

Abstract
Cancer patients often confront the decision of whether to continue high dose chemotherapy at the expense of cumulative toxicities. Reducing the dose of chemotherapy regimens while preserving efficacy is sorely needed to preserve the performance status of these vulnerable patients, yet has not been prioritized. Here, we introduce a dual pronged approach to modulate the microenvironment of desmoplastic pancreatic tumors and enable significant dose de-escalation of the FDA-approved chemotherapeutic nanoliposomal irinotecan (nal-IRI) without compromising tumor control. We demonstrate that light-based photodynamic priming (PDP) coupled with vitamin D3 receptor (VDR) activation within fibroblasts increases intratumoral nal-IRI accumulation and suppresses pro-tumorigenic CXCL12/CXCR7 crosstalk. Combined photodynamic and biochemical modulation of the tumor microenvironment enables a 75% dose reduction of nal-IRI while maintaining treatment efficacy, resulting in improved tolerability. Modifying the disease landscape to increase the susceptibility of cancer, via preferentially modulating fibroblasts, represents a promising and relatively underexplored strategy to enable dose de-escalation. The approach presented here, using a combination of three clinically available therapies with non-overlapping toxicities, can be rapidly translated with minimal modification to treatment workflow, and challenges the notion that significant improvements in chemotherapy efficacy can only be achieved at the expense of increased toxicity.
Introduction
Patients with advanced pancreatic ductal adenocarcinoma (PDAC) present with significant co-morbidities and poor performance status. Improved survival outcomes for these patients are contingent upon their ability to withstand prolonged toxic treatment regimens (1). Deteriorating performance status coupled with treatment induced toxicities prevents many of these patients from initiating or completing preferred regimens. Even the most recent clinical trials for this disease, including those utilizing stromal depletion, continue to yield unfavorable outcomes at the expense of significant toxicity (2), highlighting the dire need to translate strategies that improve the tolerability of anticancer regimens while also preserving their efficacy (1).
We introduce a multi-modal approach comprised of photodynamic priming (PDP) and vitamin D receptor (VDR) activation to modulate the microenvironment of PDAC and increase its susceptibility to cytotoxic therapies. We demonstrate that sub-cytotoxic PDP, a photochemistry-based therapeutic modality, combined with the vitamin D analogue Calcipotriol (CAL) suppresses pro-tumorigenic intratumoral paracrine signaling interactions and permeabilizes the tumor-associated microvasculature and parenchyma to significantly improve the anticancer effects of nanoliposomal irinotecan (nal-IRI). This priming regimen, which consists of two clinically approved agents with distinct mechanisms of action and non-overlapping toxicities, enables a 75% dose de-escalation of nal-IRI while maintaining durable anticancer efficacy and tolerability.
Shortcomings in treatment response are increasingly attributed to tumor heterogeneity, poor drug delivery, and disease-sustaining intratumoral paracrine interactions that involve the microenvironment (3). For advanced PDAC, only modest improvements in survival have been achieved despite the administration of consecutive multi-modal chemotherapies at the maximum tolerated dose, in part because the tumor microenvironment is not addressed (4,5). Shifting focus away from consecutive high dose chemotherapeutics towards less toxic strategies that modify the disease landscape may enable chemotherapy dose reduction without negatively impacting efficacy. Photodynamic therapy (PDT) is a photochemistry-based modality in which a photosensitizer is activated by light of a specific wavelength to produce local reactive molecular species (RMS), and is currently being evaluated clinically as an adjuvant to surgery and chemotherapy in advanced PDAC patients (6,7)(NCT03033225). PDP is a companion effect of PDT that occurs when the RMS produced by photodynamic activation do not surpass the cytotoxic threshold. Counterintuitively, subthreshold RMS can actually be advantageous and have been shown to modulate cancer cells and the surrounding stroma to sensitize tumors to chemotherapy (8,9), improve chemotherapeutic delivery by inducing intravascular and intratumoral permeabilization (10,11), yield significant anticancer immune stimulatory effects (12), and avoid dangerous evolutionary selection pressures imparted by high dose chemotherapy regimens (10,13). PDP is therefore an attractive strategy to enhance the efficacy of conventional chemotherapeutic regimens (13,14).
Targeting the interactions between tumor cells and their microenvironment may enhance patient outcomes. For pancreatic ductal carcinoma (PDAC), the tumor associated microenvironment is known to comprise a majority of the tumor, and these stromal components are a major driver of disease progression (15). Abundant within the PDAC desmoplasia are cancer associated fibroblasts (CAFs), which have been confirmed in numerous reports to contribute to poor treatment outcomes by promoting fibrosis, altering tumor metabolism, increasing metastasis, and suppressing immunogenicity (4,16). CAL has been shown to effectively modulate the PDAC associated microenvironment by inducing a variety of epigenetic and transcriptomic changes in cancer associated pancreatic stellate cells (CAPSCs), a CAF precursor, consistent with their “reprogramming” to a non-cancer associated, or quiescent, state (17,18). Based on this strategy, multiple clinical trials utilizing vitamin D analogues in combination with conventional chemo- or immunotherapies are ongoing for patients with resectable or metastatic PDAC (NCT03331562, NCT03300921, NCT03520790, NCT03519308).
Vitamin D (17) and PDP (19,20) have been shown to modulate a number of chemokines in various disease contexts including the C-X-C and C-C pathways, several of which have been implicated in PDAC progression (21). In one example, secretion of the C-X-C motif chemokine 12 (CXCL12, also known as stromal derived factor 1, or SDF-1) by stromal cells, namely stellate cells and fibroblasts, triggers a variety of aggressive phenotypes in PDAC cells, including epithelial-mesenchymal transition, chemoresistance, proliferation, angiogenesis, and suppression of antitumor T-cell activity, following its interaction with C-X-C motif receptors 4 and 7 (CXCR4 and CXCR7) (22,23). We demonstrate that combined PDP and CAL treatment reduces CXCL12 secretion by CAFs, suppressing this pro-tumorigenic signaling axis.
We present a quickly translatable approach that integrates both efficacy and tolerability for patients by combining PDP with biochemical stromal reprogramming as a priming strategy for chemotherapy. Simultaneous modulation of the tumor-associated vasculature and stroma, in part via suppression of the CXCL12/CXCR4/CXCR7 signaling, is demonstrated to markedly enhance the efficacy and tolerability of a widely used chemotherapy, and enable significant dose de-escalation while maintaining efficacy and tolerability.
Materials and Methods
Cell Culture
All cells were cultured in L-Glutamine containing DMEM (Corning 10-013-CV) supplemented with 10% FBS and 100 U/mL penicillin, 100μg/mL streptomycin (Gibco, ThermoFisher), and maintained in a humidified CO2 atmosphere at 37 °C. MIA-PaCa-2, AsPC-1, and MRC-5 were purchased from the ATCC. SCCAFs were obtained from the Department of Dermatology, MGH, following isolation from discarded squamous cell carcinoma tissue as approved by the IRB. PCAFs were isolated from tumors of patients who underwent surgical resection at the University of Michigan Hospital (24). All cells were confirmed negative for mycoplasma (Lonza MycoAlert) prior to initial use and every 3 months thereafter.
Orthotopic mouse model, treatments, and toxicology studies
All treatments, procedures, and handling of animals were in accordance with the protocol approved by MGH IACUC. MIA-PaCa-2 cells and/or PCAFs were orthotopically implanted into the pancreas of 4-6 week old male Swiss nude mice (20-25 g). Animals were anesthetized with ketamine/xylazine, and a small left abdominal flank incision was made to exteriorize the pancreas. A 50 μL media+Matrigel® (BD Biosciences) suspension of 1 x 106 MIA-PaCa-2, or 5x105 MIA-PaCa-2 and 5 x 105 PCAFs (co-implantation model) was injected into the pancreas. Calcipotriol (100 μg/kg, intraperitoneal, Tocris) was administered daily on days 5-8 and 10-13 post implantation in 1 mL sterile saline. Injections of Visudyne (liposomal verteporfin, 0.25 mg/kg, i.v.) and/or nal-IRI (5, 10 or 20 mg/kg, i.v., Ipsen) were performed on day 9 in 200 μL sterile phosphate-buffered saline, simultaneously when applicable. One hour after the injection of Visudyne, PDP was performed to the exteriorized pancreas using a 690 nm diode laser (High Power Devices) delivered at an irradiance of 100 mW/cm2 to achieve a fluence of 75 J/cm2. A one-hour drug light interval was selected because there is a balanced distribution of photosensitizer in the tumor associated microvasculature and parenchyma at this time point. Mice were randomized into treatment groups, and tumor growth was longitudinally monitored using ultrasound imaging as described previously (25). Mouse blood was collected 48 hours post PDP and/or nal-IRI treatment, and analyzed by staff at the MGH CCM Clinical Pathology Laboratory using a DriChem analyzer.
Immunofluorescence imaging of CXCL12, CXCR7, CXCR4, and Vitamin D Receptor (VDR)
Anti-CXCL12 (R&D systems, Alexa Fluor 488 conjugated, clone 79018), CXCR7 (R&D systems, PE-conjugated, clone 358426) and CXCR4 antibodies (R&D systems, PE-conjugated, clone 12G5) were used for immunofluorescence experiments. Orthotopic pancreatic tumors were harvested immediately following treatment (15 days after implantation) or 76 days after treatment (90 days after tumor implantation), and prepared as previously described (10). Confocal fluorescence imaging was performed using an Olympus FluoView 1000 confocal microscope with a 20x 0.75NA objective. Excitation of DAPI, Alexa Fluor 488, APC, and Alexa Fluor 647 fluorophores was carried out using 405-, 405-, and 635-nm lasers, respectively, with appropriate filters. 6-10 images, evenly distributed across the tumor cross-section, were collected from at least 2 tumor samples for each condition. Fluorescence was quantified as previously described (10). For analysis of VDR activation, cells were cultured in a glass bottom 24 well plate for 72 hours and treated with 1 μM calcipotriol. 24 hours later, cells were fixed with 1:1 acetone:methanol for 5 min, washed with phosphate-buffered saline (PBS), and incubated with blocking solution (DAKO protein block) for 30 minutes followed by anti-VDR antibody (1:1000, Abcam, ab3508) overnight at 4 °C. Secondary goat anti-rabbit IgG (Abcam, 1:500, Alexa Fluor 488 conjugated, ab150077) was applied, cells were washed with PBS, mounted (Invitrogen SlowFade Gold with 4′,6-diamidino-2- phenylindole, DAPI) with a round glass coverslip, and imaged with an Olympus FluoView 1000 confocal microscope with a 40x objective. 5 images, evenly distributed across each well were taken per well, with 15 images taken per condition. The dynamic range for each laser line described was established with an unstained background control.
In vitro CXCL12 secretion and conditioned medium assays
CXCL12 secretion from PDAC and fibroblast lines was evaluated by ELISA (Abcam, ab100637). Cells were cultured for 96 hours in serum free DMEM, at which point wells were 50-80% confluent. Supernatant was collected and processed per kit specifications and standard curves were generated using recombinant CXCL12 provided by the manufacturer. Treatment was performed 24 hours after plating. For PDP, cells were incubated with 0.25 μM BPD for 90 minutes in culture medium. Medium was then replaced and cells were irradiated with 690 nm light (Intense Ltd.) at an irradiance of 150 mW/cm2, to achieve a light dose of 0.125 J/cm2. The stock concentration of BPD in DMSO was determined by spectroscopy immediately prior to each experiment. Upon receipt, calcipotriol was resuspended in DMSO, aliquoted, and stored at −20 °C per manufacturer’s specifications. DMSO concentration in cell culture media did not exceed 0.1% for all treatments. CAL was thawed immediately prior to each experiment and diluted in culture medium to the appropriate concentration. For conditioned medium assays, fibroblasts were cultured in serum free DMEM. Treatments were performed 24 hours post plating, and serum free medium was refreshed 96 hours post plating. Supernatant was collected 144 hours post plating, filtered through a 0.2 micron cellulose filter, and applied to MIA-PaCa-2 or AsPC-1 cells that were plated 24 hours prior. PDAC cells were ~80% confluent at this time point. MTT and cell counts were performed on the PDAC cells 96 hours post plating, and normalized metabolic activity was calculated by dividing the corrected NADPH oxidase activity signal (MTT, adjusted to background control) by the average number of cells per well for the group and normalized to the fresh medium control.
Histopathology, αSMA Immunohistochemistry, and Masson’s trichrome staining
Untreated MIA-PaCa-2 and MIA-PaCa-2+PCAF tumors were excised 90 days after implantation and embedded in optimal cutting temperature compound and frozen at −80°C. H&E, immunohistochemistry staining of αSMA, and Masson’s trichrome were performed on 20 μm-thick cryosections by the MGH Wellman Center Photopathology Core. A pancreatic pathologist reviewed H&E, Masson’s trichrome and αSMA stains of MIA-PaCa-2 alone (n = 3 tumors) and MIA-PaCa-2+PCAF (n = 3 tumors) tumors and semi quantitatively evaluated the extent (grade 0-3) of desmoplastic fibrosis in each tumor. The pathologic grading criteria to evaluate desmoplasia were as follows: Grade 0 indicated no established desmoplasia; Grade 1 indicated few foci with desmoplastic stroma, 10% or fewer of viable tumor nests are associated with desmoplastic stroma; Grade 2 indicated that 10-50% of viable tumor nests were associated with or surrounded by desmoplastic stroma; and Grade 3 indicated that >50% of viable tumor nests are associated with/surrounded by desmoplastic stroma. For IHC, briefly, permeabilization was performed with 0.1% Triton X-100 TBS for 15 min, tissue sections were blocked with BLOXALL (VectorLabs, cat# SP-6000) for 15 min to block Endogenous Alkaline Phosphatase (AP) and blocked with Protein Block buffer (DAKA, cat# X090930-2). Tissue sections were then incubated with anti-alpha Smooth Muscle Actin (Abcam, cat# ab5694) antibody overnight at 4°C and incubated with MACH 2 Rabbit AP-Polymer Detection antibody (BioCare Medical, Cat# RALP525-G) for 30min. Vulcan Fast Red was used for color detection of the antigen-antibody reaction and hematoxylin was used as nuclei counterstain. A whole slide imaging system (Hamamatsu NanoZoomer 2.0-RS) was used to image IHC and Masson’s Trichrome stained slides. Analysis of αSMA (shown in red) from the IHC slides, and collagen (shown in blue) from the trichrome slides was performed as previously described (8). Three tumors were excised per condition, and slices were taken evenly across the tumor section. At least six images were taken per condition. Representative images of Masson’s trichrome and IHC were evenly color balanced using the Nanozoomer viewer software prior to exporting. Only unmodified images were used for quantitative analyses.
Statistical analyses
All results are reported as mean ± standard error of the mean (SEM) unless otherwise specified. Statistical tests were carried out using GraphPad Prism Version 8 (GraphPad Software), in consultation with the MGH Biostatistics Core. Specific tests are indicated in the figure captions. All reported P values are two-tailed unless otherwise specified. In vivo results, including fluorescence intensity analyses, (Fig. 1C-E; Fig. 2C-J; Fig. 3C-E; Fig. S1C, F; Fig. S2A-C; Fig. S3B, C; Fig. S4B-D; Fig. S6; Fig. S7B-D) were performed using nonparametric tests (Mann-Whitney U test or Kruskal–Wallis one-way ANOVA with appropriate post hoc test, Dunn’s test was used for multiple comparisons); the D’Agostino & Pearson omnibus (α=0.05) and Shapiro-Wilk (α=0.05) normality tests identified deviations from normality. All statements of significance in the results were based on a threshold of p<0.05. No exclusion criteria were used, and no data points or animals were excluded from analysis. Investigators were blinded to experimental groups during tumor volume monitoring.
Figure 1.
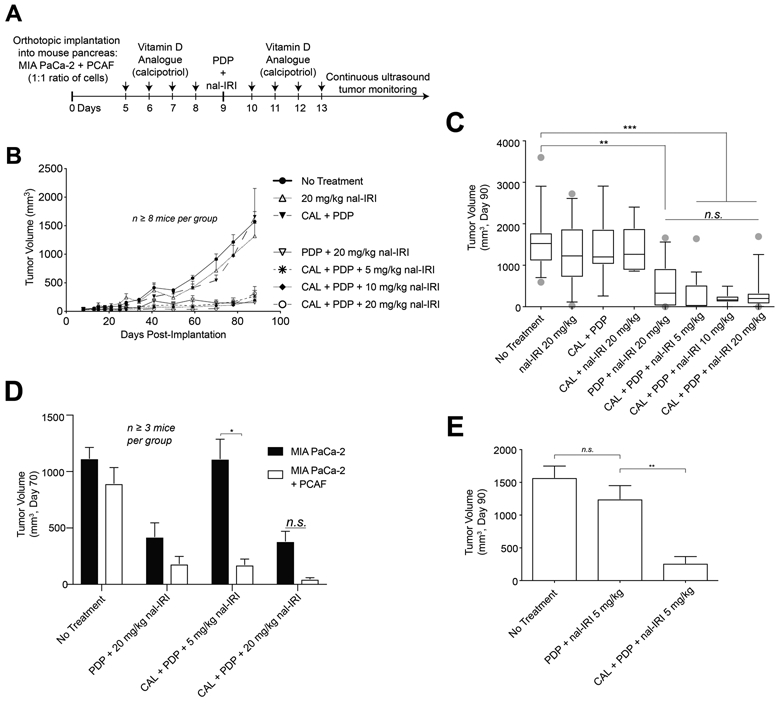
CAL+PDP enable a 75% dose de-escalation of nal-IRI without compromising efficacy.
(A) MIA-PaCa-2 or MIA-PaCa-2 + PCAF were implanted orthotopically into the pancreas of nude mice. CAL treatments were performed daily on days 5-8 and 10-14 post-implantation. PDP and/or nal-IRI were administered on day 9. (B) MIA-PaCa-2 + PCAF tumors were longitudinally monitored via non-invasive ultrasound imaging. (C) Tumor volumes were measured near day 90 prior to humane euthanization. Box and whisker plots denoting the 10th – 90th percentiles are shown. Mice treated with CAL+PDP+20 mg/kg nal-IRI exhibited prolonged tumor control for up to 90 days following implantation compared to no treatment controls (314.7 ± 120.0 mm3 vs. 1573 ± 175.8 mm3, n ≥ 14 mice per condition, p < 0.001, Kruskal-Wallis test with Dunn’s multiple comparison test). There was no difference in the tumor volume of mice treated with CAL+PDP+20 mg/kg nal-IRI vs CAL+PDP+5 mg/kg nal-IRI 90 days post-implantation (314.7 ± 120.0 vs. 265.3 ± 101.0 mm3 , n ≥ 14 mice per condition, Kruskal-Wallis test with Dunn’s multiple comparison test). (D) Seventy days following implantation, MIA PaCa-2 only tumors treated with CAL+PDP+5 mg/kg nal-IRI were significantly larger than MIA-PaCa-2 + PCAF tumors treated with the same regimen (1113 ± 174.5 mm3 vs.174.5 ± 50.2 mm3; n ≥ 3 mice per condition, two tailed t-test, p < 0.05). (E) CAL is necessary for durable low dose triple therapy in mice harboring MIA-PaCa-2 + PCAF xenografts.
Figure 2.
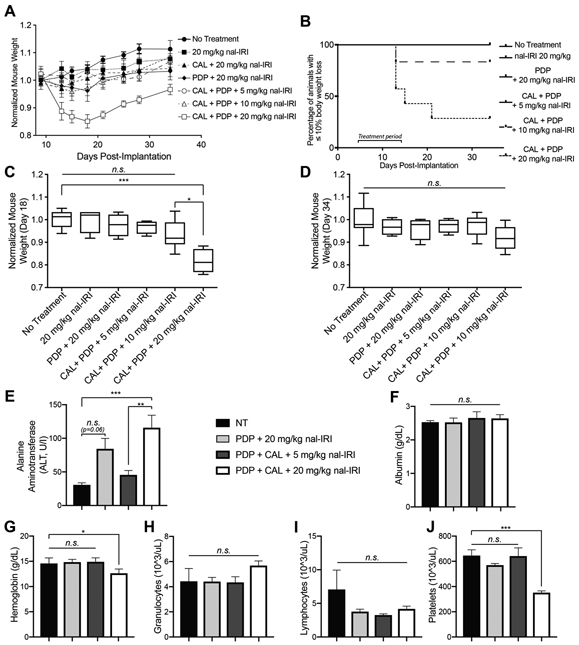
Dose de-escalation of nal-IRI from 20 mg/kg to 5 mg/kg results in decreased toxicity and improved tolerability of triple therapy.
(A) Animal weights were longitudinally monitored every 2-4 days from day 9 to 34 post-implantation. (B) The percentage of animals whose weight loss exceed 10% of the initial body mass is plotted (n ≥ 6 animals per group). For the groups in which animal weight loss did not exceed this threshold, a single representative black line is shown (No Treatment, 20 mg/kg nal-IRI, PDP+20 mg/kg nal-IRI, and CALal+PDP+5 mg/kg nal-IRI). Normalized animal body mass is plotted 18 days (C) and 34 days (D) post implantation. Box and whisker plots denoting the range of the data are shown. The normalized weight of animals treated with CAL+PDP+20 mg/kg nal IRI was significantly lower than those treated with CAL+PDP+5 mg/kg nal-IRI and untreated animals (0.8176 ± 0.023 vs. 1.000 ± 0.015 and 0.9679 ± 0.029 respectively; n ≥ 5 animals per group, p<0.05, Kruskal-Wallis with Dunn’s multiple comparisons test). (E) The serum alanine aminotransferase (ALT) (E), albumin (F), hemoglobin (G), granulocyte (H), lymphocyte (I), and platelet (J) levels were determined via analysis of mouse blood. The ALT levels of mice treated with CAL+PDP+20 mg/kg nal-IRI was significantly greater than those treated with CAL+PDP+5 mg/kg nal-IRI and untreated animals (115.5 ± 18.9 U/I vs. 45.4 ± 6.7 U/I and 30.6 ± 3.3 U/I respectively; n ≥ 5 animals per group, p<0.01, Kruskal-Wallis with Dunn’s multiple comparisons test). The ALT of animals treated with CAL+PDP+5 mg/kg nal-IRI was not significantly different from untreated animals (45.4 ± 6.7 U/I vs. 30.6 ± 3.3 U/I; n ≥ 5 animals per group, Kruskal-Wallis with Dunn’s multiple comparisons test). Animals in the PDP+20 mg/kg nal-IRI group experienced elevations in ALT, though not significantly different from untreated animals (84.20 ± 15.6 vs. 30.6 ± 3.3; n ≥ 5 animals per group, p=0.06). The hemoglobin and platelet counts of animals treated with CAL+PDP+20 mg/kg nal-IRI were significantly lower than those treated with CAL+PDP+5 mg/kg nal-IRI and untreated animals (n ≥ 4 animals per group, p<0.05, Kruskal-Wallis with Dunn’s multiple comparisons test for each platelet count and hemoglobin count).
Figure 3.

Cal + PDP reduces CXCL12 and CXCR7 expression in vivo.
(A) PCAFs secrete significantly more CXCL12 than MIA-PaCa-2 cells in vitro (1103 ± 47.1 pg/mL vs. 275.5 ± 33.7 pg/mL; n=7 wells per group, p<0.001). (B) Representative immunofluorescence images are shown of CXCL12 (yellow, top row), CXCR7 (green, bottom row), and DAPI in MIA-PaCa-2 No Treatment, and MIA PaCa-2 + PCAF tumors subjected to (i) No Treatment, (ii) PDP, (iii) CAL, (iv) CAL+PDP, (v) CAL+PDP+20 mg/kg, nal-IRI 14 days after implantation (corresponding to the end of the treatment period). Scale bars are 50 μm. To quantify immunofluorescence intensities, 6-10 images evenly across the tumor cross section were recorded from at least 2 tumor samples per condition. The total CXCL12, CXCR7, and CXCR4 fluorescence for each image was normalized to the DAPI area from the same image. (C) Relative CXCL12 intensity was found to be significantly higher in the +PCAF NT condition compared to MIA-PaCa-2 NT 14 days after implantation (2.81 ± 0.29 vs 1.01 ± 0.11; n ≥ 20 images from ≥ 2 animals per group and ≥ 6 slides per animal, p<0.001, Kruskal-Wallis with Dunn’s multiple comparisons test). Relative CXCL12 intensity for +PCAF CAL+PDP and +PCAF CAL+PDP+nal-IRI was significantly lower than +PCAF NT at the same time point (0.551 ± 0.12 and 1.254 ± 0.15 respectively vs. 2.805 ± 0.29, n ≥ 12 images, p < 0.05, Kruskal-Wallis with Dunn’s multiple comparisons test). (D) Relative CXCR7 intensity was found to be significantly higher in the +PCAF NT condition compared to MIA-PaCa-2 NT 14 days after implantation (2.39 ± 0.32 vs. 1.01 ± 0.09, n ≥14 images, p<0.001, One-way ANOVA with Tukey’s multiple comparison test). (D) Relative CXCR7 intensity for +PCAF CAL+PDP was significantly lower than +PCAF NT at the same time point (1.06 ± 0.15 vs. 2.39 ± 0.32, n ≥ 14 images, p < 0.05, One-way ANOVA with Tukey’s multiple comparison test). (E) No statistical differences were detected in the CXCR4 intensity at this time point (n ≥ 14 images, p = 0.77, Kruskal-Wallis). Box and whisker plots denoting the 10th – 90th percentile of all data are shown in C-E.
Results:
PDP + VDR activation enables durable low dose nal-IRI in vivo
We have previously reported that PDP and nal-IRI synergistically interact to control PDAC tumor burden (10). We hypothesized that a triple therapy comprising VDR activation, PDP, and nal-IRI could achieve efficient long-term tumor control at lower nal-IRI doses. To better recapitulate the pancreatic ductal adenocarcinoma (PDAC) microenvironment in vivo, an orthotopic xenograft model of PDAC consisting of patient derived pancreatic cancer associated fibroblasts (PCAF; 24) and MIA-PaCa-2 cells was established (Fig. S1). PCAFs express functional VDR that translocates to the nucleus following incubation with CAL in vitro, indicating VDR activation (Fig. S2, 26). Mice bearing PCAF-PDAC xenografts exhibited more established desmoplasia as determined by a pancreatic pathologist’s evaluation (grade 2-3 vs. grade 0-1), and significantly greater collagen (Masson’s Trichrome) and αSMA area compared to a PDAC-only model (Fig. S1), suggesting that the in vivo co-implantation model may better recapitulate desmoplasia observed in clinical PDAC (4). Mice bearing MIA-PaCa-2+PCAF xenografts, or MIA-PaCa-2 alone, were treated with CAL, PDP, and/or nal-IRI (Fig. 1A). Co-implantation mice treated with CAL+PDP+20 mg/kg nal-IRI exhibited prolonged tumor control for 90 days following implantation compared to untreated controls (Fig. 1B, S3B. All controls not shown in Fig. 1B are shown in Fig. S3A). Remarkably, dose de-escalation of nal-IRI from 20 to 5 mg/kg did not compromise the efficacy of triple therapy in co-implantation mice (Fig. 1B and C). While mice treated with the double therapy PDP+20 mg/kg nal-IRI also exhibited prolonged tumor control (Fig. 1C), their average tumor mass following excision were not significantly different from untreated mice (Fig. S3B). Because mice treated with CAL+20 mg/kg nal-IRI did not exhibit a reduction in tumor volume compared to untreated controls, the addition of PDP is necessary to yield therapeutic efficacy (Fig. 1C). Treatment efficacy was lost following dose de-escalation from 20 to 5 mg/kg nal-IRI in MIA-PaCa-2 alone mice treated with triple therapy, suggesting that CAL+PDP sustains treatment efficacy by modulation of the stromal compartment rather than the PDAC cells themselves (Fig. 1D). Low dose double therapy, PDP+5 mg/kg nal-IRI, was ineffective by itself, while the addition of CAL to this regimen resulted in durable tumor control (Fig. 1E). Taken together, these data suggest that combined PDP and VDR activation increases the susceptibility of stroma-rich tumors to nal-IRI, and that these therapies act via the tumor stroma to enable a 75% dose de-escalation of nal-IRI without compromising long-term tumor control.
Dose de-escalation of nal-IRI in the triple therapy regimen significantly decreases morbidity and toxicity
Given the similar treatment response between the triple therapy at 5 and 20 mg/kg nal-IRI, we examined whether this reduction in chemotherapy dose improved treatment tolerability. Animal body mass was monitored from days 8 to 34 post tumor implantation (Fig. 2). Triple therapy with 20 mg/kg nal-IRI caused an 18% reduction in mouse body mass by day 18 compared to untreated animals (Fig. 2A and C). Approximately 60% of high dose triple therapy animals exhibited treatment limiting toxicity by day 21, compared to only 16% of triple therapy animals treated with 10 mg/kg nal-IRI and 0% of animals with 5 mg/kg (Fig. 2B). Treatment limiting toxicity was defined as a reduction in body mass of >10%, which meets clinical criteria for cachexia and would be grounds to discontinue treatment (27). Dose de-escalation of nal-IRI from 20 to 10 and 5 mg/kg in triple therapy animals resolved this loss of body mass (Fig. 2C). By day 34, all animals exhibited body mass that was not significantly different from untreated animals (Fig. 2D).
To more sensitively evaluate the toxicity profile of these treatments, the hematologic laboratories alanine aminotransferase (ALT), albumin, hemoglobin (Hb), granulocyte, lymphocyte, and platelet levels were evaluated in untreated animals and those treated with PDP+20 mg/kg nal-IRI, CAL+PDP+5 mg/kg nal-IRI, and CAL+PDP+20 mg/kg nal-IRI. These biomarkers were selected based on the clinical toxicity profile of nal-IRI (28). High dose triple therapy resulted in a significant elevation in ALT 48 hours after treatment, which resolved following dose de-escalation of nal-IRI to 5 mg/kg (Fig. 2E). Furthermore, animals in the PDP+20 mg/kg nal-IRI group also experienced elevations in ALT, though not significantly different from untreated animals (Fig. 2E). There were no changes in the albumin levels in any of the groups following treatment, suggesting that hepatic synthetic function remained intact in all groups despite the transaminitis (Fig. 2F). Triple therapy at 20 mg/kg nal-IRI resulted in a mild but significant drop in Hb and platelet counts, but dose de-escalation to 5 mg/kg resolved this issue (Fig. 2G and J). Although no significant changes were detected in either the granulocyte or lymphocyte counts of in any of the groups (Fig. 2H and I), these data are confounded by the immunocompromised athymic swiss nu/nu mice used in the study.
Dual-targeted stromal modulation suppresses the CXCL12/CXCR7 signaling axis in vivo
Although we found that CAL+20 mg/kg nal-IRI does not inhibit tumor growth, activation of VDR is hypothesized to modulate intratumoral paracrine interactions by reprogramming the tumor associated stroma from an activated to quiescent state at the epigenetic and transcriptomic levels (17,18). One of the many signaling interactions in the tumor microenvironment is the CXCL12/CXCR7/CXCR4 signaling axis, which has been implicated in the disease progression of several cancers, including PDAC (22,29).
The impact of PDP and VDR activation on modulation of this axis was evaluated in MIA-PaCa-2 alone and MIA-PaCa-2+PCAF xenografts (Fig. 3). Consistent with previous reports (16,30), PCAFs secreted significantly more CXCL12 in vitro compared to MIA-PaCa-2 cells (Fig. 3A). CXCR4 and CXCR7 receptors have been shown to be present on MIA-PaCa-2 cells and are responsive to CXCL12. (29).
Expression levels of CXCL12, CXCR4, and CXCR7 in orthotopic tumors were assessed by immunofluorescence ex vivo at the end of the treatment period (day 14, Fig. 3). Significantly more CXCL12 and CXCR7 were detected in untreated animals bearing MIA-PaCa-2+PCAF xenografts compared to MIA-PaCa-2 alone, suggesting that the presence of CAFs increases intratumoral CXCL12/CXCR7 (Fig. 3B-D). Neither CAL nor PDP individually decreased intratumoral CXCL12 nor CXCR7 in co-implantation mice, whereas their combination led to significantly lower expression of both compared to untreated co-implantation animals (Fig. 3C and D). No appreciable differences in CXCR4 expression were observed in any of the treatment groups at day 14 (Fig. 3E). By day 90, statistically significant differences in CXCL12 and CXCR4 expression were detected between untreated MIA-PaCa-2+PCAF and MIA-PaCA-2 alone tumors (Fig. S4), suggesting that the PCAFs chronically stimulate this protumorigenic pathway. Furthermore, all treatment groups in MIA-PaCa-2+PCAF animals exhibited statistically similar CXCL12 compared to the untreated animals at this time, suggesting that multi-cycle treatment may be necessary to sustain CXCL12/CXCR7 inhibition.
PDP+CAL interferes with CAF-PDAC paracrine interactions
A conditioned medium (CM) assay was designed to evaluate the effect of CAL and PDP treatment on paracrine interactions between MIA-PaCa-2 and PCAF cells (Fig. 4A). Secretion of CXCL12 following treatment with benzoporphyrin-derivative (BPD) based-PDP at 0.03 μM*J/cm2 and CAL at 1μM by PDAC, CAF, and normal fibroblasts (MRC5) was quantified by ELISA assays. Consistent with previous reports of CAF paracrine secretion, untreated PCAF and squamous cell carcinoma associated fibroblasts (SCCAF) exhibited a baseline CXCL12 secretion that was approximately 5-fold higher than MRC5 and MIA-PaCa-2 cells (Fig. 4B) (16,30). Notably, PDP was sub-cytotoxic for all three fibroblast lines at this dose, whereas CAL induced mild cytotoxicity in MRC5 and SCCAF (Fig. S5). Importantly, CAL+PDP was sub-cytotoxic in the PCAFs (Fig. S5). Following CAL+PDP, CXCL12 secretion was reduced by approximately 65% in SCCAFs and 49% in PCAFs compared to untreated controls, correlating with the similar acute reduction observed in vivo, while MRC5 and both PDAC lines were unaffected (Fig. 4B). Treatment with CAL or PDP individually did not significantly reduce CXCL12 secretion in any of the lines examined here.
Figure 4.
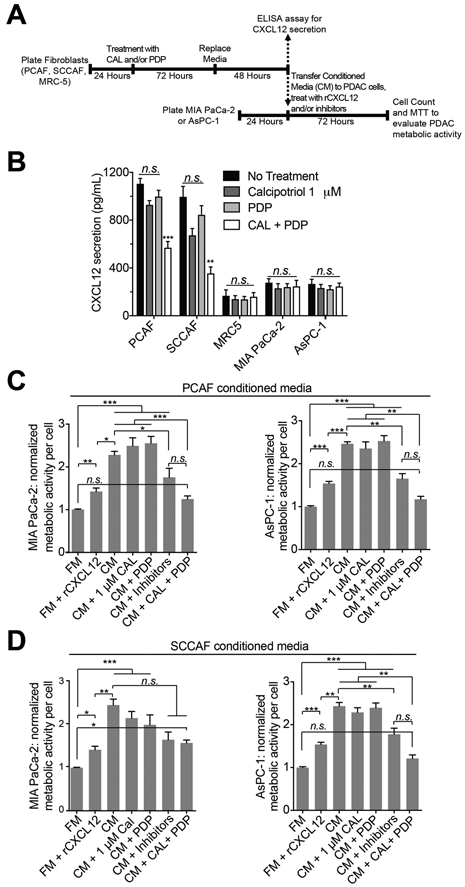
Cal + PDP abrogates CAF induced changes in metabolic activity in PDAC cells, partially through the CXCL12/CXCR4/CXCR7 signaling axis
(A) A conditioned medium assay was designed to evaluate the impact of secreted paracrine factors from CAFs on PDAC cell metabolic activity. (B) PDP+CAL was found to significantly reduce the amount of CXCL12 secreted by CAFs, but not normal fibroblasts or PDAC cells (n = 7 wells per condition, p < 0.001, one-way ANOVA with Tukey’s test, ‘PCAF Cal + PDP’ vs. ‘PCAF NT,’ ‘SCCAF Cal + PDP’ vs. ‘SCCAF NT’). The impact of recombinant CXCL12 (rCXCL12), PCAF CM (C) and SCCAF CM (D) on the metabolic activity of MIA PaCa-2 (left) and AsPC-1 (right) cells is plotted. CM from both PCAF and SCCAF cells increased the normalized metabolic activity of MIA-PaCa-2 and AsPC1 cells by over two-fold (PCAF-MIA-PaCA-2: 2.56 ± 0.16 vs. 1.00 ± 0.014, PCAF-AsPC-1: 2.46 ± 0.049 vs. 1.00 ± 0.020, SCCAF-MIA-PaCa-2: 2.47 ± 0.138 vs. 1.00 ± 0.014, SCCAF-AsPC-1: 2.43 ± 0.086 vs. 1.00 ± 0.02; n ≥ 12 wells per condition, p<0.001, Kruskal-Wallis with Dunn’s multiple comparison test). rCXCL12 and CAF CM induced approximately a 1.5-fold increase in PDAC cell metabolic activity, although CAF CM induced a significantly greater increase in PDAC cell metabolic activity than rCXCL12 (n ≥ 12 wells per condition; p<0.01, Kruskal-Wallis with Dunn’s multiple comparisons test). Treatment of CAFs with CAL+PDP abrogated the CM induced metabolic activity in PDAC cells (n ≥ 12 wells per condition, p<0.001, Kruskal-Wallis with Dunn’s multiple comparisons test, ‘CM + Cal + PDP’ vs. ‘CM’ for all plots) with the exception of MIA-PaCa-2 cells treated with SCCAF CM (n ≥ 12 wells per condition, difference in rank sum = 57.00, Kruskal-Wallis with Dunn’s multiple comparisons test, ‘CM + Cal + PDP’ vs. ‘CM’).
To evaluate the impact of CAF secreted paracrine factors on the metabolic phenotype of PDAC cells, CM from PCAFs and SCCAFs was applied to MIA-PaCa-2 or AsPC-1 cells and metabolic activity per cell was measured. Downstream activity of CXCR4 and CXCR7 has been reported to increase the metabolic activity of PDAC cells (29). CM from both PCAF and SCCAF cells increased the metabolic activity of MIA-PaCa-2 and AsPC-1 cells by two-fold (Fig. 4C and D). Although treatment of PCAFs with CAL+PDP abrogated this effect in both PDAC lines, treatment of SCCAFs with this combination was only effective in AsPC-1 cells (Fig. 4C and D), suggesting that the modulation and effects of secreted paracrine factors is dependent on the CAF origin, consistent with previous reports (31). Notably, the metabolic activity of PDAC cells was unchanged from baseline following exposure to media from MRC5 cells, indicating that any factors secreted by these normal fibroblasts do not grossly affect tumor cell metabolism (Fig. S6). To probe whether the observed increase in PDAC metabolic activity was due to the presence of secreted CXCL12 in the CM, 1 ng/mL recombinant CXCL12 (rCXCL12) was applied to PDAC cells in serum-free fresh medium (Fig. 4C and D). Consistent with previous reports, metabolic activity of both PDAC cell lines increased by approximately 1.5-fold (Fig. 4C and D; 32,33). However, this effect was significantly less pronounced than CM from CAFs, suggesting that other secreted factors also contribute to altered cancer cell metabolism (Fig. 4C and D). To evaluate the specificity of this CXCL12 mediated increase in PDAC metabolic activity and verify its dependency on the canonical CXCL12 signaling axis, PDAC cells were exposed to PCAF or SCCAF CM, AMD3100 and anti-CXCR7 (Fig. 4C and D), well established inhibitors of CXCR4 and CXCR7, respectively (34). PCAF CM+inhibitors caused a significant reduction in the metabolic activity of both PDAC lines compared to PCAF CM alone (Fig. 4C), whereas SCCAF CM+inhibitors caused a significant reduction in only AsPC-1 metabolic activity (Fig. 4D). These results further support the conclusion that the effects of CXCL12 on PDAC metabolic activity is dependent on the CAF origin. Additionally, because there was not a significant difference between the CM+inhibitors and CM+CAL+PDP groups in either PDAC line, it is possible that upstream suppression of CXCL12 induced by CAL+PDP may at least partially phenocopy downstream inhibition of CXCR4 and CXCR7. However, changes in other paracrine interactions also likely occur.
Discussion
Our study suggests that mechanistically diverse approaches, with nonoverlapping toxicities, may be an effective strategy to increase chemotherapy potency. Particularly for PDAC, existing chemotherapeutic regimens require high doses to only modestly extend survival, yet do so at the expense of significant and often life-threatening treatment-induced side effects (2,35,36). First line therapy for locally advanced or metastatic PDAC entails either a combination of 5-fluorouracil (5-FU), leucovorin, oxaliplatin, and irinotecan (FOLFIRINOX), or the less toxic combination of gemcitabine plus abraxane, depending on the patient’s performance status (36). Nanoliposomal irinotecan (nal-IRI), a second line agent used in combination with 5-FU and leucovorin in the metastatic and gemcitabine refractory settings, yields a median overall survival of 6 months despite toxicities such as neutropenia, fatigue, diarrhea, vomiting, nausea, asthenia and abdominal pain (28). Up to 50% of patients treated with nal-IRI and 5-FU may experience severe toxicities, and up to one-third of patients subsequently undergo dose de-escalation to attenuate them, likely at the expense of therapeutic efficacy (28). Strategies that enable reduction of the chemotherapy dose without compromising efficacy hold significant promise to increase the clinical utility of these regimens. The data presented here suggest that this may be accomplished via multi-modal targeting of the tumor microenvironment utilizing agents with distinct mechanisms of action.
Photodynamic and biochemical modulation of the tumor microenvironment maintains the efficacy of nal-IRI despite a 75% dose reduction. Production of RMS following photodynamic activation of Visudyne (PDP) permeabilizes the tumor associated microvasculature (Supplemental methods - Tumor vasculature and Dil5-nal-IRI fluorescence imaging, Fig. S7) and parenchyma to increase intratumoral nal-IRI (10), while PDP+VDR activation transiently suppress pro-tumorigenic CXCL12/CXCR7/CXCR4 signaling. This combined microenvironmental modulation likely increases the susceptibility of primary PDAC tumors to chemotherapy, enabling substantial dose de-escalation while retaining sub-acute (90 day) disease control (Fig. 5).
Figure 5.

VDR activation and PDP modulate the tumor microenvironment for effective low-dose chemotherapy
Pancreatic cancer is often detected at a late stage, after activation of fibroblasts and significant desmoplasia. This protumorigenic microenvironment is immunosuppressive, imparts chemo and radioresistance, and promotes disease progression and metastasis. Standard chemotherapy regimens do not address this altered microenvironment, requiring high chemotherapy doses and subsequent toxicity while providing limited non-sustained benefit. Photodynamic and biochemical reprogramming of the tumor stroma results in a disease state that is more susceptible to chemotherapy, allowing lower doses, while maintaining significant and lasting treatment response.
Although CAL+PDP+nal-IRI, henceforth referred to as triple therapy, with high dose nal-IRI (20 mg/kg) induces cachexia, ALT transaminitis, decreased hemoglobin and platelet counts, it also affords durable tumor control for up to 90 days post implantation. The toxicities associated with high dose triple therapy, which are consistent with those seen clinically, were eliminated following a 75% reduction in the administered nal-IRI dose to 5 mg/kg, while 90 day tumor control was maintained. These results were non-inferior to those seen with PDP+20 mg/kg nal-IRI, which also demonstrated durable 90 day tumor control. Whether PDP+CAL+5 mg/kg nal-IRI represents a superior strategy to PDP+20 mg/kg nal-IRI from the standpoint of efficacy was not explored in this study. An extended (> 90 days) multi-cycle study that includes an evaluation of metastasis would be required to delineate differences between these groups, as the modulatory effects of CAL+PDP are transient in nature and would likely require multiple cycles to fully capture their benefit. While still amenable for rapid translation, further preclinical study of CAL (or related Vitamin D analogs) + PDP is required for optimization of cycle and dosing parameters. However, our study serves as a proof-of-concept that the combination of CAL+PDP induces beneficial modulatory effects in the tumor and microenvironment which substantially increases the potency of chemotherapy, and lays the groundwork for such follow up studies.
The most toxic regimen evaluated in this study was PDP+CAL+20 mg/kg nal-IRI These toxicities were eliminated following dose de-escalation to 5 mg/kg, and by all measures investigated in this study, PDP+CAL+5 mg/kg nal-IRI is as tolerable as PDP+20 mg/kg nal-IRI. In the clinical setting, early phase trials of nal-IRI showed that patients who received lower doses of nal-IRI experience fewer adverse effects than those who received higher doses (37,38). Because incidence and severity of these side effects are dose dependent, the dual-priming strategy presented here may increase the therapeutic options available for toxicity-sensitive patients who would otherwise be ineligible for full-dose conventional chemotherapy regimens (28). Compared to the standard clinical dose of 70 mg/m2, the priming strategy described here enables a human-equivalent dose reduction from approximately 60 mg/m2 to 15 mg/m2 (20 mg/kg to 5 mg/kg in the mouse) (39). Since both paricalcitol, a normocalcemic Vitamin D analog, and Visudyne-PDT are both FDA approved and under separate clinical investigations for PDAC (PDT: NCT03033225; Paracalcitol: NCT03331562, NCT03300921, NCT03520790, NCT03519308), this triple therapy represents a rapidly translatable approach for the management of late stage PDAC. Further study in the phase 1 setting would be required to fully characterize the cumulative toxicity that may be associated with PDP + vitamin D, and weigh this against the beneficial modulatory effects of this regimen.
PDP+nal-IRI for PDAC is based on preclinical evidence suggesting that this combination targets nonoverlapping survival pathways to cooperatively and synergistically reduce tumor burden in vivo (8). Our findings confirm this synergism in a model that incorporates CAFs, and further demonstrates that VDR activation maintains the efficacy of PDP+nal-IRI despite reducing the chemotherapy dose. Importantly, low-dose triple therapy exhibited minimal efficacy in a PDAC model comprised only of MIA-PaCa-2 cells, suggesting that the improved efficacy observed in the co-implantation model is facilitated by PDP+VDR activation within the stromal compartment. This hypothesis is supported by the fact that PCAF cells exhibit approximately 4-fold more VDR than MIA-PaCa-2 cells (Fig. S2B). In addition, VDR activation has been shown to dramatically reduce desmoplasia and increase intratumoral concentrations of chemotherapy in a genetically engineered mouse model of PDAC, a finding not recapitulated in the present study perhaps due to limited desmoplasia at the time of treatment (17). Furthermore, the immunocompromised model used in this study may underreport the beneficial immune stimulatory effects that have separately been shown to occur following VDR activation and PDP (12,16), and further investigation of this CAL+PDP reprogramming strategy is merited in an immunocompetent PDAC model.
Interactions between PDAC cells and the surrounding stromal components have proven to be exceptionally complex. Depletion of the PDAC associated stroma via hyaluronidase or agents that are cytotoxic to CAFs can induce significant treatment-related toxicities (2) and hyperplasia of primary tumor cells with accelerated disease progression, despite suppression of the surrounding stromal desmoplasia (40). Based on a landmark 2009 preclinical study by Olive et al. (41), three clinical studies evaluating the safety and efficacy of hedgehog pathway (sHH) inhibition (via the smoothened inhibitors saridigeb and vismodegib) reported that saridigeb + gemcitabine slightly worsened survival in patients with PDA compared with gemcitabine alone (42-44). Similarly, vismodegib + gemcitabine did not improve survival compared to gemcitabine + placebo in two separate trials. Most recently, a clinical trial evaluating the use of pegylated hyaluronidase (PEGPH20) showed that not only does PEGPH20 + mFOLFIRINOX result in a shorter overall survival and progression free survival compared to mFOLFIRINOX alone, PEGPH20 imparted significant adverse events including thromboembolism and gastrointestinal toxicity. The high rate of toxicity associated with this stromal depletion strategy necessitated significant dose delays and reductions, leading to a lower overall mFOLFIRINOX exposure and reduced efficacy (2). A parallel clinical trial that evaluated the efficacy of gemcitabine/nab-paclitacxel with or without PEGPH20 demonstrated that stromal depletion plus chemotherapy acheived prolonged progression free survival compared to chemotherapy alone only for patients with tumors containing significant amounts of hyaluronic acid. Furthermore, this benefit occurred at the expense of significant thromboembolic events that required anticoagulation prophylaxis (45). These observations support the idea that stromal depletion or ablation could be deleterious. Instead, minimally toxic strategies that modulate the PDAC microenvironment from an activated, or cancer-promoting phenotype, to a quiescent phenotype, without directly killing or depleting stroma, could be more effective in simultaneously reducing toxicity and improving efficacy of anti-PDAC regimens (17,18).
Since CAL+PDP+5 mg/kg nal-IRI was equivalent to PDP+20 mg/kg nal-IRI in terms of efficacy and toxicity, we evaluated the global transcriptomic profiles of tumors following these two regimens (day 14 post implantation) to investigate whether there were differences treatment induced biomolecular changes (supplemental methods – RNA extraction and sequencing). A selected group of significantly altered genes of interest are shown in Figure S8. In general, tumors of mice treated with PDP+20 mg/kg nal-IRI expressed lower levels of tumor suppressor genes, such as microRNA-145, Cyclin Dependent Kinase Inhibitor (CDKN2A), Gasdermin D (GSDMD), Leucine zipper, down-regulated in cancer 1 (LDOC1) and Forkhead Box S1 (FOXS1), while PDP+CAL+5 mg/kg nal-IRI had a minimal effect on expression of these genes. Furthermore, PDP+20 mg/kg nal-IRI treated tumors exhibited higher expression of tumor promoters, such as Denticleless E3 Ubiquitin Protein Ligase Homolog (DTL) and E2F Transcription Factor 7 and 8 (E2F7/E2F8), as well as genes involved in DNA repair, including Breast cancer type 1 susceptibility protein 1 and 2 (BRCA1/BRCA2), DNA Polymerase Theta (POLQ), and X-Ray Repair Cross Complementing 2 (XRCC2). Similarly, PDP+CAL+5 mg/kg nal-IRI had a minor suppressive effect on the expression of these genes. Importantly, these transcriptomic profiles represent the surviving cell populations following treatment, as the majority of cell death has likely occurred by this time. As a whole, the addition of CAL and de-escalation of nal-IRI appears to alter the global transcriptomic profile of stroma-containing tumors in such a way that may make them less aggressive and more susceptible to further cytotoxic therapy in the acute setting.
Sherman et. al. (17) demonstrated through in vitro studies and animal models that CAL reprogrammed the stromal compartment to a quiescent phenotype, reduced desmoplasia, and potentiated gemcitabine efficacy. Given the immense complexity of VDR activation and its multifactorial effects on downstream cellular pathways, the specific consequences of VDR activation on PDAC-stromal paracrine crosstalk are incompletely understood (18). In our model, CAL by itself or in combination with nal-IRI was insufficient for long-term tumor control. Durable tumor control could only be achieved when CAL+PDP were administered in combination with chemotherapy, which correlated with acute suppression of CXCL12 both in vivo and in vitro. To our knowledge, this is the first study to provide evidence that VDR activation in combination with PDP suppresses PDAC-stromal CXCL12/CXCR7 signaling, and that this suppression, perhaps secondary to stromal reprogramming, correlates with improved tumor control in vivo.
Pancreatic cancer cell lines and resected tumors have been shown to express CXCR4 and CXCR7, and exhibit context dependent interactions with CXCL12 (22,29). AMD3100, an antagonist of CXCR4, is currently in clinical trials for a variety of cancers due to its ability to suppress this signaling pathway (NCT03277209, NCT03746080). Although promising, suppression of CXCR4 alone is unlikely to completely eliminate the protumorigenic effects of CXCL12 due to compensatory CXCL12/CXCR7 signaling, as well as recent evidence suggesting that AMD3100 may allosterically agonize CXCR7 (46). The present study demonstrates that PDP+VDR activation transiently suppresses CXCL12 and CXCR7 in vivo, whereas CXCR4 expression remains unaffected. Our in vitro data suggest that PDP+VDR activation directly modulates CAF secretion of CXCL12, upstream from the receptor target of AMD3100 and related therapies. Given that recombinant CXCL12 does not increase PDAC metabolic activity to the same extent as CAF conditioned medium, there are likely additional paracrine factors secreted by CAFs that induce an aggressive phenotype in PDAC cells. This hypothesis is supported by the finding that pretreatment of PDAC cells with inhibitors to CXCR4 and 7 (AMD3100 and anti-CXCR7) does not totally abrogate the effect of CAF conditioned medium on cancer cells, an observation that has also been described previously (47). However, since PDP+CAL does prevent CAF conditioned medium from increasing PDAC cell metabolic activity, we postulate that the modulatory effects of this combination extend beyond the CXCL12/CXCR4/CXCR7 signaling axis. Since the biological consequences of Vitamin D and PDP are extensive, it is likely that the biomolecular mechanisms for improved chemotherapy efficacy observed in this study are due to modulation of multiple converging pathways. While our mechanistic observations are consistent with the treatment response outcomes, it is exciting to envision a more in-depth study at the genomic/transcriptomic level that will elucidate the relationships between these molecular pathways and treatment outcomes.
In summary, we propose that advanced stage tumors, which require complex disease sustaining interactions with stroma to facilitate progression and drug resistance, can be more humanely managed with multi-modal strategies that prime the microenvironment for enhanced tumor responsiveness to chemotherapy. The rapidly translatable strategy presented in this study leverages a mechanistically diverse approach, based on PDP and stromal reprogramming, to offer a new perspective on simultaneously prioritizing treatment and tolerability. This strategy challenges the prevailing paradigm that increasing both the efficacy and tolerability of an anticancer regimen are mutually exclusive endpoints.
Supplementary Material
Acknowledgments:
This work was conducted with support from the Photopathology Center of the Wellman Center for Photomedicine and the Biostatistics Core of Massachusetts General Hospital. We thank Gian-Paulo Dotto and Sandro Goruppi of the MGH Cutaneous Biology Research Center for their generous gift of squamous cell carcinoma associated fibroblasts.
Funding: This work was supported by National Institutes of Health Grants P01CA084203(T. Hasan), UH3CA189901(T. Hasan), and S10ODO1232601(T. Hasan), K99CA194269(H.C. Huang), R00CA194269(H.C. Huang) and R00CA175292(I. Rizvi). S. Anbil was a Howard Hughes Medical Institute Medical Research Fellow.
Footnotes
Competing interests: The authors declare no potential conflicts of interest.
Data and materials availability: We thank Merrimack Pharmaceuticals for providing nanoliposomal irinotecan (nal-IRI).
References:
- 1.Tempero M, Malafa M, Al-Hawary M, Asbun H, Behrman SW, Benson III AB, et al. National Comprehensive Cancer Network Guidelines: Pancreatic Adenocarcinoma, Version 1 [Internet]. 2019.
- 2.Ramanathan RK, McDonough SL, Philip PA, Hingorani SR, Lacy J, Kortmansky JS, et al. Phase IB/II Randomized Study of FOLFIRINOX Plus Pegylated Recombinant Human Hyaluronidase Versus FOLFIRINOX Alone in Patients With Metastatic Pancreatic Adenocarcinoma: SWOG S1313. J. Clin. Oncol 2019;37:1062–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cirri P, Chiarugi P. Cancer-associated-fibroblasts and tumour cells: a diabolic liaison driving cancer progression. Cancer Metastasis Rev. 2012;31:195–208. [DOI] [PubMed] [Google Scholar]
- 4.Neesse A, Michl P, Frese KK, Feig C, Cook N, Jacobetz MA, et al. Stromal biology and therapy in pancreatic cancer. Gut. 2011;60:861–8. [DOI] [PubMed] [Google Scholar]
- 5.Spadi R, Brusa F, Ponzetti A, Chiappino I, Birocco N, Ciuffreda L, et al. Current therapeutic strategies for advanced pancreatic cancer: A review for clinicians. World J. Clin. Oncol 2016;7:27–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huggett MT, Jermyn M, Gillams A, Illing R, Mosse S, Novelli M, et al. Phase I/II study of verteporfin photodynamic therapy in locally advanced pancreatic cancer. Br. J. Cancer 2014;110:1698–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.DeWitt JM, Sandrasegaran K, O’Neil B, House MG, Zyromski NJ, Sehdev A, et al. Phase 1 study of EUS-guided photodynamic therapy for locally advanced pancreatic cancer. Gastrointest. Endosc 2019;89:390–8. [DOI] [PubMed] [Google Scholar]
- 8.Huang H-CH-C, Mallidi S, Liu J, Chiang C-TC-T, Mai Z, Goldschmidt R, et al. Photodynamic Therapy Synergizes with Irinotecan to Overcome Compensatory Mechanisms and Improve Treatment Outcomes in Pancreatic Cancer. Cancer Res. 2016;76:1066–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yu C-H, Yu C-C. Photodynamic Therapy with 5-Aminolevulinic acid (ALA) Impairs Tumor Initiating and Chemo-Resistance Property in Head and Neck Cancer-Derived Cancer Stem Cells. PLoS One. 2014;9:e87129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang H-C, Rizvi I, Liu J, Anbil S, Kalra A, Lee H, et al. Photodynamic Priming Mitigates Chemotherapeutic Selection Pressures and Improves Drug Delivery. Cancer Res. 2018;78:558–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Snyder JW, Greco WR, Bellnier DA, Vaughan L, Henderson BW. Photodynamic therapy: a means to enhanced drug delivery to tumors. Cancer Res. 2003;63:8126–31. [PubMed] [Google Scholar]
- 12.Mroz P, Hashmi JT, Huang Y-Y, Lange N, Hamblin MR. Stimulation of anti-tumor immunity by photodynamic therapy. Expert Rev. Clin. Immunol 2011;7:75–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Spring BQ, Rizvi I, Xu N, Hasan T. The role of photodynamic therapy in overcoming cancer drug resistance. Photochem. Photobiol. Sci 2015;14:1476–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Spring BQ, Bryan Sears R, Zheng LZ, Mai Z, Watanabe R, Sherwood ME, et al. A photoactivable multi-inhibitor nanoliposome for tumour control and simultaneous inhibition of treatment escape pathways. Nat. Nanotechnol 2016;11:378–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Erkan M, Hausmann S, Michalski CW, Fingerle AA, Dobritz M, Kleeff J, et al. The role of stroma in pancreatic cancer: diagnostic and therapeutic implications. Nat. Rev. Gastroenterol. Hepatol 2012;9:454–67. [DOI] [PubMed] [Google Scholar]
- 16.Feig C, Jones JO, Kraman M, Wells RJB, Deonarine A, Chan DS, et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc. Natl. Acad. Sci. U. S. A 2013;110:20212–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sherman MH, Yu RT, Engle DD, Ding N, Atkins AR, Tiriac H, et al. Vitamin D Receptor-Mediated Stromal Reprogramming Suppresses Pancreatitis and Enhances Pancreatic Cancer Therapy. Cell. 2014;159:80–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sherman MH, Yu RT, Tseng TW, Sousa CM, Liu S, Truitt ML, et al. Stromal cues regulate the pancreatic cancer epigenome and metabolome. Proc. Natl. Acad. Sci. U. S. A 2017;114:1129–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gollnick SO, Evans SS, Baumann H, Owczarczak B, Maier P, Vaughan L, et al. Role of cytokines in photodynamic therapy-induced local and systemic inflammation. Br. J. Cancer 2003;88:1772–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kammerer R, Buchner A, Palluch P, Pongratz T, Oboukhovskij K, Beyer W, et al. Induction of Immune Mediators in Glioma and Prostate Cancer Cells by Non-Lethal Photodynamic Therapy. PLoS One. 2011;6:e21834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Domanska UM, Kruizinga RC, Nagengast WB, Timmer-Bosscha H, Huls G, de Vries EGE, et al. A review on CXCR4/CXCL12 axis in oncology: No place to hide. Eur. J. Cancer 2013;49:219–30. [DOI] [PubMed] [Google Scholar]
- 22.Sun X, Cheng G, Hao M, Zheng J, Zhou X, Zhang J, et al. CXCL12 / CXCR4 / CXCR7 chemokine axis and cancer progression. Cancer Metastasis Rev. 2010;29:709–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guo J-C, Li J, Zhou L, Yang J-Y, Zhang Z-G, Liang Z-Y, et al. CXCL12-CXCR7 axis contributes to the invasive phenotype of pancreatic cancer. Oncotarget. 2016;7:62006–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Waghray M, Yalamanchili M, Dziubinski M, Zeinali M, Erkkinen M, Yang H, et al. GM-CSF Mediates Mesenchymal-Epithelial Cross-talk in Pancreatic Cancer. Cancer Discov 2016;6:886–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pigula M, Huang H-C, Mallidi S, Anbil S, Liu J, Mai Z, et al. Size-dependent Tumor Response to Photodynamic Therapy and Irinotecan Monotherapies Revealed by Longitudinal Ultrasound Monitoring in an Orthotopic Pancreatic Cancer Model. Photochem. Photobiol. 2019;95:378–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Racz A, Barsony J. Hormone-dependent translocation of vitamin D receptors is linked to transactivation. J. Biol. Chem 1999;274:19352–60. [DOI] [PubMed] [Google Scholar]
- 27.Davis MP, Dickerson ED. Cachexia and anorexia: cancer’s covert killer. Support. Care Cancer 2000;8:180–7. [DOI] [PubMed] [Google Scholar]
- 28.Ipsen. Onivyde (package insert). 2017.
- 29.Heinrich EL, Lee W, Lu J, Lowy AM, Kim J. Chemokine CXCL12 activates dual CXCR4 and CXCR7-mediated signaling pathways in pancreatic cancer cells. J. Transl. Med 2012;10:68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Demir IE, Kujundzic K, Pfitzinger PL, Saricaoglu ÖC, Teller S, Kehl T, et al. Early pancreatic cancer lesions suppress pain through CXCL12-mediated chemoattraction of Schwann cells. Proc. Natl. Acad. Sci. U. S. A 2017;114:E85–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sazeides C, Le A. Metabolic Relationship between Cancer-Associated Fibroblasts and Cancer Cells. Adv. Exp. Med. Biol 2018. page 149–65. [DOI] [PubMed] [Google Scholar]
- 32.Shen B, Zheng M-Q, Lu J-W, Jiang Q, Wang T-H, Huang X-E. CXCL12-CXCR4 Promotes Proliferation and Invasion of Pancreatic Cancer Cells. Asian Pacific J. Cancer Prev 2013;14:5403–8. [DOI] [PubMed] [Google Scholar]
- 33.Singh S, Srivastava SK, Bhardwaj A, Owen LB, Singh AP. CXCL12–CXCR4 signalling axis confers gemcitabine resistance to pancreatic cancer cells: a novel target for therapy. Br. J. Cancer 2010;103:1671–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hatse S, Princen K, Bridger G, De Clercq E, Schols D. Chemokine receptor inhibition by AMD3100 is strictly confined to CXCR4. FEBS Lett. 2002;527:255–62. [DOI] [PubMed] [Google Scholar]
- 35.Conroy T, Desseigne F, Ychou M, Bouché O, Guimbaud R, Bécouarn Y, et al. FOLFIRINOX versus Gemcitabine for Metastatic Pancreatic Cancer. N. Engl. J. Med 2011;364:1817–25. [DOI] [PubMed] [Google Scholar]
- 36.Ryan D Chemotherapy for advanced exocrine pancreatic cancer - UpToDate. 2019.
- 37.Chen L, Chang T, Cheng A, Yang C, Shiah H, Chang J, et al. Phase I study of liposome encapsulated irinotecan (PEP02) in advanced solid tumor patients. J. Clin. Oncol. 2008;26:2565–2565. [Google Scholar]
- 38.Ko AH, Tempero MA, Shan YS, Su WC, Lin YL, Dito E, et al. A multinational phase 2 study of nanoliposomal irinotecan sucrosofate (PEP02, MM-398) for patients with gemcitabine-refractory metastatic pancreatic cancer. Br. J. Cancer 2013;109:920–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.USFDA Guidance for Industry: Estimating the Maximum Safe Starting Dose in Initial Clinical Trials for Therapeutics in Adult Healthy Volunteers [Internet]. 2005.
- 40.Özdemir BC, Pentcheva-Hoang T, Carstens JL, Zheng X, Wu C-C, Simpson TR, et al. Depletion of Carcinoma-Associated Fibroblasts and Fibrosis Induces Immunosuppression and Accelerates Pancreas Cancer with Reduced Survival. Cancer Cell. 2014;25:719–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D, et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science. 2009;324:1457–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ko AH, LoConte N, Tempero MA, Walker EJ, Kate Kelley R, Lewis S, et al. A Phase I Study of FOLFIRINOX Plus IPI-926, a Hedgehog Pathway Inhibitor, for Advanced Pancreatic Adenocarcinoma. Pancreas. 2016;45:370–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim EJ, Sahai V, Abel E V, Griffith KA, Greenson JK, Takebe N, et al. Pilot Clinical Trial of Hedgehog Pathway Inhibitor GDC-0449 (Vismodegib) in Combination with Gemcitabine in Patients with Metastatic Pancreatic Adenocarcinoma. Clin Cancer Res. 2014;20:5937–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Catenacci DVT, Junttila MR, Karrison T, Bahary N, Horiba MN, Nattam SR, et al. Randomized Phase Ib/II Study of Gemcitabine Plus Placebo or Vismodegib, a Hedgehog Pathway Inhibitor, in Patients With Metastatic Pancreatic Cancer. J. Clin. Oncol 2015;33:4284–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hingorani SR, Zheng L, Bullock AJ, Seery TE, Harris WP, Sigal DS, et al. HALO 202: Randomized Phase II Study of PEGPH20 Plus Nab-Paclitaxel/Gemcitabine Versus Nab-Paclitaxel/Gemcitabine in Patients With Untreated, Metastatic Pancreatic Ductal Adenocarcinoma. J. Clin. Oncol 2018;36:359–66. [DOI] [PubMed] [Google Scholar]
- 46.Kalatskaya I, Berchiche YA, Gravel S, Limberg BJ, Rosenbaum JS, Heveker N. AMD3100 is a CXCR7 ligand with allosteric agonist properties. Mol. Pharmacol 2009;75:1240–7. [DOI] [PubMed] [Google Scholar]
- 47.Gao Z, Wang X, Wu K, Zhao Y, Hu G. Pancreatic Stellate Cells Increase the Invasion of Human Pancreatic Cancer Cells through the Stromal Cell-Derived Factor-1/CXCR4 Axis. Pancreatology. 2010;10:186–93. [DOI] [PubMed] [Google Scholar]
- 48.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. Sequence analysis STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Anders S, Pyl PT, Huber W. HTSeq - a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Robinson MD, Mccarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.