

Summary
Leigh Syndrome (LS) is a mitochondrial disorder defined by progressive focal neurodegenerative lesions in specific regions of the brain. Defects in NDUFS4, a subunit of complex I of the mitochondrial electron transport chain, cause LS in humans; the Ndufs4 knockout mouse (Ndufs4(KO)) closely resembles the human disease. Here, we probed brain region-specific molecular signatures in pre-symptomatic Ndufs4(KO) to identify factors which underlie focal neurodegeneration. Metabolomics revealed that free amino acid concentrations are broadly different by region, and glucose metabolites are increased in a manner dependent on both region and genotype. We then tested the impact of the mTOR inhibitor rapamycin, which dramatically attenuates LS in Ndufs4(KO), on region specific metabolism. Our data revealed that loss of Ndufs4 drives pathogenic changes to CNS glutamine/glutamate/α-ketoglutarate metabolism which are rescued by mTOR inhibition Finally, restriction of the Ndufs4 deletion to pre-synaptic glutamatergic neurons recapitulated the whole-body knockout. Together, our findings are consistent with mTOR inhibition alleviating disease by increasing availability of α-ketoglutarate, which is both an efficient mitochondrial complex I substrate in Ndufs4(KO) and an important metabolite related to neurotransmitter metabolism in glutamatergic neurons.
Keywords: Mitochondria, genetics, metabolism, rapamycin, reactive oxygen species, mouse, Leigh Syndrome
Introduction
Mitochondrial diseases can arise from any of a number of distinct genetic defects in either the nuclear or mitochondrial genomes, with an overall incidence over 1 in 5,000 live births [1]. In humans, the severe pediatric mitochondrial disease Leigh syndrome (LS) causes progressive bilateral focal lesions in specific regions of the brain and typically leads to mortality within the first decade of life [2–6]. No effective clinical therapies have been demonstrated for LS. Mutations in components of the electron transport chain (ETC) can cause LS, many affecting ETC complex I (CI). One gene associated with LS in humans is the nuclear gene Ndufs4, encoding a subunit involved in the assembly and stability of ETC CI [7–9]. The phenotype of the Ndufs4 knockout mouse model (Ndufs4(KO)) closely resembles the human disease, including focal progressive central nervous system (CNS) lesions and a dramatically shortened lifespan; Ndufs4(KO) mice die at a median age of about 55 days after birth (P55) [10–12]. Ndufs4(KO) mice display near normal behavior and weight gain until approximately post-natal day 35 [11]. After ~ P35, neurological dysfunction presents and progressively worsens with forelimb clasping, loss of coordination, ataxia, and abnormal breathing [11, 13].
LS CNS lesions, become prominent in the olfactory bulb, cerebellum, and brainstem of Ndufs4(KO) mice after onset of disease, while the cortex is generally devoid of overt degeneration during the lifespan of this mouse [11, 12]. These appear as focal areas of necrosis with astrocyte and microglial accumulation. Mechanisms underlying this regional specificity of CNS lesions are unknown. Deletion of Ndufs4 driven by Nestin promoter driven Cre, which is largely specific to the nervous system, results in mice which recapitulate the major neurological symptoms and deteriorate and die with the same time course as the full body Ndufs4(KO) [12] This indicates that the CNS lesions, as well as the other major pathological features of LS, arise from neuronal dysfunction.
Loss of NDUFS4 has a marked impact on mitochondrial respiratory capacity, reducing maximum CI dependent oxidative phosphorylation in isolated brain mitochondria to about 60 percent of normal values when pyruvate or glutamate are provided as substrates [10, 14]. Intriguingly, Ndufs4(KO) brain mitochondria maintain a normal respiratory rate when α-ketoglutarate is provided as a CI specific substrate [14]. The mechanistic basis for this difference is not clear. Synaptosomal mitochondria from lesion sensitive areas show disease-associated respiration defects, in contrast to synaptosomal mitochondria from disease resistant CNS. Non-synaptosome mitochondria are not regionally distinct. The data suggest that synaptosome mitochondria may play a role in lesion formation [14].
While no clinical treatments for LS currently exist, two interventions have been shown to dramatically attenuate disease in the Ndufs4(KO) mouse: exposure to chronic hypoxia (or related oxygen reducing strategies) and inhibition of the mechanistic Target of Rapamycin (mTOR) [10, 15]. mTOR inhibition with the macrolide rapamycin more than doubles survival time and significantly suppresses neurological features of disease in the Ndufs4(KO) mice [10, 16]. Rapamycin has recently been shown to rescue primary cell models of human mitochondrial disease, and also appears to benefit mitochondrial disease patients [17]. Critically, attenuation of mitochondrial disease pathology by rapamycin occurs without rescue of ETC CI assembly, stability, supercomplex formation, mitochondrial respiratory capacity, changes to mitochondrial biomass or heteroplasmy [10, 17]. Rapamycin inhibits the key nutrient sensing signaling complexes mTORC1 and mTORC2 (the mTOR complexes 1 and 2), leading to broad downstream anabolic and catabolic shifts [18, 19]. Several mechanisms have been proposed to contribute to the beneficial effects of mTOR inhibition in the presence of mitochondrial dysfunction, including energetic rescue via reduced protein synthesis, activation of mitophagy, and metabolic rewiring to compensate for the primary ETC defects [10, 20, 21]. The precise mechanism or mechanisms underlying the benefits of mTOR inhibition in mitochondrial disease remain undefined.
Here, we aimed to identify causes for the focal CNS deterioration seen in Ndufs4(KO) mice by defining metabolic and energetic factors which define the sensitive regions of the CNS. We focused on pre-symptomatic mice at post-natal day 25 to 30 to attempt to identify factors which may lead to focal neurodegeneration, as opposed to examining tissue which already harbors sequelae of disease. In addition, we examined the effects of a short daily course of rapamycin treatment in this context in order to identify putative metabolic mechanisms by which inhibition of mTOR may alleviate CNS deterioration in Ndufs4(KO) mice. Our results provide direct evidence that the ɑ-ketoglutarate/glutamate/glutamine metabolic axis is dysfunctional in the Ndufs4(KO) mice and indicate that mTOR inhibition may act through restoration of these key metabolites.
Results
Regional Mitochondrial ETC CIV Levels
We first considered the possibility that distinct requirements for mitochondrial oxidative phosphorylation driven energy production may underlie the region specificity of CNS lesions in LS. Neurodegeneration in the Ndufs4(KO) would, in this model, occur in regions with the highest ETC demand. We assessed regional mitochondrial energy demand by staining brain slices using cytochrome C oxidase (COX, ETC Complex IV) histochemical staining. Histochemical staining for COX activity is a well-established technique which takes advantage of the COX dependent oxidation of diaminobenzidine, which produces a brown product visible by light microscopy [22, 23] (Figure 1A–B). The level of COX is generally accepted to reflect overall mitochondrial oxidative phosphorylation, and local capacity is generally expected to reflect local mitochondrial energetic demands.
Figure 1 – Regional Mitochondrial Energetic Capacity.
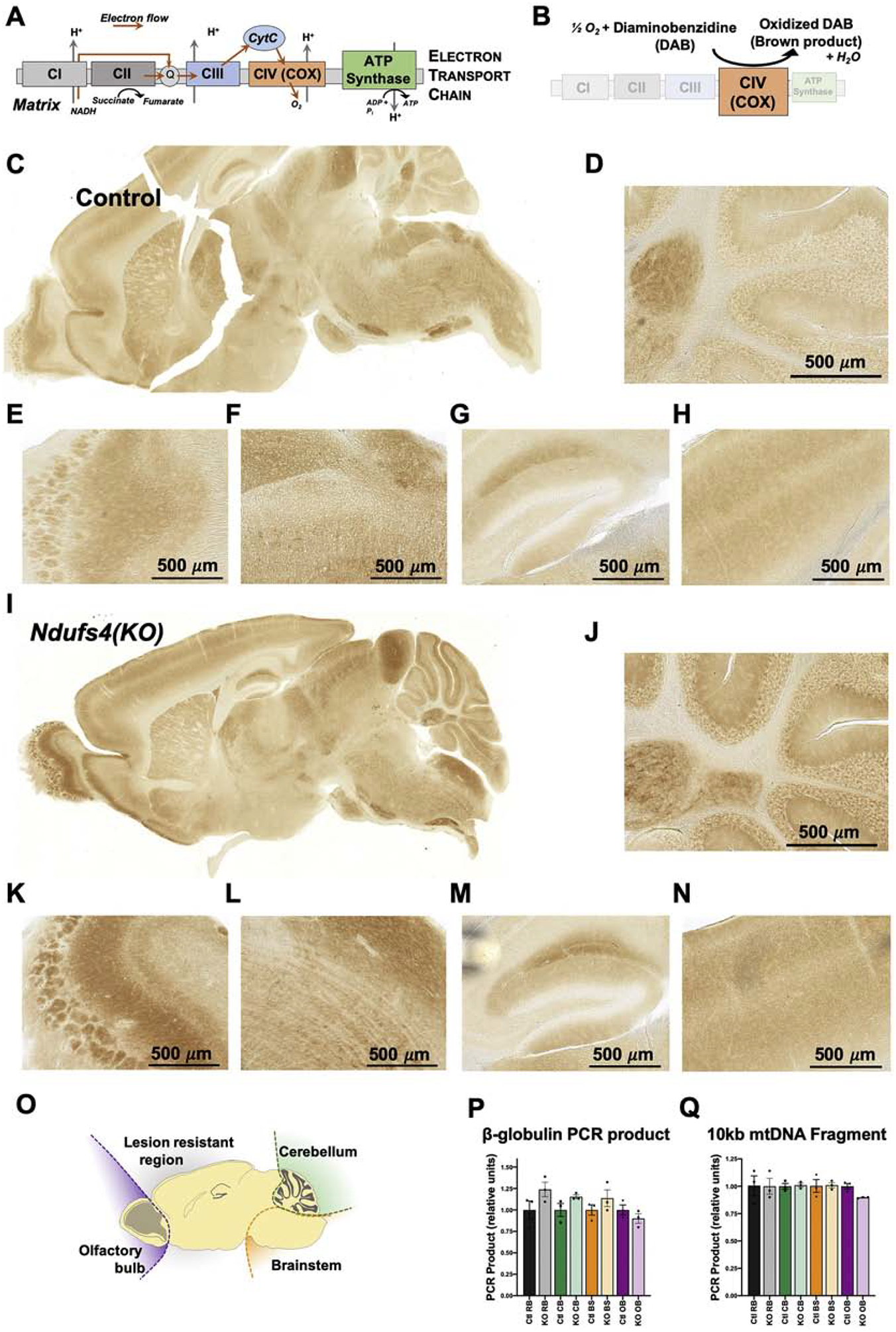
(A) Schematic of the mitochondrial electron transport chain (ETC). (B) Schematic of the histochemical staining method for ETC complex IV (CIV) activity using the DAB reaction. (C) Representative sagittal brain section from a 28-day old control mouse stained for ETC CIV activity. (D-H) Magnified images of cerebellum (D), olfactory bulb (E), brainstem (F), hippocampus (G), and cortex (H) from the section in (C). (I) Representative sagittal brain section from a 28-day old Ndufs4(KO) mouse stained for ETC CIV activity. (J-N) Magnified images of cerebellum (J), olfactory bulb (K), brainstem (L), hippocampus (M), and cortex (N) from the section in (I). (O) Schematic of brain sectioning for DNA damage assessment by qPCR. (P) Quantification of long-range PCR of the nuclear gene beta-globulin in DNA isolated from key brain regions in control and Ndufs4(KO) mice. All results are normalized to the mean values for control samples from the respective regions. (Q) Quantification of long-range qPCR targeting a mitochondrial DNA encoded 10kb fragment in DNA isolated from key brain regions in control and Ndufs4(KO) mice. All results are normalized to the mean values for control samples from the respective regions. (P-Q) Data presented are the means of 3 technical repeats for each of 3 biological samples per genotype. No statistically significant differences observed between control and Ndufs4(KO) samples. In this figure and all subsequent figures; Ctl RB= control resistant brain region, KO RB= knockout resistant brain region, Ctl CB = control cerebellum, KO CB = knockout cerebellum, Ctl BS = control brainstem, KO BS= knockout brainstem, Ctl OB= control olfactory bulb, KO OB= knockout olfactory bulb. Values are given as the means + standard errors of the mean (SEM).
COX staining of pre-disease control (Figure 1C–H) and Ndufs4(KO) (Figure 1I–N) brain samples revealed significant variance in mitochondrial ETC capacity across the brain. However, the observed regional differences did not reveal any predictive pattern regarding the region specificity of lesion formation. Increased staining was observed in a subset of cells in the granular layer of the cerebellum (Figure 1D, J), the glomeruli of the olfactory bulb (Figure 1E, K), and in vestibular nuclei and inferior olive of the brainstem (Figure 1F, L), all reported to present with necrotic lesions in the Ndufs4(KO) mouse [12, 24]. Dark staining was also observed, however, in the hippocampus and inferior colliculus of the cortex (Figure 1G, M), in the pons (Figure 1C, I), and in various additional brain regions not known to degenerate. We observed no differences in stain distribution between control and Ndufs4(KO) mice at this age. We conclude that increased regional reliance on mitochondrial oxidative phosphorylation may be necessary but is not sufficient to define lesion sites.
Oxidative stress – DNA damage
Reactive oxygen species (ROS) are one by-product of respiratory metabolism, and ROS can lead to oxidative stress, accumulated damage, and pathology. Oxidative damage has been reported to be increased in settings of mitochondrial dysfunction, and can lead to neuropathology, including CNS cell death [25, 26]. Protein carbonylation, one form of oxidative damage, has previously been shown to be increased in the brain of Ndufs4(KO) mice, but levels were only assessed in whole-brain homogenates in overtly diseased animals [12]. In contrast, regional differences in lipid oxidative damage by 4-hydroxynonenal staining have been reported to be unchanged in Ndufs4(KO) globally or in vulnerable brain regions from Ndufs4 [14]. A regional assessment of other forms of oxidative damage in Ndufs4(KO) compared to control brains prior to overt inflammatory neuropathology has been lacking.
DNA oxidative damage induced lesions can interfere with DNA polymerase activity, and accumulated damage can, therefore, be assessed using long range quantitative PCR (LR-qPCR), as the efficiency of amplification from equimolar templates is determined by the proportion of undamaged copies in the sample. To assess whether DNA damage differs by region in the Ndufs4(KO), we performed LR-qPCR on DNA isolated from individual regions. Using nuclear DNA from vulnerable and resistant brain regions, we found no differences in LR-qPCR of the beta-globulin gene when comparing genotypes in any given region (Figure 1O,P).
Due to its proximity to the source of ROS at the ETC and limited repair capacity versus the nuclear genome, mitochondrial DNA (mtDNA) is especially vulnerable to oxidative damage. To determine whether oxidative damage to mtDNA is increased in Ndufs4(KO) mice compared to controls, or is higher in lesion-associated brain regions, we performed LR-qPCR of a 10 kb mtDNA PCR product. Within a given brain region we found no differences in amplification efficiency between Ndufs4(KO) and control animals. (Figure 1O,Q, see discussion).
To further assess whether overt oxidative damage is present in the brain of Ndufs4(KO) mice compared to controls in a region-specific manner prior to disease onset, we assessed 8-Oxo-2’-deoxyguanosine (8-oxo-dG) levels using antibody-based staining (see Methods). 8-oxodG is an ROS derived damage to nucleic acids commonly used to assess oxidative stress. We found no evidence for oxidative damage in pre-disease Ndufs4(KO) brain, relative to control animal, in any region (Figure S1).
Region Specific mTOR Activity in Pre-Lesion CNS
Inhibition of mTOR with the specific inhibitor rapamycin significantly prolongs survival and suppresses neurodegeneration in the Ndufs4(KO) model, and is being considered as a therapy for human mitochondrial disease [10, 16, 17, 27]. We therefore questioned whether mTOR activity was increased in vulnerable regions in Ndufs4(KO). Phosphorylation of ribosomal protein S6 (rpS6) is a well-established downstream effect of increased mTOR complex 1 (mTORC1) activity. Elevated levels of phospho-rpS6 (p-rpS6) were reported in whole brain homogenates from older (P>45 days) untreated Ndufs4(KO) mice which already displayed neurologic symptoms, indicating mTOR is hyperactive in the setting of overt disease in these mice [10]. Since inhibition of mTORC1 suppresses formation of brain lesions in the KO, we next considered whether focal neurodegeneration may be driven by local hyperactivity of mTORC1 prior to overt disease. Using tissue lysates extracted from individual brain regions, we assessed mTORC1 activity in pre-symptomatic mice (P29–30) by western blotting for phospho- and total rpS6 (Figure 2A–D). The ratio of p-rpS6/rpS6, although more variable in Ndufs4(KO) mice compared to controls did not differ between genotypes. As expected, rapamycin treatment reduced pS6 in both Ndufs4(KO) and control mice in each brain region (Figure 2A–D).
Figure 2 – Region Specific mTOR Activity in Pre-Lesion CNS .
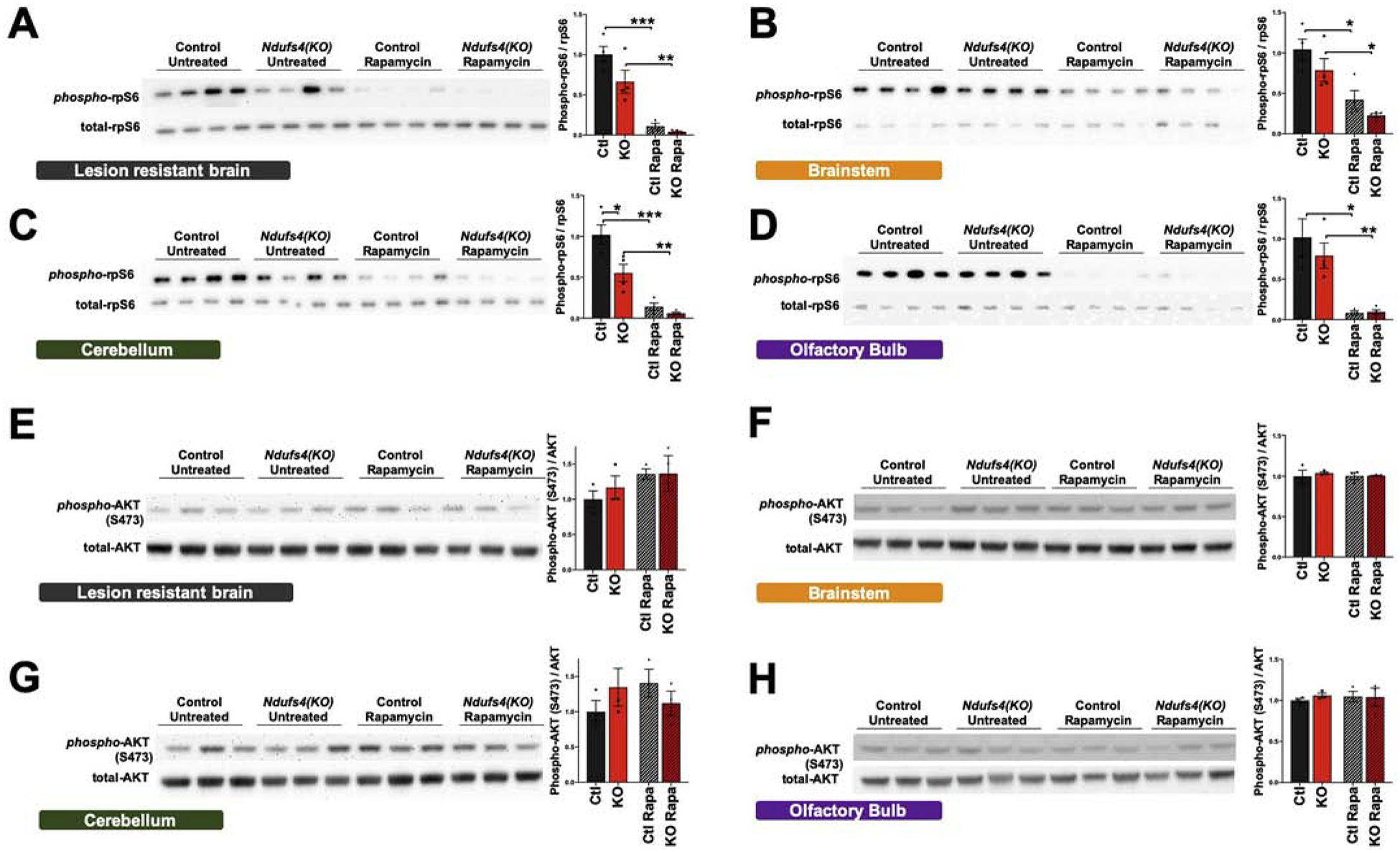
Western blots and quantification of the phosphorylation of ribosomal protein S6 (rpS6), a downstream target of mTORC1, in the lesion resistant brain region (A), brainstem (B), cerebellum (C), and olfactory bulb (D) in ad libitum fed pre-disease onset (P25–28) animals. Western blots and quantification of the phosphorylation of AKT at serine 473 (S473), a downstream target of mTORC2, in the lesion resistant brain region (E), brainstem (F), cerebellum (G), and olfactory bulb (H). For histograms, N=4 in all cases. Ctl = wild type without rapamycin, KO = Ndufs4(KO) without rapamycin, Ctl Rapa = wildtype treated with rapamycin, KO Rapa = Ndufs4(KO) treated with rapamycin. Dots in histograms represents 4 biological samples (even where the adjacent blot shows only 3 of the samples). (A) p<0.0001, one-way ANOVA. (B) p=0.001, one-way ANOVA. (C) p<0.0001, one-way ANOVA. (D) p=0..0007, one-way ANOVA. (E-H) p>0.05, one-way ANOVA, groups not significantly different. (A-H) * designates p<0.05. ** p<0.005, ***p≤0.0005 by pairwise t-test.
mTOR participates in two distinct multi-protein complexes: mTORC1 and mTORC2. While short duration low dose rapamycin is generally thought to have a greater inhibitory effect on mTORC1, chronic or high-dose treatment with rapamycin also reduces mTORC2 activity [28]. The doses of chronic rapamycin necessary to attenuate disease suggest that mTORC2 inhibition could mediate the benefits of this therapy. To assess mTORC2 activity, we performed Western blotting for the phosphorylation state of mTORC2 substrate Akt at serine 473 (Figure 2E–H). We focused on four general CNS regions: cerebellum, olfactory bulb(OB), brainstem (BS), and the ‘lesion resistant’ region, which includes the cortex. Regional phosphorylation status of Akt did not correlate with vulnerability. Furthermore, we did not detect a difference in Akt phosphorylation as a function of rapamycin treatment at this age.
Region-Specific Metabolomics in CNS of Control and Ndufs4(KO) Mice
Loss of Ndufs4 results in markedly abnormal whole-brain metabolism [10, 14], and metabolic alterations are thought to contribute to disease pathogenesis. Multiple lines of evidence suggest that broad shifts in mTOR regulated metabolic processes play a role [10, 17, 29]. It is not known, however, how observed changes in metabolism reconcile with the region specificity of lesion formation. To address this, we performed CNS region specific metabolomics from Ndufs4(KO) and control animals collected at P29–31, an age preceding lesion formation and overt neurological dysfunction. We focused on four general CNS regions as in Figure 2 (Figure 3A).
Figure 3 – Region-Specific Metabolomics in CNS of Control and Ndufs4(KO) Mice.
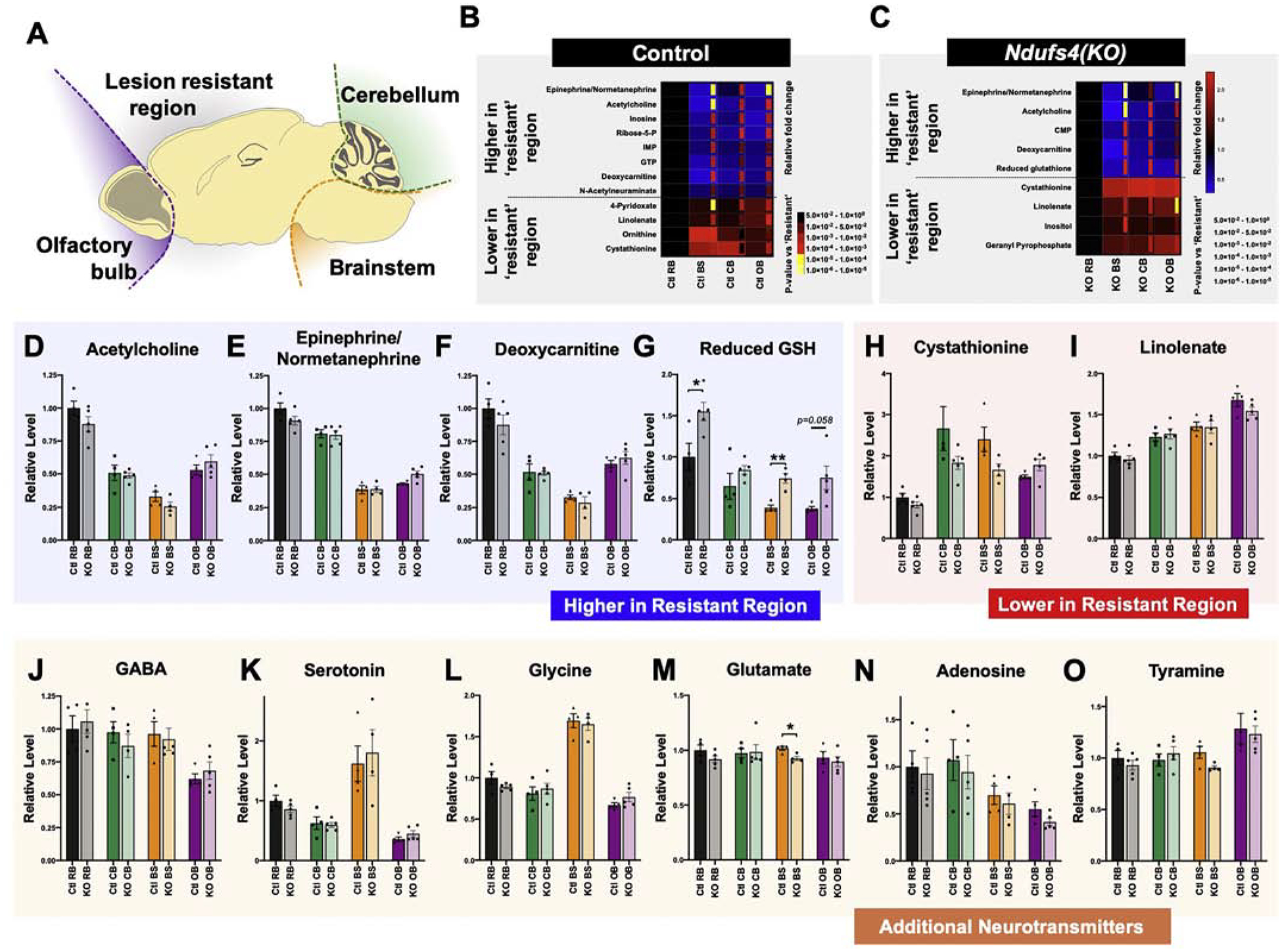
(A) Schematic of brain regions physically separated and analyzed by metabolomics. (B) Metabolites in control brain samples that show significantly different abundance between the lesion ‘resistant’ region and each of the lesion associated regions, with the same direction of change in each pairwise comparison. Relative fold change (larger squares) and nominal p-values (inset rectangles) of each comparison to the ‘resistant’ region are indicated by heat maps. (C) Metabolites in Ndufs4(KO) brain samples that show significantly different abundance between the lesion ‘resistant’ region and each of the lesion associated regions, with the same direction of change in each pairwise comparison. Relative fold change (larger squares) and nominal p-values (inset rectangles) of each comparison to the ‘resistant’ region are indicated by heat maps. (D-G) Metabolites significantly higher in the ‘resistant’ region, versus the lesion associated regions, in both the control and Ndufs4(KO) datasets (see also Supplemental Figure 2). (H-I) Metabolites significantly lower in the ‘resistant’ region, versus the lesion associated regions, in both the control and Ndufs4(KO) datasets. (J-O) Additional neurotransmitters appearing in these metabolomics datasets. (D-I) ANOVA p<0.0001 for each of these metabolites. Pairwise t-tests between each sensitive and the resistant region appear in (B-C). No significant genotype differences observed except in (G) and (M), where indicated (*p<0.05, **p<0.005 by pairwise t-test). (J) ANOVA p=0.0075, no significant differences by genotype. (M) ANOVA p=0.48, no significant differences by genotype. (N) ANOVA p=0.026, no significant differences by genotype. (O) ANOVA p=0.0119, no significant differences by genotype. For all panels (D-O) dots represent biological replicates (N>4), histograms and error bars designate median values and standard error of the mean. Ctl RB= control resistant brain region, KO RB = knockout resistant brain region, Ctl CB = control cerebellum, KO CB = knockout cerebellum, Ctl BS = control brainstem, KO BS= knockout brainstem, Ctl OB= control olfactory bulb, KO OB= knockout olfactory bulb.
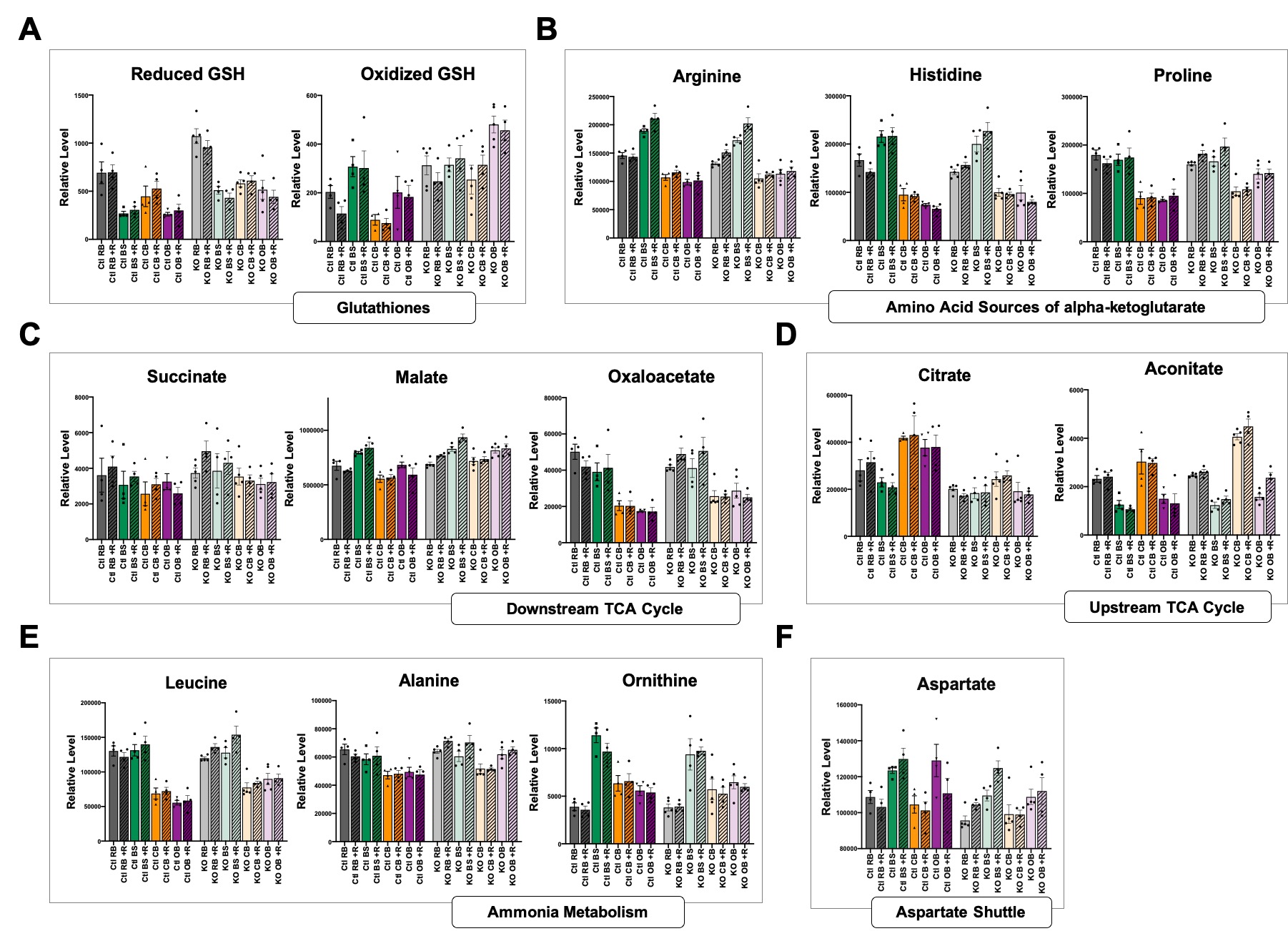
First, we assessed control (Figure 3B) and Ndufs4(KO) (Figure 3C) animals separately to identify metabolites which define the ‘lesion sensitive’ regions compared to the ‘resistant’ region. Metabolites which show significantly different levels in all three sensitive regions compared to the resistant region, and show the same directionality for each pairwise comparison against the resistant region, were identified in control (Figure 3B) and Ndufs4(KO) (Figure 3C) mice. The regional patterns of metabolite differences show substantial similarities in each individual genotype comparison. In both Ndufs4(KO) and control animals, the ‘resistant’ region is distinguished by higher levels of the neurotransmitters epinephrine and acetylcholine, as well as higher levels of deoxycarnitine (an intermediate in carnitine production) compared to sensitive regions. In contrast, lower levels of cystathionine and linolenate are found in the resistant region compared to sensitive regions in both genotypes (Figure 3D–I).
Acetylcholine, epinephrine, deoxycarnitine, and linolenate show no genotype difference in any region (Figure 3D, E, F, and I, respectively), whereas reduced glutathione is significantly higher in Ndufs4(KO) brainstem, olfactory bulb, and ‘resistant’ regions compared to the matching regions in control mice (Figure 3G, see Discussion). The overall ratio of GSH/GSSG is unchanged save for brainstem, indicating that total levels are increased (Figure S2 A–B).
While glutathione was not a focus of this study, we considered the possibilities that GSH changes could provide an early biomarker of disease or that glutathione might be dysregulated in other tissues. Analysis of whole-brain samples and liver lysates from separate mouse cohorts at age P26–30 showed no evidence that this is this case (Figure S2 C–H). Metabolomics of plasma, however, suggests that oxidized glutathione is elevated in young (P30) Ndufs4(KO) mice compared to controls and that the increase is more pronounced in animals aged P45 (reduced glutathione was not detected in this assay) (Figure S2 I–J).
The increased levels of acetylcholine and epinephrine in the ‘resistant’ region compared to vulnerable regions suggests that neuron type (defined by neurotransmitter) may be a key determining feature. For a more comprehensive picture of neurotransmitter abundance by region, we examined the level of each neurotransmitter captured in our metabolome study by both region and genotype (Figure 3J–O). We observed no significant differences in neurotransmitter level in any region when comparing genotype (Ndufs4(KO) versus control animals). By region, we found that brainstem has uniquely high levels of serotonin and glycine compared to other regions (Figure 3K, L), consistent with the role of glycine as the major inhibitory neurotransmitter in the brainstem [30]. Levels of GABA, serotonin, and adenosine are lower or trend lower in olfactory bulb, while this region shows the highest level of tyramine (Figure 3J, K, O). There are no differences in glutamate levels specific to all vulnerable regions (Figure 3M), although glutamate is significantly lower in the brainstem of Ndufs4(KO) versus controls.
Regional Metabolic Profiles of Control and Ndufs4(KO) Mice
To gain further insight into the metabolic changes associated with ETC CI deficiency pre-disease, we performed principal component analysis (PCA) and unsupervised clustering (Figure 4). The criteria for identifying metabolites in Figure 3 (statistical significance in comparisons between each of the ‘sensitive’ regions and the ‘resistant’ region) likely precludes detection of many metabolites as a result of the high stringency, while trends involving only some regions would also be overlooked. A PCA analysis of Ndufs4(KO) and control brain samples of different regions revealed, surprisingly, that the majority of variance among these samples is driven by brain region (Figure 4A–C), further highlighting the diversity of different CNS regions.
Figure 4 – Regional Metabolic Profiles of Control and Ndufs4(KO) Mice.
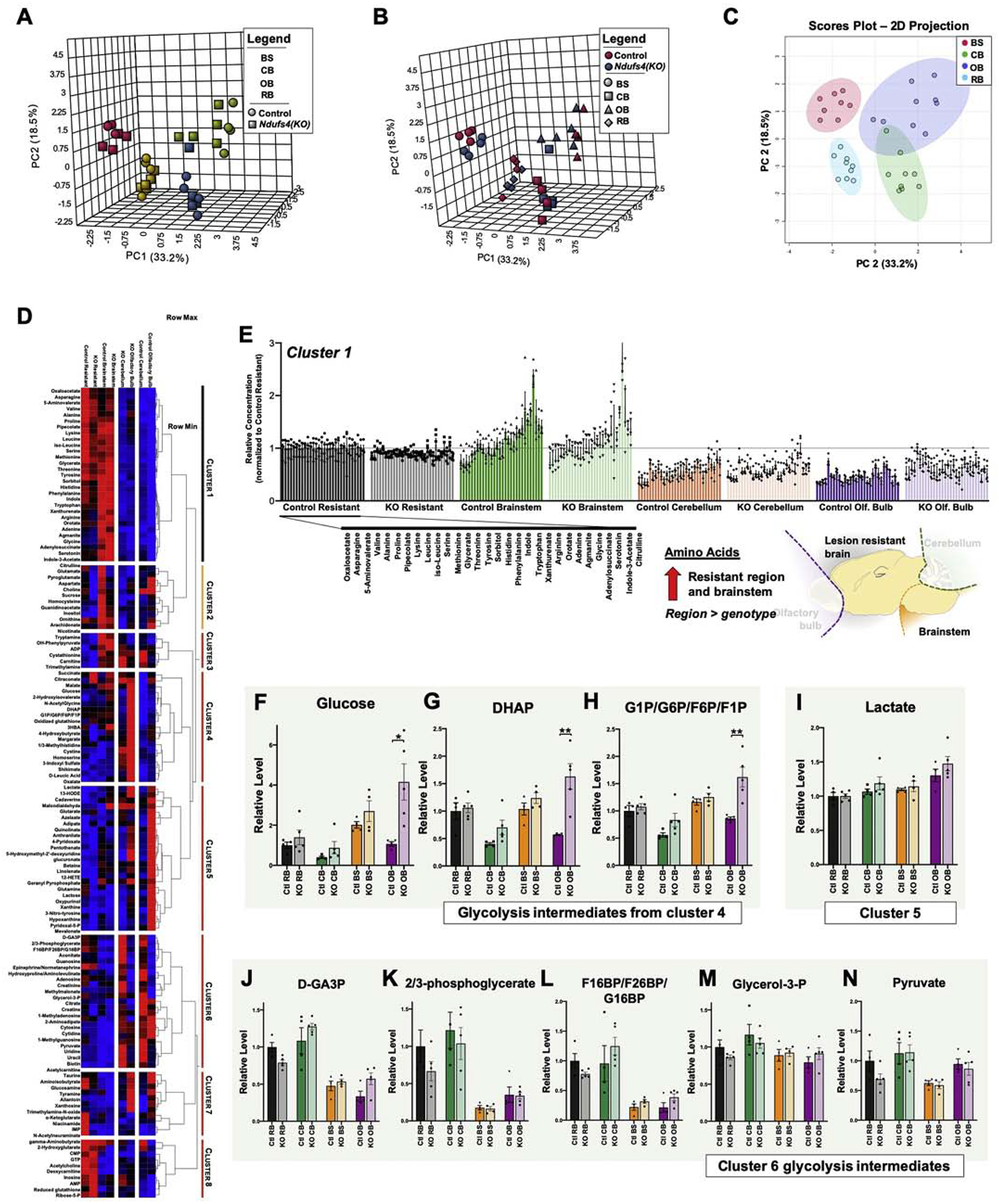
(A-B) 3-dimensional principal component analysis (PCA) of brain region metabolomics data with region (A) or genotype (B) indicated (see Methods). (C) Projection of first two major principal components with 95% confidence intervals of groups indicated (colored ellipses). Brain region is the major determinant of sample clustering by PCA. (D) Unsupervised clustering of brain region metabolite level data in control and Ndufs4(KO) mice. (E) Cluster 1, which includes the majority of amino acids. (C-E) Glycolysis intermediates appearing in cluster 4. ANOVA p-values: (F) glucose p<0.0001, (G) DHAP p<0.0001, (H) G-1-P/G-6-P/F-6-P/F-1-P p<0.0001. *p<0.05 and **p<0.005 by two-tailed pairwise t-test. (I) Lactate, which appears in cluster 5. ANOVA p=0.0008. (J-N) Glycolysis intermediates appearing in cluster 6. ANOVA p-values: (J) D-GA3P p<0.0001, (K) 2/3 phosphoglycerate p<0.0001, (L) F-1,6-BP/F-2,6-BP/G-1,6-BP, p<0.0001 (M) glycerol-3-P, Not significant, (N) pyruvate p<0.008. For all panels (F-N) individual points represent biological replicates (individual mice), N>4 per group; histograms and error bars designate median values and standard error of the mean. Ctl RB= control resistant brain region, KO RB = knockout resistant brain region, Ctl CB = control cerebellum, KO CB = knockout cerebellum, Ctl BS = control brainstem, KO BS= knockout brainstem, Ctl OB= control olfactory bulb, KO OB= knockout olfactory bulb.
We next performed unsupervised clustering of sample group medians, enabling us to identify additional region and genotype driven trends (Figure 4D, see Methods). Data-driven clustering suggests that regional differences are greater than genotype differences in brainstem and ‘resistant’ region samples, as in the PCA analyses, whereas olfactory bulb and cerebellum cluster by genotype (Figure 4D, horizontal).
We performed supervised dendrogram splitting of the data into 8 ‘clusters’ of metabolites for further analysis. The largest cluster in number of detected metabolites is composed primarily of free amino acids, which are higher in brainstem than ‘resistant’ brain, but lower in both the cerebellum and olfactory bulb (Figure 4D–E).
Intriguingly, glutamate clusters with pyroglutamate, aspartate, and ornithine, rather than other amino acids or neurotransmitters (Figure 4D, cluster 2, see next section for caveats regarding pyroglutamate detection). Cluster 2 reflects the majority of the urea cycle and shows a pattern of lower levels in the Ndufs4(KO) in every region, though in this set no individual differences rise to even nominal statistical significance.
Our previous whole-brain metabolic data indicated that glycolysis intermediates are increased in overtly diseased Ndufs4(KO) brains compared to controls [10]. Clusters 4–6 include glycolytic intermediates and lactate, which separate into distinct patterns by region and genotype. Glucose, DHAP, and G-1-P/G-6-P/F-1-P/F-6-P (indistinguishable by MS) are elevated in every brain region in Ndufs4(KO) mice compared to controls, though only reaching statistical significance in a subset of these (Figure 4D, F–H, see figure legend). Similarly, lactate modestly trends upward in knockout mice in each region, reaching statistical significance only in olfactory bulb (Figure 4I). D-GA-3-P, 2/3-bisphosphoglycerate, and F-1,6-BP/F-2,6-BP tend to be lower in brainstem and olfactory bulb, while glycerol-3-P, and pyruvate show no overall regional specificity (Figure 4J–N, see Discussion).
Region-Specific Metabolic Impact of mTOR Inhibition
We next assessed the impact of chronic mTOR inhibition on the CNS region-specific metabolomes of Ndufs4(KO) and control mice. Using samples from animals treated daily from P21 with 8 mg/kg rapamycin untilP29–30, (pre-disease onset) mice, we performed unsupervised clustering of control (vehicle only injections) (Figure 5A) and Ndufs4(KO) (Figure 5B) samples by metabolite. The impact of rapamycin treatment at this young age appears far less dramatic than in the older (overtly diseased, >P50) animal whole-brain metabolomics that we previously reported [10].
Figure 5 – Region-Specific Metabolic Impact of mTOR Inhibition.
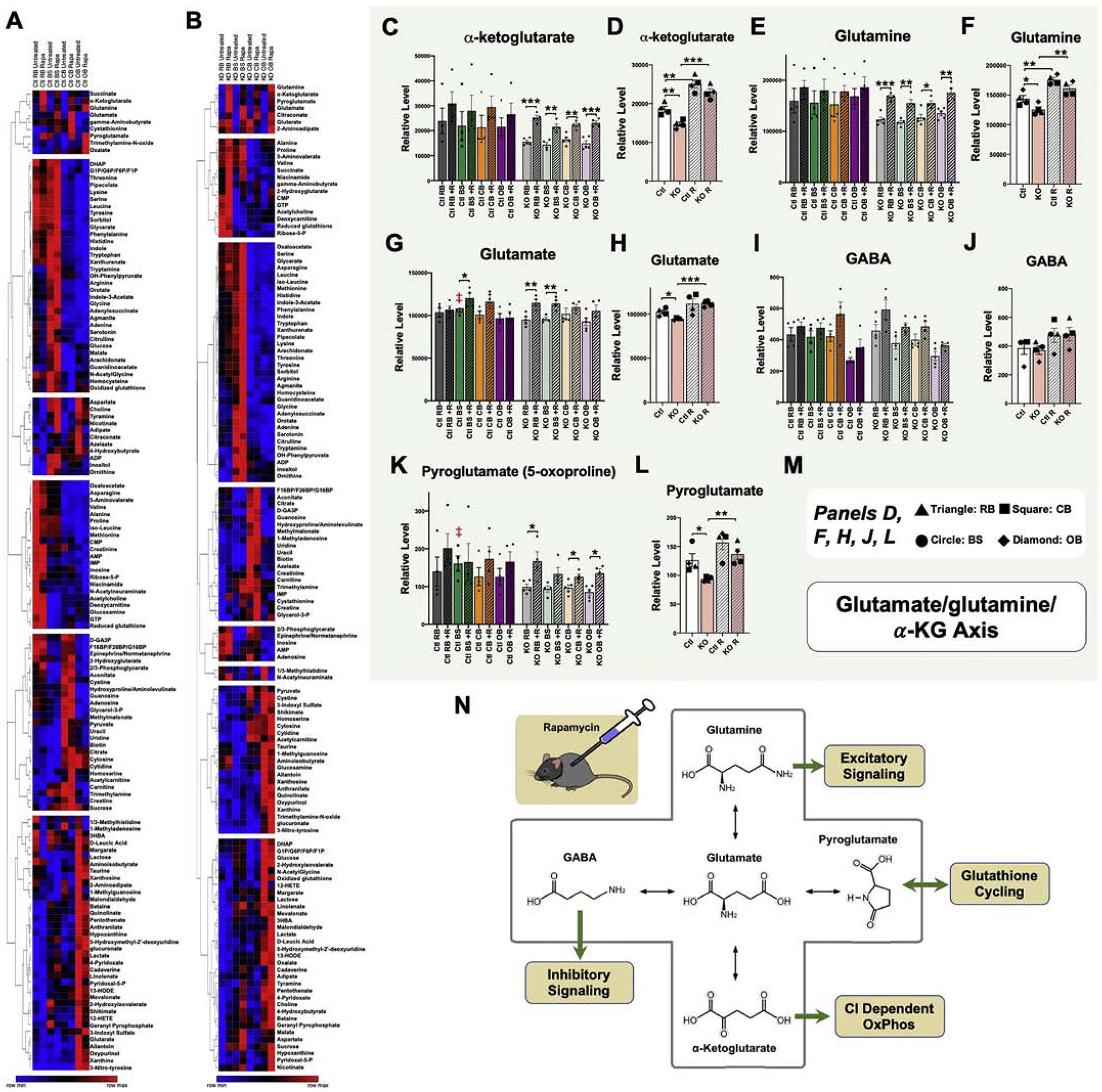
(A-B) Unsupervised clustering of brain region metabolite levels in untreated and rapamycin treated control (A) and Ndufs4(KO) (B) mice. Metabolites appearing in cluster 1 in both control and Ndufs4(KO) datasets indicate a rapamycin-responsive glutamate/glutamine/α-ketoglutarate metabolite group. (C) α-ketoglutarate levels by brain region, genotype, and treatment. ANOVA *p=0.01. (D) Data-reduced α-ketoglutarate data using median values for each brain region, revealing genotype and treatment dependent, region independent, changes. ANOVA ***p<0.0001. (E) Glutamine levels by brain region, genotype, and treatment. ANOVA **p=0.005. (F) Data-reduced glutamine data using median values for each brain region, revealing genotype and treatment dependent, region independent, changes. ANOVA ***p<0.0001. (G) Glutamate levels by brain region, genotype, and treatment. ANOVA **p<0.005. (H) Data-reduced glutamate data using median values for each brain region, revealing genotype and treatment dependent, region independent, changes. ANOVA **p=0.0055. (I) GABA levels by brain region, genotype, and treatment. ANOVA ***p<0.0001. (J) Data-reduced GABA data using median values for each brain region, revealing genotype and treatment dependent, region independent, changes. Not significantly different by ANOVA. (K) Pyroglutamate levels by brain region, genotype, and treatment. ANOVA *p=0.03. (L) Data-reduced pyroglutamate data using median values for each brain region, revealing genotype and treatment dependent, region independent, changes. ANOVA **p=0.004. (M) Symbol key for panels D, F, H, J, and L; triangles, squares, circles, and diamonds represent the median values (across biological replicates) for ‘resistant brain’ (RB), cerebellum (CB), brainstem (BS), and olfactory bulb (OB), respectively. (C, E, G, I, K) Dots represent biological replicates (N>=4); columns and error bars designate median values and standard error of the mean. (D, F, H, J, L) Dots represent median values (among biological replicates) for each brain region. Columns represent median values among regions within the same genotype and treatment, and error bars represent standard error of the mean. (C-L) pairwise t-test p-values * designates p≤0.05, ** designates p≤0.005, and *** designates p≤0.0005. (N) Schematic of the glutamine/glutamate/α-ketoglutarate metabolite group with key CNS functions of each glutamate metabolite indicated. Ctl RB= control resistant brain region, KO RB= knockout resistant brain region, Ctl CB = control cerebellum, KO CB = knockout cerebellum, Ctl BS = control brainstem, KO BS= knockout brainstem, Ctl OB= control olfactory bulb, KO OB= knockout olfactory bulb. For (D,F,H,J,L) Ctl and KO represent untreated wild type and Ndufs4(KO) averages, respectively; Ctl R and KO R represent rapamycin treated wild type and Ndufs4(KO) averages, respectively.
While rapamycin-driven effects can be seen in many individual metabolites, dendrogram splitting to eight metabolite clusters appears to be primarily driven by region-specific patterns, rather than rapamycin driven metabolic changes, confirming again that individual brain regions are highly metabolically distinct (see Figure 4). The exception is a set of metabolites which appear as cluster 1 in both control and Ndufs4(KO) sample sets (Figure 5A, B). This cluster includes the closely related metabolites glutamine, glutamate, pyroglutamate, GABA, and α-ketoglutarate (Figure 5 C–L). The cluster pattern of these metabolites appears to be driven by treatment, rather than region, and appears in both genotypes. Given the lack of regional differences, we performed data reduction by taking the median value (among the biological replicates) for each region and compared by genotype and treatment. Strikingly, the untreated Ndufs4(KO) show reduced levels of this entire cluster in all regions, while rapamycin increases levels and ameliorates deficits, for each of the key metabolites from this cluster except GABA (Figure 5 D, F, H, J, L, and M; see Discussion). We conclude that (1) Ndufs4 deficiency reduces glutamine/glutamate/α-ketoglutarate levels in a region independent manner, and (2) mTOR inhibition increases glutamine/glutamate/α-ketoglutarate levels in a region independent manner, rescuing the defects present in the Ndufs4(KO) in all regions.
It is important to note that a large fraction of pyroglutamine can be produced in the MS reaction, depending on the conditions, and crossover signal can result between glutamine, glutamate, and pyroglutamine [31]. Previously, we found that it can be produced in the sample preparation phase as well, at least for serum samples [32]. These metabolites are also closely related through in vivo metabolism, however, and correlations observed are also consistent with co-regulation of their shared metabolic pathway. Critically, given their high correlation, summing the signal (if the relative concentrations were known) would not change either the significance or the conclusions of our findings as both genotype and treatment impact these metabolites in the same manner.
Impact of NDUFS4 loss restricted to glutamatergic neurons
We previously established that VGlut2-expressing pre-synaptic excitatory glutamatergic neurons mediate the anesthesia sensitivity of Ndufs4(KO) mice, while loss of Ndufs4 in GABAergic or cholinergic neurons does not [33]. Considered together our metabolic data indicating altered glutamine/glutamate/α-ketoglutarate levels, we considered the possibility that glutamatergic neuron dysfunction may also drive CNS disease. To address this, we bred VGlut2 specific Ndufs4(KO) mice using a VGlut2 promoter driven Cre line and the floxed Ndufs4 mouse strain (see Methods, gift of A Quintana). Using these animals, we found that deletion of Ndufs4 specifically in pre-synaptic glutamatergic neurons is sufficient to recapitulate the lifespan (Figure 6A), age of disease onset and progressive weight loss (Figure 6B), and overt neurological symptoms of whole-body Ndufs4(KO) animals (Figure 6C), albeit with a delayed onset of neurologic symptoms relative to the whole-body knockout. Similar findings have also been recently independently reported by Bolea, et al. [24].
Figure 6 – Neuronal Effects of Ndufs4 Deficiency.
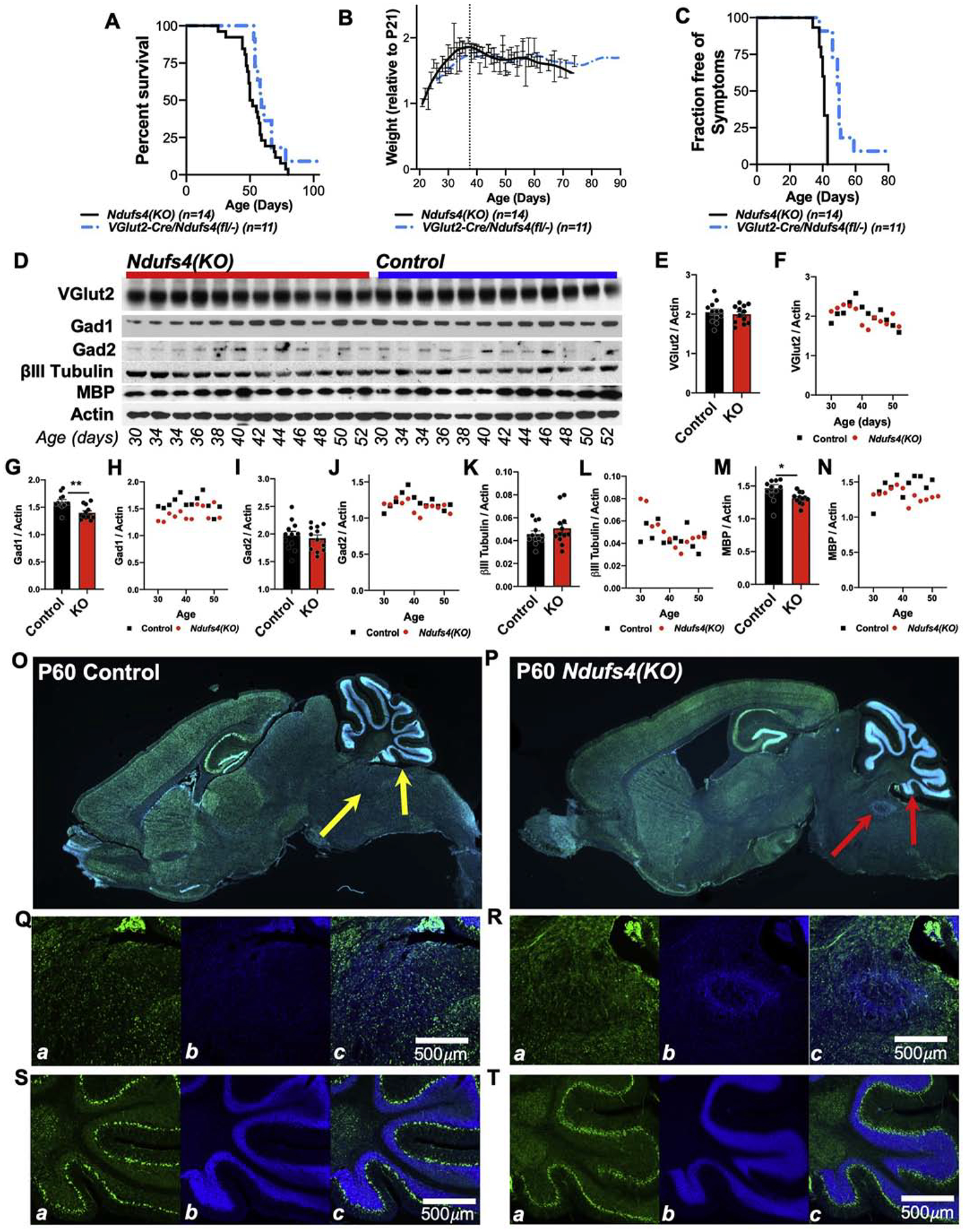
(A-C) VGlut2 specific knockout of Ndufs4 leads to shortened lifespan (A), progressive weight loss after approximately post-natal day 37, and (C) onset of neurologic symptoms by approximately P40 roughly equivalent to that observed in the whole-body Ndufs4(KO) mouse. (A) Log-rank test p=0.1. (B) No significant differences at any age. (C) Log-rank test ***p<0.0001. (D) Western blot analysis of whole-brain lysates from Ndufs4(KO) and control animals as a function of age. (E-N) Quantification of protein abundance. (E, G, I, K, M) Analysis of genotypes with all ages pooled, *p<0.05, **p<0.005 by pairwise t-test. (F, H, J, L, N) Scatter-plots of target abundance by genotype and age. GAD1 (Glutamate Decarboxylase 1); GAD2 (Glutamate Decarboxylase 2); MBP (myelin basic protein). (O-P) VGlut2-Cre promoter driven ZsGreen (Ai6 construct) expression in P60 control (O) and Ndufs4(KO) (whole-body knockout) (P) brains. Green = ZsGreen, blue = DAPI (DNA dye). Arrows indicate known lesion-associated areas of the cerebellum (right) and brainstem (left). An advanced lesion is visible in the brainstem region of the Ndusf4(KO) sample (P), as determined by the abnormal accumulation of nuclei (DAPI, blue) at this location. No overt changes to VGlut2-Cre expressing cell populations are present in the Ndufs4(KO) compared to control. Representative images, n>3 brains imaged per genotype. (Q-R) Higher magnification imaging of brainstem (Q-R) and cerebellum (S-T) in these control (Q, S) and Ndusf4(KO) (R, T) brains. In each set of panels, a-ZsGreen, b-DAPI, c-overlay.
Neuronal populations in the Ndufs4(KO) model
Given these findings, and the metabolic data, we next considered the possibility that Ndufs4 deficiency leads to a loss of specific neuron populations related to glutamine/glutamate/α-ketoglutarate metabolism as a function of disease progression. To assess this, we probed whole-brain lysates collected from Ndufs4(KO) and control mice for presynaptic markers of GABAergic and glutamatergic neurons, and pan-neuronal markers. We used a sample set including mice ranging in age from P30, pre-disease onset, to P52, where overt neurologic symptoms, weight loss, and CNS lesions are present in 100% of animals (Figure 6D–N). Ndufs4(KO) brains show no overt differences in levels of VGlut2, expressed in many pre-synaptic glutamatergic neurons, compared to controls (Figure 6E–F). GAD1 (Glutamate Decarboxylase 1, expressed by a subset of GABAergic neurons), is generally lower in Ndufs4(KO) mice compared to controls, but there is no age dependent change, suggesting that this difference is not related to the progressive CNS lesions or related symptoms which start ~P35–37 (Figure 6G–H). GAD2 (Glutamate Decarboxylase 2, expressed by a subset of GABAergic neurons) levels are not significantly lower in Ndufs4(KO) compared to controls and do not change with disease progression (Figure 6I–J). βIII-tubulin (a cytoskeletal protein broadly expressed in neurons) shows no genotype or age-dependent differences (Figure 6K–L). Finally, MBP (myelin basic protein, involved in the myelin sheath of oligodendrocytes and Schwann cells) appears to be significantly lower in Ndufs4(KO) mice compared to controls, with a possible decrease after P37 (Figure 6M–N).
While whole-brain levels of VGlut2 are not impacted by Ndufs4 deficiency, we considered the possibility that VGlut2 positive neurons are lost in a region or lesion-site specific manner. To investigate this possibility, we performed a three-way cross, generating whole-body Ndufs4(KO) expressing the VGlut2-Cre driver and carrying the Cre-activated green fluorescent protein (ZsGreen) construct Ai6 (Figure 6O–P). In these mice, Cre activates high levels of expression of ZsGreen, allowing for direct assessment of the Cre-positive population. Using these animals, we found that there are no overt changes in overall expression or localization of VGlut2 positive cells in the Ndufs4(KO) compared to controls in any brain region (Figure 6O–P). In fact, even in Ndufs4(KO) mice with overt CNS lesions, no changes to VGlut2 positive cell populations can be detected in either the brainstem (Figure 6Q–R) or cerebellum (Figure 6S–T) at the lesion sites.
Discussion
Our data demonstrate that, in young animals prior to CNS symptoms, the glutamine/glutamate/α-ketoglutarate axis is strikingly reduced in all brain regions in the Ndufs4(KO) compared to control mice, and these defects are rescued by rapamycin treatment. These data are consistent with the model that α-ketoglutarate plays an important role in disease onset and in the beneficial effects of mTOR inhibition. mTOR inhibition may act, in part, by increasing α-ketoglutarate and driving mitochondrial energetics since α-ketoglutarate is an effective ETC CI substrate even in the setting of Ndufs4 deficient mitochondria [14]. These data are supported by a recent report that α-ketoglutarate supplementation extends lifespan and alleviates encephalopathic symptoms in Ndufs4(KO) mice [34]. Alternatively, our VGlut2-specific Ndufs4 knockout data, supported by recent published findings [24], demonstrate that pre-synaptic glutamatergic neurons drive disease in this model; the defective glutamine/glutamate/α-ketoglutarate axis may lead to dysfunction of these neurons through disruption of neurotransmitter metabolism, and rescue of these metabolites by mTOR inhibition may be mediated by rescuing neuronal function. With the relative importance of the glutamine/glutamate/α-ketoglutarate in mind, we discuss our other findings below.
Regional CIV staining
In prior work, we have established that maximal respiratory capacity of isolated mitochondria through α-ketoglutarate/malate or succinate + rotenone (CII specific) is unaffected by Ndufs4 deficiency, demonstrating that capacity at CIV is not impacted by genotype, but leaving open the possibility that regional/spatial differences in capacity are related to susceptibility to lesion formation [10, 14]. Here, using complex IV (COX) staining as a proxy for local mitochondrial capacity/demand, we examined the spatial pattern of CIV to assess whether local energetic demand correlates with sensitivity to lesions. Our findings indicate that capacity at CIV is highly region specific, with some areas associated with lesions showing high CIV staining, but we found no genotype-specific differences. Thus, high regional demand may contribute to lesion formation, but is not sufficient to identify regions susceptible to degeneration. Importantly, COX activity staining is only one possible measure for local energetic demand and does not infer activities of other mitochondrial complexes; further work will be necessary to completely eliminate a causal role for energetics in the spatial specificity of CNS lesions in LS.
ROS damage
Mitochondrial disorders can increase ROS production, which in turn has been suggested to underlie neurodegeneration [35]. Our data found no correlation between regional or local ROS damage and areas of neurodegeneration in LS. In fact, we did not find any evidence of elevated nucleic acid damage in the KO compared to the control, consistent with our previous work showing a lack of increased mitochondrial or cytoplasmic 4-HNE, an indicator of ROS-induced lipid peroxidation, in the Ndufs4(KO). Taken together, these data suggest that oxidative stress is neither increased in the Ndufs4(KO) nor a predictor of regional sensitivity. Since hypoxia can lengthen lifespan of Ndufs4(KO), its effects may be other than direct reduction of ROS, or may be at regional level finer than our methods could detect.
Metabolic profiles of lesion-associated regions
Prior studies have suggested that CNS disease in the Ndufs4(KO) mouse may be driven by metabolic abnormalities [10]. Reports showing disease attenuation in this model by mTOR inhibition [10] or hypoxia [15] have suggested that these interventions may prevent disease through their effects on metabolism. Our data identify a number of intriguing region-specific metabolic differences which may pre-dispose lesion-associated brain regions to degeneration in the setting of mitochondrial dysfunction. These include differences in neurotransmitter abundance compared to ‘resistant’ brain matter, higher levels of the lipid linolenate, and lower reduced glutathione metabolites compared to ‘resistant’ brain. Of these regionally distinct factors, only GSH shows genotype-specific differences as well as regional differences, with Ndufs4(KO) animals showing higher overall glutathione in most regions compared to controls (see Results for additional glutathione data).
A very recent whole-genome CRISPR screen for modifiers of sensitivity to mitochondrial dysfunction has shown that glutathione related pathways are upregulated in the setting of mitochondrial stress and disruption of GSH related antioxidant pathways results in synthetic lethality [36]. Taken together, our brain and plasma data support the notion that glutathione may provide a biomarker of disease, and may be mechanistically important, but further study will be needed to elucidate the role of these and other redox metabolites.
Overall, the region-metabolite data suggest that differences in the relative abundance of neuron types as defined by neurotransmitter (inferred by the observed differences in acetylcholine and epinephrine) may be a defining factor in lesion susceptibility. Similarly, regional differences in GSH synthesis or levels may play a role.
mTOR activity
We previously found mTOR is hyperactive in whole-brain lysates of overtly diseased Ndufs4(KO) animals, and, more recently, have shown mTOR hyperactivity in primary patient fibroblasts derived from human mitochondrial disease patients [10, 17]. mTOR hyperactivity has also been reported in other models of mitochondrial disease [27, 30], and targeting mTOR has been shown to attenuate disease in a number of distinct mitochondrial disease paradigms. These include: the Ndufs4(KO) mouse model of Leigh syndrome; a COX IV mouse model of mitochondrial myopathy related to Leigh syndrome; the Tk2 deficient mouse model of mitochondrial DNA depletion; a Twinkle deficient mouse model of mitochondrial myopathy [10, 16, 21, 27, 37–41]; yeast with mitochondrial defects [37]; Drosophila melanogaster models of Leigh syndrome and ETC CII deficiency [42, 43]; zebrafish acyl-CoA dehydrogenase deficiency associated mitochondrial dysfunction [44]; cultured cell models for mitochondrial dysfunction [17, 20, 45]. Finally, recent clinical trials in pediatric and adult mitochondrial disease patients have suggested these benefits may extend to humans [17, 46].
Intriguingly, our data indicate that mTOR is not overtly hyperactive in Ndufs4(KO) brain pre-disease onset at resting, not feeding- synchronized, state. This may suggest that the mTOR hyperactivation we and others have previously reported either occurs at some later age (perhaps following a normal developmental change in the CNS), that observed mTOR hyperactivation is a product of neuroinflammation and astrocyte activation associated with lesions, or that synchronization of feeding status, as performed in our previous studies, uncovers defects in mTOR signaling not detected at resting state. Our goal in this study was to assess resting-state status of metabolites and signaling in control and diseased animals synchronized only by time of day. Further work will be necessary to probe the role of feeding status, nutritional and hormonal cues, and age in mediating the reported increase in mTOR signaling in the Ndufs4(KO) mice.
CNS region-specific metabolic impact of rapamycin
It was striking that, at this pre-lesion age, the metabolic impact of rapamycin treatment was relatively limited compared to the impact of rapamycin in the brain of Ndufs4(KO) at older ages where overt pathology was present [10]. The majority of significant metabolic changes in whole brain lysates of Ndufs4(KO) mice treated with rapamycin, compared to untreated Ndufs4(KO) animals, were reversals or attenuations of disease-associated changes [10]. The current pre-lesion age dataset reveals the mTOR inhibition driven metabolic changes which are independent of lesion onset and indicates which of those previously reported metabolic effects were specific to an age where overt pathology is present in untreated mice. Whether those findings reflected the metabolic signature of CNS degeneration itself or are the result of an age-related change in brain metabolism remains to be determined.
A single cluster of glutamine related metabolites was uniquely increased by rapamycin treatment in both genotypes, and in every brain region. These include glutamate and GABA, neurotransmitters previously implicated in the CNS pathologies of LS, and their precursor glutamine, as well as α-ketoglutarate, the TCA cycle intermediate produced by glutamate metabolism. As noted above, α-ketoglutarate is able to fully drive CI activity in Ndufs4(KO) mitochondria [14]. Furthermore, α-ketoglutarate also supports substrate level ATP or GTP synthesis by succinyl-CoA synthetase in the mitochondrial matrix, allowing for mitochondrial energy production without direct involvement of the ETC. Pyroglutamate, an intermediate in GSH production from glutamate, is also increased in rapamycin treated mutants. While the precise role of any of these mTOR inhibition-altered metabolites remains unclear, each have been implicated in driving the pathologies associated with mitochondrial dysfunction [34, 47, 48]. Our data suggest that each (or the sum of all) are candidates for mediating any beneficial metabolic effects of mTOR inhibition in the setting of mitochondrial disease.
Functional ETC CI consumes NADH, and CI deficiency has been shown to alter intracellular redox status by significantly increasing the NADH/NAD+ ratio [34]. The ratio of NADH to NAD+ directly impacts the function of enzymes which utilize these molecules. Among these are the NADH/NAD+ sensitive deacetylase sirtuins, which regulate metabolism, growth, and stress responses through removal of the post-translational acetyl modification in response to redox status [49, 50]. Recent reports have found decreasing NADH/NAD+ by supplementation of Ndufs4(KO) with the NAD+ precursors nicotinamide mononucleotide (NMN), or treatment with the nicotinamide phosphor-ribosyltransferase (NAMPT) activator P7C3, attenuated disease in the LS model [34]. Intriguingly, NMN increased α-ketoglutarate levels in tissues where the NADH/NAD+ ratio was attenuated by treatment, and supplementation with the cell-permeable α-ketoglutarate analog dimethylketoglutarate (DMKG) significantly increased survival in this model [34].
The precise mechanism by which increased α-ketoglutarate attenuates disease is unclear, and causality for its role in the effects of NMN are yet to be determined but may involve its role as an alternate CI substrate. We have previously shown that while complex I specific mitochondrial respiratory capacity is reduced by ~40% percent in Ndufs4(KO) brain when pyruvate and malate are used as substrates, activity is strikingly normal when α-ketoglutarate is provided instead. It is intriguing that our data shows the glutamine/glutamate/α-ketoglutarate axis to be the most notably altered set of metabolites in rapamycin treated young mouse brain. Further work is needed to resolve mechanistic underpinnings of this increase and define the precise role of α-ketoglutarate and related compounds in the mechanisms of disease rescue by rapamycin.
Neuronal impact of NDUFS4 deficiency
Ndufs4 deletion driven by Nestin promoter driven Cre, largely specific to the neurons, was reported to cause the major neurological symptoms, CNS lesions, weight loss, and shortened lifespan of full body Ndufs4(KO) [12], indicating that the major pathological features of LS arise from neuronal origins. In previous work, we found that VGlut2 positive pre-synaptic excitatory glutamatergic neurons are responsible for the anesthesia sensitivity in the Ndufs4(KO) [33]. Given our metabolic findings, we tested the possibility that neurons with unique functional requirements for glutamine/glutamate/α-ketoglutarate, such as glutamate neurotransmitter metabolism, may be uniquely impacted by loss of NDUFS4. Here, we found that knockout of Ndufs4 restricted to VGlut2 expressing cells recapitulates the overall disease features of the whole-body Ndusf4(KO) and Nestin-Cre driven knockout, findings which have also recently been reported by Bolea et al [24]. Importantly, while loss of NDUFS4 in VGlut2 expressing cells drives disease, we found no evidence for glutamatergic neuron loss in Ndufs4(KO) brain compared to controls at any age, as a function of disease progression, or in lesion-associated sites (Fig. 6). This suggests that glutamatergic neurons drive lesion formation and neurologic disease in the setting of NDUFS4 deficiency through mechanisms other than neuronal death.
Conclusions
We have uncovered a direct link between Ndusf4 deficiency and decreased glutamine/glutamate/α-ketoglutarate levels, which is rescued by inhibition of mTOR through rapamycin treatment. Together, our data support the model that mTOR inhibition attenuates neurological disease in the Ndufs4(KO) mouse specifically by rescuing glutamine/glutamate/α-ketoglutarate levels. Rescue of these metabolites may attenuate disease by providing α-ketoglutarate as a functional mitochondrial complex I substrate to support oxidative phosphorylation, through rescue of neuron specific metabolic pathways related to neurotransmitter metabolism, or through a combination of both of these mechanisms. Future efforts will be aimed at testing these models.
Material and Methods
Animals
All animal experiments were approved by the IACUC of Seattle Children’s Research Institute (protocol 13416) and University of Washington (protocol 4359–03.) Mice were housed with 12 hours dark-light cycle at 22°C, with water and standard rodent diet available ad libitum. The Ndufs4(KO) mice were generated as previously described [11, 12, 14]. For experimental animals, genotypes were always determined by genotyping by polymerase chain reaction and agarose-gel electrophoresis. Heterozygotes animals have no overt phenotypes, have NDUFS4 protein levels similar to the wild-type (Ndufs4+/+), and show no defects in mitochondrial function, both heterozygous and wild-type animals were used as controls in these experiments, as done previously, and are simply referred to as controls [11]. Similarly, male and female animals were used at approximately equal numbers as disease relevant phenotypes are indistinguishable by sex [11, 12]. Ndufs4(KO) mice were kept with control littermates and received supplemental moistened food pellets and hydration gel if mobility became limited. At designated ages, mice were euthanized by cervical dislocation, followed by decapitation, and brains were removed and processed as detailed below.
VGlut2-Cre specific knockout mice were generated as previously described [33], with a breeding scheme compatible with VGlut2-specific deletion of Ndufs4. Briefly, these animals were generated by crossing VGlut2-Cre (Slc17a6-promoter driven Cre, Jackson laboratories stock #028867) positive Ndufs4(+/−) animals to female Ndufs4(fl/fl) mice, which are homozygous for the LoxP site flanked exon 2 allele of Ndufs4 (Jackson Laboratories strain # 026963, backcrossed into the C57Bl/6 line over a dozen times), from which the Ndufs4(KO) line was derived. VGlut2-Cre+::Ndufs4(-/fl) offspring provide VGlut2-specific knockout animals. VGlut2-Cre/Ai6 reporter mice were generated by crossing VGlut2-Cre and Ai6 / RCL-ZsGreen (a Cre-activated GFP reporter, Jackson Laboratories stock # 007906) into the Ndufs4(+/−) background and maintaining this line as Ndusf4(+/−)::VGlut2-Cre(+/+)::Ai6(+/+).
All genotyping protocols performed as described by Jackson Laboratories strain information.
Cytochrome oxidase (COX) cytochemistry
Whole brains were fixed in 1X phosphate buffered saline (PBS) / 4% formalin at 4°C overnight, cryoprotected by soaking in 1XPBS / 30% sucrose at 4°C for 3 days, embedded in Tissue-Tec O.C.T. reagent and frozen on dry ice, and stored at −80°C. Brains were sectioned into 40 μm sagittal sections at −25°C with a Leica CM3050 S cryostat. Slices were mounted on Superfrost Plus slides (Fisher Scientific). COX staining was performed as described by Wong-Riley [51]. Briefly: sections were washed with 0.1 M potassium phosphate buffer pH7.4 (KPi), then incubated at 37°C in the dark with gentle rotational movement in staining solution consisting of 2.6 mM diaminobezidine (Sigma), 20 μM cytochrome c (type III, Sigma), 130 mM sucrose in KPi. Color development was stopped by washing in 1XPBS. Air dried sections were mounted with DPX (Sigma) and covered. Slides were imaged with a brightfield microscopy (Keyence BZX710) using identical settings for all samples. Control sections, treated with staining solution lacking either DAB or Cytochrome C, did not stain.
Assessment of DNA damage by long range PCR
Assessment of DNA damage by PCR was performed as described by Furda et al. [52]. Briefly, QIAGEN Genomic-tips 20/G were used to extract total DNA. DNA was quantified by PicoGreen (Quant-iT dsDNA quantification kit, Life technologies/Molecular Probes) fluorescence (excitation 485nm/emission 525nm) in a SpectraMax M3 (Molecular Devices) plate reader. Long range PCR reactions contained 1U Phusion HotStartII High Fidelity DNA polymerase (Thermo Scientific F-549), 0.2mM of each dNTP, 0.4μM of forward and reverse primer in 50μl Phusion HF Buffer. qPCR was performed on a StepOne Plus (Applied Biosystems) thermocycler, with PicoGreen fluorescence measured after each cycle to quantify dsDNA product accumulation. Linearity of the reactions within the range of samples was validated by performing a dilution series; with 27 cycles of amplification, 15ng template DNA yielded twice (2.1 ± 0.1) as much product as 7.5ng for all samples.
The relative copy numbers for mtDNA were determined with a real time qPCR delta CT experiment using a StepOne Plus (Applied Biosystems) Thermocycler. 30μl reactions were prepared with 15ng template DNA, 0.4μM of both primers mtND6-F and -R, 1x “DyNAmo SYBR Green 2step qPCR Mastermix” and 1x ROX DyNAmo Kit (Thermo Scientific). The reported results are based on CT values for 15ng template DNA. For each sample, CT values were averaged over 3 technical replicates and then deducted from the average CT value of an arbitrarily picked sample (wildtype #78, P25, region C) used as reference for the whole data set. The relative copy number for each sample was determined as 2−(CTsample − mean (CTreference)); relative copy number for the group was 2−(mean(CTgroup) − mean(CTreference)).
qPCR primers
β-globin 8.7 kbp forward primer:
TTGAGACTGTGATTGGCAATGCCT.
β-globin 8.7 kbp reverse primer:
CCTTTAATGCCCATCCCGGACT.
10 kbp mitochondrial forward primer:
GCCAGCCTGACCCATAGCCATAATAT.
10 kbp mitochondrial reverse primer:
GAGAGATTTTATGGGTGTAATGCGG.
Thermal cycler programs
Amplicon; initial step; cycles; final elongation; storage temp Nuclear (β-globin) 8.7kb; 98°C, 45s; 27 × [98°C, 8s; 72°C, 340s], 72°C,120s, 4°C Mito. 10kb; 98°C, 30s; 20 × [98°C, 7.5s; 72°C, 400s], n/a, 4°C
8-oxo-dG staining
Sagittal cryosections were obtained as described for COX stain. Antigen retrieval was performed by incubating slices for 15min in 10 mM citrate, pH 6.0, at 98°C. Slices were washed 3X 5-min in 1XPBS, then permeabilized with 0.2% Triton X-100 in PBS for 30min. Following 3X additional 5-min washes in 1XPBS, slices were blocked with 10% goat serum (GS) and incubated overnight at 4°C with anti 8-oxo-dG antibody (Abcam ab62623) diluted 1:1000 in 5% GS. The following day, slides were washed at room temperature twice with PBS/0.05% Tween-20 and twice with PBS, followed by secondary antibody incubation at room temperature for 1-hour with goat anti mouse IgG conjugated to AlexaFluor 488 (Molecular Probes A11029) diluted 1:800 in 5% GS. Slides were again washed 3X in 1X PBS and covered with glass coverslips and Fluoromount-G (Thermo Fisher) containing DAPI to counterstain nuclei. Fluorescence in the green (488/GFP) channel was assessed with identical exposure settings for all samples using a Keyence BZ-X710 microscope. Tests slices incubated without antibodies or with secondary antibody only, revealed background fluorescence varied with the anatomical structures. Genotypes were compared by manually outlining regions of interest in ImageJ [53].
Assessment of mTOR1/2 activity by Immuno-Western blotting
Brain region specimens aged P25–28 were flash frozen in liquid nitrogen upon collection and stored at −80° C. Tissues were ground into RIPA buffer (140 mM NaCl, 10 mM Tris-HCl pH 8.0, 1 mM EDTA, 1 % (w/v) Triton X100, 0.1 % (w/v) SDS, 0.1 % (w/v) Na-Deoxycholate) containing Halt Protease and Phosphatase Inhibitor Cocktail (Fisher Scientific) using a Douncehomogenizer (Kortes) until fully homogenized. Protein concentration was determined with the DC assay (BioRad). Samples were reduced with 50mM DTT before denaturing SDS-PAGE on NuPAGE 4–12% Bis-Tris Midi Protein Gels in NuPAGE MOPS SDS Running buffer (both ThermoFisher). A (BioRad) Trans-Blot Turbo was used for semidry protein transfer to nitrocellulose membrane. Membranes were blocked for >30min with 5% non-fat dry milk in TBS-0.1% Tween-20 and incubated over night at 5°C in primary antibody diluted into TBS-T 1% dry milk.
The following antibodies were used: S6 Ribosomal Protein (54D2) Mouse mAb (Cell Signaling Technology #2317, diluted 1:1000); Phospho-S6 Ribosomal Protein (Ser235/236) (D57.2.2E) XP® Rabbit mAb (Cell Signaling Techn. #4858, diluted 1:1000); Akt (pan) (C67E7) Rabbit mAb (Cell Signaling Techn. #4691, diluted 1:2000); Phospho-Akt (Ser473) (D9E) XP® Rabbit mAb (Cell Signaling Techn. #4060, diluted 1:2000). After washing with 4 changes TBS-T 5 min each, blots were incubated with the appropriate secondary antibody, either goat anti-(mouse-IgG) HRP (Santa Cruz sc2054) or goat anti-(rabbit-IgG(H+L)) HRP (Invitrogen 626520) diluted 1:10,000. SuperSignal West Pico Chemiluminescent Substrate (Thermo Fisher) was used as substrate for HRP and chemiluminescence was recorded with a ChemiDocIt2 Imager (UVA). Fluorescence of the β-Actin mouse-IgG-Alexa Fluor680 (mAbcam 8226, diluted 1:10,000) on dried blots was imaged with an Odyssey (Lycor) near-infrared scanner. Chemiluminescence and fluorescence was quantitated using ImageJ software.
Metabolomics
In these experiments, samples from postnatal days 25–28 (P25–28; n=4 for each group) were compared. Mice were treated with daily intraperitoneal injections with 8mg/kg/day rapamycin or vehicle (1.2% DMSO, 5% PEG-400, 5% Tween-20) solution starting at weaning (P21). They were sacrificed between 9 and 11:00 in the mornings by cervical dislocation and decapitated into ice cold 1X PBS. The brains were dissected into 4 regions: olfactory bulb (O), cerebellum (C), brainstem (B) and the remainder referred to as “rest” brain (R) consisting of midbrain and the entire forebrain except olfactory bulb as previously described [14]. These sections were snap frozen in liquid nitrogen and submitted to the metabolomics core of the University of Washington.
Sample preparation:
Brain samples were thawed at 4°C. Tissue samples (2–5 mg) were homogenized in 200 μL PBS, after which 800 μL of methanol containing 53.1 μM 13C-arginine and 50.1 μM 13C-glucose (Sigma-Aldrich, Saint Louis, MO) was added, and the samples were then incubated for 30 min at −70 ° C. The suspension was sonicated in an ice bath for 10 min and then centrifuged at 20,800g for 10 min at 0–4 ° C. Supernatants (600 μL) were collected and dried for 60 min using an Eppendorf Vacufuge Drier (Happauge, NY), then each reconstituted in 600 μL 40% Solution A and 60% Solution B, also containing 5.13 μM 13C-tyrosine and 22. 6 μM 13C-lactate. Solution A was 30 mM ammonium acetate in 85% H2O/ 15% acetonitrile and 0.2% acetic acid. Solution B was 15% H2O/ 85% acetonitrile and 0.2% acetic acid. Reagents for these solutions were purchased from Fisher Scientific (Pittsburgh, PA). All samples were then filtered through Millipore PVDF filters (Pittsburgh, PA) immediately prior to chromatography.
Chromatography conditions:
The LC system was composed of two Agilent 1260 binary pumps, an Agilent 1260 auto-sampler and Agilent 1290 column compartment containing a column-switching valve (Agilent Technologies, Santa Clara, CA). Each sample was injected twice, 10 μL for analysis using negative ionization mode and 2 μL for analysis using positive ionization mode. Both chromatographic separations were performed in HILIC mode on two parallel Waters XBridge BEH Amide columns (150 × 2.1 mm, 2.5 μm particle size, Waters Corporation, Milford, MA). While one column was performing the separation, the other column was reconditioned and ready for the next injection. The flow rate was 0.300 mL/min, the auto-sampler temperature was kept at 4 ° C, and the column compartment was set at 40 ° C, and total separation time for both ionization modes was 20 min. The gradient conditions for both separations were identical and consisted of 10% Solvent A from 0–2 min, a ramp to 50% during min 2–5, continued 50% Solvent A from 5–9 min, a ramp back to 10% from 9–11 min, and then 10% Solvent A from 11–20 min.
Mass spectrometry (MS) conditions:
After the chromatographic separation, MS ionization and data acquisition was performed using an AB Sciex QTrap 5500 mass spectrometer (AB Sciex, Toronto, ON, Canada) equipped with an electrospray ionization (ESI) source. The instrument was controlled by Analyst 1.5 software (AB Sciex, Toronto, ON, Canada). Targeted data acquisition was performed in multiple-reaction-monitoring (MRM) mode. We monitored 121 and 80 MRM transitions in negative and positive mode, respectively (201 transitions total). The source and collision gas was N2 (99.999% purity). The ion source conditions in negative mode were: Curtain Gas (CUR) = 25 psi, Collision Gas (CAD) = high, Ion Spray Voltage (IS) = −3.8KV, Temperature (TEM) = 500 ° C, Ion Source Gas 1 (GS1) = 50 psi and Ion Source Gas 2 (GS2) = 40 psi. The ion source conditions in positive mode were: CUR = 25 psi, CAD = high, IS = 3.8KV, TEM = 500 ° C, GS1 = 50 psi and GS2 = 40 psi.
For the data presented the supplemental figures, the samples were sent to Creative Proteomics (as flash-frozen samples) and analyzed by HPLC-MS. (https://www.creative-proteomics.com/services/gsh-and-gssg.htm).
MS data processing:
The extracted MRM peaks were integrated using MultiQuant 2.1 software (AB Sciex, Toronto, ON, Canada). Metabolites were identified by LC-QQQ MS in MRM mode targeting a total of 201 possible compounds. For quality control, samples were spiked with 13C-labeled reference metabolites and run randomized and interspersed with reference samples. The average coefficient of variance for metabolites in the QC samples was 5.5%. Results were normalized to total protein, determined by BCA assay. For each metabolite, normalized counts were averaged over the biological repeats; pairwise comparisons between genotypes or regions performed using the Student’s t-test (2 tails, unequal variance) unless otherwise specified.
Principal component analysis (PCA), hierarchical clustering, and statistics
Primary component analysis was performed using the on-line analysis tool Metaboanalyst4.0 (https://www.metaboanalyst.ca), which is maintained by the Xia Lab at McGill University. Metabolites with incomplete datasets were excluded from analysis. Data were log-transformed and range-scaled as input for PCA, with protein-content normalized metabolite values used for analysis.
Unsupervised hierarchical clustering of metabolomic data and dendrogram/heat-map generation of cluster data was performed using Morpheus, a publicly available tool provided by the Broad Institute (https://software.broadinstitute.org/morpheus). Clustering was performed using the one minus Pearson correlation metric and complete linkage.
Statistical comparisons of more than two groups were performed using the ordinary one-way ANOVA test. Pairwise t-tests, including post-hoc pairwise comparisons, between two groups were performed using the students t-test. T-test comparisons were always performed using an unpaired, two-tailed approach. Statistical significance was determined by p-value. All statistical analyses were performed using GraphPad Prism analytical and plotting software. Graphical plots of data were prepared using GraphPad Prism.
Fluorescent brain imaging
Brains in Figure 6 were fixed in 4% PFA for 48 hours, cryoprotected, sectioned at 70 μm, mounted to charged slides, stained with 5 μg/mL DAPI for 5 min, rinsed with 1XPBS 3X, mounted with aqueous mounting media Fluoromount (Thermo Fisher), and stored at 4°C. Whole-brain section composite imaging was performed on a Zeiss AxioObserver using automated tiling. Confocal images were collected on a Zeiss LSM710 confocal laser scanning microscope using 405 nm (DAPI) and 488 nm (ZsGreen) excitation lasers. Images were collected using identical parameters, but channel intensity was not quantified for these experiments, so panels in Figure 6 were adjusted to best display important features (nuclei/DAPI).
Supplementary Material
(A) Schematic of a sagittal section of murine whole brain, with key regions highlighted. (B) 8-oxo-dG immunohistochemical staining of the lesion resistant brain region, including hippocampus (Hip) and cortex (Ctx). (C) 8-oxo-dG immunohistochemical staining of cerebellum and adjacent brainstem, including the cerebellar lobes (Cb10, Cb9) and ventricular nucleus (VN). (D) 8-oxo-dG immunohistochemical staining of the caudal brainstem, including the inferior olive (IO), reticulotegmental nucleus (RtTg), and pons (Pn). (B-D) Inset boxes (left panels) are approximately 500 μm in width and shown expanded in center (yellow rectangle) and right (red rectangle) panels. (E) Quantification of 8-oxo-dG staining in resistant brain regions in control and Ndufs4(KO) mice. Number of slices evaluated: Ctx 8, Hip 8. (F) Quantification of 8-oxo-dG staining in vulnerable brainstem regions in control and Ndufs4(KO) mice. (N: IO 4, Pons 6, VB 8) (G) Quantification of 8-oxo-dG staining in vulnerable cerebellar brain regions in control and Ndufs4(KO) mice (N: 9Cb 7, 10Cb 7). (H) Higher-resolution imaging with DAPI co-staining to indicate cellular localization of 8-oxo-dG signal. Vestibular nuclei in the brainstem of a P25 Ndufs4 KO mouse immunostained with antibody specific for 8-OHG and counterstained for DNA with DAPI. Arrows point to neurons where nuclei (arrows) show low reactivity, indicating little oxidative DNA damage is present. Secondary antibody only staining displayed only very dim background (not shown).
{kind=link}
(A) Oxidized glutathione in brain regions by genotype, normalized to control resistant brain region. ANOVA ***p=0.002. (B) Ratio of reduced to oxidized glutathione (GSH/GSSG) in brain regions by genotype. ANOVA *p<0.05. (A-B) Ctl RB= control resistant brain region, KO = knockout resistant brain region, Ctl CB = control cerebellum, KO CB = knockout cerebellum, Ctl BS = control brainstem, KO BS= knockout brainstem, Ctl OB= control olfactory bulb, KO OB= knockout olfactory bulb. (C-E) Quantitative assessment of oxidized glutathione (C), reduced glutathione (D), and the ratio of reduced to oxidized glutathione (E) in whole brain samples. For all bar graphs. (F-H) Quantitative assessment of oxidized glutathione (F), reduced glutathione (G), and the ratio of reduced to oxidized glutathione (H) in liver homogenates. (I) Plasma oxidized glutathione levels in young (P30) control and Ndufs4(KO) mice. p=0.07 by pairwise t-test. (J) Plasma oxidized glutathione levels in young (P30) control and Ndufs4(KO) mice. *p=0.01 by pairwise t-test. (A-J) dots represent biological replicates (n≥4), histograms and error bars designate median values and standard error of the mean.
{kind=link}
Analyses of potential tissue intrinsic sources of increased glutamate and related metabolites from figure 6. (A) reduced and oxidized glutathione. (B) Amino acid sources of α-ketoglutarate through anaplerotic reactions. (C) Metabolites in the TCA cycle downstream of α-ketoglutarate. (D) Metabolites in the TCA cycle upstream of α-ketoglutarate. (E) Metabolites involved in ammonia metabolism. (F) The amino acid aspartate, involved in the malate-aspartate shuttle. (A-F) Dots represent biological replicates (N>4); histograms and error bars designate median values and standard error of the mean. Ctl RB= control resistant brain region, KO RB = knockout resistant brain region, Ctl CB = control cerebellum, KO CB = knockout cerebellum, Ctl BS = control brainstem, KO BS= knockout brainstem, Ctl OB= control olfactory bulb, KO OB= knockout olfactory bulb.
{kind=link}
Acknowledgments.
SCJ was supported in part by NIH grant K99GM126147/ R00GM126147. EBK, MMS and PGM were supported in part by NIH grant R01 GM105696. MK was supported in part by NIH grant R01 NS098329. EBK, MMS, SCJ, and PGM were also supported by the NW Mitochondrial Research Guild. The authors would like to thank Dr. Beatrice Predoi for her technical assistance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests. The authors declare no competing interests.
References
- 1.Schaefer AM, Taylor RW, Turnbull DM, and Chinnery PF (2004). The epidemiology of mitochondrial disorders--past, present and future. Biochim Biophys Acta 1659, 115–120. [DOI] [PubMed] [Google Scholar]
- 2.Leigh D (1951). Subacute necrotizing encephalomyelopathy in an infant. J Neurol Neurosurg Psychiatry 14, 216–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lake NJ, Compton AG, Rahman S, and Thorburn DR (2016). Leigh syndrome: One disorder, more than 75 monogenic causes. Ann Neurol 79, 190–203. [DOI] [PubMed] [Google Scholar]
- 4.Gerards M, Sallevelt SC, and Smeets HJ (2016). Leigh syndrome: Resolving the clinical and genetic heterogeneity paves the way for treatment options. Mol Genet Metab 117, 300–312. [DOI] [PubMed] [Google Scholar]
- 5.Bonfante E, Koenig MK, Adejumo RB, Perinjelil V, and Riascos RF (2016). The neuroimaging of Leigh syndrome: case series and review of the literature. Pediatr Radiol 46, 443–451. [DOI] [PubMed] [Google Scholar]
- 6.Chourasia N, Adejumo RB, Patel RP, and Koenig MK (2017). Involvement of Cerebellum in Leigh Syndrome: Case Report and Review of the Literature. Pediatr Neurol 74, 97–99. [DOI] [PubMed] [Google Scholar]
- 7.Ortigoza-Escobar JD, Oyarzabal A, Montero R, Artuch R, Jou C, Jimenez C, Gort L, Briones P, Muchart J, Lopez-Gallardo E, et al. (2016). Ndufs4 related Leigh syndrome: A case report and review of the literature. Mitochondrion 28, 73–78. [DOI] [PubMed] [Google Scholar]
- 8.Kahlhofer F, Kmita K, Wittig I, Zwicker K, and Zickermann V (2017). Accessory subunit NUYM (NDUFS4) is required for stability of the electron input module and activity of mitochondrial complex I. Biochim Biophys Acta Bioenerg 1858, 175–181. [DOI] [PubMed] [Google Scholar]
- 9.Calvaruso MA, Willems P, van den Brand M, Valsecchi F, Kruse S, Palmiter R, Smeitink J, and Nijtmans L (2012). Mitochondrial complex III stabilizes complex I in the absence of NDUFS4 to provide partial activity. Hum Mol Genet 21, 115–120. [DOI] [PubMed] [Google Scholar]
- 10.Johnson SC, Yanos ME, Kayser EB, Quintana A, Sangesland M, Castanza A, Uhde L, Hui J, Wall VZ, Gagnidze A, et al. (2013). mTOR inhibition alleviates mitochondrial disease in a mouse model of Leigh syndrome. Science 342, 1524–1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kruse SE, Watt WC, Marcinek DJ, Kapur RP, Schenkman KA, and Palmiter RD (2008). Mice with mitochondrial complex I deficiency develop a fatal encephalomyopathy. Cell Metab 7, 312–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Quintana A, Kruse SE, Kapur RP, Sanz E, and Palmiter RD (2010). Complex I deficiency due to loss of Ndufs4 in the brain results in progressive encephalopathy resembling Leigh syndrome. Proc Natl Acad Sci U S A 107, 10996–11001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Quintana A, Zanella S, Koch H, Kruse SE, Lee D, Ramirez JM, and Palmiter RD (2012). Fatal breathing dysfunction in a mouse model of Leigh syndrome. J Clin Invest 122, 2359–2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kayser EB, Sedensky MM, and Morgan PG (2016). Region-Specific Defects of Respiratory Capacities in the Ndufs4(KO) Mouse Brain. PLoS One 11, e0148219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jain IH, Zazzeron L, Goli R, Alexa K, Schatzman-Bone S, Dhillon H, Goldberger O, Peng J, Shalem O, Sanjana NE, et al. (2016). Hypoxia as a therapy for mitochondrial disease. Science 352, 54–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Johnson SC, Yanos ME, Bitto A, Castanza A, Gagnidze A, Gonzalez B, Gupta K, Hui J, Jarvie C, Johnson BM, et al. (2015). Dose-dependent effects of mTOR inhibition on weight and mitochondrial disease in mice. Front Genet 6, 247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Johnson SC, Martinez F, Bitto A, Gonzalez B, Tazaerslan C, Cohen C, Delaval L, Timsit J, Knebelmann B, Terzi F, et al. (2019). mTOR inhibitors may benefit kidney transplant recipients with mitochondrial diseases. Kidney Int 95, 455–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF, Markhard AL, and Sabatini DM (2006). Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell 22, 159–168. [DOI] [PubMed] [Google Scholar]
- 19.Kim DH, Sarbassov DD, Ali SM, King JE, Latek RR, Erdjument-Bromage H, Tempst P, and Sabatini DM (2002). mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 110, 163–175. [DOI] [PubMed] [Google Scholar]
- 20.Zheng X, Boyer L, Jin M, Kim Y, Fan W, Bardy C, Berggren T, Evans RM, Gage FH, and Hunter T (2016). Alleviation of neuronal energy deficiency by mTOR inhibition as a treatment for mitochondria-related neurodegeneration. Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Civiletto G, Dogan SA, Cerutti R, Fagiolari G, Moggio M, Lamperti C, Beninca C, Viscomi C, and Zeviani M (2018). Rapamycin rescues mitochondrial myopathy via coordinated activation of autophagy and lysosomal biogenesis. EMBO Mol Med 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reith A, and Schuler B (1972). Demonstration of cytochrome oxidase activity with diaminobenzidine. A biochemical and electron microscopic study. J Histochem Cytochem 20, 583–589. [DOI] [PubMed] [Google Scholar]
- 23.Ross JM (2011). Visualization of mitochondrial respiratory function using cytochrome c oxidase/succinate dehydrogenase (COX/SDH) double-labeling histochemistry. J Vis Exp, e3266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bolea I, Gella A, Sanz E, Prada-Dacasa P, Menardy F, Bard AM, Machuca-Marquez P, Eraso-Pichot A, Modol-Caballero G, Navarro X, et al. (2019). Defined neuronal populations drive fatal phenotype in a mouse model of Leigh syndrome. Elife 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hahn A, and Zuryn S (2019). Mitochondrial Genome (mtDNA) Mutations that Generate Reactive Oxygen Species. Antioxidants (Basel) 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Douiev L, Soiferman D, Alban C, and Saada A (2016). The Effects of Ascorbate, NAcetylcysteine, and Resveratrol on Fibroblasts from Patients with Mitochondrial Disorders. J Clin Med 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Johnson SC (2014). Translational Medicine. A target for pharmacological intervention in an untreatable human disease. Science 346, 1192. [DOI] [PubMed] [Google Scholar]
- 28.Martin-Perez M, Ito TK, Grillo AS, Valente AS, Han J, Entwisle S, Huang HZ, Kim D, Yajima M, Kaeberlein M, Villen J (2019). Protein kinase C is a key target for attenuation of Leigh syndrome by rapamycin. bioRxiv. [Google Scholar]
- 29.Ito TK, Lu C, Khan J, Nguyen Q, Huang HZ, Kim D, Phillips J, Tan J, Lee Y, Nguyen T, et al. (2017). Hepatic S6K1 Partially Regulates Lifespan of Mice with Mitochondrial Complex I Deficiency. Front Genet 8, 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gundersen RY, Vaagenes P, Breivik T, Fonnum F, and Opstad PK (2005). Glycine--an important neurotransmitter and cytoprotective agent. Acta Anaesthesiol Scand 49, 1108–1116. [DOI] [PubMed] [Google Scholar]
- 31.Purwaha P, Silva LP, Hawke DH, Weinstein JN, and Lorenzi PL (2014). An artifact in LC-MS/MS measurement of glutamine and glutamic acid: in-source cyclization to pyroglutamic acid. Anal Chem 86, 5633–5637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nagana Gowda GA, Gowda YN, and Raftery D (2015). Massive glutamine cyclization to pyroglutamic acid in human serum discovered using NMR spectroscopy. Anal Chem 87, 3800–3805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zimin PI, Woods CB, Quintana A, Ramirez JM, Morgan PG, and Sedensky MM (2016). Glutamatergic Neurotransmission Links Sensitivity to Volatile Anesthetics with Mitochondrial Function. Curr Biol 26, 2194–2201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee CF, Caudal A, Abell L, Nagana Gowda GA, and Tian R (2019). Targeting NAD(+) Metabolism as Interventions for Mitochondrial Disease. Sci Rep 9, 3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cowan K, Anichtchik O, and Luo S (2019). Mitochondrial integrity in neurodegeneration. CNS Neurosci Ther. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.To TL, Cuadros AM, Shah H, Hung WHW, Li Y, Kim SH, Rubin DHF, Boe RH, Rath S, Eaton JK, et al. (2019). A Compendium of Genetic Modifiers of Mitochondrial Dysfunction Reveals Intra-organelle Buffering. Cell 179, 1222–1238 e1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schleit J, Johnson SC, Bennett CF, Simko M, Trongtham N, Castanza A, Hsieh EJ, Moller RM, Wasko BM, Delaney JR, et al. (2013). Molecular mechanisms underlying genotype-dependent responses to dietary restriction. Aging Cell 12, 1050–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yu AK, Datta S, McMackin MZ, and Cortopassi GA (2017). Rescue of cell death and inflammation of a mouse model of complex 1-mediated vision loss by repurposed drug molecules. Hum Mol Genet 26, 4929–4936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Felici R, Buonvicino D, Muzzi M, Cavone L, Guasti D, Lapucci A, Pratesi S, De Cesaris F, Luceri F, and Chiarugi A (2017). Post onset, oral rapamycin treatment delays development of mitochondrial encephalopathy only at supramaximal doses. Neuropharmacology 117, 74–84. [DOI] [PubMed] [Google Scholar]
- 40.Siegmund SE, Yang H, Sharma R, Javors M, Skinner O, Mootha V, Hirano M, and Schon EA (2017). Low-dose rapamycin extends lifespan in a mouse model of mtDNA depletion syndrome. Hum Mol Genet 26, 4588–4605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Khan NA, Nikkanen J, Yatsuga S, Jackson C, Wang L, Pradhan S, Kivela R, Pessia A, Velagapudi V, and Suomalainen A (2017). mTORC1 Regulates Mitochondrial Integrated Stress Response and Mitochondrial Myopathy Progression. Cell Metab 26, 419–428 e415. [DOI] [PubMed] [Google Scholar]
- 42.Wang A, Mouser J, Pitt J, Promislow D, and Kaeberlein M (2016). Rapamycin enhances survival in a Drosophila model of mitochondrial disease. Oncotarget 7, 80131–80139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fan F, Sam R, Ryan E, Alvarado K, and Villa-Cuesta E (2019). Rapamycin as a potential treatment for succinate dehydrogenase deficiency. Heliyon 5, e01217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim SH, Scott SA, Bennett MJ, Carson RP, Fessel J, Brown HA, and Ess KC (2013). Multi-organ abnormalities and mTORC1 activation in zebrafish model of multiple acyl-CoA dehydrogenase deficiency. PLoS Genet 9, e1003563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Peng M, Ostrovsky J, Kwon YJ, Polyak E, Licata J, Tsukikawa M, Marty E, Thomas J, Felix CA, Xiao R, et al. (2015). Inhibiting cytosolic translation and autophagy improves health in mitochondrial disease. Hum Mol Genet 24, 4829–4847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sage-Schwaede A, Engelstad K, Salazar R, Curcio A, Khandji A, Garvin JH Jr., and De Vivo DC (2019). Exploring mTOR inhibition as treatment for mitochondrial disease. Ann Clin Transl Neurol 6, 1877–1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Takahashi Y, Kioka H, Shintani Y, Ohki A, Takashima S, Sakata Y, Higuchi T, and Saito S (2019). Detection of increased intracerebral lactate in a mouse model of Leigh syndrome using proton MR spectroscopy. Magn Reson Imaging 58, 38–43. [DOI] [PubMed] [Google Scholar]
- 48.Iuso A, Scacco S, Piccoli C, Bellomo F, Petruzzella V, Trentadue R, Minuto M, Ripoli M, Capitanio N, Zeviani M, et al. (2006). Dysfunctions of cellular oxidative metabolism in patients with mutations in the NDUFS1 and NDUFS4 genes of complex I. J Biol Chem 281, 10374–10380. [DOI] [PubMed] [Google Scholar]
- 49.Sultani G, Samsudeen AF, Osborne B, and Turner N (2017). NAD(+) : A key metabolic regulator with great therapeutic potential. J Neuroendocrinol 29. [DOI] [PubMed] [Google Scholar]
- 50.Yang Y, and Sauve AA (2016). NAD(+) metabolism: Bioenergetics, signaling and manipulation for therapy. Biochim Biophys Acta 1864, 1787–1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wong-Riley M (1979). Changes in the visual system of monocularly sutured or enucleated cats demonstrable with cytochrome oxidase histochemistry. Brain Res 171, 11–28. [DOI] [PubMed] [Google Scholar]
- 52.Furda AM, Bess AS, Meyer JN, and Van Houten B (2012). Analysis of DNA damage and repair in nuclear and mitochondrial DNA of animal cells using quantitative PCR. Methods Mol Biol 920, 111–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rueden CT, Schindelin J, Hiner MC, DeZonia BE, Walter AE, Arena ET, and Eliceiri KW (2017). ImageJ2: ImageJ for the next generation of scientific image data. BMC Bioinformatics 18, 529. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Schematic of a sagittal section of murine whole brain, with key regions highlighted. (B) 8-oxo-dG immunohistochemical staining of the lesion resistant brain region, including hippocampus (Hip) and cortex (Ctx). (C) 8-oxo-dG immunohistochemical staining of cerebellum and adjacent brainstem, including the cerebellar lobes (Cb10, Cb9) and ventricular nucleus (VN). (D) 8-oxo-dG immunohistochemical staining of the caudal brainstem, including the inferior olive (IO), reticulotegmental nucleus (RtTg), and pons (Pn). (B-D) Inset boxes (left panels) are approximately 500 μm in width and shown expanded in center (yellow rectangle) and right (red rectangle) panels. (E) Quantification of 8-oxo-dG staining in resistant brain regions in control and Ndufs4(KO) mice. Number of slices evaluated: Ctx 8, Hip 8. (F) Quantification of 8-oxo-dG staining in vulnerable brainstem regions in control and Ndufs4(KO) mice. (N: IO 4, Pons 6, VB 8) (G) Quantification of 8-oxo-dG staining in vulnerable cerebellar brain regions in control and Ndufs4(KO) mice (N: 9Cb 7, 10Cb 7). (H) Higher-resolution imaging with DAPI co-staining to indicate cellular localization of 8-oxo-dG signal. Vestibular nuclei in the brainstem of a P25 Ndufs4 KO mouse immunostained with antibody specific for 8-OHG and counterstained for DNA with DAPI. Arrows point to neurons where nuclei (arrows) show low reactivity, indicating little oxidative DNA damage is present. Secondary antibody only staining displayed only very dim background (not shown).
(A) Oxidized glutathione in brain regions by genotype, normalized to control resistant brain region. ANOVA ***p=0.002. (B) Ratio of reduced to oxidized glutathione (GSH/GSSG) in brain regions by genotype. ANOVA *p<0.05. (A-B) Ctl RB= control resistant brain region, KO = knockout resistant brain region, Ctl CB = control cerebellum, KO CB = knockout cerebellum, Ctl BS = control brainstem, KO BS= knockout brainstem, Ctl OB= control olfactory bulb, KO OB= knockout olfactory bulb. (C-E) Quantitative assessment of oxidized glutathione (C), reduced glutathione (D), and the ratio of reduced to oxidized glutathione (E) in whole brain samples. For all bar graphs. (F-H) Quantitative assessment of oxidized glutathione (F), reduced glutathione (G), and the ratio of reduced to oxidized glutathione (H) in liver homogenates. (I) Plasma oxidized glutathione levels in young (P30) control and Ndufs4(KO) mice. p=0.07 by pairwise t-test. (J) Plasma oxidized glutathione levels in young (P30) control and Ndufs4(KO) mice. *p=0.01 by pairwise t-test. (A-J) dots represent biological replicates (n≥4), histograms and error bars designate median values and standard error of the mean.
Analyses of potential tissue intrinsic sources of increased glutamate and related metabolites from figure 6. (A) reduced and oxidized glutathione. (B) Amino acid sources of α-ketoglutarate through anaplerotic reactions. (C) Metabolites in the TCA cycle downstream of α-ketoglutarate. (D) Metabolites in the TCA cycle upstream of α-ketoglutarate. (E) Metabolites involved in ammonia metabolism. (F) The amino acid aspartate, involved in the malate-aspartate shuttle. (A-F) Dots represent biological replicates (N>4); histograms and error bars designate median values and standard error of the mean. Ctl RB= control resistant brain region, KO RB = knockout resistant brain region, Ctl CB = control cerebellum, KO CB = knockout cerebellum, Ctl BS = control brainstem, KO BS= knockout brainstem, Ctl OB= control olfactory bulb, KO OB= knockout olfactory bulb.