL, Debeaumont R, Saito RM, Hyberts SG, Yang S et al (2006) An ARC/Mediator subunit required for SREBP control of cholesterol and lipid homeostasis. Nature 442: 700–704 [DOI] [PubMed] [Google Scholar]
- 9. Im S, Yousef L, Blaschitz C, Liu JZ, Edwards RA, Young SG, Raffatellu M, Osborne TF (2011) Article linking lipid metabolism to the innate immune response in macrophages through sterol regulatory element binding protein‐1a. Cell Metab 13: 540–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shimano H, Sato R (2017) SREBP‐regulated lipid metabolism : convergent physiology — divergent pathophysiology. Nat Rev Endocrinol 13: 710–730 [DOI] [PubMed] [Google Scholar]
- 11. Lee JN (2004) Proteolytic activation of sterol regulatory element‐binding protein induced by cellular stress through depletion of insig‐1 *. J Biol Chem 279: 45257–45265 [DOI] [PubMed] [Google Scholar]
- 12. Lauressergues E, Bert E, Duriez P, Hum D, Majd Z, Staels B, Cussac D (2012) Neuropharmacology does endoplasmic reticulum stress participate in APD‐induced hepatic metabolic dysregulation ? Neuropharmacology 62: 784–796 [DOI] [PubMed] [Google Scholar]
- 13. Kammoun HL, Ferré P, Foufelle F, Kammoun HL, Chabanon H, Hainault I, Luquet S (2009) GRP78 expression inhibits insulin and ER stress – induced SREBP‐1c activation and reduces hepatic steatosis in mice. J Clin Invest 119: 1201–1215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bensinger SJ, Tontonoz P (2008) Integration of metabolism and inflammation by lipid‐activated nuclear receptors. Nature 454: 470–477 [DOI] [PubMed] [Google Scholar]
- 15. Janssens S, Pulendran B, Lambrecht BN (2014) Emerging functions of the unfolded protein response in immunity. Nat Immunol 15: 910–919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Grootjans J, Kaser A, Kaufman RJ, Blumberg RS (2016) The unfolded protein response in immunity and inflammation. Nat Rev Immunol 16: 469–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Silk JD, Salio M, Brown J, Jones EY, Cerundolo V (2008) Structural and functional aspects of lipid binding by CD1 molecules. Annu Rev Cell Dev Biol 24: 369–395 [DOI] [PubMed] [Google Scholar]
- 18. Sugita M, Porcelli SA, Brenner MB (1997) Assembly and retention of CD1b heavy chains in the endoplasmic reticulum. J Immunol 169: 2358–2365 [PubMed] [Google Scholar]
- 19. Kang S, Cresswell P (2002) Calnexin, calreticulin, and ERp57 cooperate in disulfide bond formation in Human CD1d heavy chain *. J Biol Chem 277: 44838–44844 [DOI] [PubMed] [Google Scholar]
- 20. De Silva AD, Park J, Matsuki N, Stanic AK, Brutkiewicz RR, Edward M, Joyce S, De Silva AD, Park J, Matsuki N et al (2002) Lipid protein interactions: the assembly of CD1d1 with cellular phospholipids occurs in the endoplasmic reticulum. J Immunol 168: 723–733 [DOI] [PubMed] [Google Scholar]
- 21. Park J, Kang S, De Silva AD, Stanic AK, Casorati G, Hachey DL, Cresswell P, Joyce S (2004) Lipid – protein interactions : biosynthetic assembly of CD1 with lipids in the endoplasmic reticulum is evolutionarily conserved. Proc Natl Acad Sci USA 101: 1022–1026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dougan SK, Salas A, Rava P, Agyemang A, Kaser A, Morrison J, Khurana A, Kronenberg M, Johnson C, Exley M et al (2005) Microsomal triglyceride transfer protein lipidation and control of CD1d on antigen‐presenting cells. J Exp Med 202: 529–539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. De Libero G, Mori L (2005) Recognition of lipid antigens by T cells. Nat Rev Immunol 5: 485–496 [DOI] [PubMed] [Google Scholar]
- 24. Joyce S, Woods AS, Yewdell JW, Bennink JR, De Silva D, Boesteanu A, Balk SP, Cotter RJ, Randy R, Joyce S et al (2016) Natural ligand of mouse CD1d1: cellular glycosylphosphatidylinositol. Science 279: 1541–1544 [DOI] [PubMed] [Google Scholar]
- 25. Bendelac A, Savage PB, Teyton L (2007) The biology of NKT cells. Annu Rev Immunol 25: 297–336 [DOI] [PubMed] [Google Scholar]
- 26. Van Kaer L (2004) Natural killer T cells as targets for immunotherapy of autoimmune diseases. Immunol Cell Biol 82: 315–322 [DOI] [PubMed] [Google Scholar]
- 27. Morita M, Motoki K, Akimoto K, Natori T, Sakai T, Sawa E, Yamaji K, Koezuka Y, Kobayashi E, Fukushima H (1995) Structure‐activity relationship of a‐galactosylceramides against B16‐bearing mice. J Med Chem 38: 2176–2187 [DOI] [PubMed] [Google Scholar]
- 28. Lee AH, Glimcher LH (2009) Intersection of the unfolded protein response and hepatic lipid metabolism. Cell Moelcular Life Sci 66: 2835–2850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Basseri S, Austin RC (2012) Endoplasmic reticulum stress and lipid metabolism: mechanisms and therapeutic potential. Biochem Res Int 2012: 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ozcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi NN, Ozdelen E, Tuncman G, Görgün C, Glimcher LHHG, Hotamisliqil GS (2004) Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 306: 457–461 [DOI] [PubMed] [Google Scholar]
- 31. Kawano T, Cui J, Koezuka Y, Toura I, Kaneko Y, Motoki K, Ueno H, Nakagawa R, Sato H, Kondo E et al (1997) CD1d‐restricted and TCR‐mediated activation of valpha14 NKT cells by glycosylceramides. Science 278: 1626–1629 [DOI] [PubMed] [Google Scholar]
- 32. Eberl G, MacDonald HR (1998) Rapid death and regeneration of NKT cells in anti‐CD3e ‐ or IL‐12‐treated mice: a major role for bone marrow in NKT cell Homeostasis. Immunity 9: 345–353 [DOI] [PubMed] [Google Scholar]
- 33. Brigl M, Raju V, Leadbetter EA, Barton N, Cohen NR, Hsu F, Besra GS, Brenner MB (2011) Innate and cytokine‐driven signals rather than microbial antigens dominate in natural killer T cell activation during microbial infection. J Exp Med 208: 1163–1177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zeng L, Lu M, Mori K, Luo S, Lee AS, Zhu Y, Shyy JY (2004) ATF6 modulates SREBP2‐mediated lipogenesis. EMBO J 23: 950–958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wang C, Huang Z, Du Y, Cheng Y, Chen S, Guo F (2010) ATF4 regulates lipid metabolism and thermogenesis. Cell Res 20: 174–184 [DOI] [PubMed] [Google Scholar]
- 36. Rutkowski DT, Wu J, Back S, Callaghan MU, Ferris SP, Iqbal J, Clark R, Miao H, Hassler JR, Fornek J et al (2008) Article UPR pathways combine to prevent hepatic steatosis caused by ER stress‐mediated suppression of transcriptional master regulators. Dev Cell 15: 829–840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bobrovnikova‐marjon E, Hatzivassiliou G, Grigoriadou C, Romero M, Cavener DR, Thompson CB, Diehl JA (2008) PERK‐dependent regulation of lipogenesis during mouse mammary gland development and adipocyte differentiation. Proc Natl Acad Sci USA 105: 16314–16319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sriburi R, Jackowski S, Mori K, Brewer JW (2004) XBP1: a link between the unfolded protein response, lipid biosynthesis, and biogenesis of the endoplasmic reticulum. J Cell Biol 167: 35–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Cross BCS, Bond PJ, Sadowski PG, Jha BK, Zak J, Goodman JM, Silverman RH, Neubert TA, Baxendale IR, Ron D et al (2012) The molecular basis for selective inhibition of unconventional mRNA splicing by an IRE1‐binding small molecule. Proc Natl Acad Sci USA 109: E869–E878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Axten JM, Medina JR, Feng Y, Shu A, Romeril SP, Grant SW, Li WHH, Heerding DA, Minthorn E, Mencken T et al (2012) Discovery of 7‐methyl‐5‐(1‐{[3‐(trifluoromethyl)phenyl]acetyl}‐2,3‐dihydro‐ 1H‐indol‐5‐yl)‐7H‐pyrrolo[2,3‐d]pyrimidin‐4‐amine (GSK2606414), a potent and selective first‐in‐class inhibitor of protein kinase R (PKR)‐like endoplasmic reticulum kinase (PERK). J Med Chem 55: 7193–7207 [DOI] [PubMed] [Google Scholar]
- 41. Gallagher CM, Walter P (2016) Ceapins inhibit ATF6α signaling by selectively preventing transport of ATF6α to the Golgi apparatus during ER stress. Elife 5: 1–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Folch J, Lees M, Sloane Stanley GH (1957) A simple method for the isolation and purification of total lipides from animal tissues. J Biol Chem 226: 497–509 [PubMed] [Google Scholar]
- 43. Bodennec J, Koul O, Aguado I, Brichon G, Zwingelstein G, Portoukalian J (2000) A procedure for fractionation of sphingolipid classes by solid‐phase extraction on aminopropyl cartridges. J Lipid Res 41: 1524–1531 [PubMed] [Google Scholar]
- 44. Chiu BY, Jayawardena J, Weiss A, Lee D, Park S, Dautry‐varsat A, Bendelac A (1999) Distinct subsets of CD1d‐restricted T cells recognize self‐antigens loaded in different cellular compartments. J Exp Med 189: 103–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Spada BFM, Koezuka Y, Porcelli SA (1998) CD1d‐restricted recognition of synthetic glycolipid antigens by human natural killer T cells. J Exp Med 188: 1529–1534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Prigozy TI, Naidenko O, Qasba P, Elewaut D, Khurana A, Kronenberg M (2001) Glycolipid antigen processing for presentation by CDld molecules. Science 291: 664–667 [DOI] [PubMed] [Google Scholar]
- 47. Meissner F, Scheltema RA, Mollenkopf HJ, Mann M (2013) Direct proteomic quantification of the secretome of activated immune cells. Science 340: 475–478 [DOI] [PubMed] [Google Scholar]
- 48. Schröder M (2008) Endoplasmic reticulum stress responses. Cell Mol Life Sci 65: 862–894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kain L, Webb B, Anderson BL, Deng S, Holt M, Constanzo A, Zhao M, Self K, Teyton A, Everett C et al (2014) The identification of the endogenous ligands of natural killer T cells reveals the presence of mammalian α‐linked glycosylceramides. Immunity 41: 543–554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Porcelli S, Brenner MB, Greenstein JL, Balk SP, Terhorst CBP (1989) Recognition of cluster of differentiation 1 antigens by human CD4‐CD8‐cytolytic T lymphocytes. Nature 341: 447–450 [DOI] [PubMed] [Google Scholar]
- 51. Bendelac A (1995) Positive selection of mouse NK1+ T cells by CD1‐expressing cortical thymocytes. J Exp Med 182: 2091–2096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Iwakoshi NN, Pypaert M, Glimcher LH (2007) The transcription factor XBP‐1 is essential for the development and survival of dendritic cells. J Exp Med 204: 2267–2275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Tavernier SJ, Osorio F, Vandersarren L, Vetters J, Vanlangenakker N, Van Isterdael G, Vergote K, De Rycke R, Parthoens E, Van De Laar L et al (2017) Regulated IRE1‐dependent mRNA decay sets the threshold for dendritic cell survival. Nat Cell Biol 19: 698–710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Osorio F, Tavernier SJ, Hoffmann E, Saeys Y, Martens L, Vetters J, Delrue I, De Rycke R, Parthoens E, Pouliot P et al (2014) The unfolded‐protein‐response sensor IRE‐1α regulates the function of CD8α+ dendritic cells. Nat Immunol 15: 248–257 [DOI] [PubMed] [Google Scholar]
- 55. Osorio F, Lambrecht BN, Janssens S (2018) Antigen presentation unfolded: identifying convergence points between the UPR and antigen presentation pathways. Curr Opin Immunol 52: 100–107 [DOI] [PubMed] [Google Scholar]
- 56. Cubillos‐Ruiz JR, Silberman PC, Rutkowski MR, Chopra S, Perales‐Puchalt A, Song M, Zhang S, Bettigole SE, Gupta D, Holcomb K et al (2015) ER stress sensor XBP1 controls anti‐tumor immunity by disrupting dendritic cell homeostasis. Cell 161: 1527–1538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lyttonsg J, Westlins M, Hanleyll R (1991) Thapsigargin inhibits the sarcoplasmic or endoplasmic reticulum Ca‐ATPase family of calcium pumps *. J Biol Chem 266: 17067–17071 [PubMed] [Google Scholar]
- 58. Heifetz A, Keenan RW, Elbein AD (1979) Mechanism of action of tunicamycin on the UDP‐GlcNAc:dolichyl‐phosphate GlcNAc‐1‐phosphate transferase. Biochem J 18: 2186–2192 [DOI] [PubMed] [Google Scholar]
- 59. Sag D, Kronenberg M, Wingender G, Sag D, Krause P, Hedrick CC, Kronenberg M, Wingender G (2014) IL‐10 – producing NKT10 cells are a distinct regulatory invariant NKT cell subset Find the latest version : IL‐10 – producing NKT10 cells are a distinct regulatory invariant NKT cell subset. J Clin Invest 124: 3725–3740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kronenberg M, Gapin L (2002) The unconventional lifestyle of NKT cells. Nat Rev Immunol 2: 557–568 [DOI] [PubMed] [Google Scholar]
- 61. Godfrey DI, Le Nours J, Andrews DM, Uldrich AP, Rossjohn J (2018) Unconventional T cell targets for cancer immunotherapy. Immunity 48: 453–473 [DOI] [PubMed] [Google Scholar]
- 62. Fischer HC, Hauck TS, Gómez‐Aristizábal A, Chan WC (2010) Exploring primary liver macrophages for studying quantum dot interactions with biological systems. Adv Mater 22: 2520–2524 [DOI] [PubMed] [Google Scholar]
- 63. Govindarajan S, Gaublomme D, Van Der Cruyssen R, Verheugen E, Gassen V, Saeys Y, Tavernier S, Iwawaki T, Bloch Y, Savvides SN et al (2018) Stabilization of cytokine mRNAs in iNKT cells requires the serine‐threonine kinase IRE1alpha. Nat Commun 9: 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Brigl M, Bry L, Kent SC, Gumperz JE, Brenner MB (2003) Mechanism of CD1d‐restricted natural killer T cell activation during microbial infection. Nat Immunol 4: 1230–1237 [DOI] [PubMed] [Google Scholar]
- 65. Brennan PJ, Brigl M, Brenner MB (2013) Invariant natural killer T cells: an innate activation scheme linked to diverse effector functions. Nat Rev Immunol 13: 101–117 [DOI] [PubMed] [Google Scholar]
- 66. Starck SR, Tsai JC, Chen K, Shodiya M, Wang L, Yahiro K, Martins‐Green M, Shastri N, Walter P (2016) Translation from the 5′ untranslated region shapes the integrated stress response. Science 351: aad3867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Natori T, Morita M, Akimoto K, Koezuka Y (1994) Agelasphins, novel antitumor and immunostimulatory cerebrosides from the marine sponge Agelas mauritianus . Tetrahedron 50: 2771–2784 [Google Scholar]
- 68. Im JS, Yu KOA, Illarionov PA, Leclair KP, Storey JR, Kennedy MW, Besra GS, Porcelli SA (2004) Direct measurement of antigen binding properties of CD1 proteins using fluorescent lipid probes *. J Biol Chem 279: 299–310 [DOI] [PubMed] [Google Scholar]
- 69. Bedard M, Shrestha D, Priestman DA, Wang Y, Schneider F, Matute JD, Iyer SS, Gileadi U, Prota G, Kandasamy M et al (2019) Sterile activation of invariant natural killer T cells by ER‐stressed antigen‐presenting cells. Proc Natl Acad Sci USA 116: 23671–23681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Ahamd S (2007) iGb3: to be or not to be? Nat Rev Immunol 7: 2082 [Google Scholar]
- 71. Amantini C, Farfariello V, Cardinali C, Beatrice M, Marinelli O, Nabissi M, Santoni M, Bonfili L, Cecarini V, Eleuteri AM et al (2017) The TRPV1 ion channel regulates thymocyte differentiation by modulating autophagy and proteasome activity. Oncotarget 8: 90766–90780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Govindarajan S, Elewaut D, Drennan M (2015) An optimized method for isolating and expanding invariant natural killer T cells from mouse spleen. J Vis Exp 105: 1–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Venken K, Decruy T, Aspeslagh S, Van S, Lambrecht BN, Elewaut D (2013) Bacterial CD1d − restricted glycolipids induce IL‐10 production by human regulatory T cells upon cross‐talk with invariant NKT cells. J Immunol 191: 2174–2183 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Table EV1
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Data Availability Statement
No primary datasets have been generated and deposited.