

Abstract
Communication between and inside cells as well as their response to external stimuli relies on elaborated systems of signal transduction. They all require a directional transmission across membranes, often realized by primary messenger docking onto external receptor units and subsequent internalization of the signal in form of a released second messenger. This in turn starts a cascade of events which ultimately control all functions of the living cell. Although signal transduction is a fundamental biological process realized by supramolecular recognition and multiplication events with small molecules, chemists have just begun to invent artificial models which allow to study the underlying rules, and one day perhaps to rescue damaged transduction systems in nature. This review summarizes the exciting pioneering efforts of chemists to create simple models for the basic principles of signal transduction across a membrane. It starts with first attempts to establish molecular recognition events on liposomes with embedded receptor amphiphiles and moves on to simple transmembrane signaling across lipid bilayers. More elaborated systems step by step incorporate more elements of cell signaling, such as primary and secondary messenger or a useful cellular response such as cargo release.
Keywords: signal transduction, messengers, liposomes, fluorescence, cell membranes
Although signal transduction is a fundamental biological process realized by supramolecular recognition and multiplication events with small molecules, chemists have just begun to invent artificial models which allow to study its underlying rules. This review summarizes the exciting pioneering efforts of chemists to create simple models for the basic principles of signal transduction across a membrane.

1. Introduction
1.1. Signal Transduction in Nature
In living cells, communication is a prerequisite for viability and for appropriate response to external stimuli. Signals often travel a long distance from their origin to their final destination, in the form of messenger molecules. In the course of these events the signal must cross at least one membrane, i. e. the one of the target cell. Inside the cell a second messenger is generally released and triggers a whole cascade of biochemical reactions, which ultimately turn on gene expression and control all functions of the cell. The signaling process starts with docking of the primary messenger onto the extracellular receptor site, and is passed on to the intracellular area by elegant transduction mechanisms.
Two prominent ways of signal transduction have evolved, i. e., the principle of receptor tyrosine kinases (RTK) and the principle of G‐protein coupled receptors (GPCRs).
a) The first principle (RTKs)1, 2, 3, 4 requires two (or more) transmembrane elements, which carry recognition sites on their top end and signaling sites on their bottom end. Incoming signals, e. g., from growth factors, bind to the receptor sites of two neigbouring transmembrane (TM) units and force them into close spatial proximity, a process called ligand‐induced dimerization. This recognition event triggers the action of kinases which phosphorylate multiple tyrosine OH groups in the intracellular region of the receptor dimer (or cluster). The resulting phosphotyrosines function as specific sites for the assembly of downstream signaling molecules that are recruited to the receptor and activated in response to growth factor stimulation. Therefore, an activated RTK can be thought of as a node in a complex signaling network that transmits information from the exterior to the interior of the cell.
b) A totally different approach was realized by GPCRs.5, 6, 7, 8 Here a receptor protein (e. g., the adrenalin receptor) is embedded in the cell membrane and presents a shallow groove for primary messenger docking on its exterior. As soon as the messenger binds (in a noncovalent, reversible process), it triggers a series of massive conformational changes inside the GPCR. These conformational changes activate heterotrimeric G‐proteins, which execute the downstream signaling pathways through the recruitment and activation of cellular enzymes. More specifically, exchange of GDP for GTP separates the α‐subunit, which dissociates from the ensemble and binds to another effector protein nearby, e. g. the adenylyl cyclase. As a result, c‐AMP is released into the cytosol as secondary messenger, which in turn starts a cascade of biochemical reactions and ultimately leads to efficient expression of a target gene – the cellular answer on the incoming signal! (e. g., release of thousands of glucose molecules).
Both pathways represent key elements of control, and hot points for potential interference, e. g., with drugs (agonists and/or antagonists). The structural as well as mechanistic elucidation of the GPCR signaling pathway has been rewarded with the Nobel Prize for Chemistry in 2012.
Until today, there is a surprising lack of signal transduction models despite their fundamental importance and ubiquitiuos application in all living organisms. Problems come from very low local concentrations on lipid bilayers and indirect analytical proof. The related field of supramolecular membrane transport is covered by many groups and has been summarized in excellent reviews.9, 10, 11, 12 Recent years have witnessed the creation of numerous ion carriers, membrane spanning or self‐assembled ion channels as well as membrane pores. Starting from modified gramicidines,13 more and more entirely artificial nonpeptidic ion channels have been constructed, which even allow ligand gating. Prominent examples are the rigid oligo(p‐phenylene) rods from the Matile group, which aggregate into potassium‐selective π‐slides and subsequent variants of synthetic multifunctional pores (SMPs).14 Webb et al. used tailored pyridyl cholates and porphyrins which self‐assembled into ion channels in the presence of Pd2+ ions.15 Ligand gating was achieved by bolaamphiphiles from the Kinbara group which adopt an M‐conformation inside the membrane.16 Finally Hennig et al. recently reported on amphiphilic p‐sulfonatocalix4arenes, which allow enzyme‐regulated ligand gating of cell‐penetrating peptide transport across membranes, a concept which could be used in label‐free enzyme assays.17
However, a transmembrane signal per se requires primary and secondary messenger, often coupled to the above‐mentioned self‐reinforcing cascade of events, resulting in expression of important cellular factors. This poses a great challenge to chemists: Can we imitate this process with an entirely artificial system? The article will present the development of this young field until today, beginning with initial experiments on one face of bilayers over simple transduction systems to more elaborate processes with built‐in molecular recognition and signaling events as well as the release of secondary messengers.
1.2. Model Membranes
Throughout the living world, the cell membrane primarily separates the interior of the cell from its external environment. A real biological membrane is a complex mixture of embedded proteins, lipids, cholesterol, receptors and ion channels, which all play a pivotal role in regulation of molecular transport and signaling. The large number of membrane functions are thus laterally segregated and physically decoupled, and the incorporated components can partition into nanometer‐scale domains termed lipid rafts.18, 19, 20, 21 These domains participate in many key cell signaling processes.22, 23, 24, 25
As the natural membrane is a very complex construct, many detailed studies on single cell functions, for example, on signal transduction, cannot be carried out in living cells.26, 27 Therefore, numerous different model systems have been created that retain the essential lipid bilayer structure with individual components, but in a more simplified composition. This allows to study natural signaling pathways as well as provides the opportunity to imitate this fundamental biological process with synthetic compounds.
In this review, we focus on entirely artificial signaling systems with incorporated synthetic transducers in simplified model membranes, which were designed by chemists. We start with a brief introduction into phospholipid‐based model membranes and give a short overview about metal receptor recognition on vesicle surfaces, which can be regarded as pioneering work towards true artificial signal transduction.
1.2.1. Liposomes
In 1965, Bangham et al. first described artificial lipid vesicles (also called liposomes).28 Generally, liposomes can be formed from a diverse set of lipids including cholesterol, natural (nontoxic) and synthetic phospholipids.29 Hence, the properties of liposomes vary considerably with lipid composition, surface charge, size, and the method of preparation. Here we focus on systems which employ phospholipids as key component.
Phospholipids represent the main elements of all cellular membranes (over 50 %), and are prominent for their biocompatibility and their amphiphilic chemical structure. With respect to their glycerol or sphingosine backbone, phospholipids can be further subdivided into glycerophospholipids (main phospholipids in eukaryotic cells) and sphingomyelins (SMs).30 The artificial signaling systems within this review were all created in liposomes from glycerophospholipids and our brief introduction is therefore limited to a few examples of this kind.
A finer subdivision depends on the chemical structure of the head and tail group. If the head group of a glycerophospholipid is a choline moiety, the lipid is called phosphatidylcholine (PC). The hydrophobic tail of the fatty acid is connected to the glycerol‐backbone via esterification. Its length and degree of saturation leads to different properties:30 thus saturated phospholipids with long acyl chains such as dipalmitoylphosphatidylcholine (DPPC) form a rigid, rather impermeable bilayer structure, whereas unsaturated PC species from natural sources like eggs or soybeans provide much more permeable, more fluidic and less stable bilayers.30, 31 In addition, liposome properties are also defined by their phase transition temperature (Tc or Tm), which is the temperature at which phospholipids transit from the gel to the liquid crystalline state. Some phospholipids adopt the liquid crystalline state already at ambient temperature; two of these are depicted in Figure 1: 1,2‐dioleoyl‐sn‐glycero‐3‐phosphocholine (DOPC, 18 : 1 (Δ9‐Cis)) and 1‐palmitoyl‐2‐oleoyl‐glycero‐3‐phosphocholine (POPC). In general, the Tc of phosphatidylcholines increases with length and saturation of the hydrocarbon chain. PCs with longer hydrocarbon chains such as DPPC (16 : 0, Tc(DPPC) 41 °C ), have a significantly higher Tc than those with shorter ones (DMPC, 14 : 0, Tc(DMPC) 23.6 °C). In PCs of the same aliphatic chain, a higher saturation degree results in a higher Tc value. It is noticeable that the phase transition temperature is used for liposome preparation.
Figure 1.

(a) Schematic representation of a lipid bilayer. (b) Chemical structure of the lipids, which are used for signaling assemblies below. The structural elements of the amphiphilic molecules are colored: The glycerol backbone (light orange) connects hydrophilic head group (light green) to hydrophobic acyl chains (light blue).
1.2.2. Liposome Preparation
Amphiphilic phospholipids form multilamellar assemblies of closed spherical structure, when they are hydrated in aqueous solution. Such vesicles are characterized by stacks of phospholipid bilayers, which give them an onion‐like structure and they encapsulate only a small water volume. The polar lipid head groups are orientated towards the interior and exterior aqueous phase. Multilamellar vesicles (MLV) are typically transformed by downsizing into small (SUV<100 nm), large (LUV 100–1000 nm) and giant (GUV >1000 nm) unilamellar vesicles by a mechanical dispersion (SUVs and LUVs) or electro‐formation method, which is frequently used for GUVs. The resulting model membranes consist of only one bilayer, i. e., they are unilamellar.
In the literature several procedures are described for the controlled size reduction of MLVs.32, 33 A standard procedure for disrupting MLV suspensions involves several freeze‐thaw cycles followed by extrusion through polycarbonate filters with a large pore size in the range of 0.2–1.0 μm.34 This method yields unilamellar vesicles with average diameters determined by the filter pore size. This procedure must be performed above the phospholipid phase transition temperature (Tc). Gel state bilayers are too rigid and liposomes below the Tc can hardly pass through the filter pores. All the model membranes which were used for the artificial signaling systems discussed below were prepared by extrusion.
1.3. Early Supramolecular Approaches and Artificial Liposome‐Bound Receptor Systems
In the late 1970s, chemists started to investigate supramolecular host‐guest interactions of small molecules in order to imitate natural molecular recognition events in solution.35, 36 However, these were approaches without a membrane. In 1995, Sasaki and Arnold incorporated a pyrene‐labeled lipid (5 %) with a metal chelating iminodiacetic acid (IDA) head group for divalent metal (e. g. CuII‐ion) recognition into liposomes of distearoyl phosphatidylcholine (DSPC).37 These bilayer assemblies are believed to have domains of pyrene‐rich aggregates, which are observed by an intense pyrene excimer fluorescence emission at 470 nm. When CuII‐ions are added into the vesicle suspension, they bind to the IDA head groups and the pyrene‐lipid aggregates are dispersed. This is strongly supported by a drastic decrease of the pyrene excimer emission intensity concomitant with a marked increase in monomer emission intensity at 377 nm. In 1999, Kikuchi et al. presented another example of an artificial recognition event on the liposome surface.38, 39 His group demonstrated that metal ions can be coordinated to vesicle‐embedded synthetic receptor ligands. The receptor molecule, an amphiphilic amino‐functionalized bile acid derivative, was incorporated into a liposome vesicle together with lactate dehydrogenase (LDH), which could be inhibited by external CuII‐ion addition. A chemical transformation of the extra‐vesicular amino head group into a chelating Schiff base 1 subsequently translocated all copper ions to the new artificial metal ion‐binding site and restored the LDH activity (structure 1 is illustrated in Figure 2).
Figure 2.

(a) Examples of artificial receptor systems for selective molecular recognition on the vesicle surface. (b) Chemical structure of the employed amphiphilic receptor molecules. (c) Selective binding partners and cationic lipid for vesicle recognition: Illustration of copper(II) ion, coumarin ligand 4 and cationic lipid (DOTAP) 5.
Related examples of this kind relied on the formation of unusual receptor‐metal complexes: In 2003, Hunter and Williams published a dansyl ethylenediamine ligand 2 for selective and cooperative binding of CuII‐ions on the external vesicular face.40 The formation of (common) Cu(2)2 and (unusual) Cu(2)4 complexes was controlled by the concentration of embedded receptor ligand 2 and monitored by stepwise quenching of the dansyl fluorescence at 538 nm upon consecutive addition of copper(II) chloride. The experiment impressively demonstrated that the metal affinity was much higher for the vesicle‐bound receptor compared to the free ligand in solution.
Several other groups investigated molecular recognition at bilayer vesicles,41, 42, 43, 44, 45, 46, 47 including Lehn and coworkers, who studied the aggregation and fusion of interacting vesicles induced by specific metal ligand interactions on the vesicle surface.48, 49
Smith and Jiang studied the coordination of Zn2+‐dipicolylamine (DPA) receptor complex 3 to a coumarin‐based dye 4,[50] in part comparable to Hunters system. However, Smith pointed out, that cationic lipids like DOTAP 5 enhanced the association between 4 and 3, and explained it by induced clustering of receptor 4 in the presence of 5.
Thus the early days witnessed the generation of an embedded transition metal receptor system for efficient and selective recognition of copper(II) ions (Kikuchi), a receptor concentration‐dependent copper(II) complexation motif with different coordination numbers of the chelating receptor (Hunter) and a lipid composition‐based enhancement for a Zn2+ receptor ligand interaction (Smith). In addition, metal‐induced vesicle fusion was realized (Lehn).
Although these investigations gave deeper insight into recognition events at membranes, they all lacked the transmission of the binding event across the lipid bilayer. In the biological sense, a receptor does not only bind a primary messenger, but more importantly transmits the chemical input signal to the opposite face of the membrane, where it releases the second messenger molecule and finally initiates a signal cascade.51, 52
1.4. True artificial Signal Transduction Systems and Present Supramolecular Approaches
In 2002, Hunter et. al. started with the first entirely artificial signal transduction system, a chemical model with synthetic compounds that mimicked the signaling principle of receptor tyrosine kinases (RTK) and operated across a liposome vesicle.53 This idea offered an incentive for the development of more advanced signaling systems over the past 15 years (Figure 3). Very recently, a remarkable example was published that even holds promise for application in medicine: A single transducer was developed for an abiotic signaling mechanism across a lipid bilayer, which performs multiple hydrolysis reactions (output signal amplification) and finally triggers cargo release out of the vesicle, potentially suitable for on‐demand drug delivery.54, 55, 56 This system also allows reversible ON‐ and OFF‐switching.
Figure 3.
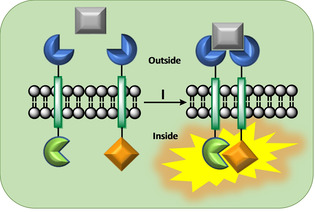
General RTK signaling principle: (I) A primary messenger (grey) docks onto two embedded receptor sites (dark blue), bridges both receptor units and initiates a chemical reaction inside the vesicle.
Back in 2006, the Schrader group added novel unsymmetrical transmembrane units to the RTK principle57 and realized unidirectional signaling, induced by molecular recognition of a primary messenger on the vesicle surface.58, 59, 60
Until today it remains a formidable challenge to synthetic and supramolecular chemists to realize all steps of natural signal transduction. Meanwhile chemists have developed a synthetic reaction cascade with certain elements of the signaling pathway, which can perform modifications on post‐assemblies within a reaction flask, albeit without starting from model membranes.61
In general, evidence for a successful signal transduction event across a vesicle membrane is not at all trivial and often requires the use of optical detection methods. For this reason, most of the signaling systems known today operate with labeled reactive head or tail groups, that provide a specific spectroscopic readout, most often by changes in UV/Vis absorption or fluorescence emission.
In the concept of the artificial RTK signaling system an almost general structure of TM building blocks was developed, with minor changes in the transmembrane unit itself, but with a large variety of head and tail functional groups as will be described below. The mechanistic pathway is comprised by three key steps: In the sensing event, two transducers are dimerized by an incoming primary messenger which docks onto the exterior side of the lipid bilayer. Through this molecular recognition event, both TM units are brought into close proximity by the spatial information from the compact primary messenger. Inside the vesicle, the reactive tail moieties of two neighboring TM units also approach each other and trigger a chemical reaction on the intracellular side of the membrane, i. e., an intra‐vesicular response.
2. Artificial RTK‐Signaling Deteced by UV/Vis Absorption
2.1. Concept and Design of The First Signaling Model
In the earliest prototype of Hunters artificial RTK approach, two symmetrical and lipophilic transmembrane (TM) units were embedded inside PC liposomes.53 Each TM molecule bore two cholenic acid moieties that were dimerized via a propargylic diynene linker. Cholenic acid is a cholesterol derivative and inserts well into the model membrane. These dimers offer the advantage of a relatively rigid structure, which leads to the desired transmembrane orientation, when they are embedded in a liposome vesicle. As illustrated in Figure 4 the cholenic acid dimer acts as a spacer which is able to span the entire lipid bilayer from the exterior to the interior side of the liposome (∼30 Å) and thus connects the head and the tail group responsible for sensing and signaling. Hunter's TM unit 6 carried on both sides a cysteinyl moiety, one designed as a reactive receptor head and the other one as a potential nucleophilic tail for the envisaged, intra‐vesicular, nucleophilic substitution reaction (SN2). The other transducer 7 was functionalized with a cysteine‐2‐pyridinyl disulfide on both ends. When both transducers were embedded inside the lipid bilayer, external reduction and subsequent oxidative formation of a disulfide bond between both head groups was intended to dimerize the two transducers inside the membrane (sensing event). This step should bring both reactive tail groups inside the membrane into close spatial proximity and lead to a thiol‐disulfide displacement, which should trigger the release of 2‐mercaptopyridine (8) into the inner volume of the liposome. In this final step, called signaling event, a UV/Vis‐detectable signal at 341 nm indicated the release of the second messenger 8. The mechanism of this signaling system is depicted in Figure 4 and may suggest that sensing and signaling event had the same diagnostic signal (because 8 was released on both sides). However, in this special case, the sensing event was realized through consecutive reduction and oxidation at the outer vesicle surface and was not a consequence of TM dimerization, but a prerequisite brought about by external deprotection of the thiol‐pyridine head group of compound 7. The result was a free thiol reactive head group of a now unsymmetrical TM unit 9. Thus only the second thiol‐pyridine absorption can be regarded as indication for the signaling event.
Figure 4.

Illustration of the signaling pathway: Structure of synthetic transducers (6, 7 and 9) which are embedded in a lipid bilayer. (I) External reduction of cysteine‐2‐pyridinyl disulfide (7) gives 9 with a thiol head group. (II) Oxidation of both thiol head moieties (in 6 and 9) by potassium ferricyanide bridges both TM units outside the liposome and initiates the thiol‐disulfide reaction (SN2‐reaction) inside the membrane. (III) Triggered release of pyridine‐2‐thiol as a UV/Vis active compound (341 nm).
In the experimental design, transducers 6 and 7 were incorporated in equimolar ratio inside the bilayer vesicle, which was assembled from egg yolk L‐α‐phosphatidylcholine as the lipid source. The vesicles, with an overall 2.5 mol % dotation by both TM units, were prepared by the extrusion method to give a large unilamellar vesicle (LUV) of 200 nm in diameter.
Figure 4 shows how in the first step, external addition of the reducing agent tris(3‐sulfonatophenyl)phosphane reduced the symmetrical transducer 7 to the unsymmetrical thiol 9 with a free thiol‐reactive head group. At this stage, the first UV/Vis absorption indicated the release of mercaptopyridine (341 nm), triggered through the external chemical reaction. Exposure of both thiol head groups to an oxidizing reagent (∼ primary messenger), potassium ferricyanide, covalently dimerized both TM units at the external side, and thus brought both internal reactive groups into spatial proximity for the signaling event. The internal SN2‐type reaction furnishes the macrocyclic TM unit 10 and releases the second messenger 8 over a period of 10 min, as the “intracellular” response.
The main drawback of this simple symmetric system lies in the absence of a molecular recognition event, because receptor dimerization proceeds via a covalent chemical reaction.
2.2. Introducing Signaling Triggered by a Primary Messenger
A somehow related signal transduction system with identical tail‐reactive groups was published in 2012 by Bernitzki and Schrader, but this model system contained suitable head groups for a true molecular recognition event at the liposome surface.59 The receptor moieties of both transducers 11 und 12 were m‐xylylene bisphosphonate dianions which were used for an efficient intermolecular complexation by the compact diethylene triammonium cation (DET, primary messenger) in the sensing event. This complex stability in buffer lies in the low micromolar range.
The tail group of 11 was a cysteinyl residue, whereas the tail moiety of 12 was a pyridinyl‐disulfide. The transmembrane section of the transducers had a slightly different chemical structure compared to Hunter's original: Instead of cholenic acid and propargylic diyne esters the authors used propargylic diyne amides to connect two lithocholic acid derivatives.60 Figure 5a illustrates the signaling mechanism with the novel head groups for recognition of the primary messenger (DET); minor deviations within the hydrophobic part are highlighted in green.
Figure 5.

(a) Cartoon of the signaling pathway with both transmembrane units in a lipid bilayer. (I) The general concept shows the designed release of thiopyridine from the precursor 12. (II) Docking of the primary messenger followed by the induced SN2‐type displacement of thiopyridine results in disulfide‐bridged TM unit 13. (b) External reduction of oppositely orientated disulfide 11 by a water‐soluble phoshine. (c) Background‐, signal‐, and reduction‐dependence on the composition of the lipid. (d) Key experiments in which both TM units are embedded in the mixed DMPC/DPPC vesicles: the double absorption increase is a result of the consecutive addition of the primary messenger and the reducing agent. (e) Messenger‐filled liposomes are exposed to ninhydrin (no messenger penetration for 45 min) and to Triton X (lysis of the vesicle). (f) Conceivable inter‐vesicle reaction by DET crosslink.
In the sensing event, DET (blue) is bridging two TM units, which is leading to the formation of a chelating complex outside the membrane. This in turn brings both transducers into spatial proximity and favors the thiol‐disulfide reaction at the interior of the vesicle. Release of 2‐thiopyridine 8 as second messenger is again observed by UV/Vis absorption at 341 nm and imitates the cellular response towards the incoming primary messenger (DET).
If the signaling event occurred exclusively inside the liposome, all the unproductive orientations of TM units with disulfide groups pointing outside the vesicle should still be intact. As a direct proof, these were subsequently reduced by addition of a water‐soluble phosphine, giving rise to a second thiopyridine 8 release detectable in the UV/Vis spectrum (Figure 5b,d).
A detailed study was performed on this signaling system, to prove the postulated mechanism and to learn about the influence of experimental parameters, lipid composition and the nature of the primary messenger. It also revealed some drawbacks as challenges for the future.
Liposomes were prepared by extrusion to yield LUV's of 200 nm in diameter and were subsequently doped with 2.5 mol % of each TM unit. In order to optimize the signal transduction system, the lipid composition was varied: vesicles from egg yolk L‐α‐phosphatidylcholine (PC; Tc=−7 °C; 18 : 1–16 : 0) turned out to be unstable after addition of the primary messenger (DET), so that in the absence of TM units precipitation occurred upon addition of DET to the liposome suspension.
For this reason another source of lipids were tested, one with a more rigid and the other with a more fluidic property: The preparation of the model membrane was performed with 1,2‐dipalmitoyl‐sn‐glycero‐3‐phosphocholine (DPPC, Tc=41 °C; 16 : 0) or 1,2‐dimyristoyl‐sn‐glycero‐3‐phosphocholine (DMPC; Tc=24 °C; 14 : 0) or mixtures of both. Figure 5c compares the signal transduction efficiency as a function of the liposome composition in a bar diagram. The experimental results revealed that at room temperature (rt=25 °C) a 3 : 1 DMPC/DPPC mixture was the best compromise, keeping the background signal at a minimum (20 %) and the induced signal at a maximum (80 %). Variation of experimental parameters indicated that the signaling efficiency strongly depended on both temperature and lipid fluidity. In pure DMPC‐liposomes spontaneous chemical reaction of the tail groups occurred, which leads to a high background signal (completed signaling after 2 min). The authors explained this result with a high lateral mobility of TM units inside the most fluidic membrane. By contrast, pure DPPC (gel phase) suppressed the background signal, as well as the induced signal. Surprisingly, the signaling process reached saturation after 10 min in DPPC‐ but also in 3 : 1 DMPC/DPPC‐liposomes; perhaps indicating the formation of signaling clusters within the DPPC‐containing vesicle before DET addition. Receptor clustering is also discussed in RTK signaling.
A main drawback of unsymmetrical transducers is that just 50 % (statistical distribution) of all bisphosphonates are oriented correctly towards the vesicle exterior. Only those TM units are available for signal transduction which begins with primary messenger capture (Figure 5a). This in turn means that all TM units with opposite orientation do not take part in the signaling process, and their pyridine‐disulfide moiety on the liposome exterior could be reduced to the cysteinyl residue. Indeed, addition of the water‐soluble phosphine trisodium 3,3’,3’’‐ triphenylphosphine trisulfonate after completion of the first signal, resulted in a second saturation curve (Figure 5d). A control experiment demonstrated that the signaling process was exclusively working, when both TM units were incorporated in the vesicle membrane and DET (50 equivs with respect to the overall concentration of both TM units) was added externally. Importantly, both the signaling and reduction process were shown to proceed independently of each other and thus occurred on opposite faces of the lipid bilayer: thus reducing agent and DET could be added in both orders, and each time produced two typical consecutive saturation curves (Figure 5d).
In a separate control experiment the authors also demonstrated that DET cannot penetrate the liposome membrane made from DMPC/DPPC; this is important because leakage of the messenger inside the vesicle could trigger a signal in the opposite direction and would destroy unidirectionality. To exclude this, DET was encapsulated in mixed DMPC/DPPC liposomes and exposed to externally added ninhydrin, which forms a purple color with free amino groups in aqueous solution (Figure 5e). No purple color appeared for 45 min, so that the unwanted DET leakage could be excluded. However, external addition of the nonionic surfactant Triton X immediately disrupted the membrane and lead to an instant purple color.
Finally a potential inter‐vesicle reaction (recognition and signaling induced by DET between different vesicles) was excluded by preparation of two LUVs each of which contained only one kind of TM unit (Figure 5f). Both units were mixed and DET was added to induce a potential vesicle bridging with concomitant external thiopyridine displacement. However, no UV/Vis absorption occurred at 341 nm, testifying that no inter‐vesicle reaction took place.
3. Artificial RTK‐Signaling Deteced by Fluorescence Emission
3.1. Bidirectional Responsive Signaling
In 2007, Hunter et. al. introduced the first signaling system with a fluorescence response (Figure 6). A pair of two identical synthetic transmembrane units were shown to bind a primary messenger and subsequently transmit binding information across the liposome bilayer – as a mimicry of biological signaling.62 On both ends, the transducer 14 carried a dansyl unit as an ethylenediamine sulfonamide (fluorescence dye). In this very early model system, the ethylenediamine moieties of the exterior fluorescence probes were titrated with copper(II) ions. Since copper(II) ions prefer tetraccordination, square planar complexes were formed between two neighboring dansyl ethylendiamine receptors on the outside. Another copper ion equivalent freely crossed the membrane bilayer and in turn bound to both dansyl ethylene diamine units at the respective tail moieties inside. Tail dimerization was found to be much faster than head dimerization, as could be expected from positive cooperativity. Since signaling occurred on both sides of the membrane the authors called the process bidirectional signaling, in which the spectroscopic readout is the same for sensing and signaling event and therefore indistinguishable. In the experiment, a two step dansyl fluorescence quenching (λex=337 nm; λem=520 nm) indicated indeed an efficient copper‐induced dimerization of two TM units 14.
Figure 6.

(a) Illustration of the signaling concept with embedded transmembrane unit 14 in a lipid bilayer. (I) External addition of CuCl2 leads to the formation of a Cu(14)2 complex and quenches the initial dansyl fluorescence on the exterior side of the lipid bilayer. (II) Penetration of a copper(II) ion across the model membrane results in subsequent coordination of CuII to a preorganized tetradentate ligand Cu(14)2 (cooperative binding event) inside the vesicle (second fluorescence quenching). (b) Transition‐metal receptor system for selective copper(II) ion recognition. (I) Similarly, formation of the Cu(2)2 complex is quenching the dansyl fluorescence at the exterior vesicle surface. (II) Diffusion of a CuII ion through the phospholipid bilayer is now leading to a non‐cooperative binding event and the formation of another quenched Cu(2)2 complex inside the vesicle.
For the experimental setup, LUV's of 800 nm diameter were prepared from egg yolk phospatidylcholine (PC) and doped with transducer 14 (0.50, 1.25 and 2.50 mol %). In titration experiments, the authors found out that the affinity of the vesicle for copper(II) ions increased with transducer (14) loading. This result was also observed in studies with an non‐membrane spanning receptor 2 and indicated that copper(II) ion‐induced receptor dimerization was the same on both vesicle surfaces. The authors explained that copper(II) ion bound to a higher effective receptor concentration, because the receptors could form aggregates within the constrained volume of the liposome membrane. However, it was emphasized that the membrane spanning unit 14 showed more efficient quenching of the final dansyl fluorescence than the half unit 2. The first complexation event at the liposome exterior led to TM dimerization and in turn also increased the effective ethylenediamine concentration within the vesicle. It was expected that in the second binding event, CuII ions within the liposome would coordinate to a preformed tetradentate bis(ethylendiamie) moiety. The authors pointed out, that such a cooperativity of coupled binding events across the lipid bilayer was only possible for a membrane spanning unit.
It was concluded that these studies might illustrate how nature is controlling aggregation of signaling clusters and the transmission of binding information from one side of a membrane to the other, and may shed light on the far more complex biological signaling processes.
3.2. Unidirectional Responsive Signaling
In 2009, Schrader et. al. published a new pair of unsymmetrical TM units for a unidirectional signaling pathway.58 Here, the TM units were constructed in a similar way to the UV/Vis responsive system and both carried the bisphosphonate dianion receptor head group for intermolecular recognition of DET as the primary messenger. At their opposite end, the transducers were functionalized with two different fluorescence dyes, capable of producing a FRET effect (FRET=fluorescence resonance energy transfer). Receptor dimerization was thus intended to give an intra‐vesicular response in form of a sensitive and specific change in fluorescence emission intensities as a quantitative spectroscopic readout. When both transducers were incorporated into LUV's from 3 : 1 DMPC/DPPC and both tail groups could be brought into close proximity, a FRET effect would lead to a significant decrease in tryptophan (donor) fluorescence emission intensity coupled to a simultaneous increase in dansyl (acceptor) emission intensity (Figure 7). Certainly, both TM units were again intended to be noncovalently dimerized by external docking of the primary messenger DET. Indeed, the postulated FRET effect was observed on irradiation into the absorption maximum of tryptophan (280 nm), while the TM units were complexed by DET. As a special feature of the FRET phenomenon, an average donor‐acceptor distance of 1.0 nm could be estimated from the FRET efficiency. This spectroscopic readout is depicted in Figure 7a, showing a significant decrease in tryptophan (yellow arrow) and also a strong increase in dansyl (green arrow) emission intensity. In principle, this signaling process may be reversible with a possible ON and OFF switching, because a strong competing DET binder added after signaling would initiate receptor dissociation and thereby turn off the FRET.
Figure 7.

(a) Left: Fluorescence emission spectrum showing the induced FRET effect after DET addition to the doped vesicles. Right: cartoon of the signaling complex inside the lipid bilayer. (b) Potential unwanted U‐shaped conformation of bisamphiphilic TM units 15 and 16 – no signaling. (c) Left: Fluorescence spectrum of the multi FRET system before (black curve) and after DET addition (red curve). The black curve demonstrates an eosin‐induced permanent multi‐FRET effect (excitation 280 nm) on the vesicle surface. DET addition triggers a new FRET effect inside the vesicle (cellular response, excitation 280 nm), with the typical donor emission decrease and acceptor emission increase, but without any enhancement of the eosin emission intensity expected from U‐shaped TM units (enhanced external multi‐FRET). Therefore, the U‐shaped complex could be excluded. Center: Cartoon of the signaling FRET (I) and the permanent multi‐FRET complex (II) observed in the ternary fluorophore system. Right: Vesicle suspension with embedded TM‐units (15 and 16) and free eosin: Cuvette before and after DET addition – signaling visible with the naked eye.
Due to the small Förster distance (2.1 nm) the authors excluded an unwanted FRET effect between oppositely oriented transmembrane units across the lipid, because lipid bilayers from DMPC and DPPC have a diameter between 3.2–3.8 nm, much too far for a transmembrane FRET effect.
Another potential pitfall in transmembrane signaling would arise from a U‐shape conformation of the bisamphiphiles 15 and 16 inside the membrane. Such an arrangement would be produced by rotation about the diyne center‐piece connection (green) inside the membrane (Figure 7b). In this case the whole process from the sensing to signaling would take place at the exterior face of the vesicle without transmission of binding information across the lipid bilayer.
Therefore, a control experiment had to be developed, which is able to distinguish between an induced DET signaling inside the vesicle and an induced FRET outside. To this end, the authors took advantage of the fact that almost 50 % of all the building blocks were oppositely oriented and pointed with their fluorophore tail groups out of the vesicle. Here, an additional permanent multi‐FRET system (trp‐dan‐eosin) was installed by external addition of eosin, which creates an additional FRET with the dansyl moiety (Figure 7c), and thereby alters the fluorescence emission intensities of all incorporated fluorophores. Spectroscopically, the emission intensity of tryptophan and dansyl decrease, whereas the fluorescence intensity of eosin strongly increases. Hypothetically, if the transducers 15 and 16 adopt a U‐shaped conformation inside the lipid bilayer, DET addition should greatly enhance the multi‐FRET outside the vesicle resulting in a significantly elevated eosin emission intensity. In the experiment, however, vesicles with equimolar 15/16 showed no further increase in eosin fluorescence emission upon addition of DET. Instead they produced the typical fluorescence decrease of the tryptophan donor with concomitant increase of the dansyl acceptor (Figure 7c). This experiment provides evidence for true signaling and excludes the presence of U‐shaped TM units inside the membrane.
As a bonus, receptor dimerization also led to a hypsochromic shift in the dansyl emission so that the color of the whole vesicle suspension changed from yellow to green when the primary messenger was added – signal transduction visible with the naked eye (Figure 7c).
A critical evaluation of this system must admit that the intensity changes of donor and acceptor are moderate (25–30 %), largely due to the presence of 50 % of unproductive TM units with the wrong orientation. However, there is a high potential to establish a switchable signaling mechanism, since both processes, DET complexation and FRET, are reversible.
4. A new Signaling Principle with Controlled Intra‐Vesicular Translocation and Output Signal Amplification
4.1. Transducer Translocation
The above‐described biomimetic, entirely artificial signal transduction systems imitated the principle of receptor tyrosine kinases. However, recently a new abiotic mechanism for a signaling model was introduced by the Hunter group. The whole signaling process required only one single non‐membrane spanning synthetic transducer, which was carrying a sensing head and a reactive tail.
The mechanistic pathway of this new approach comprises three steps: A chemical change in the sensor head group (sensing event) leads to the translocation of the transducer from the exterior to the interior side of the vesicle (signal transmission), where its pro‐catalyst tail group is activated for ester hydrolysis of an encapsulated non‐fluorescent substrate. The product of this catalytic reaction is a detectable fluorescent compound (signaling event), whose turned‐on fluorescence represents the intra‐vesicular response of the signal transduction process (Figure 8). The strength of this concept is its simplicity as well as the signal amplification. Thus a 5‐fold amplification of the input signal was observed, because the internal catalytic reaction produces multiple copies of the second messenger.
Figure 8.

Cross‐section of a supramolecular assembly for transmembrane signal transduction and amplification, showing the controlled translocation of a membrane‐embedded transducer. An external input signal (orange) is recognized on the receptor head group and initiates the translocation process of the transducer to the inner aqueous volume, where the pro‐catalyst (red) becomes active (green) for multiple conversions of an encapsulated substrate (grey) to the second messenger – the intra‐vesicular response (yellow). (Langton et al., Nat. Chem. 2017)
Although subsequent modifications of this system were mainly directed at creating alternative sensing opportunities at the vesicle interface, the amplified second messenger release also bears a great potential as a novel drug delivery system.63
Two different molecular recognition methodologies for signal initiation and transmission were recently presented in successive publications, and these could be finally coupled to a cargo release process.
4.2. A Signaling Pathway Relying on pH‐Responsive Translocation
This signaling approach54 relies on two switchable functional elements on both ends of a short transducer (Figure 9 a,b). It carries a pH sensitive morpholine head group (blue, purple), which can be switched between a charged (protonated) and uncharged (deprotonated) state. In the starting position, the morpholine moiety is protonated (pH 7; OFF state), and therefore acts as an anchor fixing the transducer at the external aqueous phase, because the polar head group is too hydrophilic for entering the lipid bilayer. The central element of the transducer is a short lithocholic acid derivative (grey), which keeps the pro‐catalyst tail moiety inside the lipid bilayer, where it remains unreactive. Once the external pH is changed from 7 to 9, the morpholine head group becomes deprotonated and compatible with the lipid bilayer, leading to a translocation of the synthetic molecule from the exterior to the interior side of the vesicle. This shuttling movement allows the pro‐catalyst (pink), a neutral pyridine‐oxime group, to reach the inner aqueous solution, where it coordinates to an encapsulated zinc(II)‐cation. The activated Zn(II)‐pyridine‐oxime catalyst (green, ON state) is then hydrolyzing the encapsulated acetyl group of the water‐soluble pyrene‐derivative 18 (charcoal grey) to give 19 (yellow) as fluorescent product. Its fluorescence emission is detectable at 510 nm (excited at 415 nm) and the emission intensity is proportional to the conversion of 18 within the vesicle.
Figure 9.

(a) The translocation pathway: In the OFF state the receptor head group (blue) is charged and membrane‐impermeable. (I) The chemical input signal (OH−) deprotonates the head group to a neutral species and renders the transducer membrane‐permeable (purple). (II) Translocation of the transducer is followed by complex formation between the pro‐catalyst (pink) and an encapsulated zinc(II)‐cation leading to the active catalyst (green). (III) The intra‐vesicular non‐fluorescent substrate (grey) is hydrolyzed assisted by zinc(II)‐catalysis to give a fluorescent product (yellow). (b) The chemical structure of the transducer carries a morpholine head and a pyridine oxime tail moiety. The central part consists of a lithocholic acid derivative (grey) and acts as a lipophilic anchor inside the lipid bilayer. 17⋅H+ represents the transducer in the OFF state, while 17⋅Zn2+ depicts the ON state. (Langton et al., Nat. Chem. 2017)
Under these conditions, the output fluorescence signal of the released hydrolysis product reached a fivefold concentration amplification relative to the input chemical signal (sodium hydroxide). The magnitude of the amplified signal depends on the input signal and the encapsulated substrate concentration, but the reaction rate increases with growing transducer concentration. As an inherent limitation, the orientation of the receptor head group is subjected to a statistical distribution, so that only 50 % of the transducers are pointing outside where they may be deprotonated.
In a control experiment the authors compared the signal‐transduction system to a control system. A vesicle without incorporated transducer 17, but with encapsulated substrate 18, only produced a very small fluorescence increase at an external pH of 7 over a period of hours, indicating slow solvolysis of 18 (Figure 10a, black data). The result was identical for the signaling system (with embedded transducer 17), when the external pH was maintained at 7 (Figure 10a, red data). However, a sharp increase in fluorescence emission was achieved for the signaling system by raising the external pH to 9 (Figure 10a, green data).
Figure 10.

Detection of signal transduction by pH‐responsive translocation: (a) Time‐dependent changes in fluorescence emission at 510 nm (excited at 415 nm): Signaling assembly with incorporated transducer 17⋅H+ in the OFF state (red curve); signaling system with embedded 17⋅Zn2+ in the ON state (green curve). Liposome vesicle with encapsulated zinc chloride and substrate 18, but without transducer 17 (black curve). (b) Fluorescent vesicles detectable by TIRF microscopy: top: ON state; bottom: OFF state. (Langton et al., Nat. Chem. 2017)
An important experiment was to visualize the output signal of the fluorescent vesicles as a proof for the amplified hydrolysis inside by total internal reflection fluorescence microscopy (TIRFM). Spherical regions with a fluorescence emission at 510 nm were observed without leakage of substrate 18 und product 19 (Figure 10b).
It was also demonstrated that the translocation process across the bilayer is reversible. External addition of hydrochloric acid turned off the catalyzing machinery by restoring the initial neutral pH value outside the vesicles. No fluorescence increase was observed within one hour, until the process was turned on again by addition of sodium hydroxide.
4.3. A Signaling Pathway Relying on Transition Metal responsive Translocation
In 2017, Hunter et al extended the already existing signal transduction system by a metal‐chelating receptor unit.55 For this approach, the authors used the identical lithocholic acid transducer as before, but instead of a pH‐sensitive moiety they employed a phenanthroline head group for selective recognition of CuII‐cations. The tail moiety of the transducer (a pyridine‐oxime), the lipid composition (DOPC/DOPE 1 : 1) of the vesicle and the substrate (acetylated pyranine 18) remained the same. At first glance, the changes in the system appear to be minimal, but they replace a simple deprotonation by a real complexation event with a true primary messenger.
The modified signaling pathway across the lipid bilayer is illustrated in Figure 11, which depicts the embedded synthetic transducer in the OFF state, when copper(II) is bond to the phenanthroline head group. Addition of ethylenediaminetetraacetate (EDTA) leads to quantitative dissociation of this phenanthroline‐cupper(II) complex, which loses the CuII cation and becomes neutral. This in turn enables the translocation mechanism at the vesicle interior and subsequent activation of the tail pro‐catalyst to the ON state. As a consequence, the zinc catalyst is again hydrolyzing the acetylated pyranine substrate 18 into the fluorescent alcohol 19.
Figure 11.

(a) Time‐dependent changes in fluorescence emission: Fluorescence increase at 510 nm (excitation 415 nm) in the presence (+) und absence (−) of 20 and EDTA. (b) Background metal cation binding equilibria (top) and assumed mechanism for the signal transduction (bottom): (I) Negligible Cu2+‐phenanthroline complex dissociation in absence of EDTA. (II) Hardly any translocated transducers 20 are turned to the ON state for catalyzed substrate conversion. (III) External addition of EDTA immediately traps Cu2+ ions from the phenanthroline moiety and enables translocation of the neutral transducer. (IV) Zn2+ binding to the pyridine oxime tail switches the transducer to the ON‐state. (c) Chemical structure of the transducer as CuII‐phenanthroline 20⋅Cu2+ (initial OFF‐state) and zinc‐pyridine oxime complex 20⋅Zn2+ (ON‐state). (Langton et al., J. Am. Chem. Soc. 2017)
At this point it should be mentioned that the similarity of both translocation systems is also reflected in the output signal. Again, the fluorescence emission of pyranine 19 at 510 nm (excited at 415 nm) increases fivefold, depending again on the amount of encapsulated substrate and the concentration of 20⋅Zn2+ inside the vesicle, which are identical in both systems.
To prove the importance of the input signal, this experiment was also carried out in the absence of EDTA. In this case, due to the low dissociation rate of the phenanthroline‐copper complex, only small amounts of transducer were switched to the ON state, which were neglectable. This experiment demonstrates the necessity of a quantitative complex formation after external addition of the primary messenger (Figure 11a, b). The influence of a tuned complex stability is also illustrated by the occurrence of multiple interdependent metal coordination equilibria at the in‐ and outside: Thus, the phenanthroline‐copper complex (log K=9.0) is favored in aqueous solution and dominates in equilibrium over the zinc complex (log K=4.6), until external addition of EDTA captures the CuII ions and initiates the signaling ON‐state. As an important prerequisite for a functioning signaling, the intra‐vesicular zinc concentration must be 50 times higher than the copper concentration (5 μM) at the vesicle exterior, because the ZnCl2 loading is a crucial factor for the efficiency of catalytic ester cleavage.
The authors could further show, that the above‐described translocation process could be switched back and forth between the ON‐ and OFF‐state at will by successive addition of EDTA and CuCl2 (Figure 12). Reversible diffusion of the transducer between the vesicle exterior and interior occurred, until the substrate 18 was completely consumed in the vesicle. The ability to reset the mechanism to its initial state hinges on the reversibility of zinc complex formation inside the vesicle.
Figure 12.

Input signal‐controlled reversible translocation mechanism: successive additions of EDTA and CuCl2 to the extra‐vesicle solution switch signaling transducer between its OFF and ON state. (Langton et al., J. Am. Chem. Soc. 2017)
4.4. A pH‐Responsive Signaling Pathway for Controlled Cargo Release
The original pH‐responsive signaling vesicle system could very recently be extended to include cargo release (Figure 13).56 Vesicle design was very similar to the original (mixed DOPC/DOPE‐liposomes, zinc(II)‐cation cofactor and transducer 17, Figure 13a), but this time the encapsulated substrate was the naphthoylated pyranine‐derivative 21. The inner vesicle solution was first loaded with a water‐soluble fluorophore, a calcein‐derivative 22, which is self‐quenched at a concentration of 70 mM. Control experiments secured that both the pyranine‐ and calcein‐derivative are membrane‐impermeable and fluorescence‐silent in the initial state.
Figure 13.

Signal transduction mechanism which triggers cargo release from vesicles. (a) Illustration of the signaling pathway. The primary messenger switches the state of the external transducer head group from polar (blue) to apolar (purple), allowing its translocation to the inside of the liposome. Zn2+‐binding to the inner tailgroup (rose) activates the catalyst (green). (b) Catalytic surfactant generation. The catalyst hydrolyzes 21 (grey) and generates the surfactant 23 (yellow). (c) Cargo release. The surfactant enters the lipid bilayer which in turn enhances the permeability of the membrane for polar solutes, enabling cargo release (pink). (Langton et al., J. Am. Chem. Soc. 2017)
In the ON‐state the activated zinc‐pyridine oxime complex catalyzed the hydrolysis of 21, which was generating a low concentration of a hydrophobic carboxylic acid, 2‐naphthoic acid 23, as a secondary messenger. 23 is a known hydrotrope und membrane‐permeable. It migrated into the lipid bilayer, where it favored the membrane permeability for hydrophilic solutes, like calcein, by its surfactant properties and without destructing the vesicle (Figure 13c). This triggered the release of calcein into the extra‐vesicle phase. Its diffusion was observable by an increase in fluorescence emission intensity as a result from fluorophore dilution. After 800 min, the signaling system achieved a 15 times amplified signal outcome (calcein release), when compared to the amount of added “primary messenger” (sodium hydroxide).
Pyranine 19, the other product of ester cleavage, enables monitoring of surfactant generation, its quantification and acts as a fluorescence sensor for local pH change. Exclusively in the ON‐state, the released fluorophore indicated a pH change from 7 to 8. This could be explained by two different scenarios: 1) enhanced membrane permeability for H+/OH− and equilibration between the external pH of 9 and the internal pH of 7; 2) enhanced membrane permeability for encapsulated 19, which diffused out of the vesicle. The authors answered this question by removing all non‐encapsulated molecules from the bulk solution by size exclusion chromatography, and observed a decreased emission intensity for 19. It thus turned out that the formation of surfactant 23 also caused an efflux of 19 out of the vesicle. The purified vesicles were also measured with dynamic light scattering (DLS) and 1H NMR experiments which confirmed identical size and unaltered phospholipid concentration before and after signaling, testifying the all vesicles were still intact after cargo release.
5. Conclusions and Outlook
This review has summarized the research efforts in imitating natural signal transduction by artificial means, specifically by creating new receptor molecules capable of recognizing an incoming message and sending out a signal at the intracellular space of a liposome. Most of these systems follow the example of RTKs, but a new artificial mechanism involving translocation of a small TM unit has recently also been discovered. The incoming signal was initially a simple oxidation reaction, but has gradually matured into reversible noncovalent interactions with small primary messenger molecules which exploit well‐defined receptor sites on TM units.
In most cases, however, it is unclear, if this results in the formation of just dimers or, as it is also discussed in cellular signalling events, in receptor clustering.
The signal was produced as a released secondary messenger molecule, which could be detected septroscopically, as released cargo from the liposome interior. Alternatively it could merely be a FRET signal resulting from spatial proximity of 2 TM units with fluorescent donors and acceptors. Thus, a proof‐of‐principle was now established for the feasibility of sending a signal across a membrane relying only on non‐natural elements.
However, many open questions are still unsolved and the subsequent cascade of events in the cell remains a great challenge. No group has hitherto been able to embed the receptor modules in the correct uniform orientation with recognition sites outside and signaling sites inside. The unproductive combination of TM units in wrong orientation severely limits the signaling intensity and efficiency of synthetic signaling systems.
Another challenge is to avoid premature signaling in the absence of primary messengers. Caging of a reactive functionality, e. g. by means of o‐nitrobenzyl capping, may be a solution, but the release of the masked functionality requires another photochemical irradiation prior to to messenger injection. Finally, biological signaling events are so effective because the released second messenger triggers a cascade of programmed events with strongly mutually reinforcing and multiplying character – here the triggered formation of transition metal complexes as catalysts for simple hydrolysis reactions inside the vesicle is a first promising step. Kinetics is another challenge: a signal should be built up in seconds rather than in hours, and it should be entirely reversible. All these fascinating aspects of biological signaling await a manmade synthetic alternative or model. Research in this direction will teach us a lot about the prerequisites for efficient membrane signaling, and will perhaps one day allow us to implement artificial signaling systems into living cells and to mildly influence existing pathways. If damaged signaling can thus be restored or pathological signaling be prevented/suppressed, these systems may even find therapeutic application.
Conflict of interest
The authors declare no conflict of interest.
Biographical Information
Robert Bekus received his B. Sc. degree in Chemistry from Ruhr University Bochum in Germany. He pursued his graduate studies at the same university and wrote his Master thesis on “artificial signal transduction” supervised by Gerald Dyker and Thomas Schrader (M. Sc. degree in 2015). Subsequently, he moved to the University of Duisburg‐Essen and joined the group of Thomas Schrader. In 2016 he was awarded a PhD fellowship from the Professor Werdelmann foundation. He is currently developing highly efficient synthetic signaling systems that imitate the principle of receptor tyrosine kinases.

Biographical Information
Thomas Schrader studied Chemistry at Bonn University and obtained his PhD with Wolfgang Steglich. Subsequently, he joined the group of E. C. Taylor at Princeton University, USA, as a postdoc. Back in Germany, he pursued his habilitation work associated to the group of Günter Wulff at Düsseldorf University. He received a call in 2000 to become Associate Professor at Marburg University. 6 years later he moved to the University of Duisburg‐Essen as a Full Professor. Thomas Schrader is the speaker of the Collaborative Research Center 1093 “Supramolecular Chemistry on Proteins”. His research focuses on the development of Supramolecular Ligands tailored for Proteins, Nucleic Acids and Membranes, which are able to interfere with their biological function. His molecular tweezers are in the preclinical stage as new drugs against neurodegenerative diseases (AD/PD).

Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
R. Bekus, T. Schrader, ChemistryOpen 2020, 9, 667.
References
- 1. Lemmon M. A., Schlessinger J., Cell 2010, 141, 1117–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Simons K., Toomre D., Nat. Rev. Mol. Cell Biol. 2000, 1, 31–41. [DOI] [PubMed] [Google Scholar]
- 3. Cebecauer M., Spitaler M., Sergé A., Magee A. I., J. Cell Sci. 2010, 123, 309–320. [DOI] [PubMed] [Google Scholar]
- 4. Ségaliny A., Tellez-Gabriel M., Heymann M.-F., Heymann D., J. Bone Oncol. 2015, 4, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hanlon C. D., Andrew D. J., J. Cell Sci. 2015, 128, 3533–3542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Granier S., Kobilka B., Nat. Chem. Biol. 2012, 8, 670–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lefkowitz R. J., Angew. Chem. Int. Ed. 2013, 52, 6366–6378; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 6494–6507. [Google Scholar]
- 8. Hilger D., Masureel M., Kobilka B. K., Nat. Struct. Mol. Biol. 2018, 25, 4–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Vargas Jentzsch A., Hennig A., Mareda J., Matile S., Acc. Chem. Res. 2013, 46, 2791–2800. [DOI] [PubMed] [Google Scholar]
- 10. Fuertes A., Juanes M., Granja J. R., Montenegro J., Chem. Commun. 2017, 53, 7861–7871. [DOI] [PubMed] [Google Scholar]
- 11. Gale P. A., Davis J. T., Quesada R., Chem. Soc. Rev. 2017, 46, 2497–2519. [DOI] [PubMed] [Google Scholar]
- 12. Wu X., Howe E. N. W., Gale P. A., Acc. Chem. Res. 2018, 51, 1870–1879. [DOI] [PubMed] [Google Scholar]
- 13. Pfeifer J. R., Reiß P., Koert U., Angew. Chem. Int. Ed. 2006, 45, 501–504; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 515–518. [Google Scholar]
- 14. Tedesco M. M., Ghebremariam B., Sakai N., Matile S., Angew. Chem. Int. Ed. 1999, 38, 540–543; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 1999, 111, 523–526. [Google Scholar]
- 15. Wilson C. P., Boglio C., Ma L., Cockroft S. L., Webb S. J., Chem. Eur. J. 2011, 17, 3465–3473. [DOI] [PubMed] [Google Scholar]
- 16. Muraoka T., Endo T., Tabata K. V., Noji H., Nagatoishi S., Tsumoto K., Li R., Kinbara K., J. Am. Chem. Soc. 2014, 136, 15584–15595. [DOI] [PubMed] [Google Scholar]
- 17. Peng S., Barba-Bon A., Pan Y.-C., Nau W. M., Guo D.-S., Hennig A., Angew. Chem. Int. Ed. 2017, 56, 15742–15745; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 15948–15951. [Google Scholar]
- 18. Lingwood D., Simons K., Science 2010, 327, 46–50. [DOI] [PubMed] [Google Scholar]
- 19. Levental I., Veatch S. L., J. Mol. Biol. 2016, 428, 4749–4764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sezgin E., Levental I., Mayor S., Eggeling C., Nat. Rev. Mol. Cell Biol. 2017, 18, 361–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Simons K., Ehehalt R., J. Clin. Invest. 2002, 110, 597–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Varshney P., Yadav V., Saini N., Immunology 2016, 149, 13–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Milovanovic D., Honigmann A., Koike S., Göttfert F., Pähler G., Junius M., Müllar S., Diederichsen U., Janshoff A., Grubmüller H., et al., Nat. Commun. 2015, 6, 5984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yang S.-T., Kiessling V., Simmons J. A., White J. M., Tamm L. K., Nat. Chem. Biol. 2015, 11, 424–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mapstone M., Cheema A. K., Fiandaca M. S., Zhong X., Mhyre T. R., MacArthur L. H., Hall W. J., Fisher S. G., Peterson D. R., Haley J. M., et al., Nat. Med. 2014, 20, 415–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Keller H., Worch R., Schwille P., Protein-Ligand Interactions, Springer, 2013, pp. 417–438. [Google Scholar]
- 27. Chan Y.-H. M., Boxer S. G., Curr. Opin. Chem. Biol. 2007, 11, 581–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bangham A., Standish M., Watkins J., J. Mol. Biol. 1965, 13, 238–52. [DOI] [PubMed] [Google Scholar]
- 29. Harayama T., Riezman H., Nat. Rev. Mol. Cell Biol. 2018, 19, 281–296. [DOI] [PubMed] [Google Scholar]
- 30. Li J., Wang X., Zhang T., Wang C., Huang Z., Luo X., Deng Y., Asian J. Pharm. Sci. 2015, 10, 81–98. [Google Scholar]
- 31. Akbarzadeh A., Rezaei-Sadabady R., Davaran S., Joo S. W., Zarghami N., Hanifehpour Y., Samiei M., Kouhi M., Nejati-Koshki K, Nanoscale Res. Lett. 2013, 8, 102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Torchilin V., Weissig: V. Liposomes: a practical approach , 2nd ed., Oxford University Press, Oxford, 2003. [Google Scholar]
- 33. Patil Y. P., Jadhav S., Chem. Phys. Lipids 2014, 177, 8–18. [DOI] [PubMed] [Google Scholar]
- 34.
- 34a. Patty P. J., Frisken B. J., Biophys. J. 2003, 85, 996–1004; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34b. MacDonald R. C., MacDonald R. I., Menco B. P., Takeshita K., Subbarao N. K., Hu L. R., Biochim. Biophys. Acta 1991, 1061, 297–303. [DOI] [PubMed] [Google Scholar]
- 35. Schneider H. J., Yatsimirsky A., Principles and Methods in Supramolecular Chemistry, New York: John Wiley & Sons, 2000. [Google Scholar]
- 36. Späth A., König B., Beilstein J. Org. Chem. 2010, 6, No. 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sasaki D. Y., Shnek D. R., Pack D. W., Arnold F. H., Angew. Chem. Int. Ed. 1995, 34, 905–907; [Google Scholar]; Angew. Chem. 1995, 107, 994–996. [Google Scholar]
- 38. Kikuchi J., Ariga K., Ikeda K., Chem. Commun. 1999, 547–548. [Google Scholar]
- 39. Kikuchi J., Ariga K., Sasaki Y., Ikeda K., J. Mol. Catal. B 2001, 11, 977–984. [Google Scholar]
- 40. Doyle E. L., Hunter C. A., Phillips H. C., Webb S. J., Williams N. H., J. Am. Chem. Soc. 2003, 125, 4593–4599. [DOI] [PubMed] [Google Scholar]
- 41. Kolusheva S., Zadmard R., Schrader T., Jelinek R., J. Am. Chem. Soc. 2006, 128, 13592–13598. [DOI] [PubMed] [Google Scholar]
- 42. Pincus J. L., Jin C., Huang W., Jacobs H. K., Gopalan A. S., Song Y., Shelnutt J. A., Sasaki D. Y., J. Mater. Chem. 2005, 15, 2938–2945. [Google Scholar]
- 43. Gruber B., Stadlbauer S., Späth A., Weiss S., Kalinina M., König B., Angew. Chem. Int. Ed. 2010, 49, 7125–7128; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 7280–7284. [Google Scholar]
- 44. Gruber B., Stadlbauer S., Woinaroschy K., König B., Org. Biomol. Chem. 2010, 8, 3704–3714. [DOI] [PubMed] [Google Scholar]
- 45. Müller A., König B., Chem. Commun. 2014, 50, 12665–12668. [DOI] [PubMed] [Google Scholar]
- 46. Banerjee S., Bhuyan M., König B., Chem. Commun. 2013, 49, 5681–5683. [DOI] [PubMed] [Google Scholar]
- 47. Banerjee S., König B., J. Am. Chem. Soc. 2013, 135, 2967–2970. [DOI] [PubMed] [Google Scholar]
- 48. Marchi-Artzner V., Brienne M.-J., Gulik-Krzywicki T., Dedieu J.-C., Lehn J.-M., Chem. Eur. J., 2004, 10, 2342–2350. [DOI] [PubMed] [Google Scholar]
- 49. Richard A., Marchi-Artzner V., Lalloz M.-N., Brienne M. J., Artzner F., Gulik-Krzywicki T., Guedeau Boudeville M.-A., Lehn J.-M., Proc. Mont. Acad. Sci., 2004, 101, 15279–15284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Jiang H., Smith B. D., Chem. Commun., 2006, 1407–1409. [DOI] [PubMed] [Google Scholar]
- 51. Bourne H. R., Curr. Opin. Cell Biol. 1997, 9, 134–142. [DOI] [PubMed] [Google Scholar]
- 52. Ji T. H., Grossmann M., Ji I., J. Biol. Chem. 1998, 273, 17299–17302. [DOI] [PubMed] [Google Scholar]
- 53. Barton P., Hunter C. A., Potter T. J., Webb S. J., Williams N. H., Angew. Chem. Int. Ed. 2002, 41, 3878–3881; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2002, 114, 4034–4037. [Google Scholar]
- 54. Langton M. J., Keymeulen F., Ciaccia M., Williams N. H., Hunter C. A., Nat. Chem. 2017, 9, 426–430. [DOI] [PubMed] [Google Scholar]
- 55. Langton M. J., Williams N. H., Hunter C. A., J. Am. Chem. Soc. 2017, 139, 6461–6466. [DOI] [PubMed] [Google Scholar]
- 56. Langton M. J., Scriven L. M., Williams N. H., Hunter C. A., J. Am. Chem. Soc. 2017, 139, 15768–15773. [DOI] [PubMed] [Google Scholar]
- 57. Schrader T., Maue M., Ellermann M. J. Recept. Signal Transduction 2006, 26, 473–485. [DOI] [PubMed] [Google Scholar]
- 58. Bernitzki K., Schrader T., Angew. Chem. Int. Ed. 2009, 48, 8001–8005; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 8145–8149. [Google Scholar]
- 59. Bernitzki K., Maue M., Schrader T., Chem. Eur. J. 2012, 18, 13412–13417. [DOI] [PubMed] [Google Scholar]
- 60. Maue M., Bernitzki K., Ellermann M., Schrader T., Synthesis 2008, 2247–2256. [Google Scholar]
- 61. Pilgrim B. S., Roberts D. A., Lohr T. G., Ronson T. K., Nitschke J. R., Nat. Chem. 2017, 9, 1276–1281. [Google Scholar]
- 62. Dijkstra H. P., Hutchinson J. J., Hunter C. A., Qin H., Tomas S., Webb S. J., Williams N. H., Chem. Eur. J. 2007, 13, 7215–7222. [DOI] [PubMed] [Google Scholar]
- 63. Sercombe L., Veerati T., Moheimani F., Wu S. Y., Sood A. K., Hua S., Front. Pharmacol. 2015, 6, 286. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary