

Abstract
Citrus species are some of the most valuable and widely consumed fruits globally. The genome sequences of representative citrus (e.g., Citrus clementina, C. sinensis, C. grandis) species have been released but the research base for mandarin molecular breeding is still poor. We assembled the genomes of Citrus unshiu and Poncirus trifoliata, two important species for citrus industry in Japan, using hybrid de novo assembly of Illumina and PacBio sequence data, and developed the Mikan Genome Database (MiGD). The assembled genome sizes of C. unshiu and P. trifoliata are 346 and 292 Mb, respectively, similar to those of citrus species in public databases; they are predicted to possess 41,489 and 34,333 protein-coding genes in their draft genome sequences, with 9,642 and 8,377 specific genes when compared to C. clementina, respectively. MiGD is an integrated database of genome annotation, genetic diversity, and Cleaved Amplified Polymorphic Sequence (CAPS) marker information, with these contents being mutually linked by genes. MiGD facilitates access to genome sequences of interest from previously reported linkage maps through CAPS markers and obtains polymorphism information through the multiple genome browser TASUKE. The genomic resources in MiGD (https://mikan.dna.affrc.go.jp) could provide valuable information for mandarin molecular breeding in Japan.
Keywords: Citrus unshiu, Poncirus trifoliata, hybrid genome assembly, Illumina sequencing-by-synthesis technology, PacBio single-molecule sequencing technology, genome annotation database, CAPS marker
Introduction
Citrus species are some of the most valuable fruits globally and are widely cultivated in all suitable subtropical and tropical climates, comprising various species such as sweet oranges, mandarins, grapefruits, limes, lemons, and so on. More than 25 species of citrus are cultivated in Japan and the satsuma mandarin (Citrus unshiu Marc.) has been predominantly cultivated for over 100 years in Japan (Hodgson 1967). Numerous promising cultivars such as the ‘Kiyomi’, ‘Shiranuhi’, ‘Harumi’, ‘Setoka’, ‘Kanpei’ and so on have been released through the conventional breeding programs of public research organizations. These new cultivars immensely benefit the citrus industry and their cultivation area has been growing; however, it cannot match that of the satsuma mandarin. Satsuma mandarin offers many superior characteristics such as seedless-ness, easy peeling ability, early maturing, disease resistance, and high and stable productivity, which facilitates its cultivation and consumption. Most citrus trees are grafted on trifoliate orange (Poncirus trifoliata (L.) Raf.) rootstock in the orchard. The trifoliate orange is closely related to the genus Citrus although its flowering habit is deciduous against the evergreen habit of general citrus species. It is quite suitable for satsuma mandarin and other citrus trees, and the grafted citrus trees generally form a compact canopy with high productivity and high fruit quality (Kawase et al. 1987). In addition, trifoliate orange is a cold-hardy citrus and is resistant to phytophthora root and collar rot caused by Phytophthora citrophthora, citrus tristeza virus (CTV) and citrus nematode (Tylenchulus semipenetrans), which cause severe damage to citrus trees. The grafted citrus trees can tolerate these biotic and abiotic stresses. Thus, these two citrus and relative cultivars are important resources for sustainability and expansion in the citrus industry in Japan, as well as for elucidating the genetic composition and molecular mechanisms of agronomically important traits and the isolation of related genes. Recently, the Japanese citrus industry has faced various obstacles such as lack of labor due to high-aging of industrial carriers, declining domestic demand, and an increase in low-priced imported fruits due to the progress of global economization. The development of new citrus cultivars with a top level of fruit quality in the world have contributed to protect the Japanese citrus industry; therefore, sustainable development of a new cultivar is indispensable to maintain the present international advantage of the Japanese brand. Owing to the long period required for conventional cross breeding, introduction of genome information assisted molecular breeding technology is required to efficiently generate a new superior cultivar.
Recently, the second-generation sequencing technology (e.g., Illumina HiSeq system) has drastically accelerated the whole genome sequencing process of all living organisms. Furthermore, long read information generated from the third-generation sequencing technology (e.g., PacBio RS II and Oxford Nanopore sequencers) promises to significantly improve the quality of genome assembly. However, despite decreasing the turnaround time, the costs of third-generation sequencing are at least an order of magnitude more expensive than those of Illumina sequencing. Several hybrid de novo assembly methods using both short (Illumina) and long (PacBio/Nanopore) read information, such as PacBioToCA, SPAdes, and DBG2OLC, have been reported using the advantageous points of both second- and third-generation sequencing technologies (Antipov et al. 2016, Koren et al. 2012, Ye et al. 2016). The de novo assembly of highly heterozygous genomes is still a complex and challenging task. Recently, several genome assemblers and analysis pipelines, such as Platanus and Redundans, that are specifically designed for the assembly of highly heterozygous genomes have been developed (Kajitani et al. 2014, Pryszcz and Gabaldón 2016). The International Citrus Genome Consortium (ICGC, composed of researchers from Australia, Brazil, China, France, Israel, Italy, Japan, Spain, and USA) was established in 2003 to sequence the genomes of sweet orange (C. sinensis L.) and clementine mandarin (C. clementina Hort ex Tan). The genome sequences of sweet orange (diploid) and mandarin (haploid) have been determined (Wu et al. 2014), and their draft sequences are now available in Phytozome (https://phytozome.jgi.doe.gov). General citrus cultivars are diploids with nine pairs of chromosomes and genome size varies among citrus species; the genomes of mandarin (C. reticulata Blanco) and sweet orange are approximately estimated to be 360 Mb and 367 Mb, respectively (Arumuganathan and Earle 1991, Ollitrault et al. 1994). Therefore, the assembled sequences of clementine mandarin (301.4 Mb in JGI ver. 1.0) and sweet orange (319.2 Mb in JGI ver. 1.0) cover 83.7% and 87.0% of this estimated genome size, respectively. In addition, various citrus genomes, such as that of Ponkan mandarin (C. reticulata Blanco) and Chandler pummelo (C. grandis (L.) Osbeck), have been sequenced and compared to understand the complex citrus phylogeny and sequence-directed genetic improvement (Wu et al. 2014). In a recent study, a high-quality haploid pumelo genome was assembled using single-molecule sequences generated by the PacBio RS II platform, and the draft genomes of three heterozygous Citrinae species were assembled using Illumina reads (Wang et al. 2017). In addition, the draft genome of satsuma mandarin was assembled to understand the structural features of this major Japanese mandarin species (Shimizu et al. 2017). These advances in genome research have extended to molecular breeding and the isolation of agronomically important genes, resulting in many worldwide reviews and publications on citrus genomics, genetics, and breeding (Gmitter et al. 2007, 2012, Khan 2007, Talon and Gmitter 2008). On the contrary, information integration between the various genetic linkage maps reported previously and these assembled genome sequences is an upcoming task and is desired to access the genomic regions responsible for agronomically important traits through linkage and phylogenetic DNA markers.
Herein, to enforce the progress of mandarin molecular breeding in Japan, we developed an integrated genome database named as Mikan Genome Database (MiGD) (https://mikan.dna.affrc.go.jp), which comprises the genome annotation database of C. unshiu and P. trifoliata, a genome diversity database among nine citrus species, and a Cleaved Amplified Polymorphic Sequence (CAPS) marker database. MiGD could facilitate connection of interesting genetic loci on the genetic linkage map to the genome sequences of C. clementina and C. unshiu through the CAPS marker information. The newly assembled genome sequence of P. trifoliata and enrichment of the C. unshiu genome sequences by re-sequencing are valuable genetic resources to explore the genes responsible for agriculturally important traits underlying the two major cultivated species, satsuma mandarin and trifoliate orange, in Japan.
Materials and Methods
Plant material and genome sequencing of C. unshiu and P. trifoliata
“Miyagawa wase”, one of the major cultivated satsuma mandarin cultivars (NIAS Genebank registration number: 117351 (https://www.gene.affrc.go.jp/databases-plant_search_detail.php?jp=117351)) and trifoliate orange (NIAS Genebank registration number: 113401 (https://www.gene.affrc.go.jp/databases-plant_search_detail.php?jp=113401)), grown at Okitsu Citrus Research Station of NIFTS in Japan were used as the genetic sources for genome sequencing of C. unshiu and P. trifoliata. Genomic DNA was extracted from fresh and fully expanded leaves of these cultivars, according to the method described by Dellaporta et al. (1983). For C. unshiu, two Illumina paired-end (PE) libraries with average insert sizes of 350 and 550 bp were constructed using the TruSeq DNA PCR-Free Library Preparation Kit. Each library was sequenced using a one-lane of a flow cell in the Illumina HiSeq 2000 system. The read lengths were 101 and 151 bp for each library, respectively. For PacBio RSII sequencing, the DNA sequencing library constructed by the standard protocol was sequenced using P6C4 chemistry and eight SMART cells on the PacBio RS II system. For P. trifoliata, one Illumina paired-end and three mate-pair (MP) libraries with average insert sizes of 3, 5, and 8 kb were constructed using the standard protocol. All the libraries were sequenced using the Illumina HiSeq 2000 system with a read length of 101 bp. PacBio RS II sequencing was performed in the same manner as for Satsuma mandarin. All the sequence data are available in DRA/ERA/SRA (DRA008432).
Hybrid de novo genome assembly
Low-quality bases and Illumina sequencing adapters were trimmed using Trimmomatic v.0.36 (ILLUMINACLIP:TruSeq3-PE-2.fa:2:30:10 LEADING:20 TRAILING:20 SLIDINGWINDOW:10:15 MINLEN:50) and NxTrim (--minlength 50) for Illumina PE and MP reads, respectively (Bolger et al. 2014, O’Connell et al. 2015). The Illumina PE reads were assembled using Platanus v1.2.4 with the following parameters: -k 61 -u 1.0 for satsuma mandarin and -k 41 -u 1.0 for trifoliate orange (Kajitani et al. 2014). High quality consensus sequences were constructed with the contigs assembled with Platanus and PacBio subreads using the hybrid de novo genome assembler DBG2OLC of November 1, 2016 (Ye et al. 2016). Alternative heterozygous contigs were removed using the Redundans pipeline (Pryszcz and Gabaldón 2016). The PacBio subreads were used for scaffolding with the SSPACE-LongRead package and two rounds of gap-closing with PBJelly implemented in PBSuite v15.8.24 (Boetzer and Pirovano 2014, English et al. 2012). For P. trifoliata, an additional scaffolding process with Illumina MP reads using the SSPACE-STANDARD package v.3.0 was performed before gap-closing (Boetzer et al. 2011). Finally, all Illumina PE reads were aligned to the scaffolds using BWA-MEM v.0.7.15, sequencing errors (single nucleotide polymorphisms (SNPs) and indels) were detected and filtered using the HaplotypeCaller in GATK v.3.7, and errors were corrected using in-house scripts (DePristo et al. 2011, Li and Durbin 2009). The genome sizes of the two species were estimated using MaSuRCA v3.2.2 (Zimin et al. 2013). Quality assessment and calculation of statistics for the genome assembly was performed using in-house scripts and QUAST v4.2 with the C. clementina genome and gene annotation (v1.0) downloaded from Phytozome (https://phytozome.jgi.doe.gov) as a reference (Gurevich et al. 2013). Sequence comparisons between the published C. unshiu draft genome and assembled scaffolds in this study were performed using MUMmer (v4.0.0) with the following parameters: the default parameters for nucmer, “-r -q -i 98.0 -l 1000” for delta-filter and “-c -r” for show-coords. Dot plot of the draft genome and assembled scaffolds were drawn using the R script “dotPlotly (https://github.com/tpoorten/dotPlotly)”. LTR_retriever v2.5 was used to calculate LTR Assembly Index (LAI) (Ou et al. 2018).
Genome annotation
Genome annotation on the assembled C. unshiu and P. trifoliata genome sequences was conducted with a web-based annotation system MEGANTE (Numa and Itoh 2014). C. sinensis was selected as the parameter for query sequences since the amount of available EST data for C. sinensis is the largest among citrus species in MEGANTE. For annotation, full-length cDNAs (FLcDNAs) and ESTs of C. sinensis obtained from INSDC (updated on Apr 2016) and UniProtKB (plant division of Swiss-Prot and TrEMBL in release 2016_03) were used as references for the transcript and protein sequence alignment. InterPro v56.0 was used for the functional domain search. To simply compare the gene annotation among three citrus species (C. clementina, C. unshiu and P. trifoliata), we also carried out re-annotation of the C. clementina genome (only for scaffold 1-9 of JGI v1.0 genome) using MEGANTE.
Gene conservation and orthologous relationships among the three citrus species were examined by an all-against-all blastp (NCBI Blast v2.6.0+) search of protein sequences and clustering using OrthoFinder v1.1.4 with the default parameters (Camacho et al. 2009, Emms and Kelly 2015). GO and InterPro enrichment analyses were performed using Fisher’s exact tests in combination with False discovery rate (FDR) correction.
Mapping of CAPS markers on the C. unshiu genome assembly
The molecular DNA marker information (e.g., primer sequences, STS, positions on C. clementina scaffolds, and AGI genetic linkage map) applicable for C. unshiu was collected from a previous study (Shimada et al. 2014). AGI genetic map was constructed as a standard genetic map to progress molecular breeding of mandarin in Japan. The positions of DNA markers on the C. unshiu draft genome were determined using MFEprimer v2.0 (Qu et al. 2012). The best marker positions were selected using the following criteria: 1) a primer pair coverage (PPC) score more than 30, 2) Tm values of both forward and reverse primers more than 35°C, and 3) expected product size in the range of 0.5–1.5-fold of the experimentally validated product size.
Genetic diversity among C. unshiu, P. trifoliata, and other citrus species
The aforementioned Illumina PE data of C. unshiu and P. trifoliata, and the recently published genome resequencing data of the representative citrus species: C. reticulata (SRR3747540), C. sinensis (SRR4240447), C. grandis (SRR4294213, SRR4294216), C. ichangensis (SRR4007116, SRR4006763, SRR4006743, SRR4006657), C. medica (SRR4010249, SRR4009988), and A. buxifolia (SRR4254787, SRR4254698) were downloaded from DRA/ERA/SRA (Wang et al. 2017). After trimming the low-quality bases and sequencing adapters using Trimmomatic, the reads were aligned to the C. clementina reference genome (v1.0) using BWA-MEM v.0.7.15. The SNVs were detected using the GATK HaplotypeCaller command according to the best practice protocols for SNP and indel discovery in whole genome sequences from the GATK website (https://software.broadinstitute.org/gatk/best-practices/). For sliding window analysis of genome-wide nucleotide diversity between C. clementina and other citrus genomes, the rates of sequence variations were calculated for each 200 kb window with 100 kb sliding.
Development of CAPS markers
During construction of the mandarin genetic standard map with 708 gene-based markers (Shimada et al. 2014), more than 4,000 CAPS markers were designed with reference to the expression sequencing tags (ESTs) of various cDNA libraries. Their primers were designed using Oligo ver. 5.0 (National Bioscience, Inc. Plymouth, MN, USA). Most of them were eliminated in the screening due to no polymorphism between parent lines (A255 and G434) and for technical reasons such as no amplification, unstable PCR fragment pattern, and so on. These CAPS markers without technical problems are valuable to advance mandarin molecular breeding in Japan; however, their information has been unpublished so far. A total of 2,696 CAPS markers were thus selected and deposited in the CAPS marker database.
Construction of MiGD
Draft genome sequences and gene annotation of C. unshiu and P. trifoliata were obtained from JBrowse v1.12.1 (Buels et al. 2016). BLAST DB of nucleotide transcript and protein sequences were constructed using ncbi-blast-2.6.0+. The BLAST search function against the genome and transcriptome sequences of C. unshiu and P. trifoliata was implemented in SequenceServer v1.0.9 (https://www.sequenceserver.com). All genome sequences and the genomic feature information (e.g., genes, repeats, and DNA markers) are available on the download page (https://mikan.dna.affrc.go.jp/data_download/index.html). To demonstrate the genome-wide variations between C. clementina and other citrus species, the Multiple Genome Browser: TASUKE version 1.5.3 (Kumagai et al. 2013) was installed in the MiGD server. The MiGD website is implemented on a Linux server with a CentOS and Apache web server. All CAPS marker data are stored in the MariaDB database. The user-interface and functions of the CAPS marker database were developed using PHP and JavaScript.
Results
Genome sequencing and hybrid de novo assembly of C. unshiu and P. trifoliata
We sequenced the genomes of C. unshiu (satsuma mandarin) and P. trifoliata (trifoliate orange), which are agronomically important species in Japan. For C. unshiu, 97 Gb of Illumina paired-end reads (288x coverage) and 8 Gb of PacBio RS II subreads (23x coverage) were obtained (Table 1). For P. trifoliata, 34 Gb of Illumina paired-end reads (113x coverage) and 10 Gb of PacBio RS II subreads (34x coverage) were obtained. Furthermore, Illumina mate-pair reads were used for scaffolding. Using k-mer analysis with the Illumina PE reads, the genome sizes of two species were estimated to be 336 Mb and 296 Mb for satsuma mandarin and trifoliate orange, respectively. K-mer spectra also showed the high heterozygosity of two genomes (Supplemental Fig. 1). In our assembly strategy, Illumina PE reads were assembled using Platanus followed by the hybrid de novo genome assembly of Platanus contig sequences and PacBio subreads using DBG2OLC (Fig. 1) (Kajitani et al. 2014, Ye et al. 2016). To remove the alternative heterozygous contigs, we used the Redundans pipeline (Pryszcz and Gabaldón 2016). At this step, we obtained a 336.2 Mb C. unshiu genome assembly containing 5,269 contigs (N50 = 114,572) and a 289.4 Mb P. trifoliata genome assembly containing 3,328 contigs (N50 = 176,694) without any unambiguous bases (Ns) in the assemblies. Assembled contigs were scaffolded with PacBio subreads by SSPACE-LongRead. In addition, for P. trifoliata, Illumina mate-pair (MP) reads were also used for further scaffolding processes. The obtained scaffolds would still have sequencing errors derived from the high sequencing error rate in the PacBio subreads. We corrected the sequencing errors (both SNPs and indels) detected by the alignment of Illumina PE reads and the GATK pipeline. Finally, the total lengths of the assembled scaffolds of C. unshiu and P. trifoliata were 346.4 Mb and 291.9 Mb, which comprised 3,151 and 1,313 scaffolds for C. unshiu and P. trifoliata, respectively (Table 2). The scaffold N50 of the final assemblies were 206 kb and 424 kb for C. unshiu and P. trifoliata, respectively. The 2-fold higher sequence contiguity for the P. trifoliata assembly might largely be due to the additional Illumina Mate-pair sequences. The sequence contiguities of the two assemblies were lower than those of the recently published high-quality reference genome of the haploid pummelo C. grandis (N50 = 4.2 Mb), but were comparable to the draft genomes of primitive citrus A. buxifolia (N50 = 1,074 kb), C. ichangensis (N50 = 504 kb) and citron C. medica (N50 = 367 kb) (Wang et al. 2017). The sizes of the assembled genomes were very close to the length estimated by k-mer analysis. The assembled genome sizes of C. clementina (301 Mb), C. sinensis (321 Mb), C. grandis (345 Mb), C. ichangensis (335 Mb), C. medica (368 Mb), and C. unshiu (346 Mb) were relatively larger than those of A. buxifolia (288 Mb) and P. trifoliata (292 Mb). The GC content values were similar among the citrus species and are 34.0 and 33.8 for C. unshiu and P. trifoliata, respectively (Table 2). The quality of the C. unshiu assembly was evaluated using the recently proposed measurement, the LTR assembly index (LAI), estimates the ratio of intact LTR retrotransposons in a genome assembly (Ou et al. 2018). The LAI of the C. unshiu assembly was estimated to be 11.93 (reference quality) and higher than 8.18 (draft quality) of the published C. unshiu draft genome (Shimizu et al. 2017).
Table 1.
Sequence data used for hybrid de novo genome assembly
| Species | Type | # of reads | # of bases | Coverage (x)a |
|---|---|---|---|---|
| C. unshiu | Illumina, PE101 | 532,000,280 | 54,540,028,280 | 162 |
| Illumina, PE151 | 279,626,232 | 42,223,561,032 | 126 | |
| PacBio, P6C4 | 1,099,957 | 7,705,209,842 | 23 | |
| P. trifoliata | Illumina, PE101 | 332,587,118 | 33,591,298,918 | 113 |
| Illumina, MP3k | 97,164,256 | 9,813,589,856 | 33 | |
| Illumina, MP5k | 96,069,378 | 9,703,007,178 | 33 | |
| Illumina, MP8k | 131,459,878 | 13,277,447,678 | 45 | |
| PacBio, P6C4 | 900,552 | 10,097,873,768 | 34 |
a Estimated genome sizes of 336 Mb for C. unshiu and 296 Mb for P. trifoliata were used for the calculation.
Fig. 1.
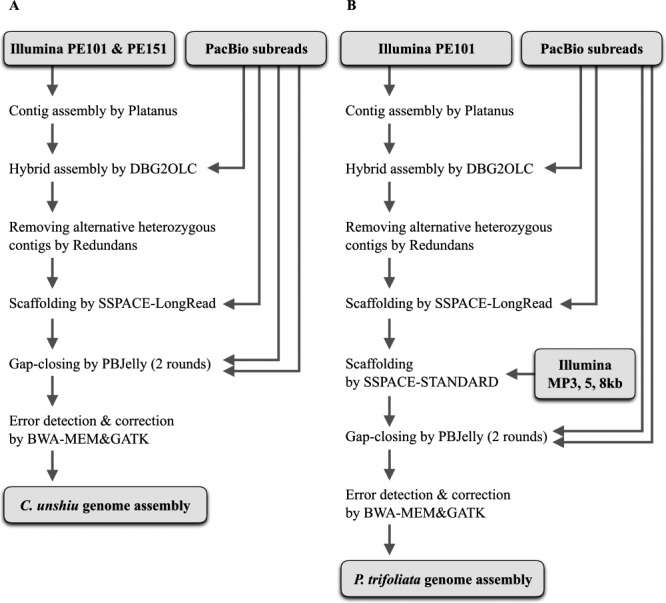
The workflow for hybrid de novo assembly. The C. unshiu (A) and P. trifoliata (B) genome was assembled using Illumina paired-end reads and PacBio subreads using the hybrid de novo genome assembly strategy. Furthermore, for P. trifoliata, Illumina mate-pair reads were used for scaffolding of the contigs.
Table 2.
Statistics of genome assemblies and annotations
|
C. clementina
(JGI v1.0) |
C. sinensis
(HZAU v2) |
C. unshiu
(Shimizu et al. 2017) |
C. unshiu
(MiGD) |
P. trifoliata
(MiGD) |
|
|---|---|---|---|---|---|
| Number of scaffolds | 1,398 | 10 | 20,470b | 3,151 | 1,313 |
| Total scaffold length (bp) | 301,386,998 | 327,944,670 | 359,692,661 | 346,435,163 | 291,927,159 |
| Mean scaffold length (bp) | 215,584 | 32,794,467 | 17,571.7 | 109,944.5 | 222,336 |
| Largest scaffolds (bp) | 51,050,279 | 88,947,451 | 27,672,184 | 2,674,519 | 2,044,385 |
| N50 | 31,410,901 | 30,837,053 | 14,331,940 | 206,057 | 424,026 |
| Number of Ns (%) | 6,218,033 (2.1) | 26,794,082 (8.2) | 28,235,341 (7.9) | 2,333,253 (0.7) | 1,743,151 (0.6) |
| %GC | 35.0 | 34.6 | 33.9 | 34.0 | 33.8 |
| LTR assembly index (LAI) | 18.61 | 1.99 | 8.18 | 11.93 | 11.65 |
| Repeat bases, Mb (%) | 134.5 (44.6) | 128.0 (39.0) | 145.3 (40.4) | 149.7 (43.2) | 116.8 (40.0) |
| - SINEs, Kb (%) | 122.3 (0.04) | 120.6 (0.04) | 128.8 (0.04) | 154.1 (0.04) | 134.0 (0.05) |
| - LINEs, Kb (%) | 5,278.1 (1.75) | 5,661.3 (1.73) | 6,015.3 (1.67) | 6,481.0 (1.87) | 4,946.5 (1.69) |
| - LTR elements, Kb (%) | 61,011.0 (20.24) | 57,240.4 (17.45) | 66,211.2 (18.41) | 68,770.9 (19.85) | 52,632.3 (18.03) |
| - DNA elements, Kb (%) | 14,867.3 (4.93) | 15,470.9 (4.72) | 17,115.4 (4.76) | 18,097.9 (5.22) | 13,179.5 (4.51) |
| - Unclassified repeats, Kb (%) | 52,053.7 (17.27) | 48,327.7 (14.74) | 54,497.3 (15.15) | 54,737.5 (15.80) | 44,786.4 (15.34) |
| Number of genes | 24,533 (36,163)a | 29,655 | 29,024 | 41,489 | 34,333 |
| Number of transcripts | 33,929 (37,570)a | 44,275 | 37,970 | 42,913 | 35,291 |
a Number of genes and transcripts in the parentheses are based on the re-annotation data using MEGANTE.
b Satsuma pseudomolecule sequences (9 chromosomes and 20,461 unanchored scaffolds) was used.
The accuracy of the C. unshiu assembly was evaluated by comparing the positions of publicly available DNA markers on the C. unshiu assembly and the integrated genetic linkage map (AGI) (Supplemental Table 1). In all, 403 markers could be uniquely placed on 266 C. unshiu scaffolds and 75 of those scaffolds had two or more markers. For each scaffold, we examined whether the DNA markers on a certain C. unshiu scaffold were assigned to a single linkage group or a C. clementina chromosome. As a result, only 8 C. unshiu scaffolds had inconsistent orders of the markers with both the C. clementina genome and AGI map, suggesting that the C. unshiu scaffolds was almost properly assembled overall. However, the number of currently available DNA markers was not sufficient for anchoring and sorting the scaffolds into pseudomolecules. Further development of marker information and additional long-read sequences are thus needed for refining our genome assemblies. By mapping of the scaffold sequences against the published C. unshiu draft genome (Shimizu et al. 2017), 62.0% (1,954/3,151) of the scaffolds can be anchored on the chromosome 1-9 (Supplemental Fig. 2, Supplemental Table 2).
Genome annotation of C. unshiu and P. trifoliata
Annotation of the assembled genome was carried out using the web-based annotation pipeline MEGANTE. The result indicted that 41,489 and 34,333 protein-coding genes with 42,913 and 35,291 transcripts were predicted for C. unshiu and P. trifoliata, respectively. The transcripts had an average length of 1,130.6 and 1,135.6, and the mean amino acid sequence sizes of 341.3 and 347.2 aa for C. unshiu and P. trifoliata, respectively. To compare these statistics of gene annotation with C. clementina, we re-annotated the C. clementina scaffold 1–9 which covered more than 96% sequence of total scaffolds using MEGANTE with the same parameters. As a result, 36,163 protein-coding genes with 37,570 transcripts were predicted in the C. clementina genome and the average length of transcript and amino acid sequences was 1,252.9 bp and 378.7 aa, respectively. These results suggest that the number and length distribution of genes was comparable among the three citrus species.
Evolutionary conservation of genes and gene families among the three species was examined by a similarity search and clustering analysis of protein sequences. All protein sequences of three species were clustered into 51,615 gene groups (Fig. 2). Of those groups, 7,499, 9,639 and 8,355 were specifically observed in the C. clementina, C. unshiu, and P. trifoliata genomes, respectively (species-specific gene groups) (Supplemental Tables 3–5). Furthermore, 4,706 gene groups were observed only in two mandarin species (mandarin-specific gene groups) (Supplemental Table 6). Among three species, large gene families were observed in C. unshiu and P. trifoliata including terpene synthase, cytochrome P450, protein kinase, leucine-rich repeat protein, UDP-glucosyltransferase, ribonuclease H-like domain protein, pectin esterase inhibitor domain protein, zinc finger protein, ankyrin repeat proteins, and so on. Enrichment analyses of Gene Ontology and InterPro domains revealed that viral movement proteins (IPR028919) were significantly (FDR = 0.01) enriched in P. trifoliata specific genes (Supplemental Tables 7, 8). On the contrary, genes required for the maintenance of basic or “housekeeping” functions (e.g., regulation of transcription, protein phosphorylation) were depleted among P. trifoliata specific genes.
Fig. 2.
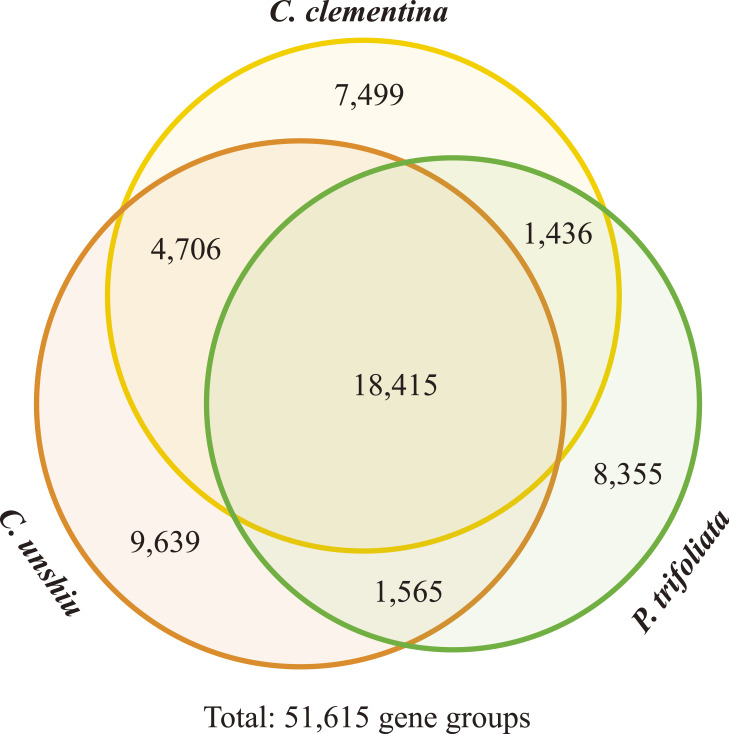
Gene conservation among three citrus species. Orthologous gene groups were defined based on the all-against-all blastp search and clustering by OrthoFinder. The numbers of gene groups that were conserved in all, two, and a specific species are represented in the Venn diagram.
The gene set in this study was compared with those in the previously published draft genome (Shimizu et al. 2017) using the same similarity search and clustering analyses. As a result, 17,218 (41.5%) protein-coding genes were annotated specifically in our genome assembly and 1,289 (7.5%) of those specific genes were supported by EST or mRNA sequences.
Database contents and functions of MiGD
The MiGD comprises three databases as follows: genome and annotation database of C. unshiu and P. trifoliata, genetic diversity database of nine citrus and relative species, and the CAPS marker database (Fig. 3A). The genome browser “JBrowse” provides the genome sequences and gene annotation data for C. unshiu and P. trifoliata (Fig. 3B). The SequenceServer system provides a BLAST search service against the genome, transcript, and protein sequences of C. clementina (JGI v1.0), C. sinensis (HZAU v2), C. unshiu (Shimizu et al. 2017), C. unshiu (MiGD), and P. trifoliata (MiGD) (Fig. 3C). All information regarding genome sequences in the FASTA format and gene annotations in GFF file are available on the download page. In JBrowse, users can search a gene with a transcript ID and obtain gene annotation data, such as description, GO terms, InterPro domains, and nucleotide sequences.
Fig. 3.

Genome annotation on JBrowse and the BLAST service in MiGD. (A) The top page of MiGD provides links to the genome browser JBrowse, BLAST, the multiple genome browser TASUKE, CAPS marker database, data download page, and so on. (B) Gene and genome annotation of C. unshiu and P. trifoliata are provided through JBrowse. (C) SequenceServer allows users to perform a sequence similarity search against the genome and gene sequences of C. unshiu, P. trifoliata, C. clementina, and C. sinensis.
Genome-wide sequence polymorphisms (SNPs and indels) among nine citrus and relative species (C. clementina, C. sinensis, C. reticulata, C. unshiu, C. grandis, C. medica, C. ichangensis, P. trifoliata and A. buxifolia) are provided through the multiple genome browser “TASUKE” (Kumagai et al. 2013) (Fig. 4). The main panel indicates the sequence alignment information among them, and the distribution and frequencies of SNPs and indels can be feasibly displayed around any regions on the nine chromosomes against the referencing genome of C. clementina (JGI ver. 1.0). The top menu bar helps users to search functions to find identifiers or genomic positions. At the most precise level, individual nucleotides and translated amino acids can be displayed. By turning on the “snpEff” function for each polymorphism, users can obtain variant effect information on whether the sequence variation triggers silent substitution, evolutionary substitution, and frame shift changes (Cingolani et al. 2012). A total of 1,225,594 homozygous SNPs (4.6 SNPs/kb) and 168,907 indels (0.6 indels/kb) were detected in C. unshiu against the reference genome of C. clementina, whereas a total of 4,681,886 SNPs (21.6 SNPs/kb) and 720,315 indels (3.3 indels/kb) were detected in P. trifoliata against the reference. A total of heterozygous 3,079,344 SNPs (11.7 SNPs/kb) and 467,683 indels (1.8 InDels/kb) were detected in C. unshiu against the reference genome of C. clementina, while, a total of 2,450,018 SNPs (11.3 SNPs/kb) and 401,371 indels (1.9 indels/kb) were detected in P. trifoliata against the reference. The number of SNPs in C. unshiu was almost similar to that in C. sinensis (3.6 SNPs/kb) (Xu et al. 2013) whereas many SNPs in P. trifoliata suggested that P. trifoliata was diverged from the general citrus species.
Fig. 4.

Genomic variations between C. clementina and nine citrus and related species on TASUKE. Sequence variant information (frequencies of SNPs and indels against the C. clementina genome) within each 30 bp block are represented in the top panel. The detailed information of each variant is shown by clicking a block as the bottom panel.
The CAPS marker database possesses the following information on 2,696 CAPS markers: PCR product size, primer sequence, electrophoresis pattern of representative citrus cultivars, the genetic locus of the previously reported genetic maps (Omura et al. 2003, Shimada et al. 2014), track records of the 384 SNP array genotyping (Fujii et al. 2013), cultivar identification (Ninomiya et al. 2015, Nonaka et al. 2017), and transcript IDs of C. clementina and C. unshiu draft genomes, although some of this information is not available (Fig. 5A).
Fig. 5.

CAPS marker information in MiGD. (A) All marker information is listed in the table. (B) The detailed information of each marker is shown by clicking the “Locus name” in the table.
In citrus, various type of DNA markers, such as simple sequence repeat (SSR), CAPS, SNP and indel, have been developed and have been applied to phylogenetic studies, linkage analysis, GWAS and so on (Fang et al. 2018, Fujii et al. 2013, Luro et al. 2008, Minamikawa et al. 2017, Shimada et al. 2014, Shimizu et al. 2016). Out of these DNA markers, we selected CAPS markers for DNA marker database in MiGD because our developed CAPS markers were gene-based markers that generated from highly conservative ESTs among citrus and related species, moreover, it was confirmed that PCR fragments were stably amplified among major citrus and related species. Therefore, our developed CAPS markers could be linked feasibly to the other databases in MiGD through transcription ID and it is promised to apply them to a wide range of citrus and related species. By clicking the icons on the left side of each transcript ID, users directly jump on the locus on the C. clementina and C. unshiu draft genomes in JBrowse and TASUKE of MiGD as well as in Phytozome (https://phytozome.jgi.doe.gov). The order of CAPS markers could be sorted by each linkage group of the AGI map or each scaffold of C. clementina and C. unshiu genome sequences, providing feasible access to the genome sequence around the target genetic locus through CAPS markers. In addition, genotyping data of 37 representative citrus cultivars for 213 CAPS markers, the graphical genotyping map of 35 representative citrus cultivars using 158 CAPS markers, and the genotyping data of 48 citrus cultivar identification using 26 CAPS markers are also available in the database (Fig. 5B). The graphical genotyping map is easily interpretable to understand the features of homogenous and heterozygous loci among the examined citrus cultivars. For example, LG7, which corresponded to scaffold 4 of C. clementina, revealed frequent homogenous loci in the graphical map of C. clementina and C. unshiu, whereas LG2 (scaffold 7 of C. clementina) and LG5 (scaffold 3 of C. clementina) revealed frequent heterozygous loci. These genotyping data among major citrus cultivars are helpful to select the optimal polymorphic CAPS markers for the user’s experimental purposes such as cultivar identification, construction of a genetic map, development of a linkage marker, and so on.
Discussion
MiGD is an integrated database of genome annotation, genetic diversity, and CAPS marker information and each database is mutually connected by the transcript ID, which could feasibly compare the locus of the DNA marker in a genetic linkage map with the corresponding locus in the assembled genome sequences of C. clementina and C. unshiu. This relational database is the first attempt among public citrus databases and MiGD could help users to link the information from the 5 previously generated genetic linkage maps of AGI (Shimada et al. 2014), KmMg (Omura et al. 2003), JtTr (Omura et al. 2003), KmO41 (Nakano et al. 2003), and RpSa (Ohta et al. 2011) with the draft genome sequences of C. clementina and C. unshiu. In the genetic diversity database, whole genome sequence data of nine citrus and related species were analyzed, and the detected nucleotide variations and depth of coverages were stored in the TASUKE browser. Citrus species are known to have highly heterozygous genomes with numerous SNPs and indels diverged among species and cultivars. Therefore, we considered that sequence comparison among nine citrus and related species would help determining the functional SNP and indel information around the target locus by removing the null sequence variation and to develop a new DNA marker aiming for mapped based cloning of agronomically important traits. The TASUKE browser has high scalability and we will add additional sequence information of various agronomically important cultivars in the future. Moreover, the optimal DNA marker can be easily selected for cultivar identification of a new cultivar and for the detection of chimera and hybrid embryos in citrus breeding based on the information of genotyping data on CAPS markers among major citrus cultivars. Thus, MiGD would be a useful database to extract the necessary information for the advancing mandarin molecular breeding and cultivar identification for protecting the breeder’s rights in Japan, without browsing multiple public databases.
In this study, we reported the first draft genome sequence of P. trifoliata assembled by a hybrid de novo assembly strategy using Illumina and PacBio data. The total size of the assembled scaffold of P. trifoliata is 292 Mb and it almost covers the entire genome sequence as the estimated genome size of P. trifoliata is 296 Mb. Interestingly, the assembled genome size of P. trifoliata (292 Mb) is smaller than that of C. clementina (301 Mb), C. sinensis (321 Mb), C. grandis (345 Mb), C. ichangensis (335 Mb), C. medica (368 Mb), and C. unshiu (346 Mb), suggesting that the genomic contents of P. trifoliata might be similar to those of the wild citrus, A. buxifolia (288 Mb). It is clear that the genome structure of P. trifoliata is different from cultivated citrus species, particularly in the non-coding region. Through evaluation of genome assemblies by QUAST, the assembled scaffolds of C. unshiu and P. trifoliata can be aligned to 76.8% and 30.8% of the C. clementina reference genome and 93.1% (19,667 + 3,174 partial) and 63.1% (10,628 + 4,862 partial) of 24,533 C. clementina reference genes were covered. Enrichment analyses of Gene Ontology and InterPro domains could not fully capture the unique features of P. trifoliata specific genes. Therefore, high diversity in the non-coding region is likely to explain the unique responses to biotic and abiotic stresses in P. trifoliata. Elucidation of whole-genome sequences is a key milestone for research to explore agronomically and academically important traits specific to the trifoliate orange such as various disease resistances for Phytophthora citrophthora, CTV, Tylenchulus semipenetrans, cold-hardiness, the aptitude of root stock, and so on. Recently, various research efforts are underway to elucidate the molecular mechanisms and identify the causative gene for cold stress tolerance (Wang et al. 2015), Huanglongbing tolerance (Rawat et al. 2017), Phytophthora disease resistance (Dalio et al. 2018) and so on. The genomic resources of P. trifoliata in MiGD can contribute to find functional mutations on the genome structure and the specific genes responsible for them.
The first genome assembly of C. unshiu was published in 2017 (Shimizu et al. 2017). We conducted re-sequencing and re-assembly of the C. unshiu genome for enrichment of the draft sequence to develop highly accurate DNA markers. Compared with previously published C. unshiu scaffold sequences, the number of scaffolds and N-ratio were clearly reduced to approximately 1/10, whereas the ratio of repeat bases was slightly increased. Enrichment of the draft sequence would be achieved by improving the assembly method by which PacBio subreads were used for contig assembly, scaffolding, and gap-closing steps in the hybrid de novo assembly. The genomes of citrus species are known to be highly heterogenous and have an admixture structure. The graphical genotyping maps also showed that numerous heterozygous loci existed throughout the linkage groups among the examined cultivars. Therefore, in the process of hybrid genome assembling, it is predicted that important sequence information located on one side of the haplotype might have fallen off. For example, most polyembryonic citrus cultivars show a heterogenous genotype on the polyembryonic locus and have polyembryonic and monoembryonic alleles (Shimada et al. 2018). The genomic structures of the polyembryonic and monoembryonic alleles are very similar except for a miniature inverted-repeat transposable element (MITE) inserted in the promoter region responsible for the transcription of CitRKD1. MITE comprises 185 bps of a short sequence element and possibly happened to fall off in the hybrid genome assembly in the previously published draft genome sequences. Recently, the molecular mechanism of various agronomically important traits in fruit trees is becoming apparent owing to the advances in deciphering the genome. Anthocyanin accumulation in blood orange (Butelli et al. 2012), grape (Kobayashi et al. 2004), and apple (Zhang et al. 2019), columnar tree type in apple (Okada et al. 2016), and non-melting flesh in stony hard peaches (Tatsuki et al. 2018) is controlled by transcriptional regulation mediated by insertion or deletion of transposons. Therefore, the advancement of technologies in assembling hybrid genomes and long sequence reads is extremely important to assign transposons, retrotransposons, and repeat elements to accurate positions in the draft sequences.
The most economically important citrus cultivars originate from repeated natural interspecific hybridization among four ancestral taxa (C. reticulata, C. grandis, C. medica and C. micrantha) and possess complex interspecific mosaic genomes (García-Lor et al. 2012). Therefore, modern breeding utilizing these conventional cultivars is hampered by these complex heterozygous genomic structures and the typical phenotypes observed in conventional cultivars are frequently broken in their progenies by the admixture of genomes through sexual hybridization (Curk et al. 2014). This observation is also applicable to the Japanese breeding program, as the present citrus economic cultivars and breeding lines are originally derived from a limited genetic source of 14 ancestral citrus cultivars by pedigree analysis (Imai et al. 2017). Therefore, the most desirable phenotypic traits in economic cultivars do not come only from a specific gene of a specific cultivar but also arise from a desirable allelic combination of commonly possessed genes among ancestral cultivars. Considering this background of the complex citrus genomic structures, whole genome sequencing and structural comparison among various citrus cultivars could be a powerful approach to decipher the admixture genomic structure of current cultivars and would enable us to understand how to build superior traits among them to advance molecular breeding efficiently. The draft sequences of C. unshiu and P. trifoliata genomes in this study would be useful to explore the genes responsible for the agronomically important traits originated from these major cultivated citruses in Japan. MiGD could accelerate the molecular breeding in citrus to renew major cultivation of the satsuma mandarin.
Author Contribution Statement
Dr. Yoshihiro Kawahara assembled the genome sequence of satsuma mandarin and trifoliate orange to develop genome annotation database, and managed Mikan Genome Database. Dr. Tomoko Endo conducted trifoliate orange genome sequencing. Dr. Mitsuo Omura developed 2,696 CAPS marker and genotyped the major citrus cultivars by CAPS markers. Ms. Yumiko Teramoto developed Mikan Genome Database. Dr. Takeshi Itoh analyzed genome sequence data of satsuma mandarin and trifoliate orange. Dr. Hiroshi Fujii annotated the information of transcription ID, linkage map locus etc. for 2,696 CAPS markers. He is a corresponding author to organize Mikan Genome Database. Dr. Takehiko Shimada conducted satsuma mandarin genome sequencing and is a corresponding author to organize the manuscript.
Supplementary Material
Acknowledgments
We gratefully acknowledge the support of Mr. Hisataka Numa in genome annotation using MEGANTE. This work was supported by a grant from the Ministry of Agriculture, Forestry, and Fisheries of Japan (Genomics-based Technology for Agricultural Improvement, GMO-1003). This research was supported by the Advanced Analysis Center Research Supporting Program of NARO. Large-scale sequence analyses were performed on the cluster computing system at NARO Advanced Analysis Center (NAAC). PacBio RSII sequencing was performed at Okinawa Institute of Advanced Sciences, supported by the “Okinawa Intellectual Cluster Program” of Okinawa Science and Technology Promotion Center.
Literature Cited
- Antipov D., Korobeynikov A., McLean J.S. and Pevzner P.A. (2016) hybridSPAdes: an algorithm for hybrid assembly of short and long reads. Bioinformatics 32: 1009–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arumuganathan K. and Earle E.D. (1991) Nuclear DNA content of some important plant species. Plant Mol. Biol. Rep. 9: 208–218. [Google Scholar]
- Boetzer M., Henkel C.V., Jansen H.J., Butler D. and Pirovano W. (2011) Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 27: 578–579. [DOI] [PubMed] [Google Scholar]
- Boetzer M. and Pirovano W. (2014) SSPACE-LongRead: scaffolding bacterial draft genomes using long read sequence information. BMC Bioinformatics 15: 211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger A.M., Lohse M. and Usadel B. (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30: 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buels R., Yao E., Diesh C.M., Hayes R.D., Munoz-Torres M., Helt G., Goodstein D.M., Elsik C.G., Lewis S.E., Stein L. et al. (2016) JBrowse: a dynamic web platform for genome visualization and analysis. Genome Biol. 17: 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butelli E., Licciardello C., Zhang Y., Liu J., Mackay S., Bailey P., Reforgiato-Recupero G. and Martin C. (2012) Retrotransposons control fruit-specific, cold-dependent accumulation of anthocyanins in blood oranges. Plant Cell 24: 1242–1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camacho C., Coulouris G., Avagyan V., Ma N., Papadopoulos J., Bealer K. and Madden T.L. (2009) BLAST+: architecture and applications. BMC Bioinformatics 10: 421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cingolani P., Platts A., Wang L.L., Coon M., Nguyen T., Wang L., Land S.J., Lu X. and Ruden D.M. (2012) A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 6: 80–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curk F., Ancillo G., García-Lor A., Luro F., Perrier X., Jacquemoud-Collet J.-P., Navarro L. and Ollitrault P. (2014) Next generation haplotyping to decipher nuclear genomic interspecific admixture in Citrus species: analysis of chromosome 2. BMC Genet. 15: 152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalio R.J.D., Máximo H.J., Oliveira T.S., Azevedo T.M., Felizatti H.L., Campos M.A. and Machado M.A. (2018) Molecular basis of Citrus sunki susceptibility and Poncirus trifoliata resistance upon Phytophthora parasitica attack. Mol. Plant Microbe Interact. 31: 386–398. [DOI] [PubMed] [Google Scholar]
- Dellaporta S.L., Wood J. and Hicks J.B. (1983) A plant DNA minipreparation: Version II. Plant Mol. Biol. Rep. 1: 19–21. [Google Scholar]
- DePristo M.A., Banks E., Poplin R., Garimella K.V., Maguire J.R., Hartl C., Philippakis A.A., del Angel G., Rivas M.A., Hanna M. et al. (2011) A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 43: 491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emms D.M. and Kelly S. (2015) OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 16: 157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- English A.C., Richards S., Han Y., Wang M., Vee V., Qu J., Qin X., Muzny D.M., Reid J.G., Worley K.C. et al. (2012) Mind the gap: upgrading genomes with Pacific Biosciences RS long-read sequencing technology. PLoS ONE 7: e47768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang Q., Wang L., Yu H., Huang Y., Jiang X., Deng X. and Xu Q. (2018) Development of species-specific InDel markers in citrus. Plant Mol. Biol. Rep. 36: 653–662. [Google Scholar]
- Fujii H., Shimada T., Nonaka K., Kita M., Kuniga T., Endo T., Ikoma Y. and Omura M. (2013) High-throughput genotyping in citrus accessions using an SNP genotyping array. Tree Genet. Genomes 9: 145–153. [Google Scholar]
- García-Lor A., Luro F., Navarro L. and Ollitrault P. (2012) Comparative use of indel and SSR markers in deciphering the interspecific structure of cultivated citrus genetic diversity: a perspective for genetic association studies. Mol. Genet. Genomics 287: 77–94. [DOI] [PubMed] [Google Scholar]
- Gmitter, Jr. F.G., C. Chen, M.N. Rao and J.R. Soneji (2007) Citrus fruits. In: Kole, C. (ed.) Genome mapping and molecular breeding in plants Volume 4 Fruits and Nuts, Springer, pp. 265–279. [Google Scholar]
- Gmitter F.G., Chen C., Machado M.A., de Souza A.A., Ollitrault P., Froehlicher Y. and Shimizu T. (2012) Citrus genomics. Tree Genet. Genomes 8: 611–626. [Google Scholar]
- Gurevich A., Saveliev V., Vyahhi N. and Tesler G. (2013) QUAST: quality assessment tool for genome assemblies. Bioinformatics 29: 1072–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgson, R.W. (1967) CHAPTER 4. Horticultural varieties of citrus. In: Reuther, W., H.J. Webber and L.D. Bachelor (eds.) The Citrus Industry Volume I, University of California Press, pp. 431–611. [Google Scholar]
- Imai A., Kuniga T., Yoshioka T., Nonaka K., Mitani N., Fukamachi H., Hiehata N., Yamamoto M. and Hayashi T. (2017) Genetic background, inbreeding, and genetic uniformity in the national citrus breeding program, Japan. Hort. J. 86: 200–207. [Google Scholar]
- Kajitani R., Toshimoto K., Noguchi H., Toyoda A., Ogura Y., Okuno M., Yabana M., Harada M., Nagayasu E., Maruyama H. et al. (2014) Efficient de novo assembly of highly heterozygous genomes from whole-genome shotgun short reads. Genome Res. 24: 1384–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawase K., Iwagaki I., Takahara T., Ono S. and Hirose K. (1987) Rootstock studies for citrus varieties in Japan. Jpn. Agric. Res. Q. 20: 253–259. [Google Scholar]
- Khan, I.A. (2007) Citrus genetics, breeding and biotechnology. CABI, Wallingford, pp. 141–150. [Google Scholar]
- Kobayashi S., Goto-Yamamoto N. and Hirochika H. (2004) Retrotransposon-induced mutations in grape skin color. Science 304: 982. [DOI] [PubMed] [Google Scholar]
- Koren S., Schatz M.C., Walenz B.P., Martin J., Howard J.T., Ganapathy G., Wang Z., Rasko D.A., McCombie W.R., Jarvis E.D. et al. (2012) Hybrid error correction and de novo assembly of single-molecule sequencing reads. Nat. Biotechnol. 30: 693–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumagai M., Kim J., Itoh R. and Itoh T. (2013) TASUKE: a web-based visualization program for large-scale resequencing data. Bioinformatics 29: 1806–1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. and Durbin R. (2009) Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25: 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luro F.L., Costantino G., Terol J., Agrout X., Allario T., Wincker P., Talon M., Ollitrault P. and Morillon R. (2008) Transferability of the EST-SSRs developed on nucles clementine (Citrus clementina Hort ex Tan) to other Citrus species and their effectiveness for genetic mapping. BMC Genomics 9: 287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minamikawa M.F., Nonaka K., Kaminuma E., Kajiya-Kanegae H., Onogi A., Goto S., Yoshioka T., Imai A., Hamada H., Hayashi T. et al. (2017) Genome-wide association study and genomic prediction in citrus: Potential of genomics-assisted breeding for fruit quality traits. Sci. Rep. 7: 4721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano, M., H. Nesumi, T. Yoshioka, M. Omura and T. Yoshida (2003) Linkage analysis between male sterility of citrus and STS markers. Proceedings of the International Society of Citriculture IX Congress, pp. 179–180. [Google Scholar]
- Ninomiya T., Shimada T., Endo T., Nonaka K., Omura M. and Fujii H. (2015) Development of Citrus Cultivar Identification by CAPS Markers and Parentage Analysis. Hort. Res. (Japan) 14: 127–133. [Google Scholar]
- Nonaka K., Fujii H., Kita M., Shimada T., Endo T., Yoshioka T. and Omura M. (2017) Identification and parentage analysis of citrus cultivars developed in Japan by CAPS markers. Hort. J. 86: 208–221. [Google Scholar]
- Numa H. and Itoh T. (2014) MEGANTE: a web-based system for integrated plant genome annotation. Plant Cell Physiol. 55: e2 (1–8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connell J., Schulz-Trieglaff O., Carlson E., Hims M.M., Gormley N.A. and Cox A.J. (2015) NxTrim: optimized trimming of Illumina mate pair reads. Bioinformatics 31: 2035–2037. [DOI] [PubMed] [Google Scholar]
- Ohta S., Endo T., Shimada T., Fujii H., Shimizu T., Kuniga T., Yoshioka T., Nesumi H., Yoshida T. and Omura M. (2011) PCR primers for marker assisted backcrossing to introduce a CTV resistance gene from Poncirus trifoliata (L.) Raf. into Citrus. J. Japan. Soc. Hort. Sci. 80: 295–307. [Google Scholar]
- Okada K., Wada M., Moriya S., Katayose Y., Fujisawa H., Wu J., Kanamori H., Kurita K., Sasaki H., Fujii H. et al. (2016) Expression of a putative dioxygenase gene adjacent to an insertion mutation is involved in the short internodes of columnar apples (Malus × domestica). J. Plant Res. 129: 1109–1126. [DOI] [PubMed] [Google Scholar]
- Ollitrault P., Dambier D., Luro F. and Duperray C. (1994) Nuclear genome size variations in Citrus. Fruits 49: 390–393. [Google Scholar]
- Omura, M., T. Ueda, T. Shimada, T. Endo, H. Fujii, H. Nesumi and T. Yoshida (2003) Graphical genotype of citrus cultivars by co-dominant CAPS markers. Abst. Plant & Animal genome XI Conference, p. 22. JAN 11–15, San Diego, CA. [Google Scholar]
- Ou S., Chen J. and Jiang N. (2018) Assessing genome assembly quality using the LTR Assembly Index (LAI). Nucleic Acids Res. 46: e126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pryszcz L.P. and Gabaldón T. (2016) Redundans: an assembly pipeline for highly heterozygous genomes. Nucleic Acids Res. 44: e113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu, W., Y. Zhou, Y. Zhang, Y. Lu, X. Wang, D. Zhao, Y. Yang and C. Zhang (2012) MFEprimer-2.0: a fast thermodynamics-based program for checking PCR primer specificity. Nucleic Acids Res. 40 (Web Server issue): W205–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawat N., Kumar B., Albrecht U., Du D., Huang M., Yu Q., Zhang Y., Duan Y.-P., Bowman K.D., Gmitter F.G. et al. (2017) Genome resequencing and transcriptome profiling reveal structural diversity and expression patterns of constitutive disease resistance genes in Huanglongbing-tolerant Poncirus trifoliata and its hybrids. Hortic. Res. 4: 17064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada T., Fujii H., Endo T., Ueda T., Sugiyama A., Nakano M., Kita M., Yoshioka T., Shimizu T., Nesumi H. et al. (2014) Construction of a citrus framework genetic map anchored by 708 gene-based markers. Tree Genet. Genomes 10: 1001–1013. [Google Scholar]
- Shimada T., Endo T., Fujii H., Nakano M., Sugiyama A., Daido G., Ohta S., Yoshioka T. and Omura M. (2018) MITE insertion-dependent expression of CitRKD1 with a RWP-RK domain regulates somatic embryogenesis in citrus nucellar tissues. BMC Plant Biol. 18: 166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu T., Kitajima A., Nonaka K., Yoshioka T., Ohta S., Goto S., Toyoda A., Fujiyama A., Mochizuki T., Nagasaki H. et al. (2016) Hybrid origins of citrus varieties inferred from DNA marker analysis of nuclear and organelle genomes. PLoS ONE 11: e0166969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu T., Tanizawa Y., Mochizuki T., Nagasaki H., Yoshioka T., Toyoda A., Fujiyama A., Kaminuma E. and Nakamura Y. (2017) Draft sequencing of the heterozygous diploid genome of satsuma (Citrus unshiu Marc.) using a hybrid assembly approach. Front. Genet. 8: 180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talon M. and Gmitter F.G. (2008) Citrus genomics. Int. J. Plant Genomics 2008: 528361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatsuki M., Soeno K., Shimada Y., Sawamura Y., Suesada Y., Yaegaki H., Sato A., Kakei Y., Nakamura A., Bai S. et al. (2018) Insertion of a transposon-like sequence in the 5ʹ-flanking region of the YUCCA gene causes the stony hard phenotype. Plant J. 96: 815–827. [DOI] [PubMed] [Google Scholar]
- Wang M., Zhang X. and Liu J.-H. (2015) Deep sequencing-based characterization of transcriptome of trifoliate orange (Poncirus trifoliata (L.) Raf.) in response to cold stress. BMC Genomics 16: 555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Xu Y., Zhang S., Cao L., Huang Y., Cheng J., Wu G., Tian S., Chen C., Liu Y. et al. (2017) Genomic analyses of primitive, wild and cultivated citrus provide insights into asexual reproduction. Nat. Genet. 49: 765–772. [DOI] [PubMed] [Google Scholar]
- Wu G.A., Prochnik S., Jenkins J., Salse J., Hellsten U., Murat F., Perrier X., Ruiz M., Scalabrin S., Terol J. et al. (2014) Sequencing of diverse mandarin, pummelo and orange genomes reveals complex history of admixture during citrus domestication. Nat. Biotechnol. 32: 656–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Q., Chen L.-L., Ruan X., Chen D., Zhu A., Chen C., Bertrand D., Jiao W.-B., Hao B.-H., Lyon M.P. et al. (2013) The draft genome of sweet orange (Citrus sinensis). Nat. Genet. 45: 59–66. [DOI] [PubMed] [Google Scholar]
- Ye C., Hill C.M., Wu S., Ruan J. and Ma Z.S. (2016) DBG2OLC: Efficient assembly of large genomes using long erroneous reads of the third generation sequencing technologies. Sci. Rep. 6: 31900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L., Hu J., Han X., Li J., Gao Y., Richards C.M., Zhang C., Tian Y., Liu G., Gul H. et al. (2019) A high-quality apple genome assembly reveals the association of a retrotransposon and red fruit colour. Nat. Commun. 10: 1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimin A.V., Marçais G., Puiu D., Roberts M., Salzberg S.L. and Yorke J.A. (2013) The MaSuRCA genome assembler. Bioinformatics 29: 2669–2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.