

Abstract
The latency associated with bone metastasis emergence in castrate‐resistant prostate cancer is attributed to dormancy, a state in which cancer cells persist prior to overt lesion formation. Using single‐cell transcriptomics and ex vivo profiling, we have uncovered the critical role of tumor‐intrinsic immune signaling in the retention of cancer cell dormancy. We demonstrate that loss of tumor‐intrinsic type I IFN occurs in proliferating prostate cancer cells in bone. This loss suppresses tumor immunogenicity and therapeutic response and promotes bone cell activation to drive cancer progression. Restoration of tumor‐intrinsic IFN signaling by HDAC inhibition increased tumor cell visibility, promoted long‐term antitumor immunity, and blocked cancer growth in bone. Key findings were validated in patients, including loss of tumor‐intrinsic IFN signaling and immunogenicity in bone metastases compared to primary tumors. Data herein provide a rationale as to why current immunotherapeutics fail in bone‐metastatic prostate cancer, and provide a new therapeutic strategy to overcome the inefficacy of immune‐based therapies in solid cancers.
Keywords: bone metastasis, dormancy, immune evasion, prostate cancer, type I interferon
Subject Categories: Cancer, Immunology, Signal Transduction
Tumor‐intrinsic type I IFN is lost upon outgrowth of dormant prostate cancer cells in bone, driving metastasis. Therapeutic reversal of tumor‐intrinsic IFN loss enhances tumor cell visibility and the effectiveness of systemic immunomodulatory agents against bone‐metastasis.
Introduction
Bone metastases are characteristic of lethal prostate cancer (PCa), occurring in up to 90% of men who develop treatment‐refractory disease 1, 2, invariably leading to death within 12–24 months of detection 3. Treatment of prostate cancer that has spread to bone primarily involves androgen deprivation, chemotherapy, and radiation, all of which may be coupled to bone‐targeted therapies. However, in castrate‐resistant prostate cancer (CRPC), while bone‐targeted and conventional treatments have been shown to enhance quality of life and delay the onset of skeletal‐related events, they have failed to prevent bone‐metastatic outgrowth or impact long‐term survival 4, 5, 6. This, along with success in metastatic melanoma 7, sparked interest in the use of checkpoint‐targeted immunotherapeutics for advanced PCa. Yet, to date, this approach has been met with underwhelming results with few long‐term responses 8, 9, 10. Therefore, the dissection of processes that drive bone metastasis, including mechanisms of immune regulation or tumor‐driven escape, is crucial to the development of new strategies for improving therapeutic response in bone‐metastatic PCa.
Several studies suggest that upward of 60% of primary PCa patients harbor disseminated tumor cells (DTCs) in bone, independent of PCa stage, and in the absence of detectable metastases 11, 12, 13. Moreover, distinct evolutionary events that precede key metastasis‐associated alterations in the primary tumor have been observed in metastatic lesions 14. These findings suggest that tumor cell spread to bone is an early event in PCa progression. Still, overt skeletal lesions often remain undetected for many years. Dormancy, in which tumor cell proliferation is restricted by cell‐intrinsic or cell‐extrinsic means for up to 20 years from initial local diagnosis 15, is proposed to govern DTC indolence and the delay often observed in PCa recurrence. In addition to cellular and angiogenic dormancy 16, 17, immune regulation of tumor cell fate in bone has been proposed as a driver of dormancy 18, 19. However, to date, little evidence exists of direct immune cell involvement in dormant cell persistence or eventual escape and outgrowth in the bone tumor microenvironment (TME). While tumor‐associated alterations that promote immune evasion at secondary soft‐tissue sites have been frequently reported, few studies have explored the contribution of immune regulatory processes to bone metastasis. Likewise, cancer cell‐intrinsic immunomodulatory signaling has been largely ignored in the exploration of tumor progression in bone. As such, there exists a gap in our understanding of the fundamental mechanisms that underpin bone‐metastatic outgrowth, which may impede development of new approaches aimed at targeting bone‐avid cancers.
In this study, a labeling technique was developed to detect and isolate dormant and proliferating tumor cells from bone using an immune‐competent, androgen receptor‐positive yet androgen‐independent mouse model of PCa, to uncover critical pathways that regulate bone‐metastatic progression. Single‐cell transcriptomics revealed that suppression of tumor‐intrinsic type I interferon (IFN)—a class of immunomodulatory cytokines previously linked to breast cancer metastasis 20—was strongly associated with dormant tumor cell outgrowth in bone and the acceleration of disease. Importantly, similar loss of tumor‐intrinsic type I IFN was observed in macrometastatic lesions in CRPC patients. We investigate the consequences of this loss in the osteoimmune niche and uncover a mechanism by which tumor‐derived IFN signals prevent bone‐metastatic outgrowth. Moreover, we reveal a new therapeutic approach to enhance tumor‐intrinsic IFN signaling using an epigenetic modulator to restore cancer cell immunogenicity and promote bone metastasis‐free survival in CRPC.
Results
Tumor‐intrinsic IFN signaling is enriched in dormant PCa cells in bone
To investigate transcriptional alterations that facilitate dormancy induction and subsequent outgrowth, we utilized the RM1 syngeneic model of bone‐metastatic PCa, which is the only immunocompetent model of CRPC that reliably yields skeletal lesions in vivo 21, 22. We coupled this model to a membrane‐dye (PKH26) retention system that permits fluorescent discrimination of dormant and proliferating cells (Appendix Fig S1A–C). Following intracardiac (IC) injection of PKH26‐labeled RM1 cells expressing cerulean (eCFP) and luciferase (luc2) into C57BL/6 mice, dormant (occurring at a rate of ~ 2 in 3 × 107 bone marrow [BM] cells in ~ 1 in eight tumor‐bearing mice) and proliferating single RM1 cells were FACS‐isolated from bone metastases on average at day 16 (Fig 1A). Single‐cell RNA‐seq of 28 dormant (PKH+) and 32 proliferating (PKH−) cells was performed, and BASiCS 23 normalization was employed. Pre‐ and post‐normalization of all samples revealed no significant batch effects, and potential technical noise was corrected after normalization (Appendix Fig S1D). Further analysis of RM1 markers (eCFP and luc2) demonstrated the authenticity of more than 83% of single tumor cells (Appendix Fig S1E). Of 2,410 differentially expressed (DE) genes in dormant cells, 1,609 were identified by BASiCS as having no residual overdispersion within dormant and proliferating groups 24, validating the absence of spurious signals in mean gene expression shifts. Enrichment of gene ontologies (GOs) defining cell cycle and mitotic processes was observed in the proliferating group (Fig 1B). Conversely, robust overrepresentation of GO biological processes defining immune effector responses in dormant cells (Fig 1C) prompted unbiased INTERFEROME 25 analysis of all DEGs. It was revealed that 39% of genes enriched in dormant cells were predicted interferon‐regulated genes (IRGs), largely type I (IFN‐α/β; 459 DEG; Fig 1D), represented by heatmap (Fig 1E). This included interferon regulatory factor (Irf) 7, a master transcription factor that mediates type I IFN production and IRG expression 26, which was expressed threefold higher in dormant than in proliferating tumor cells. A subpopulation of robustly enriched type I IRGs evident in ~ 11 single PKH+ cells was identified, eight of which cluster together (Appendix Fig S1F). Subsequent non‐negative matrix factorization (NMF) 27 analysis of all DEGs (Fig EV1A) identified three distinct groups of single cells consisting of two dominant dormant cell clusters (C1 and C2) and one proliferating cluster (C3) with a cophenetic correlation of 0.9995. Analysis of genes contributing to these three metagene clusters (Fig EV1B) revealed that C1 was enriched for genes associated with positive regulation of immune responses (Irf7, Mx1, and Unc93b1) and antiapoptotic processes (Casp7 and Bcl2a1a/b/d), while C2 was enriched for genes involved in metabolic processes (B3gat3, Hs6st1, and Man1b1) along with immune activation (B7‐H3, H2‐M3, CD80, and Mr1), suggesting that, when compared to C2, C1 may embody a more fully manifest dormant state (Fig EV1C). Indeed, it is known that tumor dormancy encompasses a range of stages that can rapidly evolve in response to TME dynamics 28 and may be influenced by bone niche‐specific pressure and co‐existing tumor growth 29. This is supported by C1 enrichment of Bhlhe41, previously linked to dormancy in breast and prostate cancers 29, 30 and more recently associated with bone‐endosteal niche dormancy in metastatic breast cancer 31. Unsurprisingly, metagene C3 was enriched for genes associated with cell proliferation (Cenpe, Bub1, and Nuf2). Stratification of type I IRGs enriched in dormant cells compared to proliferating cells isolated from the same hosts (Appendix Fig S1G) confirmed that differential IRG expression did not result from unequal representation of dormant cells derived from a single mouse. Rather, they encompass unbiased population‐level changes evident across multiple tumor‐bearing animals. Importantly, an overlap of more than 60% of type I IRGs with no residual overdispersion was observed between DEGs enriched in all dormant cells compared to those enriched in hosts from which both dormant and proliferating RM1 cells were isolated, with the highest fold changes predominantly occurring in shared IRGs (Appendix Fig S1H). Moreover, the biological processes enriched in dormant compared to proliferating cells from an isolated host (Appendix Fig S1I) were nearly identical to those enriched across all dormant cells as shown in Fig 1C.
Figure 1. Type I IFN signaling is retained in dormant PCa cells in bone metastases.
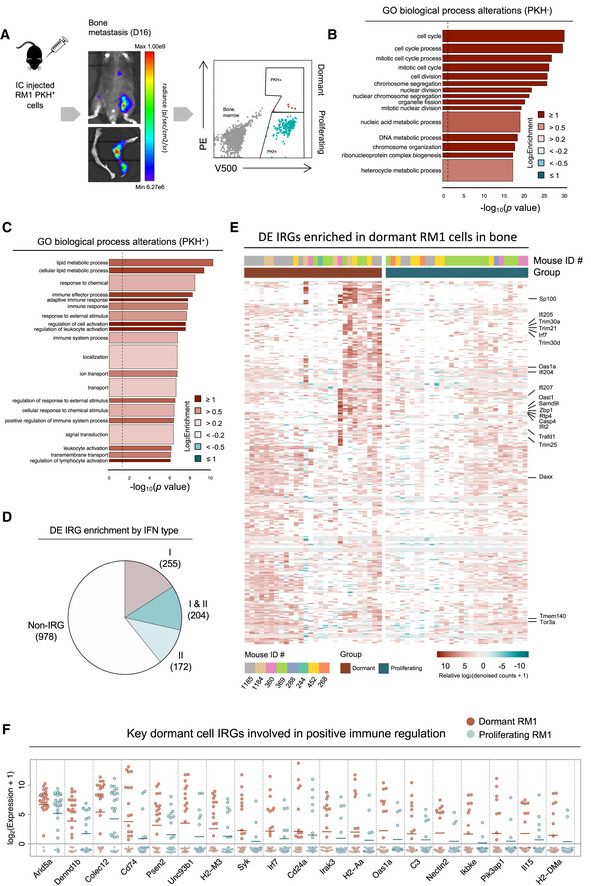
- PKH26 (PE)‐labeled RM1 (V500/eCFP) cells were injected intracardiacally (IC) into C57BL/6 mice and FACS‐isolated from bones with evident tumor burden (eight individual mice across five independent experiments) at ∼ day 16, and individual dormant and proliferating cells were isolated for scRNA‐seq. Representative tumor burden at the whole mouse level and in bone shown by bioluminescence with representative FACS plots of PKH+ (PE/V500) and PKH− (V500) RM1 cells and bone marrow cells (gray).
- goana gene ontology (GO) analysis (limma) of all DE genes enriched in proliferating (PKH−, n = 32) cells compared to dormant cells (PKH+, n = 28). Gene sets appear in order of significance (P‐value) with color representing fold enrichment and bar width indicating the number of genes in each process.
- goana GO analysis (limma) of all DE genes uniquely enriched in PKH+ compared to PKH− cells. Gene sets appear in order of significance (P‐value) with color representing fold enrichment and bar width indicating the number of genes in each process.
- INTERFEROME database classification of differentially expressed (DE) genes retained in dormant cells into predicted type I and/or II IFN‐regulated genes (IRGs).
- Heatmap of relative BASiCS‐derived log2 (denoised counts + 1) of type I IRGs (upregulated > twofold in the INTERFEROME in at least one dataset) that are differentially expressed between all dormant and active cells with no residual overdispersion. Mouse IDs for each sample are indicated, along with type I IRGs that are identified as such in at least 30 INTERFEROME datasets. Genes are displayed if detected in at least 10 samples per population. Samples with zero counts for individual genes are represented in white.
- Dot plot of BASiCS‐derived log2(denoised counts + 1) of key DE IRGs expressed in > 5 dormant cells associated with immune‐activatory processes (among one of the most enriched types of biological pathway determined by GO analyses) present for all single cells ranked by ExpLogFC (Fig 1C). Open circles are zero counts. Bars indicate mean expression.
Source data are available online for this figure.
Figure EV1. Type I IFN signaling is retained in dormant PCa cells in bone metastases.
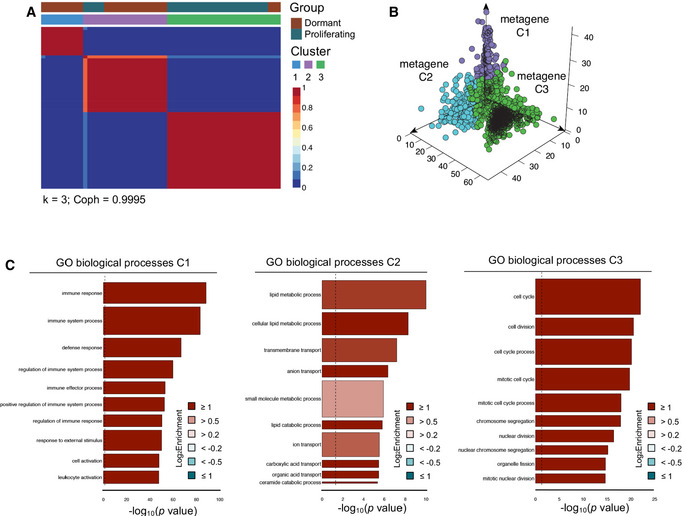
- Consensus matrix of non‐negative matrix factorization analysis of all DEGs expressed between dormant and proliferating cells defines two dominant dormant (C1, C2) and one chiefly proliferating (C3) clusters (k = 3; Coph = 0.9995).
- Distribution of all genes contributing to each of the dominant three cluster metagenes (C1–3).
- goana gene ontology (GO) analysis (limma) showing the top 10 biological processes for all genes contributing to C1, C2, and C3 in order of fold enrichment. Gene sets appear in order of significance (P‐value) with color representing fold enrichment (FE) and bar width indicating the number of genes in each process.
Interestingly, IRGs enriched in our dormant cells included several genes previously linked to dormancy 32 in other bone‐avid cancers (Appendix Fig S1J) that had not been explicitly defined as IRGs in literature, including the aforementioned Bhlhe41 30 along with Gas6 33, reinforcing our isolated PKH+ cells as dormant. While type I IRGs are known to regulate a myriad of biological processes 34, the most significantly enriched genes in dormant tumor cells were those associated with positive regulation of immune cells, lymphocyte activation, and antigen processing and presentation, with critical immune‐activatory genes (shown in Fig 1F), including major histocompatibility complex (MHC) molecules H2‐M3, H2‐DMa, and H2‐Aa. Based on these findings, we postulated that tumor cells may be growth‐restricted in bone by immune‐activatory means. Moreover, we hypothesized that loss of tumor‐intrinsic IFN signaling may release tumor cells from dormancy and promote metastatic outgrowth through loss of IFN‐regulated tumor cell immunogenicity.
Loss of intrinsic type I IFN in proliferating PCa cells occurs in bone
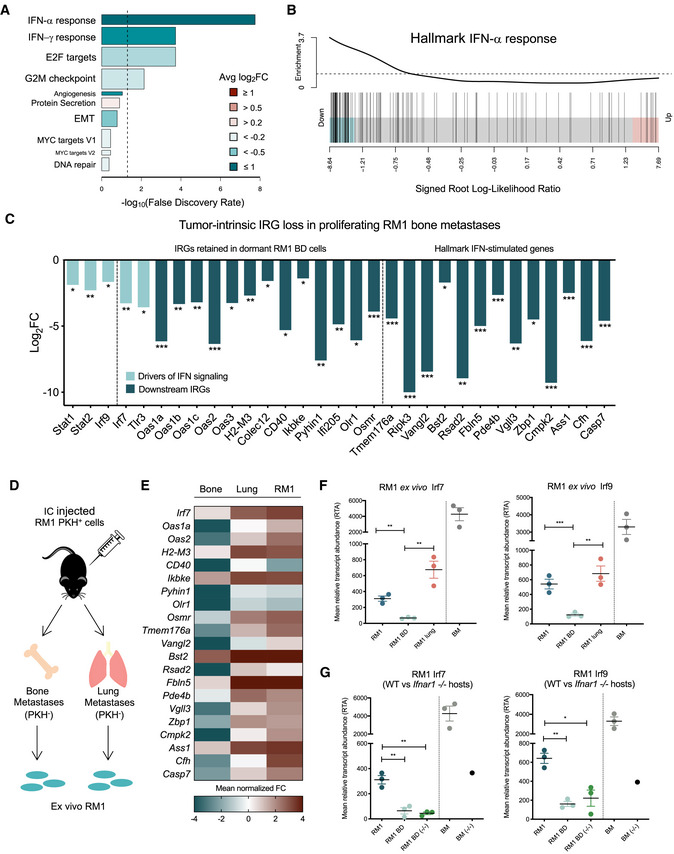
Based on our findings that dormant tumor cells have higher IFN signaling than proliferating PCa cells in bone, we questioned whether IFN signaling was being upregulated in dormant cells or alternatively lost in proliferating cells from the time of inoculation. Comparison of parental RM1 and bulk‐sorted bone‐derived proliferating (PKH−) cells revealed robust and homogenous tumor‐intrinsic suppression of IFN in proliferating tumor cells from bone metastases, with 44% of all downregulated genes defined as IRGs by INTERFEROME allocation (74 IFN‐α/β, 40 IFN‐γ, and 100 type I and II IFN target genes; Appendix Fig S2A). Gene set testing using the Hallmark gene set collection 35 revealed a robust enrichment of type I (IFN‐α) response genes that were suppressed in bone metastases (Figs 2A and B). Pathway analysis also revealed that the most suppressed biological pathways in proliferating tumor cells were those associated with IFN responsiveness and immune cell recruitment (Appendix Fig S2B), with enrichment of processes associated with positive immune regulation (Appendix Fig S2C). Among the DEGs implicated in IFN modulation were direct mediators of IFN‐α/β production, including Stat1, Stat2, and Irf9 (all critical components of the IFN‐stimulated gene factor 3 complex, ISGF3), that directly regulate Irf7, and several key IRGs retained in single dormant RM1 cells (shown in Fig 1) and multiple classic type I IFN targets 36 (Fig 2C). Further transcriptional analysis of proliferating bone‐derived RM1 (RM1 BD) cells compared to those derived from lung metastasis (Fig 2D) revealed that IRG loss was specific to bone (Fig 2E). As with comparisons between dormant and proliferating cells in bone, genes suppressed in bone‐derived tumor cells compared to lung were enriched in IFN response genes (Appendix Fig S2D). Orthogonal validation of preliminary findings confirmed that tumor‐intrinsic IFN loss was more profound in bone metastases, as evidenced through quantitative real‐time–PCR (qRT–PCR) analysis of Irf7 and Irf9 (both robust representative markers of IFN pathway activity 37) expression in RM1 BD cells compared to parental cells and RM1 cells from lung metastases derived from independent animals (Fig 2F). Interestingly, Irf7 and Irf9 expression in naïve BM was revealed to be high, reflecting public transcriptomic datasets 38, which is potentially due to the presence of megakaryocytes that express high Irf7 39, indicating bone‐specific tumor cell loss did not result from low paracrine IFN signaling in surrounding stroma. Pathway suppression was also independent of host responsiveness to type I IFN, evidenced through tumor‐intrinsic Irf7 and Irf9 loss in cells derived from bone metastases in mice deficient in the IFN‐α receptor 1 (Ifnar1 −/−; Fig 2G). As such, tumor‐intrinsic IFN suppression in bone may be induced through niche‐specific mechanisms that subsequently facilitate unchecked metastatic progression.
Figure 2. Intrinsic type I IFN pathways are specifically suppressed in proliferating PCa cells from bone.
-
A–CBulk RNA‐seq analysis of DE genes (DEG) significantly suppressed (FDR < 0.05; generalized linear model extraction (GLM) by edgeR) in proliferating RM1 bone‐derived (BD) cells compared to parental RM1 and lung metastases. Tumor‐intrinsic type I IFN signaling is suppressed in proliferating bone lesions (n = 1) compared to parental RM1 (n = 2) cells as shown by (A) camera analysis of Hallmark gene set responses associated with bone metastasis (bar length is the –log10FDR, false discovery rate). Color indicates mean log2fold change (FC) of all genes in gene set. Bar width is relative gene set size. Dashed line shows FDR = 0.05; (B) barcode plot showing enrichment of the Hallmark IFN‐α response gene set in genes suppressed in bone metastases, using the signed log‐likelihood ratio statistic (EdgeR). Bars indicate value of the statistic for each gene in the gene set; (C) log2FC values of key downregulated IRGs, with genes retained in dormant cells and classical downstream IFN signaling targets indicated. Altered IRGs directly involved in IFN‐α/β production (light blue) compared to downstream IFN targets (dark blue) are segregated. P‐values represented as * < 0.05, ** < .005, and *** < 0.0005; GLM by edgeR.
-
DSchematic of proliferating RM1 cell isolation from bone and lung metastases following IC injection for preliminary RNA‐seq and orthogonal validation by qRT–PCR.
-
EHeatmap of mean normalized voom expression of IRGs suppressed in RM1 bone metastases (n = 1) compared to lung metastases (n = 1) and parental cells (n = 2).
-
FqRT–PCR validation of Irf7 and Irf9 downregulation in RM1 cells from bone metastases (RMI BD) compared to parental RM1 cells, lung metastases (RM1 lung), and naïve bone marrow (BM) (n = 3 mice per group). P‐values represented as ** < 0.005, and *** < 0.0005 (Student's t‐test).
-
GqRT–PCR of Irf7 and Irf9 downregulation in parental RM1 cells and RM1 cells from bone metastases (RM1 BD) in WT and Ifnar1‐deficient (−/−) mice, with naïve BM from WT and Ifnar −/− animals for reference. (n = 3 mice per group, n = 1 for Ifnar −/− BM control). P‐values were represented as * < 0.05 and ** < 0.005 (Student's t‐test).
Tumor‐intrinsic type I IFN suppression is inducible and can be reversed and blocked through epigenetic targeting using the HDACi MS275
In order to assess the feasibility of targeting tumor‐intrinsic type I IFN, we needed to determine whether the loss we observed in bone metastases was indeed reversible. First, the stability of IFN suppression in proliferating RM1 cells derived from bone metastases in three independent animals was assessed by serial passage ex vivo. We revealed that with restricted passage (less than ~ 8) in culture, IFN loss was maintained in RM1 cells derived from bone, which we henceforth refer to as RM1 BD Irf− (Figs 3A and EV2A). Notably, one RM1 BD line showed initial IFN loss during early passage (EP), yet at late passage (LP; > 13), reverted back to parental IFN expression levels, which we henceforth label RM1 BD REV. As such, RM1 BD Irf− cells served as biological knockdowns for further experimentation under restricted culture. ELISA confirmed that the differential expression of Irf7 and Irf9 between parental RM1 cells and RM1 BD Irf− and RM1 BD REV ex vivo cell lines directly correlated with their capacity to produce IFN‐α when stimulated with the TLR3 agonist, poly I:C 40 (Fig 3B). Notably, in vitro poly I:C treatment also revealed that RM1 BD Irf− cells were unresponsive to IFN pathway activation by this known systemic IFN‐inducing agent.
Figure 3. Loss of tumor‐intrinsic type I IFN is inducible by bone marrow cells and is reversed by HDACi.
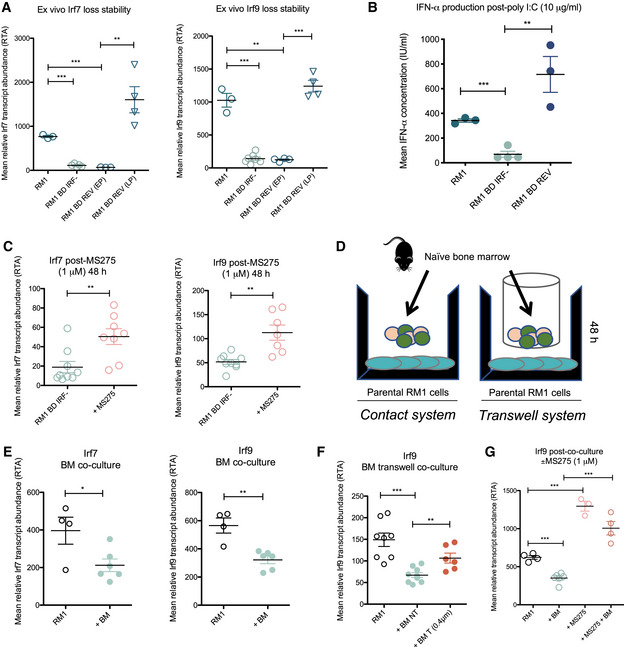
- Stability of Irf7 and Irf9 mRNA suppression by qRT–PCR in ex vivo bone‐derived cells (RM1 BD Irf−, n = 7) in culture compared to a bone‐derived line that showed initial loss during early passage (RM1 BD REV (EP), n = 3), then reverted to parental expression levels at late passage (RM1 BD REV (LP), n = 4) compared to parental RM1 (n = 3).
- ELISA of IFN‐α production by RM1 parental (n = 3), RM1 bone‐derived Irf‐low (RM1 BD Irf−, n = 4), and RM1 BD REV (n = 3) cells subsequent to poly I:C stimulation.
- qRT–PCR analysis of Irf7 and Irf9 expression in RM1 BD Irf− cells ± 48 h treatment with MS275 (1 μM) (n = 7–9).
- Schematic of contact and transwell co‐culture systems.
- qRT–PCR analysis of Irf7 and Irf9 expression in parental RM1 cells (n = 4) 48 h post‐contact culture with naïve BM (n = 6).
- qRT–PCR analysis of Irf9 expression in parental RM1 cells ± 48 h co‐culture with naïve BM under contact (non‐transwell; NT) and transwell (0.4‐μm filters that prevent cell contact) conditions (n = 6–8 per condition).
- qRT–PCR analysis of Irf9 expression in parental RM1 cells ± 48 h contact co‐culture with naïve BM ± MS275 (1 μM) (n = 3–6 per condition).
Figure EV2. Loss of tumor‐intrinsic type I IFN is inducible by bone marrow cells and is reversed through HDACi modulation.
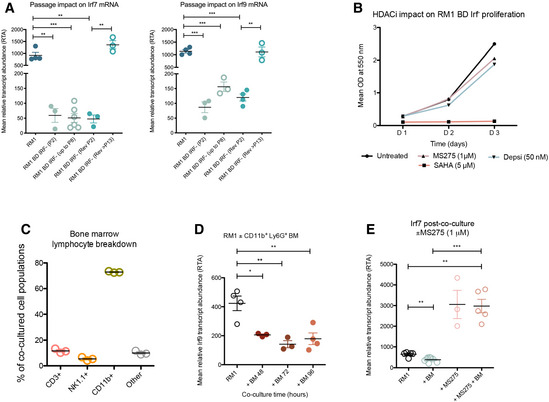
- Assessment of tumor‐intrinsic IFN suppression stability over passage (P; number indicated) in culture by qRT–PCR analysis of mean Irf7 and Irf9 mRNA expression in bone‐derived RM1 Irf‐low (RM1 BD Irf−) cells and a reverted (REV) bone‐derived cell line compared to RM1 parental cells. Values are means ± SEM of three independent experiments.
- HDACi impact on RM1 BD Irf− proliferation over time by SRB assay. Mean OD at 550 nm (n = 3).
- Flow cytometry characterization of bone marrow lymphocyte (%) populations (n = 3).
- qRT–PCR of Irf9 expression in parental RM1 cells 48, 72, and 96 h post‐contact co‐culture with FACS‐isolated naïve CD11b+ Ly6G+ BM cells (n = 3 mice per time point).
- qRT–PCR of Irf7 expression in RM1 parental cells ± co‐culture with naïve BM ± 48 h treatment with MS275 (n = 3–6). P‐values represented as * < 0.05, ** < 0.005, and *** < 0.0005 (Student's t‐test). All error bars ± SEM.
Given that histone acetylation is a well‐known and integral component of IRG regulation 41, 42, 43 and that IFN pathway loss was moderately stable in tumor cells outside of the bone TME, this led us to question whether IFN suppression could be relieved through chromatin modulation. Interrogation of in‐house microarray data revealed that a panel of histone deacetylase inhibitors (HDACi) could induce expression of type I IFN pathway mediators in HCT116 colon cancer cells (Appendix Fig S3A). As such, we investigated the capacity of various pan‐selective (i.e., SAHA [vorinostat]) and class‐selective (i.e., depsipeptide, MS275) HDACi to induce intrinsic IFN signaling in RM1 BD Irf− cells (Fig 3C; Appendix Fig S3B and C). These analyses revealed that the class I‐specific HDACi MS275 (entinostat) robustly increased tumor‐intrinsic Irf7 and Irf9 in RM1 BD Irf− at a concentration that did not impact tumor proliferation (Fig EV2B), eliminating HDACi‐induced growth inhibition as a confounding means of tumor regression. We then asked whether tumor‐intrinsic IFN suppression we observed in bone could be mimicked in vitro and whether MS275 would be sufficient to prevent this loss from occurring. While in vivo systems yield important information about the metastatic process, exploration of live stromal interactions in bone is notoriously difficult to adequately model and focally manipulate in mice. As such, an ex vivo co‐culture system was devised (Fig 3D) to assess the inducibility, timing, and potential epigenetic influence over tumor‐intrinsic type I IFN signaling downregulation. Interestingly, co‐culture of RM1 parental with naïve BM cells revealed that IFN loss could be induced in tumor cells within 48 h of BM contact (Fig 3E) and that this rapid loss is BM contact‐dependent, as demonstrated by retained tumor cell expression under non‐contact conditions (Fig 3F). Moreover, we show that the ubiquitous bone‐resident myeloid population (Fig EV2C) involved in IFN loss may be CD11b+ Ly6G+ cells, which are included in the granulocytic myeloid‐derived suppressor cell (MDSC) subset 44 and which were able to suppress key members of the IFN pathway in RM1 cells for up to 96 h (Fig EV2D). Interestingly, CD11b+ Ly6G+ cells have been previously linked to metastatic PCa progression 45. Moreover, such cells have been associated with acetylation events in the TME (reviewed in Ref. 46) that promote tumor cell expansion and immune repression. Most importantly, however, we reveal that addition of MS275 to the bone co‐culture system blocked BM‐induced IFN pathway loss in parental RM1 cells and further enhanced tumor‐intrinsic IFN signaling (Figs 3G and EV2E). Collectively, these results suggest that epigenetic agents may reverse tumor‐intrinsic IFN suppression in bone and thus prevent metastatic outgrowth by upregulating tumor‐intrinsic immune signals.
Tumor‐intrinsic IFN signaling regulates metastatic potential, dormancy status, and bone remodeling processes to prolong bone metastasis‐free survival through immune activation
We next asked how differential tumor‐intrinsic type I IFN signaling impacts tumor progression in vivo, and whether stable restoration of IFN signaling to bypass possible epigenetic suppression in tumor cells can indeed block bone metastasis. Firstly, we compared the metastatic potential of three cell lines (RM1 parental, RM1 BD Irf−, and RM1 BD REV), each with different IFN production capacity yet no difference in in vitro growth (Appendix Fig S4A). Subsequent to IC injection, mice were individually monitored for the first signs of bone metastasis, most commonly hindlimb paralysis. Survival analysis revealed that mice bearing RM1 BD tumors with low Irf expression (RM1 BD Irf−) succumbed to fatal bone metastasis faster (median survival day 13) than mice bearing tumors in which IFN signaling was intact at the time of injection, regardless of whether they were previously derived from bone or not (RM1 BD REV and RM1 parental, respectively; median survival > 24 days; Fig 4A).
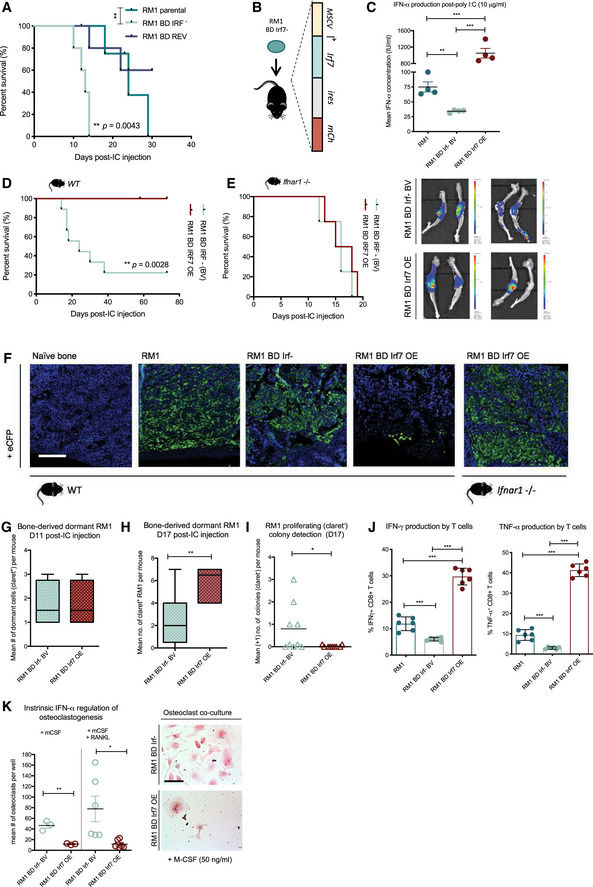
Figure 4. Molecular restoration of tumor‐intrinsic type I IFN suppression inhibits bone‐metastatic outgrowth and modulates osteoimmunity.
-
ABone metastasis‐free survival in WT C57BL/6 mice harboring RM1 parental, RM1 BD Irf−, and RM1 BD REV (LP) tumors (n = 5 per group; **P = 0.0043 by log‐rank (Mantel–Cox) test).
-
BSchematic of enforced Irf7 expression in RM1 BD Irf− cells under exogenous promoter control.
-
CELISA of IFN‐α production by RM1 parental, RM1 BD Irf− base vector (BV), and RM1 BD Irf7 overexpressing (OE) with 24‐h poly I:C stimulation (n = 4).
-
DBone metastasis‐free survival in WT C57BL/6 mice subsequent to IC inoculation of RM1 BD Irf− BV (n = 9) and RM1 BD Irf7 OE (n = 7) cells (**P = 0.0028 by log‐rank (Mantel–Cox) test).
-
EBone metastasis‐free survival in Ifnar1 −/− mice harboring RM1 BD Irf− BV and RM1 BD Irf7 OE tumors (n = 4 per group) and representative bioluminescent imaging of tumor burden in leg bones (also shown in Appendix Fig S4F).
-
FImmunohistochemical (IHC) staining for cerulean (anti‐eCFP; green) on naïve WT bone, RM1 parental, RM1 BD Irf− BV, and RM1 BD Irf7 OE tumor‐bearing bones derived from WT animals; and RM1 BD Irf7 OE tumor‐bearing bones derived from Ifnar1 −/− animals at survival assay endpoints. Blue represents DAPI nuclear staining. Scale bar, 100 μm.
-
G–IFACS analysis of dormant (claret+) RM1 BD Irf− and RM1 BD Irf7 OE cells from bone at (G) day 11 post‐IC injection (n = 4 per group) and (H) day 17 (n = 4–6 mice group/time point) with (I) D17 active (claret−) colony quantitation (mean +1) determined by multiphoton imaging and IMARIS interrogation (n = 9 bones from three mice per condition). Median shown. Upper and low box hinges denote first and third quartiles. Whiskers mark value limits.
-
JFACS analysis of IFN‐γ+ and TNF‐α+ CD8+ T cells (%) post‐ICS induction of T‐cell (spleen derived from tumor‐bearing mice) activation by RM1 parental, RM1 BD Irf− BV, and RM1 BD Irf7 OE cells (n = 6 per condition/group).
-
KMean quantitation of TRAP‐stained osteoclasts differentiated with M‐CSF ± RANKL in co‐culture with RM1 BD Irf− BV and RM1 BD Irf7 OE cells with representative wells shown (n = 3 per condition M‐CSF; n = 6 per condition M‐CSF + RANKL; also shown in Appendix Fig S4J). Scale bar represents 100 μm.
Constitutive IFN signaling in RM1 BD Irf− cells was achieved through the overexpression of Irf7 in cells henceforth labeled as RM1 BD Irf7 OE (Fig 4B). A matched base vector control line (RM1 BD Irf− BV) was also generated in order to confirm that tumor‐intrinsic IFN loss was directly responsible for the acceleration of bone metastasis in animals bearing IFN‐suppressed tumors. Upregulation of Irf7, Irf9, and downstream IFN target Oas2 was confirmed in RM1 BD Irf7 OE cells at the transcript level and enhanced IFN‐α production verified by ELISA, with both lines demonstrating equal proliferative potential (Fig 4C; Appendix Fig S4B and C). Survival analysis post‐IC injection of RM1 BD Irf7 OE and Irf− BV cells confirmed that molecular restoration of tumor‐intrinsic type I IFN signaling in aggressive bone‐derived cells confers long‐term bone metastasis‐free survival (Fig 4D; Appendix Fig S4D and E). To further confirm that tumor cell IFN signaling to host cells was a critical component of metastasis suppression in RM1 BD Irf7 OE tumor‐bearing mice, bone metastasis‐free survival was assessed in animals unresponsive to type I IFN (Ifnar1 −/−). Importantly, no difference in survival was observed in Ifnar1 −/− hosts (Fig 4E; Appendix Fig S4F) with tumor burden in bone across different models shown by immunohistochemical detection of cerulean (eCFP; Fig 4F).
Additionally, we show that outgrowth from dormancy in bone is significantly impaired in mice bearing RM1 BD Irf7 OE tumors compared to mice inoculated with RM1 BD Irf− cells. Following IC injection of claret‐labeled RM cells (Appendix Fig S4G) and subsequent analysis by flow cytometry, no difference was observed at an early time point (D11) between dormant RM1 BD Irf− and Irf7 OE populations in bone (Fig 4G). However, by day 17 post‐IC injection, more Irf7 OE dormant cells were detected in bone by both FACS (Fig 4H) and multiphoton imaging (Appendix Fig S4H), along with reduced colony outgrowth (Fig 4I), indicating that high tumor cell expression of Irf7 restricts escape from dormancy in bone. Furthermore, we identified two key IFN‐regulated tumor‐driven osteoimmune processes previously linked to bone‐metastatic progression that were dramatically different between RM1 BD Irf− and RM1 BD Irf7 OE cells. Firstly, to support the requirement of host cell interactions with IFN‐producing tumor cells, we confirmed that RM1 BD Irf7 OE cells were able to enhance tumor immunogenicity to provoke a robust, functional T‐cell response ex vivo (Fig 4J; Appendix Fig S4I). Secondly, RM1 BD Irf7 OE showed impaired ability to induce osteoclast maturation, required for bone resorption, compared to Irf− BV cells, which induced osteoclastogenesis even in the absence of RANKL (Fig 4K; Appendix Fig S4J). Moreover, enhanced osteoclast activity in the endosteal region and trabecular marrow of RM1 BD Irf− tumor‐bearing bones was revealed (Fig EV3). Taken together, these results suggest that tumor‐intrinsic type I IFN plays a crucial role in tumor cell progression, immune mediation, and destabilization of bone‐homeostatic mechanisms that fuel tumor outgrowth and may determine dormant tumor cell fate.
Figure EV3. Impact of differential tumor‐intrinsic IFN signaling on metastasis, dormancy and bone remodeling.
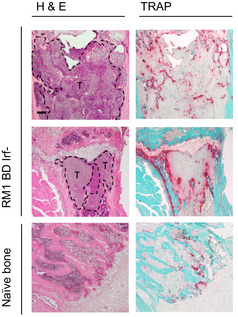
Representative H&E‐ and TRAP‐stained bone sections from naïve and RM1 BD Irf− tumor‐bearing animals. T indicates tumor regions. Scale bar, 200 μm.
HDACi induction of tumor cell type I IFN signaling blocks metastasis and enhances the efficacy of systemic immune‐based treatments
While molecular restoration of tumor‐intrinsic IFN signaling was sufficient to alter bone‐metastatic progression, it remained to be shown whether the same result could be achieved through therapeutic modulation. Following the previously unreported observation that poly I:C directly activates lymphocytes in bone (Appendix Fig S5A) in addition to standard peripheral measures, we examined whether systemic induction of type I IFN alone was sufficient to block bone metastasis. To test this, mice were inoculated by IC injection with either RM1 parental or RM1 BD Irf− cells and treated with poly I:C or saline from day 4 onward. As a single agent, poly I:C effectively decreased bone metastasis in mice bearing RM1 parental tumors, which was associated with enhanced immune activation and T‐cell cytotoxic function (Appendix Fig S5B–D). Comparatively, in mice bearing RM1 BD Irf‐ tumors, poly I:C failed to prevent bone metastasis at the day 15 endpoint. This was potentially due to TLR3 suppression in bone‐derived tumor cells with low IFN signaling (evidenced in Fig 2C), highlighting the requirement of tumor‐intrinsic stimulation to promote an early IFN‐driven antitumor immune response in addition to systemic immune‐activating agents.
Notably, pre‐treatment (48 h) of BD Irf− cells with the clinically relevant HDACi MS275 enhanced IFN‐α production by poly I:C in vitro (Fig 5A), suggesting that HDACi alleviation of tumor‐intrinsic IFN suppression may underpin optimal responsiveness to systemic immune‐based therapeutics. Furthermore, MS275 as a single‐agent upregulated surface MHC class I molecule H2‐Kb within 48 h, which was further induced through combination of MS275 and poly I:C (Fig 5B). The robust increase in tumor immunogenicity markers was again associated with enhanced CD8+ T‐cell stimulation (Fig 5C and D), with T‐cell responsiveness further augmented by the addition of poly I:C. Excitingly, the benefit of tumor‐intrinsic type I IFN induction through HDACi intervention was verified in vivo, where MS275 alone prolonged bone metastasis‐free survival in mice inoculated with aggressive RM1 BD Irf− cells (median survival extended from D14 to D24; Fig 5E). Furthermore, the inhibitory effect of MS275 on bone metastasis was enhanced in combination with poly I:C, whereby lethal bone progression was completely blocked (Fig 5E and F, and Appendix Fig S5E). Thus, tumor‐intrinsic IFN status may be a critical determinant of immunotherapeutic success in CRPC, whereby in the absence of intact or restored tumor cell IFN signaling, the tumor cells may be rendered invisible despite modulation of immune cell populations.
Figure 5. Bone metastasis is inhibited by therapeutic induction of cell‐intrinsic (HDACi) and systemic type I IFN .
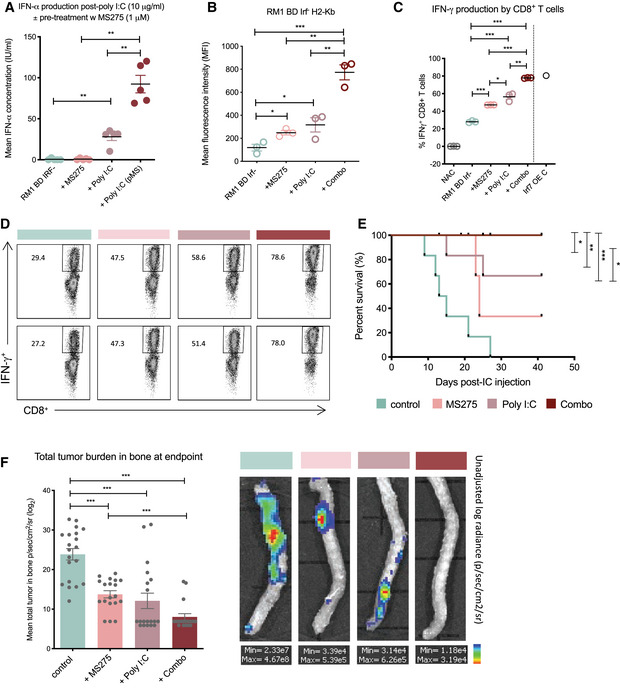
-
AELISA of IFN‐α production by RM1 BD Irf− cells ± single‐agent MS275 and poly I:C treatment or 48 h pre‐treatment with MS275 (pMS) prior to poly I:C stimulation (n = 6).
-
BMean fluorescence intensity of H2‐Kb staining on RM1 BD Irf− cells by FACS ± MS275, poly I:C, or combination treatment (n = 3).
-
C, DFACS analysis of IFN‐γ+ CD8+ T cells (%) post‐ICS induction of T‐cell (spleen derived from RM1 BD Irf− tumor‐bearing mice) activation upon re‐stimulation with RM1 cells and MS275, poly I:C, or combination treatment, with NAC (no antigen‐presenting cells) and RM1 BD Irf7 OE controls (n = 3; Irf7 OE control (C), n = 1) with (D) representative FACS plots shown.
-
EBone metastasis‐free survival in WT C57BL/6 mice harboring RM1 BD Irf− cells ± MS275, poly I:C, or combination treatment (n = 6 per group; log‐rank (Mantel–Cox) test).
-
FMean total tumor burden in bone (femurs, tibias, spine, and humerus) at endpoint by bioluminescent intensity score (log2 p/s/cm2/sr) with values < 4.0 × 104 representing zero burden (n = 24 bones per group; six mice per group) and representative bioluminescent imaging of tumor burden in spine (all shown in Appendix Fig S5E).
Tumor‐intrinsic and systemic induction of IFN signaling is required for long‐term antitumor immunity against bone‐metastatic outgrowth
The efficacy of MS275 to enhance bone metastasis‐free survival likely resulted from immune engagement. Given that our in vitro experiments revealed the requirement of at least 48 h for MS275 to induce demonstrable changes in tumor‐intrinsic type I IFN signaling, it is not surprising that systemic immune activation within the first 24 h of treatment, as evidenced through lymphocyte expression of activatory markers CD69 and NKG2D, was largely restricted to poly I:C and combination groups (Fig 6A). However, analysis of peripheral blood lymphocytes at later time points suggests that the strength of MS275 comes from its ability to increase peripheral CD8+ and CD4+ T‐cell populations and provoke a robust effector memory response without induction of immune‐suppressive Foxp3+ CD4+ regulatory T cells (Tregs)—all critical for sustained antitumor targeting (Fig 6B; Appendix Fig S6). This was demonstrated following CD8+ T‐cell harvest from metastasis‐free combination‐treated mice at endpoint (Fig 6C), in which a tumor‐specific, functional T‐cell response was evidenced by increased IFN‐γ production that directly corresponded to the degree of therapeutic tumor‐intrinsic type I IFN induction (Fig 6D). Collectively, these findings further confirm the importance of tumor‐intrinsic type I IFN modulation of tumor immunogenicity and long‐term antitumor immune activity, which modern immune‐based treatments have failed to engender in the metastatic treatment setting.
Figure 6. HDACi and systemic IFN induction provides long‐term immune protection from bone‐metastatic colonization and outgrowth.
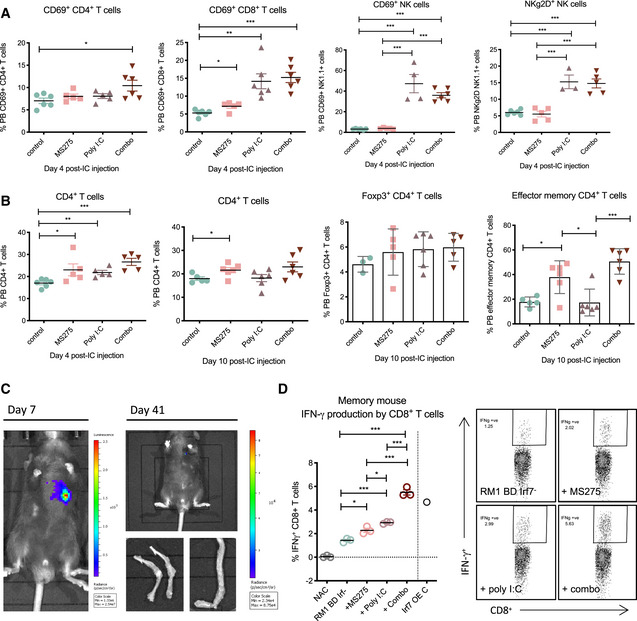
-
A, BFACS analysis of (A) peripheral blood (PB) T and NK lymphocyte representation and activation status and (B) FoxP3+ CD4+ and CD4+ effector memory T‐cell status at days 4 and 10 post‐IC tumor cell inoculation across treatment settings (n = 4–6 per group).
-
C, DBioluminescent imaging (C) of a combination group mouse with tumor clearance from days 7 to 41 (full experimental cohort shown in Appendix Fig S5E) post‐IC inoculation with (D) associated specific CD8+ memory T‐cell response. This is represented by FACS analysis of IFN‐γ+ CD8+ T cells (%) post‐ICS induction of T‐cell (spleen derived from tumor‐bearing mice) activation upon re‐stimulation with RM1 BD Irf− cells and MS275, poly I:C, or combination treatments in vitro, with NAC and RM1 BD Irf7 OE controls (n = 3; Irf7 OE control (C), n = 1). Representative FACS plots shown on right.
Loss of intrinsic type I IFN signaling and tumor immunogenicity is associated with bone metastasis in PCa patients
The unequivocal link between IFN signaling and bone metastasis identified in the murine CRPC model led us to investigate the importance of type I IFN alterations in PCa patients, given the current lack of patient responses to conventional and immune‐targeting therapies in bone‐metastatic CRPC. To address this, we compared the transcriptional profiles of bone metastases (n = 9) and matched primary tumors (n = 12) from a cohort of PCa patients. Hierarchical clustering revealed high similarity in gene expression patterns within both the primary tumor and bone metastases groups. Furthermore, overall lymphocyte composition was comparable between both sites (Fig 7A) as was epithelial content, with mean tumor cell purity > 85% in primary tumor metastasis and bone metastasis samples (Fig 7B). Consistent with the observations made in the RM1 model, genes associated with immune responses (TNF‐α and IFN responses; Appendix Fig S7A and Fig 7C.) were the most significantly downregulated in bone compared to primary tumors, with 39% of DEGs defined as IRGs (Appendix Fig S7B), from which the type I IRG subset is represented (Fig 7D).
Figure 7. IFN signaling is decreased in bone‐metastatic PCa.
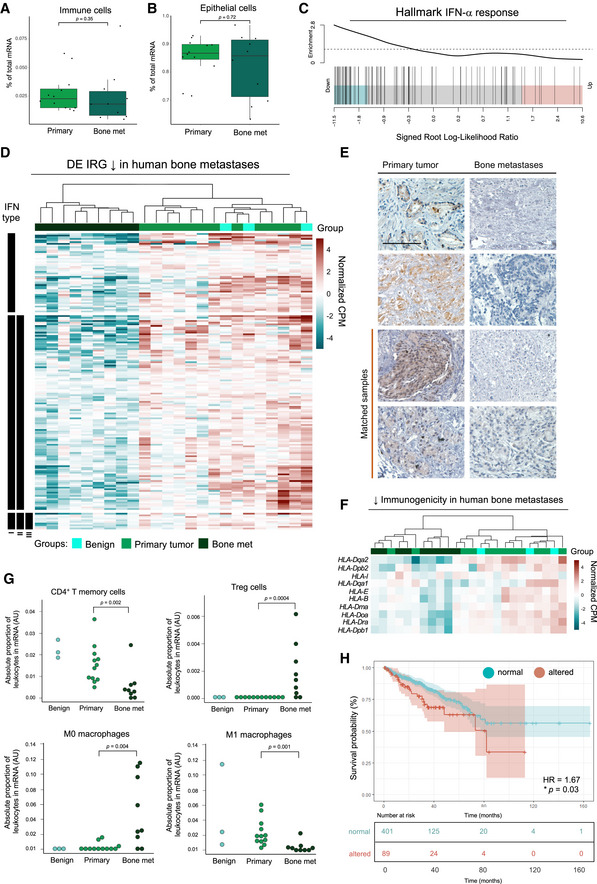
-
A–CCompositional analysis of RNA‐seq data (FDR < 0.05; GLM by edgeR) from bone metastases (n = 9) and primary tumors (n = 12) revealed (A) immune cells and (B) tumor cells were relative (% of total) across all samples, with a mean tumor purity > 85% as determined by ARMET. Median shown. Upper and low box hinges denote first and third quartiles. Upper and lower whiskers mark the values ± 1.5IQR from the hinge. Data points beyond the whiskers are shown individually. P‐values not significant by Wilcoxon rank sum test (R). Hallmark gene set responses associated with bone metastasis by camera analysis show (C) enrichment of type I (IFN‐α) IFN responses in suppressed genes (barcode plot).
-
DHeatmap of type I IFN DE IRGs (normalized log2CPM; counts per million) significantly suppressed in bone metastases (n = 9) compared to primary (n = 12) and benign (n = 3) tumors, grouped by type I IFN‐specific, and type II and III IFN co‐regulation status. Scale truncated at ± 5 relative log2CPM. GLM by edgeR.
-
EIHC for IRF9 expression in primary prostate tumors and bone metastases with matched samples indicated. IRF9 indicated by brown (DAB) staining. Scale bar represents 100 μm. Full‐face slides shown in Appendix Fig S7C.
-
FHeatmap of HLA genes (normalized log2CPM) suppressed (P < 0.05; FDR < 0.05) in bone metastases (n = 9), primary tumors (n = 12), and benign tumors (n = 3). Scale truncated at ± 5 relative log2CPM. GLM by edgeR.
-
GCIBERSORT analysis of absolute leukocyte proportions (arbitrary units, AU) in mRNA samples of bone metastases (n = 9), primary tumors (n = 12), and benign tumors (n = 3). P‐values by Mann–Whitney U‐test.
-
HKaplan–Meier curve of human prostate (n = 499 from 498 TCGA samples) biochemical recurrence based on primary tumor alterations in a core 8‐IRG signature (Appendix Fig S7D). Patient groups stratified by mRNA z‐score (RNA‐seq V2 RSEM) ± 1.5. Hazard ratio 1.674 (95% confidence interval (CI) 2.707 to 1.035); *P = 0.03 by log‐rank test.
To confirm IFN pathway loss at the protein level and that loss was indeed tumor‐intrinsic, we assessed IRF9 expression in both matched and unmatched primary tumors and bone metastases from CRPC patients, given that we have previously identified IRF9 as a robust marker of active intratumoral type I IFN signaling 37. Immunohistochemical analysis confirmed that pathway loss was localized to tumor cells in bone and occurred despite the observed intratumoral heterogeneity of IRF9 between primary cancer tissues (Fig 7E and Appendix Fig S7C). Loss of tumor‐intrinsic type I IFN in bone was associated with widespread downregulation of tumor immunogenicity, evidenced through suppression of several HLA genes (Fig 7F), which further validated murine findings. Furthermore, IFN loss in skeletal lesions compared to both benign and malignant primary tumors corresponded to leukocyte compositional alterations (Fig EV4A), in which robust increases in suppressive immune cells, such as Tregs and myeloid cells, and decreases in immune cells linked to antitumor immunity were observed (Fig 7G).
Figure EV4. IFN signaling is decreased in bone‐metastatic PCa.
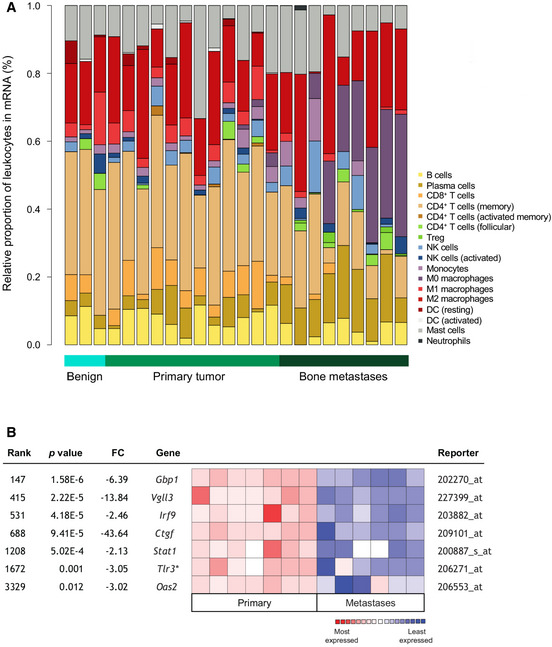
- Relative proportions (%) of leukocytes in mRNA samples from patient bone metastases (n = 9) compared to primary tumors (n = 12) and benign tumors (n = 3) by CIBERSORT. Cell subsets indicated on graph.
- Oncomine interrogation of the Varambally Prostate dataset using an 8‐IRG core signature (data unavailable for Tslp; NS for Oas1 (FC = ‐1.24; P = 0.214; Tlr3 additional)) in primary (n = 7) and metastatic (n = 6) tissues derived from Human Genome U133 Plus 2.0 Array analysis (z‐score‐normalized; log2 median‐centered intensity). Fold change (FC) values shown with corresponding P‐values (log‐rank test).
Following validation of type I IFN suppression in bone metastases and the evident heterogeneity across primary tumors despite global IFN pathway loss, we asked whether reduced signaling in primary PCa could actually predict risk of PCa progression and whether IFN‐associated progression was site‐specific in CRPC. Based on significant gene alterations common to both murine and human bone metastases, we developed an eight‐gene core signature of bone‐suppressed IRGs, including Irf9, Oas2, Gbp1, and Vgll3 (Appendix Fig S7D). This signature was used to stratify 499 PCa patients with primary tumor expression data available through The Cancer Genome Atlas (TCGA), which when probed revealed that patients with altered type I IFN expression had an increased risk of metastasis (Fig 7H). These results validate RM1 model findings that IFN pathway loss in tumor cells accelerates PCa progression and are particularly salient, given the multifocal nature of PCa 47, 48, with high heterogeneity, a key contributing factor to the current lack of biomarkers through which to effectively predict metastasis 49, 50, 51. Indeed, we provide evidence that tumor‐intrinsic Irf9 expression within high‐risk primary PCa (Gleason > 7) is indeed variable at both the protein level and transcriptional level (Appendix Fig S7E). However, we also demonstrate that low expression is largely homogenous across patients harboring bone metastases and could therefore serve as a marker of patients likely to progress, supporting further investigation of IRF9 as a prognostic marker in CRPC. Additionally, Oncomine 52 interrogation (Fig EV4B) of the Varambally prostate cancer microarray dataset 53, comprised of individual and pooled primary (n = 7) and hormone‐refractory metastatic (n = 6) tissue samples including bone, liver, and soft‐tissue metastases, revealed that the loss of bone‐suppressed IRGs is potentially shared across different metastatic sites. While this expands the clinical relevance of IFN pathway alterations in CRPC, it is widely acknowledged that visceral metastases are mostly accompanied by the presence of bone metastases in CRPC 54. As such, we cannot rule out the possibility that visceral metastases were seeded from bone lesions, as has been reported 55, 56.
Together, these findings suggest that early IFN pathway alterations at the primary site may promote successful bone colonization by tumor cells and assist in accelerated DTC expansion. Our data reiterate the critical function of type I IFN loss on PCa outgrowth to and within bone. Furthermore, we highlight the potential application of immunotherapeutic strategies that overcome intratumoral type I IFN suppression to boost tumor visibility and subsequently block bone‐metastatic progression once IFN loss has occurred, reflective of late‐stage CRPC.
Discussion
Disseminated tumor cells in bone are a common occurrence in PCa. While asymptomatic in many cases, their eventual detection and subsequent outgrowth represent an incurable and ultimately fatal stage of castrate‐resistant disease. Here, we identified tumor‐inherent IFNs as potent suppressors of bone‐metastatic outgrowth. Type I IFNs have been implicated in the regulation of processes as disparate as immune signaling to angiogenesis, cell adhesion, and migration 57. As such, the consequences of suppressed tumor‐intrinsic IFN signaling in the TME are potentially vast. Yet, the role of type I IFN in addition to general immunomodulatory processes in bone‐metastatic cancers remains gravely underexplored. Here, we show that tumor‐inherent IFN signaling may dictate the temporal development of bone metastases through osteoimmune modulation, from dormancy through to the onset of fatal skeletal events. Moreover, we demonstrate that reversal of this loss can be therapeutically induced to prevent bone‐metastatic progression in a murine model of bone‐metastatic CRPC, revealing a potential new approach to treat advanced CRPC patients, many of whom may harbor bone metastases with suppressed tumor‐intrinsic type I IFN.
The impact of tumor‐inherent signaling on PCa outgrowth in the bone TME remains poorly described, particularly in immune‐competent systems. Using the only reproducible bone‐metastatic syngeneic PCa murine line (reviewed in Ref. 21, 22), which is an unavoidable limitation of the current study, we demonstrate that loss of tumor‐intrinsic IFN signaling not only is sufficient to accelerate metastasis to bone, but also, importantly, permits tumor outgrowth. Dormant cell retention of critical IRGs, including Irf7, along with other immunoreactive markers, such as several MHC molecules 58, supports existing theories that immune mechanisms may indeed control PCa progression 59, 60. Additionally, dormant cell retention of IRGs linked to chemoresistance and quiescent cell endurance (Gas6 33) along with inhibition of apoptosis (Bhlhe41 61) suggests IRG enrichment in dormant cells may also promote effects beyond immune effector processes—the impact of which warrants further investigation. Indeed, our findings suggest dormancy may result from several concomitant processes, which are likely niche‐specific, as others have proposed. The rarity 62, 63, 64, 65, plasticity 62, 63, 64, 65, and stochastic nature of dormant cancer cells in the osteoimmune microenvironment have hindered mechanistic studies. Yet, here we show that tumor cell retention of IRGs—while potentially conferring some survival advantage—may ultimately restrict overt tumor formation and promote dormant cell eradication, likely through immune‐mediated means. Indeed, this was evidenced by the absence of bone metastases in mice inoculated with RM1 BD Irf7 OE cells at endpoints exceeding mean RM1 model survival of up to threefold.
In bone, several studies have exposed unique mechanisms of crosstalk between skeletal and immune systems that can be exploited by DTCs to initiate tumor progression 66, 67. In support of this, the current study shows that tumor‐intrinsic IFN suppression, indicated by loss of Irf7 or Irf9, occurs largely within bone and can be induced by BM in vitro in a contact‐dependent manner. Such findings may reflect site‐specific differences in stromal composition and the molecules bone‐resident cells express, particularly myeloid cells, which comprise a large percentage of the BM population 68, 69. The specific role of Ly6G+ myeloid cells in driving IFN loss and metastatic outgrowth in bone is an important area of future study and may underpin the specific loss of IFN in tumor cells in bone compared to lung. The reported accumulation of populations such as MDSCs in advanced metastatic prostate cancer 45 along with MDSC promotion of bone resorption, tumor growth, and immune suppression 20, 69, 70, 71 suggests Ly6G+ myeloid cells play a pivotal role in switching the bone TME to a permissive one. Interestingly, the class I HDACi MS275 used in this study to restore tumor‐intrinsic IFN and block metastasis has been previously shown to neutralize suppressive myeloid cell function in renal cell carcinoma 72, possibly linking epigenetic modulation by myeloid cells to the loss of IFN signaling that we observed in mice. It remains to be determined whether the CD11b+ Ly6G+ myeloid cells that induced IFN loss in tumor cells ex vivo are indeed truly suppressive and responsible for silencing tumor‐inherent IFN signaling in the bone TME. Yet, it is recognized that acetylation is an integral part of transcriptional regulation of IFN signaling 41. Therefore, further dissecting the contribution of BM populations, including Tregs and other undefined M0 macrophage populations that are enriched in human bone metastases, to tumor immune suppression may also uncover potential mechanisms of TME‐induced epigenetic modulation of cancer cells.
Our findings may have major implications for immunotherapeutic success, or lack thereof, in metastatic CRPC. It has long been established that adaptive immune cells are pivotal to tumor eradication and T‐cell activation and tumor immunogenicity are critical components of immunotherapeutic success 73. Several IRGs that regulate tumor cell visibility, antigen presentation, and immune cell function were suppressed in the bone metastases of both mice and PCa patients. Such findings support previous evidence that downregulation of antigen‐processing components occurs in the bone‐metastatic lesions of PCa patients 74, suggesting dampened immunogenicity in bone. Indeed, we showed that mice bearing cancer cells deficient in IFN signaling had impaired T‐cell function and response, suggesting bone metastases are unreactive and therefore unlikely to respond to single‐agent checkpoint‐based therapies that work best against a T cell‐inflamed tumor 75. Moreover, we showed that T cells derived from tumor‐bearing mice can become differentially cytotoxic purely based on tumor‐intrinsic IFN signaling potential. As such, tumor cell type I IFN signaling, largely ignored in solid cancers, is a key determinant of tumor cell recognition and immune memory response.
Tumor‐driven IFN‐dependent bone remodeling processes also likely contribute to tumor expansion in the osteoimmune environment. The osteomimetic properties of PCa cells, shown to express high levels of largely osteoblast and osteoclast‐restricted proteins, have been reported 76, 77. Bone resorption has been suggested to precede the emergence of osteoblastic lesions in PCa 78, with a skew toward late osteogenesis further influenced by bone‐targeted therapeutics, such as bisphosphonates 79. Expanding on these findings, we revealed high osteoclast activity within and around RM1 BD Irf− bone lesions, which recapitulate the early stage of skeletal lesion formation. Moreover, co‐cultures demonstrated that bone‐derived IFN‐deficient RM1 cells induced osteoclastogenesis, while reversal of tumor‐intrinsic IFN loss abrogated this phenomenon, supporting previous findings 80, 81 that type I IFN (IFN‐β) prevents bone loss through osteoclast inhibition. While deeper exploration into IFN‐dependent crosstalk between immune and bone cells in the TME is required, taken together, these results demonstrate the inextricable link in PCa‐driven osteoimmune modulation being intrinsic type I IFN dysregulation.
This study implicates restoration of tumor cell IFN as means for reducing metastatic burden and enhancing therapeutic response in patients with CRPC. The shortcomings of recombinant IFN as a therapy (reviewed in Ref. 34) along with recent acknowledgment of the critical influence of intact IFN signaling in the TME on therapeutic response 82, 83, 84, 85, including immune‐targeted agents, have sparked a new wave of IFN inducers being trialed in oncology, such as STING and TLR agonists. However, our data herein provide the rationale for the lack of efficacy of such immune‐activating agents, and suggest that tumor‐intrinsic IFN signaling must be intact for a treatment‐provoked antitumor response. As such, epigenetic regulators, including MS275 (entinostat), that boost tumor immunogenicity through cell‐intrinsic IFN modulation hold great promise as anticancer agents. In recent years, several HDACi have been FDA‐approved to treat hematological malignancies with many more currently undergoing clinical testing 86, 87. Notably, in addition to exerting proapoptotic and antiproliferative effects, varied classes of HDACi have also demonstrated activity in conjunction with other immunotherapeutics 88, 89. While early broad‐spectrum HDACi, such as Trichostatin A, have been shown to suppress IRG expression in non‐metastatic cancer cells in culture 90, here we demonstrate that the selective class I HDACi MS275 is able to boost tumor cell immunogenicity through cell‐intrinsic IFN induction and imparts a synergistic effect combined with immune‐activatory poly I:C to completely abrogate bone metastasis. In contrast to reports using the HDACi SAHA (vorinostat) 91, class I HDACi MS275 prevented Treg cell accumulation. Moreover, treatment promoted sustained effector T‐cell responses through CD4+ and CD8+ T‐cell expansion and enhanced T‐cell memory function, which directly correlated with IFN‐induced MHC expression in bone‐derived tumor cells. As previously reported 92, in our hands MS275 does not impact innate immune populations. As such, the additive effect of MS275 and poly I:C on bone progression may result from the induction of both innate and adaptive arms of immunity, both implicated in acute and long‐term antitumor activity. Future spatial exploration using quantitative visual analysis platforms 93, 94 may help expand our understanding of the direct effect of MS275 in bone on tumor–immune cell interactions and the mechanisms through which HDACi shape T‐cell memory in the osteoimmune niche.
Here, we report for the first time that tumor‐intrinsic type I IFN loss occurs during dormancy outgrowth and that this is perhaps one mechanism that facilitates accelerated bone‐metastatic progression in PCa. Moreover, we highlight that the complex tri‐regulation that may occur between tumor, immune, and bone cells to promote bone metastasis underscores the importance of looking beyond isolated interactions to target a highly heterogenous and bone‐avid disease. Critically, we demonstrate that in an aggressive cancer context that mimics CRPC, lost tumor‐inherent IFN can be restored using a HDACi. Use of this or other agents, including HDACi not explored in this study, may provide new therapeutic opportunities to restore inherent IFN signaling in tumor cells to enhance immunogenicity and stimulate T‐cell memory should dormant tumor cells awaken in bone. We demonstrate that low tumor cell expression of IRGs in PCa patients is associated with bone‐metastatic progression and may be important for subsequent or concurrent spread to other sites. Moreover, that members of this pathway, such as IRF9, may serve as robust markers through which to identify patients most at risk of metastasis and aid in early stratification of candidates most likely to benefit from the therapeutic strategies aimed at increasing the visibility of tumor cells, such as those explored in the current study. Furthermore, we reveal that tumor‐intrinsic IFN suppression in patient bone metastases may underpin the inefficacy of immunotherapeutics against CRPC progression by suppressing tumor cell induction of immune activation and engendering an immune‐unreactive TME. Our study will hopefully prompt more systematic trials aimed at utilizing the predictive and therapeutic value of PCa cell‐intrinsic IFN signaling to personalize immune‐based strategies in patients harboring an incurable stage of disease.
Materials and Methods
Patients and mice
Epworth Clinical Cohort patient samples were obtained and processed 95 with consent and institutional ethics approval from the Royal Melbourne Hospital and Epworth Hospital Human Ethics Committees following invitation to undergo voluntary biopsy. The metastatic clinical cohort comprised of primary adenocarcinomas spanning the common clinical grades and stages (Gleason scores 7–9; pathological T stages 2c, 3a, and 3b; and prostate‐specific antigen [PSA] 5–81 at time of diagnosis). The localized patient cohort consisted of primary adenocarcinomas ranging from Gleason scores 6 to 9, pathological T stages 2a to 3b, and PSA 5 to 81 at time of diagnosis. No patient had received bone‐targeted therapy at the time of surgery.
Primary prostate cancer specimens were obtained with written and informed consent through the Australian Prostate Cancer BioResource from men undergoing robotic radical prostatectomy at St Andrew's Hospital (Adelaide, Australia), with approvals from the St Andrew's and University of Adelaide Human Research Ethics Committees (HRECs).
Cancer Tissue Acquisition After Death (CASCADE; PMCC HREC Project 11/102)96 program prostate primary tumor and metastatic bone lesions were accessed from 5 enrolled patients.
St Vincent's‐Garvan Clinical Cohort primary adenocarcinoma biospecimens were obtained at time of radical prostatectomy from patients recruited and consented as approved by the St Vincent's Hospital HREC #SVH/12/231. Sample processing and expression analysis was performed in accordance with St Vincent's HREC approval #SVH/15/227. The full cohort comprised of 80 patients who, at time of surgery, had not received radiation, chemotherapy, and/or ADT, and post‐surgery had at least 24 months of clinical follow‐up. Patient characteristics at surgery included the following: Gleason scores 6 (n = 20), 7 (n = 32), 8 (n = 12), and 9 (n = 16), with median age in years 60.7 (range 46–75) and median PSA in ng/ml 8.2 (range 3.1–19.2).
Mouse experiments were approved by the La Trobe Animal Experimentation Ethics Committee. Male C57BL/6 mice (~ 8 weeks) were obtained from the Walter and Eliza Hall Institute of Medical Research (Melbourne, Vic, Australia) and C57BL/6 Ifnar1 −/− mice bred in‐house as previously described 20. For all in vivo studies, mice were age‐matched.
PCa models
The bone‐metastatic RM1 cell line sourced from Timothy Thompson (The Urology Research Laboratory, Veterans Affairs Medical Center, Houston, and Baylor College of Medicine, Department of Cell Biology, Houston, TX, USA) was engineered to express cerulean and luciferase via standard retroviral transfection and spin infection methods using Phoenix‐Eco cells to generate RM1 parental pool cells per previously outlined methods 20. Tissue‐specific RM1 libraries, including all RM1 bone‐derived Irf‐low (RM1 BD Irf−) cell lines, were generated through FACS isolation of cerulean (V500+) PKH− cells from tumor‐bearing mice and expanded in vitro with restricted passage (< 8). The reverted RM1 BD cell line (RM1 BD Rev) was further generated through extended passage (> 13) in vitro. The pMSCV‐ires‐mCherry retroviral expression vector (Clontech, Palo Alto, CA, USA) was used to enforce constitutive Irf7 expression in RM1 BD Irf− cells to create the RM1 BD Irf7 OE line by retroviral transduction. In parallel, pooled empty pMSCV‐ires‐mCherry vector‐expressing cells served as base vector controls (RM1 BD Irf− BV). Cherry/cerulean‐positive clones were single‐cell‐sorted, expanded, validated, and pooled from three individual clones. Cell lines were cultured in DMEM (10% FBS) at 37°C (5% CO2) and passaged using EDTA (0.01% w/v in PBS). Tumor lines were verified to be mycoplasma‐negative by the Victorian Infectious Diseases References Lab (Melbourne, Vic, Australia).
Tissue‐derived cell preparation
To obtain BM cell suspensions, the long bones (femurs, tibias, and humerus) and spines were resected and muscle was removed using a scalpel blade under sterile conditions. Long bones were cut at the epiphyses and flushed with 10–50 ml PBS using a 10‐ml syringe and 27‐gauge needle, and spines were sectioned and flushed accordingly. Whole BM was subjected to red blood cell (RBC) lysis (155 mM NH4Cl, 10 mM KHCO3, 0.1 mM EDTA, pH 7.3) and filtered through a 70‐μm membrane. Lung‐derived RM1 cells were isolated via whole lung mechanical disaggregation and filtration through a 70‐μm membrane with subsequent RBC lysis and re‐suspension. Tumor cells were sorted from crude cell suspensions and FACS‐purified based on cerulean detection using the FACSAria III. All tissues and stromal cells were harvested from male WT C57BL/6 mice aged ~ 8 weeks.
Membrane labeling
RM1 parental cells were labeled using the PKH26 Red Fluorescent Cell Linker Mini Kit (Sigma‐Aldrich), and RM1 BD Irf− and Irf7 OE cells were labeled using the CellVue® Claret Far Red Fluorescent Cell Linker Mini Kit (Sigma‐Aldrich) according to the manufacturer's instruction with minor modifications, including incubation times and centrifugation speeds. Cells were labeled 12–14 h prior to subsequent use, to facilitate cell recovery. Cell viability along with label intensity and homogeneity was analyzed by FACS prior to in vivo and ex vivo assays.
RNA‐seq
Single‐cell cDNA generation, library preparation, sequencing, and analysis
Live (APC−; Fixable Viability Dye eFluor 780, eBioscience) PKH+ and PKH− BD RM1 cells were single‐cell‐sorted by FACS (FACSAria III; BD Biosciences) into 384‐well plates containing RNA lysis buffer (0.05% 10× GeneAmp PCR Buffer without MgCl, Life Technologies; 0.9% SUPERase In RNase Inhibitor (20 U/μl), Life Technologies; 0.9% RNasin® Plus RNase Inhibitor (40 U/μl), Promega; Nuclease‐free Water), snap‐frozen, and stored at −80°C for processing. Single‐cell cDNA was generated using the SMARTer Ultra Low RNA Kit (Clontech) with modifications. Briefly, 1:2.5 × 106 dilution of ERCC spike‐in controls (Ambion) were incorporated during first‐strand cDNA synthesis. Subsequent steps were performed according to the manufacturer's instructions at half‐reaction volumes. cDNA quality assessment was performed using the Bioanalyzer HS DNA chip (Agilent Technologies) according to the manufacturer's instructions and with Qubit 4 (Thermo Fisher). Libraries (1 ng cDNA input) were subsequently generated in four batches (2 × 14 samples of PKH+ cells; 2 × 16 samples of PKH− cells sourced from eight independent animals at different time points of metastasis from d14 onward, with an average of d16, across independent experiments), and unique adapters were incorporated using the Nextera XT Kit (Illumina) according to the manufacturer's protocol. Libraries were validated by Bioanalyzer 2100 (Agilent) and quantitated by Qubit 4 and pooled (2 × pools of equal numbers of PKH ± in each group) for sequencing across 2 lanes on the Illumina HiSeq2500 (125‐bp paired‐end reads) on a high‐throughput mode with ~ 3 million reads per cell. Sequence adapters were trimmed and aligned to a modified version of the GRCm38/mm10 mouse genome (which also included ERCC and eCFP sequences) using the STAR aligner 97. Summarized gene transcript counts and TPMs were generated using RSEM 98. All subsequent normalization and differential expression analysis was performed using the BASiCS package according to methods previously described 23, 24.
Bulk RM1 RNA preparation and sequencing
Matched bone and lung metastases were derived from an RM1 parental tumor‐bearing mouse at d16 post‐IC injection of PKH26‐labeled RM1 cells. PKH− cells from a bone‐metastatic femur were bulk‐sorted on the FACSAria III (BD Biosciences). PKH26 ± cells were single‐cell‐sorted (n = 6 PKH+; 63 single PKH−) in parallel from the same mouse for validation of subsequent expression analysis. Cells were expanded in culture, until ~ 5 × 106 cells were pelleted and RNA was extracted using the innuPREP RNA Mini Kit (Analytik Jena). RNA was also extracted from unlabeled and ceru+/luc2+‐labeled RM1 parental cells (n = 4). RNA concentration was determined via NanoDrop 2000 (Thermo Fisher), validated by Bioanalyzer 2100 (Agilent), and quality of RNA was assessed by gel electrophoresis (1% agarose gel). Purified RNA (RIN > 6.8) was prepared using the NEBNext Ultra RNA Library Prep Kit for Illumina (NEB, USA), and RNA‐seq was performed on the HiSeq2500 (125‐bp paired‐end reads; 24 million pair reads per sample) by Novogene Bioinformatics (Hong Kong). Clean reads meeting exclusion criteria (no adaptor contamination; undecided nucleotide call > 10%; low base quality > 50% of read) were aligned to GRCm38 using TopHat (v2.1.10)99 and counted using HTSeq‐count 100. Differential expression was performed using edgeR, and statistical significance was determined by generalized linear model extraction (GLM)101.
Patient tissue processing
For metastatic samples, a coaxial bone biopsy needle with an 18G internal caliber (Bonopty Bone Biopsy System, 10‐1072, AprioMed AB, Sweden) was used and tissue cores were immediately flash‐frozen in liquid nitrogen. Tissue samples were embedded in optical cutting time compound (Sakura) at −24°C, and 5‐μm sections were cut by cryotome (CM1,900, Leica Microsystems, NSW, Australia). Sections were transferred to charged glass slides (SuperFrost Ultra Plus, Thermo Scientific), stained with hematoxylin and eosin, and assessed in real time by a pathologist for tumor content. On confirmation of malignancy or where this was considered likely but inconclusive, the optical cutting time compound‐embedded tissue samples were isolated with a scalpel and placed in RLT Plus Buffer for immediate homogenization (TissueRuptor, Qiagen, CA). DNA and RNA were simultaneously extracted using the Allprep Micro Kit (Qiagen, CA) following the manufacturer's instructions, including on‐column DNase digestion of the RNA. Genomic DNA was extracted from fresh‐frozen samples of whole blood with the DNeasy Blood & Tissue Kit (Qiagen, Maryland) following the manufacturer's instructions. RNA quantity and quality were checked by microelectrophoresis (Agilent 2100 Bioanalyzer), while DNA quantity was checked by spectrophotometry (NanoDrop 1000, Thermo Scientific), and the quality was assessed by gel electrophoresis (0.8% agarose gel).
Patient library preparation and RNA sequencing
A total of 1 μg RNA was input into the TruSeq RNA Sample Preparation Kit v2 (Illumina, RS‐122‐2001) according to the manufacturer's instructions. Second‐strand cDNA synthesis was performed using DNA Polymerase I and RNase H. The resulting cDNA fragments were end‐repaired, 3′ ends were adenylated, and indexed paired‐end adaptors were ligated. The products were purified and then enriched with PCR to create the cDNA library, validated by 2100 Bioanalyzer (Agilent). Sample libraries were normalized to 10 nM using Tris‐Cl 10 mM, pH 8.5, with 0.1% Tween‐20. Samples were multiplexed with multiple samples per lane in the flow cell and thus were pooled accordingly. Cluster generation occurred on a cBot automated cluster generation system using TruSeq PE Cluster Kit v3 (cBot‐HS, Illumina) reagents for 100‐bp paired‐end sequencing. Each flow cell was loaded onto a HiSeq2000 sequencing platform with reagents from TruSeq SBSv3 HS (200 cycles; Illumina) and 120–180 million reads per flow‐cell lane performed. Reads were aligned to GRCh37 and counted, and differential expression was performed per bulk RM1 outline.
Clustering analysis
Differentially expressed genes enriched in dormant (PKH+) compared to proliferating RM1 single cells, as identified by BASiCS, were clustered by non‐NMF using the NMF package in R.
The Cancer Genome Atlas analysis
Human survival curves were generated using the prostate adenocarcinoma TCGA provisional dataset (TCGA‐PRAD; 499 RNA‐seq samples from 498 patients; RSEM z‐Score ± 1.5) using cBioPortal 102, 103. Statistical significance was determined by log‐rank test. Hazard ratios are exp(coef) with upper and lower confidence intervals of 95%.
Oncomine analysis
The Varambally 53 prostate dataset (GSE3325) generated using the Affymetrix Human Genome U133 Plus 2.0 Array platform from individual and pooled benign (not shown) primary and metastatic prostate cancer tissues was probed for an 8‐IRG core signature for mRNA expression (z‐score normalized; log2 median‐centered ratio). Statistical significance was determined by log‐rank test.
INTERFEROME, gene set testing, and GO enrichment analyses
Differentially expressed genes high in dormant cells with no residual dispersion difference from the scRNA‐seq analysis or downregulated gens with FDR ≪ 0.05 from the bulk RNA‐seq analyses were subjected to INTERFEROME 25 interrogation to identify IRGs (with a twofold upregulation threshold).
Gene ontology (GO) overrepresentation analysis was performed on DEGs with no residual dispersion from the single‐cell data using GO biological process terms with the goana function from the limma package.
Gene set testing was performed using the cameraPR function 104 on the signed log‐likelihood ratio (LR) statistic derived from edgeR 101 differential expression results, calculated as sign(logFC) × sqrt(LR). The Hallmark gene set collection 35 and the GO biological process subset 105 of the c5 gene set collection from the Molecular Signatures Database 106 were tested.
Gene enrichment and pathway annotations of biological processes from IRG signatures were derived using the PANTHER v11 package 107, 108. Output was restricted to fold enrichment (FE) > 2; P < 0.05.
Multidimensional scaling (MDS) plot
The MDS plot was generated using the plotMDS function from the limma package using log2(denoised counts + 1) expression values.
ARMET and CIBERSORT analyses
Differential tissue composition analyses to determine tumor cell (epithelial) and immune cell content (% of total sample) of human tumor mRNA were quantified using the custom probabilistic Bayesian model ARMET (version 0.6.0) according to methods recently outlined 109. Significance was determined using the stat_compare_means function Mann–Whitney U‐test from the ggpubr R package.
For immune cell deconvolution of leukocytes in human tumor mRNA, raw counts were converted to counts per million using the edgeR cpm function. Gene symbols were obtained from Ensembl IDs using the Homo.sapiens package. Expression data for annotated genes were used for CIBERSORT analysis 110 in absolute mode with quantile normalization disabled. For each cell type, bone metastases and primary tumor groups were compared by Mann–Whitney U‐test using the wilcox.test function in R.
Sulforhodamine B proliferation assay
In vitro cell proliferation was assessed using a sulforhodamine B‐binding assay over 6 days with a seeding number of 500 cells per cell line in triplicate and measured at 550 nm using methods previously described 111.
qRT–PCR
RNA was extracted from cell pellets using the innuPREP RNA Mini Kit (Analytik Jena) according to the manufacturer's instructions. When required, cells were transfected with poly I:C (10 μg/ml) overnight or treated with HDACi (entinostat [MS275], 1 μM [Selleckchem]; romidepsin [depsipeptide], 50 nM [Selleckchem]; or vorinostat [suberanilohydroxamic acid, SAHA], 2.5 μM [Selleckchem]) for 48 h prior to RNA extraction. cDNA was generated using the iScript Reverse Transcriptase Supermix cDNA for qRT–PCR kit (Bio‐Rad). qRT PCR was performed using SsoAdvanced Universal SYBR Green Supermix (Bio‐Rad) to quantify murine Irf7, Irf9, and Oas2 transcript expression on the CFX96 (Bio‐Rad) cycler per manufacturer's guidelines. Gene expression (arbitrary units) relative to housekeeper gene Hprt was calculated as mean relative transcript abundance (RTA) by methods previously outlined 20. Primers sourced from IDT were used as follows: Irf7 fwd: 5′‐CCACACCCCCATCTTCGA‐3′; Irf7 rev: 5′‐CCTCCGAGCCCGAAACTC‐3′; Irf9 fwd: 5′‐GCTCTAGCCATAGCCAAGAGAATC‐3′; Irf9 rev: 5′‐TCCAGTAAATGTCGGGCAAAG‐3′; Oas2 fwd: 5′‐CTGTTGGAAGCAGTCCATGA‐3′; Oas2 rev: 5′‐CCCTGTGAAGGAAGTGGCTA‐3′; Hprt fwd: 5′‐GGCCAGACTTTGTTGGATTT‐3′; Hprt rev: 5′‐ACTGGCAACATCAACAGGACT‐3′.
RNA was prepared from 88 primary biopsy cores from 80 St Vincent's–Garvan PCa patients using the AllPrep DNA/RNA Mini Kit (Qiagen). cDNA generated using the QuantiTect Reverse Transcription Kit (Qiagen) was assessed for Irf9 expression, with each sample (100 ng RNA) run in triplicate using primers and probe sourced from IDT: Irf9 fwd: 5′‐CCAGCCATACTCCACAGAAT‐3′; Probe: 5′‐ACAGTGAAGATGGAGCAGGCCTTT‐3′; Rev: 5′‐GAGTCTGCTCCAGCAAGTAT‐3′ with expression relative to GAPDH (IDT).
Microarray
HCT116 colon cancer cells were ± treated with SAHA (vorinostat; 5 μM), romidepsin (50 nM), and belinostat (5 μM; Selleckchem) for 24 h. Total RNA was isolated from pelleted cells using the RNeasy kit (Qiagen). Samples were submitted to the Australian Genome Research Facility (AGRF) for processing and direct hybridization using Illumina HumanHT‐12 v4 Expression BeadChip whole‐genome microarrays containing probes for over 47,000 transcripts (Illumina). Data were analyzed using R and the Bioconductor package (limma), where raw data were background‐corrected using the normexp function, log‐transformed, and quantile‐normalized. Differential expression in treated samples was measured relative to controls.
IFN‐α enzyme‐linked immunosorbent assay (ELISA)
IFN‐α ELISA was performed using standard molecular biology techniques. Capture antibody, clone RMMA‐1, was used at 1/500 (0.16 μg/ml; PBL Interferon source) prior to detection antibody (1/500 rabbit polyclonal mouse IFN‐α 32100‐1; 4 μg/ml; PBL Interferon Source) and tertiary antibody (1/1,000 anti‐rabbit‐HRP; AP182P; Chemicon). In‐house‐generated recombinant IFN‐α gifted from the Hertzog Lab. Cells were ± pre‐treated with MS275 (1 μM) for 48 h and then seeded for same day ± poly I:C (10 μg/ml) transfection overnight.
Flow cytometry analysis
For analysis of peripheral blood lymphocytes, tail or submandibular vein blood (< 100 μl) was collected and subjected to RBC lysis. Subsequent cell suspensions were stained with panels of antibodies: CD8α‐PE‐Cy7 (53‐6.7), CD4‐APC‐Cy7 (GK1.5), CD69‐APC (H1.2F3), CD44‐FITC (IM7), CD62L‐BV421 (MEL‐14), PD‐1‐PE (J43), NK1.1‐BV421 (PK136), FOXP3‐FITC (MF23), TNF‐α‐FITC (MP6‐XT22), and IFN‐γ–PE (XMG 1.1) (all from BD Biosciences), and NKg2D‐PE‐Cy7 (CX5) (eBioscience). For characterization of bone stromal cell populations, cells and isolation of CD11b+ subsets for co‐culture cells were also stained with CD11b‐BV421/BV605 (M1/70), CD3‐PE (17A2), TCR‐β–FITC (H57‐597), and Ly6G‐PE (1A8; all BD Biosciences), CD11c‐PercP (N418; Biolegend), Ly6C‐APC (HK1.4; eBioscience), and F4/80‐PeCy7 (BM8; Invitrogen). For characterization of H2‐Kb expression, RM1 cells were stained with H2‐Kb‐PE (AF6‐88.5; BD Biosciences). Data are represented as lymphocyte percentage. All analysis was performed on the FACSAria III (BD Biosciences), and data were analyzed using FlowJo 10.5.0 software (Tree star).
Co‐cultures
Parental RM1 cells were seeded at 1–2.5 × 104 cells per well in a 24‐well plate. BM was derived as previously specified, and ~ 8 h later, 6 × 105 cells were added (±) to cultures for 48 h ± MS275 (1 μM). Excess naïve BM was stored at −80°C for RNA extraction to assess IRG expression. For transwell cultures, BM was ± seeded onto 0.4‐μm filters (Corning) in a 24‐well plate and cultured for 48 h. For CD11b+ co‐cultures, CD11b+ Ly6G+ populations were FACS‐isolated from naïve BM after staining as described. At all co‐culture endpoints, RM1 cells were pelleted for RNA extraction and qRT–PCR by methods previously outlined.
Osteoclastogenesis assay
RM1 BD Irf− BV and RM1 BD Irf7 OE were seeded in triplicate at 1 × 104 cells per well in a 48‐well plate. BM was derived as previously specified and plated independently from individual animals (n = 3), and ~ 8 h later, 3 × 105 cells were added ± M‐CSF (50 ng/ml) for 24 h, after which fresh media containing ± M‐CSF (50 ng/ml) and recombinant RANKL (60 ng/ml) were added every 2 days until day 9. Due to rapid RM1 growth, tumor cells were split 1:4 on d4 and d7 by adding 100 μl fresh media, pipetting gently, and removing 100 μl of RM1‐suspended cells. On day 9, RM1 cells were removed via gentle washing and TRAP staining was performed as previously published 112.
Survival analysis and in vivo treatment
For in vivo experiments, 3–4 × 104 cells (RM1, RM1 BD Irf−, RM1 BD Rev, RM1 BD Irf− BV, or RM1 BD Irf7 OE) were injected under isoflurane anesthesia into the left ventricle of the heart on day 0. To derive subcutaneous cell lines and investigate the impact of PKH labeling on in vivo growth, 8 × 104 RM cells were injected into the hind flank. Growth was measured using electric calipers, and tumor volume (mm3) was calculated as L (mm) × W (mm)2/2. For survival assays, mice were euthanized following paralysis or signs of metastatic distress. Metastatic burden in whole animals, viscera and bones, was confirmed in all subjects at endpoint via bioluminescence imaging (BLi) following IP injection of d‐luciferin (3 mg/mouse; Gold Biotechnology, USA) and IVIS Lumina XR III Detection (Caliper Life Science). Living Image v4.4 software was used for normalization and luminescence quantitation (Caliper Life Science).
To compare saline (0.9%) to poly I:C using RM1 and RM1 BD Irf− cells, saline or poly I:C (25 mg/mouse in saline; Sigma) was ip‐administered three times weekly for 2 weeks from d3 post‐IC injection of RM cells. To assess the efficacy of HDACi and poly I:C on bone metastasis inhibition in RM1 BD Irf− tumor‐bearing mice, the following groups (n = 6) were administered: vehicle (1 × dose of saline; 1 × dose of DSPT [2% DMSO, 0.09% saline, 30% PEG 300, and 2% Tween‐80]); MS275 (8 mg/kg in DSPT; 1 × dose saline; Selleckchem); poly I:C (25 mg/mouse in saline; 1 × dose DSPT); and Combo (1 × dose MS275; 1 × dose poly I:C). MS275 was delivered once daily (Monday–Friday) for 2 weeks from d3 post‐IC injection of PCa cells. Poly I:C was delivered three times weekly for 2 weeks from d3 post‐IC injection of RM cells.
Intracellular cytokine staining for T‐cell specificity
Antigen‐specific CD8+ T cells were identified following splenocyte isolation from RM1 BD Irf− tumor‐inoculated mice (> day 50) and T‐cell generation following co‐culture with irradiated RM1 BD Irf− cells at a 10:1 ratio in RPM1 (10% FBS; 10U IL‐2/ml) for 14 days. In vitro‐expanded RM1 BD Irf−‐specific CD8+ T cells (5 × 104) or splenocytes from memory tumor‐bearing mice (1 × 106) were re‐stimulated with 5 × 104 untreated or treated RM1 cell lines (RM1, RM1 BD Irf−, and RM1 BD Irf7 OE) in the presence of 10 μg/ml Brefeldin A for 5 h. Following incubation, cells were surface‐antibody‐stained for CD8 (30 min, 4°C) and fixed with 1% paraformaldehyde (20 min, RT), and intracellular antibody was stained for IFN‐γ and TNF‐α in the presence of 0.4% saponin (30 min, 4°C) before analysis by flow cytometry. For assessment of drug‐induced changes in intracellular cytokine staining (ICS) assays, RM1 BD IRF− cells were ± pre‐treated with 1 μM of MS275 for 48 h, then transfected with poly I:C (10 μg/ml) for a further 24 h prior to ICS inclusion.
Immunohistochemistry
Differential tumor burden in mouse bone was visualized using eCFP antibodies with the OPAL method using the 7‐Color Manual immunohistochemistry (IHC) Kit (PerkinElmer; NEL811001KT). Bones were decalcified and fixed as per conventional IHC methods mentioned above. Staining was performed according to the manufacturer's protocol with the exception of 30‐min blocking and secondary antibody incubations. Primary antibodies obtained from Abcam used were CFP (ab6556; 1 μg/ml), and incubations were performed for 1 h at RT. Secondary antibody was donkey anti‐rabbit HRP (AP182P; 1:2,000, Chemicon). After all rounds of staining, including DAPI, tissues were mounted using Vectashield hardset mounting medium (Vector Labs). Whole slide scans and multispectral imaging of sections were undertaken using the VECTRA 3.0 (PerkinElmer) at 200× magnification. Image analysis was performed using inForm software (PerkinElmer) following tissue segmentation into tumor and stromal areas.
For differential IRF9 expression in human primary tumors and bone metastases, tissues were subject to heat‐induced epitope retrieval in a decloaking chamber (110°C for 5 min) before incubating with anti‐IRF9 (ab51639; 7 μg/ml, Abcam) overnight at 4°C. Tissues were then incubated with goat anti‐rabbit secondary antibody (BA‐1000; 1:250; Vector Labs) following incubation with avidin/biotinylated enzyme complex (ABC; Vectastain) and visualized using DAB (Vectastain). Tissues were counterstained using hematoxylin.
TRAP staining of bone sections
Slides were dewaxed through histolene (3×, 3 min), 100% ETOH (3×, 1 min), 70% ETOH (1×, 1 min), and distilled water (1×, 1 min). Slides were incubated in 50 ml acetate buffer pre‐warmed to 37°C following addition of 0.5% Naphthol AS‐BI phosphate substrate (Sigma‐Aldrich) as previously described 112. A 1:1 mix of sodium nitrite solution (0.58 M) and pararosaniline dye (154 mM in 2M HCl) was prepared and added to a second Coplin jar of pre‐warmed acetate buffer 2 min prior to incubation endpoint. The slides were then transferred, and TRAP was developed for 5–12 min with periodical monitoring for optimal induction. Slides were washed three times with distilled water and counterstained with 0.02% Fast Green (Sigma‐Aldrich) for 45 s. Slides were rinsed with distilled water, dehydrated with 100% ETOH (3×, 1 min) and histolene (3×, 4 min), and coverslipped with Entellan.
Two‐photon microscopy
Whole tibias and femurs were harvested from tumor‐bearing animals. Two‐photon excitation was achieved with a Chameleon Vision II Ti:Sa laser (Coherent). Images were acquired on a Zeiss 7MP microscope with LBF 760 and BSMP 760 filters to enable far‐red detection. Non‐descanned detectors used were SP485 (blue, second‐harmonic generation and eCFP), BP 500–550 (green, eCFP), and BP 540–719 (far‐red, CellVue Claret). Imaging and cell quantitation were performed on intact whole tibias as published 113.
Quantification and statistical analysis
Unless otherwise described in the figure legends, Student's two‐tailed t‐tests were used to determine significance between groups. Dot plots and histograms are means and all error bars ± SEM with exact n described. The Mantel–Cox log‐rank tests were performed to evaluate differences in survival time from the Kaplan–Meier analyses in murine assays. GraphPad Prism software (v5.0) was used for analyses, and P‐values were deemed significant as follows: *P < 0.05, **P < 0.005, and ***P < 0.0005.
Author contributions
BSP and PIC conceived this study. BSP and KLO designed the experiments. KLO performed the experiments with assistance from DJZ, WHK, C‐LC, NKB, AJS. AP, RJL, and HMD. MM performed multiphoton imaging and Imaris analysis. KLO and DJZ analyzed and interpreted data. DLR, WHK, MC, LJG, and KLO performed bioinformatics analysis. LJG and KLO performed INTERFEROME, CIBERSORT, and pathway analysis. MKH, NMC, SS, VMH, LMB, and CH consented patients and obtained tissues. CH performed TCGA interrogation. SM performed ARMET analysis. AR verified tumor content in human sections. KLO and BSP wrote and revised the manuscript, which was edited by PIC, JMM, MKH, CH, HMD, TGP, PJH, AS, NKB, and DJZ.
Conflict of interest
The authors have no competing interests to declare. BSP obtained commercial funding for an independent project from AstraZeneca/MedImmune.
Supporting information
Appendix
Expanded View Figures PDF
Review Process File
Source Data For Figure 1
Source Data For Figure 2
Acknowledgements
The authors thank the patients who donated material for this study and the efforts of all Biobank staff. This project is supportesd by grant funding from the Prostate Cancer Foundation of Australia and Movember (Movember Revolutionary Team Award) to P.I.C., B.S.P., C.H., N.C., A.S., and V.M.H., and Fellowship funding to B.S.P. (Australian Research Council Future Fellowship, FT130100671; Victorian Cancer Agency Mid‐career fellowship, MCRF16022).
EMBO Reports (2020) 21: e50162
Data availability
The RNA‐seq datasets produced in this study and corresponding to Figs 1 and 2 are available in the following database: Gene Expression Omnibus (GSE147151; https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE147151).
References
- 1. Norgaard M, Jensen AO, Jacobsen JB, Cetin K, Fryzek JP, Sorensen HT (2010) Skeletal related events, bone metastasis and survival of prostate cancer: a population based cohort study in Denmark (1999 to 2007). J Urol 184: 162–167 [DOI] [PubMed] [Google Scholar]
- 2. Perrault L, Fradet V, Lauzon V, LeLorier J, Mitchell D, Habib M (2015) Burden of illness of bone metastases in prostate cancer patients in Québec, Canada: a population‐based analysis. Can Urol Assoc J 9: 307–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sharma NL, Massie CE, Ramos‐Montoya A, Zecchini V, Scott HE, Lamb AD, MacArthur S, Stark R, Warren AY, Mills IG et al (2013) The androgen receptor induces a distinct transcriptional program in castration‐resistant prostate cancer in man. Cancer Cell 23: 35–47 [DOI] [PubMed] [Google Scholar]
- 4. Dearnaley DP, Mason MD, Parmar MK, Sanders K, Sydes MR (2009) Adjuvant therapy with oral sodium clodronate in locally advanced and metastatic prostate cancer: long‐term overall survival results from the MRC PR04 and PR05 randomised controlled trials. Lancet Oncol 10: 872–876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fizazi K, Carducci M, Smith M, Damião R, Brown J, Karsh L, Milecki P, Shore N, Rader M, Wang H et al (2011) Denosumab versus zoledronic acid for treatment of bone metastases in men with castration‐resistant prostate cancer: a randomised, double‐blind study. Lancet (London, England) 377: 813–822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Van Acker HH, Anguille S, Willemen Y, Smits EL, Van Tendeloo VF (2016) Bisphosphonates for cancer treatment: mechanisms of action and lessons from clinical trials. Pharmacol Ther 158: 24–40 [DOI] [PubMed] [Google Scholar]
- 7. Hodi FS, Day SJO, Mcdermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Ph D, Schadendorf D, Hassel JC et al (2010) Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 363: 711–723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ylitalo EB, Thysell E, Jernberg E, Lundholm M, Crnalic S, Egevad L, Stattin P, Widmark A, Bergh A, Wikström P (2016) Subgroups of castration‐resistant prostate cancer bone metastases defined through an inverse relationship between androgen receptor activity and immune response. Eur Urol 71: 1–12 [DOI] [PubMed] [Google Scholar]
- 9. Kwon ED, Drake CG, Scher HI, Fizazi K, Bossi A, Alfons JM, Den Eertwegh V, Krainer M, Houede N, Santos R et al (2014) Ipilimumab versus placebo after radiotherapy in patients with metastatic castration‐resistant prostate cancer that had progressed after docetaxel chemotherapy (CA184‐043): a multicentre, randomised, double‐blind, phase 3 trial. Lancet 15: 700–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Reinstein Z, Pamarthy S, Sagar V, Costa R, Abdulkadir SA, Giles FJ, Carneiro BA (2017) Overcoming immunosuppression in bone metastases. Crit Rev Oncol Hematol 117: 114–127 [DOI] [PubMed] [Google Scholar]
- 11. Solakoglu O, Maierhofer C, Lahr G, Breit E, Scheunemann P, Heumos I, Pichlmeier U, Schlimok G, Oberneder R, Kollermann MW et al (2002) Heterogeneous proliferative potential of occult metastatic cells in bone marrow of patients with solid epithelial tumors. Proc Natl Acad Sci USA 99: 2246–2251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Morgan TM, Lange PH, Porter MP, Lin DW, Ellis WJ, Gallaher IS, Vessella RL (2009) Disseminated tumor cells in prostate cancer patients after radical prostatectomy and without evidence of disease predicts biochemical recurrence. Clin Cancer Res 15: 677–683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gužvić M, Braun B, Ganzer R, Burger M, Nerlich M, Winkler S, Werner‐Klein M, Czyz ZT, Polzer B, Klein CA (2014) Combined genome and transcriptome analysis of single disseminated cancer cells from bone marrow of prostate cancer patients reveals unexpected transcriptomes. Cancer Res 74: 7383–7394 [DOI] [PubMed] [Google Scholar]
- 14. Weckermann D, Polzer B, Ragg T, Blana A, Schlimok G, Arnholdt H, Bertz S, Harzmann R, Klein CA (2009) Perioperative activation of disseminated tumor cells in bone marrow of patients with prostate cancer. J Clin Oncol 27: 1549–1556 [DOI] [PubMed] [Google Scholar]
- 15. Popiolek M, Rider JR, Andrén O, Andersson SO, Holmberg L, Adami HO, Johansson JE (2013) Natural history of early, localized prostate cancer: a final report from three decades of follow‐up. Eur Urol 63: 428–435 [DOI] [PubMed] [Google Scholar]
- 16. Van der Toom EE, Verdone JE, Pienta KJ (2016) Disseminated tumor cells and dormancy in prostate cancer metastasis. Curr Opin Biotechnol 40: 9–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Klein CA (2011) Framework models of tumor dormancy from patient‐derived observations. Curr Opin Genet Dev 21: 42–49 [DOI] [PubMed] [Google Scholar]
- 18. Ghajar CM (2015) Metastasis prevention by targeting the dormant niche. Nat Rev Cancer 15: 238–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Aguirre‐Ghiso JA (2007) Models, mechanisms and clinical evidence for cancer dormancy. Nat Rev Cancer 7: 834–846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bidwell BN, Slaney CY, Withana NP, Forster S, Cao Y, Loi S, Andrews D, Mikeska T, Mangan NE, Samarajiwa SA et al (2012) Silencing of Irf7 pathways in breast cancer cells promotes bone metastasis through immune escape. Nat Med 18: 1224–1231 [DOI] [PubMed] [Google Scholar]
- 21. Hibberd C, Cossigny D, Quan G (2013) Animal cancer models of skeletal metastasis. Cancer Growth Metastasis 6: 23–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jinnah AH, Zacks BC, Gwam CU, Kerr BA (2018) Emerging and established models of bone metastasis. Cancers (Basel) 10: 1–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Vallejos CA, Richardson S, Marioni JC (2016) Beyond comparisons of means: understanding changes in gene expression at the single‐cell level. Genome Biol 17: 1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Eling N, Richard AC, Richardson S, Marioni JC, Vallejos CA (2018) Correcting the mean‐variance dependency for differential variability testing using single‐cell RNA sequencing data. Cell Syst 7: 284–294.e12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rusinova I, Forster S, Yu S, Kannan A, Masse M, Cumming H, Chapman R, Hertzog PJ (2013) Interferome v2.0: an updated database of annotated interferon‐regulated genes. Nucleic Acids Res 41: D1040–D1046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Honda K, Yanai H, Negishi H, Asagiri M, Sato M, Mizutani T, Shimada N, Ohba Y, Takaoka A, Yoshida N et al (2005) IRF‐7 is the master regulator of type‐I interferon‐dependent immune responses. Nature 434: 772–777 [DOI] [PubMed] [Google Scholar]
- 27. Kim H, Park H (2007) Sparse non‐negative matrix factorizations via alternating non‐negativity‐constrained least squares for microarray data analysis. Bioinformatics 23: 1495–1502 [DOI] [PubMed] [Google Scholar]
- 28. Yeh AC, Ramaswamy S (2015) Mechanisms of cancer cell dormancy—another hallmark of cancer? Cancer Res 75: 5014–5022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chéry L, Lam H‐M, Coleman I, Lakely B, Coleman R, Larson S, Aguirre‐Ghiso JA, Xia J, Gulati R, Nelson PS et al (2014) Characterization of single disseminated prostate cancer cells reveals tumor cell heterogeneity and identifies dormancy associated pathways. Oncotarget 5: 9939–9951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kim RS, Avivar‐Valderas A, Estrada Y, Bragado P, Sosa MS, Aguirre‐Ghiso JA, Segall JE (2012) Dormancy signatures and metastasis in estrogen receptor positive and negative breast cancer. PLoS ONE 7: 1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. McGrath J, Panzica L, Ransom R, Withers HG, Gelman IH (2019) Identification of genes regulating breast cancer dormancy in 3d bone endosteal niche cultures. Mol Cancer Res 17: 860–869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sosa MS, Bragado P, Aguirre‐Ghiso JA (2014) Mechanisms of disseminated cancer cell dormancy: an awakening field. Nat Rev Cancer 14: 611–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Shiozawa Y, Pedersen EA, Patel LR, Ziegler AM, Havens AM, Jung Y, Wang J, Zalucha S, Loberg RD, Pienta KJ et al (2010) GAS6/AXL axis regulates prostate cancer invasion, proliferation, and survival in the bone marrow niche. Neoplasia 12: 116–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Parker BS, Rautela J, Hertzog PJ (2016) Antitumour actions of interferons: implications for cancer therapy. Nat Rev Cancer 16: 131–144 [DOI] [PubMed] [Google Scholar]
- 35. Liberzon A, Birger C, Thorvaldsdóttir H, Ghandi M, Mesirov JP, Tamayo P (2015) The molecular signatures database (MSigDB) hallmark gene set collection. Cell Syst 1: 417–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Li W, Hofer MJ, Songkhunawej P, Jung SR, Hancock D, Denyer G, Campbell IL (2017) Type I interferon‐regulated gene expression and signaling in murine mixed glial cells lacking signal transducers and activators of transcription 1 or 2 or interferon regulatory factor 9. J Biol Chem 292: 5845–5859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Brockwell NK, Rautela J, Owen KL, Gearing LJ, Deb S, Harvey K, Spurling A, Zanker D, Chan C‐L, Cumming HE et al (2019) Tumor inherent interferon regulators as biomarkers of long‐term chemotherapeutic response in TNBC. NPJ Precis Oncol 3: 21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Fagerberg L, Hallstro M, Oksvold P, Kampf C, Djureinovic D, Odeberg J, Habuka M, Tahmasebpoor S, Danielsson A, Edlund K et al (2014) Analysis of the human tissue‐specific expression by genome‐wide integration of transcriptomics and antibody‐based. Mol Cell Proteomics 13: 397–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Negrotto S, Giusti CJDE, Lapponi MJ, Etulain J, Rivadeneyra L, Pozner RG (2011) Expression and functionality of type I interferon receptor in the megakaryocytic lineage. J Thromb Haemost 9: 2477–2485 [DOI] [PubMed] [Google Scholar]
- 40. Bernardo AR, Cosgaya JM, Aranda A, Jiménez‐Lara AM (2013) Synergy between RA and TLR3 promotes type I IFN‐dependent apoptosis through upregulation of TRAIL pathway in breast cancer cells. Cell Death Dis 4: e479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nusinzon I, Horvath CM (2005) Unexpected roles for deacetylation in interferon‐ and cytokine‐induced transcription. J Interferon Cytokine Res 25: 745–748 [DOI] [PubMed] [Google Scholar]
- 42. Ning S, Pagano JS, Barber GN (2011) IRF7: activation, regulation, modification and function. Genes Immun 12: 399–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ivashkiv LB, Donlin LT (2014) Regulation of type I interferon responses. Nat Rev Immunol 14: 36–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wesolowski R, Markowitz J, Carson WE (2013) Myeloid derived suppressor cells – a new therapeutic target in the treatment of cancer. J Immunother Cancer 1: 10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mehra N, Seed G, Lambros M, Sharp A, Fontes MS, Crespo M, Sumanasuriya S, Yuan W, Boysen G, Riisnaes R et al (2016) Myeloid‐derived suppressor cells (MDSCs) in metastatic castration‐resistant prostate cancer (CRPC) patients (PTS). Ann Oncol 27: 243–265 [Google Scholar]
- 46. Zhang C, Wang S, Liu Y, Yang C (2016) Epigenetics in myeloid derived suppressor cells: a sheathed sword towards cancer. Oncotarget 7: 57452–57463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Andreoiu M, Cheng L (2010) Multifocal prostate cancer: biologic, prognostic, and therapeutic implications. Hum Pathol 41: 781–793 [DOI] [PubMed] [Google Scholar]
- 48. Abeshouse A, Ahn J, Akbani R, Ally A, Amin S, Andry CD, Annala M, Aprikian A, Armenia J, Arora A et al (2015) The molecular taxonomy of primary prostate cancer. Cell 163: 1011–1025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Werner‐klein M, Scheitler S, Hoffmann M, Hodak I, Dietz K, Lehnert P, Naimer V, Polzer B, Treitschke S, Werno C et al (2018) Genetic alterations driving metastatic colony formation are acquired outside of the primary tumour in melanoma. Nat Commun 9: 1–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Gundem G, Van Loo P, Kremeyer B, Alexandrov LB, Tubio JMC, Papaemmanuil E, Brewer DS, Kallio HML, Högnäs G, Annala M et al (2015) The evolutionary history of lethal metastatic prostate cancer. Nature 520: 353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Gupta GP, Massagué J (2006) Cancer metastasis: building a framework. Cell 127: 679–695 [DOI] [PubMed] [Google Scholar]
- 52. Rhodes DR, Kalyana‐Sundaram S, Mahavisno V, Varambally R, Yu J, Briggs BB, Barrette TR, Anstet MJ, Kincead‐Beal C, Kulkarni P et al (2007) Oncomine 3.0: genes, pathways, and networks in a collection of 18,000 cancer gene expression profiles. Neoplasia 9: 166–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Varambally S, Yu J, Laxman B, Rhodes DR, Mehra R, Tomlins SA, Shah RB, Chandran U, Monzon FA, Becich MJ et al (2005) Integrative genomic and proteomic analysis of prostate cancer reveals signatures of metastatic progression. Cancer Cell 8: 393–406 [DOI] [PubMed] [Google Scholar]
- 54. Shah RB, Mehra R, Chinnaiyan AM, Shen R, Ghosh D, Zhou M, MacVicar GR, Varambally S, Harwood J, Bismar TA et al (2004) Androgen‐independent prostate cancer is a heterogeneous group of diseases: lessons from a rapid autopsy program. Cancer Res 64: 9209–9216 [DOI] [PubMed] [Google Scholar]
- 55. Mangiola S, Hong MKH, Cmero M, Kurganovs N, Ryan A, Costello AJ, Corcoran NM, Macintyre G, Hovens CM (2016) Comparing nodal versus bony metastatic spread using tumour phylogenies. Sci Rep 6: 33918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Hong MKH, Macintyre G, Wedge DC, Van Loo P, Patel K, Lunke S, Alexandrov LB, Sloggett C, Cmero M, Marass F et al (2015) Tracking the origins and drivers of subclonal metastatic expansion in prostate cancer. Nat Commun 6: 6605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Hertzog PJ, Williams BRG (2013) Fine tuning type I interferon responses. Cytokine Growth Factor Rev 24: 217–225 [DOI] [PubMed] [Google Scholar]
- 58. Andrews DM, Sullivan LC, Baschuk N, Chan CJ, Berry R, Cotterell CL, Lin J, Halse H, Watt SV, Poursine‐Laurent J et al (2012) Recognition of the nonclassical MHC class I molecule H2‐M3 by the receptor Ly49A regulates the licensing and activation of NK cells. Nat Immunol 13: 1171–1177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Bryant G, Wang L, Mulholland DJ (2017) Overcoming oncogenic mediated tumor immunity in prostate cancer. Int J Mol Sci 18: 1542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Silvestri I, Cattarino S, Aglianò AM, Collalti G, Sciarra A (2015) Beyond the immune suppression: the immunotherapy in prostate cancer. Biomed Res Int 2015: 794968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Li P, Jia Y‐F, Ma X‐L, Zheng Y, Kong Y, Zhang Y, Zong S, Chen Z‐T, Wang Y‐S (2016) DEC2 suppresses tumor proliferation and metastasis by regulating ERK/NF‐κB pathway in gastric cancer. Am J Cancer Res 6: 1741–1757 [PMC free article] [PubMed] [Google Scholar]
- 62. Cabezas‐wallscheid N, Buettner F, Sommerkamp P, Rieger MA, Stegle O, Cabezas‐wallscheid N, Buettner F, Sommerkamp P, Klimmeck D, Ladel L et al (2017) Vitamin A‐retinoic acid signaling regulates hematopoietic stem cell dormancy article vitamin A‐retinoic acid signaling regulates hematopoietic stem cell dormancy. Cell 169: 807–823 [DOI] [PubMed] [Google Scholar]
- 63. Plaks V, Kong N, Werb Z (2015) The cancer stem cell niche: how essential is the niche in regulating stemness of tumor cells? Cell Stem Cell 16: 225–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Yang X, Liang X, Zheng M, Tang Y (2018) Cellular phenotype plasticity in cancer dormancy and metastasis. Front Oncol 8: 505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Yadav AS, Pandey PR, Butti R, Radharani NNV, Roy S, Bhalara SR, Gorain M, Kundu GC, Kumar D (2018) The biology and therapeutic implications of tumor dormancy and reactivation. Front Oncol 8: 72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Galon J, Angell H, Bedognetti D, Marincola F (2013) The continuum of cancer immunosurveillance: prognostic, predictive, and mechanistic signatures. Immunity 39: 11–26 [DOI] [PubMed] [Google Scholar]
- 67. Takayanagi H (2007) Osteoimmunology: shared mechanisms and crosstalk between the immune and bone systems. Nat Rev Immunol 7: 292–304 [DOI] [PubMed] [Google Scholar]
- 68. Youn J‐I, Gabrilovich DI (2010) The biology of myeloid‐derived suppressor cells: the blessing and the curse of morphological and functional heterogeneity. Eur J Immunol 40: 2969–2975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Sawant A, Deshane J, Jules J, Lee CM, Harris BA, Feng X, Ponnazhagan S (2013) Myeloid‐derived suppressor cells function as novel osteoclast progenitors enhancing bone loss in breast cancer. Cancer Res 73: 672–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Law AMK, Lim E, Ormandy CJ, Gallego‐Ortega D (2017) The innate and adaptive infiltrating immune systems as targets for breast cancer immunotherapy. Endocr Relat Cancer 24: R123–R144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. OuYang L‐Y, Wu X‐J, Ye S‐B, Zhang R, Li Z‐L, Liao W, Pan Z‐Z, Zheng L‐M, Zhang X‐S, Wang Z et al (2015) Tumor‐induced myeloid‐derived suppressor cells promote tumor progression through oxidative metabolism in human colorectal cancer. J Transl Med 13: 47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Orillion A, Hashimoto A, Damayanti N, Shen L, Adelaiye‐Ogala R, Arisa S, Chintala S, Ordentlich P, Kao C, Elzey B et al (2017) Entinostat neutralizes myeloid‐derived suppressor cells and enhances the antitumor effect of PD‐1 inhibition in murine models of lung and renal cell carcinoma. Clin Cancer Res 23: 5187–5201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Blankenstein T, Coulie PG, Gilboa E, Jaffee EM (2012) The determinants of tumour immunogenicity. Nat Rev Cancer 12: 307–313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Ylitalo EB, Thysell E, Jernberg E, Lundholm M, Crnalic S, Egevad L, Stattin P, Widmark A, Bergh A, Wikström P (2017) Subgroups of castration‐resistant prostate cancer bone metastases defined through an inverse relationship between androgen receptor activity and immune response. Eur Urol 71: 776–787 [DOI] [PubMed] [Google Scholar]
- 75. Tang H, Wang Y, Chlewicki LK, Zhang Y, Guo J, Liang W, Wang J, Wang X, Fu Y‐X (2016) Facilitating T cell infiltration in tumor microenvironment overcomes resistance to PD‐L1 blockade. Cancer Cell 29: 285–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Koeneman K, Yeung F, Chung LWK (2014) Osteomimetic properties of prostate cancer cells : a hypothesis supporting the predilection of prostate cancer metastasis and growth in the bone environment. Prostate 39: 10344214 [DOI] [PubMed] [Google Scholar]
- 77. Huang WC, Xie Z, Konaka H, Sodek J, Zhau HE, Chung LWK (2005) Human osteocalcin and bone sialoprotein mediating osteomimicry of prostate cancer cells: role of cAMP‐dependent protein kinase A signaling pathway. Cancer Res 65: 2303–2313 [DOI] [PubMed] [Google Scholar]
- 78. Keller ET, Brown J (2004) Prostate cancer bone metastases promote both osteolytic and osteoblastic activity. J Cell Biochem 91: 718–729 [DOI] [PubMed] [Google Scholar]
- 79. Morrissey C, Roudier MP, Dowell A, True LD, Ketchanji M, Welty C, Corey E, Lange PH, Higano CS, Vessella RL (2013) Effects of androgen deprivation therapy and bisphosphonate treatment on bone in patients with metastatic castration‐resistant prostate cancer: results from the University of Washington Rapid Autopsy Series. J Bone Miner Res 28: 333–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Wilkison M, Gauss K, Ran Y, Searles S, Taylor D, Meissner N (2012) Type 1 interferons suppress accelerated osteoclastogenesis and prevent loss of bone mass during systemic inflammatory responses to Pneumocystis lung infection. Am J Pathol 181: 151–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Zhang J, Zhao H, Chen J, Xia B, Jin Y, Wei W, Shen J, Huang Y (2012) Interferon‐β‐induced miR‐155 inhibits osteoclast differentiation by targeting SOCS1 and MITF. FEBS Lett 586: 3255–3262 [DOI] [PubMed] [Google Scholar]
- 82. Sistigu A, Yamazaki T, Vacchelli E, Chaba K, Enot DP, Adam J, Vitale I, Goubar A, Baracco EE, Remédios C et al (2014) Cancer cell–autonomous contribution of type I interferon signaling to the efficacy of chemotherapy. Nat Med 20: 1301 [DOI] [PubMed] [Google Scholar]
- 83. Legrier M‐E, Bièche I, Gaston J, Beurdeley A, Yvonnet V, Déas O, Thuleau A, Château‐Joubert S, Servely J‐L, Vacher S et al (2016) Activation of IFN/STAT1 signalling predicts response to chemotherapy in oestrogen receptor‐negative breast cancer. Br J Cancer 114: 177–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Jacquelot N, Roberti MP, Enot DP, Rusakiewicz S, Ternès N, Jegou S, Woods DM, Sodré AL, Hansen M, Meirow Y et al (2017) Predictors of responses to immune checkpoint blockade in advanced melanoma. Nat Commun 8: 592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Brockwell NK, Owen KL, Zanker D, Spurling A, Rautela J, Duivenvoorden HM, Baschuk N, Caramia F, Loi S, Darcy PK et al (2017) Neoadjuvant interferons: critical for effective PD‐1 based immunotherapy in TNBC. Cancer Immunol Res 5: 871–884 [DOI] [PubMed] [Google Scholar]
- 86. Li Y, Seto E (2017) HDACs and HDAC inhibitors in cancer development and therapy. Cold Spring Harb Perspect Med 6: 1–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. West AC, Johnstone RW (2014) New and emerging HDAC inhibitors for cancer treatment. J Clin Invest 124: 30–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Booth L, Roberts JL, Poklepovic A, Kirkwood J, Dent P (2017) HDAC inhibitors enhance the immunotherapy response of melanoma cells. Oncotarget 8: 83155–83170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Terranova‐Barberio M, Thomas S, Ali N, Pawlowska N, Park J, Krings G, Rosenblum MD, Budillon A, Munster PN (2017) HDAC inhibition potentiates immunotherapy in triple negative breast cancer. Oncotarget 8: 114156–114172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Nusinzon I, Horvath CM (2003) Interferon‐stimulated transcription and innate antiviral immunity require deacetylase activity and histone deacetylase 1. Proc Natl Acad Sci USA 100: 14742–14747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Zhang X, Han S, Kang Y, Guo M, Hong S, Liu F, Fu S, Wang L, Wang QX (2012) SAHA, an HDAC inhibitor, synergizes with tacrolimus to prevent murine cardiac allograft rejection. Cell Mol Immunol 9: 390–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Bridle BW, Chen L, Lemay CG, Diallo J‐S, Pol J, Nguyen A, Capretta A, He R, Bramson JL, Bell JC et al (2013) HDAC inhibition suppresses primary immune responses, enhances secondary immune responses, and abrogates autoimmunity during tumor immunotherapy. Mol Ther 21: 887–894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Ihle CL, Provera MD, Straign DM, Smith EE, Edgerton SM, Van Bokhoven A, Lucia MS, Owens P (2019) Distinct tumor microenvironments of lytic and blastic bone metastases in prostate cancer patients. J Immunother Cancer 7: 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Cabrita R, Lauss M, Sanna A, Donia M, Skaarup Larsen M, Mitra S, Johansson I, Phung B, Harbst K, Vallon‐Christersson J et al (2020) Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature 577: 561–565 [DOI] [PubMed] [Google Scholar]
- 95. Hong MKH, Sapre N, Phal PM, MacIntyre G, Chin X, Pedersen JS, Ryan A, Kerger M, Costello AJ, Corcoran NM et al (2014) Percutaneous image‐guided biopsy of prostate cancer metastases yields samples suitable for genomics and personalised oncology. Clin Exp Metastasis 31: 159–167 [DOI] [PubMed] [Google Scholar]
- 96. Alsop K, Thorne H, Sandhu S, Hamilton A, Mintoff C, Christie E, Spruyt O, Williams S, McNally O, Mileshkin L et al (2016) A community‐based model of rapid autopsy in end‐stage cancer patients. Nat Biotechnol 34: 1010–1014 [DOI] [PubMed] [Google Scholar]
- 97. Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR (2013) STAR: ultrafast universal RNA‐seq aligner. Bioinformatics 29: 15–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Li B, Dewey CN (2011) RSEM: accurate transcript quantification from RNA‐Seq data with or without a reference genome. BMC Bioinformatics 12: 323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL (2013) TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14: R36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Anders S, Pyl PT, Huber W (2015) HTSeq—a Python framework to work with high‐throughput sequencing data. Bioinformatics 31: 166–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Robinson MD, McCarthy DJ, Smyth GK (2010) edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26: 139–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E et al (2013) Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 6: pl1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E et al (2012) The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2: 401–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Wu D, Smyth GK (2012) Camera: a competitive gene set test accounting for inter‐gene correlation. Nucleic Acids Res 40: e133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT et al (2000) Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 25: 25–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES et al (2005) Gene set enrichment analysis: a knowledge‐based approach for interpreting genome‐wide expression profiles. Proc Natl Acad Sci USA 102: 15545–15550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Mi H, Muruganujan A, Casagrande JT, Thomas PD (2013) Large‐scale gene function analysis with the PANTHER classification system. Nat Protoc 8: 1551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Mi H, Huang X, Muruganujan A, Tang H, Mills C, Kang D, Thomas PD (2017) PANTHER version 11: expanded annotation data from Gene Ontology and Reactome pathways, and data analysis tool enhancements. Nucleic Acids Res 45: D183–D189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Mangiola S, Stuchbery R, McCoy P, Chow K, Kurganovs N, Kerger M, Papenfuss A, Hovens CM, Corcoran NM (2019) Androgen deprivation therapy promotes an obesity‐like microenvironment in periprostatic fat. Endocr Connect 8: 518–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, Hoang CD, Diehn M, Alizadeh AA (2015) Robust enumeration of cell subsets from tissue expression profiles. Nat Methods 12: 453–457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Houghton P, Fang R, Techatanawat I, Steventon G, Hylands PJ, Lee CC (2007) The sulphorhodamine (SRB) assay and other approaches to testing plant extracts and derived compounds for activities related to reputed anticancer activity. Methods 42: 377–387 [DOI] [PubMed] [Google Scholar]
- 112. Edgington‐Mitchell LE, Rautela J, Duivenvoorden HM, Jayatilleke KM, van der Linden WA, Verdoes M, Bogyo M, Parker BS (2015) Cysteine cathepsin activity suppresses osteoclastogenesis of myeloid‐derived suppressor cells in breast cancer. Oncotarget 6: 8–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Lawson MA, McDonald MM, Kovacic N, Hua Khoo W, Terry RL, Down J, Kaplan W, Paton‐Hough J, Fellows C, Pettitt JA et al (2015) Osteoclasts control reactivation of dormant myeloma cells by remodelling the endosteal niche. Nat Commun 6: 8983 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Review Process File
Source Data For Figure 1
Source Data For Figure 2
Data Availability Statement
The RNA‐seq datasets produced in this study and corresponding to Figs 1 and 2 are available in the following database: Gene Expression Omnibus (GSE147151; https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE147151).