

Abstract
Fibroblast growth factor receptors (FGFR) are commonly altered in non-small cell lung cancer (NSCLC). FGFRs activate multiple pathways including RAS/RAF/MAPK, PI3K/AKT and STAT, which may play a role in the cellular response to radiation. We investigated the effects of combining the selective FGFR 1–3 tyrosine kinase inhibitor AZD4547 with radiation in cell line and xenograft models of NSCLC. NSCLC cell lines were assessed with proliferation, clonogenic survival, apoptosis, autophagy, cell cycle, and DNA damage signaling and repair assays. In vivo xenografts and immunohistochemistry were used to confirm in vitro results. NSCLC cell lines demonstrated varying degrees of FGFR protein and mRNA expression. In vitro clonogenic survival assays showed radiosensitization with AZD4547 in two NSCLC cell lines. In these two cell lines, an increase in apoptosis and autophagy was observed with combined radiation and AZD4547. The addition of AZD4547 to radiation did not significantly affect γH2AX foci formation. Enhanced xenograft tumor growth delay was observed with the combination of radiation and AZD4547 compared to radiation or drug alone. Immunohistochemistry results revealed inhibition of pMAPK and pS6 and demonstrated an increase in apoptosis in the radiation plus AZD4547 group. This study demonstrates that FGFR inhibition by AZD4547 enhances the response of radiation in FGFR expressing NSCLC in vitro and in vivo model systems. These results support further investigation of combining FGFR inhibition with radiation as a clinical therapeutic strategy.
Keywords: FGFR, non-small cell lung cancer, radiation, radiosensitization
Introduction
Non-small cell lung cancer (NSCLC) can be categorized into distinct molecular subtypes driven by specific genomic alterations1. In lung adenocarcinomas, molecularly-targeted therapeutics have considerably improved outcomes in patients harboring EGFR, BRAF and ERBB2 mutations as well as those with ALK, RET, and ROS1 translocations2. In contrast to adenocarcinomas, lung squamous cell carcinomas (SCC) have fewer genomic driver alterations and thus limited targeted therapy options1.
One of the most common and promising targets in lung SCC are the fibroblast growth factor receptors (FGFR). The FGFR family consists of four transmembrane receptor tyrosine kinases: FGFR1, FGFR2, FGFR3, and FGFR43. These receptors activate multiple pathways involved in tumor growth and invasion including the RAS/RAF/MAPK, PI3K/AKT and STAT pathways3. Amplification of FGFR1 at chromosomal region 8p12 is a frequent event seen in 10–20% of lung SCC1,4,5 and mutations of all four FGFR kinases have also been reported1. FGFR1 amplifications have been linked to former and current smokers and have been associated with increased metastatic potential and worse disease-free survival6–9. Preclinical studies have shown that NSCLC harboring focal amplification of FGFR1 are dependent on FGFR1 activity for cell growth5 and are sensitive to small-molecule FGFR kinase inhibitors4,5,10. Multiple clinical studies are now assessing the utility of these small molecule inhibitors in patients with NSCLC harboring aberrations in the FGFR pathway11,12.
Radiation plays an important role in treating locally advanced NSCLC. With a 5-year overall survival rate around 15–30%13, locally advanced NSCLC remains a difficult entity to treat and combining radiation with molecular targeted therapies is one strategy to improve outcomes. Emerging data has shown that the FGFR signaling pathway plays a role in the cellular response to radiation14,15. Given the high rate of FGFR1 amplification in lung SCC, we tested the ability of the pan-FGFR inhibitor AZD454716 to augment radiation response across a series of NSCLC cell lines and xenografts. We found that AZD4547 increased the radiosensitivity in NSCLC FGFR expressing cell lines and xenograft models. AZD4547 is being investigated in multiple clinical trials and combining it with radiotherapy may be a therapeutic strategy for patients whose tumors have FGFR aberrations.
Materials and Methods
Cell lines and drug
Seven NSCLC cell lines and two immortalized non-cancerous cell lines were obtained from indicated sources, cultured in standard conditions, kept at low passage and identity confirmed via short tandem repeat testing (Supplemental Table 1). Cell lines included two squamous cell carcinomas (NCI-H226 and NCI-H520), four adenocarcinomas (NCI-H2228, NCI-H3122, NCI-H358 and A549), one large cell carcinoma (NCI-1581), a human bronchial epithelial cell line (BeasB2) and a human tonsillar epithelial cell line (HTE). AZD4547, provided by AstraZeneca, was reconstituted in dimethyl sulfoxide (DMSO) and stored at −70 °C for in vitro experiments.
Western blot analysis
Cells were harvested and washed with PBS, lysed with RIPA buffer supplemented 1% (v/v) with phosphatase/protease inhibitor complex (Cell Signaling Technologies #5872) and sonicated. Equal amounts of protein were analyzed by SDS-PAGE, transferred to polyvinylidene diflouride membranes, and probed with specific primary antibodies. Targets were detected with NIR-conjugated anti-mouse and anti-rabbit secondary antibodies (LiCOR) and imaged on a LiCOR Odyssey FC. Specific antibodies and sources are listed in Supplementary Table 2.
Quantitative real time-polymerase chain reaction (qRT-PCR)
RNA was extracted from cultured cells using Allprep RNA Mini Kit (Qiagen, Valencia, CA) and measured by Nanodrop. First strand cDNA synthesis was performed using the SensiFAST kit (Bioline). Pre-designed TaqMan gene specific primers (Life Technologies, Carlsbad, CA) were used (Supplemental Methods). qPCR reactions were set up on an automated robot platform (Gilson, Inc Middleton, WI, USA) and PIPETMAX qPCR assistant. Samples were normalized to normal human lung tissue. All reactions were performed in triplicate from RNA isolated from three independent biological experiments.
DNA-copy number by qRT-PCR
DNA was isolated from cultured cells using DNeasy Mini Kit (Qiagen) and quantified by Nanodrop to 5 μg per sample. FGFR1 and FGFR2 copy number variation was determined by the TaqMan® Copy Number Assay (Life Technologies #Hs04933308_cn). RT-PCR was performed using the 7500 Fast Real-Time PCR System (Life Technologies). Samples were normalized to a positive control, RNAseP, known to have two copies. All reactions were performed in quadruplicate from DNA isolated from three independent biological experiments.
Proliferation assay
Cells were plated in 96-well plates at densities ranging from 2,000 to 10,000 cells/well according to cell type growth rate. Twenty-four hours post-plating, cells were treated with indicated doses of AZD4547 or DMSO control and incubated for 48–72 hours. Once control wells neared full confluence, Cell Counting Kit-8 (CCK8) reagent was added (Dojindo Molecular Technologies) and absorbance measured at 450 nm on a SpectraMax i3 plate reader (Molecular Devices). The absorbance of treated wells was normalized to control wells and the half-maximal inhibitory concentration (IC50) values calculated.
Irradiation
Cells were irradiated with a Xstrahl X-ray System, Model RS225 (Xstrahl, UK) at a dose rate of 3.27 Gy/min at 30 cm FSD, tube voltage of 195 kV, current of 10 mA and filtration with 3 mm Al. Animals were irradiated with a Precision Xray XRAD 320 with 1 Gy/minute delivered at 320 kV/12.5 mA at 50 cm FSD with a beam hardening filter with half-value layer of 4 mm Cu. The delivered dose rate was confirmed by ionization chamber. Mice were shielded with custom-built lead jigs to limit radiation exposure to the rear quarter of the body.
Clonogenic survival assay
Cells were seeded into 6-well plates at specific densities, incubated overnight, and treated with AZD4547 or DMSO. Colony counts from treated wells were normalized to DMSO control and IC50 values were calculated. For the radiation survival clonogenic assay, cells were irradiated as indicated after 1 hour of AZD4547 treatment, and media was refreshed 23 hours later. Once colonies averaged 50 or more cells (12–20 days) in the control wells, plates were fixed and stained with 1% (w/v) crystal violet in methanol, imaged, and colonies of 50 or more cells were counted. Survival curves were generated after normalizing for the amount of AZD4547-induced cell death. The clonogenic survival curve for each condition was fitted to a linear quadratic model (Y = e−[A * × + B * X2]) according to a least squares fit, weighted to minimize the relative distances squared. Each point represents the mean surviving fraction calculated from three independent experiments done in triplicate for each treatment condition. Radiation dose enhancement factors were calculated at 10% survival levels by dividing the mean radiation dose for control conditions by the mean radiation dose after drug exposure. A value > 1.0 indicates enhancement of radiosensitivity.
Apoptotic cell death
Cells were seeded into 6-well plates at defined densities, incubated overnight, exposed to DMSO or 0.1 μM of AZD4547, and irradiated with 3 Gy one hour later. Seventy-two hours post-treatment, cells were trypsinized, washed twice with cold PBS, and stained with FITC Annexin V and propidium iodide according to manufacturer instructions (BD Biosciences #556547). Staining was detected with the Countess II FL Automated Cell Counter (Thermo Fisher Scientific) utilizing GFP and RFP EVOS Light Cubes.
Autophagy
Cells were plated, incubated overnight, and treated with DMSO or 0.1 μM AZD4547 one hour before irradiation with 3 Gy. Seventy-two hours post-treatment, cells were washed with PBS and stained with acridine orange (Sigma-Aldrich #318337) at a concentration of 1 μg/mL in PBS for 15 minutes at 37°C. After staining, cells were washed with PBS, trypsinized, and harvested to create a single cell suspension. Staining was detected with the Countess II FL Automated Cell Counter utilizing GFP and RFP EVOS Light Cubes. Acidic autophagic vesicles were stained bright orange/red, which contrasts with green stained nuclei and dim red stained cytoplasm. An RFP channel intensity threshold was established for each cell line using cells grown in serum (negative control) and serum-free conditions (positive control) in order to differentiate autophagic activity from cytoplasmic staining. Cells meeting or exceeding this intensity threshold were quantified and presented as a percentage of total detected cells in the GFP channel.
Cell cycle analysis
Cells were plated in 6-well plates at densities ranging from 200,000–300,000 cells/well depending on the cell type and growth rate. Twenty-four hours post-plating, cells were treated with the indicated dose of AZD4547 or DMSO control, irradiated with 3 Gy, trypsinized and collected at the indicated time points. Cells were centrifuged at 500 × g for 10 minutes, washed with PBS, pelleted again, and a single cell suspension was created in 0.5 ml PBS. 4.5 ml chilled 70% methanol was forcefully added to each tube and cells were stored in 4°C. Twenty-four hours later, cells were centrifuged at 500 × g for 10 minutes, and a single cells suspension was created in 1 ml solution of propidium iodide (Molecular probes # P3566), Triton X-100 (0.1%) (Sigma #T9284), and RNase A (Thermo Scientific #EN0531) in PBS. The samples were analyzed using the Attune NxT flow cytometer (Thermo Scientific), and the cell cycle distribution was calculated using ModFIT software (Verity Software House, Top-sham, ME).
γH2AX immunofluorescence
Cells were plated in 8-well chamber slides at densities ranging from 5,000 to 9,000 cells/well. Twenty-four hours after plating, they were treated with the indicated dose of AZD4547 or DMSO, irradiated with 3 Gy after 1 hour, and fixed with 70% methanol in PBS at indicated time points. Cells were permeabilized with 0.1% Triton-X 100 in TBST for 15 minutes, blocked with SuperBlock™ (Thermofisher #37515) for 1 hour in 25°C, and incubated with anti-phospho-H2AX primary antibody overnight at 4°C (Supplementary Table 2). Cells were then probed with Alexa Fluor 555 conjugated secondary antibody (CST #4413) for 1 hour at 25°C in the dark, and cover-slipped with Fluoromount G™ containing DAPI.
For in vivo γH2AX experiments, deparaffinized 5 μm slides were washed in 1X PBS, blocked with SuperBlock™ (PBS) (Thermofisher #37515) for 1 hour and incubated with anti-phospho-H2AX primary antibody overnight at 4°C (Supplementary Table 2). Slides were washed thrice with 1X PBS the next day and probed with Alexa Fluor 555 (Life Technologies: A27039) secondary antibody at 1:300 dilution in SuperBlock™ Blocking Buffer for 1 hour at room temperature. Slides were washed thrice with 1X PBS and tissues were mounted using Fluoromount-G™ with DAPI (Invitrogen: 00–4959-52). In vivo and in vitro cells were imaged at 60x magnification using a Nikon A1RS inverted point scanning confocal microscope system. γH2AX foci were counted and data presented as number of foci per cell. For each treatment condition, γH2AX foci per cell were counted in at least 150 cells from three independent experiments.
In vivo immunohistochemistry
5 μm sections from formalin fixed paraffin embedded samples were deparaffinized with Xylene and hydrated through graded solutions of ethanol. Antigen retrieval was conducted in sodium citrate retrieval buffer (pH 6.0) followed by washing in running water. Slides were washed in PBS and then incubated with 0.3% hydrogen peroxide solution. Blocking was carried out using 10% goat serum in PBS and then incubated with the primary antibody (Supplemental Table 2) diluted in 1% goat serum in PBS containing 0.1% Triton X-100 overnight at 4°C. Slides were washed with PBS next day; secondary antibody was used (SignalStain® Boost IHC Detection Reagent (HRP, Rabbit) CST #8114). Staining was detected using diaminobenzidine (Vector Laboratories, Inc. #SK-4100). The slides were counterstained with 1:10 hematoxylin (Thermo Scientific #TA-125-MH) solution for 2 minutes, then dehydrated in ethanol and xylene solutions and sections were covered with coverslip with Cytoseal (Thermo Scientific #8312–4). Three sections were taken from three independent tumors for each control and experimental condition. For the phosph-MAPK and phosph-S6 analysis, images were obtained at 10x magnification and quantified in a blinded manner with ImageJ software (http://rsb.info.nih.gov/ij). The immunohistochemistry (IHC) Image Analysis Toolbox Plug-tool was first used to identify areas of positive staining.17 Images were then converted to the RGB scale and the threshold tool was used to calculate the percentage area positively stained. Cleaved caspase 3–positive cells were manually counted by a blinded individual in 2 high-powered 20x fields for two independent tumors.
Xenograft growth delay studies
Five to six week old female Hsd:athymic Nude-Foxn1nu mice (Envigo) were used for growth delay studies. Mice were kept in the Association for Assessment and Accreditation of Laboratory Animal Care-approved Wisconsin Institute for Medical Research Animal Care Facility. Animals were housed in specific pathogen-free rooms, and their clinical health was evaluated weekly. Studies involving the mice were carried out in accordance with an animal protocol approved by the University of Wisconsin.
NCI-H1581 and NCI-H226 tumor cells were mixed 1:1 with Matrigel (BD Biosciences) and injected subcutaneously into bilateral flanks of nude mice at 1 × 106 cells/site. Tumor volume was measured twice weekly with Vernier calipers and calculated according to the relationship . Once average tumor size reached 200 mm3, mice were randomized into treatment groups (8–10 mice).
Statistical analyses
In vitro experiments were repeated three times and statistical analyses were carried out using a Student’s t-test or one-way ANOVA. Data are presented as the mean ± standard error of the mean (SEM). A p value of <0.05 was considered statistically significant. All graphs and in vitro analyses were performed and graphed using GraphPad Prism version 8.0 (GraphPad Software, San Diego, CA). To analyze the longitudinal tumor growth data (volume) resulting from the xenograft studies, linear mixed models were fit to the log-transformed data using the ‘lme4’ package in R (V 3.6.1).18 The fixed-effect model matrices were parameterized such that all main and interactive effects between treatment groups (vehicle, radiation, AZD4547 and AZD4547+radiation) and time were estimated, while the random-effect structure accommodated for intra-tumor correlation via random intercepts. All models were estimated using the restricted maximum likelihood estimation (REML) criterion, and resulting p-values were calculated using Satterthwaite’s approximation. Model assumptions (normality of error and random effects) were assessed via graphical analysis. The synergistic effect between the two treatments was assessed via inference about the three-way interaction between time, AZD4547, and radiation. Note that an initial volume for a single tumor in the NCI-H226 xenograft was measured below the limit of quantification, this observation was omitted from the statistical model.
Results
FGFR expression varies among NSCLC cell lines
Western blot analysis determined that FGFR1 and FGFR2 proteins were expressed to varying degrees under cell culture conditions in our panel of seven NSCLC cell lines (Figure 1A) and non-cancerous cell lines (Supplemental Figure 1). FGFR3 expression was absent (Figure 1A). FGFR1, FGFR2 and FGFR3 mRNA expression in the NSCLC cell lines varied compared to a normal human lung tissue (Figure 1B). As previously reported, our copy number analysis revealed that NCI-H1581 harbors both FGFR1 and FGFR2 amplification19 and NCI-H520 harbors FGFR1 amplification.20,21 The other five cell lines did not show FGFR1 or FGFR2 amplification (Figure 1C).
Figure 1:
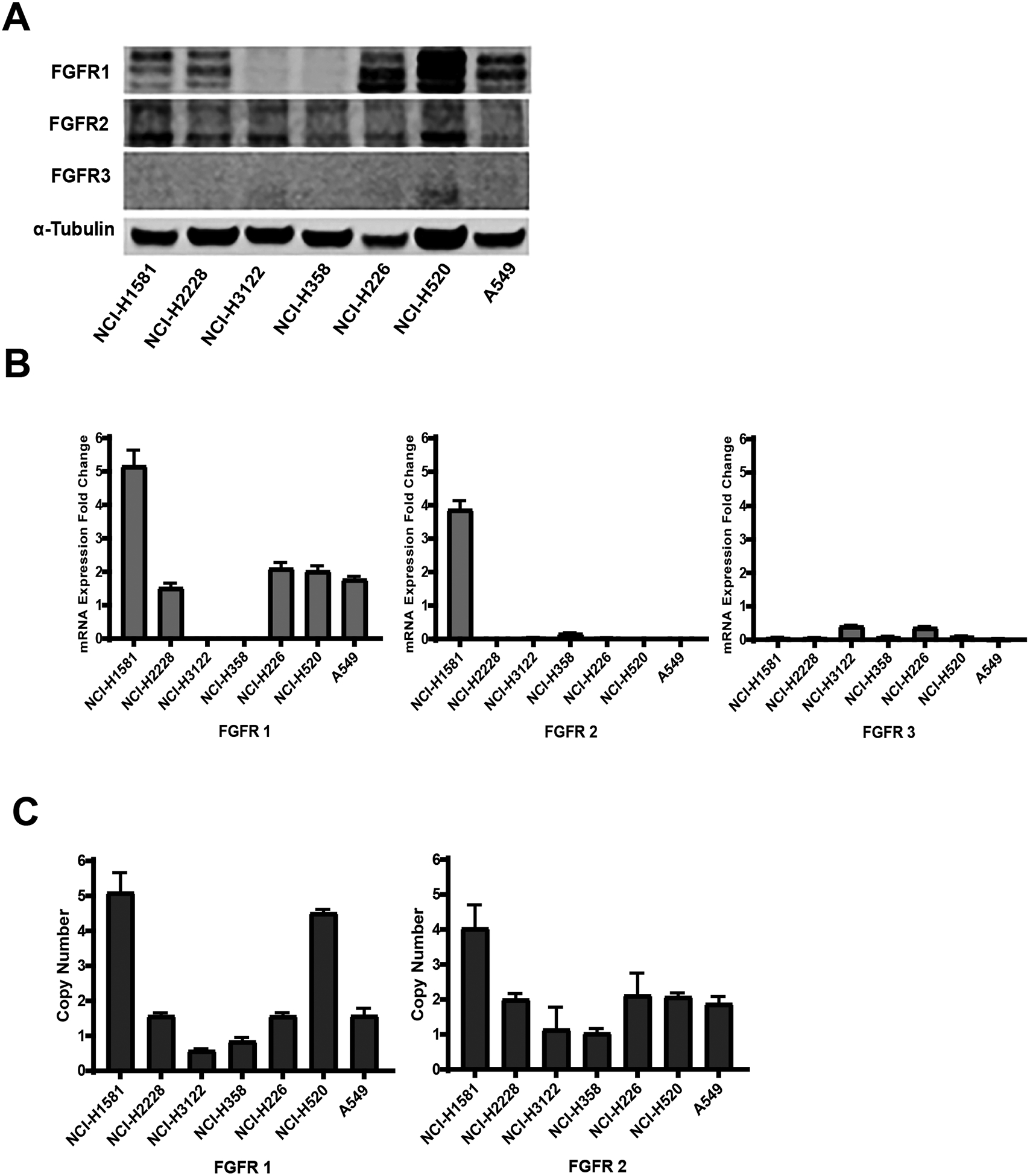
FGFR expression in a panel of seven NSCLC cell lines. (A) Western blot showing FGFR1, FGFR2 and FGFR3 protein levels, (B) FGFR1, FGFR2 and FGFR3 mRNA expression – fold change from normal lung tissue expression (C) FGFR1 and FGFR2 copy number analysis. Columns, mean; bars, SEM (n=3).
AZD4547 inhibits cell growth in vitro
To determine sensitivity to FGFR inhibition, a cell viability assay was performed on the panel of NSCLC cell lines after 72-hour exposure to varying doses of AZD4547. Figure 2 A–G shows the full dose response curves and Figure 2H shows the proliferation IC50 for the seven cell lines. Four cell lines were sensitive to AZD4547 with NCI-H1581 and NCI-H226 being the most sensitive to micromolar concentrations of AZD4547 (Figure 2H). Colony formation assays were performed with NCI-H1581 and NCI-H226 as well as NCI-H520 to confirm sensitivity and determine appropriate drug concentrations for radiosensitization studies. NCI-H520 was included given its known FGFR1 amplification and previous reports showing limited response to AZD4547 in proliferation assays yet higher sensitivity in colony formation assays.20 The colony formation IC50 was 0.014 μM for NCI-H1581, 0.144 μM for NCI-H226 and 2.11 μM for NCI-H520 (Figure 2I).
Figure 2:
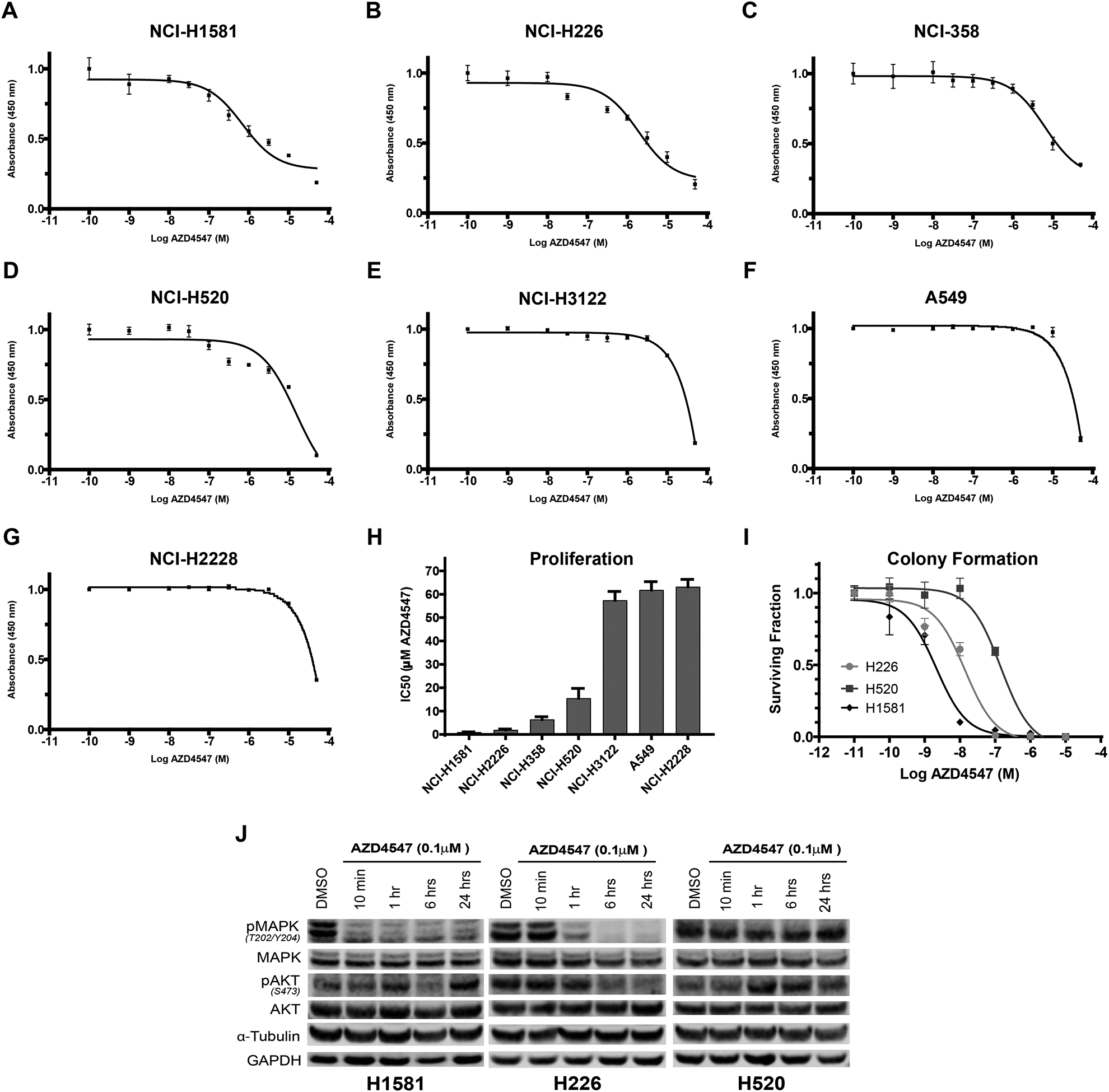
Response to FGFR inhibitor AZD4547 in seven NSCLC cell lines.
AZD4547 proliferation dose response curves for: A) NCI-H1581, (B) NCI-H226, (C) NCI-H358, (D) NCI-H520, (E) NCI-3122, (F) A549, (G) NCI-H2228. (H) Cell line IC50 sensitivity determined by proliferation assay, (I) clonogenic survival following varying doses of AZD4547 of three high-FGFR expressing cell lines, (J) Western blot downstream signaling analysis of AZD4547 treated NSCLC cell lines. NCI-H1581, NCI-H226 and NCI-H520 cells were treated for 10 minutes, 1 hour, 6 hours and 24 hours with 0.1 μM AZD4547. Columns, mean; bars, SEM (n=3).
To assess the effect of AZD4547 on FGFR-associated signaling transduction, we measured phospho-MAPK (Thr202/Tyr204) and phospho-AKT (S473) protein levels in NCI-H1581, NCI-H226, and NCI-H520. AZD4547 at 0.1 μM strongly inhibited phospho-MAPK and a lesser degree phospho-AKT activation in NCI-H1581 and NCI-H226 within 1 hour of exposure. NCI-H520 showed limited inhibition (Figure 2J). Supplemental Figure 2 shows quantification of the western blots.
AZD4547 enhances NSCLC response to radiation
Clonogenic survival assays were used to analyze the effect of AZD4547 treatment on radiation sensitivity in our panel of seven NSCLC lung cancer and two non-cancerous cell lines. AZD4547 radiosensitized both NCI-H1581 and NCI-H226 (p<0.001, Figure 3 A, B). The dose enhancement factor (DEF) at a surviving fraction of 0.1 was 1.31 for NCI-H1581, 1.29 for NCI-H226 and 1.11 for NCI-H520. The other cell lines, including two non-cancerous cell lines, showed minimal to no difference in survival with the combination therapy when compared to radiation treatment alone (Figure 3 C–I and Supplemental Table 3).
Figure 3:
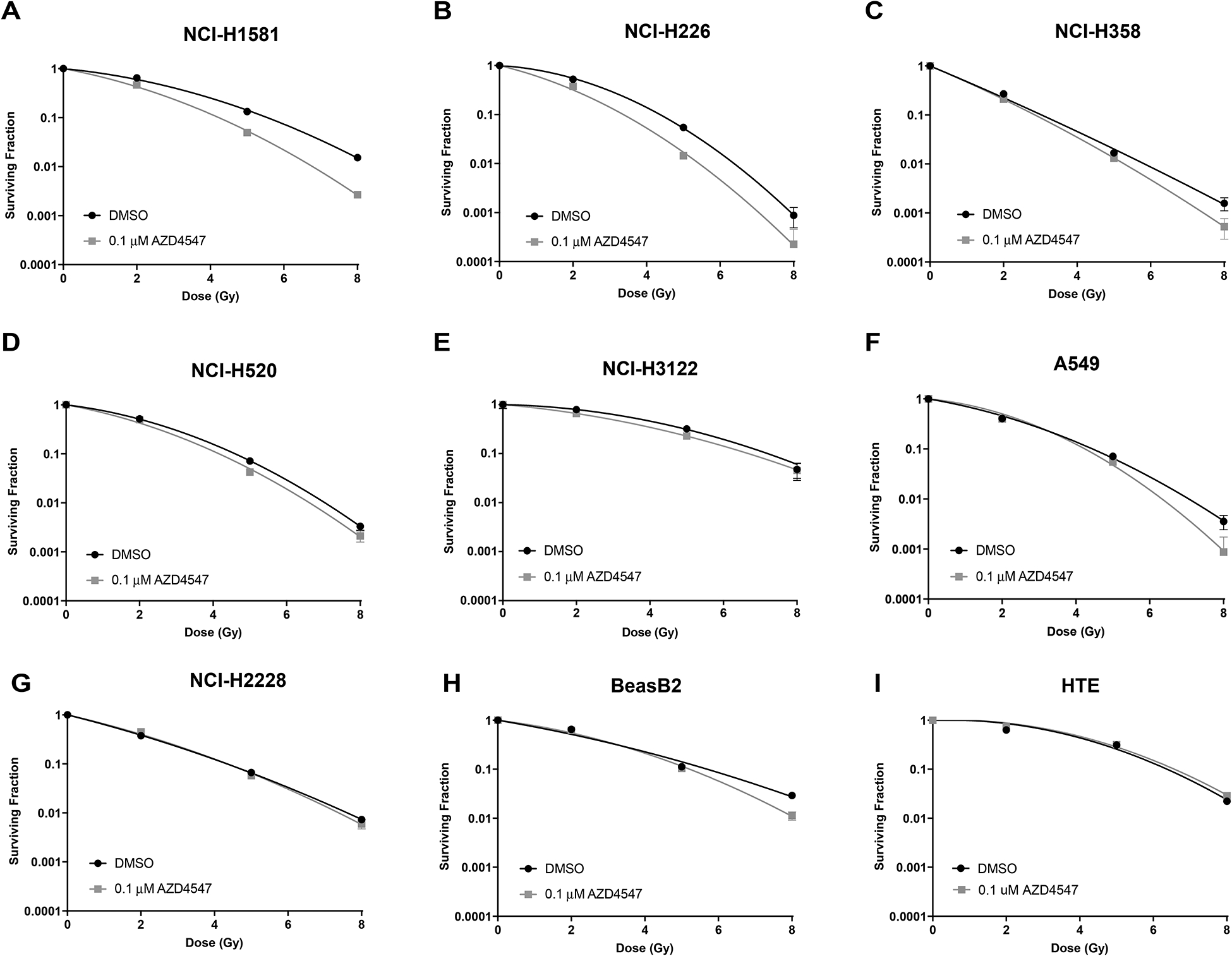
Radiosensitizing effects of AZD4547 on NSCLC cell lines grown in vitro. The radiosensitizing effects of AZD4547 on (A) NCI-H1581, (B) NCI-H226, (C) NCI-H358, (D) NCI-H520, (E) NCI-3122, (F) A549, (G) NCI-H2228, (H) BeasB2, (I) HTE cells. Cell cultures were exposed to 0.1 μM of AZD4547 for 1 h before irradiation and maintained in the medium after irradiation. Colony-forming efficiency was determined 12–20 days later and survival curves were generated after normalizing for AZD4547 cytotoxicity. Points, mean; bars, SEM (n=3).
Apoptosis and autophagy are increased after combined radiation and AZD4547 treatment
To understand the mechanism of cell death following AZD4547 and radiation combination therapy in NCI-H1581 and NCI-H226, markers of apoptosis and autophagy were studied (Figure 4 A, B). AZD4547 alone has been reported to induce apoptosis in several cell lines18. Our results using Annexin V and propidium iodide staining indicate treatment with AZD4547 alone (p=0.0069 for NCI-H1581 and p=0.0053 for NCI-H226) and radiation alone (p=0.001 for NCI-H1581 and p=0.01 for NCI-H226) significantly increased apoptosis in both cell lines compared to DMSO controls. In NCI-H1581, combination therapy significantly increased apoptosis compared to radiation treatment alone (p=0.0154) or drug alone (p=0.0014). Similar trends were seen in NCI-H226 although not statistically significant (Figure 4 A). These results suggest that enhanced apoptosis may play a role in the radiosensitizing effects of AZD4547. Acridine orange staining was used to study autophagy in the two responding cell lines. While AZD4547 alone and radiation alone increase autophagy in both cell lines, the combination therapy significantly increased autophagy in both cell lines when compared to radiation alone (Figure 4 B, p<0.01 for both). Increased autophagy was confirmed in the adherent NCI-H226 cell line using LC3B immunofluorescence (Supplemental Figure 3). These results suggest that enhanced autophagy plays a role in mediating the radiosensitization effects of AZD4547.
Figure 4:
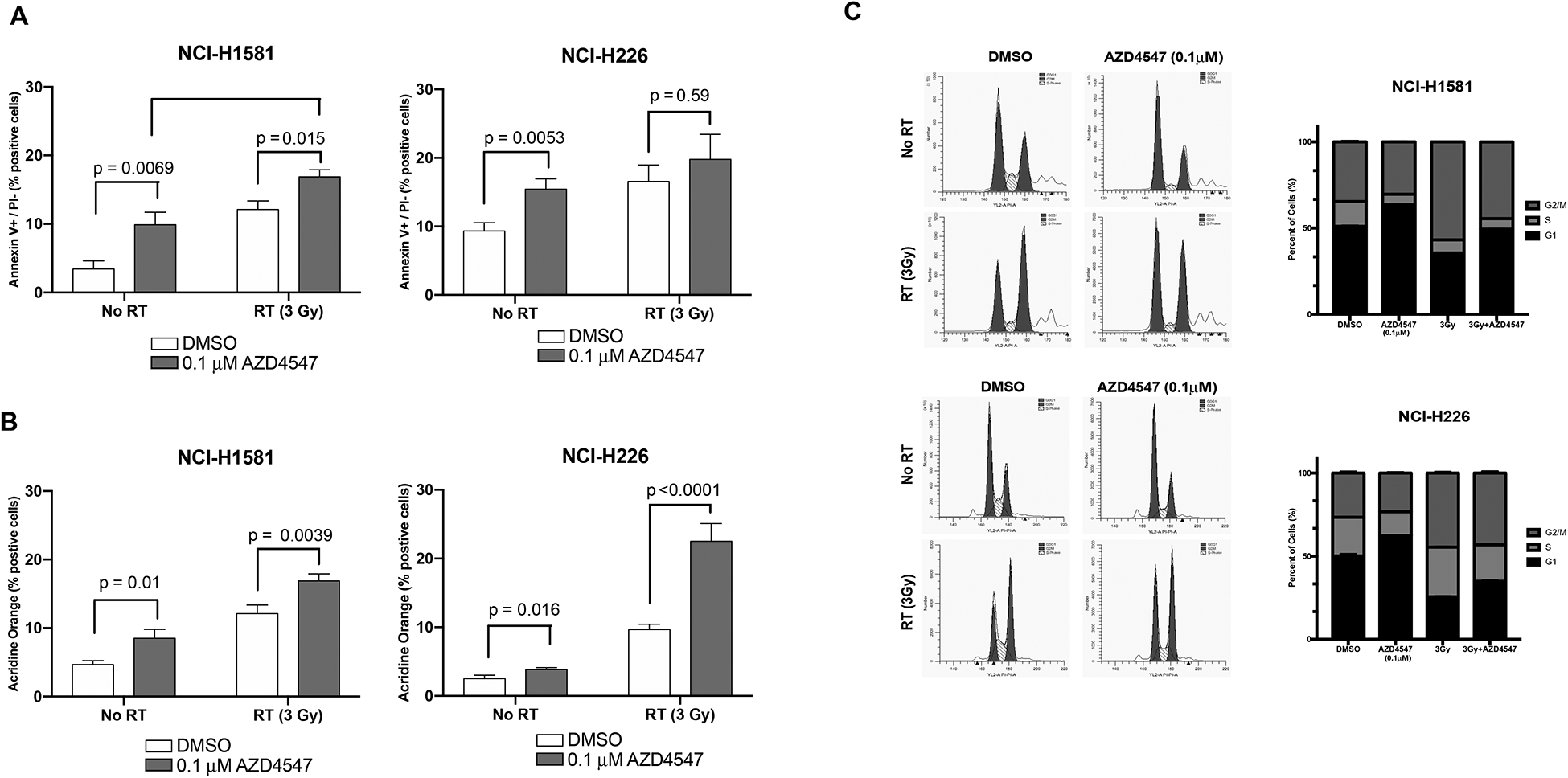
Mechanism of cell death after combined radiation and AZD4547 treatment in NCI-H226 and NCI-H1581 cell lines. Cells were treated with DMSO, AZD4547 (0.1 μM), radiation (3 Gy), or combined AZD4547 (0.1 μM) and radiation (3 Gy) and analyzed at designated time points. (A) Annexin V apoptosis, (B) acridine orange autophagy, and (C) cell cycle arrest analysis. Columns, mean; bars, SEM (n=3). *, P < 0.01, according to t test (irradiation versus AZD4547 plus irradiation).
Redistribution of cell cycle with AZD4547 and radiation treatment
Because redistribution of cells in the cell cycle can affect radiosensitivity, flow cytometry was used to determine the cell cycle phase distribution of cells exposed to 0.1 μM AZD4547, radiation (3 Gy) or both AZD4547 and radiation at 15 hours after treatment. NCI-H1581 and NCI-H226 cells treated with AZD4547 alone showed increased G1 arrest (p<0.001), as has been previously reported.16 Radiation alone caused an accumulation of the G2 population (p<0.001) compared to non-treated cells (Figure 4 C). There was a decrease in the G2 population in NCI-H1581 cells when treated with AZD4547 plus radiation compared to radiation alone (p<0.001). In NCI-H226 cells treated with the combination therapy there was no difference in G2 arrest compared to radiation alone, (p=0.61) (Figure 4 C). These results indicate that redistribution of cells into a radiosensitive phase of the cell cycle is not the main mechanism of radiation-induced cell killing observed in Figure 3.
γH2AX foci after combined AZD4547 and radiation treatment
To determine whether AZD4547 inhibited DNA damage repair, γH2AX expression was evaluated in both the in vitro and in vivo setting. In vitro γH2AX assessment was performed with only NCI-H226 cells as NCI-H1581 cells are a suspension cell line and do not adhere to slides. Both cell lines were used for the in vivo analysis. In the NCI-H226 in vitro experiment, treatment with AZD4547 alone did not enhance induction of γH2AX foci. The combination of AZD4547 plus irradiation did not lead to a statistically significant increase in the number of foci at 3, 8 or 24 hours (Figure 5 A). These findings were confirmed in our in vivo assessment of γH2AX (Figure 5 B). NCI-H1581 also demonstrated no significant difference in foci expression at 24 hours in the AZD4547 plus radiation-treated tumors compared to the radiation alone treated tumors (Figure 5 C).
Figure 5:
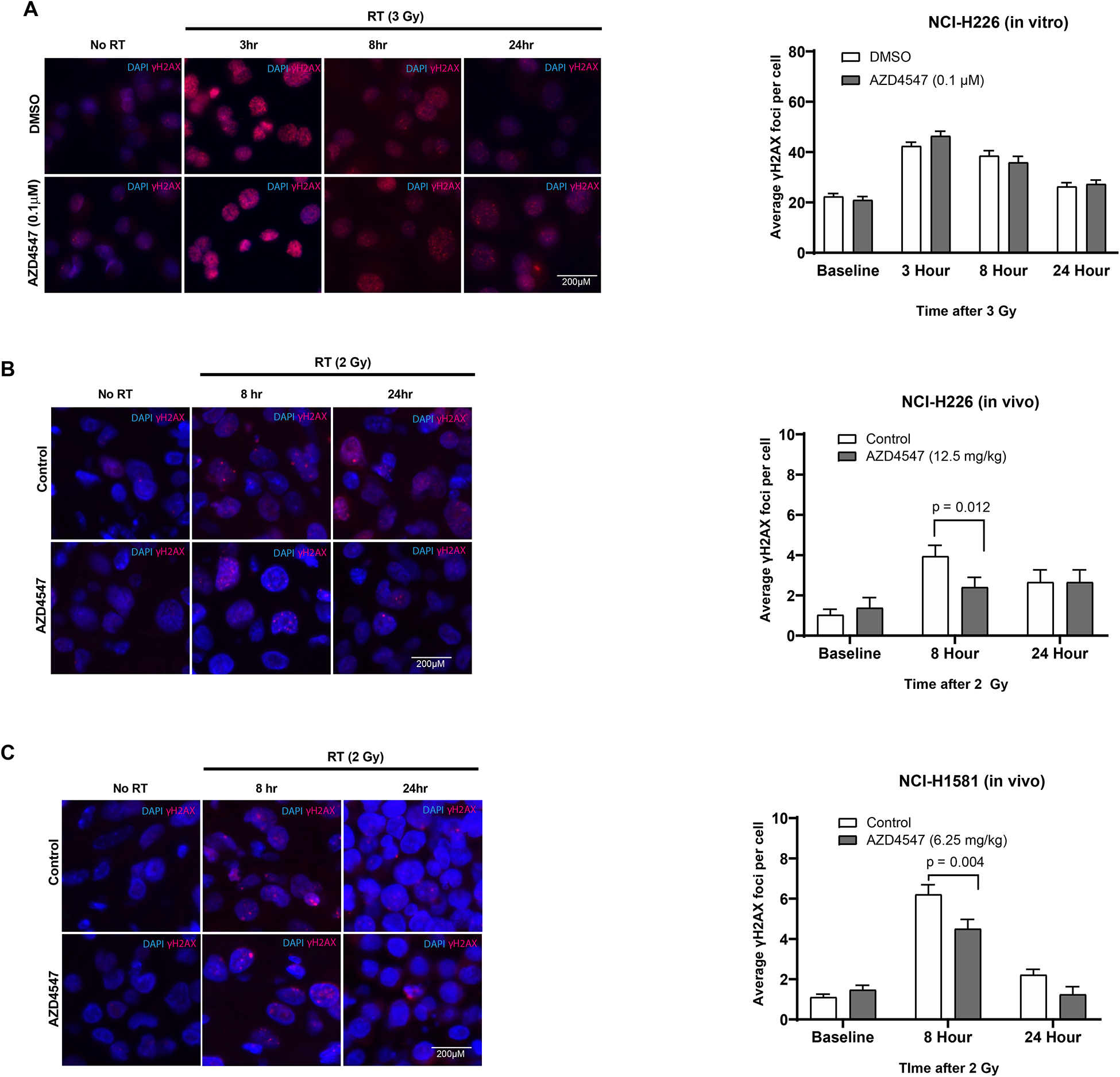
In vitro and in vivo γH2AX analysis after combined radiation and AZD4547 treatment in NCI-H226 and NCI-H1581 cell lines. Representative immunofluorescence γH2AX foci images and foci quantification for (A) NCI-H226 cells grown in vitro, (B) NCI-H226 cells grown in vivo and (C) NCI-H1581 cells grown in vivo. In vitro NCI-H226 cells were treated with DMSO, AZD4547 (0.1 μM), radiation (3 Gy) or combined AZD4547 (0.1 μM) and radiation (3 Gy) and analyzed at designated time points. NCI-H1581 and NCI-H226 xenografts were treated with DMSO, AZD4547 (6.25 or 12.5 mg/kg), radiation (2 Gy single dose) or combined AZD4547 (6.25 or 12.5 mg/kg) and radiation (2 Gy single dose). Columns, mean; bars, SEM (n=3).
AZD4547 enhances radiation response in NSCLC tumor xenografts
Given our in vitro findings, we assessed radiosensitization by AZD4547 using cell line xenografts. Mice bearing NCI-H1581 and NCI-H226 tumors were randomized into four groups: vehicle, AZD4547, radiation, or AZD4547 plus radiation. The mice were given AZD4547 (6.25 mg/kg or 12.5 mg/kg) by p.o. gavage 1 h before local tumor irradiation (2 Gy × 5 or 2 Gy × 10 daily fractions). The combination of AZD4547 and radiation delayed the growth of NCI-H1581 and NCI-H226 tumors compared to vehicle control, AZD4547 or radiation alone (Figure 6 A & B). In both experiments, the growth delay following the combination treatment was more than the sum of the growth delays caused by individual treatments.
Figure 6:
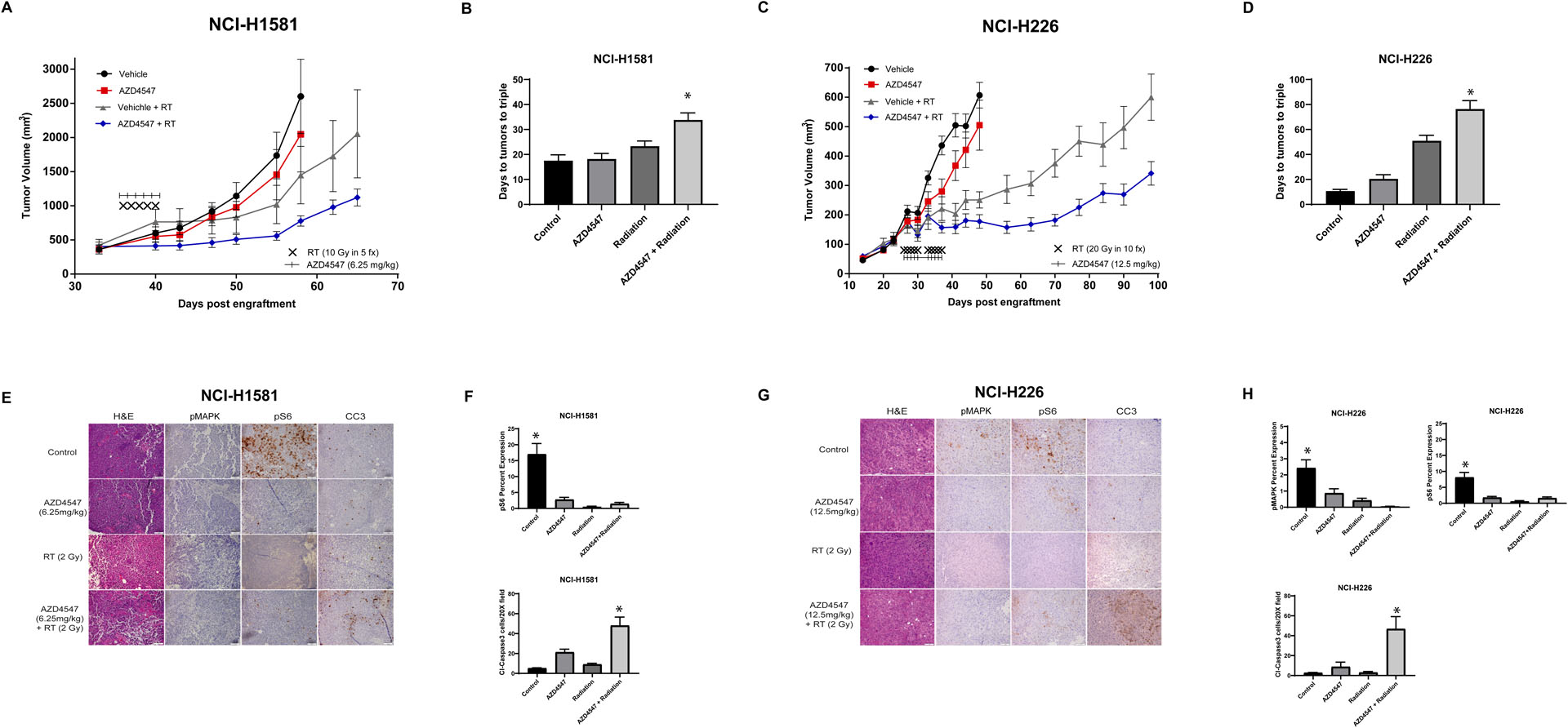
The effects of AZD4547 on radiation-induced xenograft tumor growth delay. Tumor growth delay curves of (A) NCI-H1581 and (C) NCI-H226 subcutaneous flank xenografts treated with vehicle, AZD4547 (6.26 mg/kg or 12.5 mg/kg), radiation (2 Gy × 5 or 2 Gy x10) or AZD4547 (6.25 mg/kg or 12.5 mg/kg) plus radiation (2 Gy × 5 or 2 Gy x10). AZD4547 was delivered as p.o. gavage at 1 h before radiation fraction. (B & D) Time from start of treatment for tumor volumes to triple. IHC analysis of (E) H1581 and (G) NCI-H226 tumor sections showing modulation of FGFR-downstream signaling (pMAPK and pS6) and apoptotic markers (CC3). pMAPK and pS6 analysis were conducted at 8 hours following a single dose of AZD4547 (6.25 mg/kg or 12.5 mg/kg), radiation (2 Gy) or AZD4547 plus radiation whereas CC3 data were collected at 48 hours post-treatment, respectively. (F and H) Percent staining for pMAPK, pS6 at 8 hours post treatment and number of CC3 positive cells after 48 hours of treatment. Each stain was objectively quantified in three high-powered fields for three independent tumors. No pMAPK expression was observed in the NCI-H1581 tumors. The scale bar represents 50 μm. Points, mean tumor volume in mice after treatment; bars, SEM, *, P < 0.05, according to one-way ANOVA.
The mean time for NCI-H1581 xenografts tumors to triple in size from start of treatment was 17.5 days for vehicle treated mice; 18.2 days for AZD4547 treated mice and 23.3 days for irradiated mice. In mice that received AZD4547 plus irradiation, the time for tumors to triple increased to 33.8 days, which was statistically significant compared to the other groups (p< 0.01, Figure 6B). The absolute growth delay (time in days for tumors in treated mice to triple minus the time in days for tumors to reach the same size in vehicle treated mice) were 0.7 days for AZD4547 alone and 5.8 days for irradiation alone, whereas the tumor growth delay induced by the AZD4547 plus irradiation treatment was 16.3 days. In the NCI-H1581 tumors, the model fit of the data did not show evidence of a synergistic effect in regards to the growth curves (p=0.99), however the slope of the combination treatment group was significantly lower than that of the AZD4547 treated group (p<0.0001).
For NCI-H226 tumor xenografts, mean time to triple was 10.7 days for vehicle treated mice; 20.2 days for AZD4547 treated mice and 50.9 days for irradiated mice. In mice that received the AZD4547 plus irradiation, the time for tumors to triple was 75.7 days, which was statistically significant compared to the other groups. (p<0.001, Figure 6D). The absolute growth delay was 9.5 days for AZD4547 alone and 40.2 days for irradiation alone and 65.0 days for AZD4547 plus irradiation treatment. No synergistic effect between radiation and AZD4547 (p=0.88) was observed in the NCI-H226 tumors. Although the slope of the combination treatment group was significantly lower than both radiation treatment (p<0.0001) and AZD4547 alone treatment groups (p<0.0001).
The DEF, obtained by dividing the normalized tumor growth delay in mice treated with both AZD4547 and radiation by the absolute growth delay in mice treated with radiation alone, was 1.8 in the NCI-H1581 tumors and 1.4 in the NCI-H226 tumors. These results indicate that AZD4547 enhances the radiation-induced tumor growth delay in these xenografts.
Markers of downstream FGFR signaling, phospho-MAPK and phospho-S6, were assessed immunohistochemically and quantified at 8 hours post-treatment (Figure 6E and G). Both NCI-H1581 and NCI-H226 tumors demonstrated statistically significant inhibition of pS6 after AZD4547 was given with or without radiation compared to tumors receiving no treatment (Figure 6F and 6H). Significant reduction in p-MAPK was only seen in treated NCI-H226 tumors compared to no-treatment tumors (Figure 6F). p-MAPK expression was not evident in any of the NCI-H1581 tumor samples (Figure 6E). At 48 hours post treatment Cleaved Caspase 3 (CC3), a marker of apoptosis, was most pronounced in the combined AZD4547 and radiation-treated tumors compared to other treatments (Figure 6F, H). When quantified, there was a statistically significant increase in cleaved caspase expression in the radiation and AZD4547 combination group compared to controls (p<0.001 for NCI-H1581 and p=0.029 for NCI-H226), AZD4547 alone (p=0.029 for NCI-H1581 and p=0.084 for NCI-H226) and radiation alone (p=0.003 for NCI-H1581 and p=0.03 for NCI-H226) (Figure 6F and 6H).
Discussion
Radiotherapy in combination with chemotherapy remains the backbone of treatment in unresectable locally advanced NSCLC. Even with this aggressive therapy, overall survival is poor and local recurrence remains high.13 Finding ways to improve the therapeutic ratio of radiotherapy with molecular directed therapies is an ongoing endeavor. In this study, we have focused on FGFR as a target for radiosensitization. We have demonstrated that inhibition of FGFR by the selective tyrosine kinase inhibitor AZD4547 resulted in radiosensitization in both a FGFR expressing and a FGFR amplified NSCLC preclinical model.
Efforts to exploit the high expression and amplification of FGFRs in lung SCC patients are ongoing. Multiple preclinical studies have evaluated several FGFR inhibitors in NSCLC models.19 Wynes et al. tested the sensitivity of 58 lung cancer cell lines to the FGFR inhibitor ponatinib and showed that 15 were sensitive to ponatinib; FGFR1 mRNA and protein expression and not gene copy number predicted for response.19 Alternatively, Kotani et al assessed the response of seven lung cancer cell lines to three FGFR inhibitors, BGJ398, ponatinib and AZD4547, and found FGFR1-amplified and high FGFR1 protein-expressing lung cancers were sensitive to FGFR inhibitor monotherapy and FGFR inhibition resulted in suppression of ERK signaling.22 They also reported a discrepancy between FGFR1 amplification and FGFR1 protein expression in a number of these cells, and the cell lines with low FGFR1 expression were uniformly resistant to the different FGFR inhibitors.22 Zhang et al. established five FGFR1-amplified squamous NSCLC patient-derived tumor xenograft models and demonstrated that AZD4547 induced tumor growth delay in four of five models that harbored FGFR1 amplification.10 These encouraging preclinical results have led to the initiation of clinical studies testing FGFR inhibitors in metastatic lung SCC.
The pan-FGFR inhibitor AZD4547 has been investigated in both a phase Ib and phase II study of patients with lung SCC. The phase Ib trial enrolled 15 patients with previously treated stage IV FGFR1-amplified lung SCCs (NCT00979134).11 In this study, AZD4547 was tolerable at a dosage of 80 mg oral twice a day and showed modest anti-tumor activity.11 The phase II study of AZD4547 was conducted under the LungMAP National Clinical Trials Network umbrella trial for previously treated lung SCC.12 Eligibility for this study included patients with metastatic lung SCC who had progression after one line of systemic therapy and harbored a FGFR1, 2, 3 amplification, mutation or fusion. Forty-three patients enrolled and 28 received AZD4547. FGFR alterations included FGFR1 amplification, defined as ≥6 estimated copies, in 38 patients (86%); FGFR3 S249C mutation in four patients (9%); FGFR3 amplification in 3 patients (7%); and FGFR3 fusion in two patients (5%). Nine patients (26%) had more than one FGFR alteration. The study reported minimal activity of AZD4547 and was closed at interim analysis.12 Besides AZD4547, two other pan-FGFR inhibitors, BGJ398 (infigratinib)23 and dovitinib24, have showed modest clinical response in a few number of patients with FGFR1-amplified lung SCC determined by FISH. These early studies with FGFR inhibitors reported skin and gastrointestinal toxicity, which raises the concern for an increase in esophageal toxicity when combined with thoracic radiotherapy.23,24 However, AZD4547 was well tolerated in the LungMAP study with only one grade 3 gastrointestinal event of oral mucositis being reported.12
Overall, the limited responses seen with these drugs highlight the need for better predictive biomarkers. FGFR1 amplification by FISH appears not to correlate with mRNA expression or be a good predictor for drug response. This could be because of the heterogeneity of the 8p11–12 amplicon, raising the possibility that FGFR1 is not actually being amplified and thus overexpressed.25
While the status of FGFR monotherapy remains unclear in FGFR amplified or expressing lung SCC, combining FGFR inhibition with radiation remains a potential therapeutic strategy. The initial data supporting the FGF/FGFR signaling pathway in regulation of the cellular response to radiation focused on FGFR activation to prevent radiation-induced apoptosis in normal tissues. Work by Fuks and colleagues demonstrated FGF2 protected endothelial cells from radiation-induced apoptosis and administration of FGF2 protected against the development of fatal radiation pneumonitis in mice.14,26 Additional studies have shown FGF2 to mitigate the effects of radiation on gastrointestinal crypt cells and hematopoietic cells through reduction in apoptosis, stem cell renewal, progenitor cell differentiation, and epithelial proliferation.27,28
In tumor cells, aberrant FGFR signaling activates pro-survival pathways including anti-apoptotic pathways through activation of either RAF/MAPK and PI3K/AKT.3 This in turn leads to chemotherapy and radiation resistance.15 Two transcriptome analyses have identified FGFRs as targets for radiosensitization21,29 and two studies have demonstrated radiosensitization with FGFR inhibitors in glioblastoma cells15,30. In our study, the addition of AZD4547 resulted in inhibition of phospho-MAPK and radiation enhancement in two FGFR expressing NSCLC cell lines. This includes NCI-H1581 cells, which harbor FGFR1 amplification and are known to be dependent on FGFR1 signaling, and NCI-H226 cells that do not have FGFR1 amplification but overexpresses FGFR1. We observed an increase in apoptosis and autophagy with the addition of AZD4547 to radiation but did not observe a difference in prolongation of γH2AX or a significant shift in cell G2 cell cycle arrest. These results suggest that radiation-enhancing effect of AZD4547 is not through direct action on DNA repair but rather inactivation of FGFR signaling, leading to enhanced apoptosis and autophagy. The MAPK/ERK pathway appears to be the predominant pathway inhibited by AZD4547 and is responsible for AZD4547-induced apoptosis.16 Reactivation of the canonical MAPK/ERK signaling cascade can lead to AZD4547 resistance.31 AZD4547 also induces autophagy and in our study, when combined with radiation this effect was increased. Others have reported that induction of autophagy in NCI-H1581 and H520 cells by AZD4547 or FGFR1 short hairpin RNA is mediated through inhibition of the MAPK/ERK pathway and not through the AKT pathway.32 While autophagy is commonly thought to play a pro-survival role, in cancer the effects of autophagy are likely more complex and its role in radiosensitization is not fully understood33; further work will be needed to define its role when combining AZD4547 with radiation.
In summary, we have demonstrated in vitro and in vivo radiation enhancement by combining the selective FGFR inhibitor AZD4547 with clinically meaningful doses of radiotherapy in FGFR amplified and expressing NSCLC tumors. This combination holds translational implications given the well-established use of radiotherapy in the treatment of locally advanced NSCLC. These data provide the rationale for continuation of studies combining radiotherapy with molecular agents that inhibit the FGFRs. However, further work is needed to identify those who would benefit from this combination.
Supplementary Material
Funding:
This project was supported in part by grants from the American Cancer Society (RSG-16-091-01-TBG), UW Paul P. Carbone Young Investigator Award and the University of Wisconsin Carbone Cancer Center Support Grant (P30 CA014520), and the Wisconsin Head and Neck Cancer SPORE Grant through the NIH National Institute for Dental and Craniofacial Research (NIDCR) and National Cancer Institute (NCI, P50DE026787). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Footnotes
Disclosures: None
Conflict of Interest: The authors declare no potential conflicts of interest
References
- 1.Cancer Genome Atlas Research N. Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012;489(7417):519–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cancer Genome Atlas Research N. Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511(7511):543–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Turner N, Grose R. Fibroblast growth factor signalling: from development to cancer. Nature reviews Cancer. 2010;10(2):116–129. [DOI] [PubMed] [Google Scholar]
- 4.Weiss J, Sos ML, Seidel D, et al. Frequent and focal FGFR1 amplification associates with therapeutically tractable FGFR1 dependency in squamous cell lung cancer. Science translational medicine. 2010;2(62):62ra93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dutt A, Ramos AH, Hammerman PS, et al. Inhibitor-sensitive FGFR1 amplification in human non-small cell lung cancer. PloS one. 2011;6(6):e20351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goke F, Franzen A, Menon R, et al. Rationale for treatment of metastatic squamous cell carcinoma of the lung using fibroblast growth factor receptor inhibitors. Chest. 2012;142(4):1020–1026. [DOI] [PubMed] [Google Scholar]
- 7.Kim HR, Kim DJ, Kang DR, et al. Fibroblast growth factor receptor 1 gene amplification is associated with poor survival and cigarette smoking dosage in patients with resected squamous cell lung cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2013;31(6):731–737. [DOI] [PubMed] [Google Scholar]
- 8.Cihoric N, Savic S, Schneider S, et al. Prognostic role of FGFR1 amplification in early-stage non-small cell lung cancer. British journal of cancer. 2014;110(12):2914–2922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Seo AN, Jin Y, Lee HJ, et al. FGFR1 amplification is associated with poor prognosis and smoking in non-small-cell lung cancer. Virchows Archiv : an international journal of pathology. 2014. [DOI] [PubMed] [Google Scholar]
- 10.Zhang J, Zhang L, Su X, et al. Translating the therapeutic potential of AZD4547 in FGFR1-amplified non-small cell lung cancer through the use of patient-derived tumor xenograft models. Clinical cancer research : an official journal of the American Association for Cancer Research. 2012;18(24):6658–6667. [DOI] [PubMed] [Google Scholar]
- 11.Paik PK, Shen R, Berger MF, et al. A Phase Ib Open-Label Multicenter Study of AZD4547 in Patients with Advanced Squamous Cell Lung Cancers. Clinical cancer research : an official journal of the American Association for Cancer Research. 2017;23(18):5366–5373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aggarwal C, Redman MW, Lara PN Jr., et al. SWOG S1400D (NCT02965378), a Phase II Study of the Fibroblast Growth Factor Receptor Inhibitor AZD4547 in Previously Treated Patients With Fibroblast Growth Factor Pathway-Activated Stage IV Squamous Cell Lung Cancer (Lung-MAP Substudy). J Thorac Oncol. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bradley JD, Paulus R, Komaki R, et al. Standard-dose versus high-dose conformal radiotherapy with concurrent and consolidation carboplatin plus paclitaxel with or without cetuximab for patients with stage IIIA or IIIB non-small-cell lung cancer (RTOG 0617): a randomised, two-by-two factorial phase 3 study. The Lancet Oncology 2015;16(2):187–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fuks Z, Persaud RS, Alfieri A, et al. Basic fibroblast growth factor protects endothelial cells against radiation-induced programmed cell death in vitro and in vivo. Cancer Res. 1994;54(10):2582–2590. [PubMed] [Google Scholar]
- 15.Ma J, Benitez JA, Li J, et al. Inhibition of Nuclear PTEN Tyrosine Phosphorylation Enhances Glioma Radiation Sensitivity through Attenuated DNA Repair. Cancer Cell. 2019;35(3):504–518 e507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gavine PR, Mooney L, Kilgour E, et al. AZD4547: an orally bioavailable, potent, and selective inhibitor of the fibroblast growth factor receptor tyrosine kinase family. Cancer Res. 2012;72(8):2045–2056. [DOI] [PubMed] [Google Scholar]
- 17.Shu J, Dolman GE, Duan J, Qiu G, Ilyas M. Statistical colour models: an automated digital image analysis method for quantification of histological biomarkers. Biomed Eng Online. 2016;15:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bates D, Machler M, Bolker BM, Walker SC. Fitting Linear Mixed-Effects Models Using lme4. J Stat Softw. 2015;67(1):1–48. [Google Scholar]
- 19.Wynes MW, Hinz TK, Gao D, et al. FGFR1 mRNA and protein expression, not gene copy number, predict FGFR TKI sensitivity across all lung cancer histologies. Clinical cancer research : an official journal of the American Association for Cancer Research. 2014;20(12):3299–3309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rooney C, Geh C, Williams V, et al. Characterization of FGFR1 Locus in sqNSCLC Reveals a Broad and Heterogeneous Amplicon. PloS one. 2016;11(2):e0149628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ishigami T, Uzawa K, Higo M, et al. Genes and molecular pathways related to radioresistance of oral squamous cell carcinoma cells. Int J Cancer. 2007;120(10):2262–2270. [DOI] [PubMed] [Google Scholar]
- 22.Kotani H, Ebi H, Kitai H, et al. Co-active receptor tyrosine kinases mitigate the effect of FGFR inhibitors in FGFR1-amplified lung cancers with low FGFR1 protein expression. Oncogene. 2016;35(27):3587–3597. [DOI] [PubMed] [Google Scholar]
- 23.Nogova L, Sequist LV, Perez Garcia JM, et al. Evaluation of BGJ398, a Fibroblast Growth Factor Receptor 1–3 Kinase Inhibitor, in Patients With Advanced Solid Tumors Harboring Genetic Alterations in Fibroblast Growth Factor Receptors: Results of a Global Phase I, Dose-Escalation and Dose-Expansion Study. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2017;35(2):157–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lim SH, Sun JM, Choi YL, et al. Efficacy and safety of dovitinib in pretreated patients with advanced squamous non-small cell lung cancer with FGFR1 amplification: A single-arm, phase 2 study. Cancer. 2016;122(19):3024–3031. [DOI] [PubMed] [Google Scholar]
- 25.Malchers F, Dietlein F, Schottle J, et al. Cell-autonomous and non-cell-autonomous mechanisms of transformation by amplified FGFR1 in lung cancer. Cancer Discov. 2014;4(2):246–257. [DOI] [PubMed] [Google Scholar]
- 26.Haimovitz-Friedman A, Balaban N, McLoughlin M, et al. Protein kinase C mediates basic fibroblast growth factor protection of endothelial cells against radiation-induced apoptosis. Cancer Res. 1994;54(10):2591–2597. [PubMed] [Google Scholar]
- 27.Houchen CW, George RJ, Sturmoski MA, Cohn SM. FGF-2 enhances intestinal stem cell survival and its expression is induced after radiation injury. Am J Physiol. 1999;276(1 Pt 1):G249–258. [DOI] [PubMed] [Google Scholar]
- 28.Zhang L, Sun W, Wang J, et al. Mitigation effect of an FGF-2 peptide on acute gastrointestinal syndrome after high-dose ionizing radiation. International journal of radiation oncology, biology, physics. 2010;77(1):261–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Skinner HD, Giri U, Yang L, et al. Proteomic Profiling Identifies PTK2/FAK as a Driver of Radioresistance in HPV-negative Head and Neck Cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2016;22(18):4643–4650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ader I, Delmas C, Skuli N, et al. Preclinical evidence that SSR128129E--a novel small-molecule multi-fibroblast growth factor receptor blocker--radiosensitises human glioblastoma. Eur J Cancer. 2014;50(13):2351–2359. [DOI] [PubMed] [Google Scholar]
- 31.Kas SM, de Ruiter JR, Schipper K, et al. Transcriptomics and Transposon Mutagenesis Identify Multiple Mechanisms of Resistance to the FGFR Inhibitor AZD4547. Cancer Res. 2018;78(19):5668–5679. [DOI] [PubMed] [Google Scholar]
- 32.Yuan H, Li ZM, Shao J, Ji WX, Xia W, Lu S. FGF2/FGFR1 regulates autophagy in FGFR1-amplified non-small cell lung cancer cells. J Exp Clin Cancer Res. 2017;36(1):72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ondrej M, Cechakova L, Durisova K, Pejchal J, Tichy A. To live or let die: Unclear task of autophagy in the radiosensitization battle. Radiother Oncol 2016;119(2):265–275. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.