

Abstract
Mitochondria are cellular power plants that supply ATP to power various biological activities essential for neuronal growth, survival, and function. Due to extremely varied morphological features, neurons face exceptional challenges to maintain energy homeostasis. Neurons require specialized mechanisms distributing mitochondria to distal synapses where energy is in high demand. Axons and synapses undergo activity-dependent remodeling, thereby altering mitochondrial distribution. The uniform microtubule polarity has made axons particularly useful for exploring mechanisms regulating mitochondrial transport. Mitochondria alter their motility under stress conditions or when their integrity is impaired. Therefore, research into the mechanisms regulating mitochondrial motility in healthy and diseased neurons is an important emerging frontier in neurobiology. In this chapter, we discuss the current protocols in the characterization of axonal mitochondrial transport in primary neuron cultures isolated from embryonic rats and adult mice. We also briefly discuss new procedures developed in our lab in analyzing mitochondrial motility patterns at presynaptic terminals and evaluate their impact on synaptic vesicle release.
1. INTRODUCTION
Neurons are polarized cells consisting of a relatively small cell body, dendrites with multiple branches, and a thin long axon that can extend up to a meter in some peripheral nerves. Due to these morphological compartments and complex geometry of neurons, it is an exceptional challenge to maintain energy homeostasis throughout cells. Specialized mechanisms are required for neurons to efficiently distribute mitochondria to far distal areas where energy is in high demand, such as synaptic terminals, growth cones, and axonal branches (Fig. 5.1; Morris & Hollenbeck, 1993; Ruthel & Hollenbeck, 2003; also see review by Sheng, 2014). Axonal branches and synapses undergo dynamic remodeling during neuronal development and in response to synaptic activity, thereby changing mitochondrial trafficking and distribution. Neurons are postmitotic cells surviving for the lifetime of the organism. A mitochondrion needs to be removed when it becomes aged or dysfunctional. Mitochondria also alter their motility and distribution under certain stress conditions or when their integrity is impaired (Cai, Zakaria, Simone, & Sheng, 2012; Chang & Reynolds, 2006; Miller & Sheetz, 2004). Therefore, efficient regulation of mitochondrial transport and anchoring is essential to: (1) recruit and redistribute mitochondria to meet altered metabolic requirements and (2) remove aged and damaged mitochondria and replenish healthy ones at distal terminals. Research into complex regulation of mitochondrial trafficking and anchoring in healthy and diseased neurons is thus a very important frontier in neurobiology (see recent in-depth reviews by Birsa, Norkett, Higgs, Lopez-Domenech, & Kittler, 2013; MacAskill & Kittler, 2010; Saxton & Hollenbeck, 2012; Schwarz, 2013; Sheng, 2014; Sheng & Cai, 2012).
Figure 5.1.
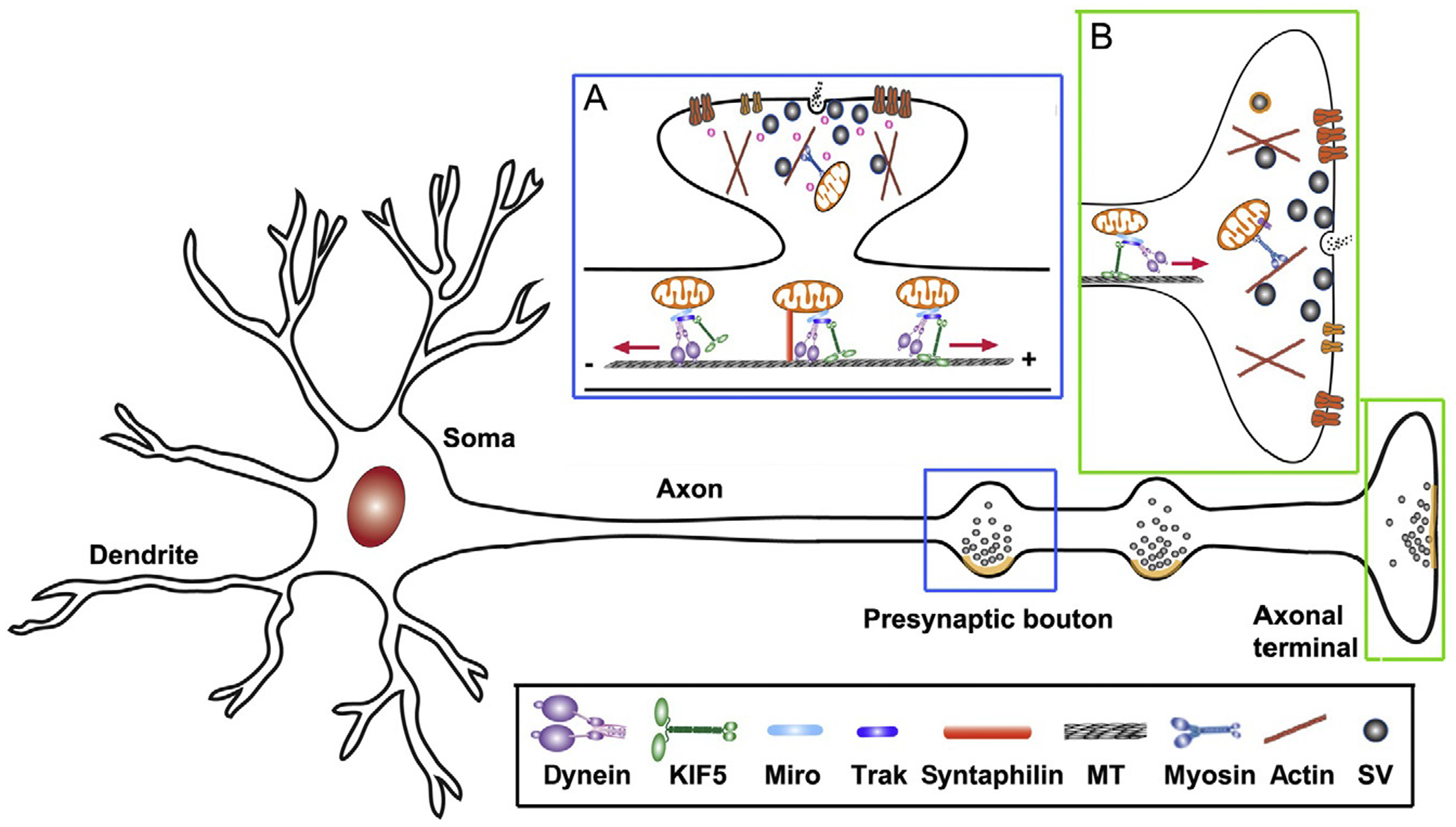
Mitochondrial transport in neurons. Mitochondria are transported to synaptic terminals (A) and distal axonal regions (B) where metabolic homeostatic capacity is in a high demand. MT-based long-distance transport relies on MT polarity. In axons, MT’s plus-ends (+) are oriented toward axonal terminals while minus-ends (−) are directed toward the soma. Thus, KIF5 motors drive mitochondria for anterograde transport to distal synaptic terminals, whereas dynein motors return mitochondria to the soma. The motor adaptor Trak-Miro protein complexes mediate KIF5- and dynein-driven transport of axonal mitochondria. Myosin motors likely drive short-range mitochondrial movement at presynaptic terminals where enriched actin filaments constitute cytoskel-etal architecture. Motile mitochondria can be recruited into stationary pools via dynamic anchoring interactions between syntaphilin and MTs (Kang et al., 2008). Such anchoring mechanisms ensure neuronal mitochondria adequately distributed along axons and at synapses, where constant energy supply is crucial. Adapted from Sheng (2014).
Long-range mitochondrial transport from the soma to distal axonal and dendritic regions depends upon the polarity and organization of neuronal microtubules (MTs). Axonal MTs are arranged so that their plus-ends (+) are directed distally and the minus-ends (−) are directed toward the soma. Thus, in axons, cytoplasmic dynein motors are responsible for returning mitochondria to the soma, whereas KIF5 motors drive anterograde mitochondrial transport to distal regions and synaptic terminals (Fig. 5.1). For dendritic processes where MTs exhibit mixed polarity in proximal regions, KIF5 and dynein motors can transport mitochondria in either anterograde or retrograde direction depending on the MT polarity. Mobile axonal mitochondria can also be recruited to stationary pools via the docking receptor syntaphilin and MTs (Chen & Sheng, 2013; Kang et al., 2008). Mitochondrial docking mechanisms ensure that stationary mitochondria are adequately distributed within axons and at synapses.
Neuronal mitochondria display complex motility patterns, which can be visualized by time-lapse imaging in live primary neuronal cultures. Mitochondria are labeled by expressing DsRed-Mito, a mitochondria-targeted fluorescent protein, or by loading fluorescent dyes such as MitoTracker Green or MitoTracker® Red CMXRos, the latter stains mitochondria in live cells depending upon membrane potential. Mitochondria move bidirectionally over long distances along processes, pause briefly, and move again, frequently changing direction. Motile mitochondria can become stationary and stationary ones can be remobilized and redistributed in response to changes in metabolic status and synaptic activity. Given frequent pauses and bidirectional movements, the mean velocities of mitochondrial transport are highly variable (Macaskill et al., 2009; Morris & Hollenbeck, 1995). In mature neurons, approximately 20–30% of axonal mitochondria are motile, some of which pass by presynaptic terminals, while the remaining two-thirds are stationary (Sun, Qiao, Pan, Chen, & Sheng, 2013).
Impaired mitochondrial transport is one of key pathogenic changes in aging-associated neurodegenerative diseases (Sheng & Cai, 2012). For example, reduced transport of axonal mitochondria was consistently reported in amyotrophic lateral sclerosis (ALS)-linked hSOD1G93A mice (Bilsland et al., 2010; De Vos et al., 2007; Magrane & Manfredi, 2009; Marinkovic et al., 2012; Song et al., 2013; Zhu & Sheng, 2011). Live imaging in primary cultured embryonic neurons provides an important cell model to study mechanisms regulating mitochondrial transport. However, this culture system has some significant limitations for the study of late-onset disease mechanisms. Therefore, establishing a neuron culture system isolated from adult mice at disease stages will provide ideal models to investigate into the mitochondrial transport defects underlying adult-onset neuronal pathogenesis. An ex vivo study of mitochondrial transport in acute nerve explants was developed in transgenic mice, in which neuronal mitochondria are labeled with mito-CFP or YFP-mito (Misgeld, Kerschensteiner, Bareyre, Burgess, & Lichtman, 2007). Given that the morphology and conditions of neurons grown in culture dishes differ from that of neurons in vivo, an in vivo transport study would better reflect physiological and pathological conditions.
2. ANALYSES OF MITOCHONDRIAL TRANSPORT IN EMBRYONIC NEURON CULTURES
Primary cultured hippocampal and cortical neurons are widely used to study the molecular mechanisms underlying regulation of mitochondrial transport. MTs are uniformly arranged in axons. Such a uniform polarity has made axons particularly useful to study mechanisms regulating bidirectional transport. Axonal mitochondrial movement can be imaged by transfection with DsRed-Mito. By analyzing 2137 axonal mitochondria from over 100 neurons, we recently revealed that most axonal mitochondria (76%) are small vesicular or short vesicular-tubular structures with a diameter <2 μm. We also found that almost all DsRed-Mito signals target mitochondria labeled by cytochrome c along axons (99.17±0.22%, n = 1078 mitochondria from 17 neurons), thus further confirming DsRed-Mito as a mitochondrial marker (Sun et al., 2013).
2.1. Preparation of embryonic neuron cultures
2.1.1. Materials
Poly-l-ornithine (Sigma, P4957)
Fibronectin (Sigma, F1141–5MG)
Ca2+/Mg2+-free PBS (Invitrogen, 10010–023)
Ca2+/Mg2+-free HBSS buffer (Hanks’ Balanced Salt Solution, Invitrogen, 14175–095)
HBSS buffer (Invitrogen, 14025–092)
Papain (Worthington, LK003176)
DNase (Worthington, LK003170)
Ovomucoid inhibitor (Worthington, 3182)
BSA (Sigma, A9647)
Trypsin inhibitor (Sigma, T9253)
Neurobasal medium (Invitrogen, 21103–049)
50 × B27 supplement (Invitrogen, 17504–044)
GlutaMAX™-I (Invitrogen, 25030–061)
Insulin (Sigma, I5500)
Fetal bovine serum (FBS, heat inactive at 57 °C for 30 min, stored at 4 °C)
Cytosine arabinoside (Ara-C, Sigma, C6645)
HEPES (Invitrogen, 15630)
100 × penicillin/streptomycin (Invitrogen, 15140–122)
2.1.2. Solutions
(All solutions should be prepared freshly and filter-sterilized using a 0.22-μm filter).
Dissection buffer: 500 ml of Ca2+/Mg2+-free HBSS buffer containing 0.1 M HEPES and 1 × penicillin/streptomycin. Keep on ice.
Papain solution: 5 ml of HBSS buffer containing 100 U of papain and 500 U of DNase.
1/1 solution: 5 ml of HBSS buffer containing 5 mg ovomucoid inhibitor and 500 units of DNase.
10/10 solution: 10 ml of HBSS containing 100 mg of BSA and 100 mg trypsin inhibitor. Mix well and incubate at 37 °C to dissolve.
Plating medium: Neurobasal medium supplemented with 10% FBS, 1 × B27, 0.5 mM GlutaMAX™-I, 0.1% 2-mercaptoethanol, and 25 μg ml−1 insulin. Prewarm in the 37 °C incubator.
Feeding medium: Neurobasal medium supplemented with 1 × B27 and 0.5 mM GlutaMAX™-I. Prewarm in the 37 °C incubator.
2.1.3. Procedures
-
Before the day of dissection, coat the clean coverslips with 25 μg ml−1 poly-l-ornithine in Ca2+/Mg2+-free PBS at 37 °C for overnight.
(Coverslips must be cleaned, rinsed, and sterilized properly. It is essential to get a high quality primary neuronal culture for live imaging).
Wash the coverslips extensively with Ca2+/Mg2+-free PBS three times, 15 min each.
Coat the coverslips with 1 μg ml−1 fibronectin in Ca2+/Mg2+-free PBS at 37 °C for 1 h before use.
Remove day 18 rat embryos from euthanatized pregnant rats and decapitate by sharp scissors.
Dissect out the brains and place in a 35-mm dish with 5 ml ice-cold dissection buffer.
Hemi-dissect the brain and remove the brain stem and diencephalon from cerebral hemispheres. Carefully peel away meninges and blood vessels from cerebrum by forceps under the dissecting scope.
Dissect out hippocampi from cerebrum by curved forceps, then collect and wash with 5 ml ice-cold Dissection Buffer.
Cut the hippocampi into thin pieces and transfer the sliced hippocampi into a 15-ml tube.
Digest the hippocampi with 5 ml papain solution at 37 °C for 45 min with gentle shake every 10 min.
Gently dissociate tissue by pipetting up and down 10–15 times with a 5-ml pipette. Do it slowly to avoid bubbles. Centrifuge at 300 × g for 5 min. (Over digestion and trituration will damage neurons.)
Remove Digestion solution and resuspend the pellet with 5 ml 1/1 solution.
Allow larger tissue pieces to settle down for 5 min and then layer the suspension on 10 ml 10/10 solution. Spin at 300 × g for 10 min.
Discard supernatant and resuspend pellet with 5 ml of prewarmed plating medium. Determine the cell density by hemocytometer.
Plate hippocampal neurons at a density of 50,000 cells cm−2 onto poly-l-ornithine/fibronectin coated coverslips.
After 24 h, remove half of the plating medium and replace with the same amount of neuronal feeding medium. At days in vitro (DIV3), apply Ara-C at a final concentration 3 μM to stop glia proliferation. (All sequential feedings are done with medium with 3 μM Ara-C.)
Feed the cells every 3 days by aspirating half of the medium and replace with the same amount of neuronal feeding medium.
2.2. Transfection of primary hippocampal neurons
Transfect neurons with various construct at DIV6–8 by using calcium phosphate method (Jiang & Chen, 2006). Live neurons are imaged at DIV9–10 by confocal microscopy.
2.2.1. Materials
Dulbecco’s modified Eagle’s medium (DMEM) medium (Invitrogen, 11995–065)
2.5 M CaCl2, filter-sterilized, aliquot, and stored in frozen (stable for at least 6 months)
2 × HeBS (274 mM NaCl, 10 mM KCl, 1.4 mM Na2HPO4, 15 mM d-glucose, 42 mM HEPES, pH 7.07). Filter-sterilized using a 0.22-μm filter. Aliquot and stored in frozen. (Avoid freeze and thaw. The pH of 2× HeBS must be between 7.01 and 7.12.)
2.2.2. Procedures
Transfer hippocampal neurons to a 35-mm dish containing prewarmed DMEM medium, and keep the original conditioned medium.
Prepare two sterilized microtubes. To one set of the tube, mix the DNA (4–6 μg),4 μl of 2.5 M CaCl2, and sterile H2O to a total volume of 40 μl. To another tube, add 40 μl of 2 × HeBS. (No more than 10 μg of total DNA for a 35-mm dish.)
Slowly drip the DNA/CaCl2 mixture to 2× HeBS and mix by vortex.
Keep the DNA mixture in dark at room temperature for 25 min.
Add dropwise the precipitated DNA to the cells, while gently swirling the culture dish.
Incubate cells with the precipitate for 25 min in the 5% CO2 incubator at 37 °C. Check under microscopy, the sand-like DNA/Ca2+ precipitates should cover cells after 25 min incubation.
Transfer cells to the DMEM medium, which is preequilibrated in 16% CO2 incubator at 37 °C for 30 min. (The 16% CO2 will make the solution more acidic. This step helps to dissolve the precipitates, which are toxic to cells.)
Incubate cells in the 5% CO2 incubator at 37 °C for 10 min. Transfer cells back to the original dish containing conditioned medium. Image neurons 2–4 days after transfection.
2.3. Image acquisition and analysis in live neurons
2.3.1. Image acquisition
In live imaging experiments, axons can be recognized by their unique morphology characteristics: long, thin, uniform in diameter, growth cones, and sparse branches (Banker & Cowan, 1977). Axonal mitochondria are generally small and vesicular in structure, and are sparsely distributed throughout axons. In contrast, dendritic mitochondria are elongated tubular structures and occupy a large proportion of the neurite (Chang, Honick, & Reynolds, 2006; Popov, Medvedev, Davies, & Stewart, 2005).
Time-lapse imaging is recorded in the proximal or distal segment of axons, and only select single processes distinct from others in the field. (Segments with crossing or fasciculation are excluded from analysis.)
Transfer transfected hippocampal neurons to Tyrode’s buffer (10 mM HEPES, 10 mM glucose, 1.2 mM CaCl2, 1.2 mM MgCl2, 3 mM KCl, and 145 mM NaCl, pH 7.4). Maintain temperature at 37 °C with an air stream incubator.
Image cells with a 40× objective oil-immersion lens on a LSM 510 Zeiss confocal microscope, using 543 nm for DsRed and TMRE. To minimize laser-induced photobleaching while maximizing pinhole opening, time-lapse images at 10-s intervals are acquired with 1% laser intensity.
Start recording 6 min after the coverslip is placed in the chamber. Time-lapse imaging of labeled mitochondria is acquired at a 512 × 512-pixel resolution (8 bit) for total of 100 frames (Kang et al., 2008).
2.3.2. Image analysis
Image stacks are imported into the NIH ImageJ software and convert to QuickTime movies. Kymographs are created from an extra ImageJ plugin (Kang et al., 2008; Miller & Sheetz, 2004). The “LSM Reader” plugin is used to read Zeiss* lsm images, curved axons are straightened via the “Straighten” plugin, resliced time-lapse images are z-axially projected with the “Grouped ZProjector” plugin, and time-stamped images/frames are produced with the “Time Stamper” plugin. The x-axis of kymographs represents the length (μm) of imaged axon, while the γ-axis represents recording time. Hundred frames for each recording are averaged to ensure accurate representation of transport events (Cai et al., 2012; Kang et al., 2008). Vertical lines in the kymograph represent stationary mitochondria, while slanted lines or curves represent motile mitochondria and direction of movement (Fig. 5.2). The relative mobility is defined by comparing the percentage of mitochondria undergoing retrograde and anterograde movements versus stationary ones. For a mitochondrion to be considered stationary, it must remain immotile (displacement ≤5 μm) for the entire recording period. To quantify mitochondrial velocity, average the distance displaced per second for anterograde and retrograde movements. Mitochondrial flux is quantified by counting the number of anterograde and retrograde events per minute.
Figure 5.2.
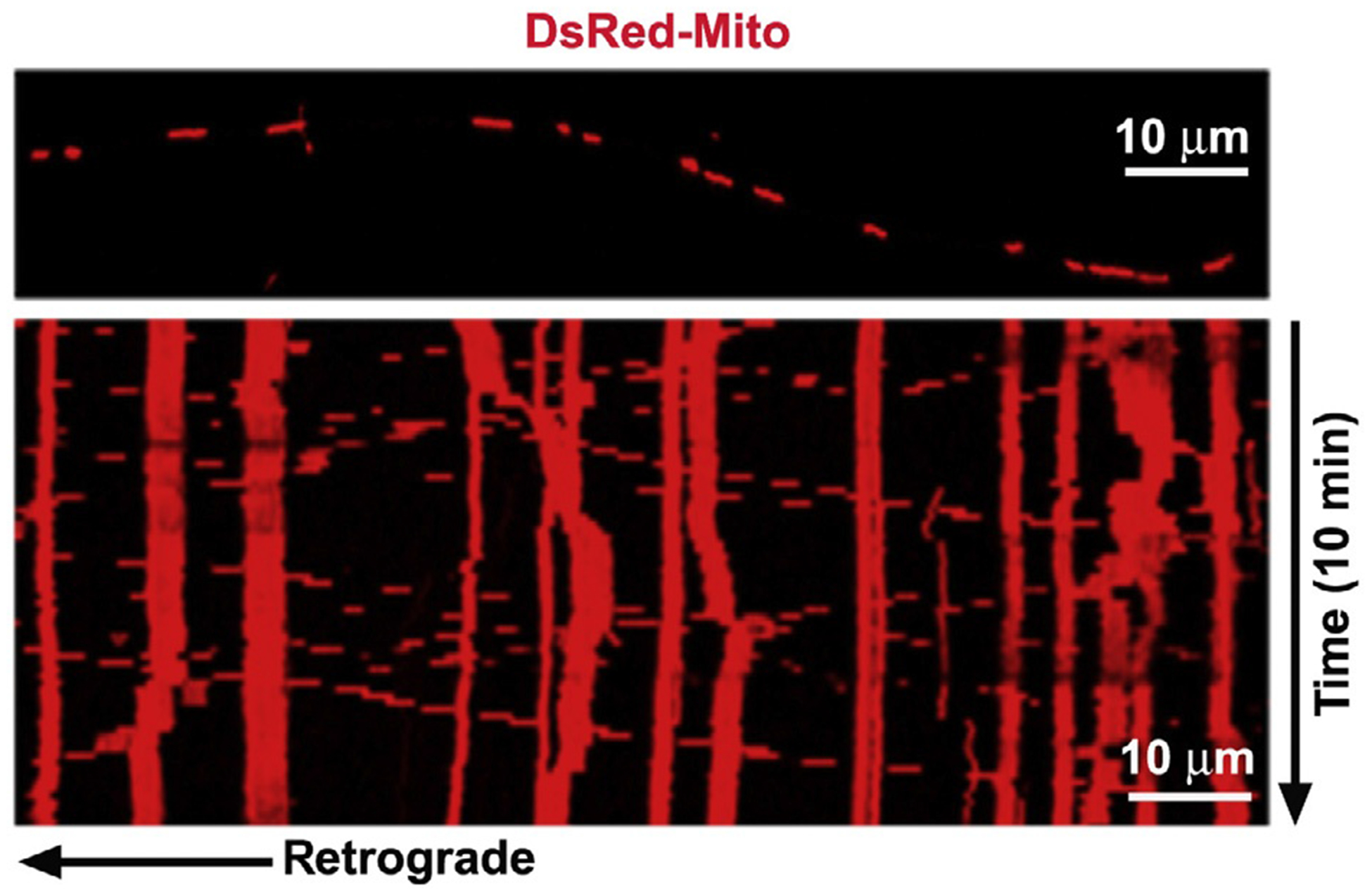
Analysis of mitochondrial motility. Representative axonal image shows DsRed-labeled mitochondria (upper), and the corresponding time-lapse imaging of mitochondrial movement is converted into kymograph (lower). In kymographs, vertical lines represent stationary mitochondria, while slanted or curved lines indicate mobile mitochondria. To quantify relative motility, a mitochondrion is considered stationary if it remained immotile (displacement ≤5 μm) during the entire recording period (10 min). The total number of mitochondria is defined as the number of mitochondria in each frame.
3. ANALYSES OF MITOCHONDRIAL TRANSPORT IN ADULT NEURON CULTURES
Mitochondrial pathology is one of the most notable hallmarks in major age-associated neurodegenerative diseases. Proper removal of damaged mitochondria from distal synapses may serve as an early neuroprotective mechanism. Cultured embryonic sensory and motor neurons (MNs) have been widely used for investigating MN degeneration such as ALS-linked pathogenesis. However, these cell models have their significant limitations in revealing altered cellular pathways associated with adult-onset pathogenesis. Studying these dynamic cellular processes in live neurons isolated from disease-stage adult mice will provide reliable models for delineating defects of mitochondrial transport underlying adult-onset pathogenesis. Sensory and MNs can be isolated from adult mice and maintained in culture medium for weeks (Fig. 5.3). Sensory neurons can be isolated from dorsal root ganglias (DRGs), which are located in the intervertebral foramen along the spinal column. Spinal MNs are enriched in the ventral horn of spinal cord. Isolated sensory and MNs can be used for characterizing axonal transport of various cargoes, including mitochondria and endosomes. Isolated adult neurons enable molecular manipulation by expressing transgenes and mitochondria can be labeled by expressing DsRed-Mito or by loading fluorescent dyes to monitor organelles under physiological and pathological conditions. Compared to embryonic neuron cultures, a key advantage in using adult neuron cultures is that it bypasses early development and provides an ideal cell model for late-onset neurodegenerative disease. The downside of adult neuron cultures is the limited number of cells that can be acquired, thus preventing from biochemical analyses of cell lysates.
Figure 5.3.
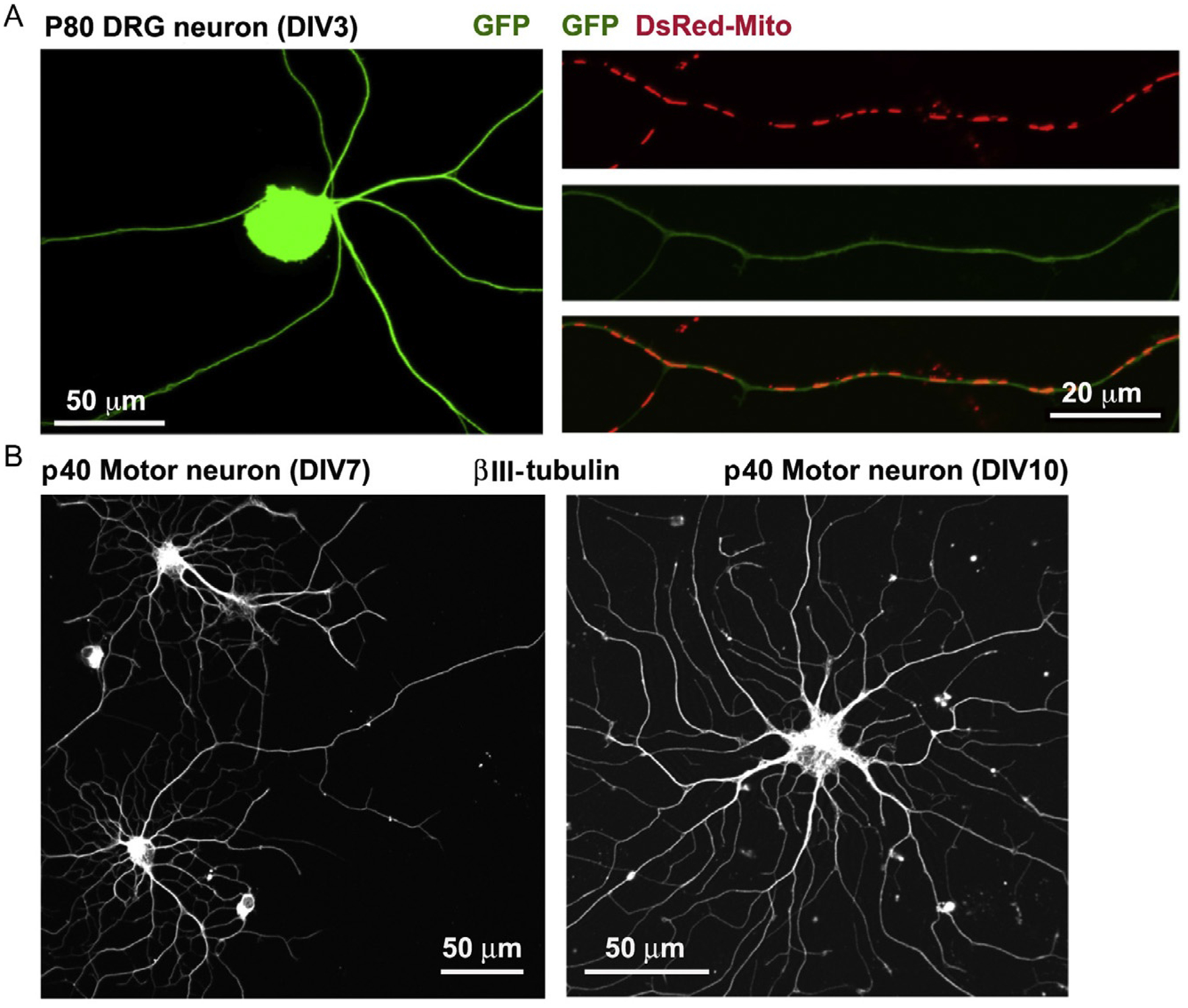
Cultured DRG neurons and motor neurons from adult mice. (A) Representative images of cultured DRG neurons at DIV3 isolated from p80 adult mouse. Right panels show DsRed-Mito-labeled mitochondria along axonal segment for time-lapse imaging analysis. (B) Representative images of cultured spinal motor neurons at DIV7 (left) or DIV10 (right) isolated from p40 adult mouse. Neurons are stained with neuron-specific βIII-tubulin.
3.1. Preparation of adult DRG neuron cultures
3.1.1. Materials
Poly-l-ornithine (Sigma, P4957)
Laminin (Roche, 11243217001)
Collagenase type 2 (Worthington, LS004176)
Dispase II (Roche, 04942078001)
HBSS (Invitrogen, 14025–092)
HBSS Ca2+/Mg2+ free (Invitrogen, 14175–095)
Penicillin/streptomycin (Invitrogen, 15140–122)
PBS Ca2+/Mg2+ free (Invitrogen, 10010–023)
12 mm coverslips, No. 1 (Fisher, 12-545-82)
70-μm cell strainer (Fisher, 08-771-2)
Amaxa basic neuron SCN nucleofector kit (Lonza, VSPI-1003)
3.1.2. Solutions
(All reagents are sterilized with 0.22-μm filters).
Dispase/Collagenase (2.5 U ml−1 of Dispase and 100 U ml−1 Collagenase, made with HBSS with Ca2+/Mg2+, aliquot, and store in −70 °C up to 12 months).
Adult DRG culture medium: Neurobasal A supplement with 2 mM glutamine MAX, 2% B27 supplement, and 1% penicillin/streptomycin.
3.1.3. Procedures
Prepare clean coverslips. After extensive wash, the coverslips were stored in 80% ethanol. Dry the coverslips by flaming with alcohol burner under culture hood. Coverslips are then coated with 25 μg ml−1 poly-l-ornithine in PBS at 37 °C for overnight.
On 2nd day, wash the coverslips in dishes three times with Ca2+/Mg2+-free PBS, 15 min each. Wash one more time with ddH2O.
Coat the coverslips with 5 μg ml−1 laminin at 37 °C for 1 h before use. The laminin stock is stored at −20 or −70 °C (avoid repeat thaw–freeze cycles). Prepare adult DRG culture medium. Wash the coverslips with culture medium once and keep coverslips in medium until cell plating.
Anesthetize mice by avertin i.p. (0.4–0.5 ml per mouse). Once mouse are not respond to hind paw squeezing, spray with 70% ethanol. Remove the back skin and expose the whole spinal regions. Along the spine, clip off bone on both sides of vertebral canal until the column roof is removed and DRGs are exposed down the length of the spinal cord. Trim off excess bone when necessary. (Avoid hair contamination.)
Make an incision with scissors from rostral to caudal, cut down the length of the cord to the sacrum close to the spinal cord, repeat on the both side. Then, cut transversely to remove the spinal column from the animal.
Starting ventrally, grasp the edge of tissue and cut vertebra and muscle off. Place spinal column in iced Ca2+/Mg2+-free HBSS followed by briefly wash.
Put ice pack on dissecting microscope with sterilized filter paper on top. Place spinal column on filter paper. Starting with rostral direction, grasp the dorsal or ventral root with forceps and pull out the DRGs. Use forceps tip to turn the cord to the opposite side to expose the DRGs. Transfer the ganglia to Ca2+/Mg2+-free HBSS in petri dish. Proceed down to the caudal end of the spinal cord until all DRGs are collected.
Trim off the excess roots and blood from DRGs with microscissors, transfer trimmed DRG to a 1.5-ml tube with 1 ml Dispase/Collagenase.
- Dissociate DRGs.
- Incubate DRGs in Dispase/Collagenase at 37 °C for 30 min and then put it on rotor for 30–35 min at room temperature.
- Centrifuge cells at 1000 × g for 2 min. Remove solution and resuspend the pellet with 1 ml adult DRG culture medium.
- Triturate with 200-μl pipette tips for 10–15 times. Transfer the cell suspension to new tubes through the cell strainer. Spin at 200 × g for 3 min.
Count and plate DRG: for mice age between p40 and p80, resuspend pellets and plate cells at 1000 cells cm−2. For mice older than p150, plate the neuron at 2000 cells cm−2. Replace the medium 2 h after, and then again next morning. After 3 days, glia start growing, add 3 μM Ara-C to curb the glial cells for proliferation.
3.2. Preparation of adult MN cultures
Adult MN cultures and density gradient centrifugation are prepared according to previous descriptions (Brewer & Torricelli, 2007; Montoya et al., 2009) with some modifications.
3.2.1. Materials
C2C12 mouse myoblast cell line (ATCC, CRL-1772)
Poly-l-ornithine (Sigma, P4957)
Laminin (Roche, 11243217001)
DMEM (Invitrogen, 10564–011)
DMEM (Invitrogen, 12430–054)
Hibernate A (Invitrogen, A12475–01)
Neurobasal A (Invitrogen, 10888–022)
50× B27 (Invitrogen, 17504–044)
100× Insulin-Transferrin-Selenium (Invitrogen, 41400–045)
Papain (Worthington, LS003124)
GlutaMAX™-I (Invitrogen, 25030–061)
OptiPrep density 1.32 (Axis-Shield, Oslo)
Pasteur pipettes (VWR, 14672–412)
FBS
Horse serum (HS)
Growth factors: BDNF (Invitrogen, 10908–010). GDNF (Invitrogen, PHC7045). FGF2 (Invitrogen, PMG0034). cAMP analog: 8-(4-chlorophenylthio)adenosine 3′,5′-cyclic monophosphate sodium salt (Sigma, C3912)
3.2.2. Solutions
(All reagents are sterilized with 0.22-μm filters).
C2C12 cell proliferation medium: DMEM (10564), 10% FBS, and 1% penicillin/streptomycin.
C2C12 cell differentiate medium: DMEM (12430), 2% HS, and 1% penicillin/streptomycin.
C2C12 condition medium: Neurobasal A plus 2% B27, 0.5 mM Glu-taMAX™-I, 1% penicillin/streptomycin.
10× ACSF stock: 25 mM KCl, 12.5 mM NaH2PO4, 13 mM MgSO4. Store at 4 °C.
1× ACSF: Sequentially add 10× ACSF, H2O, 26 mM NaHCO3, 2.5 mM CaCl2, 11 mM glucose, 202 mM sucrose, bring the final volume to 100 ml per spinal cord isolation. Bubble the ACSF solution for 30 min with 95% O2/5% CO2 on ice.
Hibernate A complete medium (HA-c): Hibernate A, 2% B27, 0.5 mM GlutaMAX™-I, 1% penicillin/streptomycin, store at 4 °C. Prepare 100 ml per spinal cord isolation.
Papain solution: 10 ml of HA-c containing 40 U ml−1 papain, 10 U ml−1 DNase. Warm at 37 °C to fully dissolved.
Dissociation solution: 6 ml of HA-c with 10 U ml−1 DNase, 1% FBS.
First OptiPrep 1.32 density gradient: Prepare 6% solution by mixing 0.5 ml OptiPrep 1.32 stock solution with 4.5 ml HA-c in a 50-ml tube.
Second OptiPrep 1.32 density gradient: Prepare OptiPrep fraction solution from bottom to top 10.5%, 7.5%, 6%, 4.5%, 2 ml each in a 15-ml tube.
Motor neuron feed medium (MFM): 75% Neurobasal A plus 25% C2C12 condition medium, 1 ng ml−1 BDNF, 0.1 ng ml−1 GDNF, and 125 mM cAMP. 2% B27, 0.5 mM GlutaMAX™-I, 1% penicillin/streptomycin. Prepare 25 ml per spinal cord isolation.
3.2.3. Procedures
The low passage C2C12 cells are maintained in C2C12 proliferation medium. The cells are split at 50–70% confluence. When the cells reach 90% confluence, 10 ml C2C12 differentiate medium is added to T75 flask. After 3 days, cells are briefly washed with Neurobasal A once and then replaced by condition medium. After 2 days, collect the condition medium from C2C12 cultures, aliquot and store in −70 °C.
Coverslips are prepared and coated as described in adult DRG culture procedure. Dilute and coat laminin at 1 μg ml−1 for the second coating.
Prepare 1× ACSF, HA-c, MFM, papain, and dissociation solution.
Anesthetize the mouse by avertin i.p (0.4–0.5 ml per mouse). Once mouse does not respond to hind paw squeezing, spray with 70% ethanol. Perfuse the mice with 30 ml iced ACSF, then decapitate by a guillotine. Remove the back skin and expose spinal column. (Avoid hair contamination.)
At the caudal end of column, cut transversely of spinal column at 2 cm from tail base. Then insert a 201/2G needle capped syringe that filled with 20 ml ACSF into spinal canal and extrude the cord from the decapitated cervical end. Collect and immerse the spinal cord to iced HA-c buffer.
Cut the spinal cord in small pieces in 0.5 mm by McIlwain tissue chopper for better cell digestion.
Transfer tissue pieces to a 15-ml tube with 10 ml papain solution. Then incubate in a rotate at 30 °C for 30 min.
During the incubation, fire polishes 2-ml Pasteur pipettes with large diameter. Coat the glass pipette by aspirating FBS one time. Prepare first and second OptiPrep 1.32 gradient solution in 50-ml and 15-ml tubes, respectively.
Centrifuge the tissue at 1000 × g for 2 min. Remove the supernatant and resuspend the pellet with 2 ml dissociation solution.
Gently and carefully triturate the tissue ten times with Pasteur pipettes. Then settle the homogenate for 1 min and transfer the supernatant to a 15-ml tube. Repeat this process for two more times with no bubble introduced. The resulting 6 ml of dissociated homogenate is slowly loaded on the top of 5 ml of 6% OptiPrep in a 50-ml tube.
Centrifuge the cells at 800 × g for 10 min. The supernatant is carefully removed and the pellet is resuspended by 6 ml HA-c, then cells are passed through a 70-μm cell strainer to remove undigested debris.
Add 6 ml of the cell suspension to the top of second gradient. Centrifuge the cell at 800 × g for 15 min. The 7.5%, 6%, and 4.5% fractions are collected, add 8 ml HA-c and mix well. Centrifuge at 300 × g for 3 min.
Discard the supernatant and resuspend pellet with 240 μl MFM. Place 20 μl of cell suspension (~1000 cells) in the middle of each 12-mm glass coverslip.
After incubate at 37 °C for 1 h, the additional MFM is added. The cells are fed by half changed fresh medium the day after plating, and every other 3 days until the end of the experiment.
3.3. Mitochondria labeling and image
- Mitochondria are labeled by expressing DsRed-Mito or by loading fluorescent dye MitoTracker Green or TMRE.
- For transfection of DsRed-Mito by electroporation: neuron pellets in DRG culture are resuspended by 20 μl electroporation buffer with 0.5 μg endotoxin free plasmid. The cells are nucleofected with SCN Basic Neuro Program 6. Replace with fresh medium 2 h after plating. Alternatively, DsRed-Mito can be introduced to DRG or MNs by lentivirus infection. The lentivirus particles (MOI = 10–50) are incubated 4–24 h with neurons after cell attachment. Replace half the medium with fresh medium after infection.
- For labeling mitochondria by loading MitoTracker Green dye: adult neurons are incubated with 15 nM MitoTracker Green dye for 30 min and washed with culture medium for 30 min. Alternatively, mitochondria can be also stained with 15 nM TMRE for 30 min in culture medium, followed by brief wash before image.
Once the mitochondria are labeled, the coverslips are transferred to Hibernate A low fluorescence medium (BrainBits, HA-lf), plus 2% B27, and other supplements when needed.
The image acquisition and analysis are similar to procedure described in embryonic neuron culture (see Section 2.3.2). However, the mitochondria motility in adult MNs is relatively lower; thus, longer recording time (15–20 min) is required.
4. CHARACTERIZATION OF MITOCHONDRIAL TRANSPORT AT SYNAPSES
Mitochondrial transport and spatial distribution in axons and at synapses are correlated with synaptic activity (see reviews by MacAskill & Kittler, 2010; Schwarz, 2013; Sheng, 2014; Sheng & Cai, 2012). Mitochondria are often recruited to and retained within presynaptic terminals and dendritic spines in response to elevated intracellular Ca2+ during sustained synaptic activity. Elevated Ca2+ influx, either by activating voltage-dependent calcium channels at presynaptic terminals or by activating NMDA receptors at postsynaptic terminals, arrests mitochondrial movement (Chang et al., 2006; Chen & Sheng, 2013; Macaskill et al., 2009; Rintoul, Filiano, Brocard, Kress, & Reynolds, 2003; Saotome et al., 2008; Wang & Schwarz, 2009; Yi, Weaver, & Hajnoczky, 2004). Upon removal of the Ca2+ signal, the cargo-loaded motor proteins can be quickly reactivated to move mitochondria away to find new active synapses.
4.1. Characterizing mitochondrial motility patterns at presynaptic boutons
To investigate the mobility pattern of axonal mitochondria at presynaptic boutons, cultured hippocampal neurons are cotransfected with DsRed-Mito and synaptic marker GFP-Synapsinla at DIV7–9 using calcium phosphate method (see Section 2.2), followed by dual-channel time-lapse imaging at DIV12–14 (see Section 2.3). Mitochondrial mobility is presented with a kymograph, which is created using ImageJ plugins (NIH) as previously described (Kang et al., 2008). We recently characterized the relative populations of axonal mitochondria in hippocampal neurons into five distinct motility patterns during a 16-min recording (Fig. 5.4; Sun et al., 2013): (1) stationary mitochondria sitting out of presynaptic boutons (54.07%±2.53%), (2) stationary mitochondria sitting at presynaptic sites (16.29%±1.66%), (3) motile mitochondria passing by boutons (14.77%±1.58%), (4) motile mitochondria briefly pausing (<200 s) at presynaptic sites (7.01%±1.29%), and (5) motile mitochondria pausing for an extended time (>200 s) at presynaptic sites (8.30%±1.52%). These findings are consistent with a previous study in primary cortical neurons (Chang et al., 2006).
Figure 5.4.

Complex motility patterns of axonal mitochondria at presynaptic boutons. Representative imaging (A), kymograph (B), and quantitative analysis (C) showing multiple motility patterns of axonal mitochondria (red) pausing at or passing by synapses (green). Hippocampal neurons were cotransfected with DsRed-Mito and GFP-Synapsin1a at DIV9, followed by time-lapse imaging at DIV12. Motility is presented in kymograph (B) in which vertical lines (red) represent stationary mitochondria; slanted lines or curves indicate motile ones. Relative populations of axonal mitochondria were quantified as (1) stationary mitochondria out of synaptic sites (Out syn) (arrowheads in A), (2) stationary mitochondria at synaptic sites (In syn) during a 16-min recording time (arrows in A), (3) moving mitochondria quickly passing by boutons (white arrows along green vertical lines in B), (4) moving mitochondria pausing at synaptic sites for a short-time period (<200 s) (two small white boxes in B), and (5) moving mitochondria pausing at synaptic sites for a longer time period (>200 s) (a large box in B). Data were quantified from numbers of axonal mitochondria indicated within bars pooled from total 10 axons. Scale bar: 10 μm. Error bars: SEM. Adapted from Sun et al. (2013).
4.2. Characterizing impact of motile mitochondria on presynaptic activity
Mitochondria are commonly found within synaptic terminals, where they power neurotransmission by supplying ATP. A stationary mitochondrion within presynaptic terminals provides stable and continuous ATP supply and maintains energy homeostasis at synapses. Conversely, a motile mitochondrion passing by presynaptic boutons dynamically alters local ATP levels, thus influencing various ATP-dependent functions at synapses. At any given time, presynaptic boutons along the same axons will display different patterns of mitochondrial distribution and motility; mitochondria move either in or out of boutons or pass through boutons, thus impacting synaptic activity. To examine presynaptic variability at single-bouton levels, we recently examined two dynamic events by dual-channel live imaging of axonal mitochondria labeled with DsRed-Mito and synaptic vesicle (SV) release using synapto-pHluorin (spH). spH is a SV-targeted and pH-sensitive GFP whose fluorescence is quenched by intraluminal acidic pH, but increases upon exocytosis when it is exposed to the neutral pH of the extracellular medium. Thus, changes in spH fluorescence reflect the relative strength of SV release and recycling (Sankaranarayanan & Ryan, 2000). Because of technical limitation in resolving single action potential (AP)-induced spH responses, we instead used trains of stimulation (20 Hz, 10 s) to provide a sufficient signal-to-noise ratio (Sun et al., 2010). Therefore, applying this dual-channel live imaging enables us to investigate the contribution of motile axonal mitochondria to synaptic function at the single-bouton levels.
Neurons are cotransfected with spH and DsRed-Mito at DIV7–9, followed by imaging at DIV12–14 (see Sections 2.2 and 2.3). The coverslips are mounted in a stimulation chamber (RC-21BRFS chamber, Warner Instruments), and neurons are evoked by APs by passing a 1-ms current pulse of 20 mA with platinum electrodes in the chamber. The amount of required current depends on the imaging chamber and how much imaging solution is used.
To establish a sufficient signal-to-noise ratio, four repeated trains of stimulation (20 Hz, 10 s with 100-s intervals) are applied to induce activity-dependent spH responses. To correlate synaptic activity with mitochondrial distribution and transport, images of both spH, and DsRed-Mito are simultaneously acquired at 1 Hz using the Zeiss LSM 510 META confocal microscope with a 40×, 1.3 numerical aperture oil-immersion objective.
All functionally visible varicosities along axons are selected for analysis by testing their response to stimulation. Region of interest (ROI) over each synapse is centered and analyzed. ROIs should be as small as possible to maximize the signal-to-noise ratio, but large enough to contain both sites of exo- and endocytosis to avoid fluorescence decay caused by faster diffusive processes (Granseth, Odermatt, Royle, & Lagnado, 2006).
To determine how axonal mitochondrial mobility affects SV release, spH fluorescence intensity is analyzed during four successive train stimulations at boutons (1) with a stationary mitochondrion, (2) without a stationary mitochondrion, and (3) with a motile mitochondrion. Our study demonstrates that SV release remains constant at boutons with a stationary mitochondrion, whereas SV release is reduced at boutons lacking a mitochondrion. Interestingly, mitochondrial transport significantly impacts SV release, which is quickly depleted when a mitochondrion moves out of the boutons or increased when a mitochondrion passes by boutons during stimulations (Fig. 5.5; Sun et al., 2013).
Figure 5.5.
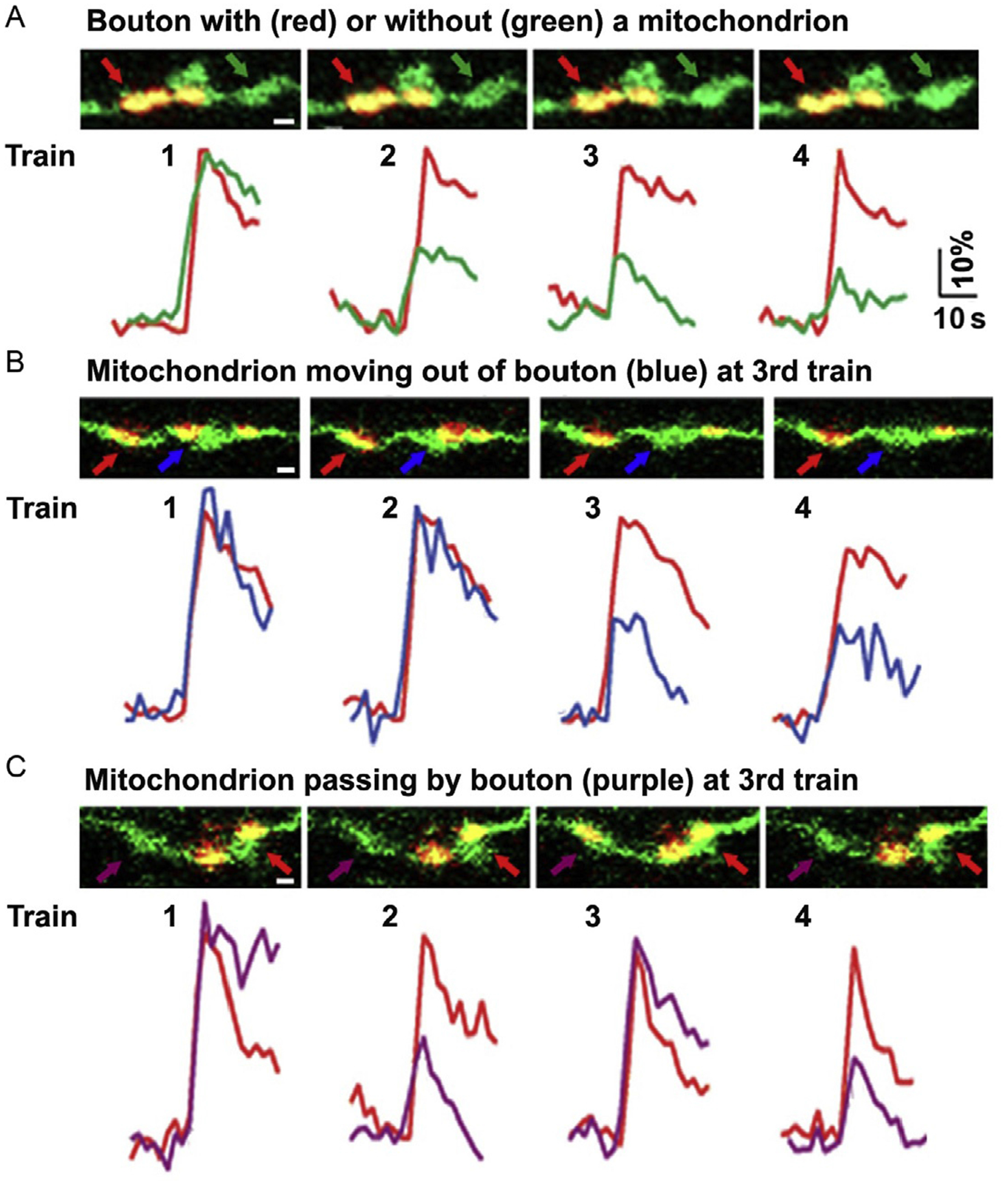
Mitochondrial motility influences SV release at single-bouton levels. (A-C) Dual-channel live imaging showing the distribution and motility of axonal mitochondria at presynaptic boutons and corresponding spH traces during four trains of stimulation (20 Hz at 10 s with 100-s interval). Note that SV exocytosis remained robust and stable at boutons with a stationary mitochondrion (A-C, red arrows/traces), while exocytosis diminished starting at the 2nd train at mitochondrion-free boutons (A, green arrows/traces) or when a mitochondrion is moving out of the bouton at the 3rd train (B, blue arrows/traces). A mitochondrion passing by bouton during the 3rd train rescued SV release (C, purple arrows/traces). Adapted from Sun et al. (2013).
5. SUMMARY
This chapter discusses the current approaches to characterize neuronal mitochondrial transport by focusing on the axon as its uniform MT polarity. We first describe the routine procedures for monitoring axonal mitochondrial transport in embryonic hippocampal neuron cultures. In the second part of this chapter, we provide detailed methods currently used in our laboratory to characterize mitochondrial dynamics in DRG sensory neurons and spinal MNs isolated from adult mouse. In addition, we also discuss our recent study analyzing mitochondrial motility at presynaptic terminals and their impact on synaptic function. The recent discoveries of proteins involved in regulation of mitochondrial transport has boosted our understanding of the mechanisms that regulate mitochondrial transport and anchoring in healthy and diseased neurons. Studying these dynamic cellular processes in live neurons isolated from disease-stage adult mice will provide reliable models for delineating defects of mitochondrial transport underlying adult-onset pathogenesis.
ACKNOWLEDGMENTS
The authors thank the colleagues in the Sheng laboratory and other laboratories who contributed to the research procedures described in this chapter. Work of the Sheng laboratory is supported by the Intramural Research Program of NINDS, NIH.
ABBREVIATION
- AP
action potential
- DIV
days in vitro
- DRG
dorsal root ganglia
- MN
motor neuron
- MT
microtubule
- SV
synaptic vesicle
- spH
synapto-pHluorin
- WT
wild type
REFERENCES
- Banker GA, & Cowan WM (1977). Rat hippocampal neurons in dispersed cell culture. Brain Research, 126, 342–397. [DOI] [PubMed] [Google Scholar]
- Bilsland LG, Sahai E, Kelly G, Golding M, Greensmith L, & Schiavo G (2010). Deficits in axonal transport precede ALS symptoms in vivo. Proceedings of the National Academy of Sciences of the United States of America, 107, 20523–20528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birsa N, Norkett R, Higgs N, Lopez-Domenech G, & Kittler JT (2013). Mitochondrial trafficking in neurons and the role of the Miro family of GTPase proteins. Biochemical Society Transactions, 41, 1525–1531. [DOI] [PubMed] [Google Scholar]
- Brewer GJ, & Torricelli JR (2007). Isolation and culture of adult neurons and neurospheres. Nature Protocols, 2, 1490–1498. [DOI] [PubMed] [Google Scholar]
- Cai Q, Zakaria HM, Simone A, & Sheng ZH (2012). Spatial parkin translocation and degradation of damaged mitochondria via mitophagy in live cortical neurons. Current Biology, 22, 545–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang DT, Honick AS, & Reynolds IJ (2006). Mitochondrial trafficking to synapses in cultured primary cortical neurons. The Journal of Neuroscience, 26, 7035–7045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang DT, & Reynolds IJ (2006). Mitochondrial trafficking and morphology in healthy and injured neurons. Progress in Neurobiology, 80, 241–268. [DOI] [PubMed] [Google Scholar]
- Chen Y, & Sheng ZH (2013). Kinesin-1-syntaphilin coupling mediates activity-dependent regulation of axonal mitochondrial transport. The Journal of Cell Biology, 202, 351–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vos KJ, Chapman AL, Tennant ME, Manser C, Tudor EL, Lau KF, et al. (2007). Familial amyotrophic lateral sclerosis-linked SOD1 mutants perturb fast axonal transport to reduce axonal mitochondria content. Human Molecular Genetics, 16, 2720–2728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granseth B, Odermatt B, Royle SJ, & Lagnado L (2006). Clathrin-mediated endocytosis is the dominant mechanism of vesicle retrieval at hippocampal synapses. Neuron, 51, 773–786. [DOI] [PubMed] [Google Scholar]
- Jiang M, & Chen G (2006). High Ca2+-phosphate transfection efficiency in low-density neuronal cultures. Nature Protocols, 1, 695–700. [DOI] [PubMed] [Google Scholar]
- Kang JS,Tian JH, Pan PY, Zald P, Li C, Deng C, et al. (2008). Docking of axonal mitochondria by syntaphilin controls their mobility and affects short-term facilitation. Cell, 132, 137–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacAskill AF, & Kittler JT (2010). Control of mitochondrial transport and localization in neurons. Trends in Cell Biology, 20, 102–112. [DOI] [PubMed] [Google Scholar]
- Macaskill AF, Rinholm JE, Twelvetrees AE, Arancibia-Carcamo IL, Muir J, Fransson A, et al. (2009). Miro1 is a calcium sensor for glutamate receptor-dependent localization of mitochondria at synapses. Neuron, 61, 541–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magrane J, & Manfredi G (2009). Mitochondrial function, morphology, and axonal transport in amyotrophic lateral sclerosis. Antioxidants & Redox Signaling, 11, 1615–1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marinkovic P, Reuter MS, Brill MS, Godinho L, Kerschensteiner M, & Misgeld T (2012). Axonal transport deficits and degeneration can evolve independently in mouse models of amyotrophic lateral sclerosis. Proceedings of the National Academy of Sciences of the United States of America, 109, 4296–4301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller KE, & Sheetz MP (2004). Axonal mitochondrial transport and potential are correlated. Journal of Cell Science, 117, 2791–2804. [DOI] [PubMed] [Google Scholar]
- Misgeld T, Kerschensteiner M, Bareyre FM, Burgess RW, & Lichtman JW (2007). Imaging axonal transport of mitochondria in vivo. Nature Methods, 4, 559–561. [DOI] [PubMed] [Google Scholar]
- Montoya GJV, Sutachan JJ, Chan WS, Sideris A, Blanck TJ, & Recio-Pinto E (2009). Muscle-conditioned media and cAMP promote survival and neurite outgrowth of adult spinal cord motor neurons. Experimental Neurology, 220, 303–315. [DOI] [PubMed] [Google Scholar]
- Morris RL, & Hollenbeck PJ (1993). The regulation of bidirectional mitochondrial transport is coordinated with axonal outgrowth. Journal of Cell Science, 104, 917–927. [DOI] [PubMed] [Google Scholar]
- Morris RL, & Hollenbeck PJ (1995). Axonal transport of mitochondria along microtubules and F-actin in living vertebrate neurons. The Journal of Cell Biology, 131, 1315–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popov V, Medvedev NI, Davies HA, & Stewart MG (2005). Mitochondria form a filamentous reticular network in hippocampal dendrites but are present as discrete bodies in axons: A three-dimensional ultrastructural study. The Journal of Comparative Neurology, 492, 50–65. [DOI] [PubMed] [Google Scholar]
- Rintoul GL, Filiano AJ, Brocard JB, Kress GJ, & Reynolds IJ (2003). Glutamate decreases mitochondrial size and movement in primary forebrain neurons. The Journal of Neuroscience, 23, 7881–7888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruthel G, & Hollenbeck PJ (2003). Response of mitochondrial traffic to axon determination and differential branch growth. The Journal of Neuroscience, 23, 8618–8624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankaranarayanan S, & Ryan TA (2000). Real-time measurements of vesicle-SNARE recycling in synapses of the central nervous system. Nature Cell Biology, 2, 197–204. [DOI] [PubMed] [Google Scholar]
- Saotome M, Safiulina D, Szabadkai G, Das S, Fransson A, Aspenstrom P, et al. (2008). Bidirectional Ca2+-dependent control of mitochondrial dynamics by the Miro GTPase. Proceedings of the National Academy of Sciences of the United States of America, 105, 20728–20733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxton WM, & Hollenbeck PJ (2012). The axonal transport of mitochondria. Journal of Cell Science, 125, 2095–2104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz TL (2013). Mitochondrial trafficking in neurons. Cold Spring Harbor Perspectives in Biology, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng ZH (2014). Mitochondrial trafficking and anchoring in neurons: New insight and implications. The Journal of Cell Biology, 204, 1087–1098. 10.1083/jcb.201312123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng ZH, & Cai Q (2012). Mitochondrial transport in neurons: Impact on synaptic homeostasis and neurodegeneration. Nature Reviews. Neuroscience, 13, 77–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y, Nagy M, Ni W, Tyagi NK, Fenton WA, Lopez-Giraldez F, et al. (2013). Molecular chaperone Hsp110 rescues a vesicle transport defect produced by an ALS-associated mutant SOD1 protein in squid axoplasm. Proceedings of the National Academy of Sciences of the United States of America, 110, 5428–5433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun T, Qiao H, Pan PY, Chen Y, & Sheng ZH (2013). Motile axonal mitochondria contribute to the variability of presynaptic strength. Cell Reports, 4, 413–419. 10.1016/jxelrep.2013.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun T, Wu XS, Xu J, McNeil BD, Pang ZP, Yang W, et al. (2010). The role of calcium/calmodulin-activated calcineurin in rapid and slow endocytosis at central synapses. The Journal of Neuroscience, 30, 11838–11847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, & Schwarz TL (2009). The mechanism of Ca2+-dependent regulation of kinesin-mediated mitochondrial motility. Cell, 136, 163–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi M, Weaver D, & Hajnoczky G (2004). Control of mitochondrial motility and distribution by the calcium signal: A homeostatic circuit. The Journal of Cell Biology, 167, 661–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu YB, & Sheng ZH (2011). Increased axonal mitochondrial mobility does not slow amyotrophic lateral sclerosis (ALS)-like disease in mutant SOD1 mice. The Journal of Biological Chemistry, 286, 23432–23440. [DOI] [PMC free article] [PubMed] [Google Scholar]