

Abstract
Acquired chromosomal DNA copy gains are a feature of many tumors; however, the mechanisms that underpin oncogene amplification are poorly understood. Recent studies have begun to uncover the importance of epigenetic states and histone lysine methyltransferases (KMTs) and demethylases (KDMs) in regulating transient site-specific DNA copy number gains (TSSGs). In this study, we reveal a critical interplay between a myriad of lysine methyltransferases and demethylases in modulating H3K4/9/27 methylation balance in order to control extrachromosomal amplification of the EGFR oncogene. This study further establishes that cellular signals (hypoxia and epidermal growth factor) are able to directly promote EGFR amplification through modulation of the enzymes controlling EGFR copy gains. Moreover, we demonstrate that chemical inhibitors targeting specific KMTs and KDMs are able to promote or block extrachromosomal EGFR amplification, which identifies potential therapeutic strategies for controlling EGFR copy number heterogeneity in cancer, and in turn, drug response.
Introduction
Chromosomal instability is a hallmark of cancer cells (1). These abnormalities can include entire chromosome events or they can be localized to site-specific chromosomal regions (2). For example, the chromosome 1q12–25 (1q12–25) region is regularly amplified in tumors (3–9). This amplification event is often associated with drug resistance as a number of drug resistant genes (e.g., MCL1, CKS1B) reside within this chromosomal region (3–9). Amplification of these regions can occur as frequently as the well documented oncogene amplifications MYC and epidermal growth factor receptor (EGFR) in certain tumor types (e.g., 1q21.3 at 21% versus MYC at 26% in liver cancer; (10)). However, it is important to note that amplifications are not always permanently integrated (2). A recent study estimated that approximately 50% of tumors contain extrachromosomal DNA (ecDNA) amplifications for the EGFR and MYC genes (11). The extrachromosomal nature of these copy gains provides the cell an opportunity to either select for or against these amplifications, which will impact cell growth and drug response. For example, extrachromosomal amplification of EGFR results in increased sensitivity to targeted therapies. However, following prolonged treatment with an EGFR inhibitor, the ecDNA copies of EGFR are reduced, leading to therapy resistance (12). In the case of methotrexate therapy, the dihydrofolate reductase (DHFR) gene is amplified and provides resistance (13–16). DHFR amplifications can occur as integrated and/or extrachromosomal events (13–16). Therefore, extrachromosomal amplifications promote tumor heterogeneity and tumor adaptation, both of which are major contributors to drug resistance (2,11). Elucidating the cellular physiology and molecular mechanisms that promote oncogene-associated extrachromosomal events will have a profound impact on our understanding of tumor heterogeneity and drug resistance.
The mechanisms by which extrachromosomal amplification events occur are still poorly understood; however, recent studies have demonstrated a critical role for epigenetic states and chromatin modifying enzymes in controlling site-specific rereplication, and in turn, DNA copy number amplification (10,17–19). For example, overexpression or stabilization of the H3K9/36 tri-demethylase KDM4A, and the direct modulation of chromatin states (H3K9 and K36 methylation) promotes transient site-specific DNA copy gains (TSSGs) within the Chr1q12–21 region (17–20). These DNA copy gains are transiently generated during S phase and are lost in late S or early G2 phase of the cell cycle (18). Indeed, KDM4A interacts with components of the replication machinery, facilitating rereplication at the TSSG sites (18). Consistent with these findings, we illustrated that targeting KDM4 family members through H3K4 methylation can result in TSSGs (10). This study reveals that lysine methyltransferases and demethylases have a high degree of specificity and work in concert to modulate site-specific DNA copy gains in the genome. These studies highlight the possibility that clinically relevant oncogenes exhibiting plasticity in their copy number gains (i.e., EGFR; (12)) could be regulated by a comparable mechanism. It also leads one to question whether extrinsic cellular cues are also able to facilitate these oncogenic amplifications (21). Elucidating mechanisms by which chromatin modulators and epigenetic states regulate oncogenes would identify new therapeutic avenues to control copy number heterogeneity and drug responses.
EGFR DNA amplification tends to result in poor prognosis for patients with EGFR-amplified cancer (22). EGFR-targeted therapies have been developed in recent years (23) and EGFR amplifications have been shown to associate with varying degrees of patient response across various amplified tumors (24–29). EGFR DNA amplification is prevalent across a number of different cancer types, with up to 54% of patients exhibiting amplification in some tumor types (e.g., glioblastoma multiforme) (10). An important clinical challenge with EGFR amplification is the plasticity of the amplification (12). Therefore, there is a major clinical need to resolve the mechanisms driving EGFR amplification.
In this study, we demonstrate that chromatin modifying enzymes and their associated epigenetic states control amplification of the EGFR locus. Specifically, we demonstrate that directly interfering with H3K9 and H3K27 methylation promotes EGFR amplification. Furthermore, we establish a critical interplay between H3K4/9/27 lysine methyltransferases and demethylases in either promoting or blocking EGFR amplification. For example, KDM4A overexpression promotes EGFR copy gains in conjunction with three H3K4 methyltransferases: KMT2A/MLL1, SETD1A and SETD1B. In addition, we demonstrate that suppression of specific H3K9 KMTs and the H3K27 KMT EZH2 generates EGFR amplification. Consistent with these genetic experiments, we demonstrate for the first time that chemical inhibitors targeting KMT-KDMs are able to rheostat EGFR copy number, and in turn, growth factor and EGFR inhibitor responses. Lastly, we demonstrate that extrinsic cellular cues [hypoxia and Epidermal Growth Factor (EGF)] promote EGFR amplification by modulating the KMT-KDM network that controls EGFR copy number. Taken together, our study uncovers both chromatin modifiers and extracellular signals that control EGFR amplification and demonstrate that epigenetic therapies could hold a key to modulating EGFR copy number heterogeneity in cancer, which has significant clinical implications.
Results
K9 and K27 methylation interference promotes EGFR amplification.
Previous analysis demonstrated that up to 54% of primary tumors across the pancancer TCGA dataset harbour EGFR amplifications of which some were shown to harbour extrachromosomal amplification (10,11). To further explore EGFR amplification heterogeneity across and within tumors, we assessed the range of EGFR DNA copy gains and the associated EGFR RNA expression levels in each of the tumors in the pancancer TCGA dataset (7069 samples; Figure 1A). The analyses revealed significant plasticity in EGFR DNA copy number across tumor types, ranging from 2.4–8 copies (red) to more than 8 copies (blue) (Figure 1A). We also observed tumors with a loss of EGFR (black; Figure 1A). As the DNA copy number increased there was an increase in EGFR RNA levels (Figure 1A). Therefore, promoting more EGFR DNA copies tends to associate with increased EGFR transcripts in tumors.
Figure 1. H3K9/27 methylation controls EGFR amplification.

A) Scatter plot comparing EGFR gene expression (Y-axis) to EGFR DNA copy number (X-axis) from the pan-cancer TCGA data set (7069 patients spanning 21 tumor types). Expression is shown in units of transcripts per million (TPM), converted to log2 values. Copy number is shown as number of copies. B) Representative DNA FISH images of RPE nuclei from cells transduced with H3.3 Wild Type (H3.3 WT), K4M, K9M, K27M or K36M variants. EGFR (red), DAPI (blue) and merge are shown. C) RPE cells transduced with H3.3 K9M or H3.3 K27M variants exhibit EGFR copy gains. D) RPE cells transduced with H3.3 K9M or H3.3 K27M variants do not exhibit 7p tel copy gains. E) RPE cells transduced with H3.3 K9M or H3.3 K27M variants do not exhibit IKZF1 copy gains. F) Representative DNA FISH images of RPE nuclei from cells transduced with H3.3 K9M or K27M variants with more than 4 DNA copies of EGFR (red) are shown. G) RPE cells transduced with H3.3 K9M or H3.3 K27M variants have a higher percentage of cells with 3 to 4 copies and 5 or more copies of EGFR DNA. H) RPE cells transduced with H3.3 K9M or H3.3 K27M variants have an increase in EGFR transcripts compared to H3.3 wild type-transduced cells, as measured by quantitative polymerase chain reaction (qPCR). I) Input-normalized ChIP-seq tracks of H3K36me3, H3K27me3 an H3K9me3 density in the megabase vicinity of the EGFR gene, aligned with the Hi-C map of chromatin interactions. J-L) Input-normalized ChIP-seq tracks of H3K36me3, H3K27me3 an H3K9me3 density in the megabase vicinity of the EGFR gene, aligned with the Hi-C map of chromatin interactions in human HMEC (J) and K562 (L) cells as well as mouse B-lymphoblasts (CH12-LX) (L) (34,35). M) H3K27me3 ChIP-seq enrichment tracks from (36) in the vicinity of the EGFR gene in wild type and K27M-expressing 293 T-REx cells. Error bars represents S.E.M. The * represents p=≤0.05 by two-tailed Student’s t-test. The arrowheads mark DNA FISH foci. The scale bars are 5um.
Since there is a range of EGFR DNA copy gains across tumors (Figure 1A) and others have observed amplification plasticity (12), we assessed whether perturbation of epigenetic states associated with transient site-specific DNA amplifications could promote EGFR DNA copy gains (10,18). Prior studies demonstrated that the introduction of histone 3.3 lysine to methionine (H3.3 K-M) mutants into cells could interfere with the associated methylation at that specific lysine (30). In fact, we demonstrated that transducing cells with different H3.3 K-M mutants could interfere with associated methylation, and in turn, promote amplification at specific regions in the genome (18,30). For example, 1q12h was copy gained with the introduction of H3.3K9M and H3.3K36M (18), while 1p32.3 was amplified with only H3.3K36M (10). These data illustrate the localized impact that modifying specific lysine methylation states have on the predilection of regions to undergo amplification versus whole genome instability and amplification (10,18). For these reasons, we tested whether introduction of H3K-M mutants into the immortalized retinal pigmental epithelial (RPE) cells that have a nearly diploid genome (18,31) and no documented mutations in the EGFR gene could allow us to identify residues important in repressing EGFR amplification (Figure S1A). Since cell cycle arrest (e.g., G1/S; (18)) has been shown to block copy gains driven by epigenetic perturbation, we evaluated cell cycle profiles for each mutant tested (Figure S1B). Our analyses did not reveal major changes in cell cycle profiles between cells expressing the different H3.3 K-M mutants (Figure S1B); therefore, the cells were subsequently processed for DNA fluorescence in situ hybridization (DNA FISH) to determine the copy number status of EGFR.
Individually H3.3 K9M and H3.3 K27M resulted in significant increases in EGFR DNA copy number, without changing the copy number of chromosome 7 and 8 centromeres (7C and 8C, respectively; Figure 1B, C), the 7p telomere (7p tel, 7p22.2; Figure 1D and Figure S1C) or at a region adjacent to EGFR ( IKZF1, 7p12.2; Figure 1E and Figure S1D). These data suggest that EGFR is undergoing site-specific copy gain as opposed to a whole chromosome 7 or chromosome 7p arm copy gain events, which is consistent with prior studies illustrating that exposure to K9M could promote site-specific amplifications on chromosome 1 (10,18). To date, no other region undergoing site-specific copy gain has been shown to be controlled by H3K27M (10,18). We also observed that ~1–2% of cells in the K9M and K27M mutants had higher EGFR amplification levels than wild type H3.3 (≥5 copies; Figure 1F, G). Consistent with this increase in EGFR copy number, these cells also had a modest increase in EGFR transcript levels when compared to cells expressing a wild type H3 (Figure 1H). Of interest, H3.3K4M and H3.3K36M did not promote EGFR copy gains. H3.3K36M has been shown to promote copy gains of other regions (e.g., 1q12h, 1q21.2, 1p32.3) (10,18), which further highlights the specificity of epigenetic states in modulating amplification sensitivity in the genome.
Consistent with these observations, our ChIP-sequencing (ChIP-seq) coupled with publicly available Hi-C data in RPE cells (32) suggest that the genomic vicinity of the EGFR locus has a specific chromosome structure and pattern of chromatin modifications (Figure 1I). The EGFR gene body corresponds to a subdomain within a larger 500 kilobase (Kbp) 3D interaction domain, that also includes another gene, SEC61G. Both gene bodies are enriched in H3K36me3 (Figure 1I). This chromatin interaction domain is flanked by two wide ~100 Kbp regions enriched in H3K27me3 (Figure 1I, marked with blue shadow). The H3K27me3 region immediately adjacent to the 3’ end of the EGFR gene forms a chromatin interaction domain reminiscent of the small Polycomb-generated domains described by Kundu et al (33). This subdomain on one boundary of the EGFR-containing domain interacts with the H3K27me3 region on the opposite boundary of the EGFR-containing domain, resulting in the streak of interactions above this domain in the Hi-C map (Figure 1I). This looping interaction between distant H3K27me3 regions is similar to the Polycomb-mediated looping interactions previously described (33). Thus, EGFR resides within a 3D interaction domain whose boundaries are marked by two wide regions of H3K27me3 enrichment that form a looping interaction with each other (Figure 1I).
At a larger scale, the EGFR-containing domain, together with the adjacent H3K27me3-enriched boundary regions and two other interaction domains to the 3’ of EGFR are part of ~1 megabase (Mb) region that is flanked on both sides by wide areas of H3K9me3 enrichment (Figure 1I). In order to assess the significance of these associations, we evaluated a panel of cells analysed by ENCODE (34,35). In all cases, they contain similar configurations of H3K9me3 and H3K27me3 at the noted locations in the RPE cells (Figure 1I–L). Cells that did not express EGFR had broader H3K27me3 in between the domains we documented in RPE cells (Figure 1K, L). Furthermore, this pattern was conserved at the modification and organizational level when analyzing a mouse lymphoblast cell line C12-LX (Figure 1L) (34,35). Lastly, we analysed publicly available H3K27me3 data in cells expressing H3.3K27M (36) and observed a loss of the H3K27me3 domains that flank EGFR (Figure 1M), which was consistent with a direct effect on the locus, and in turn, amplification observed in Figure 1B, C. Taken together, these epigenomic profiles and our genetic experiments using mutated histones are consistent with the possibility that this topological structure and H3K9/27 methylation are suppressing the propensity of the EGFR locus to undergo amplification and increased expression.
KDM4A overexpression promotes EGFR copy number gains.
The KDM4 family of histone lysine demethylases catalyze H3K9 and K36 demethylation (37,38). In addition, this enzyme family directly regulates DNA amplifications associated with H3K9 and K36 demethylation (10,18). Therefore, we tested whether overexpression of the KDM4 family of enzymes could phenocopy the increase in EGFR copy number observed upon H3K9 methylation interference. The GFP-tagged KDM4 family members were transiently overexpressed for 24 hours followed by DNA FISH analysis for EGFR DNA copy number (Figure S2A–F). Of the overexpressed KDM4 family members, only transient KDM4A overexpression promoted increased EGFR DNA copy number, which highlights the specificity of KDM4A in regulating the EGFR locus (Figure 2A). Furthermore, catalytically inactive KDM4A (H188A; (38)) and a mutant lacking the Tudor domains that recognize H3K4 methylation (Tudor Del; (39)) were unable to promote EGFR copy gains (Figure 2B and Figure S2G–I). These data were also confirmed with RPE cells that stably overexpressed KDM4A (Figure 2C, D and Figure S2J, K). Chromosome 7 or 8 centromere loci, 7p Tel as well as other previously tested loci were not impacted by KDM4A overexpression (Figure 2A, D and Figure S2I; (10,18)).
Figure 2. KDM4A overexpression promotes EGFR copy gains.
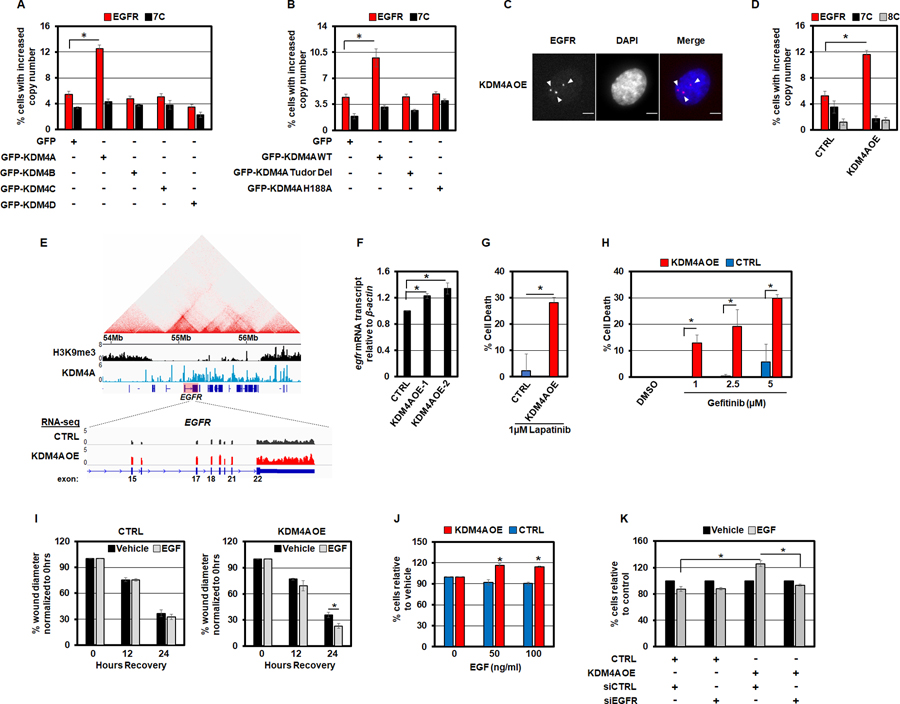
A) Transient overexpression of GFP-tagged KDM4A drives EGFR copy number gain in RPE cells. B) Catalytic activity of KDM4A and the Tudor domains are required for EGFR copy gains. C) Representative DNA FISH image of a stable KDM4A overexpressing RPE nucleus with EGFR DNA copy number gain (red). D) RPE cells with stable KDM4A overexpression have increased EGFR DNA copies. E) Upper panel: Analysis of publicly available ChIP-sequencing data reveals that KDM4A is recruited to the EGFR locus (40). Lower panel: RNA sequencing analysis showed increased EGFR transcripts in RPE cells stably overexpressing KDM4A. F) KDM4A overexpressing RPE cells have increased EGFR transcripts as measured by qPCR. G) KDM4A overexpressing RPE cells have increased sensitivity to the EGFR-family inhibitor, Lapatinib, as measured by trypan blue exclusion assay. H) KDM4A overexpressing RPE cells have a dose-dependent increase in sensitivity to the specific EGFR inhibitor, Gefitinib, as measured by trypan blue exclusion assay. I) KDM4A overexpressing RPE cells migrate faster following 24 hours of 50ng/ml EGF stimulation as measured by scratch assays. J) KDM4A overexpressing RPE cells proliferate faster in response to a 48 hour treatment with 50ng/ml EGF. K) siRNA-mediated depletion of EGFR prevents increased EGF-stimulated (50ng/ml) cell growth in KDM4A overexpressing RPE cells. Error bars represents S.E.M. The * represents p=≤0.05 by two-tailed Student’s t-test. The arrowheads mark DNA FISH foci. The scale bars are 5um.
Consistent with these findings, analysis of available KDM4A ChIP-seq data (40) revealed that KDM4A was enriched across the EGFR locus between the H3K9me3 blocks (1.6 Mb; Figure 2E, upper panel: KDM4A in blue and H3K9me3 in black). These observations were consistent with the previous analyses that noted KDM4A occupied the EGFR promoter region (40), which occurs within this larger binding domain that we have identified. In addition to promoting copy gains, KDM4A overexpression also increased EGFR transcripts as determined by both RNA-sequencing and quantitative RT-PCR (Figure 2E, F). These findings are entirely consistent with a recent report that demonstrated a role for KDM4A in regulating EGFR gene expression (40). Taken together, these data illustrate that KDM4A directly regulates this locus and plays a fundamental role in generating EGFR DNA copy gains and in regulating EGFR expression.
Since KDM4A overexpression promoted EGFR copy gains and increased RNA expression, we assessed the impact that KDM4A overexpression had on EGFR inhibitor sensitivity. We observed increased sensitivity to both Lapatinib and Gefitinib in KDM4A overexpressing cells when compared to control cells (Figure 2G, H). Based on these data, we further reasoned that EGF supplementation would promote cell proliferation and migration in KDM4A overexpressing cells. Consistent with this hypothesis, KDM4A overexpressing cells had a modest but significant increase in cell migration as compared to control cells in scratch assays and increased cell proliferation in response to EGF (Figure 2I, J and Figure S2L). Furthermore, the increased cell proliferation upon EGF-treatment in KDM4A overexpressing cells was completely suppressed by siRNA-mediated depletion of EGFR (Figure 2K and Figure S2M), which emphasized the importance of increased EGFR gene expression in order to observe the increased cell proliferation.
Since EGFR amplifications are observed in lung tumors (10,22) and cancer cell lines (i.e., HCC827; (41,42)), we tested whether KDM4A could be contributing to the observed EGFR amplifications in lung cancer cells. We used both KDM4A siRNA depletion and chemical inhibition (KDM4i (QC6352); (43)) of the KDM4 family to assess the impact of KDM4A on EGFR amplification. Specifically, we used DNA FISH to test whether the classically used EGFR amplified HCC827 lung cancer cells would have reduced amplifications upon KDM4A depletion or inhibition. HCC827 cells, in addition to exhibiting extensive EGFR amplification, have an acquired activating mutation within the EGFR tyrosine kinase domain (Exon 19 deletion) (41). As previously noted, HCC827 cells had very high EGFR DNA amplification levels that form large EGFR gene cluster clouds (Figure 3A). The amplifications are so abundant that they appear as clouds in the interphase nuclei (42). However, KDM4A depletion significantly reduced the size of EGFR amplified clouds in the HCC827 cells (Figure 3 A,B and Figure S3A, B). Furthermore, KDM4 family inhibition significantly reduced the extent of EGFR amplification in these cells as well (Figure 3C, D). Based on these observations and the fact that EGFR DNA amplification has been shown to correlate with better EGFR inhibitor responses in patients (24–29), we hypothesized that KDM4 inhibitor treatment would reduce the sensitivity of HCC827 cells to the EGFR inhibitor, Gefitinib. As anticipated, a reduction in EGFR DNA amplification directly correlated with a reduced response to Gefitinib (Figure 3E, right panel). Taken together, these data highlight a functional and significant role for KDM4A in modulating EGFR amplifications, expression and response to both EGFR inhibitors and growth factors in both diploid, non-transformed cells and EGFR amplified cancer cells.
Figure 3. KDM4A controls EGFR amplification in HCC827 cells.

A) Representative DNA FISH images of HCC827 lung cancer cells, treated with either siRNA control or siRNA targeted to KDM4A. EGFR (red), DAPI (blue) and merge are shown. B) siRNA-mediated depletion of KDM4A (red), reduces the size of EGFR amplified gene cluster clouds in HCC827 lung cancer cells. C) Representative DNA FISH images of HCC827 lung cancer cells, treated with either DMSO or a KDM4 family inhibitor. EGFR (red), DAPI (blue) and merge are shown. D) Inhibition of KDM4 family (red) reduces the size of EGFR amplified gene cluster clouds in HCC827 lung cancer cells. E) KDM4 family inhibitor treatment reduces the efficacy of the EGFR inhibitor, Gefitinib, in HCC827 lung cancer cells (right panel).
H3K9 KMTs regulate EGFR copy number
Since K9 methylation interference and catalytically active KDM4A overexpression promoted EGFR amplification, we tested whether each of the H3K9 lysine methyltransferases (K9 KMT) were equally capable of inhibiting EGFR copy gains or whether there was enzyme specificity. Specifically, RPE cells were siRNA depleted with at least two independent siRNAs for each K9 KMT. The knockdowns were confirmed and cell cycle profiles were generated for each siRNA, which ensures no overt arrests occurred, and in turn, interfere with EGFR copy gains (Figure S4A–F). Samples were then analyzed by EGFR DNA FISH. With the exception of G9a/KMT1C, depletion of all other K9 KMTs resulted in a significant increase in EGFR DNA copy number (Figure 4A). These data coupled with the methylation interference and demethylase activity requirement, strongly suggest that maintaining the degree of H3K9me1/2/3 methylation at this locus is critical for preventing amplification. Consistent with this notion, H3K9me1/2/3 ChIP-seq illustrated that the higher ordered organized domains that contained the EGFR locus were enriched for H3K9me1/2 between the H3K9me3 flanking blocks (Figure 4B). Taken together, these data imply the need to balance H3K9 methylation in order to prevent or promote EGFR copy gains.
Figure 4. H3K9/K27 KMTs regulate EGFR amplification.

A) DNA FISH analysis of EGFR (red), 7c (black) and 8c (grey) following siRNA-mediated depletion of K9 KMT family members. B) Input-normalized ChIP-seq tracks of H3K9me1–3, H3K4me1–3, H3K27me3 (34) and EZH2 (50) density in the megabase vicinity of the EGFR gene, aligned with the Hi-C map of chromatin interactions. C) EZH2 depletion promotes EGFR DNA copy gains that are KDM4A-dependent. D) Representative DNA FISH images of RPE nuclei from cells treated with DMSO, 1µM EZH2i (EZH2i Gain) or 3µM EZH2i (EZH2i Amp). EGFR (red) and DAPI (blue) are shown. E) EGFR DNA copy gains occur in a dose-dependent manner in response to EZH2 inhibitor (1, 3 and 5µM) treatment. F) EGFR copy number gains return to baseline 24 hours after EZH2 inhibitor (3µM) drug removal (+ washout). G) RPE cells have increased EGFR transcripts following 24 hours of EZH2i treatment at higher doses (3 and 5µM), as measured by qPCR. H) Spike-in normalized H3K27me3 ChIP-seq data from previously published dataset (44). PC9 cells were treated for 5 days with EZH2 inhibitor GSK126. I) Transient overexpression of the K27 tri-demethylases, KDM6A and KDM6B, promote EGFR DNA copy gains in RPE cells. J) RPE cells pre-treated for 24 hours with 3µM of EZH2i proliferate faster upon stimulation with 50ng/ml EGF. K) siRNA-mediated depletion of EZH2 caused increased cell proliferation in response to 50ng/ml EGF, which is completely rescued by co-depletion with EGFR in RPE cells. Error bars represents S.E.M. The * represents p=≤0.05 by two-tailed Student’s t-test. The arrowheads mark DNA FISH foci. The scale bars are 5um.
EZH2 and KDM6 enzymes regulate EGFR copy number
In addition to H3K9 methylation interference causing EGFR amplification, we also observed that H3K27 methylation interference increased EGFR copy number (Figure 1B, C). Therefore, we sought to further explore the importance of K27 methylation in modulating EGFR amplification. First, we compared a publicly available EZH2 ChIP-seq dataset (44) to our H3K27me3 profiles. We observed an overlap between EZH2 occupancy and the H3K27me3 domains that flank the EGFR locus (Figure 4B). These binding profiles and the fact H3K27 methylation interference promoted EGFR amplification (Figure 1B, C) and reduced H3K27me3 flanking the EGFR locus (Figure 1M) prompted us to then test whether EZH2 siRNA-mediated depletion or chemical inhibition would phenocopy H3K27 methylation interference-induced EGFR copy gains.
Using two independent siRNAs against EZH2 in RPE cells, we observed a significant increase in EGFR copy number while not changing copy number of other chr7 regions (Figure 4C and Figure S4G–K). We further demonstrated that a specific EZH2 pharmacological inhibitor (EZH2i- C24; (45)) was able to promote EGFR amplification in a dose-dependent manner while not altering copy number of other loci in RPE cells (Figure 4D, E and Figure S4L,M). EGFR copy number was also increased in colorectal cancer cell lines that have either low (HCT-15) or high (HT-29) EGFR copy number when treated with the EZH2i (Figure S4N–Q). Furthermore, removing EZH2i from RPE cells (referred to as washout) allowed EGFR copy gains to return to baseline, which highlights their transient nature (Figure 4F and Figure S4M). At the higher doses of EZH2 inhibition, EZH2i also resulted in a higher number of copies within some of the copy gained nuclei and increased EGFR transcript (Figure 4G), which phenocopies H3K27M transduced cells (Figure 1F-H). Furthermore, ChIP-seq analyses of PC9 cells (lung adenocarcinoma) treated with an EZH2 inhibitor (44) also showed a reduction in the H3K27me3 domains flanking EGFR (Figure 4H). In order to further explore the importance of H3K27me3 in preventing the EGFR locus from amplifying, we overexpressed each of the H3K27 demethylases- KDM6A and KDM6B- individually and then conducted EGFR FISH (Figure 4I and S4R, S). In contrast to the specificity observed with KDM4 family members, both KDM6 members promoted EGFR copy gains (Figure 4I). Taken together, these results demonstrate a critical role for H3K27 methylation and the associated enzymes in preventing the EGFR locus from undergoing amplification and expression.
Since EZH2 inhibition promoted EGFR amplification and increased expression levels, we tested whether cells treated with the EZH2i or EZH2 siRNA depletion would respond differently to EGF supplementation. Specifically, cells were pre-treated with EZH2 inhibitor for 24 hours or depleted with two independent EZH2 siRNAs in order to promote increases in EGFR copy number before being supplemented with DMSO or EGF. Both EZH2 inhibitor and EZH2 siRNA treated cells demonstrated a significantly increased proliferation in response to exogenous supplementation with EGF compared to DMSO-treated cells (Figure 4J,K). Moreover, EGFR/EZH2 siRNA co-depletion completely blocked the increased response to EGF (Figure 4K and Figure S4T). These data are consistent with a prior report noting that combined inhibition of EZH2 and EGFR were able to increase the sensitivity of colorectal cancer cells when compared to either drug alone (46). Our data suggest that this observation could be in part through EZH2i-induced EGFR DNA amplification. In support of this hypothesis, EZH2i treatment in two different colorectal cancer cell lines (HCT-15 and HT-29) resulted in increases in EGFR DNA copy number in these cells (Figure S4N–Q), which paralleled their sensitivity (46). Taken together, these data illustrate a critical role for K27 methylation, EZH2 and KDM6 in modulating EGFR locus amplification, expression, and in turn, cellular response to either EGFRi or growth factors.
H3K4 and H3K27 methylation controls EGFR amplification
In a recent study, we demonstrated that H3K4 methylation enrichment at specific genomic loci was sufficient to recruit KDM4 family members to chromatin, and in turn, promote copy number gains on chromosome 1 (e.g., 1q12h and 1p32.3; (10)). Upon evaluating H3K4me1/2/3 across the EGFR locus, we observed an inverse relationship between H3K4 methylation states and H3K27me3, which appears to flank the regions containing H3K4 methylation (Figure 4B). Therefore, we hypothesized that inhibition of EZH2 promoted the KDM4A driven EGFR copy gains through H3K4 methylation. Consistent with this hypothesis, RPE cells transduced with H3.3 K4M did not generate EGFR copy gains upon EZH2 inhibition (Figure 5A and Figure S5A, B), which highlights the importance of this amino acid in promoting EGFR copy gains downstream of EZH2 depletion or inhibition. Furthermore, KDM4A was required for EZH2 depletion to generate EGFR amplification (Figure 4C).
Figure 5. H3K4/27 methylation controls EGFR amplification.
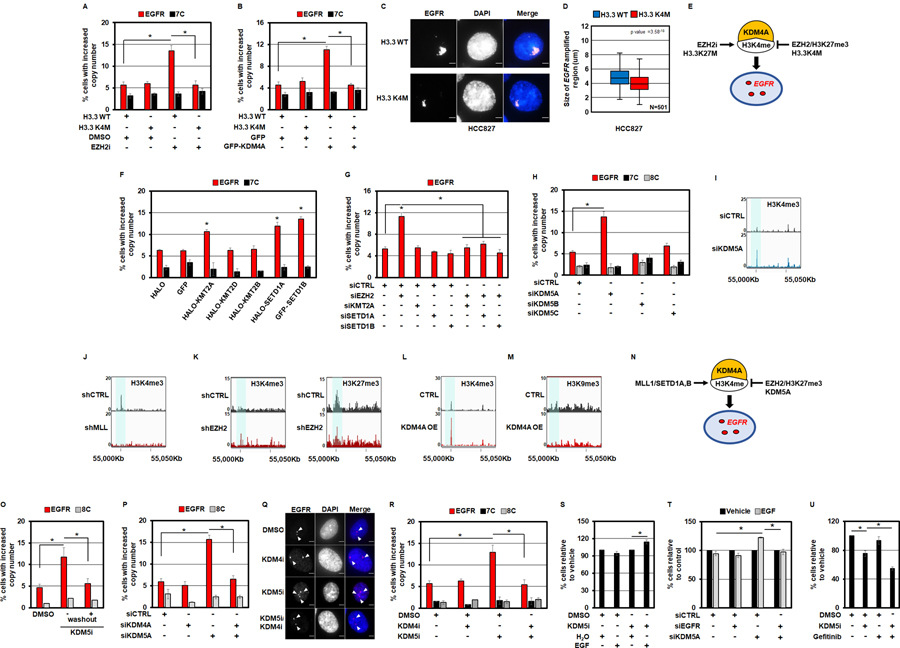
A) RPE cells transduced with H3.3 K4M completely inhibit EZH2i-mediated DNA copy gains of EGFR. B) RPE cells transduced with H3.3 K4M completely inhibit GFP-KDM4A overexpression-mediated DNA copy gains of EGFR. C) Representative DNA FISH images of HCC827 lung cancer cells transduced with H3.3 wild type (WT) or H3.3 K4M. EGFR (red) and DAPI (blue) are shown in merge. D) H3.3 K4M transduced HCC827 cells (red), reduces the size of EGFR amplified gene cluster clouds when compared to H3.3 WT transduced cells (blue). E) A model depicting the impact EZH2, H3K4M and H3K27M have on EGFR copy gains through KDM4A and H3K4 methylation based on the genetic experiments in panels A-D. F) Transient overexpression of HALO-tagged KMT2A and SETD1A as well as GFP-tagged SETD1B promote EGFR DNA copy gains in RPE cells. G) siRNA-mediated co-depletion of KMT2A, SETD1A or SETD1B with EZH2 completely blocks EZH2-depletion mediated EGFR copy gains in RPE cells. H) siRNA-mediated depletion of the H3K4 tri-demethylase KDM5A promoted EGFR copy number gains. I-M) A candidate control intergenic locus in the vicinity of the EGFR region. Input-normalized ChIP-seq tracks of H3K4me3 density near the EGFR locus (chr7:55Mbp) are highlighted in control cells (top tracks) versus cells treated with: siKDM5A (I), shMLL (J) (49), shEZH2 (K)(50), and KDM4A overexpression (L) in bottom tracks. H3K27me3 density is shown in the shEZH2 treated cells (bottom tracks) compared to control cells (top tracks) (50). H3K9me3 density is shown in the KDM4A overexpressing RPE cells (bottom tracks) compared to control RPE cells (top tracks). N) A model depicting the interplay between KMT2 enzymes (MLL1/SETD1A, B), KDM5A and EZH2 in regulating EGFR copy gains through KDM4A and H3K4 methylation based on the genetic and epigenomic experiments in panels F-M. O) EGFR copy number gains return to baseline 24hrs after KDM5i (1µM) removal (+washout). P) siRNA-mediated co-depletion of KDM4A with KDM5A, blocks KDM5A-depletion mediated EGFR copy gains. Q) Representative DNA FISH images of RPE nuclei from cells treated with KDM4i (1nM), KDM5i (1µM) or pre-treated with KDM4i (1nM) followed by KDM5i (1µM) treatment. EGFR (red), DAPI (blue) and merge are shown. R) A 24 hour pre-treatment of RPE cells with KDM4i (1nM) completely blocks KDM5i -mediated DNA copy gains of EGFR. S) RPE cells pre-treated with KDM5i (1µM) for 24 hours proliferate faster in response to a 24 hour stimulation with 50ng/ml EGF (compared to the respective vehicle control). T) KDM5A depleted RPE cells proliferate faster in response to a 24 hour stimulation with 50ng/ml EGF (compared to KDM5A-depleted cells treated with vehicle), which is rescued by co-depletion of EGFR. U) Co-treatment of RPE cells with KDM5i (1µM) and Gefitinib (2.5µM) reduces the percentage of cells relative to controls and single agent treatment as measured by trypan blue exclusion assay. Error bars represents S.E.M. The * represents p=≤0.05 by two-tailed Student’s t-test. The arrowheads mark DNA FISH foci. The scale bars are 5um.
Since H3K4 methylation is key to KDM4A binding and copy gain generation at other specific sites in the genome (10,18), we also tested whether H3K4M would block KDM4A promoted EGFR copy gains. Indeed, H3K4M blocked KDM4A driven EGFR amplification (Figure 5B and Figure S5C, D). Consistent with these observations, HCC827 cells containing the EGFR amplification clouds that were reduced in size upon KDM4A depletion and inhibition (Figure 3) also had a significant reduction EGFR cloud size with H3K4M transduction (Figure 5C,D). A similar reduction was also observed with H3K4M transduction in the HT-29 cells that contain EGFR copy gains (Figure S5E, F). Furthermore, the Tudor domains that recognize H3K4 methylation within KDM4A (39) were required to generate EGFR copy gains (Figure 2B). Taken together, these data collectively emphasize the importance of H3K4 methylation for generating the EGFR amplifications and highlight the need to balance K4/27 methylation states in order to modulate EGFR copy gains through KDM4A (Figure 5E).
H3K4 KMTs were recently shown to be important in controlling the predilection of regions to amplify downstream of KDM4 members. For example, overexpression of each KMT2 family member promoted copy gains of specific chromosome 1 TSSG loci (10). Therefore, we overexpressed each H3K4 KMT (MLL1/KMT2A, MLL2/KMT2B, MLL4/KMT2D, SETD1A and SETD1B; (47)) to determine whether all or select KMTs promote EGFR amplification (Figure S5G–I). Only KMT2A, SETD1A and SETD1B were able to significantly promote EGFR copy gains (Figure 5F). To date, the EGFR loci was the only target identified for SETD1A. Since EZH2 and the MLL family of KMTs oppose one another and their associated methylation states (48), we tested whether EGFR amplifications that occur downstream of EZH2 depletion are dependent on these H3K4 KMTs. When EZH2 and each KMT2 member (KMT2A, SETD1A, SETD1B) were co-depleted, the EGFR copy gains were blocked, which highlights the importance of H3K4 methylation upon EZH2 depletion (Figure 5G and Figure S5J, K).
Since specific H3K4 KMTs controlled EGFR amplification, we tested whether the same was true for the H3K4 KDMs. The KDM5 enzymes are H3K4 tri-demethylases and have been shown to impact other TSSG sites (10). Depletion of the H3K4 tri-demethylases will increase H3K4 methylation, thereby promoting copy gains of EGFR if copy gains of this locus are indeed dependent on H3K4 methylation. Therefore, we depleted each KDM5 member with at least two independent siRNAs before conducting cell cycle profiles and EGFR FISH (Figure S5L, M). Only KDM5A depletion generated EGFR copy gains, while other genomic loci were unaffected (Figure 5H and Figure S5N–P; (10)). Consistent with these genetic experiments, we observed enrichment for H3K4me3 upon ChIP-seq at an intergenic region within the EGFR structural domain that is 87 Kb upstream of the EGFR transcription start site (TSS) (Figure 5I, blue shaded region). Furthermore, we observed a loss of this H3K4me3 peak upon shMLL1 treatment in a published dataset (Figure 5J; (49)) and enrichment upon shEZH2 treatment in a public dataset that was accompanied with reduced H3K27me3 (Figure 5K; (50)). Moreover, RPE cells that overexpress KDM4A exhibited increases in H3K4me3 as well as a reduction in H3K9me3 at this same region (Figure 5L, M, respectively). Taken together, these data suggest that KMT2 enzymes, KDM5A and EZH2 modulate the balance of H3K4/27 methylation at the EGFR locus, and in turn, EGFR copy gains (Figure 5N).
Our studies suggest that promoting H3K4 methylation through KDM5A depletion or inhibition would promote EGFR amplification through KDM4A. First, we evaluated the impact of KDM5 inhibitor (KDM5i) treatment on EGFR amplification (KDM5i – C70; (51,52)). KDM5i treatment promoted EGFR copy gains, however, they returned to baseline upon KDM5i washout (Figure 5O and Figure S5Q), which emphasizes the reversibility of the amplifications as observed with EZH2i washout. We then used both genetic and chemical inhibition of KDM4 enzymes (KDM4i; (43)) to determine whether this would block EGFR amplifications generated upon either KDM5A siRNA depletion or KDM5i treatment (Figure 5P-R and Figure S5R–U). In fact, the use of KDM5 and KDM4 inhibitors can be used to generate and prevent EGFR copy gains (Figure 5Q, R), which highlights plasticity of the copy gains and the ability to therapeutically control DNA amplification with drugs targeting epigenetic factors.
Having therapeutic control of key growth factor receptors could have a profound impact on the ability to control cell proliferation and drug response when delivering inhibitors. Therefore, we tested whether KDM5i would alter cell proliferation when treated with supplemental EGF. Consistent with the previous experiments demonstrating that factors promoting EGFR copy gains increased EGF associated proliferation, KDM5i increased cell proliferation with supplemented EGF when compared to KDM5i alone (Figure 5S), which was also observed with KDM5A siRNA depletion, and in turn, was blocked with EGFR siRNA depletion (Figure 5T and Figure S5V). Lastly, we were also able to demonstrate that KDM5i increased the sensitivity to Gefitinib when compared to KDM5i alone (Figure 5U), which was consistent with a previous report (53). Taken together, these data highlight the ability to modulate H3K4 methylation in order to harness control of EGFR DNA copy levels, and in turn, growth factor and drug response.
Hypoxia and Epidermal Growth Factor induce EGFR amplification
Previous work from our laboratory demonstrated that hypoxia directly promoted TSSG formation of chromosome 1 associated loci (e.g., 1q12h) via stabilization of the KDM4A protein, through reduced association with the SKP1– Cul1–F-box (SCF) ubiquitin ligase complex (17). Consistent with this previous observation, 24 hours of exposure to hypoxia was able to stabilize KDM4A in RPE cells and promote EGFR copy gains that required KDM4A (Figure 6A-D and Figure S6A–D). The hypoxia-induced copy gains of EGFR were transient in nature, in a similar manner to those observed with KDM5i and EZH2i. Indeed, moving cells back to normoxia following 24 hours of hypoxia exposure completely restored EGFR copy number to baseline levels (Figure 6E, F). In agreement with the observation that KDM4A is required for hypoxia-induced EGFR DNA copy gains, pre-treatment of cells with KDM4 family inhibitor prior to hypoxic exposure also blocks hypoxia-induced EGFR copy gains (Figure 6G and Figure S6E). Furthermore, H3.3 K4M mutant expressing cells did not generate the hypoxia-induced EGFR amplifications (Figure 6H and Figure S6F,G). Taken together, these data demonstrate that hypoxia promotes EGFR copy gains in a similar fashion to KDM4A overexpression in that the amplifications require increased KDM4A levels and proper targeting via H3K4 methylation (Figure S6H).
Figure 6. Hypoxia and EGF induce EGFR amplification.
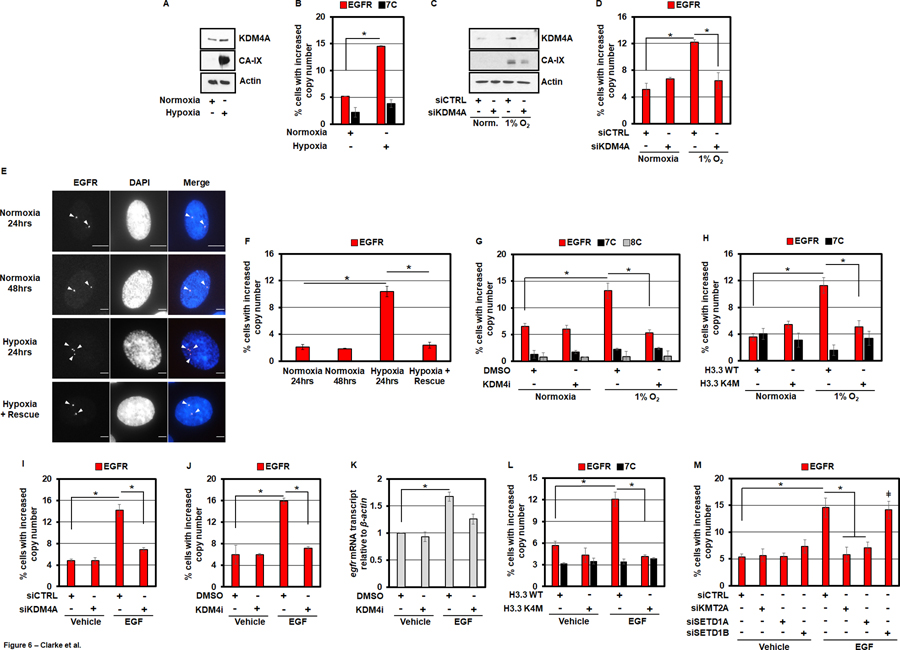
A) KDM4A protein levels increase in RPE cells after being cultured in hypoxia (1% O2) for 24 hours. B) RPE cells cultured in hypoxia for 24 hours have increased EGFR copy gains. C) KDM4A protein levels increase with hypoxia (1% O2 for 24 hours) and are depleted with siKDM4A. D) Hypoxia induces EGFR DNA copy gains in a KDM4A-dependent manner. E) Representative EGFR DNA FISH images of RPE nuclei from cells treated with normoxia (24 and 48 hours), hypoxia (24 hours) or hypoxia (24 hours) and transferred to normoxia (24 hours). EGFR (red) and DAPI (blue) are shown in the merge. F) Hypoxia induced EGFR DNA copy gains are rescued with transfer to normoxia. G) A 24 hour pre-treatment of RPE cells with KDM4i (1nM) completely blocks hypoxia-induced EGFR amplification. H) RPE cells transduced with H3.3 K4M do not exhibit EGFR DNA copy gains when cultured in hypoxia for 24 hours. I) RPE cells treated with 50ng/ml EGF for 24 hours exhibit EGFR amplification that are KDM4A-dependent. J) A 24 hour pre-treatment of RPE cells with KDM4i (1nM) completely blocks EGF-induced EGFR DNA copy gains. K) 50ng/ml EGF for 24 hours increases EGFR transcripts in RPE cells, which is partially KDM4-dependent. L) Following 24 hour stimulation with 50ng/ml EGF, RPE cells transduced with H3.3 K4M do not exhibit increases in EGFR DNA copy number. M) siRNA-mediated depletion of KMT2A and SETD1A blocks EGF-induced EGFR DNA copy gains.
While investigating hypoxia modulation of EGFR amplification, we also tested whether EGF supplementation would impact EGFR copy gains. A recent report demonstrated that cancer cells with EGFR amplification required EGF supplementation in the media to propagate the copy gains, which raised the possibility that EGF could directly promote EGFR copy gains (54). Consistent with this possibility, we demonstrated that treating cells for 24 hours with 50ng/ml EGF, the preferred ligand of EGFR, results in significant copy number gains of the EGFR locus (Figure 6I, J). Furthermore, these gains were entirely dependent on KDM4A as both siRNA-mediated depletion and pharmacological inhibition of the KDM4 family was able to completely suppress these gains (Figure 6I, J and Figure S6I–K). We also observed increased EGFR transcripts that were reduced upon KDM4 inhibition (Figure 6K). Even though KDM4A was required for both the copy gains and increased expression, we did not observe increased KDM4A protein levels (Figure S6L), which suggest another pathway could be promoting KDM4A-dependent copy gains.
Given the importance of H3K4 methylation in targeting KDM4A so that TSSGs occur, we hypothesized that EGF was promoting the TSSGs via the H3K4 KMTs, and in turn, H3K4 methylation. Therefore, we first tested whether H3K4M would block EGF-induced EGFR copy gains. We observed that expression of the methyl-deficient H3K4M mutant was sufficient to block these growth factor-induced copy gains (Figure 6L and Figure S6M, N). Therefore, we then depleted with two independent siRNAs each of the H3K4 methyltransferases that generated amplification of the EGFR locus (KMT2A, SETD1A, SETD1B; Figure 5F and Figure S6O, P). Following depletion, cells were treated with EGF (50ng/ml) for 24 hours and their EGFR copy number was assessed by DNA FISH. Depletion of KMT2A (MLL1) and SETD1A were able to completely inhibit EGF-induced copy gains of EGFR, whereas depletion of SETD1B had no impact on the amplifications (Figure 6M). Thus, EGF treatment appears to promote EGFR copy gains through KMT2A, SETD1A and H3K4 methylation, which illustrates how extrinsic cues can promote selective copy gains via specific methyltransferases. Taken together, these data suggest that two different extrinsic cues (hypoxia and EGF) are promoting EGFR copy gains through similar but distinct epigenetic mechanisms (Figure S6H).
Epigenetic dysregulation combined with hypoxia or increased EGF induce higher EGFR copy number
In this study, we have identified a network of chromatin regulators and physiological signals that influence EGFR copy gains. In the case of hypoxia, KDM4A stabilization mirrors overexpression and promotes EGFR amplification, while EGF appears to promote EGFR amplification through KMT2A/SETD1A and targeting KDM4A (Figure S6H). Therefore, multiple extracellular cues could promote parallel triggers for the amplifications, which raises the question as to whether the combined signals elicit stronger amplification events. In order to test this possibility, cells were exposed to hypoxia for an initial 24 hours, followed by supplementation with EGF (50ng/ml) under hypoxia conditions for an additional 24 hours. At this point, cells were harvested and their EGFR copy number assessed by DNA FISH. Hypoxia, in combination with EGF, resulted in a modest but additive increase in EGFR DNA copy number (Figure 7A and Figure S7A). Moreover, an increased percentage of these cells demonstrated higher EGFR DNA copies per nucleus (≥5 copies; Figure 7B, C). This observation suggests that hypoxia-induced KDM4A stabilization in concert with EGF-stimulated increases in H3K4 methylation promote EGFR locus plasticity so higher DNA copy numbers are achieved. To strengthen this model, RPE cells that stably overexpress KDM4A, thus mimicking the phenotype observed upon hypoxic exposure, were also treated with EGF (Figure S7B). EGF treatment of KDM4A overexpressing cells phenocopied the increase in EGFR amplification levels and increased DNA copies per nucleus that were observed in cells treated with hypoxia and EGF (Figure 7A, D–F and Figure S7C). Moreover, in further support of this model, inhibition of KDM5 or EZH2, both of which increase H3K4me3 at the EGFR locus (Figure 5I, K), enhance EGFR amplification when combined with hypoxic exposure (Figure S7D–I). Taken together, our data supports a model by which physiological triggers such as increased EGF concentration and/or hypoxia function in combination with epigenetic perturbation to directly modulate chromatin states and determine whether site-specific low or high copy DNA amplifications occur (Figure S7C).
Figure 7. Combination of epigenetic dysregulation, hypoxia and EGF induce higher EGFR amplification.

A) RPE cells cultured in hypoxia (24 hours) and then treated for 24 hours with 50ng/ml EGF in continued hypoxia results in a higher percentage of cells with EGFR copy gains. B) Representative DNA FISH images of RPE nuclei from cells treated with hypoxia, EGF or a combination of hypoxia and EGF. EGFR (red), DAPI (blue) and merge are shown. C) Graph illustrating the % of cells with >4 and >5 EGFR DNA copies from panel A. D) RPE cells stably overexpressing KDM4A exhibit additive increases in EGFR DNA copy number when stimulated with 50ng/ml EGF for 24 hours. E) Graph illustrating the % of cells with >4 and >5 EGFR DNA copies from panel D. F) Representative DNA FISH images of higher EGFR DNA copies in KDM4A overexpressing cells treated with 50ng/ml EGF for 24 hours. EGFR (red), DAPI (blue) and merge are shown. Error bars represents S.E.M. The * represents p=≤0.05 by two-tailed Student’s t-test. The arrowheads mark DNA FISH foci. The scale bars are 5um.
Discussion
To date, little knowledge exists about the molecular mechanisms that promote specific oncogene amplifications. In this study, we uncover epigenetic regulators and physiologic cues that facilitate amplification of the oncogene EGFR. Moreover, we provide some of the first evidence of the ability to rheostat an oncogenic amplification through therapeutic intervention. These data illustrate a molecular basis for EGFR amplification and establish that the extra cellular microenvironment can directly contribute to DNA amplification heterogeneity in both normal and tumor cells. Furthermore, we demonstrate that these copy gains are transient and that combined cues and/or epigenetic factor manipulation are sufficient to promote higher copy number amplifications. Overall, this study describes a series of key observations that demonstrate oncogenic amplification is hardwired into cells, providing a definable basis for cellular plasticity for EGFR copy number in both normal and cancer cells, which has significant clinical implications.
Specificity, Crosstalk and Methylation States
Prior studies have illustrated that both somatic and tumor cells have extrachromosomal DNA (ecDNA; (11,55)) with key oncogenes such as MYC and EGFR occurring as ecDNA in as many as 50% of tumors (11). Early studies on extrachromosomal MYC demonstrated that the ecDNA harbored epigenetic states associated with active gene expression (56). Consistent with these observations, a recent study in somatic cells illustrated that ecDNAs were observed in gene-rich chromosomal regions (55), which suggested a relationship between their generation and actively marked loci. Our study provides additional evidence to these prior correlative observations by being able to directly promote or block such modifications through manipulation of histones, histone modifying enzymes and their upstream regulators such as hypoxia. The data presented within this manuscript illustrates a critical role for KMT-KDMs in balancing the methylation states controlling the repressive state (H3K9/27 methylation) and more accessible, active states (H3K4 methylation) so that EGFR amplification is either blocked or promoted.
Methylation states appear to control the predilection of a region to amplify, however, not all enzymes controlling those states are responsible for generating the EGFR amplifications. For example, KDM4A and KDM5A were the only members within their lysine demethylase enzyme families to promote EGFR. However, the KDM6 family members, KDM6A and KDM6B, were both sufficient to generate EGFR amplification upon overexpression, which suggest that these enzymes could have functional redundancy at this locus in controlling H3K27me3 balance. These data highlight that enzyme families could have unique targets, and in certain cases, overlapping specificity.
Similar observations were also true for KMTs targeting H3K4/9 methylation. For example, KMT2A/MLL1, SETD1A and SETD1B promoted EGFR amplification (Figure 5). SETD1A was not responsible for previously identified TSSGs (10), highlighting the specificity for KMT2 family members in controlling genomic regions undergoing TSSG. Similarly, all H3K9 KMTs except KMT1C/G9a/EHMT2 promoted EGFR copy gains when depleted from cells. The EGFR locus has a clear pattern for the H3K9me1/2/3 distribution across the locus, which suggest an important arrangement for these methylation states and their associated KMTs. In fact, H3K9M introduction promoted EGFR low and higher level copy gains (Figure 1). Therefore, future studies need to interrogate the dependencies of regions on the various members and establish regions that have unique targets or overlapping control.
Data presented here implies that KDM4A utilizes the same mechanism to generate EGFR amplification as other previously mapped regions undergoing copy gains (e.g., 1q21.3- CKS1B; (10,17,18)). KDM4A and KDM4B were shown to recruit the replication machinery and facilitate rereplication (10,18). Our previous studies also demonstrated that H3K4 methylation was key to the recruitment of KDM4 family members and the modulation of chromosome 1 targets, which was driven by select H3K4 KMTs (10). These previous studies did not observe a role for H3K27 methylation in controlling the TSSG formation (10,18). However, H3K27me3 was a key modulator of EGFR amplification. Indeed, EZH2 depletion and chemical inhibition promoted EGFR copy gains. EZH2 occupies the blocks of H3K27me3 that flank H3K4 methylation within the EGFR locus. Furthermore, H3K4 methylation interference or depletion of the KMT2 enzymes that control EGFR amplification completely blocked the EGFR copy gains generated by EZH2 suppression or inhibition. These data are consistent with the collection of studies illustrating an antagonistic relationship between these methylation states and the associated enzymes promoting H3K4/27 methylation balance (48). Our data is also consistent with a recent report demonstrating cross-talk between EZH2 and KMT2A disrupts H3K27 methylation balance, resulting in resistance to EZH2i monotherapy (57). Therefore, common principles assigned to gene regulation could also be true for TSSG regulation, which has direct clinical implications.
Future studies need to evaluate the impact that combinations of histone modifications have on enzyme targeting and TSSG regulation. Studies should also consider the contribution of other genomic features (e.g., insulator elements, enhancers, CpG islands, etc.) in controlling these events. Based on HiC data and larger H3K9me3 and H3K27me3 blocks, we suspect that in addition to localized affects, higher order structure is also key to regulating site-specific copy gains. Future studies need to explore the impact of altering higher order genome organization at regions undergoing site-specific amplification. The EGFR locus would serve as an ideal region to evaluate these future questions.
Cues, Epigenetics and Targeting Heterogeneity
Previous studies have illustrated EGFR amplification plasticity (12). EGFR copy gains can range from few in number to large clouds in the nuclei of cancer cells (see Figure 3; (42)). Glioblastoma patients that received EGFRi have been shown to develop resistance because ecDNA copies of EGFR disappear. However, upon drug removal EGFR ecDNA recurs, re-establishing sensitivity to targeted therapy (12). This plasticity has also been illustrated in cell culture as EGFR copy gains disappear in some cell lines (e.g., GBM cells). A recent study demonstrated however, that supplementing EGF enables propagation of the EGFR amplifications (54). Therefore, there is a critical need to understand the mechanisms that promote or suppress this amplification event.
Consistent with the prior body of work suggesting certain extracellular cues (hypoxia and EGF) associate with tumor cells harboring EGFR amplification, we have now demonstrated that these stimuli directly control EGFR DNA copy number (Figures 6,7). We observe that hypoxia-induced stabilization (17) and the catalytic activity of KDM4A is a central component in generating EGFR amplification. In fact, KDM4 chemical inhibition was sufficient to reduce this oncogenic amplification in hypoxia-stimulated cells as well as lung cancer cells harboring EGFR ecDNA. This observation is critical because it suggests that targeting KDM4A with small molecules could serve as a novel approach to control EGFR amplification and heterogeneity observed in hypoxic tumors. In addition, amplification of wild type EGFR has been shown to drive acquired resistance to mutation-selective EGFR tyrosine kinase inhibitors, identifying an additional therapeutic arena for potentially deploying KDM4i therapy (58).
EGF treatment does not impact KDM4A levels but rather triggers EGFR amplification through two specific H3K4 KMTs- KMT2A/MLL1 and SETD1A. While both KMT2A and SETD1A controlled EGF-induced copy gains, SETD1B was dispensable, which highlights that enzymes can be selectively required under certain physiological conditions to generate DNA copy gains. Future studies should address whether other cellular signals or stresses could serve as important triggers to selectively activate or repress the enzymes required to generate amplification at EGFR and other TSSG sites. Understanding the triggers for amplifications will provide insights into tumor heterogeneity and uncover novel biomarkers and drug targets in controlling amplification.
Consistent with these two pathways (hypoxia and EGF) working in parallel, when hypoxia is combined with KDM4A, there is little change in the low copy number; however, when combined with increased H3K4 methylation there are higher copy number gains of EGFR per nucleus. The same is true when EGF is combined with KDM4A overexpression. These data illustrate that combining extracellular cues and epigenetic factor alterations promotes EGFR copy number generation and the degree of amplification. Given the range of EGFR amplification across tumors (Figure 1A), these observations could be important when considering variables impacting DNA copy number plasticity in tumors and when considering ways to therapeutically intervene.
Data presented within this manuscript is suggestive of two populations of EGFR DNA amplifications. For example, in cancer cell lines which exhibit significant DNA amplification of EGFR (e.g. HCC827 or HT-29 cells), these amplifications are reduced by modulation of K4 methylation via the introduction of a H3.3 K4M methyl-deficient mutant, or inhibition/depletion of KDM4A. However, despite a reduction in EGFR DNA amplification under these conditions, significant levels of EGFR amplification remain. Future studies will need to investigate this in more detail, but these results could indicate a balance between integrated DNA copy number amplification and transient extrachromosomal amplifications of EGFR, the latter of which appears targetable with compounds directed towards epigenetic modifiers.
In closing, we have uncovered both chromatin modifiers and extracellular signals that control EGFR amplification and demonstrate that epigenetic therapies could hold a key to modulating EGFR copy number heterogeneity in cancer and associated diseases, which could have significant clinical implications in the future.
Materials and Methods
Cell Culture:
Retinal pigment epithelial (RPE) and 293T cells were cultured in DMEM-high glucose (Sigma) media supplemented with 10% heat-inactivated fetal bovine serum (FBS), 100U/ml penicillin, 100ug/ml streptomycin, and 2mM L-glutamine. HCC827 cells (lung adenocarcinoma), HCT-15 (colorectal adenocarcinoma) and HT-29 (colorectal adenocarcinoma) were cultured in RPMI 1640 Glutamax media (Gibco) supplemented with 10% heat-inactivated FBS, 100U/ml penicillin, 100ug/ml streptomycin, 1% sodium pyruvate and 1% glucose. Cell line identities were authenticated by STR analysis and mycoplasma tested using the MycoAlert Detection Kit (Lonza - LT07–218).
Hypoxic Conditions:
Cells were plated in cell culture dishes and allowed to adhere for 16–20 hours in normoxia (5% CO2, 21% O2, 74% N2). For hypoxic treatment, cells were maintained in a HERA Cell 150 incubator (Thermo Scientific) flushed with 5% CO2 and 1% O2 and balanced with N2 for the duration of the experiment. Cells were cultured in hypoxia for between 24 and 48 hours, prior to harvesting and downstream experimental processing. Hypoxic culture conditions were validated by immunoblot detection of carbonic anhydrase IX (CA-IX) expression, a well-established hypoxic biomarker signature (17).
Transfection Procedure(s):
Cells were plated in cell culture dishes and allowed to adhere for 16–20 hours. Cell culture medium was removed and cells rinsed with phosphate buffered saline (PBS) prior to siRNA transfections (5nM-10nM/transfection) being performed using Lipofectamine 3000 transfection reagent (Life Technologies) in OPTI-MEM medium (Life Technologies). Transfections were changed to complete cell culture media after 4 hr of transfection, and cells were collected 72 hr post transfection. For co-transfection experiments both siRNA sequences were transfected at the same time. For experiments involving hypoxia or drug treatment, cells were exposed to these conditions at least 48 hours post-siRNA transfection.
Transient overexpression transfections were performed using Lipofectamine 3000 transfection reagent and P3000 reagent (Life Technologies) in OPTI-MEM medium for 4 hrs, followed by changing to complete media. Silencer select negative controls and siRNAs were purchased from Life Technologies. Their sequences and unique identification numbers are tabulated in supplementary table 1. At least two different siRNAs against every gene were used for each experiment.
Transduction with Histone H3.3 variants:
Plasmids for H3.3 wild type (WT), K4M, K9M, K27M and K36M mutants were provided by Peter Lewis (University of Wisconsin). Virus was generated by co-transfection of specific plasmids along with the packaging plasmids (AmphoPAK and VSVG) in 293T cells, using Lipofectamine 3000 transfection reagent (Life Technologies). DNA transfection was performed in Opti-MEM medium (Life Technologies) overnight. The virus containing supernatant was collected after 24 hrs. RPE cells were infected in the presence of 8 mg/ml polybrene for 12 hrs with the viral supernatant (18). Cells were washed two times with DMEM, prior to being cultured in complete medium for 48 hours, before harvesting. For hypoxia, drug or growth factor treatment and overexpression experiments involving H3.3 K4M, RPE cells were infected as described above and cultured for 24 hours post infection before additional treatment (hypoxia, drug or growth factor treatment, or KDM4A transient-overexpression). Cells were harvested 24 hours later. For all experiments involving Histone H3.3 variants, RPE, HT-29 and HCC827 cells were harvested 48 hours after viral infection for western blot, cell cycle profiles and DNA FISH analysis.
RNA extraction and quantitative real-time PCR:
Cells were washed and collected by trypsinization, followed by washing in PBS two times. Cell pellet was resuspended in Qiazol reagent (QIAGEN) and stored at −80°C before further processing. Total RNA was extracted using miRNAeasy Mini Kit (QIAGEN) with an on-column DNase digestion according to the manufacturer’s instructions. RNA was quantified using NanoDrop 2000 (Thermo Scientific). Single strand cDNA was prepared using Super Script IV first strand synthesis kit (Invitrogen) using random hexamers. Expression levels were analyzed using FastStart Universal SYBR Green Master (ROX) (Roche) according to the manufacturer’s instructions on a LightCycler 480 PCR machine (Roche). Samples were normalized to β-actin. Primer sequences are provided in supplementary table 2.
Immunoblotting:
Cells were trypsinized and washed two times with PBS before resuspending in RIPA lysis buffer [50mM Tris pH 7.4, 150mM NaCl, 0.25% Sodium Deoxycholate, 1% NP40, 1mM EDTA, 10% Glycerol] freshly supplemented with protease inhibitor and PhosSTOP phosphatase inhibitor cocktails (Roche). Cells were lysed on ice for 15 min and stored at −80°C until further processing. Lysates were sonicated for 15 min (30sec ON and 30sec OFF cycle) at 70% amplitude in QSonica Q700 sonicator (Qsonica) followed by centrifugation at 12,000rpm for 15min. Cell lysate was transferred to a fresh tube and protein estimations were performed with Pierce BCA reagent (Thermo Scientific). Equal amounts of proteins were separated by SDS gel electrophoresis and transferred on nitrocellulose membrane (BioTrace NT, Pall Life Sciences) for at least 3 hr at a constant current. The membranes were blocked for at least 1 hr in 5% BSA-PBST (1X PBS with 0.5% Tween-20) or 5% milk-PBST and probed over night with specific antibodies as follows at the following dilutions: anti-GFP (Neuro mAb) (1:100); anti-β-Actin (1:10,000); anti-KDM4A (Neuro mAb) (1:100); anti-Flag (Sigma Aldrich) (1:500); anti-KDM5A (abcam) (1:500); anti-HALO (Promega) (1:1000); anti-Carbonic Anhydrase IX (Abcam) (1:1000). Catalog numbers for all antibodies used in this study can be found in supplementary table 3. Membranes were washed three times in PBST the next day, incubated with goat anti-mouse IgG peroxidase conjugated secondary antibody (170–6516, Biorad) or goat anti-rabbit peroxidase conjugated secondary antibody (A00167, GenScript) at 1:2500 in 5% milk-PBST for at least 1hr at room temperature, washed 3 times with PBST and incubated in Lumi-Light western blotting substrate (12015200001, Roche) or SuperSignal West Pico PLUS Chemiluminiscent substrate (34577, ThermoScientific) for 2–4 min(s). Membranes were developed with Lumi-Film Chemiluminiscent detection film (11666657001, Roche). The western blot images displayed in the figures have been cropped and auto-contrasted.
Cell Cycle Analysis:
Samples were washed with PBS, centrifuged at 1400rpm for 5 min, and permeabilized with 500mL PBS containing 0.5% Triton X-100 for 30 min. After this incubation, cells were washed with PBS and centrifuged at 1400rpm for 5 min. Samples were then stained with 1:100 dilutions of 1mg/mL PI solution and 0.5M EDTA with 100 mg RNase A, overnight at 4°C. Cell cycle distribution was analysed by flow cytometry using the LSRII flow cytometry system (BD Biosciences).
DNA Fluorescent In Situ Hybridization (FISH):
The FISH protocol was performed as described previously in Black et al. (2013)(18). Briefly, cell suspensions were fixed in ice-cold methanol:glacial acetic acid (3:1) solution for a minimum of four hours, before being spun onto 8 Chamber Polystyrene vessel tissue culture treated glass slides (Falcon, Fisher Scientific), using a centrifuge at 900rpm. The slides were air-dried and incubated in 2X SSC buffer for 2 min, followed by serial ethanol dilution (70%, 85% and 100%) incubations for 2 min each, for a total of 6 min. Air-dried slides were hybridized with probes that were diluted in appropriate buffer overnight at 37°C, following a 4 minute incubation on a heat block at 78°C. The slides were washed the next day for 3 to 4 mins in appropriate wash buffers at 69°C with 0.4X SSC for Cytocell probes or commercially available Agilent wash buffer 1 followed by washing in 2X SSC with 0.05% Tween-20 (Cytocell probes) or commercially available Agilent wash buffer 2 (Agilent probes). The slides were incubated in 1mg/mL DAPI solution made in 1% BSA-PBS, followed by a final 1X PBS wash. After the wash, the slides were mounted with ProLong Gold antifade reagent (Invitrogen).
FISH images were acquired using an Olympus IX81 or Olympus IX83 spinning disk microscope at 40X magnification and analyzed using Slidebook 6.0 softwares. A minimum of 25 z-planes with 0.5um step size was acquired for each field. Copy number gains for EGFR, 7 centromere (7C), 7p telomere (7p22.3), IKZF1 (7p12.2) and 8 centromere (8C) were scored in RPE cells as three or more foci. A minimum of 100 nuclei are scored for each independent experiment.
HCC827 cells have many EGFR amplifications that present as large EGFR amplification clouds (42). Therefore, the length of the EGFR DNA amplification cloud(s) was measured at its longest point, using the measuring tool within the Slidebook software. If multiple amplification clouds were present in a single nucleus, each cloud was measured. Each measurement was plotted and comparisons made between the overall size of the amplification cloud (um) in cells treated with siRNA to KDM4A,an inhibitor to the KDM4 family or transduced with either H3.3 WT or K4M. The analysis represents data from more than 400 nuclei from two-independent experiments with two different siRNAs to KDM4A, or across three independent experiments for the KDM4 inhibitor treatment and H3.3 transduction experiments. Each treatment condition is compared to either a non-targeting siRNA control or a DMSO vehicle control.
Lapatinib Treatment:
Control or stable KDM4A overexpression RPE cells were plated in 24 well tissue culture plates at a density of 5×103. Cells were allowed to adhere for approximately 16 hours before Lapatinib (Abcam) (dissolved in DMSO) was supplemented to media to a final concentration of 1µM. Cells were cultured in Lapatinib for a total of 48 hours before harvesting. Cells were stained with trypan blue solution (Sigma Aldrich) to assess cell viability and counted using a haemocytometer. Each condition was plated in triplicate wells and each well was counted in duplicate. An average was taken of all triplicates and used as a representative total. Data is displayed in Figure 2G, in a comparable manner to Nathanson et al (2014) (12).
Gefitinib Treatment:
Control or stable KDM4A overexpression RPE cells were plated in 24 well tissue culture plates at a density of 8×103. Cells were allowed to adhere for approximately 16 hours before Gefitinib (Abcam) (dissolved in DMSO) was supplemented to media to a final concentration of 1, 2.5 or 5µM. Cells were cultured in Gefitinib for a total of 48 hours before harvesting. Cells were stained with trypan blue solution (Sigma Aldrich) to assess cell viability and counted using a haemocytometer. Each condition was plated in triplicate wells and each well was counted in duplicate. An average was taken of all triplicates and used as a representative total. Data is displayed in Figure 2H, in a comparable manner to Nathanson et al (2014) (12).
Cell Proliferation Assay:
Control, KDM4A overexpression or wild type parental RPE cells were plated at a density of 5×103 per well in a 6 well tissue culture plate. Each condition was plated in triplicate for each independent experiment. Cells were allowed to adhere for 16 hours before fresh complete media was added containing a final concentration of 50ng/ml human recombinant EGF (Abcam). For control and KDM4A overexpression RPE cells, cells were harvested after 48 hours of EGF treatment and cell number calculated using a haemocytometer.
For combinatorial drug experiments using parental RPE cells, EGF was added for 24 hours, following a 24 hour drug treatment with either KDM5i (1µM) or EZH2i (3µM).
For siRNA conditions, parental RPE cells were plated in 10cm tissue culture plates at a density of 3×105. Cells were allowed to adhere for approximately 16 hours, prior to siRNA transfection (as previously described). 24 hours post siRNA transfection, cells were re-plated in triplicate into 24 well tissue culture plates at a density of 8×103. Remaining cells were re-plated and harvested 24 hours later for RNA extraction and qPCR transcript analysis. Cells were allowed to adhere in the 24 well plates for 24 hours before media was supplemented with human recombinant EGF to a final concentration of 50ng/ml EGF. Cells were cultured in EGF for 24 hours before harvesting and counting using a haemocytometer, as described previously.
Scratch assay:
Control or stable KDM4A overexpression RPE cells were plated in 6 well tissue culture plates at a density of 2×105. Cells were allowed to adhere to plates for 24 hours. After 24 hours in culture, a p200 pipette tip was used to introduce a scratch wound in the centre of the well from the 12 o’clock to 6 o’clock position. Following the induction of the scratch wound, media was removed from each well and 1ml of DMEM was used to rinse the wells, removing any cellular debris. After this wash, 3ml of DMEM supplemented with vehicle or human recombinant EGF (Abcam) to a final concentration of 50ng/ml, was added to each well. Cells were imaged at 0hrs, 12hrs and 24hrs, using the EVOS imaging platform at 4x magnification. Scratch wound measurements were performed using the EVOS software with a minimum of 5 measurements taken at various locations, per scratch wound. All measurements were averaged. Each condition was performed in triplicate for each independent experiment.
H3.3 WT vs K4M with EGF treatment:
Human recombinant EGF was added to histone H3.3 WT or K4M expressing RPE cells, 24 hours after viral transduction. Cells were cultured in EGF for 24 hours before harvesting for RNA, protein, cell cycle and DNA FISH analysis.
HCC827 KDM4 inhibitor treatment:
HCC827 cells were treated at approximately 80% confluency with KDM4 inhibitor at a final concentration of 5nM for 48 hours before being harvested for RNA, protein and DNA FISH analysis.
For hypoxia, EGF and KDM5i combinatorial experiments, RPE cells were pre-treated with 1nM of KDM4 inhibitor exactly 24 hours prior to hypoxic exposure or EGF treatment. Immediately before transferring cells to hypoxia, or before drug or growth factor treatment, KDM4 inhibitor was supplemented to cells again at a concentration of 1nM (double spike). Cells were then cultured in the respective conditions for 24 hours prior to harvesting for RNA, protein, cell cycle and DNA FISH analysis.
KDM5 inhibitor treatment:
RPE cells were treated with KDM5 inhibitor (C70) at a final concentration of 1µM for a total treatment time of 48 hours.
For experiments involving combination treatment with KDM4i, cells are pre-treated with KDM4i (1nM) for 24 hours. After this 24 hour treatment, KDM5 inhibitor is supplemented along with an additional dose of KDM4 inhibitor at doses of 1µM and 1nM, respectively. Cells are harvested 48 hours after combination treatment.
For experiments involving combinations of KDM5 inhibitor and hypoxic exposure, RPE cells are treated with 1µM KDM5 inhibitor for 24 hours. After this 24 hour treatment, cells are transferred to hypoxic conditions (1% O2) for an additional 24 hours prior to harvesting for RNA, protein, cell cycle and DNA FISH analysis.
KDM5i and Gefitinib combinatorial treatment:
Parental RPE cells are plated in 24 well tissue culture plates, in triplicate at a density of 5×103. Cells are allowed to adhere for approximately 16 hours before KDM5 inhibitor and Gefitinib alone or in combination are supplemented to each well at a final concentration of 1µM and 2.5µM, respectively. Cells are cultured for a total of 72 hours under drug treatment conditions before being harvested, stained with trypan blue and counted using a haemocytometer.
EZH2 inhibitor treatment:
Parental RPE cells were treated with 1, 3 or 5µM of EZH2 inhibitor for a total treatment duration of 72 hours before being harvested for RNA, protein, cell cycle and DNA FISH analysis. HT-29 and HCT-15 cells were treated with 3µM EZH2i for 48 hours before being harvested for RNA, protein, cell cycle and DNA FISH analysis.
EZH2i + EGF Growth Assay:
Parental RPE cells were plated in 24 well tissue culture plates, in triplicate at a density of 5×103. Cells were allowed to adhere for approximately 16 hours before media was supplemented with EZH2 inhibitor (C24) at a final concentration of 3µM. After an initial 24 hour treatment, human recombinant EGF was supplemented to each well at a final concentration of 50ng/ml. After 48 hours of EGF treatment, cells were harvested and counted using a haemocytometer.
EZH2i and H3.3 WT vs K4M:
Parental RPE cells were virally transduced with either histone H3.3 wild type or K4M constructs, as previously described. 24 hours after viral transduction, EZH2 inhibitor was supplemented to cells at a final concentration of 3µM. Cells were treated under these conditions for 24 hours, followed by harvesting for RNA, protein, cell cycle and DNA FISH analysis.
EZH2i + Hypoxia:
Parental RPE cells were pre-treated with EZH2 inhibitor at a final concentration of 3µM for 24 hours, prior to being transferred to hypoxic conditions for an additional 24 hours. Cells were then harvested and processed for RNA, protein, cell cycle and DNA FISH analysis.
RNA-Sequencing:
Cells were incubated with Hoechst 33342 (ThermoFisher Scientific H3570) at 1/1000 directly into the media for 1h at 37°C degrees. Cells were then trypsinized and resuspended in media containing Hoechst at 1/1000. Cells were sorted with a BD FACS Fusion using the laser BV421-A into Qiazol, based on DNA content. Late S phase RNA was purified from cells using the Qiagen miRNeasy kit including a DNAse treatment. Total RNA sequencing libraries were prepped using the TruSeq Stranded Total RNA Sample Preparation with Ribo-Zero kit (Illumina). Libraries were paired-end sequenced (150 cycles each way) using a NextSeq500 (Illumina). STAR (59) aligner was used to map sequencing reads to transcripts in human hg19 reference genome. Read counts for individual transcripts were produced with HTseq-count (60), followed by the estimation of expression values and detection of differentially expressed transcripts using edgeR (61).
ChIP-sequencing:
Cells were incubated with Hoechst 33342 (ThermoFisher Scientific H3570) at 1/1000 directly into the media for 1h at 37°C degrees. Cells were then trypsinized and resuspended in media containing Hoechst at 1/1000 before crosslinking with 1% formaldehyde for 13min at 37°C degrees and quenching with 0.125M glycine. Cells were washed with 1X PBS and resuspended in media containing Hoechst (1/1000). Cells were sorted with a BD FACS Fusion using the laser BV421-A based on DNA content. For siKDM5A ChIP, cells were harvested as previously described (10,18). Sonication of chromatin was done with the Qsonica Q800R2 system (Qsonica). 0.5–10ug of chromatin were used based on DNA content (nanodrop concentrations) with the following antibodies: H3K4me1 (Abcam ab8895), H3K4me2 (Abcam ab32356), H3K4me3 (Millipore 07–473), H3K9me1 (Abcam ab8896–100), H3K9me2 (Abcam ab1220), H3K9me3 (Abcam ab8898). ChIP sequencing libraries were prepped using the TruSeq ChIP Sample Preparation kit (Illumina). Libraries were single-end sequenced (75 cycles) using a NextSeq500 (Illumina).
ChIP-seq analysis:
ChIP-seq data for the cells with KDM4A overexpression and the corresponding controls were based on merged samples for multiple points of cell cycle. Datasets for MLL knockdown (GSE81795) (49) and EZH2 knockdown samples (50) with their respective controls were retrieved from GEO. Sequencing reads were aligned against the human hg19 reference genome using BWA (62). Alignments were filtered for uniquely mapped reads and duplicates were removed. Input-normalized ratio coverage tracks were generated using Deeptools (63).
The ENCODE ChIP-seq data on histone modification enrichment were downloaded from the ENCODE website (www.encodeproject.org). These data were normalized by the ENCODE pipeline (64). Other public ChIP-seq datasets were downloaded as fastq files from GEO (GSE64243, GSE118954, GSE81795) (65), followed by mapping to the hg19 reference genome using BWA (66) and the generation of input-normalized coverage tracks using Deeptools (63).
EGFR copy number and expression analysis:
Data from The Cancer Genome Atlas (TCGA) was obtained from the Broad Institute’s Genomic Data Analysis Center (GDAC) Firehose (http://gdac.broadinstitute.org). A set of 7069 tumors spanning 21 tumor types was analyzed. Expression values for EGFR were extracted from the database, in units of transcripts per million (TPM). Copy number values for EGFR were also extracted from the database. Figure 1A shows these EGFR expression and copy number values for each tumor analyzed.
Statistical Analysis:
All pairwise comparisons were done using two-tailed Student’s t-test unless otherwise stated. Significance was determined if the p value was ≤ 0.05. All FISH experiments were carried out with at least two independent siRNAs unless otherwise stated and at least 100 nuclei per replicate per experiment were counted for all the FISH studies conducted. All error bars represent the SEM.
Supplementary Material
Statement of significance.
This study identifies a network of epigenetic factors and cellular signals that directly control EGFR DNA amplification. We demonstrate that chemical inhibitors targeting enzymes controlling this amplification can be used to rheostat EGFR copy number, which uncovers therapeutic opportunities for controlling EGFR DNA amplification heterogeneity and the associated drug response.
Acknowledgements
We are grateful to Ravi Mylvaganam and the MGH Flow Cytometry Core for assistance with flow cytometry. We thank Peter Lewis (University of Wisconsin) for the H3K-M constructs. We also thank Reuben Duttweiler for technical contributions. We would like to thank Drs. Margie Clapper, Jon Chernoff and Hossein Borghaei for comments on the manuscript. Work related to this study is supported by the R01GM097360 (J.R.W.) and the NIH/NCI Cancer Center Support Grant P30 CA006927. J.R.W. was a Tepper Family Massachusetts General Hospital Scholar and Leukemia and Lymphoma Scholar, as well as a recipient of an American Lung Association Lung Cancer Discovery Award. R.S. was supported by NIH P30 DK040561. J.J. acknowledges the support by the grant R01GM122749 from the U.S. National Institutes of Health. T.L.C. is supported by an EMBO long term fellowship (EMBO ALTF 449–2017). S.M. was supported by a Senior Research Training Fellowship from the American Lung Association and the MGH ECOR Tosteson Postdoctoral Training Fellowship. D.C. is a recipient of an award from the Ovarian Cancer Research Fund Alliance (Ann and Sol Schreiber Mentored Investigator Award-543667).
Footnotes
Competing Interest.
JRW has received funding from AstraZeneca, previously consulted for Celgene and served on Salarius Pharmaceuticals advisory board. JRW consults for Qsonica.
References.
- 1.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011;144(5):646–74 doi 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 2.Mishra S, Whetstine JR. Different Facets of Copy Number Changes: Permanent, Transient, and Adaptive. Mol Cell Biol 2016. doi 10.1128/MCB.00652-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Diskin SJ, Hou C, Glessner JT, Attiyeh EF, Laudenslager M, Bosse K, et al. Copy number variation at 1q21.1 associated with neuroblastoma. Nature 2009;459(7249):987–91 doi 10.1038/nature08035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fonseca R, Van Wier SA, Chng WJ, Ketterling R, Lacy MQ, Dispenzieri A, et al. Prognostic value of chromosome 1q21 gain by fluorescent in situ hybridization and increase CKS1B expression in myeloma. Leukemia 2006;20(11):2034–40 doi 10.1038/sj.leu.2404403. [DOI] [PubMed] [Google Scholar]
- 5.Goeze A, Schluns K, Wolf G, Thasler Z, Petersen S, Petersen I. Chromosomal imbalances of primary and metastatic lung adenocarcinomas. J Pathol 2002;196(1):8–16 doi 10.1002/path.1009. [DOI] [PubMed] [Google Scholar]
- 6.Inoue J, Otsuki T, Hirasawa A, Imoto I, Matsuo Y, Shimizu S, et al. Overexpression of PDZK1 within the 1q12-q22 amplicon is likely to be associated with drug-resistance phenotype in multiple myeloma. The American journal of pathology 2004;165(1):71–81 doi 10.1016/S0002-9440(10)63276-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kudoh K, Takano M, Koshikawa T, Hirai M, Yoshida S, Mano Y, et al. Gains of 1q21-q22 and 13q12-q14 are potential indicators for resistance to cisplatin-based chemotherapy in ovarian cancer patients. Clinical cancer research : an official journal of the American Association for Cancer Research 1999;5(9):2526–31. [PubMed] [Google Scholar]
- 8.Petersen S, Aninat-Meyer M, Schluns K, Gellert K, Dietel M, Petersen I. Chromosomal alterations in the clonal evolution to the metastatic stage of squamous cell carcinomas of the lung. Br J Cancer 2000;82(1):65–73 doi 10.1054/bjoc.1999.0878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, et al. The landscape of somatic copy-number alteration across human cancers. Nature 2010;463(7283):899–905 doi 10.1038/nature08822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mishra S, Van Rechem C, Pal S, Clarke TL, Chakraborty D, Mahan SD, et al. Cross-talk between Lysine-Modifying Enzymes Controls Site-Specific DNA Amplifications. Cell 2018;174(4):803–17 e16 doi 10.1016/j.cell.2018.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Turner KM, Deshpande V, Beyter D, Koga T, Rusert J, Lee C, et al. Extrachromosomal oncogene amplification drives tumour evolution and genetic heterogeneity. Nature 2017;543(7643):122–5 doi 10.1038/nature21356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nathanson DA, Gini B, Mottahedeh J, Visnyei K, Koga T, Gomez G, et al. Targeted therapy resistance mediated by dynamic regulation of extrachromosomal mutant EGFR DNA. Science 2014;343(6166):72–6 doi 10.1126/science.1241328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Biedler JL, Spengler BA. A novel chromosome abnormality in human neuroblastoma and antifolate-resistant Chinese hamster cell lives in culture. Journal of the National Cancer Institute 1976;57(3):683–95. [DOI] [PubMed] [Google Scholar]
- 14.Biedler JL, Spengler BA. Metaphase chromosome anomaly: association with drug resistance and cell-specific products. Science 1976;191(4223):185–7. [DOI] [PubMed] [Google Scholar]
- 15.Alt FW, Kellems RE, Bertino JR, Schimke RT. Selective multiplication of dihydrofolate reductase genes in methotrexate-resistant variants of cultured murine cells. The Journal of biological chemistry 1978;253(5):1357–70. [PubMed] [Google Scholar]
- 16.Haber DA, Schimke RT. Unstable amplification of an altered dihydrofolate reductase gene associated with double-minute chromosomes. Cell 1981;26(3 Pt 1):355–62. [DOI] [PubMed] [Google Scholar]
- 17.Black JC, Atabakhsh E, Kim J, Biette KM, Van Rechem C, Ladd B, et al. Hypoxia drives transient site-specific copy gain and drug-resistant gene expression. Genes & development 2015;29(10):1018–31 doi 10.1101/gad.259796.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Black JC, Manning AL, Van Rechem C, Kim J, Ladd B, Cho J, et al. KDM4A lysine demethylase induces site-specific copy gain and rereplication of regions amplified in tumors. Cell 2013;154(3):541–55 doi 10.1016/j.cell.2013.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Black JC, Zhang H, Kim J, Getz G, Whetstine JR. Regulation of Transient Site-specific Copy Gain by MicroRNA. J Biol Chem 2016;291(10):4862–71 doi 10.1074/jbc.M115.711648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Black JC, Whetstine JR. Too little O2 Too much gain. Cell cycle 2015;14(18):2869–70 doi 10.1080/15384101.2015.1076659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Junttila MR, de Sauvage FJ. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature 2013;501(7467):346–54 doi 10.1038/nature12626. [DOI] [PubMed] [Google Scholar]
- 22.Hirsch FR, Varella-Garcia M, Bunn PA Jr., Di Maria MV, Veve R, Bremmes RM, et al. Epidermal growth factor receptor in non-small-cell lung carcinomas: correlation between gene copy number and protein expression and impact on prognosis. J Clin Oncol 2003;21(20):3798–807 doi 10.1200/JCO.2003.11.069. [DOI] [PubMed] [Google Scholar]
- 23.Sullivan I, Planchard D. Next-Generation EGFR Tyrosine Kinase Inhibitors for Treating EGFR-Mutant Lung Cancer beyond First Line. Front Med (Lausanne) 2016;3:76 doi 10.3389/fmed.2016.00076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hirsch FR, Varella-Garcia M, McCoy J, West H, Xavier AC, Gumerlock P, et al. Increased epidermal growth factor receptor gene copy number detected by fluorescence in situ hybridization associates with increased sensitivity to gefitinib in patients with bronchioloalveolar carcinoma subtypes: a Southwest Oncology Group Study. J Clin Oncol 2005;23(28):6838–45 doi 10.1200/JCO.2005.01.2823. [DOI] [PubMed] [Google Scholar]
- 25.Maron SB, Alpert L, Kwak HA, Lomnicki S, Chase L, Xu D, et al. Targeted Therapies for Targeted Populations: Anti-EGFR Treatment for EGFR-Amplified Gastroesophageal Adenocarcinoma. Cancer Discov 2018;8(6):696–713 doi 10.1158/2159-8290.CD-17-1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang J, Fan Q, Lu P, Ying J, Ma C, Liu W, et al. Icotinib in Patients with Pretreated Advanced Esophageal Squamous Cell Carcinoma with EGFR Overexpression or EGFR Gene Amplification: A Single-Arm, Multicenter Phase 2 Study. J Thorac Oncol 2016;11(6):910–7 doi 10.1016/j.jtho.2016.02.020. [DOI] [PubMed] [Google Scholar]
- 27.Luber B, Deplazes J, Keller G, Walch A, Rauser S, Eichmann M, et al. Biomarker analysis of cetuximab plus oxaliplatin/leucovorin/5-fluorouracil in first-line metastatic gastric and oesophago-gastric junction cancer: results from a phase II trial of the Arbeitsgemeinschaft Internistische Onkologie (AIO). BMC Cancer 2011;11:509 doi 10.1186/1471-2407-11-509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Petty RD, Dahle-Smith A, Stevenson DAJ, Osborne A, Massie D, Clark C, et al. Gefitinib and EGFR Gene Copy Number Aberrations in Esophageal Cancer. J Clin Oncol 2017;35(20):2279–87 doi 10.1200/JCO.2016.70.3934. [DOI] [PubMed] [Google Scholar]
- 29.Herbst RS, Redman MW, Kim ES, Semrad TJ, Bazhenova L, Masters G, et al. Cetuximab plus carboplatin and paclitaxel with or without bevacizumab versus carboplatin and paclitaxel with or without bevacizumab in advanced NSCLC (SWOG S0819): a randomised, phase 3 study. Lancet Oncol 2018;19(1):101–14 doi 10.1016/S1470-2045(17)30694-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lewis PW, Muller MM, Koletsky MS, Cordero F, Lin S, Banaszynski LA, et al. Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science 2013;340(6134):857–61 doi 10.1126/science.1232245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jiang XR, Jimenez G, Chang E, Frolkis M, Kusler B, Sage M, et al. Telomerase expression in human somatic cells does not induce changes associated with a transformed phenotype. Nat Genet 1999;21(1):111–4 doi 10.1038/5056. [DOI] [PubMed] [Google Scholar]
- 32.Darrow EM, Huntley MH, Dudchenko O, Stamenova EK, Durand NC, Sun Z, et al. Deletion of DXZ4 on the human inactive X chromosome alters higher-order genome architecture. Proc Natl Acad Sci U S A 2016;113(31):E4504–12 doi 10.1073/pnas.1609643113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kundu S, Ji F, Sunwoo H, Jain G, Lee JT, Sadreyev RI, et al. Polycomb Repressive Complex 1 Generates Discrete Compacted Domains that Change during Differentiation. Mol Cell 2017;65(3):432–46 e5 doi 10.1016/j.molcel.2017.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Consortium EP. An integrated encyclopedia of DNA elements in the human genome. Nature 2012;489(7414):57–74 doi 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rao SS, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 2014;159(7):1665–80 doi 10.1016/j.cell.2014.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stafford JM, Lee CH, Voigt P, Descostes N, Saldana-Meyer R, Yu JR, et al. Multiple modes of PRC2 inhibition elicit global chromatin alterations in H3K27M pediatric glioma. Sci Adv 2018;4(10):eaau5935 doi 10.1126/sciadv.aau5935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Black JC, Van Rechem C, Whetstine JR. Histone lysine methylation dynamics: establishment, regulation, and biological impact. Mol Cell 2012;48(4):491–507 doi 10.1016/j.molcel.2012.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Whetstine JR, Nottke A, Lan F, Huarte M, Smolikov S, Chen Z, et al. Reversal of histone lysine trimethylation by the JMJD2 family of histone demethylases. Cell 2006;125(3):467–81 doi 10.1016/j.cell.2006.03.028. [DOI] [PubMed] [Google Scholar]
- 39.Huang Y, Fang J, Bedford MT, Zhang Y, Xu RM. Recognition of histone H3 lysine-4 methylation by the double tudor domain of JMJD2A. Science 2006;312(5774):748–51 doi 10.1126/science.1125162. [DOI] [PubMed] [Google Scholar]
- 40.Metzger E, Stepputtis SS, Strietz J, Preca BT, Urban S, Willmann D, et al. KDM4 Inhibition Targets Breast Cancer Stem-like Cells. Cancer Res 2017;77(21):5900–12 doi 10.1158/0008-5472.CAN-17-1754. [DOI] [PubMed] [Google Scholar]
- 41.Amann J, Kalyankrishna S, Massion PP, Ohm JE, Girard L, Shigematsu H, et al. Aberrant epidermal growth factor receptor signaling and enhanced sensitivity to EGFR inhibitors in lung cancer. Cancer Res 2005;65(1):226–35. [PubMed] [Google Scholar]
- 42.Varella-Garcia M Stratification of non-small cell lung cancer patients for therapy with epidermal growth factor receptor inhibitors: the EGFR fluorescence in situ hybridization assay. Diagn Pathol 2006;1:19 doi 10.1186/1746-1596-1-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen YK, Bonaldi T, Cuomo A, Del Rosario JR, Hosfield DJ, Kanouni T, et al. Design of KDM4 Inhibitors with Antiproliferative Effects in Cancer Models. ACS Med Chem Lett 2017;8(8):869–74 doi 10.1021/acsmedchemlett.7b00220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Egan B, Yuan CC, Craske ML, Labhart P, Guler GD, Arnott D, et al. An Alternative Approach to ChIP-Seq Normalization Enables Detection of Genome-Wide Changes in Histone H3 Lysine 27 Trimethylation upon EZH2 Inhibition. PLoS One 2016;11(11):e0166438 doi 10.1371/journal.pone.0166438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang X, Li F, Konze KD, Meslamani J, Ma A, Brown PJ, et al. Structure-Activity Relationship Studies for Enhancer of Zeste Homologue 2 (EZH2) and Enhancer of Zeste Homologue 1 (EZH1) Inhibitors. J Med Chem 2016;59(16):7617–33 doi 10.1021/acs.jmedchem.6b00855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Katona BW, Liu Y, Ma A, Jin J, Hua X. EZH2 inhibition enhances the efficacy of an EGFR inhibitor in suppressing colon cancer cells. Cancer Biol Ther 2014;15(12):1677–87 doi 10.4161/15384047.2014.972776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shilatifard A The COMPASS family of histone H3K4 methylases: mechanisms of regulation in development and disease pathogenesis. Annu Rev Biochem 2012;81:65–95 doi 10.1146/annurev-biochem-051710-134100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schuettengruber B, Bourbon HM, Di Croce L, Cavalli G. Genome Regulation by Polycomb and Trithorax: 70 Years and Counting. Cell 2017;171(1):34–57 doi 10.1016/j.cell.2017.08.002. [DOI] [PubMed] [Google Scholar]
- 49.Rickels R, Hu D, Collings CK, Woodfin AR, Piunti A, Mohan M, et al. An Evolutionary Conserved Epigenetic Mark of Polycomb Response Elements Implemented by Trx/MLL/COMPASS. Mol Cell 2016;63(2):318–28 doi 10.1016/j.molcel.2016.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Koubi M, Poplineau M, Vernerey J, N’Guyen L, Tiberi G, Garciaz S, et al. Regulation of the positive transcriptional effect of PLZF through a non-canonical EZH2 activity. Nucleic Acids Res 2018;46(7):3339–50 doi 10.1093/nar/gky080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Johansson C, Velupillai S, Tumber A, Szykowska A, Hookway ES, Nowak RP, et al. Structural analysis of human KDM5B guides histone demethylase inhibitor development. Nat Chem Biol 2016;12(7):539–45 doi 10.1038/nchembio.2087. [DOI] [PubMed] [Google Scholar]
- 52.Horton JR, Liu X, Gale M, Wu L, Shanks JR, Zhang X, et al. Structural Basis for KDM5A Histone Lysine Demethylase Inhibition by Diverse Compounds. Cell Chem Biol 2016;23(7):769–81 doi 10.1016/j.chembiol.2016.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sharma SV, Lee DY, Li B, Quinlan MP, Takahashi F, Maheswaran S, et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell 2010;141(1):69–80 doi 10.1016/j.cell.2010.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.William D, Mokri P, Lamp N, Linnebacher M, Classen CF, Erbersdobler A, et al. Amplification of the EGFR gene can be maintained and modulated by variation of EGF concentrations in in vitro models of glioblastoma multiforme. PLoS One 2017;12(9):e0185208 doi 10.1371/journal.pone.0185208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Moller HD, Mohiyuddin M, Prada-Luengo I, Sailani MR, Halling JF, Plomgaard P, et al. Circular DNA elements of chromosomal origin are common in healthy human somatic tissue. Nat Commun 2018;9(1):1069 doi 10.1038/s41467-018-03369-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Smith G, Taylor-Kashton C, Dushnicky L, Symons S, Wright J, Mai S. c-Myc-induced extrachromosomal elements carry active chromatin. Neoplasia 2003;5(2):110–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Huang X, Yan J, Zhang M, Wang Y, Chen Y, Fu X, et al. Targeting Epigenetic Crosstalk as a Therapeutic Strategy for EZH2-Aberrant Solid Tumors. Cell 2018;175(1):186–99 e19 doi 10.1016/j.cell.2018.08.058. [DOI] [PubMed] [Google Scholar]
- 58.Nukaga S, Yasuda H, Tsuchihara K, Hamamoto J, Masuzawa K, Kawada I, et al. Amplification of EGFR Wild-Type Alleles in Non-Small Cell Lung Cancer Cells Confers Acquired Resistance to Mutation-Selective EGFR Tyrosine Kinase Inhibitors. Cancer Res 2017;77(8):2078–89 doi 10.1158/0008-5472.CAN-16-2359. [DOI] [PubMed] [Google Scholar]
- 59.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 2013;29(1):15–21 doi 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Anders S, Pyl PT, Huber W. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 2015;31(2):166–9 doi 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010;26(1):139–40 doi 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010;26(5):589–95 doi 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ramirez F, Ryan DP, Gruning B, Bhardwaj V, Kilpert F, Richter AS, et al. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res 2016;44(W1):W160–5 doi 10.1093/nar/gkw257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Consortium EP. The ENCODE (ENCyclopedia Of DNA Elements) Project. Science 2004;306(5696):636–40 doi 10.1126/science.1105136. [DOI] [PubMed] [Google Scholar]
- 65.Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res 2002;30(1):207–10 doi 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009;25(14):1754–60 doi 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.