

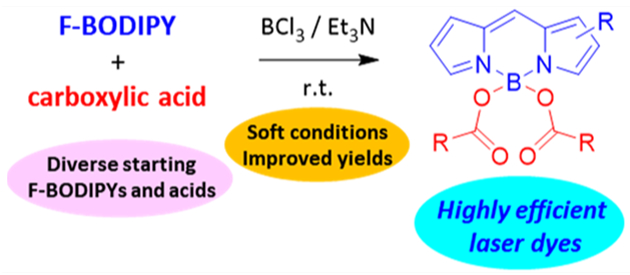
Abstract
A general and straightforward method for the synthesis of COO-BODIPYs from F-BODIPYs and carboxylic acids is established. The method is based on the use of boron trichloride to activate the involved substitution of fluorine, which leads to high yields through rapid reactions under soft conditions. This mild method opens the way to unprecedented laser dyes with outstanding efficiencies and photostabilities, which are difficult to obtain by the current methods.
Graphical Abstract
INTRODUCTION
BODIPYs (BOron DIPYrromethenes) are a fascinating class of small organic dyes, which spotlight their excellent (photo)-physical properties and a well-known chemistry.1 BODIPY chemistry allows the access to a broad structural diversity and makes possible the fine modulation of key properties (chemical, physical, (chir)optical, biological, etc.).2 As a matter of fact, BODIPYs are extensively used as the key optically active component in a plethora of photonic applications, from energy to medicine, and are currently the subject of a myriad of research projects focused to expand their potential in photonic and optoelectronic systems and applications.3
BODIPY chemistry is well established in terms of the construction of the BODIPY core from simple precursors (e.g., pyrroles, aldehydes and boron trifluoride).4 Also, likewise, the chemistry is extensive regarding the BODIPY postfunctionalization at the dipyrromethene unit.5 However, chemistry at boron is still restricted to the formation of a small, but interesting, number of BODIPY families,6 such as the known as O-BODIPYs (involving B–O bonds), C-BODIPYs (involving B–C bonds), and N-BODIPYs (involving C–N bonds).
COO-BODIPYs constitute a subclass of O-BODIPYs involving two acyloxyl moieties hanging from the boron center (e.g., see 1 in Scheme 1). The interest of these dyes lies in two main factors: enhanced photostability and almost maintained fluorescent signatures when compared with parent F-BODIPYs. These outstanding photophysical properties have allowed the development of more robust BODIPYs for lasing and photostable water-soluble BODIPY probes for bioimaging.7 However, the synthesis of COO-BODIPYs is still not well resolved: the scope of the hitherto described methods is not general and, besides, all of them demand a high excess of a trimethylsilylated reagent in combination with high reaction temperatures and/or long reaction times (Scheme 1).
Scheme 1.
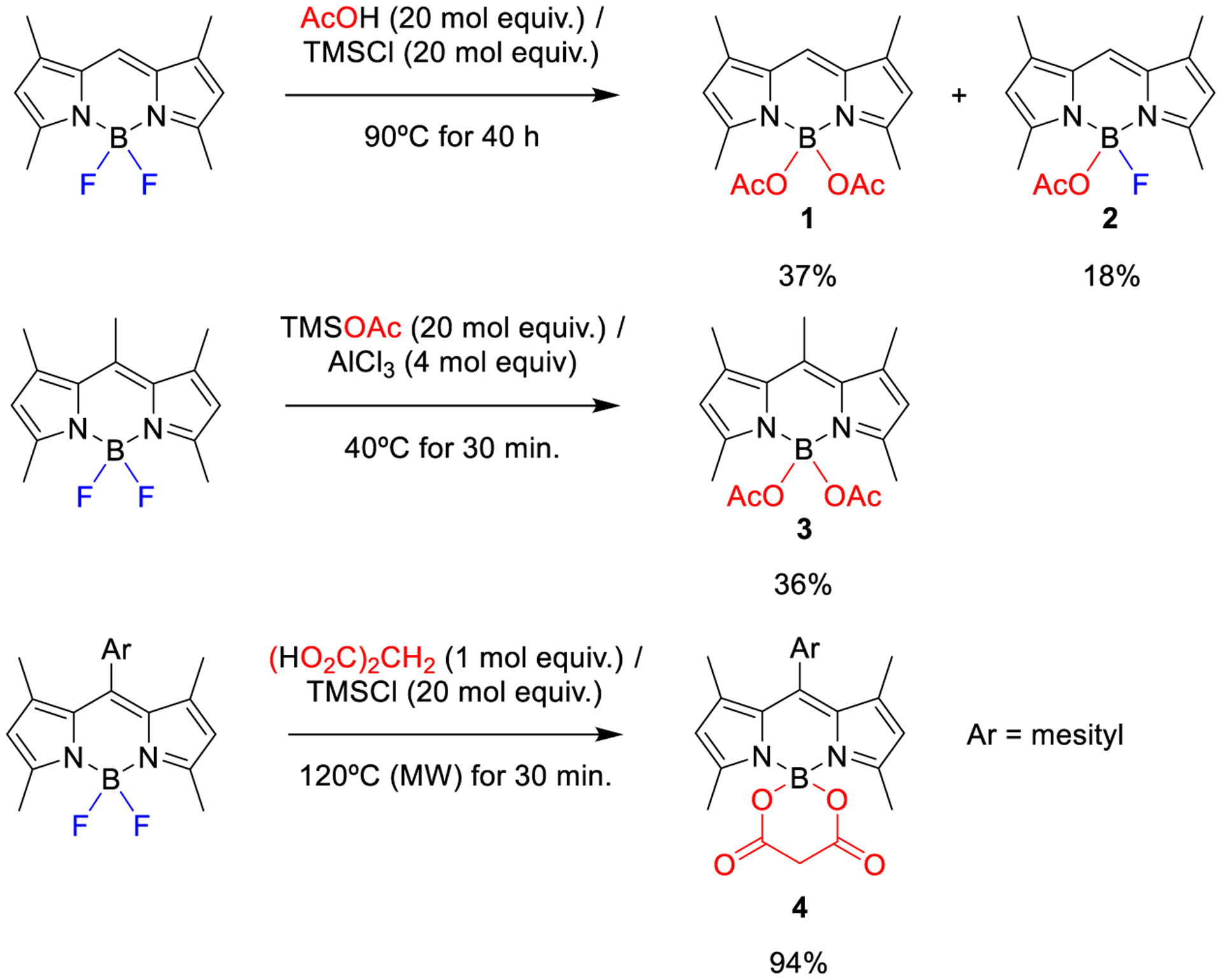
Representative Examples of the Previous Methods To Prepare COO-BODIPYs from F-BODIPYs
The first COO-BODIPY (1 in Scheme 1) was obtained by Jiang and Zhao in 2012 from the corresponding F-BODIPY by nucleophilic substitution of its fluorine atoms.8 To achieve the desired at-boron nucleophilic substitution, in situ generated trimethylsilyl acetate was used as the properly activated O-nucleophile in a 20:1 stoichiometric ratio to the F-BODIPY substrate. Besides, heating the reaction mixture at 90 °C (refluxing 1,2-dichloroethane) for 40 h was required to achieve the reaction in acceptable conversion. However, despite these harsh reaction conditions, 1 was obtained in moderate chemical yield (37%) together with monoacylated intermediate 2 (18%).8 On the other hand, lowering the O-nucleophile/F-BODIPY stoichiometric ratio to 5:1 gave place to a significant diminution of the 1:2 ratio, as well as to a significant decrease of the individual chemical yields (13% for 1; 12% for 2).8
One year later, García-Moreno and Ortiz showed that some stable-enough COO-BODIPYs, mainly diacetyl and bis-(trifluoroacetyl) derivatives, could be prepared in moderate chemical yield (from 22 to 48%) from the corresponding F-BODIPY, using trimethylsilyl ester as the O-nucleophile, in combination with excess of AlCl3 as the activating agent for the at-boron nucleophilic substitution in some cases.7a Although the new method allowed faster reactions (between 10 and 30 min) under lower reaction temperatures, the high 20:1 O-nucleophile/F-BODIPY stoichiometric ratio was maintained (e.g., see the formation of 3 in Scheme 1).7a Recently, Cerdań and Chiara have used microwave irradiation in acetonitrile at 120 °C (sealed tube) to promote the reaction of in situ generated bis(trimethylsilyl) diesters, mainly derived from oxalic or malonic acids, with F-BODIPYs to generate the corresponding spiranic COO-BODIPYs in high chemical yield (from 60 to 97%) upon short reaction times (e.g., see the formation of 4 in Figure 1), although a huge amount of trimethylsilyl chloride (ca. 20 mol equiv; e.g., see Figure 1) was still needed.7b,9 However, the method failed for sterically hindered COO-BODIPYs.7b Interestingly, Vicente used stoichiometric O,O′-bis(trimethylsilyl)ated hydroxyacetic acid (ca. 5 mol equiv) in combination with SnCl4 to obtain a spiranic O,COO-BODIPY in excellent yield (93%) from the corresponding F-BODIPY under mild reaction conditions.10 A similar procedure was used to prepare different N-BODIPYs from F-BODIPYs by using N-trimethylsilylated N-nucleophiles and SnCl4 or BCl3.10
Figure 1.
Chemical yields reached in the preparation of known COO-BODIPYs by the new method (see Supporting Information for experimental details) in comparison with those previously reported (in brackets) by using other methods.
Regarding BCl3 activation, Thompson was the first one to report the ability of this Lewis acid to activate the reaction of F-BODIPYs with O-nucleophiles, specifically metal alkoxides, and generate the corresponding O-BODIPYs.11 Later, we used BCl3 activation in the presence of triethylamine to prepare the first N-BODIPYs from F-BODIPYs and protic sulfonamides under mild reaction conditions.6b The successful synthesis of the elusive N-BODIPYs prompted us to test the combined use of BCl3 and triethylamine for the direct preparation of COO-BODIPYs from F-BODIPYs and carboxylic acids under soft reaction conditions. In fact, the group of Vicente has recently reported the direct synthesis of highly stable spiranic N,COO-BODIPYs based on N-acylglycine by using the BCl3/triethylamine approach.12
Thus, we first tried to prepare known 3 from the corresponding pentamethylated F-BODIPY (the commercial laser dye PM546), following the described procedure for the synthesis of N-BODIPYs,6b that is, 2 mol equiv of BCl3, 8 mol equiv of Et3N and 6 mol equiv of acetic acid as the nucleophile, in methylene dichloride at room temperature for 2 h. To our satisfaction, 3 could be obtained in 50% chemical yield. Upon experimentation, the amounts of triethylamine and acetic acid, as well as the reaction time, could be diminished without significant loss of yield (see Scheme 2). An excess of base was proven necessary for the reaction to progress adequately.
Scheme 2.
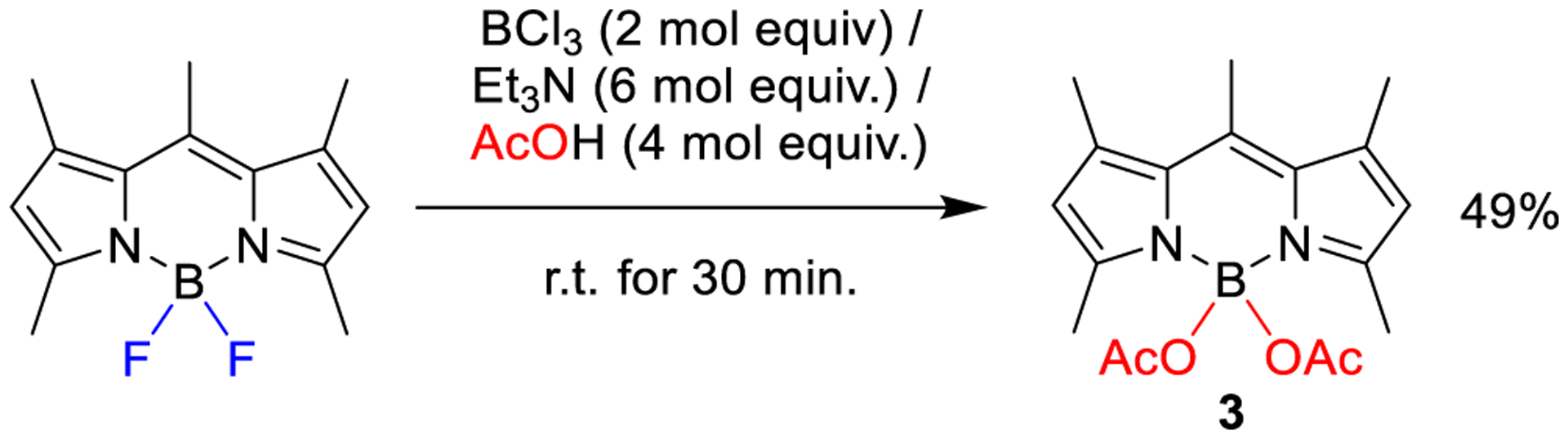
BCl3-Activated Synthesis of 3 under Optimized Reaction Conditions
Although we have not carried out a deep investigation on the mechanism of this transformation, we did try to isolate the intermediate Cl-BODIPY that has been described before.11 Thus, simple Celite filtration of the dark-purple mixture obtained after the initial treatment of the F-BODIPY solution with BCl3 (different commercial alkylated F-BODIPYs were essayed) gave place to the recovery of the starting dye (15–48%) without detection of the corresponding Cl-BODIPY (the residue was analyzed by 1H and 11B NMR). This result suggests that the exchange of F for Cl is not taking place, at least for the tested F-BODIPYs. Instead, BCl3 must just coordinate to the fluorine atoms, thus facilitating the subsequent exchange of F for the carboxylic acid.
Nonetheless, formation of highly reactive Cl-BODIPY cannot be discarded on the basis of the conducted experiments. Note that both an F-BODIPY–BCl3 complex and a Cl-BODIPY could react with free hydroxyl groups or with water present in the Celite matrix, decomposing and anchoring the dye to it. In fact, a colored band appeared in the Celite filter, which could not be eluted by using dichloromethane or more polar solvents.
RESULTS AND DISCUSSION
The obtained result brings about a significant improvement of the previous methods,7,8 not only regarding the chemical yield but also in that dealing with the reaction conditions and the reagent waste (cf. Schemes 1 and 2).
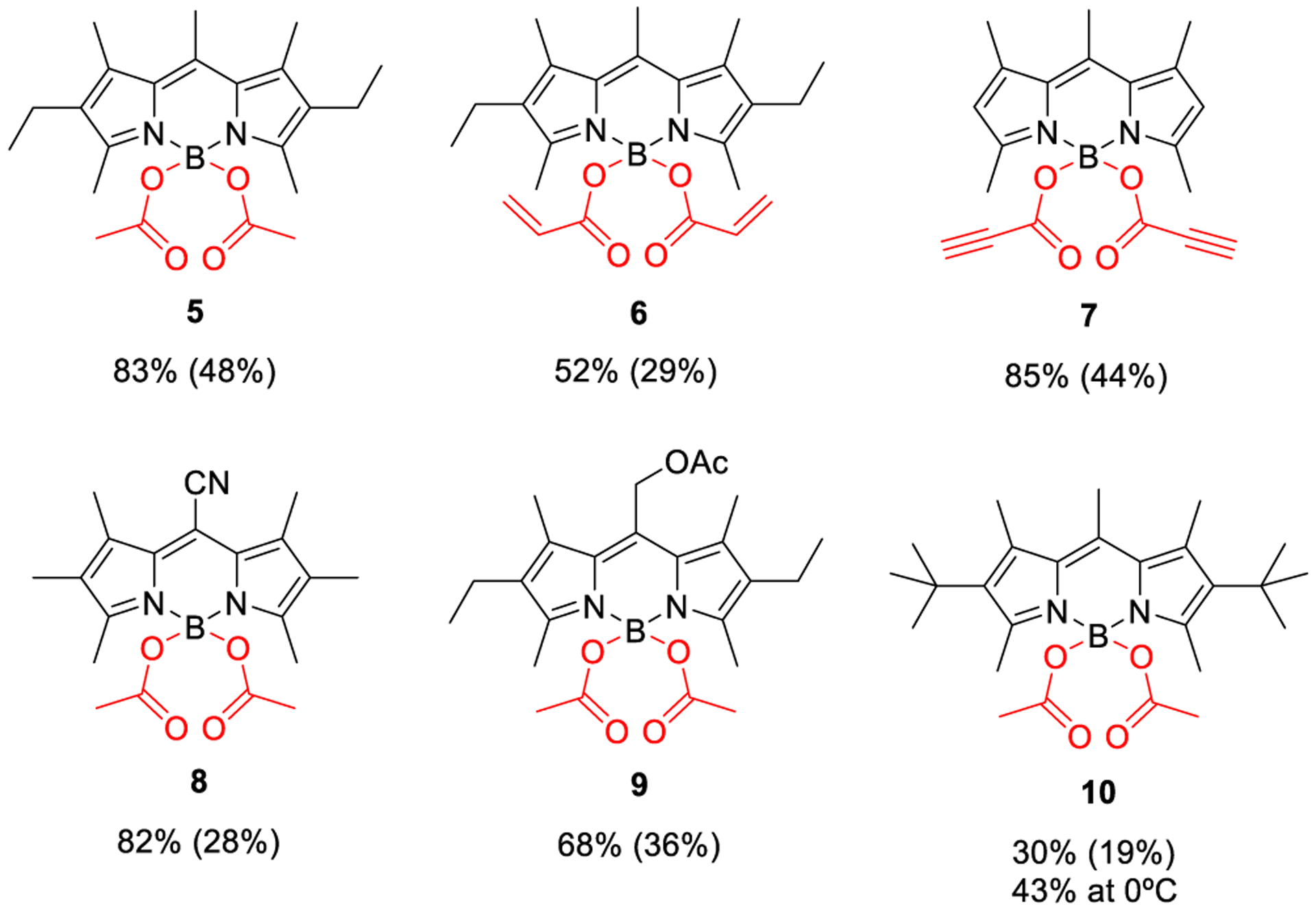
Encouraged with these findings, we decided to study the scope of the method. For such a purpose, we tackled the synthesis of a selected battery of known COO-BODIPYs (5–10 in Figure 1) from the corresponding parent F-BODIPYs and carboxylic acids, under the same experimental conditions previously optimized for 3 (see Scheme 2). Figure 1 shows the significantly improved yields in comparison with those reached by previous methodologies.7a The obtained yields support the superiority of the new method, which can be even applied to the synthesis of COO-BODIPYs involving reactive functional groups at the acyl moiety (e.g., 6 and 7) or at the BODIPY core (e.g., 8 and 9), or having unstable BODIPY cores prone to dealkylation (e.g., 10).7a
These results prompted us to expand the study on the scope of the new method, which was done by synthesizing a selected battery of unprecedented COO-BODIPYs (Figure 2). Thus, the successful direct preparation of dichlorinated 11, from the corresponding F-BODIPY (3,5-dichloro-8-mesityl-F-BODI-PY13) and acetic acid, supports the compatibility of the new method with reactive halo-F-BODIPYs able to undergo aromatic nucleophilic substitution of the halogen.14 Satisfactorily, the mild conditions of our method allow the perfect control of such a competitive reaction so that the nucleophilic substitution takes place only at boron to give 11 (possible secondary products coming from at-carbon halosubstitution were not isolated, neither detected by NMR analysis of the reaction mixture). Additionally, the high chemical yields reached in the direct preparation of 12–19 (Figure 2) from 2,6-diethyl-1,3,5,7,8-pentamethyl-F-BODIPY (i.e., the commercial laser dye PM567) and different carboxylic acids (linear, α-branched, α,β-unsaturated, aromatic, heteroaromatic, Z–E isomerizable, gem-dicarboxylic, with reactive functional groups, etc.) support the broad scope of the new method.
Figure 2.
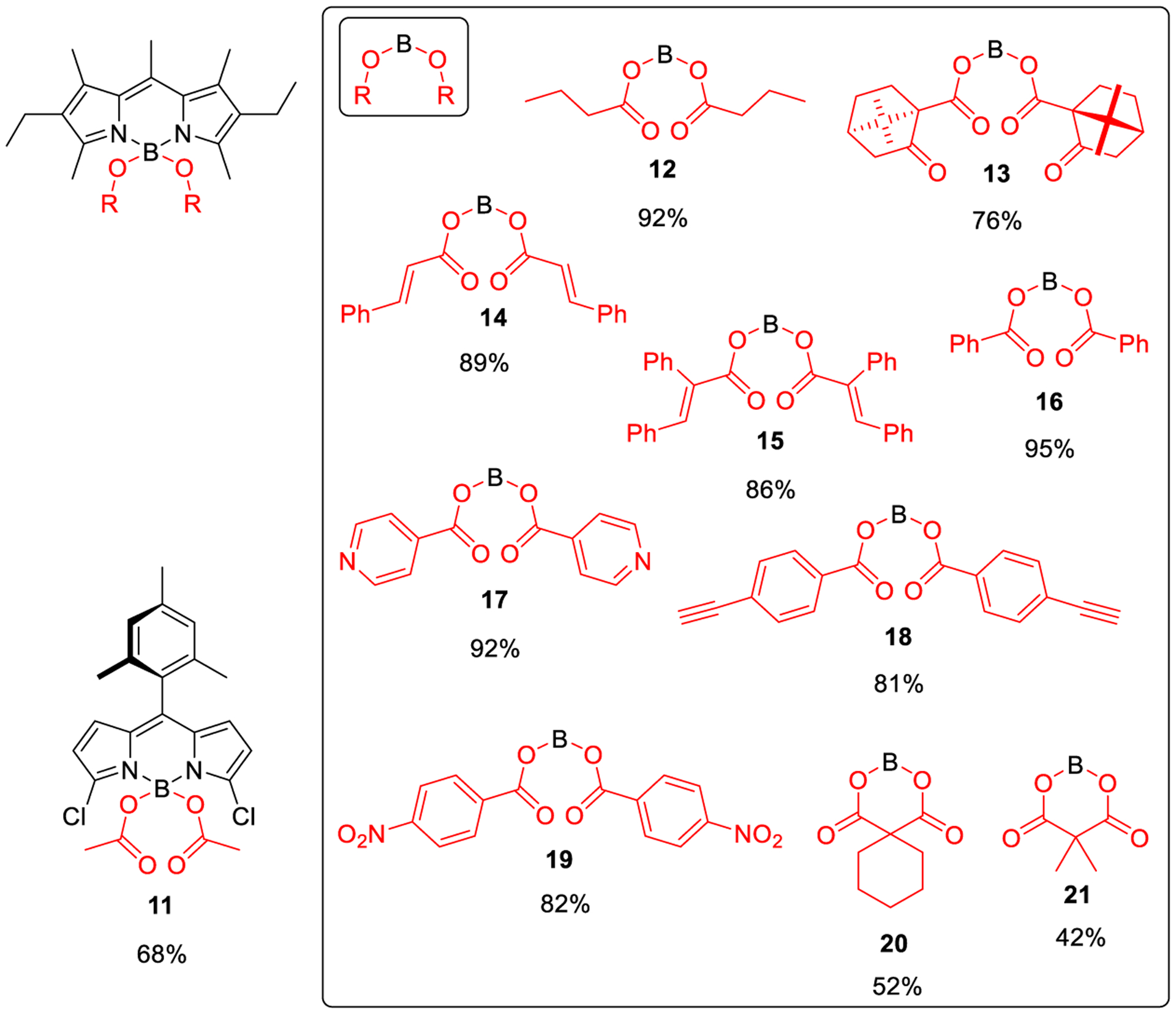
Unprecedented COO-BODIPYs obtained by the new synthetic method (see Supporting Information for experimental details).
Thus, for example, it was possible to obtain the spiranic malonic-acid derivatives 20 and 21, which could not be obtained by previous methods (a polymer had been formed instead).7b Also, the successful preparation of 13 shows that our method makes it possible to use sterically hindered (α-branched and conformationally rigid) carboxylic acids to generate the corresponding COO-BODIPYs in high chemical yield. Besides, the efficient chiral perturbation exerted by the chiral acyl moieties of 13 (based on (1S)-ketopinic acid) over the BODIPY chromophore allowed visible circularly polarized luminescence (CPL) (glum15ca. −0.001 at 565 nm in chloroform; see Figure S1 in Supporting Information), supporting the utility of the method in the rapid construction of valuable CPL-SOMs (i.e., simple organic molecules enabling circularly polarized luminescence).16
The expected capability of the novel COO-BODIPYs to act as robust laser dyes7 prompted us to study their photophysical signatures and lasing behavior in comparison with those exhibited by the corresponding F-BODIPY. The absorption and fluorescence spectral profiles of nonspiranic 11–18 fully resembled to those of the corresponding F-BODIPY (mostly PM567) and related previously reported COO-BODIPYs.7,9
Interestingly, these new dyes proved highly fluorescent (Table S1 in Supporting Information), with efficiency quantum yields surpassing 80%, independently of the solvent properties (see Figure 3, and Table S1 in Supporting Information), and even approaching 100% in some cases (11 and 14–16). However, this is not the case for 19, where the bright BODIPY-core fluorescence was virtually vanished in polar media (see Table S1 in Supporting Information), suggesting that a nonradiative channel was induced by the involved, strongly electron-withdrawing nitro groups. In fact, conducted computations (see Supporting Information) revealed that the lowest unoccupied molecular orbitals (LUMO and LUMO+1) located at the nitrophenyl moieties were intercalated within the energy gap responsible of the main BODIPY absorption transition (HOMO → LUMO+2) (see Figure S2 in Supporting Information). With this energetic landscape, the excited BODIPY electron can be transferred to the molecular orbitals located at the nitrophenyl unit through a thermodynamically enabled oxidative photoinduced electron transfer, quenching the BODIPY fluorescence.
Figure 3.
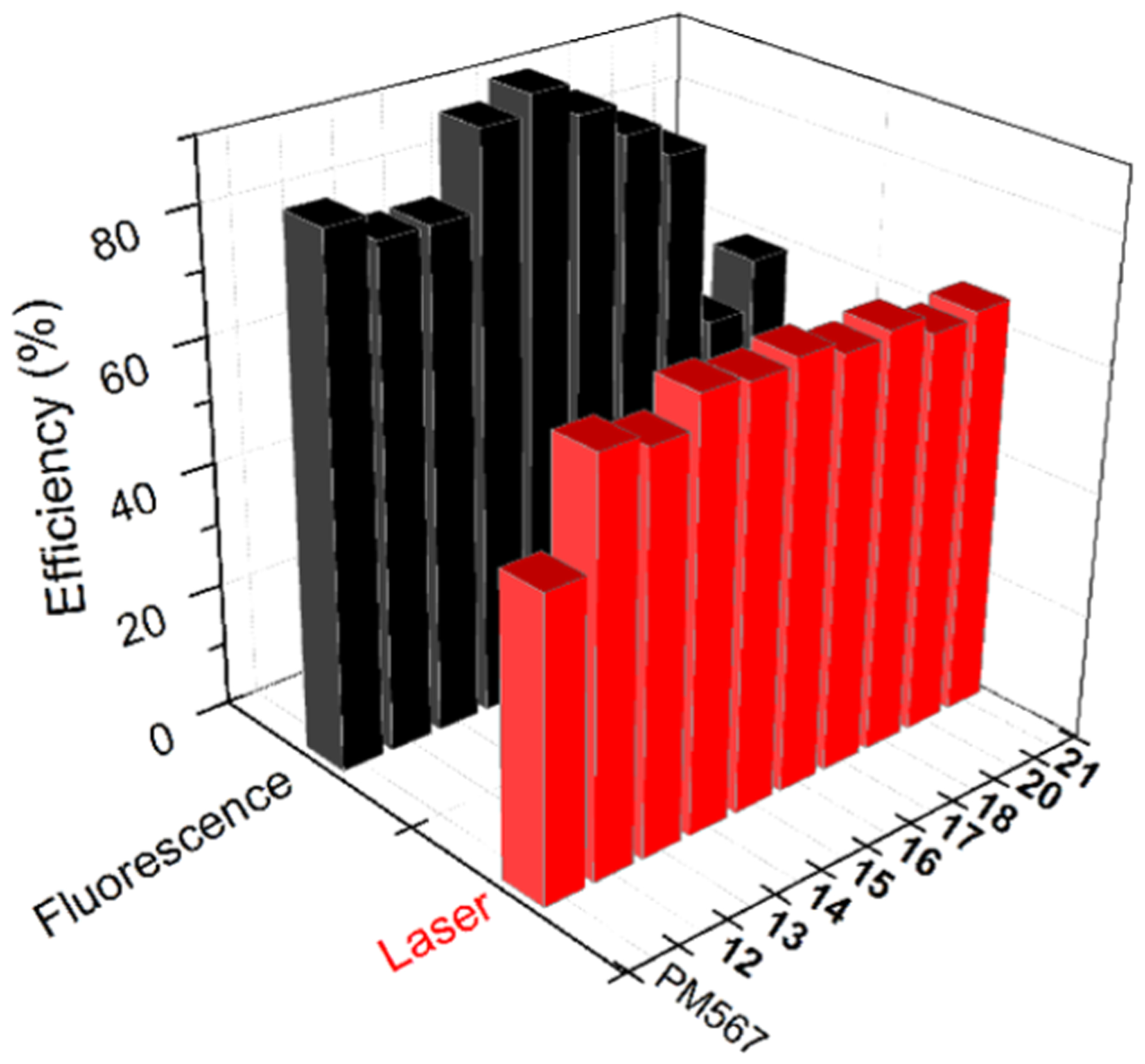
Fluorescence and laser efficiencies of PM567-derived 12–18 and 20–21 in ethyl acetate.
On the other hand, spiranic 20 and 21 displayed an unexpected red-shifted emission (see Figure S3 in Supporting Information), with lower both fluorescence efficiencies and lifetimes (see Figure 3, and Table S1 in Supporting Information). The computed optimized geometries envisage that the steric hindrance of the spiranic rings sharing the boron center distorts the BODIPY chromophore, inducing a bending along its transversal axis and a deviation from planarity of around 24°, with the boron atom 0.8 Å out of the dipyrromethene plane (see Figure S4 in Supporting Information). Such a geometrical strain entails a larger geometrical relaxation upon excitation, thereby reducing the energy gap in emission, increasing the Stokes shift (up to 1450 cm−1, three times higher than for the rest of the studied dyes) and enhancing the nonradiative pathways related with internal conversion. These fluorescence signatures fully resemble to those reported for the sterically hindered 2,6-di-tert-butyl-1,3,5,7,8-pentamethyl-F-BODIPY (commercial PM597).17
According to their photophysical properties, dyes 12–18 became highly effective laser dyes. Thus, under transversal pumping at 532 nm in ethyl acetate solution (1.5 mM), the new dyes exhibited laser action peaked at ca. 570 nm, with a pump threshold energy of 0.5 mJ, a beam divergence of 0.5 mrad, a pulse duration of 8 ns full-width at half maximum (fwhm), and lasing efficiencies as high as 68% (Figure 3), which significantly enhanced that recorded from parent PM567 (a well-known commercial laser dye) when pumped under identical experimental conditions (48%). Strikingly, 20 and 21 also showed high laser efficiencies, in spite of their lower fluorescence efficiencies (Figure 3) but red-shifted (at 585 nm) in agreement with their fluorescence signatures (see Figure S3 in Supporting Information). Likely, their higher Stokes shift implies a reduction of the deleterious impact of the re-absorption/reemission phenomena on the amplification of the stimulated emission at high optical densities, counterbalancing their lower fluorescences.
On the other hand, an important parameter for any practical application of the new laser dyes is their photostability under hard radiation conditions and long operation times. A reasonable evaluation of this parameter can be done by irradiating a small volume of the dye solution with exactly the same pumping energy and geometry as that used in the laser experiment and monitoring the evolution of the laser-induced fluorescence (LIF) intensity with respect to the number of pumping pulses (see Experimental Section for details). Under the selected experimental conditions, the studied new dyes behaved as highly photostable dyes because their laser emission remained at its initial level after 100,000 pumping pulses, enhancing that recorded from parent PM567, whose lasing emission dropped more than 80% in the same pumping period and experimental conditions.
CONCLUSIONS
In summary, we have developed a new, simple method for the straightforward preparation of valuable COO-BODIPYs from available F-BODIPYs and carboxylic acids in soft conditions and short reaction times, using BCl3 as the activating agent. The method allows higher chemical yields than previous ones, being compatible with BODIPYs having different reactive functional groups and with carboxylic acids of different stereo-electronic natures (linear, α-branched, α,β-unsaturated, aromatic, heteroaromatic, Z–E isomerizable, gem-dicarboxylic, with reactive functional groups, etc.). The unprecedented COO-BODIPYs achieved by the new method are demonstrated to maintain the excellent photophysical properties (absorption and fluorescence) of their parent well-known F-BODIPYs. Noticeably, they have been confirmed to exhibit outstanding laser properties, even surpassing those of the parent commercial laser dye PM567, with a higher laser efficiency and a much improved photostability. Finally, the method offers a possibility for the chiral perturbation of the BODIPY chromophore toward CPL, exerted by chiral acyl moieties at the boron atom, and, therefore, for the rapid development of valuable CPL-SOMs.
EXPERIMENTAL SECTION
Materials and Methods.
All reagents were used without purification. All solvents were of HPLC grade and were dried according to standard methods. Starting chemical substrates and reagents were used as commercially provided unless otherwise indicated. Thin-layer chromatography (TLC) was performed on silica gel, and the chromatograms were visualized using UV light (λ = 254 or 365 m). Flash column chromatography was performed using silica gel (230–400 mesh). 1H, 13C, and 11B NMR spectra were recorded in CDCl3 solution at 20 °C. NMR chemical shifts are expressed in parts per million (δ scale). 1H and 13C NMR spectra are referenced to residual protons of CDCl3 as the internal standard (δ = 7.26 and 77.16 ppm, respectively), and 11B NMR spectra are referenced to 15% BF3·Et2O in CDCl3 as the external standard (δ = 0.00 ppm). The type of carbon (C, CH, CH2, or CH3) was assigned by DEPT-135 NMR experiments. Additionally, complex spin-system signals were simulated by using MestReNova program version 10.0.1–14719. Fourier transform infrared (FTIR) spectra were obtained from neat samples using the attenuated total reflection technique. High-resolution mass spectrometry (HRMS) was performed using electrospray ionization (ESI) and hybrid quadrupole time-of-flight (positive ion mode) mass analyzer. Optical rotations in chloroform solution (dye concentration, c, expressed in g/100 mL, ca. 0.1 × 10−3, unless otherwise indicated) were recorded at 293 K on an Anton Paar MCP 100 polarimeter.
Synthesis of COO-BODIPYs.
Under an argon atmosphere, BCl3 (1 M in CH2Cl2, 2 mol equiv) was dropwise added over a solution of corresponding F-BODIPY (1 mol equiv) in dry CH2Cl2 (5 mL). The reaction mixture was stirred at room temperature for 5 min (disappearance of starting F-BODIPY was monitored by TLC). Then, triethylamine (6 mol equiv) was added, followed by the corresponding carboxylic acid (4 mol equiv for monoacids or 2 mol equiv for diacids), and the resulting mixture was stirred for 30 min. The reaction mixture was filtered through Celite S, washing thoroughly with CH2Cl2, and the solvent was evaporated under reduced pressure. The obtained residue was purified by flash chromatography to afford the desired product.
Synthesis of 3.
According to the described general procedure, commercial PM546 (4,4-difluoro-1,3,5,7,8-pentamethyl-BODIPY) (52 mg, 0.20 mmol) was reacted with acetic acid (47 mg, 0.78 mmol). The reaction crude was purified by flash chromatography (silica gel, CH2Cl2/EtOAc 9:1) to obtain 3 (33 mg, 49%) as an orange solid. RF = 0.26 (CH2Cl2/EtOAc 97:3). 1H NMR (CDCl3, 300 MHz): δ 6.01 (s, 2H), 2.65 (s, 3H), 2.42 (s, 6H, s), 2.41 (s, 6H), 1.99 (6H, s) ppm. The spectroscopic data were consistent with those reported in the literature.7a
Synthesis of 5.
According to the described general procedure, commercial PM567 (2,6-diethyl-4,4-difluoro-1,3,5,7,8-pentamethyl-BODIPY) (55 mg, 0.17 mmol) was reacted with acetic acid (41 mg, 0.69 mmol). The reaction crude was purified by flash chromatography (silica gel, CH2Cl2) to obtain 5 (57 mg, 83%) as an orange solid. RF = 0.28 (hexane/CH2Cl2 2:8). 1H NMR (CDCl3, 300 MHz): δ 2.67 (s, 3H), 2.38 (s, 6H), 2.35 (q, J = 7.6 Hz, 4H), 2.33 (s, 6H), 1.98 (s, 6H), 1.01 (t, J = 7.6 Hz, 6H) ppm. The spectroscopic data were consistent with those reported in the literature.7a
Synthesis of 6.
According to the described general procedure, commercial PM567 (53 mg, 0.17 mmol) was reacted with acrylic acid (48 mg, 0.67 mmol). The reaction crude was purified by flash chromatography (silica gel, hexane/CH2Cl2 1:9) to obtain 6 (36 mg, 52%) as an orange solid. RF = 0.39 (hexane/CH2Cl2 2:8). 1H NMR (CDCl3, 300 MHz): δ 6.33 (dd, J = 17.3, 2.0 Hz, 2H), 6.12 (dd, J = 17.1, 10.2 Hz, 2H), 5.70 (dd, J = 10.1, 2.0 Hz, 4H), 2.69 (s, 3H), 2.34 (s, 6H), 2.33 (s, 6H), 2.33 (q, J = 7.6 Hz, 6H), 0.99 (t, J = 7.6 Hz, 6H) ppm. The spectroscopic data were consistent with those reported in the literature.7a
Synthesis of 7.
According to the described general procedure, commercial PM546 (56 mg, 0.21 mmol) was reacted with propionic acid (60 mg, 0.85 mmol). The reaction crude was purified by flash chromatography (silica gel, hexane/CH2Cl2 2:8) to obtain 7 (66 mg, 85%) as an orange solid. RF = 0.29 (hexane/CH2Cl2 2:8). 1H NMR (CDCl3, 300 MHz): δ 6.05 (s, 2H), 2.68 (s, 2H), 2.64 (s. 3H), 2.45 (s, 6H), 2.42 (s, 6H) ppm. The spectroscopic data were consistent with those reported in the literature.7a
Synthesis of 8.
According to the described general procedure, commercial PM650 (8-cyano-4,4-difluoro-1,2,3,5,6,7-hexamethyl-BODIPY) (50 mg, 0.17 mmol) was reacted with acetic acid (40 mg, 0.67 mmol). The reaction crude was purified by flash chromatography (silica gel, CH2Cl2) to obtain 8 (52 mg, 82%) as a golden solid. RF = 0.24 (CH2Cl2). 1H NMR (CDCl3, 300 MHz): δ 2.41 (s, 6H), 2.39 (s, 6H), 2.00 (s, 6H), 1.90 (s, 6H) ppm. The spectroscopic data were consistent with those reported in the literature.7a
Synthesis of 9.
According to the described general procedure, commercial PM605 (8-acetoxymethyl-2,6-diethyl-4,4-difluoro-1,3,5,7-tetramethyl-BODIPY) (61 mg, 0.16 mmol) was reacted with acetic acid (39 mg, 0.65 mmol). The reaction crude was purified by flash chromatography (silica gel, CH2Cl2/EtOAc 9:1) to obtain 9 (47 mg, 68%) as a red solid. RF = 0.37 (CH2Cl2/EtOAc 97:3).1H NMR (CDCl3, 300 MHz): δ 5.38 (2H), 2.39 (s, 6H), 2.34 (q, J = 7.6 Hz, 4H), 2.26 (s, 6H), 2.13 (s, 3H), 1.99 (s, 6H), 1.02 (t, J = 7.6 Hz, 6H) ppm. The spectroscopic data were consistent with those reported in the literature.7a
Synthesis of 10.
According to the described general procedure, commercial PM597 (2,6-di-tert-butyl-4,4-difluoro-1,3,5,7,8-pentamethyl-BODIPY) (25 mg, 0.07 mmol) was reacted with acetic acid (16 mg, 0.27 mmol). The reaction crude was purified by flash chromatography (silica gel, hexane/CH2Cl2 1:1) to obtain 10 (9 mg, 30%) as a red solid. RF = 0.27 (hexane/CH2Cl2 2:8). 1H NMR (CDCl3, 300 MHz): δ 2.65 (s, 3H), 2.57 (s, 6H), 2.47 (s, 6H), 1.96 (s, 6H), 1.38 (s, 18H) ppm. The spectroscopic data were consistent with those reported in the literature.7a Following a variation of the general procedure, commercial PM597 (25 mg, 0.07 mmol) was reacted with acetic acid (16 mg, 0.27 mmol) at 0 °C. After work-up and purification as described above, 10 (13 mg, 43%) was obtained.
Synthesis of 11.
According to the described general procedure, 3,5-dichloro-4,4-difluoro-8-mesityl-BODIPY13 (50 mg, 0.13 mmol) was reacted with acetic acid (31 mg, 0.52 mmol). The reaction crude was purified by flash chromatography (silica gel, CH2Cl2/MeOH 99:1) to obtain 11 (41 mg, 68%) as an orange solid. RF = 0.20 (CH2Cl2). 1H NMR (CDCl3, 300 MHz): δ 6.95 (s, 2H), 6.64 (d, J = 4.4 Hz, 2H), 6.31 (d, J = 4.4 Hz, 2H), 2.35 (s, 3H), 2.20 (s, 6H), 2.11 (s, 6H) ppm. 13C{1H} NMR (CDCl3, 75 MHz): δ 172.0 (C), 145.5 (C), 142.6 (C), 139.1 (C), 137.4 (C), 136.0 (C), 130.0 (CH), 128.6 (C), 128.2 (CH), 118.6 (CH), 22.9 (CH3), 21.3 (CH3), 19.8 (CH3) ppm. 11B NMR (CDCl3, 160 MHz): δ −0.74 ppm. FTIR ν: 1724, 1569, 1265, 1191, 1113, 981 cm−1. HRMS (ESI) m/z: [M + Na]+ calcd for C22H21BCl2N2O4Na, 481.0873; found, 481.0884.
Synthesis of 12.
According to the described general procedure, commercial PM567 (50 mg, 0.16 mmol) was reacted with butanoic acid (56 mg, 0.64 mmol). The reaction crude was purified by flash chromatography (silica gel, hexane/CH2Cl2 2:8) to obtain 12 (66 mg, 92%) as a brown orange solid. RF = 0.32 (CH2Cl2). 1H NMR (CDCl3, 500 MHz): δ 2.66 (s, 3H), 2.36 (s, 6H), 2.34 (q, J = 7.6 Hz, 4H), 2.33 (s, 6H), 2.24 (t, J = 7.4 Hz, 4H), 1.54 (sext, J = 7.4 Hz, 4H), 0.99 (t, J = 7.6 Hz, 6H), 0.83 (t, J = 7.4 Hz, 6H) ppm. 13C{1H} NMR (CDCl3, 126 MHz): δ 173.8 (C), 149.4 (C), 140.7 (C), 136.5 (C), 133.3 (C), 132.0 (C), 38.2 (CH2), 18.6 (CH2), 17.4 (CH3), 17.4 (CH2), 15.0 (CH3), 14.9 (CH3), 13.9 (CH3), 12.5 (CH3) ppm. 11B NMR (CDCl3, 160 MHz): δ −0.42 ppm. FTIR ν: 1721, 1558, 1202, 981 cm−1. HRMS (ESI) m/z: [M + Na]+ calcd for C26H39BN2O4Na, 477.2905; found, 477.2918.
Synthesis of 13.
According to the described general procedure, commercial PM567 (50 mg, 0.16 mmol) was reacted with (1S)-ketopinic acid (7,7-dimethyl-2-oxobicyclo[2.2.1]heptane-1-carboxylic acid, 114 mg, 0.63 mmol). The reaction crude was purified by flash chromatography (silica gel, hexane/EtOAc 6:4) to obtain 13 (77 mg, 76%) as an orange solid. RF = 0.18 (CH2Cl2/MeOH 99.5:0.5). (c 0.039, CHCl3). 1H NMR (CDCl3, 500 MHz): δ 2.63 (s, 3H), 2.47 (dm, J = 18.1 Hz, 2H), 2.39 (s, 6H), 2.41–2.31 (m, 4H), 2.33 (s, 6H), 2.26 (ddd, J = 14.8, 11.4, 4.1 Hz, 2H), 2.02 (t, J = 4.4 Hz, 2H), 1.93 (m, 2H), 1.86 (d, J = 18.1 Hz, 2H), 1.75 (ddd, J = 14.4, 9.4, 5.0 Hz, 2H), 1.31 (ddd, J = 12.9, 9.3, 4.1 Hz, 2H), 1.22 (s, 6H), 1.06 (s, 6H), 1.03 (t, J = 7.6 Hz, 6H) ppm. 13C{1H} NMR (CDCl3, 75 MHz): δ 212.0 (C), 169.7 (C), 150.2 (C), 140.0 (C), 136.3 (C), 133.0 (C), 132.1 (C), 69.3 (C), 48.4 (C), 44.8 (CH), 44.0 (CH2), 27.0 (CH2), 26.2 (CH2), 21.8 (CH3), 20.0 (CH3), 17.4 (CH2), 17.3 (CH3), 14.9 (CH3), 14.8 (CH3), 13.4 (CH3) ppm. 11B NMR (CDCl3, 160 MHz): δ −0.34 (s) ppm. FTIR ν: 1750, 1708, 1559, 1480, 1327, 1203, 980 cm−1. HRMS (ESI) m/z: [M + Na]+ calcd for C38H51BN2O6Na, 665.3745; found, 665.3760.
Synthesis of 14.
According to the described general procedure, commercial PM567 (50 mg, 0.16 mmol) was reacted with (2E)-3-phenylprop-2-enoic acid (93 mg, 0.63 mmol). The reaction crude was purified by flash chromatography (silica gel, hexane/CH2Cl2 2:8) to obtain 14 (80 mg, 89%) as an orange-red solid. RF = 0.19 (hexane/CH2Cl2 2:8). 1H NMR (CDCl3, 300 MHz): δ 7.63 (d, J = 15.9 Hz, 2H), 7.53–7.47 (m, 4H), 7.41–7.32 (m, 6H), 6.56 (d, J = 15.9 Hz, 2H), 2.74 (s, 3H), 2.41 (s, 6H), 2.38 (s, 6H), 2.34 (q, J = 7.5 Hz, 4H), 0.99 (t, J = 7.5 Hz, 6H) ppm. 13C{1H} NMR (CDCl3, 75 MHz): δ 166.9 (C), 149.9 (C), 143.9 (CH), 140.6 (C), 136.7 (C), 135.0 (C), 133.4 (C), 132.1 (C), 129.9 (CH), 128.9 (CH), 128.1 (CH), 121.1 (CH), 17.5 (CH2), 15.0 (CH3), 14.9 (CH3), 12.6 (CH3) ppm. 11B NMR (CDCl3, 160 MHz): δ 0.09 ppm. FTIR ν: 1697, 1664, 1555, 1325, 1200 cm−1. HRMS (ESI) m/z: [M + Na]+ calcd for C36H39BN2O4Na, 597.2907; found, 597.2924.
Synthesis of 15.
According to the described general procedure, commercial PM567 (50 mg, 0.16 mmol) was reacted with (2E)-2,3-diphenylprop-2-enoic acid (141 mg, 0.63 mmol). The reaction crude was purified by flash chromatography (silica gel, hexane/CH2Cl2 2:8) to obtain 15 (98 mg, 86%) as an orange solid. RF = 0.29 (hexane/CH2Cl2 2:8). 1H NMR (CDCl3, 300 MHz): δ 7.73 (s, 2H), 7.37–7.30 (m, 6H), 7.21–7.10 (m, 10H), 7.03–6.97 (m, 4H), 2.67 (s, 3H), 2.34 (s, 6H), 2.33 (q, J = 7.6 Hz, 4H), 2.18 (s, 6H), 1.00 (t, J = 7.6 Hz, 6H) ppm. 13C{1H} NMR (CDCl3, 75 MHz): δ 166.9 (C), 150.0 (C), 140.5 (C), 138.9 (CH), 137.2 (C), 136.6 (C), 135.3 (C), 133.1 (C), 132.1 (C), 130.6 (CH), 129.8 (CH), 128.7 (CH), 128.5 (CH), 128.2 (CH), 127.3 (CH), 17.5 (CH2), 17.4 (CH3), 15.0 (CH3), 14.9 (CH3), 12.3 (CH3) ppm. 11B NMR (CDCl3, 160 MHz): δ −0.03 ppm. FTIR ν: 1695, 1554, 1264, 1203 cm−1. HRMS (ESI) m/z: [M + Na]+ calcd for C48H47BN2O4Na, 749.3535; found, 749.3563.
Synthesis of 16.
According to the described general procedure, commercial PM567 (50 mg, 0.16 mmol) was reacted with benzoic acid (77 mg, 0.63 mmol). The reaction crude was purified by flash chromatography (silica gel, hexane/CH2Cl2 2:8) to obtain 16 (76 mg, 93%) as an orange red solid. RF = 0.35 (hexane/CH2Cl2 2:8). 1H NMR (CDCl3, 300 MHz): δ 8.18 (dm, J = 7.4 Hz, 4H), 7.54 (ddt, J = 7.4, 7.2, 1.4 Hz, 2H), 7.45 (m, 4H), 2.77 (s, 3H), 2.39 (s, 6H), 2.35 (s, 6H), 2.29 (q, J = 7.6 Hz, 4H), 0.95 (t, J = 7.6 Hz, 6H) ppm. 13C{1H} NMR (CDCl3, 75 MHz): δ 166.1 (C), 150.1 (C), 140.7 (C), 136.7 (C), 133.2 (C), 132.8 (C), 132.3 (CH), 132.2 (C), 130.0 (CH), 128.3 (CH), 17.5 (CH3), 17.4 (CH2), 15.0 (CH3), 14.9 (CH3), 12.6 (CH3) ppm. 11B NMR (CDCl3, 160 MHz): δ 0.40 ppm. FTIR ν: 1706, 1556 1323, 1292, 1202, 979 cm−1. HRMS (ESI) m/z: [M + Na]+ calcd for C32H35BN2O4Na, 545.2593; found, 545.2617.
Scaled Synthesis of 16.
According to the described general procedure, commercial PM567 (320 mg, 1.01 mmol) was reacted with benzoic acid (490 mg, 4.01 mmol). The reaction crude was purified by flash chromatography (silica gel, hexane/CH2Cl2 2:8) to yield 16 (501 mg, 95%) after work-up and purification.
Synthesis of 17.
According to the described general procedure, commercial PM567 (50 mg, 0.16 mmol) was reacted with pyridine-4-carboxylic acid (77 mg, 0.63 mmol). The reaction crude was purified by flash chromatography (silica gel, CH2Cl2/MeOH 98:2) to obtain 17 (76 mg, 92%) as an orange solid. RF = 0.40 (CH2Cl2/MeOH 98:2).1H NMR (CDCl3, 300 MHz): δ 8.77 and 7.94 (AA′XX′ system, JAX = JA′X′ = 5.0 Hz, 4H), 2.77 (s, 3H), 2.39 (s, 6H), 2.31 (s, 6H), 2.30 (q, J = 7.6 Hz, 4H), 0.95 (t, J = 7.6 Hz, 6H) ppm. 13C{1H} NMR (CDCl3, 75 MHz): δ 164.4 (C), 150.6 (CH), 150.0 (C), 141.0 (C), 139.7 (C), 137.4 (C), 133.3 (C), 132.6 (C), 123.3 (CH), 17.5 (CH3), 17.3 (CH2), 14.9 (CH3), 14.8 (CH3), 12.6 (CH3) ppm. 11B NMR (CDCl3, 160 MHz): δ 0.48 ppm. FTIR ν: 1716, 1556, 1388, 1324, 1298, 1266, 981 cm−1. HRMS (ESI) m/z: [M + Na]+ calcd for C30H33BN4O4Na, 547.2498; found, 547.2519.
Synthesis of 18.
According to the described general procedure, commercial PM567 (50 mg, 0.16 mmol) was reacted with 4-ethynylbenzoic acid (92 mg, 0.63 mmol). The reaction crude was purified by flash chromatography (silica gel, hexane/EtOAc 8:2) to obtain 18 (73 mg, 81%) as a pinkish orange solid. RF = 0.32 (hexane/CH2Cl2 2:8). 1H NMR (CDCl3, 300 MHz): δ 8.10 (d, J = 8.4 Hz, 4H), 7.55 (d, J = 8.4 Hz, 4H), 3.21 (s, 2H), 2.76 (s, 3H), 2.38 (s, 6H), 2.29 (q, J = 7.6 Hz, 4H), 2.31 (s, 6H), 0.95 (t, J = 7.6 Hz, 6H) ppm. 13C{1H} NMR (CDCl3, 75 MHz): δ 165.4 (C), 150.0 (C), 140.8 (C), 136.9 (C), 133.3 (C), 132.8 (C), 132.3 (C), 132.1 (CH), 129.8 (CH), 126.1 (C), 83.2 (C), 79.7 (CH), 17.5 (CH3), 17.4 (CH2), 15.0 (CH3), 14.9 (CH3), 12.6 (CH3) ppm. 11B NMR (CDCl3, 160 MHz): δ 0.42 ppm. FTIR ν: 3285, 3242, 1704, 1554, 1479, 1326, 1289, 1201, 1117, 978 cm−1. HRMS (ESI) m/z: [M + Na]+ calcd for C36H35BN2O4Na, 593.2594; found, 593.2606.
Synthesis of 19.
According to the described general procedure, commercial PM567 (57 mg, 0.18 mmol) was reacted with 4-nitrobenzoic acid (119 mg, 0.71 mmol). The reaction crude was purified by flash chromatography (silica gel, hexane/CH2Cl2 3:7) to obtain 19 (90 mg, 82%) as a red solid. RF = 0.28 (hexane/CH2Cl2 1:1). 1H NMR (CDCl3, 500 MHz): δ 8.30 (m, 8H), 2.79 (s, 3H), 2.41 (s, 6H), 2.32 (s, 6H), 2.31 (q, J = 7.6 Hz, 4H), 0.96 (t, J = 7.6 Hz, 6H) ppm. 13C{1H} NMR (CDCl3, 75 MHz): δ 164.1 (C), 150.4 (C), 149.9 (C), 141.1 (C), 137.9 (C), 137.5 (C), 133.3 (C), 132.7 (C), 130.9 (CH), 123.6 (CH), 17.5 (CH3), 17.3 (CH2), 14.9 (CH3), 14.8 (CH3), 12.6 (CH3) ppm. 11B NMR (CDCl3, 160 MHz): δ 0.54 ppm. FTIR ν: 1716, 1557, 1527, 1480, 1328, 1293, 1203, 982 cm−1. HRMS (ESI) m/z: [M + Na]+ calcd for C32H33BN4O8Na, 635.2295; found, 635.2314.
Synthesis of 20.
According to the described general procedure, commercial PM567 (50 mg, 0.16 mmol) was reacted with cyclohexane-1,1-dicarboxylic acid (50 mg, 0.32 mmol). The reaction crude was purified by flash chromatography (silica gel, hexane/CH2Cl2 3:7) to obtain 20 (37 mg, 52%) as a red solid. RF = 0.30 (hexane/CH2Cl2 2:8).1H NMR (CDCl3, 500 MHz): δ 2.63 (s, 3H), 2.36 (q, J = 7.6 Hz, 4H), 2.321 (s, 6H), 2.316 (s, 6H), 2.10 (m, 4H), 1.81 (m, 4H), 1.56 (m, 2H), 0.99 (t, J = 7.6 Hz, 6H) ppm. 13C{1H} NMR (CDCl3, 75 MHz): δ 173.6 (C), 151.1 (C), 141.2 (C), 138.3 (C), 133.2 (C), 133.2 (C), 50.4 (C), 33.2 (CH2), 24.8 (CH2), 21.5 (CH2), 17.9 (CH3), 17.2 (CH2), 15.0 (CH3), 14.6 (CH3), 13.7 (CH3) ppm. 11B NMR (CDCl3, 160 MHz): δ 1.11 ppm. FTIR ν: 1737, 1703, 1556, 1481, 1262, 1191, 982 cm−1. HRMS (ESI) m/z: [M + Na]+ calcd for C23H31BN2O4Na, 433.2279; found, 433.2287.
Synthesis of 21.
According to the described general procedure, commercial PM567 (50 mg, 0.16 mmol) was reacted with 2,2-dimethylmalonic acid (42 mg, 0.32 mmol). The reaction crude was purified by flash chromatography (silica gel, hexane/CH2Cl2 1:1) to obtain 21 (27 mg, 42%) as a red solid. RF = 0.28 (CH2Cl2). 1H NMR (CDCl3, 500 MHz): δ 2.63 (s, 3H), 2.37 (q, J = 7.6 Hz, 4H), 2.332 (s, 6H), 2.328 (s, 6H), 1.69 (s, 6H), 1.00 (t, J = 7.6 Hz, 6H) ppm. 13C{1H} NMR (CDCl3, 75 MHz): δ 174.2 (C), 151.1 (C), 141.3 (C), 138.4 (C), 133.3 (C), 133.2 (C), 46.0 (C), 26.6 (CH3), 17.9 (CH3), 17.2 (CH2), 15.0 (CH3), 14.6 (CH3), 13.7 (CH3) ppm. 11B NMR (CDCl3, 160 MHz): δ 1.07 ppm. FTIR ν: 1742, 1707, 1560, 1482, 1384, 1318, 1194, 983 cm−1. HRMS (ESI) m/z: [M + Na]+ calcd for C23H31BN2O4Na, 433.2279; found, 433.2287.
Photophysical Properties.
Photophysical signatures were registered using quartz cuvettes with optical pathways of 1 cm in diluted solutions (around 2 × 10−6 M). These solutions were prepared taking an aliquot from a concentrated stock solution in acetone. After removing the acetone by vacuum evaporation, the dry solid was redissolved with the appropriate solvent (spectroscopic grade). Ultraviolet–visible (UV–vis) absorption and fluorescence spectra were recorded on a Varian model Cary 4E spectrophotometer and an Edinburgh Instrument spectrofluorimeter (model FLSP920), respectively. Fluorescence quantum yields (ϕ) were obtained using commercial PM546 (exciton, ϕr = 0.85 in ethanol)18 for compound 11, PM597 (exciton, ϕr = 0.43 in ethanol)18 for compounds 20 and 21, and PM567 (exciton, ϕr = 0.84 in ethanol)18 for the rest of the herein tested BODIPYs. Radiative decay curves were registered with the time-correlated single-photon counting technique, as implemented in the aforementioned spectrofluorimeter. Fluorescence emission was monitored at the maximum emission wavelength, by means of a microchannel plate detector (Hamamatsu C4878) of picosecond time-resolution (20 ps), after excitation with a Fianium pulsed laser (time resolution of around 150 picoseconds). The fluorescence lifetime (τ) was obtained after the deconvolution of the instrumental response signal from the recorded decay curves by means of an iterative method. The goodness of the exponential fit was controlled by statistical parameters (χ2) and the analysis of the residuals. Radiative (kfl) and nonradiative (knr) rate constants were calculated as follows: kfl = ϕ/τ; knr = (1 – ϕ)/τ.
The circular dichroism spectrum was recorded on a Jasco (model J-715) spectropolarimeter using standard quartz cells of 1 cm optical path length in chloroform solution at a dye concentration of ca. 3.5 × 10−5 M.
CPL and total luminescence spectra were recorded at 295 K in degassed chloroform solution, at a dye concentration of ca. 1.5 × 10−3 M, on an instrument described previously,19 operating in a differential photon-counting mode. The light source for excitation was a continuous wave 1000 W xenon arc lamp from a Spex Fluorolog-2 spectrofluorimeter, equipped with excitation and emission mono-chromators with a dispersion of 4 nm/mm (SPEX, 1681B). To prevent artefacts associated with the presence of linear polarization in the emission,20 a high-quality linear polarizer was placed in the sample compartment and aligned, so that the excitation beam was linearly polarized in the direction of emission detection (z-axis). The key feature of this geometry is that it ensures that the molecules that have been excited and that are subsequently emitting are isotropically distributed in the plane (x, y) perpendicular to the direction of emission detection. The optical system detection consisted of a focusing lens, long pass filter, and 0.22 m monochromator. The emitted light was detected by a cooled (−20 °C) EMI-9558B photomultiplier tube operating in photo-counting mode.
Computational Methods.
Ground-state energy minimizations were performed using range-B3LYP hybrid functional, within the density functional theory, using the triple valence basis set with a polarization function (6–311g*). The optimized geometries were taken as a true energy minimum using frequency calculations (no negative frequencies). The polarizable continuum model considered solvent effect (ethyl acetate) in all the calculations. All the calculations were performed in Gaussian 16, using the “arina” computational resources provided by the UPV-EHU.
Lasing Properties.
Laser efficiency was evaluated from concentrated solutions (millimolar) of dyes in ethyl acetate contained in rectangular quartz cells of 1 cm optical path length carefully sealed to avoid solvent evaporation during experiments. The liquid solutions were transversely pumped with 5 mJ, 8 ns fwhm pulses from the second harmonic (532 nm) of a Q-switched Nd:YAG laser (Lotis TII 2134) at a repetition rate of 1 Hz. The exciting pulses were line-focused onto the cell using a combination of positive and negative cylindrical lenses (f = 15 cm and f = −15 cm, respectively) perpendicularly arranged. The plane parallel oscillation cavity (2 cm length) consisted of a 90% reflectivity aluminum mirror acting as back reflector, and the lateral face of the cell acting as the output coupler (4% reflectivity). The pump and output energies were detected by a GenTec powermeter. The photostability of the dyes in methanol solution was evaluated by using a pumping energy and geometry exactly equal to that of the laser experiments. Spectroscopic quartz cuvettes with 0.1 cm optical path length were used to allow for the minimum solution volume (40 μL) to be excited. The lateral faces were ground, whereupon no laser oscillation was obtained. Information about photostability was obtained by monitoring the decrease in LIF intensity after 100,000 pump pulses and 10 Hz repetition rate to speed up the experimental running. The fluorescence emission and laser spectra were monitored perpendicular to the exciting beam, collected by an optical fiber, and imaged onto a spectrometer (Acton Research Corporation) and detected with a charge-coupled device (SpectruMM:GS128B). The fluorescence emission was recorded by feeding the signal to the boxcar (Stanford Research, model 250) to be integrated before being digitized and processed by a computer. The estimated error in the energy and photostability measurements was 10%.
Supplementary Material
ACKNOWLEDGMENTS
Financial support from Ministerio de Ciencia, Innovación y Universidades de España (MAT2017-83856-C3-1-P MAT2017-83856-C3-2-P and MAT2017-83856-C3-3-P) and Gobierno Vasco (IT912-16) is gratefully acknowledged. G.M. thanks the NIH, Minority Biomedical Research Support (1 SC3 GM089589-08) and the Henry Dreyfus Teacher-Scholar Award for financial support. C.S. thanks Comunidad de Madrid/UCM for a research contract.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.joc.9b03059.
Photophysical and computational results, including fluorescence signatures in different solvents, as well as representative absorption, fluorescence, laser, total luminescence, CD and CPL spectra; selected computed geometries and key molecular orbitals, including Cartesian coordinates and computed energies; schematic representation of the described PET; 1H NMR spectra of known compounds; and 1H NMR, 13C{1H} and 11B NMR of new compounds (PDF)
The authors declare no competing financial interest.
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.joc.9b03059
Contributor Information
Teresa Arbeloa, Departamento de Química Física, Facultad de Ciencia y Tecnología, Universidad del País Vasco-EHU, 48080 Bilbao, Spain.
Inmaculada García-Moreno, Departamento de Sistemas de Baja Dimensionalidad, Superficies y Materia Condensada, Instituto de Química-Física Rocasolano, Centro Superior de Investigaciones Científicas (CSIC), 28006 Madrid, Spain.
Santiago de la Moya, Departamento de Química Orgánica, Facultad de Ciencias Químicas, Universidad Complutense de Madrid, 28040 Madrid, Spain.
REFERENCES
- 1.Bañuelos J Chem. Rec 2016, 16, 335–348. [DOI] [PubMed] [Google Scholar]
- 2.(a) Bertrand B; Passador K; Goze C; Denat F; Bodio E; Salmain M Coord. Chem. Rev 2018, 358, 108–124. [Google Scholar]; (b) Lu H; Mack J; Nyokong T; Kobayashi N; Shen Z Coord. Chem. Rev 2016, 318, 1–15. [Google Scholar]; (c) Zhao J; Xu K; Yang W; Wang Z; Zhong F Chem. Soc. Rev 2015, 44, 8904–8939. [DOI] [PubMed] [Google Scholar]; (d) Lakshmi V; Rajeswara Rao M; Ravikanth M Org. Biomol. Chem 2015, 13, 2501–2517. [DOI] [PubMed] [Google Scholar]; (e) Fan G; Yang L; Chen Z Front. Chem. Sci. Eng 2014, 8, 405–417. [Google Scholar]; (f) Lu H; Mack J; Yang Y; Shen Z Chem. Soc. Rev 2014, 43, 4778–4823. [DOI] [PubMed] [Google Scholar]; (g) Ni Y; Wu J Org. Biomol. Chem 2014, 12, 3774–3791. [DOI] [PubMed] [Google Scholar]; (h) Bessette A; Hanan GS Chem. Soc. Rev 2014, 43, 3342–3405. [DOI] [PubMed] [Google Scholar]
- 3.(a) Zhang T; Ma C; Sun T; Xie Z Coord. Chem. Rev 2019, 390, 76–85. [Google Scholar]; (b) Kolemen S; Akkaya EU Coord. Chem. Rev 2018, 354, 121–134. [Google Scholar]; (c) Durantini AM; Heredia DA; Durantini JE; Durantini EN Eur. J. Med. Chem 2018, 144, 651–661. [DOI] [PubMed] [Google Scholar]; (d) Klfout H; Stewart A; Elkhalifa M; He H ACS Appl. Mater. Interfaces 2017, 9, 39873–39889. [DOI] [PubMed] [Google Scholar]; (e) Marfin YS; Solomonov AV; Timin AS; Rumyantsev EV Curr. Med. Chem 2017, 24, 2745–2772. [DOI] [PubMed] [Google Scholar]; (f) Kowada T; Maeda H; Kikuchi K Chem. Soc. Rev 2015, 44, 4953–4972. [DOI] [PubMed] [Google Scholar]; (g) Singh SP; Gayathri T Eur. J. Org. Chem 2014, 4689–4707. [Google Scholar]; (h) Kamkaew A; Lim SH; Lee HB; Kiew LV; Chung LY; Burgess K Chem. Soc. Rev 2013, 42, 77–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.For example, see:Loudet A; Burgess K “BODIPY dyes and their derivatives: Syntheses and spectroscopic properties”. In Handbook of Porphyrin Science; Kadish KM, Smith KM, Guilard R, Eds.; World Scientific, 2010; Vol. 8, pp 1–164.
- 5.For example, see:Boens N; Verbelen B; Dehaen W Eur. J. Org. Chem 2015, 6577–6595.
- 6.(a) Bodio E; Goze C Dyes Pigm. 2019, 160, 700–710. [Google Scholar]; (b) Ray C; Díaz-Casado L; Avellanal-Zaballa E; Bañuelos J; Cerdán L; de la García-Moreno I; Moreno F; Maroto BL; López-Arbeloa Í; Moya S Chem.—Eur. J 2017, 23, 9383–9390. [DOI] [PubMed] [Google Scholar]
- 7.(a) Durán-Sampedro G; Agarrabeitia AR; Cerdán L; Pérez-Ojeda ME; Costela A; García-Moreno I; Esnal I; Bañuelos J; López-Arbeloa I; Ortiz MJ Adv. Funct. Mater 2013, 23, 4195–4205. [Google Scholar]; (b) Manzano H; Esnal I; Marqués-Matesanz T; Bañuelos J; López-Arbeloa I; Ortiz MJ; Cerdán L; Costela A; García-Moreno I; Chiara JL Adv. Funct. Mater 2016, 26, 2756–2769. [Google Scholar]
- 8.Jiang X-D; Zhang J; Furuyáma T; Zhao W Org. Lett 2012, 14, 248–251. [DOI] [PubMed] [Google Scholar]
- 9.Blázquez-Moraleja A;Álvarez-Fernandez D; Prieto Montero R; García-Moreno I; Martínez-Martínez V; Bañuelos J; Saenz-de-Sánta-María I; Chiara MD; Chiara JL Dyes Pigm. 2019, 170, 107545. [Google Scholar]
- 10.Zhang G; Wang M; Fronczek FR; Smith KM; Vicente MGH Inorg. Chem 2018, 57, 14493–14496. [DOI] [PubMed] [Google Scholar]
- 11.Lundrigan T; Thompson AJ Org. Chem 2013, 78, 757–761. [DOI] [PubMed] [Google Scholar]
- 12.Wang M; Zhang G; Bobadova-Parvanova P; Merriweather AN; Odom L; Barbosa D; Fronczek FR; Smith KM; Vicente MGH Inorg. Chem 2019, 58, 11614–11621. [DOI] [PubMed] [Google Scholar]
- 13.Sakida T; Yamaguchi S; Shinokubo H Angew. Chem., Int. Ed 2011, 50, 2280–2283. [DOI] [PubMed] [Google Scholar]
- 14.For example, see:Satoh T; Fujii K; Kimura Y; Matano Y J. Org. Chem 2018, 83, 5274–5281.Ray C; Bañuelos J; Arbeloa T; Maroto BL; Moreno F; Agarrabeitia AR; Ortiz MJ; López-Arbeloa I; de la Moya S Dalton Trans. 2016, 45, 11839–11848.Esnal I; Duran-Sampedro G; Agarrabeitia AR; Bañuelos J; García-Moreno I; Macías MA; Peña-Cabrera E; López-Arbeloa I; de la Moya S; Ortiz MJ Phys. Chem. Chem. Phys 2015, 17, 8239–8247.
- 15.The degree of CPL is given by the luminescence dissymmetry ratio, glum(λ) = 2ΔI/I = 2(IL − IR)/(IL + IR), where IL and IR refer, respectively, to the intensity of left and right circularly polarized emissions.
- 16.(a) Sánchez-Carnerero EM; Agarrabeitia AR; Moreno F; Maroto BL; Muller G; Ortiz MJ; de la Moya S Chem.—Eur. J 2015, 21, 13488–13500. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Tanaka H; Inoue Y; Mori T ChemPhotoChem 2018, 2, 386–402. [Google Scholar]; (c) Kumar J; Nakashima T; Kawai TJ Phys. Chem. Lett 2015, 6, 3445–3452. [DOI] [PubMed] [Google Scholar]
- 17.Bañuelos Prieto J; López Arbeloa F; Martínez VM; Arbeloa López T; López Arbeloa IJ Phys. Chem. A 2004, 108, 5503–5508. [Google Scholar]
- 18.López Arbeloa F; Bañuelos J; Martínez V; Arbeloa T; López Arbeloa I Int. Rev. Phys. Chem 2005, 24, 339–374. [Google Scholar]
- 19.Brunet E; Jiménez L; de Victoria-Rodriguez M; Luu V; Muller G; Juanes O; Rodríguez-Ubis JC Microporous Mesoporous Mater. 2013, 169, 222–234 and references cited therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dekkers HPJM; Moraal PF; Timper JM; Riehl JP Appl. Spectrosc 1985, 39, 818–821. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.