

Abstract
Background:
The ADCK proteins are predicted mitochondrial kinases. Most studies of these proteins have focused on the Abc1/Coq8 subfamily, which contributes to Coenzyme Q biosynthesis. In contrast, little is known about ADCK1 despite its evolutionary conservation in yeast, Drosophila, Caenorhabditis elegans and mammals.
Results:
We show that Drosophila ADCK1 mutants die as second instar larvae with double mouth hooks and tracheal breaks. Tissue-specific genetic rescue and RNAi studies show that ADCK1 is necessary and sufficient in the trachea for larval viability. In addition, tracheal-rescued ADCK1 mutant adults have reduced lifespan, are developmentally delayed, have reduced body size, and normal levels of basic metabolites.
Conclusion:
The larval lethality and double mouth hooks seen in ADCK1 mutants are often associated with reduced levels of the steroid hormone ecdysone, suggesting that this gene could contribute to controlling ecdysone levels or bioavailability. Similarly, the tracheal defects in these animals could arise from defects in intracellular lipid trafficking. These studies of ADCK1 provide a new context to define the physiological functions of this poorly understood member of the ADCK family of predicted mitochondrial proteins.
Keywords: metabolism, mitochondria, molting, trachea
INTRODUCTION
Mitochondria are an important organelle in all eukaryotic cells. Not only are they the main source for cellular energy production, but they are also important for critical cellular functions such as calcium homeostasis, lipid homeostasis, and metabolite synthesis.1,2 Consistent with this, mitochondrial dysfunction is associated with aging and a plethora of metabolic disorders including cancer, neurodegeneration, diabetes, and inborn errors of metabolism.3,4 These important roles for mitochondria have led to extensive research using multiple model systems, providing insights into mitochondrial activities and their impact on cellular and organismal growth, development, and physiology.5 In spite of these efforts, however, about a fifth of the mitochondrial proteome remains largely uncharacterized.1,5 This includes many proteins that are highly conserved throughout eukaryotic organisms, a strong indication that they perform fundamentally important functions.
In this study, we examine the ADCK family of predicted mitochondrial kinases, represented by ADCK1–5 in humans (Figure 1). Most studies have centered on the ABC1/Coq8 subfamily, which includes ABC1/Coq8 in yeast and human ADCK3 and ADCK4.6 Coq8 is part of a multiprotein complex involved in the biosynthesis of Coenzyme Q (CoQ), which is a lipid that resides in the mitochondrial inner membrane and contributes to electron transport. Loss of ADCK3 in humans is associated with an inherited form of ataxia accompanied by CoQ deficiency.7–10 A recent biochemical study has shown that ADCK3 can bind ATP and adopt an atypical protein kinase-like fold that is blocked by autoinhibitory domains, providing initial insights into its molecular function.11 Mutations in ADCK4 are associated with steroid-resistant nephrotic syndrome in humans as well as reduced levels of CoQ.12,13 In spite of multiple studies, however, the molecular functions of Coq8 and its human counterparts remain unclear.14 In addition, ADCK2 and ADCK5 are largely uncharacterized.
FIGURE 1.

Maximum likelihood phylogeny of ADCK family members. Reciprocal BLAST searches using the amino acid sequences for the five mammalian ADCK proteins were used to obtain homologous protein sequences from yeast, Drosophila and Caenorhabditis elegans. These sequences were aligned using MUSCLE software and the aligned peptide sequences were uploaded to MEGA7 to generate a maximum likelihood phylogram. Bootstrap values are shown at the branchpoints. The lengths of the bars represent the protein sequence divergence
One study has reported a role for ADCK1 in yeast. These investigators found that overexpression of yeast ADCK1 (referred to as Mcp2 in this study) can rescue the growth defects of mutants in Mdm10, which encodes part of the ERMES complex.15 This multiprotein complex tethers the endoplasmic reticulum to mitochondria and is required to maintain mitochondrial lipid homeostasis.16 ADCK1 localizes to the inner membrane of mitochondria and does not directly associate with the ERMES complex. ADCK1 overexpression, however, can restore the membrane lipid defects in mdm10 mutants as well as rescue this phenotype in mutants for other ERMES complex members. The authors propose that ADCK1 has a redundant role with the ERMES complex in maintaining proper mitochondrial lipid levels.15 ERMES complex components, however, have not been found in metazoans to date.16 In addition, the molecular roles of ADCK1 remain uncharacterized. Here we present a genetic analysis of ADCK1 in Drosophila in an effort to define its roles during development.
We show that Drosophila ADCK1 mutants display larval lethality and double mouth hooks, which are hallmarks of molting defects and often associated with reduced levels of the steroid hormone ecdysone.17 Mutants for ADCK1 also display tracheal breaks and the re-expression of ADCK1 in tracheal tissue is sufficient to rescue their larval lethality, demonstrating an essential role for the gene in this tissue. The rescued adults, however, have a reduced lifespan, delayed development, and reduced body size, indicating that ADCK1 also has functions outside the trachea. These studies provide a foundation for understanding the roles of ADCK1 during larval and adult Drosophila development.
2 |. RESULTS
2.1 |. ADCK1 is conserved through evolution
To investigate the evolutionary conservation of ADCK family members we generated a phylogenetic tree using maximum likelihood analysis (Figure 1). The ADCK family of predicted kinases includes Ylr253, Ypl109, and ABC1/Coq8 (Ygl119) in yeast, CG3608, CG32649, and CG7616 in Drosophila, ADCK1–5 in mammals, and D2023.6, Y32H12A.7, and C35D10.4 in Caenorhabditis elegans. ABC1/Coq8 is most similar to CG32649 in Drosophila, C35D10.4 in C. elegans, and ADCK3 and ADCK4 in mammals. Ypl109 is most similar ADCK2 and Y32H12A.7 in C. elegans. ADCK5 is a homolog of CG7616 in Drosophila and more distantly related to Ylr253 in yeast. Our studies are focused on ADCK1, designated CG3608 in Drosophila. Although this protein is highly conserved through evolution (48% amino acid identity between fly and human), its function remains poorly understood. As a first step toward defining the functional role of ADCK1 in Drosophila, we generated deletions in this locus using CRISPR-Cas9.
2.2 |. Drosophila ADCK1 mutants are larval lethal
Two deletion mutations were generated in the ADCK1 locus, designated ADCK11 and ADCK12. ADCK11 consists of an ~1.5 kb deletion that removes part of the 5’ UTR, the start codon, and most of the protein coding sequence (Figure 2A). ADCK12 is an overlapping deficiency that removes the coding region for the N-terminal portion of the protein (Figure 2A). Transheterozygous mutants carrying both alleles die as second instar larvae, with approximately 85% (n = 34) of the animals displaying double mouth hooks, indicative of a molting defect (Figure 2B,C). The remaining animals complete the molt and display only the second instar larval mouth hooks. Similar lethal phenotypes are seen when ADCK1 homozygous mutants are analyzed. Additionally, mutant larvae appear to suffer from hypoxic stress and prematurely wander away from the food.18 Consistent with this, ADCK1 mutants display breaks in the dorsal tracheal trunks (Figure 2D,E). Both larval molting defects and tracheal breaks are associated with changes in ecdysone signaling, suggesting that ADCK1 may play a role in cholesterol homeostasis, sterol trafficking, or steroid hormone produc tion.17,19,20 Feeding 20-hydroxyecdysone to ADCK1 mutant larvae, however, had no effects on molting or viability, suggesting that ecdysone titers are not limiting.
FIGURE 2.
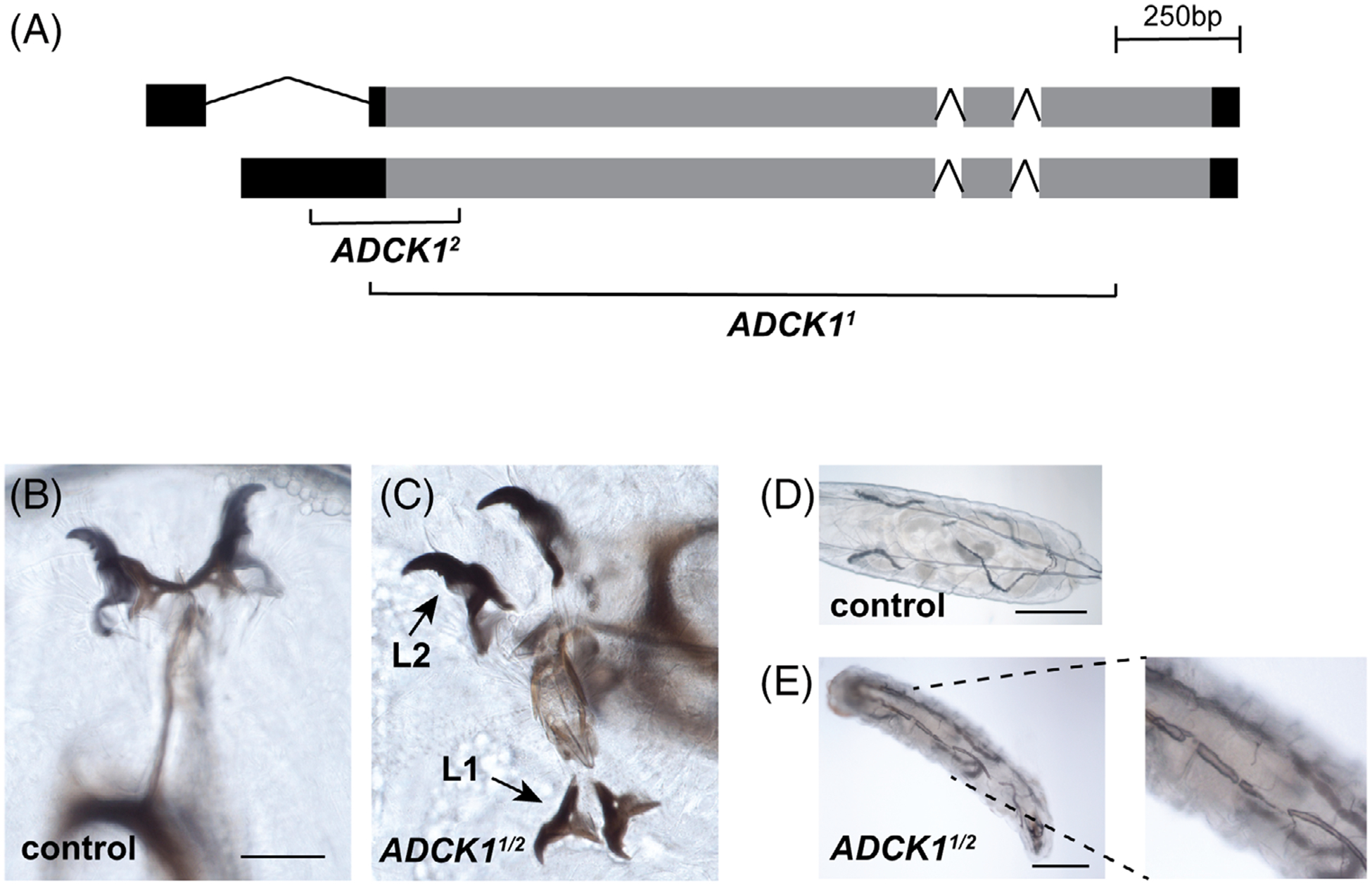
Drosophila ADCK1 mutants are larval lethal. A, The structure of the ADCK1 locus is depicted along with the locations of the two deletion alleles. The deleted regions are replaced with an eye-specific DsRed reporter in both mutants. B,C, Transheterozygotes of ADCK1/2 mutants display second instar larval lethality with double mouth hooks, indicative of a molting defect. The first and second instar mouth hooks are labeled. Scale bar: 20 μm. D,E, ADCK1/2 mutants display defects in tracheal morphology and tracheal breaks. Controls and mutants are depicted with representative images of the dorsal tracheal trunks. Panel E has an enlarged inset on the right. Scale bars: 0.25 mm (control), 0.2 mm (mutant)
2.3 |. ADCK1 is required in trachea for larval growth and development
To investigate the tissue-specific functions of ADCK1, we expressed the wild-type gene in ADCK1 mutants using the GAL4/UAS system.21 Expression of ADCK1 in the fat body (cg-GAL4, r4-GAL4), intestine (mex-GAL4, npc1b-GAL4), neurons (elav-GAL4) or prothoracic gland (phm-GAL4) had no effect on larval viability (Figure 3A). However, expressing ADCK1 ubiquitously using the da-GAL4 driver or in the tracheal system using btl-GAL4 rescued lethality through to adulthood (Figure 3A). Interestingly, the extent of rescue with these two drivers is different, with more complete rescue from the ubiquitous driver than is seen with tracheal-specific expression (Figure 3A). This suggests that, while ADCK1 expression is sufficient in the trachea for larval development, its activity is required in other tissues for complete viability. Consistent with this, ADCK1 is expressed widely in Drosophila, with higher levels in the adult intestine, fat body, and heart.22
FIGURE 3.
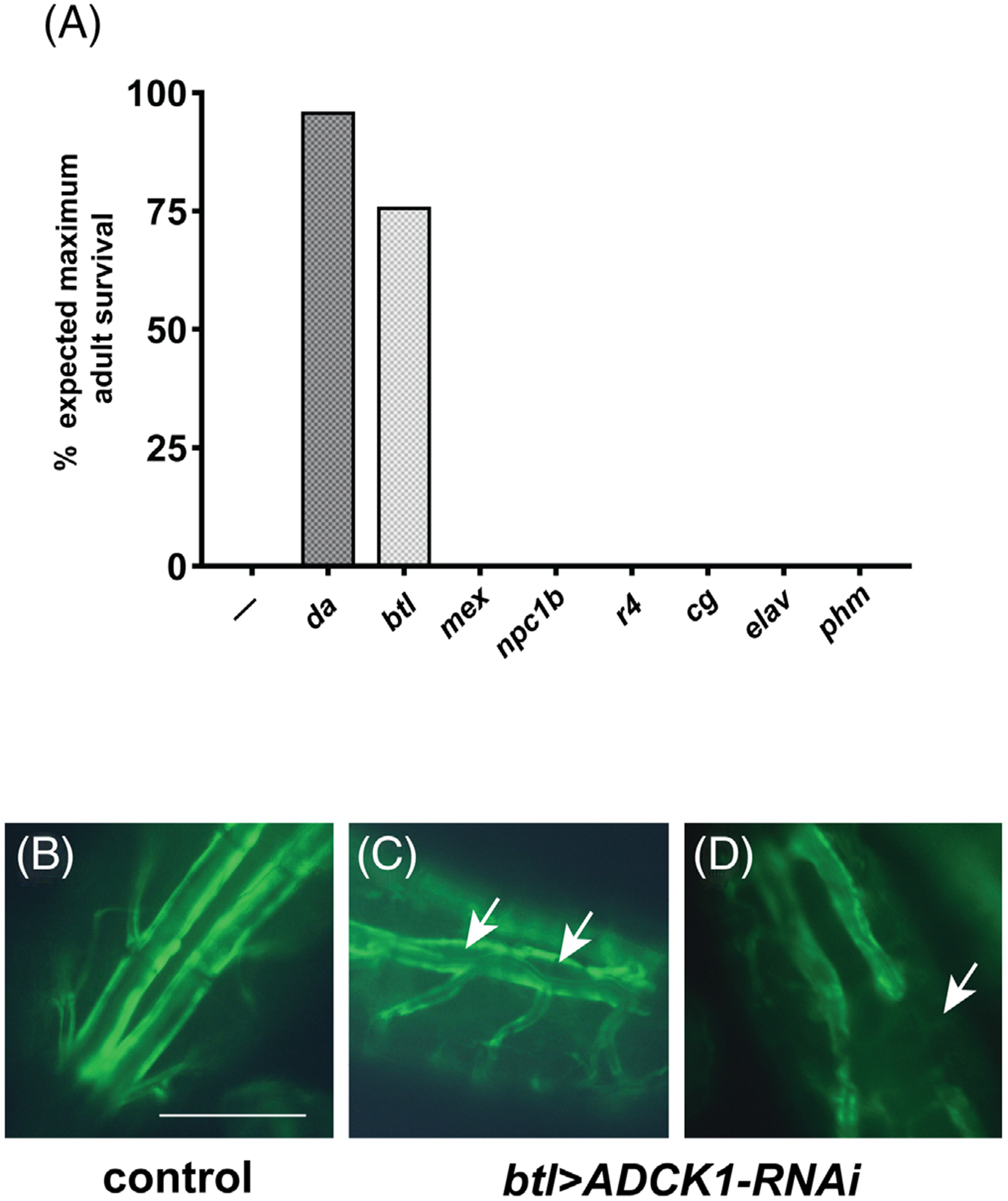
ADCK1 acts in the trachea to support larval viability. A, Tissue-specific GAL4 drivers listed on the x-axis were combined with the UAS-ADCK1 transgene in an ADCK1/2 mutant background and scored for adult viability relative to ADCK1 mutants alone (—). The efficiency of rescue is shown as a percentage of adult offspring relative to the expected value for full rescue. n ≥200. Ubiquitous (da-GAL4), fat body (cg-GAL4, r4-GAL4), intestine (mex-GAL4, npc1b-GAL4), neuronal (elav-GAL4), tracheal (btl-GAL4), and prothoracic gland (phm-GAL4) drivers were used. B-D, Dorsal tracheal trunks were imaged from offspring obtained by crossing btl-GAL4, UAS-GFP to either UAS-mCherry RNAi (control, B) or UAS-ADCK1-RNAi (C,D). The arrows mark regions that display either abnormal tracheal morphology (C) or tracheal breaks (D). Scale bar = 0.25 mm
To investigate whether ADCK1 is necessary in trachea for larval viability we performed tissue-specific RNAi studies. Consistent with our genetic rescue results, ubiquitous loss of ADCK1 using the da-GAL4 driver resulted in fully penetrant lethality (100%, n = 725). Lethality was also seen with btl-GAL4 driving ADCK1 RNAi (100%, n = 382), as well as with a different ADCK1 RNAi line (VDRC; 100%, n = 282). The btl > ADCK1-RNAi animals arrested their development as either first instar larvae (67%) or second instar larvae (33%, n = 30). They did not, however, display double mouth hooks, suggesting that this mutant phenotype arises from a loss of ADCK1 activity outside the trachea. Additionally, btl > ADCK1-RNAi larvae display tracheal abnormalities, which includes tracheal breaks and lumen separation (Figure 3B–D). Consistent with these morphological defects, these larvae prematurely wander away from the food possibly due to hypoxic stress. Taken together, our tissue-specific genetic rescue and RNAi studies suggest that ADCK1 is necessary and sufficient in the trachea for larval viability and growth as well as tracheal integrity.
2.4 |. Tracheal-rescued ADCK1 mutants are small and have a reduced lifespan
Phenotypic characterization of ADCK1 mutants is complicated by their lethality as early second instar larvae. We thus took advantage of the mutants rescued to adulthood to conduct initial functional studies of ADCK1 outside of trachea. In particular, we were interested in examining the possibility that ADCK1 might contribute to longevity and systemic metabolism given its mitochondrial localization and expression in the fat body and intestine, which play central roles in maintaining metabolic homeostasis.
As a first step toward this goal, we examined the lifespan of tracheal-rescued ADCK1 mutants. Both male and female rescued ADCK1 mutants display a reduced lifespan relative to controls (Figure 4A,B). Interestingly, the ubiquitous rescue of ADCK1 is more efficient at extending lifespan than tracheal-specific rescue. This supports our tissue-specific genetic rescue studies and suggests that ADCK1 has important activities outside of the trachea that are required for the overall health of the animal. In addition, we noticed that tracheal-rescued animals eclose approximately 2 days later than controls and this developmental delay can be rescued by ubiquitous ADCK1 expression using da-GAL4 (Figure 5A). Delayed development is often accompanied by a reduction in body size and, consistent with this, pupal body size is reduced in tracheal-rescued mutants (Figure 5B). In addition, tracheal-rescued ADCK1 mutant adults have reduced protein levels in both males and females (Figure 5C,D), further supporting a role for ADCK1 in growth during larval stages.
FIGURE 4.
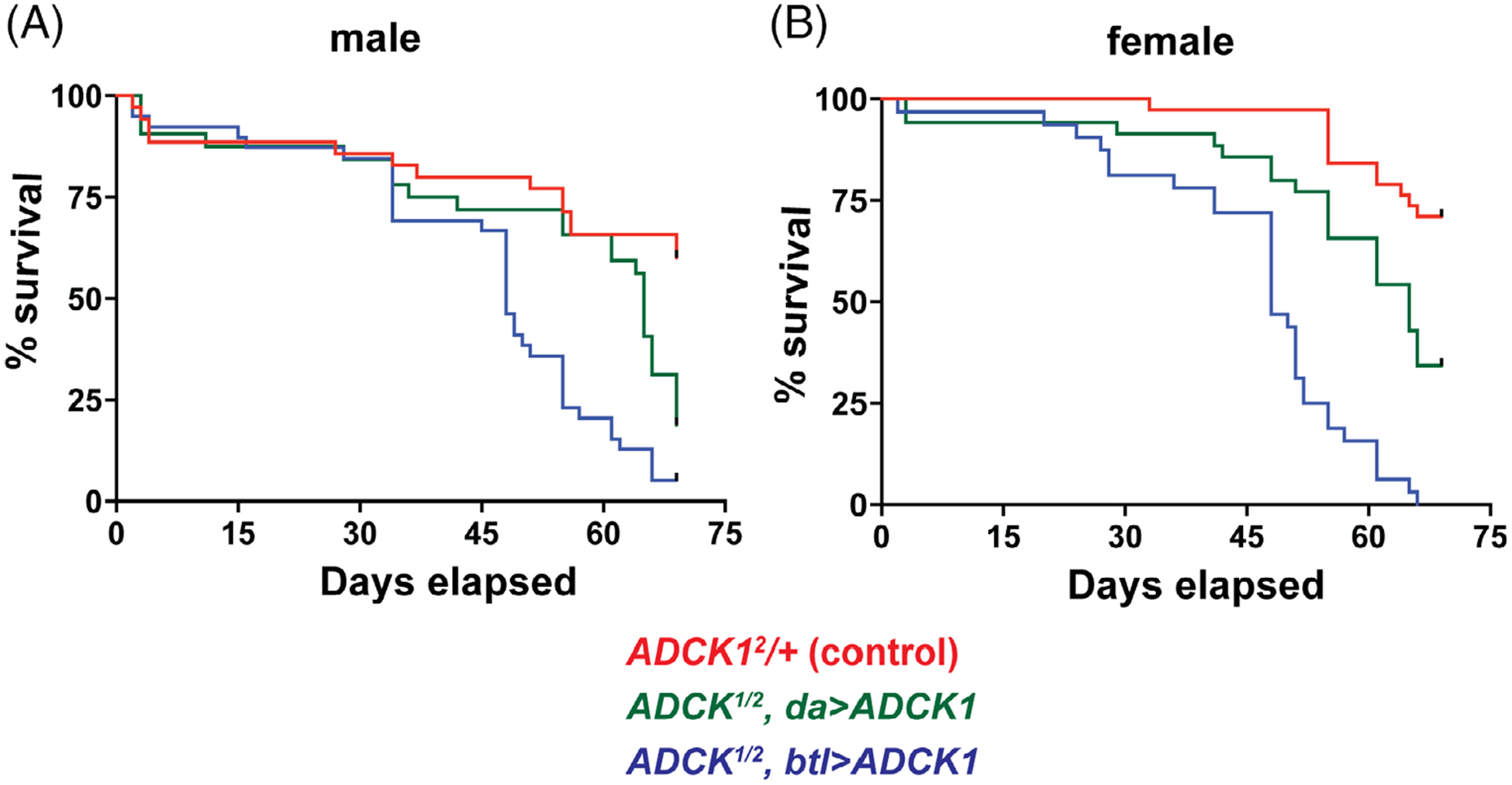
Tracheal-rescued ADCK1 mutant adults have a reduced lifespan. A,B, ADCK1 mutants were rescued to adulthood using either the ubiquitous da-GAL4 driver (green) or the tracheal-specific btl-GAL4 driver (blue) and the lifespan of males (A) or mated females (B) was determined, along with genetically-matched controls (red). P values for the lifespan curves are: tracheal-rescued males compared to controls P ≤ .0001, tracheal-rescued females compared to controls P ≤ .0001, tracheal-rescued males compared to da-rescued males P ≤ .01, tracheal-rescued females compared to da-rescued females, P ≤ .0001, da-rescued males compared to controls P ≤ .01, da-rescued females compared to controls P ≤ .01. n ≥ 35 flies for each genetic condition
FIGURE 5.
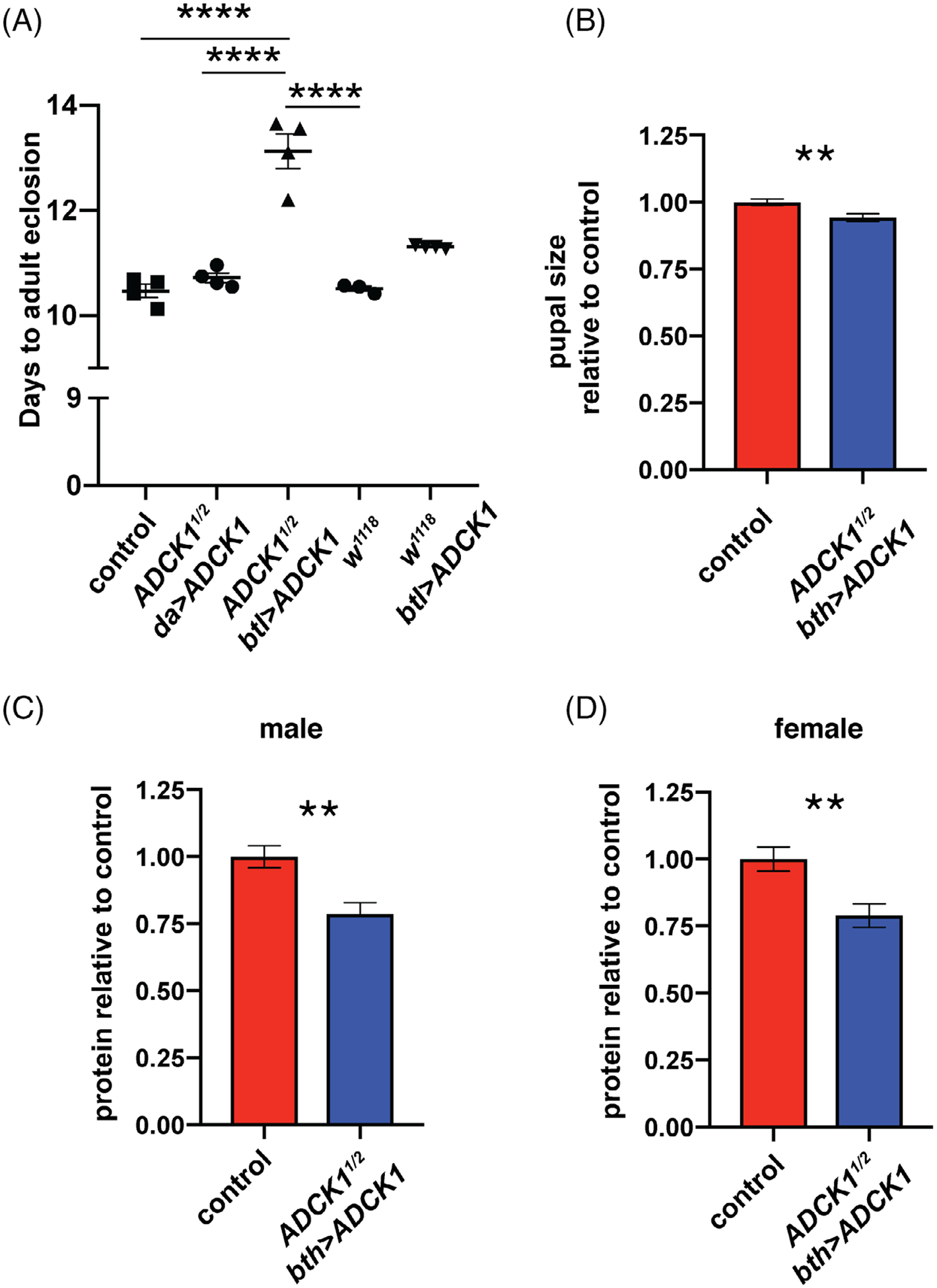
Tracheal-rescued ADCK1 mutants are developmentally delayed and have reduced body size. A, Developmental delay was calculated for w1118 animals, w1118; ADCK11/+ hemizygotes (control), ubiquitously-rescued mutants (ADCK1/2 da > UAS-ADCK1), tracheal-rescued mutants (ADCK1/2 btl > UAS-ADCK1), and control animals with tracheal-specific ADCK1 overexpression (w1118, btl > UAS-ADCK1). The time required for 50% of the population to eclose is shown on the y-axis. Each data point represents results from one collection of animals (each collection n ≥ 150) and the bars depict the mean ± SEM. P values were calculated using one-way ANOVA with Tukey’s multiple comparison test. B, Tracheal-rescued mutant pupae are smaller than controls. Pupal areas were measured and are presented as normalized to the size of w1118 control pupae. C,D, Protein levels were calculated using a Bradford assay in tracheal-rescued males (C) and females (D). Bars represent mean ± SEM, n = 8 collections of five animals for each genotype. * P ≤ .05, **P ≤ .01, ****P ≤ .0001
To address possible metabolic functions for ADCK1, we assayed the major forms of stored energy in tracheal-rescued mutants, stored carbohydrates in the form of glycogen and stored lipids as triglycerides, as well as free glucose (Figure 6A–D). None of these metabolites, however, are affected in tracheal-rescued ADCK1 mutants, suggesting that this gene does not play a critical role in maintaining adult metabolic homeostasis under normal feeding conditions.
FIGURE 6.
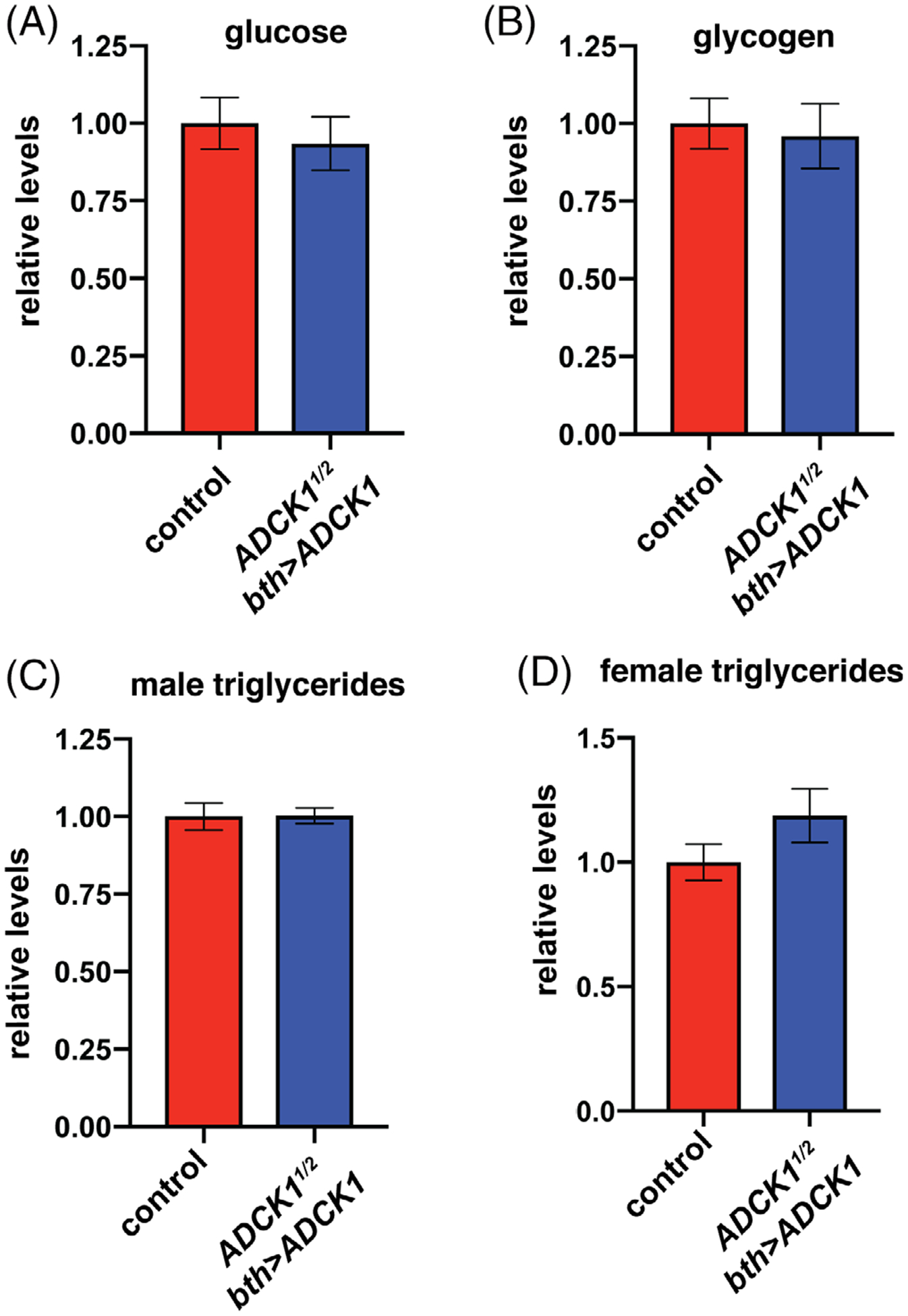
Tracheal-rescued ADCK1 mutants have normal levels of basic metabolites. A-D, Glucose (A), glycogen (B), and triglycerides from males (C) or females (D) were measured in mature tracheal-rescued ADCK1 mutant adults. n = 8 for each sample. Metabolite levels are normalized to soluble protein levels and presented relative to the controls. Bars represent mean ± SEM, n ≥ 8 (A), n ≥ 20 (B), and n ≥ 15 (C,D) collections of five animals for each genotype
3 |. DISCUSSION
In this study, we present a functional analysis of ADCK1 during development. Drosophila ADCK1 mutants die as second instar larvae with molting defects and tracheal breaks, each of which is associated with reduced signaling by the steroid hormone ecdysone.17,19,20 ADCK1 does not, however, appear to impact systemic sterol levels since we cannot rescue the mutants with dietary ecdysone, which is a standard test for phenotypes caused by a reduced ecdysone titer. In addition, specific expression of wild-type ADCK1 in the prothoracic glands of ADCK1 mutants, where ecdysone is produced, has no effect on larval viability, suggesting that this gene has no role in ecdysteroid biosynthesis. Rather, considering that ADCK1 could encode a lipid kinase like other ADCK family members, one possibility to consider is a role for ADCK1 in phosphorylating ecdysteroids. Ecdysteroid phosphates provide a major source of inactive stored hormone in the yolk granules of insect oocytes.23–25 Thus, reduced ecdysteroid phosphorylation could lead to increased rather than decreased titers of active hormone in ADCK1 mutants. Consistent with this model, increased ecdysone signaling leads to a shortened lifespan in Drosophila, similar to the effects we see on lifespan in rescued ADCK1 mutants.26,27 An alternative model to consider is that ADCK1 mutants suffer from defects in intracellular sterol trafficking. Mutants for the Drosophila Niemann-Pick Disease gene npc1a, which is required for intracellular cholesterol trafficking, die as first instar larvae with reduced ecdysteroid titers.28,29 Direct ecdysone titer measurements in ADCK1 mutants would provide a means of testing these models and gaining further insights into ADCK1 function in Drosophila.
Our tissue-specific genetic rescue and RNAi studies indicate the ADCK1 is both necessary and sufficient in the trachea for viability. It remains unclear, however, how the tracheal expression of wild-type ADCK1 can rescue the molting defects and second instar lethality since these are not known to be associated with tracheal function. It is also worth noting that ubiquitous expression of wild-type ADCK1 in mutants is more efficient at rescuing larval viability and lifespan than tracheal- specific expression, suggesting that ADCK1 has important functions in peripheral tissues. Further studies could address this in more detail, including systematic ADCK1 RNAi studies using a range of tissue-specific GAL4 drivers. More detailed phenotypic characterization of the tracheal-rescued ADCK1 mutants could address if the partial lethality and/or developmental delay seen in these animals is due to a reduced ecdysone titer arising from ADCK1 activity outside the trachea. One interesting model to consider in this regard is a potential role for Shade, which is a mitochondrial enzyme that catalyzes the conversion of ecdysone to active 20-hydroxyecdysone (20E) in peripheral tissues.30 Roles for ADCK1 in 20E production could be addressed by determining the levels of sterol intermediates in tracheal-rescued ADCK1 mutants, including ecdysone and 20E.
The requirement for ADCK1 in tracheal molting and morphology are consistent with the known roles for lipid metabolism in tracheal development. The fatty acyl-CoA reductase encoded by the waterproof locus is required for gas filling of the trachea at the molts, and lipoprotein-mediated lipid uptake is necessary for the proper structure of the tracheal lumen.31,32 In addition, lipid phosphate phosphatases encoded by the Drosophila Wunen family are required for normal tracheal development during embryogenesis.33 These enzymes are integral membrane proteins that catalyze the dephosphorylation of a wide range of lipid phosphates.34 Given that functional studies of ADCK1 in yeast have implied a role for this putative lipid kinase in intracellular lipid trafficking, it is interesting to consider that the tracheal defects in Drosophila ADCK1 mutants could arise from a defect in this process.15 Further studies are required to determine if ADCK1 contributes to lipid uptake or intracellular lipid trafficking in the trachea, and if these activities could explain the tracheal defects that we observe in ADCK1 mutant larvae. Finally, it should be noted that, as this study was in its final stages of revision, a article was published describing roles for Drosophila ADCK1 in the muscle.35 This study supports and extends the work shown here by demonstrating roles for this gene in mitochondrial function and motility.
Taken together, our work in Drosophila establishes a requirement for ADCK1 in tracheal development and larval molting, both of which are consistent with a role for this predicted mitochondrial kinase in sterol modifications and/or intracellular lipid trafficking. In addition, our characterization of tracheal-rescued ADCK1 mutant adults has revealed roles for this gene in lifespan, developmental timing, and overall growth. These studies provide a new context to define the molecular functions of ADCK1 as well as directions for future work that could enhance our understanding of this poorly studied member of the ADCK family of mitochondrial proteins.
4 |. EXPERIMENTAL PROCEDURES
4.1 |. Drosophila strains and media
Drosophila was raised at 25 °C on media containing 8% yeast, 9% sugar, and 1% agar. Larval experiments were performed on egg caps containing grape juice supplemented with yeast paste. All genetic studies were performed using a transheterozygous combination of w1118; ADCK11 and w1118; ADCK2 alleles. w1118; ADCK11/+ transheterozygotes were used as a control genotype.
4.2 |. Generation of ADCK1 deletion mutants
Mutations in Drosophila ADCK1 were generated using CRISPR-Cas9 as described.36 Our strategy resulted in two overlapping deletions in the ADCK1 locus, with the deleted region replaced by an eye-specific DsRed reporter to facilitate the identification of targeted mutants (Figure 2A). The 3XP3-DsRed marker was inserted in an opposite orientation from ADCK1. ADCK11 consists of a 1553 bp deletion and ADCK12 carries a 323 bp deletion. Guide RNAs for ADCK11: 5’ guide GTGAAAGCCCAAAGGTTAATCGG, 3’ guide GAACCGGATGACCGCCTTCTGGG. Guide RNAs for ADCK12: 5’ guide GCCAAAAATCAGTCAGCGGAGGG, 3’ guide GGGAACTTTACTACAGGGAATGG. Both ADCK11 and ADCK2 mutants were outcrossed to w1118 to provide a consistent genetic background for functional studies.
4.3 |. Generation of the UAS-ADCK1 transgenic flies
The ADCK1 coding region was amplified from the plasmid (DGRC SD09850) using the forward primer AAGCGAATT CATGCTGTTGCGTCGCGTTCTGGGC and reverse primer AAGCGGTACCCTAAATAACCTGCTTGAGAGCTTCAA G. The ADCK1 cDNA amplicon was digested with EcoR1 and Kpn1 and then inserted into the pUAST-attB plasmid digested with the same enzymes. The presence of the ADCK1 cDNA in the pUAST-attB plasmid was verified by restriction digestion and DNA sequencing. The final UAS-ADCK1 transgene was inserted into the fly genome at the attP40 site on the second chromosome.37
4.4 |. Phylogenetic reconstruction
NCBI genome sequence databases were queried to obtain the peptide sequences for human ADCK proteins as well as the homologous yeast, C. elegans and D. melanogaster ADCK protein sequences. Protein FASTA sequences were aligned using MUSCLE (MUltiple Sequence Comparison by Log Expectation) and the aligned peptide sequences were uploaded to MEGA7 to obtain a maximum likelihood phylogram.38
4.5 |. Tissue-specific genetic rescue studies
The deletion line w1118; ADCK12 was recombined with the UAS-ADCK1 rescue construct, which is also on the second chromosome, and maintained with a CyO balancer. This stock was crossed to w1118; ADCK11 /CyO animals carrying different tissue-specific GAL4 drivers in the same genetic background. To calculate the efficiency of rescue with each GAL4 driver, we compared the proportion of surviving rescued animals to the expected Mendelian ratio, assuming a complete rescue. Since the ADCK1 mutant males and females used for each cross carry a CyO balancer, the expected Mendalian ratio for complete rescue in their offspring would be 33% of the final population lacking CyO (rescued ADCK11/ADCK12 mutants), with the remaining 66% of the population carrying the CyO balancer (ADCK1/CyO or ADCK12/CyO). Eggs were collected on grape juice caps overnight and 200 eggs were transferred to standard food vials. Surviving adults were scored for the presence or absence of the CyO balancer.
4.6 |. Developmental delay
To calculate the developmental delay, each cross was allowed to lay eggs for an 8-hour window. The number of adults that eclosed in that vial was recorded daily. This data was used to calculate the number of days it took for 50% of the population to eclose, as presented in Figure 5A.
4.7 |. Pupal size determination
To quantify pupal size, control or tracheal-rescued ADCK11/ADCK12 mutant pupae were removed and placed on a cover slip with double-sided tape. Sixty pupae for each genotype were imaged and the area of each animal was calculated using ImageJ software. The sizes of tracheal-rescued ADCK1 mutants were normalized to the controls for the figure.
4.8 |. Metabolite assays
Adult flies were collected 4 days after eclosion, aged for 7 days, and samples of five flies per replicate were flash frozen in liquid nitrogen. Homogenates prepared from these animals were subjected to metabolite assays as described.39 Triglyceride measurements were performed using a coupled colorimetric assay (Sigma T2449). Glycogen and glucose concentrations were determined using the hexokinase (HK) and/or glucose oxidase (GO) assay kits (Sigma GAHK20, GAGO20). Total protein levels were determined in parallel by Bradford assay.
4.9 |. Longevity
For adult lifespan studies, 4-day-old flies were sorted and mated with the opposite sex for 7 days, after which 10–15 flies were transferred to vials containing 8% yeast, 9% sugar. Animals were scored for lethality daily and transferred to fresh food every 2 days.
4.10 |. Statistics
GraphPad PRISM 6 software was used to plot data and perform statistical analysis. Pairwise comparison P-values were calculated using a two-tailed Student’s t-test unless otherwise specified. Longevity is presented as a Kaplan Meier curve and the P-values were determined using a log-rank Mantel-Cox test. Error bars represent the SE of the mean.
ACKNOWLEDGMENTS
We thank M. Metzstein for helpful discussions and providing the btl-GAL4, UAS-GFP stock, A-F. Ruaud and J. McCormick for help with the phylogenetic analysis of ADCK family members, the Bloomington Stock Center for fly stocks (NIH P40OD018537), the Transgenic RNAi Project (TRiP), and FlyBase for informatic support. This work was supported by the NIH (R01GM094232, R01CA228346).
Funding information
National Cancer Institute, Grant/Award Number: R01CA228346; National Institute of General Medical Sciences, Grant/Award Number: R01GM094232
REFERENCES
- 1.Rutter J, Hughes AL. Power(2): the power of yeast genetics applied to the powerhouse of the cell. Trends Endocrinol Metab. 2015;26:59–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Spinelli JB, Haigis MC. The multifaceted contributions of mitochondria to cellular metabolism. Nat Cell Biol. 2018;20:745–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gray LR, Tompkins SC, Taylor EB. Regulation of pyruvate metabolism and human disease. Cell Mol Life Sci. 2014;71:2577–2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wallace DC. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu Rev Genet. 2005;39:359–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pagliarini DJ, Rutter J. Hallmarks of a new era in mitochondrial biochemistry. Genes Dev. 2013;27:2615–2627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Acosta MJ, Vazquez Fonseca L, Desbats MA, et al. Coenzyme Q biosynthesis in health and disease. Biochim Biophys Acta. 2016; 1857:1079–1085. [DOI] [PubMed] [Google Scholar]
- 7.Barca E, Musumeci O, Montagnese F, et al. Cerebellar ataxia and severe muscle CoQ10 deficiency in a patient with a novel mutation in ADCK3. Clin Genet. 2016;90:156–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cullen JK, Abdul Murad N, Yeo A, et al. AarF domain containing kinase 3 (ADCK3) mutant cells display signs of oxidative stress, defects in mitochondrial homeostasis and lysosomal accumulation. PLoS One. 2016;11:e0148213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lagier-Tourenne C, Tazir M, Lopez LC, et al. ADCK3, an ancestral kinase, is mutated in a form of recessive ataxia associated with coenzyme Q10 deficiency. Am JHum Genet. 2008;82:661–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mollet J, Delahodde A, Serre V, et al. CABC1 gene mutations cause ubiquinone deficiency with cerebellar ataxia and seizures. Am J Hum Genet. 2008;82:623–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stefely JA, Reidenbach AG, Ulbrich A, et al. Mitochondrial ADCK3 employs an atypical protein kinase-like fold to enable coenzyme Q biosynthesis. Mol Cell. 2015;57:83–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ashraf S, Gee HY, Woerner S, et al. ADCK4 mutations promote steroid-resistant nephrotic syndrome through CoQ10 biosynthesis disruption. J Clin Invest. 2013;123:5179–5189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vazquez Fonseca L, Doimo M, Calderan C, et al. Mutations in COQ8B (ADCK4) found in patients with steroid-resistant nephrotic syndrome alter COQ8B function. Hum Mutat. 2018;39: 406–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stefely JA, Licitra F, Laredj L, et al. Cerebellar ataxia and coenzyme Q deficiency through loss of unorthodox kinase activity. Mol Cell. 2016;63:608–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tan T, Ozbalci C, Brugger B, Rapaport D, Dimmer KS. Mcp1 and Mcp2, two novel proteins involved in mitochondrial lipid homeostasis. J Cell Sci. 2013;126:3563–3574. [DOI] [PubMed] [Google Scholar]
- 16.Kornmann B, Walter P. ERMES-mediated ER-mitochondria contacts: molecular hubs for the regulation of mitochondrial biology. J Cell Sci. 2010;123:1389–1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Neubueser D, Warren JT, Gilbert LI, Cohen SM. Molting defective is required for ecdysone biosynthesis. Dev Biol. 2005;280: 362–372. [DOI] [PubMed] [Google Scholar]
- 18.Wingrove JA, O’Farrell PH. Nitric oxide contributes to behavioral, cellular, and developmental responses to low oxygen in Drosophila. Cell. 1999;98:105–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fisk GJ, Thummel CS. The DHR78 nuclear receptor is required for ecdysteroid signaling during the onset of Drosophila metamorphosis. Cell. 1998;93:543–555. [DOI] [PubMed] [Google Scholar]
- 20.Lam G, Hall BL, Bender M, Thummel CS. DHR3 is required for the prepupal-pupal transition and differentiation of adult structures during Drosophila metamorphosis. Dev Biol. 1999; 212:204–216. [DOI] [PubMed] [Google Scholar]
- 21.Caygill EE, Brand AH. The GAL4 system: a versatile system for the manipulation and analysis of gene expression. Methods Mol Biol. 2016;1478:33–52. [DOI] [PubMed] [Google Scholar]
- 22.Robinson SW, Herzyk P, Dow JA, Leader DP. FlyAtlas: database of gene expression in the tissues of Drosophila melanogaster. Nucleic Acids Res. 2013;41:D744–D750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gibson J, Isaac R, Dinan L, Rees H. Metabolism of (3H)-ecdysone in Schistocerca gregaria: formation of ecdysteroid acids together with free and phosphorylated ecdysteroid acetates. Arch Insect Biochem Phys. 1984;1:385–407. [Google Scholar]
- 24.Ito Y, Yasuda A, Sonobe H. Synthesis and phosphorylation of ecdysteroids during ovarian development in the silkworm, Bombyx mori. Zoolog Sci 2008;25:721–727. [DOI] [PubMed] [Google Scholar]
- 25.Sonobe H, Ohira T, Ieki K, et al. Purification, kinetic characterization, and molecular cloning of a novel enzyme, ecdysteroid 22-kinase. J Biol Chem. 2006;281:29513–29524. [DOI] [PubMed] [Google Scholar]
- 26.Simon AF, Shih C, Mack A, Benzer S. Steroid control of longevity in Drosophila melanogaster. Science. 2003;299:1407–1410. [DOI] [PubMed] [Google Scholar]
- 27.Tricoire H, Battisti V, Trannoy S, Lasbleiz C, Pret AM, Monnier V. The steroid hormone receptor EcR finely modulates Drosophila lifespan during adulthood in a sex-specific manner. Mech Ageing Dev. 2009;130:547–552. [DOI] [PubMed] [Google Scholar]
- 28.Fluegel ML, Parker TJ, Pallanck LJ. Mutations of a Drosophila NPC1 gene confer sterol and ecdysone metabolic defects. Genetics. 2006;172:185–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huang X, Suyama K, Buchanan J, Zhu AJ, Scott MP. A Drosophila model of the Niemann-pick type C lysosome storage disease: dnpc1a is required for molting and sterol homeostasis. Development. 2005;132:5115–5124. [DOI] [PubMed] [Google Scholar]
- 30.Petryk A, Warren JT, Marques G, et al. Shade is the Drosophila P450 enzyme that mediates the hydroxylation of ecdysone to the steroid insect molting hormone 20-hydroxyecdysone. Proc Natl Acad Sci USA. 2003;100:13773–13778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Baer MM, Palm W, Eaton S, Leptin M, Affolter M. Microsomal triacylglycerol transfer protein (MTP) is required to expand tracheal lumen in Drosophila in a cell-autonomous manner. J Cell Sci. 2012;125:6038–6048. [DOI] [PubMed] [Google Scholar]
- 32.Jaspers MH, Pflanz R, Riedel D, Kawelke S, Feussner I, Schuh R. The fatty acyl-CoA reductase waterproof mediates airway clearance in Drosophila. Dev Biol. 2014;385:23–31. [DOI] [PubMed] [Google Scholar]
- 33.Ile KE, Tripathy R, Goldfinger V, Renault AD. Wunen, a Drosophila lipid phosphate phosphatase, is required for septate junction-mediated barrier function. Development. 2012;139:2535–2546. [DOI] [PubMed] [Google Scholar]
- 34.Tang X, Benesch MG, Brindley DN. Lipid phosphate phosphatases and their roles in mammalian physiology and pathology. J Lipid Res. 2015;56:2048–2060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yoon W, Hwang SH, Lee SH, Chung J. Drosophila ADCK1 is critical for maintaining mitochondrial structures and functions in the muscle. PLoS Genet. 2019;15:e1008184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gratz SJ, Ukken FP, Rubinstein CD, et al. Highly specific and efficient CRISPR/Cas9-catalyzed homology-directed repair in Drosophila. Genetics. 2014;196:961–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Venken KJ, Bellen HJ. Genome-wide manipulations of Drosophila melanogaster with transposons, Flp recombinase, and PhiC31 integrase. Methods Mol Biol. 2012;859:203–228. [DOI] [PubMed] [Google Scholar]
- 38.Kumar S, Stecher G, Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33:1870–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tennessen JM, Barry WE, Cox J, Thummel CS. Methods for studying metabolism in Drosophila. Methods. 2014;68:105–115. [DOI] [PMC free article] [PubMed] [Google Scholar]