

Abstract
Purpose
Current sequencing strategies can genetically solve 55–60% of inherited retinal degeneration (IRD) cases, despite recent progress in sequencing. This can partially be attributed to elusive pathogenic variants (PVs) in known IRD genes, including copy-number variations (CNVs), which have been shown as major contributors to unsolved IRD cases.
Methods
Five hundred IRD patients were analyzed with targeted next-generation sequencing (NGS). The NGS data were used to detect CNVs with ExomeDepth and gCNV and the results were compared with CNV detection with a single-nucleotide polymorphism (SNP) array. Likely causal CNV predictions were validated by quantitative polymerase chain reaction (qPCR).
Results
Likely disease-causing single-nucleotide variants (SNVs) and small indels were found in 55.6% of subjects. PVs in USH2A (11.6%), RPGR (4%), and EYS (4%) were the most common. Likely causal CNVs were found in an additional 8.8% of patients. Of the three CNV detection methods, gCNV showed the highest accuracy. Approximately 30% of unsolved subjects had a single likely PV in a recessive IRD gene.
Conclusion
CNV detection using NGS-based algorithms is a reliable method that greatly increases the genetic diagnostic rate of IRDs. Experimentally validating CNVs helps estimate the rate at which IRDs might be solved by a CNV plus a more elusive variant.
Key words: inherited retinal degeneration, retinitis pigmentosa, rod–cone dystrophy, cone–rod dystrophy, copy-number variation
INTRODUCTION
Over 2 million people worldwide are affected by inherited retinal degenerations (IRDs), a family of blinding diseases characterized by progressive death and dysfunction of primarily rod and cone photoreceptors.1 Pathogenic variants (PVs) in over 270 genes have been associated with IRDs, many of which were discovered recently due to advances in sequencing technologies.2 However, despite substantial progress in genetic methodologies, current strategies can genetically solve only about 55–60% of IRD cases.3–7 The remaining missing diagnoses are in part due to undiscovered IRD genes. However, PVs in each new disease gene are rare,2 suggesting that the missing genetic causality largely lies in the known IRD genes. Many of these elusive PVs are due to structural variations (SVs) such as copy-number variations (CNVs), or deep intronic variants that affect splicing,7–14 which are not readily available from the standard output of targeted next-generation sequencing (NGS) pipelines.
Our previous work analyzed 28 genetically unsolved families with exome sequencing (ES) and single-nucleotide polymorphism (SNP) and/or comparative genomic hybridization (CGH) arrays, and showed that large deletions in known IRD genes were responsible for disease in five of the families.14 In this study we applied further bioinformatic analyses that permit detection of CNVs on the panel-based NGS Genetic Eye Disease (GEDi) diagnostic test that involves sequencing the exons of all known IRD disease genes.3 To assess the accuracy of CNV calling based on the NGS read-depth we compared two algorithms, ExomeDepth15 and gCNV,16 with the SNP array–based approach.17 A subset of the CNVs were subsequently validated by quantitative polymerase chain reaction (qPCR). In addition, we specifically searched for the Alu transposable element insertion in MAK, which is a common cause of IRD in people of Ashkenazi Jewish descent.18,19 Applying these techniques improved the genetic diagnostic rate for IRD patients by 10.2%.
MATERIALS AND METHODS
Ethical guidelines
The study was approved by the institutional review board at the Massachusetts Eye and Ear (Human Studies Committee, MEE, USA) and adhered to the tenets of the Declaration of Helsinki. Informed consent was obtained from all individuals on whom genetic testing and further molecular evaluations were performed.
Clinical evaluation
Patients included in the study were recruited and clinically examined at MEE. Ophthalmic examination included best-corrected Snellen visual acuity, dynamic Goldmann visual field testing, dark adaptation testing, and full-field electroretinographic (ERG) testing with assessment of 0.5 Hz ERG amplitude and 30 Hz ERG amplitudes.
Genomic DNA extraction and targeted sequencing
DNA was extracted from venous blood using the DNeasy Blood and Tissue Kit (Qiagen, Hilden, Germany). All samples underwent Genetic Eye Disease test (GEDi) sequencing as described previously.3 The GEDi version used in this study included 266 genes known to be associated with monogenic inherited retinal degenerations2,3 (Supplementary Table S5). The capture libraries were sequenced on MiSeq (9 samples per run) or HiSeq (96 samples per run) NGS platforms (Illumina, San Diego, CA) as previously described.3 The NGS data were analyzed using Genome Analysis Toolkit (GATK) version 3 (https://software.broadinstitute.org/gatk/) and annotated using the Variant Effect Predictor (VEP) tool (https://useast.ensembl.org/info/docs/tools/vep/) with additional annotations taken from gnomAD Genomic Evolutionary Rate Profiling (GERP), SIFT, PolyPhen-2 and retinal expression.20 Rare variants were selected based on the minor allele frequency (MAF) in public databases of less than 0.3%. Dominant pathogenic variants were considered with MAF <0.01%. Variants were annotated based on the transcripts included in Supplementary Table S5.
Exon 15 of RPGR transcript NM_001034853 (called RPGR ORF15) is not fully covered by the NGS and therefore it was PCR amplified and Sanger sequenced with previously established protocols (Supplementary Methods).
CNV analysis
NGS read-depth analysis
Copy-number variation from NGS read-depth was inferred using ExomeDepth15 and gCNV from the GATK version 4.16 Samples from all MiSeq runs were processed together (193 samples), and HiSeq runs were analyzed separately (batches of 96 or 48 samples). For ExomeDepth, the samples were separated by gender; for gCNV, they were kept together. For gCNV, the GEDi-captured regions were padded by 250 bp on each side and run in COHORT mode without external control samples. CNVs present in more than 15% of samples were removed as they were considered to be either capture artifacts or common CNVs that would not lead to a rare Mendelian disorder. CNVs in the OPN1LW gene and the OPN1MW gene were removed as they were likely artifacts of poor NGS read mapping quality.
SNP array
Genomic DNA (gDNA) samples from probands were analyzed with whole-genome SNP microarray (HumanOmni2.5 BeadChip, Illumina) according to the manufacturer’s instructions. The hybridized SNP arrays were analyzed using an array reader (iScan array scanner, Illumina) and the SNP calls were made with the genotyping module of the data analysis software (GenomeStudio, Illumina). Copy-number variation from the SNP array results was detected with PennCNV using default parameters and sex information.17
CNV confirmation
Quantitative real-time PCR
Deletions were validated using quantitative real-time PCR (qPCR) on gDNA with primers specific to sequences inside the presumed CNV and flanking the CNV (Supplementary Table S1). The amplification was normalized to the ZNF80 reference gene. For each qPCR reaction 5 ng of gDNA, 200 nM of each primer and 10 µl of Fast SYBR Green master mix (Life Technologies, Grand Island, NY) were used. The amplification was performed in a qPCR system (Stratagene Mx3000P®, Agilent Technologies) using the standard thermocycling program: 95 °C for 3 minutes, 40 cycles of 95 °C for 20 seconds, 60 °C for 1 minute followed by a melting curve. Each sample was assayed in triplicate. Relative changes in genomic sequence abundance were calculated using the 2−ddCT method, and error was calculated using standard propagation of errors. Data was visualized using custom R scripts.
Mapping of the breakpoints in confirmed duplications
Experimental details are presented in the supplementary material.
RESULTS
A cohort of 500 unrelated IRD patients was sequenced with the targeted NGS panel Genetic Eye Disease test (GEDi)3 and analyzed with a comprehensive approach, which included a standard NGS analysis pipeline detecting single-nucleotide variants (SNVs) and small insertions and deletions (indels),16 interrogation of sequence reads to detect the known pathogenic MAK-Alu insertion,21 and application of NGS read-depth algorithms (ExomeDepth15 and gCNV16) to detect larger deletions and duplications, or CNVs (Fig. 1). In this study we define CNVs as deletions or duplications that range from an average size of an exon (≈50–200 bp) and that are not detectable by standard NGS pipelines, to megabases of DNA.22
Fig. 1. Flowchart of analysis strategy for inherited retinal degeneration (IRD) cohort.

In this study, 500 unrelated IRD patients underwent targeted next-generation sequencing (NGS) and subsequent analysis for potentially causal genetic variants in IRD genes. A subset of the samples underwent additional single-nucleotide polymorphism (SNP) array analysis. Samples that had predicted likely causal copy-number variations (CNVs) were further analyzed with quantitative polymerase chain reaction (qPCR) to confirm the CNVs.
The integrated analytical approach yielded pathogenic SNVs/small indels likely leading to disease in 278 IRD subjects (55.6% of the total cohort), where PVs in USH2A (11.8%), RPGR (4%), and EYS (4%) were the most common causes of disease (Fig. 2, Supplementary Table S2). In addition, MAK-Alu insertion was detected in seven patients by analyzing the NGS sequence reads.21
Fig. 2. Summary of genetic contributions to inherited retinal degeneration (IRD) .

(a) Targeted next-generation sequencing (NGS) analysis in a cohort of 500 IRD subjects reveals single-nucleotide variant (SNV)/small indel solutions in 55.6% cases, copy-number variation (CNV) solutions in 8.8% cases, and MAK-Alu insertions in 1.4% cases. (b) Breakdown of CNV solutions by gene, with the number of patients solved by SNVs in the same gene. Note that genes that commonly have SNV solutions (USH2A, EYS) also tend to have CNV solutions, a notable exception being PRPF31, in which CNVs are more common than expected based on the number of SNVs. CNV copy-number variation, IRD inherited retinal degeneration, SNV single-nucleotide variant.
CNV predictions using NGS data
In our initial CNV analysis we focused only on CNVs that would contribute to solving the case genetically, that is (1) homozygous CNVs in autosomal recessive (ar) genes, (2) heterozygous CNVs in ar genes that are in trans with likely causal SNVs or small indel in the same gene, (3) compound heterozygous CNVs in ar genes, (4) heterozygous CNVs in a haploinsufficiency gene, and (5) duplications in autosomal dominant genes, which could lead to gain-of-function variants. Applying these criteria resulted in 55 patients with 65 potentially disease-causing CNVs (Supplementary Fig. S1). These CNVs were subsequently validated by qPCR using gDNA from patients and healthy controls. qPCR confirmed the likely CNV solutions for a total of 46 CNVs in 44 patients (8.8% of the total cohort). Thirty-two of them carried heterozygous deletions, four carried homozygous deletions, three hemizygous deletions, and five carried heterozygous duplications. Only one of the patients carrying a homozygous deletion (OGI2860_004445) was reported to be from a consanguineous marriage. CNVs in PRPF31 (10), EYS (9), and USH2A (8) were the most common (Fig. 2b, Supplementary Table S3).
CNV case highlights
Most of the CNVs detected in this project were easily interpretable as they were either homozygous deletions, heterozygous deletions coupled with a deleterious allele in trans, or heterozygous deletions in the known haploinsufficiency gene PRPF31.23 However, there were a few examples of more unusual CNV contributions to disease etiology, which we highlight below.
In two patients we detected two nonconsecutive heterozygous deletions, which are thought to be due to deletions in trans (e.g., EYS deletions in subject 121–182 [simplex RP], or PCDH15 deletions in subject OGI635_001299 [Usher syndrome], Supplementary Table S3 and Fig. S1). Unfortunately, due to lack of samples from family members and a large distance between the deletions (0.86 and 0.51 Mb), the phase of these deletions could not be confirmed.
Subject OGI655_001331 carried a heterozygous deletion of almost the entire ABCA4 gene (exons 1–40). On closer examination, it was revealed that the same subject also carried a common hypomorphic missense change p.Asn1868Ile, which due to its high allele frequency (AF = 0.042 in gnomAD) was overlooked during the initial analysis. The p.Asn1868Ile missense in trans with a deleterious missense variant or loss-of-function allele was shown to lead to a late onset macular degeneration,24 which corresponds to the disease presentation in OGI655_001331, diagnosed with inherited macular degeneration/Stargardt disease at age 42.
We identified five duplications (Fig. 1b, Supplementary Table S3). We were able to confirm the breakpoints of two tandem duplications in OFD1 and ADGRV1 (Fig. 3, Table S3), and tandem duplication in RPGRIP1 was confirmed previously.25 Tandem duplications in ADGRV1 and RPGRIP1 will lead to frameshifts and loss-of-function alleles. However, a tandem duplication of exons 6–15 in OFD1 in a male subject OGI2829_004414 will likely lead to an inframe duplication of 1242 internal coding bases (414 amino acid residues), with presumed partial preservation of protein function as complete loss of function of this gene is embryonic lethal in males.26 Subject OGI2829_004414 presented with a ciliopathy phenotype (retinal degeneration, history of kidney disease, and possible cognitive dysfunction).
Fig. 3. Mapping of tandem duplication breakpoints.

(a) OFD1 ex6–15 duplication in subject OGI2829_004414 was mapped with specific primers that only amplified the duplicated allele (primer sequences in the Supplementary Methods) and Sanger sequenced to reveal 19,443-bp tandem duplication on chromosome X (chrX:13758359–13777802 dup). (b) ADGRV1 ex2–82 duplication in subject OGI731_001448 was mapped with specific primers that only amplified the duplicated allele (primer sequences in the Supplementary Methods) and Sanger sequenced to reveal 286,732-bp tandem duplication on chromosome 5 (chr5:89888235–90174967 dup). PCR polymerase chain reaction.
Despite multiple attempts we were not able to confirm the breakpoints of duplications in EYS and JAG1, therefore considering them as tandem duplications is an assumption. Subject OGI2839_004424 carried a heterozygous duplication of exons 1–25 (3199 coding base-pairs) in JAG1 gene, which is predicted to cause a frameshift leading to a loss-of-function allele. Heterozygous loss-of-function variants in JAG1 are known to lead to Alagille syndrome through haploinsufficiency of this gene.27 The major clinical features of Alagille syndrome are abnormalities in bile ducts leading to liver damage and accompanying features are congenital heart defects, vertebrae anomalies, and ocular defects including pigmentary retinopathy and optic disc drusen.28 Investigation of the clinical notes revealed that subject OGI2839_004424 was seen in the ophthalmic clinic for the retinopathy, but in addition was previously diagnosed with Alagille syndrome, therefore we consider the JAG1 duplication as the likely cause of the disease.
Comparison of NGS-based CNV predictions versus SNP array–based CNV predictions
To determine if SNP array–based detection of CNVs could replicate the findings from our NGS-based CNV analysis, we analyzed a subset of our cohort with the Illumina Omni 2.5 SNP array using the PennCNV algorithm.17 Of the 500 samples from the NGS cohort, 144 were chosen for SNP array analysis. In these 144 samples, there were 61 predicted solving CNVs by gCNV and ExomeDepth in 52 patients (three of the samples from the initial NGS-based CNV analysis were not analyzed by the SNP array, thus not included in the comparison). The remaining 92 patients were chosen from the pool of samples that did not have likely solving NGS-based CNV or SNV predictions. These samples were chosen to see if there were any potentially solving CNVs predicted by the SNP array that were missed by NGS-based methods. In these 144 samples, gCNV predicted 52 potentially solving CNVs, ExomeDepth predicted 52 potentially solving CNVs, and SA predicted 31 potentially solving CNVs, including four CNVs in three patients that were not predicted by NGS-based methods. All predicted solving CNVs were then tested by qPCR to determine if they were confirmed true positives (Fig. 4a, b, Supplementary Fig. S1A, B). Of the three methods used, gCNV showed the best performance with 44 of 52 predictions that validated compared with ExomeDepth (37/52) and SNP array (25/31). The gCNV algorithm alone predicted all of the true positive CNVs detected in this study (Fig. 4a). False positive rate was the highest (29%) in ExomeDepth (Fig. 2b).
Fig. 4. Comparison of different copy-number variation (CNV) detection methods.
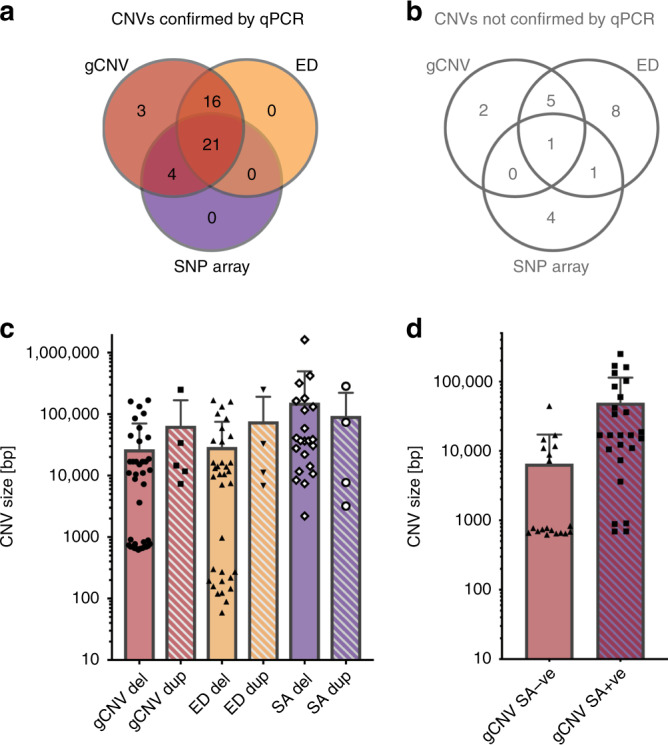
(a) Breakdown of true positive CNVs predicted by different methods. Note that all true positive CNVs were predicted by gCNV. (b) Breakdown of false positive CNVs predicted by different methods. Note that only one false positive CNV was predicted by all three CNV prediction methods. (c) The distribution of sizes of CNVs predicted by different methods. Note that in general, duplications were predicted less often, and were on average larger in size than predicted deletions. (d) Comparison of the gCNV predicted sizes of validated CNVs that were also predicted by the single-nucleotide polymorphism (SNP) array (gCNV SA + ve) versus the gCNV predicted size of validated CNVs that were not predicted by the SA (gCNV SA-ve). ED ExomeDepth, qPCR quantitative polymerase chain reaction.
Depending on the technique used, the predicted sizes of the validated CNVs ranged from 59 bp to 249 kb (ExomeDepth), 619 bp to 249 kb (gCNV), and 2.2 kb to1.6 Mb (SNP array) (Fig. 4c). Since NGS-based methods only investigate regions covered by the capture kit (i.e., IRD gene exons), and the SNP array covers the genome in a more uniform fashion, the sizes of large CNVs predicted are probably more accurately represented by the SNP array. This is because with NGS-based methods, any CNV that overlaps coding regions, but starts or ends in a noncoding region, can only be reported as the size of the coding regions plus any intervening introns. In this study, we did not attempt to map all the CNV breakpoints; however, we validated the CNVs with multiple qPCR primers inside and outside of the predicted deletions, thus chromosome coordinates of reported CNVs should be considered minimum estimates of the CNV size (Supplementary Fig. S1).
Further, we wanted to investigate why the SNP array failed to detect 19 true CNVs that were predicted by gCNV. The most plausible reason is that the CNV is not covered by a sufficient number of SNPs on the array because it is too small or it is in a region that is poorly represented on the array. First, we compared the sizes of the CNVs predicted by gCNV that were validated by the SNP array (SA + ve) and that were not validated by the SNP array (SNP-ve) (Fig. 4d). As expected CNVs that were not predicted by the SNP array were on average smaller (median = 772 bp, mean = 6.5 kb) than the CNVs that were also validated by the array (median = 17 kb, mean = 49 kb). None of the 12 CNVs that were below 1 kb had sufficient number of SNPs (at least three) on the Omni2.5 array to be detected by this method. Of the remaining seven larger CNVs (~7–44 kb) that were not detected by the SNP array, most had sufficient number of SNPs in the interval (Supplementary Table S3), therefore, failure to detect them by the SNP array was due to other undetermined experimental or analytical reasons.
Assessment of gCNV prediction scores
In the original analysis of the gCNV output we did not apply any quality filters, as the primary constraint on the data was whether a CNV fulfilled our “potentially solving” criteria. Considering only gCNV predictions, there were 55 CNVs that we attempted to validate by qPCR. Of the 46 CNVs that did validate, we noticed that some of them had low quality metrics. However, an agnostic search for true positive CNVs requires quality filters to remove false predictions, even at the expense of throwing out true positives. Therefore, we reanalyzed all CNVs in the cohort without the constraint of needing to be “potentially solving”, in order to look for CNVs that may be true structural variants that are not primary contributors to disease, or potential single hit disease-causing CNVs in genes that should be investigated further. This resulted in 3811 predicted CNVs in 500 patients, before any thresholds were applied. As an initial quality filter, we removed CNV predictions that were present in greater than 15% of samples and CNVs from the opsin gene locus on chromosome X, which due to poor NGS mapping quality generated a high rate of likely false positive CNVs. This reduced the number of predicted CNVs in the cohort to 759, and still included all 55 CNVs investigated by qPCR. By plotting the distribution of quality score metrics of these 759 CNVs, and highlighting the qPCR validated CNVs, we compared the different quality score metrics generated by gCNV (Fig. 5) to choose one for a quality score cutoff. We settled on using the score of QA >30.16 This cutoff reduced the number of CNVs in the entire cohort to 179 CNVs in 117 patients, which included 3 of 9 falsely predicted CNVs and missed 3 of 46 true positive predictions (Fig. 5).
Fig. 5. Distribution of gCNV quality metrics for rare, nonopsin copy-number variations (CNVs).
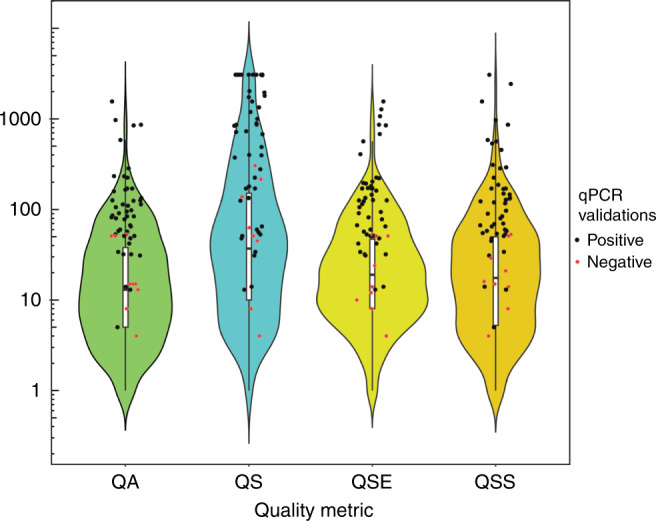
The gCNV algorithm provides four quality metrics for every predicted CNV: QA, QS, QSE, and QSS. QA is the complementary Phred-scaled probability that all points (i.e., targets or bins) in the segment agree with the segment copy-number call. QS is the complementary Phred-scaled probability that at least one point (i.e., target or bin) in the segment agrees with the segment copy-number call. QSE is the complementary Phred-scaled probability that the segment end position is a genuine copy-number changepoint. QSS is the complementary Phred-scaled probability that the segment start position is a genuine copy-number changepoint. Violin plots show the relative probability density of the distribution of each quality metric, while internal box plots show the 25th, median, and 75th percentile of distribution. The red and black data points represent CNVs within the distribution that were experimentally determined to be either true positives (black) or false positives (red) by quantitative polymerase chain reaction (qPCR) validation.
Assessment of frequency of patients with single likely pathogenic alleles in a recessive IRD gene
To assess how likely it is that the remaining genetically unsolved patients carry elusive pathogenic variants in already known IRD genes, we first evaluated how many unsolved patients have known pathogenic variants or new loss-of-function variants in recessive IRD disease genes, including CNVs. Of the 171 unsolved subjects, 44 carried stop, frameshift, essential splice-site variants or likely pathogenic CNVs and four subjects carried known pathogenic missense alleles (Supplementary Table S4). Altogether, we estimated that at least 28% of the unsolved patients (9.6% of the overall cohort) carried a single likely pathogenic allele in a recessive IRD gene.
DISCUSSION
Our results indicate that CNVs contribute significantly to the genetic causality of IRDs, and that NGS-based CNV detection methods outperform SNP array–based CNV detection. Of the 500 unrelated patients whose genetic cause of disease was investigated by panel-based NGS testing (GEDi),3 likely disease-causing CNVs were identified in 8.8% of cases. In 55.6% of cases the disease could be explained by the likely pathogenic SNVs and small indels detected by the standard NGS analysis pipeline.16 The only other structural variant investigated was a known Alu insertion in exon 9 of MAK,18,19,21 which was present in 1.4% of cases, agreeing closely with the previously reported frequency of 1.2%.18
The majority of the CNVs were heterozygous or homozygous deletions, ranging from single-exon to whole-gene deletions. Three large deletions in two genes (MERTK and TRPM1) were in regions prone to the nonallelic homologous recombination (NAHR), which is the most likely mechanism of their occurrence.14 Analysis of all CNVs reported in IRD genes performed by Van Schil and colleagues indicated that gene size, followed by the number of LINE and LTR repeats is the biggest predictor for a gene to be prone to CNVs.29 This analysis correlates well with our findings, where the largest IRD gene EYS and the third largest IRD gene USH2A, had nine and eight causal CNVs respectively. However, in the present study the most common gene to harbor CNVs was PRPF31, which is known to cause a dominant form of IRD through haploinsufficiency.23 Even though PRPF31 is known to be prone to CNVs, their contribution to IRD causality is likely still underestimated. A literature search by Van Schil and colleagues revealed that causal CNVs in PRPF31 have been reported in 14 families, and the Human Gene Mutation Database (HGMD) reports 35 SVs of the total of 196 variants (18%). In this study we detected likely causal CNVs in 10 of 22 (45%) probands with PRPF31-related disease, demonstrating a higher than previously reported CNV rate in this gene. The reasons for such high frequency of CNVs in PRPF31 is unclear, since this gene is neither large (~16.3 kb genomic length) nor does it have a high density of long interspersednuclear elements (LINE) and long terminal repeat (LTR) elements (ranked 191 of 245 genes by Van Schil and colleagues).29
Our study highlights the importance of parallel SNV and CNV analyses, as certain variants may falsely appear as homozygous when in fact they are in trans with a large deletion. This distinction is particularly important when a hypomorphic variant is involved, as in the case of p.Asn1868Ile in ABCA4 discovered in trans with a large deletion in subject OGI655_001331. The p.Asn1868Ile variant in a homozygous state is not considered pathogenic; however, when paired in trans with a severe pathogenic variant it has been shown to be causal.24 This finding also demonstrates the importance of evaluating every variant, rather than prematurely discarding variants based on certain criteria. This is becoming more feasible with the emergence of software that can rank variants, based on human-designed or machine learning algorithms.
Of 44 patients with CNVs, only five carried likely causal duplications, which may be an underestimate, as duplications are more difficult to detect, interpret, and validate. All but one of the duplications are thought to result in a loss-of-function allele. Duplication of OFD1 exons 6–15 is predicted to duplicate 414 internal amino acids of the protein and lead to a partially functioning OFD1 protein, resulting in a decreased spectrum of disease in subject OGI2829_004414, who apart from retinal disease has a history of renal failure. Variants in OFD1 may lead to a spectrum of phenotypes from an X-linked dominant oral–facial–digital type 1 syndrome, with ciliopathy phenotype in females and embryonic lethal in males, X-linked recessive Joubert syndrome to nonsyndromic IRD, depending on where the variant is located;8,26,30 however, to our knowledge no causal duplications in OFD1 have been reported to date.
NGS-based CNV prediction with the gCNV algorithm showed considerable advantage over more traditional SNP array–based prediction. This is largely because NGS-based algorithms could detect smaller CNVs as they were not restricted by the availability of SNPs. In our study, all validated CNVs detected by the SNP array were also detected by gCNV, therefore this method is an adequate replacement of the SNP array–based CNV predictions. Another accurate method of CNV detection is a customizable microarray-based comparative genomic hybridization, e.g., IRD genes array (arrEYE).11 However, this method requires an additional wet-lab assay to be applied to the samples that had already undergone NGS, which may be unnecessary if the CNVs can be detected by a robust NGS-based algorithm. In this study we used qPCR on gDNA as a validation of the CNVs, as this is a cost-effective and easily accessible method widely applied in many labs; however, other assays such as targeted locus amplification29 or genome sequencing can be applied with an added benefit of mapping CNV breakpoints.31
In a genetic diagnostic setting, filtering based on CNV prevalence in the cohort, CNV frequency per sample, and gCNV quality scores will aid in assessing the likelihood of a given CNV being true positive. In this study, we used experimental results to establish thresholds on gCNV predictions to reduce predicted CNVs to a subset with a higher probability of being true genetic variants. In future studies, more sophisticated methods can be used, such as creating a scoring method that considers multiple factors, similar to the guidelines recommended for sequence variant curation from the American College of Medical Genetics and Genomics (ACMG).32 Taking into account the population-level frequency of CNVs using publicly available data sets will also be crucial in establishing the pathogenicity of the CNVs (e.g., gnomadSV, Collins et al., bioRXiv preprint).
In conclusion, our rate of discovery for likely solving variants in IRD patients has increased from 55.6% to 65.8% by including information from CNVs and MAK-Alu insertions. This represents a significant improvement in solving genetic cases, consistent with or higher than in previous studies.11,12,33–35 Additional analysis found 9.6% of the cohort had a single potentially pathogenic SNV or CNV in a known autosomal recessive IRD gene. Although this does not constitute enough evidence to identify that gene as disease-causing, it indicates a high chance that an elusive pathogenic variant resides on the second allele, as demonstrated before for the RPGRIP1 gene.25 Searching for these elusive variants is critical for improving the discovery rate of disease causation. For example, this study did not investigate other SVs such as inversions. translocations, and other complex rearrangements including mobile element insertions, which may play an important role in IRDs.36,37 There are also examples of deep intronic variants leading to splicing aberration and untranslated region (UTR) variants contributing to retinal disease.8,9,25,38,39 The development of assays that can confirm pathogenic contributions from such variants, and their inclusion into variant analysis pipelines, will be important for better understanding the genetic contributions not just to IRDs, but to Mendelian disorders in general.
Supplementary information
Acknowledgements
This work was supported by grants from the National Eye Institute (R01EY012910 [E.A.P.], R01EY026904 [K.M.B./E.A.P.] and P30EY014104 [MEEI core support]), and the Foundation Fighting Blindness (EGI-GE-1218–0753-UCSD, K.M.B./E.A.P.). The authors thank all subjects for their participation in this study and the OGI Genomics Core members for their experimental assistance. The authors thank the Exome Aggregation Consortium, the Genome Aggregation Database (gnomAD), and the groups that provided exome variant data for comparison. A full list of contributing groups can be found at http://gnomad.broadinstitute.org/about.
Disclosure
The authors declare no conflicts of interest.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The grants funding this work are listed in the acknowledgements section.
These authors contributed equally: Erin Zampaglione and Benyam Kinde.
Supplementary information
The online version of this article (10.1038/s41436-020-0759-8) contains supplementary material, which is available to authorized users.
References
- 1.Berger W, Kloeckener-Gruissem B, Neidhardt J. The molecular basis of human retinal and vitreoretinal diseases. Prog Retin Eye Res. 2010;29:335–375. doi: 10.1016/j.preteyeres.2010.03.004. [DOI] [PubMed] [Google Scholar]
- 2.Retinal Information Network. https://sph.uth.edu/retnet/home.htm. 2019. Accessed 20 November 2018.
- 3.Consugar MB, Navarro-Gomez D, Place EM, et al. Panel-based genetic diagnostic testing for inherited eye diseases is highly accurate and reproducible, and more sensitive for variant detection, than exome sequencing. Genet Med. 2015;17:253–261. doi: 10.1038/gim.2014.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Weisschuh N, Mayer AK, Strom TM, et al. Mutation detection in patients with retinal dystrophies using targeted next generation sequencing. PLoS One. 2016;11:1–15. doi: 10.1371/journal.pone.0145951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhao L, Wang F, Wang H, et al. Next-generation sequencing-based molecular diagnosis of 82 retinitis pigmentosa probands from Northern Ireland. Hum Genet. 2015;134:217–230. doi: 10.1007/s00439-014-1512-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boulanger-Scemama E, El Shamieh S, Démontant V, et al. Next-generation sequencing applied to a large French cone and cone–rod dystrophy cohort: mutation spectrum and new genotype-phenotype correlation. Orphanet J Rare Dis. 2015;10:85. doi: 10.1186/s13023-015-0300-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Perez-Carro R, Corton M, Sánchez-Navarro I, et al. Panel-based NGS reveals novel pathogenic mutations in autosomal recessive retinitis pigmentosa. Sci Rep. 2016;6:1–10. doi: 10.1038/srep19531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Webb TR, Parfitt DA, Gardner JC, et al. Deep intronic mutation in OFD1, identified by targeted genomic next-generation sequencing, causes a severe form of X-linked retinitis pigmentosa (RP23) Hum Mol Genet. 2012;21:3647–3654. doi: 10.1093/hmg/dds194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.den Hollander AI, Koenekoop RK, Yzer S, et al. Mutations in the CEP290 (NPHP6) gene are a frequent cause of Leber congenital amaurosis. Am J Hum Genet. 2006;79:556–561. doi: 10.1086/507318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eisenberger T, Neuhaus C, Khan AO, et al. Increasing the yield in targeted next-generation sequencing by implicating CNV analysis, non-coding exons and the overall variant load: the example of retinal dystrophies. PLoS One. 2013;8:e78496. doi: 10.1371/journal.pone.0078496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Van Cauwenbergh C, Van Schil K, Cannoodt R, et al. ArrEYE: a customized platform for high-resolution copy number analysis of coding and noncoding regions of known and candidate retinal dystrophy genes and retinal noncoding RNAs. Genet Med. 2017;19:457–466. doi: 10.1038/gim.2016.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Khateb S, Hanany M, Khalaileh A, et al. Identification of genomic deletions causing inherited retinal degenerations by coverage analysis of whole exome sequencing data. J Med Genet. 2016;53:600–607. doi: 10.1136/jmedgenet-2016-103825. [DOI] [PubMed] [Google Scholar]
- 13.Braun TA, Mullins RF, Wagner AH, et al. Non-exomic and synonymous variants in ABCA4 are an important cause of Stargardt disease. Hum Mol Genet. 2013;22:5136–5145. doi: 10.1093/hmg/ddt367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bujakowska KM, Fernandez-Godino R, Place E, et al. Copy-number variation is an important contributor to the genetic causality of inherited retinal degenerations. Genet Med. 2017;19:643–651. doi: 10.1038/gim.2016.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Plagnol V, Curtis J, Epstein M, et al. A robust model for read count data in exome sequencing experiments and implications for copy number variant calling. Bioinformatics. 2012;28:2747–2754. doi: 10.1093/bioinformatics/bts526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McKenna A, Hanna M, Banks E, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;9:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang K, Li M, Hadley D, et al. PennCNV: an integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res. 2007;17:1665–1674. doi: 10.1101/gr.6861907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tucker Ba, Scheetz TE, Mullins RF, et al. Exome sequencing and analysis of induced pluripotent stem cells identify the cilia-related gene male germ cell-associated kinase (MAK) as a cause of retinitis pigmentosa. Proc Natl Acad Sci U S A. 2011;108:E569–E576. doi: 10.1073/pnas.1108918108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Özgül RK, Siemiatkowska AM, Yücel D, et al. Exome Sequencing and Cis-Regulatory Mapping Identify Mutations in MAK, Encoding a Regulator of Ciliary Length, as a Cause of Retinitis Pigmentosa Figure S1. Candidate Gene Prioritization at 34 Autosomal Recessive Retinitis Pigmentosa Loci. Am J Hum Genet. 2011;89:253–64. [DOI] [PMC free article] [PubMed]
- 20.Farkas M, Grant G, White J, Sousa M, Consugar M, Pierce E. Transcriptome analyses of the human retina identify unprecedented transcript diversity and 3.5 Mb of novel transcribed sequence via significant alternative splicing and novel genes. BMC Genomics. 2013;14:486. doi: 10.1186/1471-2164-14-486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bujakowska KM, White J, Place E, Consugar M, Comander J. Efficient in silico identification of a common insertion in the MAK gene which causes retinitis pigmentosa. PLoS One. 2015;10:e0142614.. doi: 10.1371/journal.pone.0142614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carvalho CMB, Lupski JR. Mechanisms underlying structural variant formation in genomic disorders. Nat Rev Genet. 2016;17:224–238. doi: 10.1038/nrg.2015.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Abu-Safieh L, Vithana EN, Mantel I, et al. A large deletion in the adRP gene PRPF31: evidence that haploinsufficiency is the cause of disease. Mol Vis. 2006;12:384–388. [PubMed] [Google Scholar]
- 24.Zernant J, Lee W, Collison FT, et al. Frequent hypomorphic alleles account for a significant fraction of ABCA4 disease and distinguish it from age-related macular degeneration. J Med Genet. 2017;54:404–412. doi: 10.1136/jmedgenet-2017-104540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jamshidi F, Place EM, Mehrotra S, et al. Contribution of non-coding mutations to RPGRIP1-mediated inherited retinal degeneration. Genet Med. 2019;3:694–704. doi: 10.1038/s41436-018-0104-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ferrante MI, Giorgio G, Feather Sa, et al. Identification of the gene for oral–facial–digital type I syndrome. Am J Hum Genet. 2001;68:569–576. doi: 10.1086/318802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Oda T, Elkahloun AG, Pike BL, et al. Mutations in the human Jagged1 gene are responsible for Alagille syndrome. Nat Genet. 1997;16:235–242. doi: 10.1038/ng0797-235. [DOI] [PubMed] [Google Scholar]
- 28.Kim BJ, Fulton AB. The genetics and ocular findings of Alagille syndrome. Semin Ophthalmol. 2007;22:205–210. doi: 10.1080/08820530701745108. [DOI] [PubMed] [Google Scholar]
- 29.Van Schil K, Naessens S, Van De Sompele S, et al. Mapping the genomic landscape of inherited retinal disease genes prioritizes genes prone to coding and noncoding copy-number variations. Genet Med. 2018;20:202–213. doi: 10.1038/gim.2017.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Coene KLM, Roepman R, Doherty D, et al. OFD1 is mutated in X-linked Joubert syndrome and interacts with LCA5-encoded lebercilin. Am J Hum Genet. 2009;85:465–481. doi: 10.1016/j.ajhg.2009.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Carss K, Arno G, Erwood M, et al. Comprehensive rare variant analysis via whole-genome sequencing to determine the molecular pathology of inherited retinal disease. Am J Hum Genet. 2017;100:75–90. doi: 10.1016/j.ajhg.2016.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–423. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huang X, Mao J, Huang Z, et al. Genome-wide detection of copy number variations in unsolved inherited retinal disease. Investig Ophthalmol Vis Sci. 2017;58:424–429. doi: 10.1167/iovs.16-20705. [DOI] [PubMed] [Google Scholar]
- 34.Ellingford JM, Horn B, Campbell C, et al. Assessment of the incorporation of CNV surveillance into gene panel next-generation sequencing testing for inherited retinal diseases. J Med Genet. 2018;55:114–121. doi: 10.1136/jmedgenet-2017-104791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jespersgaard C, Fang M, Bertelsen M, et al. Molecular genetic analysis using targeted NGS analysis of 677 individuals with retinal dystrophy. Sci Rep. 2019;9:1–7. doi: 10.1038/s41598-018-38007-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tavares E, Tang CY, Li S, et al. Retrotransposon insertion as a novel mutational event in Bardet-Biedl syndrome. Mol Genet Genomic Med. 2019;7:e521. doi: 10.1002/mgg3.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nishiguchi KM, Tearle RG, Liu YP, et al. Whole genome sequencing in patients with retinitis pigmentosa reveals pathogenic DNA structural changes and NEK2 as a new disease gene. Proc Natl Acad Sci U S A. 2013;110:16139–16144. doi: 10.1073/pnas.1308243110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Coppieters F, Todeschini AL, Fujimaki T, et al. Hidden genetic variation in LCA9-associated congenital blindness explained by 5’UTR mutations and copy number variations Of NMNAT1. Hum Mutat. 2015;12:1188–1196. doi: 10.1002/humu.22899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vaché C, Torriano S, Faugère V, et al. Pathogenicity of novel atypical variants leading to choroideremia as determined by functional analyses. Hum Mutat. 2019;40:31–35. doi: 10.1002/humu.23671. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.