

ABSTRACT
Centrosomes focus microtubules to promote mitotic spindle bipolarity, a critical requirement for balanced chromosome segregation. Comprehensive understanding of centrosome function and regulation requires a complete inventory of components. While many centrosome components have been identified, others yet remain undiscovered. We have used a bioinformatics approach, based on ‘guilt by association’ expression to identify novel mitotic components among the large group of predicted human proteins that have yet to be functionally characterized. Here, we identify chondrosarcoma-associated gene 1 protein (CSAG1) in maintaining centrosome integrity during mitosis. Depletion of CSAG1 disrupts centrosomes and leads to multipolar spindles, particularly in cells with compromised p53 function. Thus, CSAG1 may reflect a class of ‘mitotic addiction’ genes, whose expression is more essential in transformed cells.
KEY WORDS: Cell cycle, Spindle pole, Multipolar mitosis, Aneuploidy, Pericentriolar matrix
Summary: CSAG1, a novel regulator of mitosis that constrains the pericentriolar material to prevent spindle pole fragmentation, particularly, in cells that lack p53 function.
INTRODUCTION
Mitosis accurately and evenly divides the replicated genome into two daughter cells. During mitosis, the spindle forms from microtubules focused by centrosomes at the two opposite poles, and bundles of these microtubules attach to the kinetochores of sister chromatids. Full attachment of microtubules to kinetochores occurs as chromosomes align at the equatorial region in metaphase. Shortly thereafter, at anaphase, the chromatids of each chromosome separate into the two daughter cells (Stern and Murray, 2001; Millband et al., 2002; Bharadwaj and Yu, 2004). Formation and maintenance of the bipolar spindle is critical to ensure proper segregation of chromosomes. The formation of more than two poles during mitosis greatly compromises the fidelity of chromosome segregation at anaphase.
In vertebrate cells, centrosomes are composed of a pair of centrioles surrounded by a condensed cloud of proteins termed the pericentriolar matrix (PCM) (Woodruff et al., 2014). Multipolar mitoses are often caused by centrosome over duplication, failure to cluster extra centrosomes, premature dissociation of centrioles or PCM fragmentation during mitosis (Maiato and Logarinho, 2014). Some cancer cells with multiple centrosomes can, nevertheless, form bipolar spindles by clustering the extra centrosomes (Gergely and Basto, 2008; Pannu et al., 2015). In such cells, spontaneous or experimentally induced failure in centrosome clustering leads to multipolarity (Drosopoulos et al., 2014; Pannu et al., 2015; Mittal et al., 2016). The induction of multipolar spindles by microtubule-stabilizing drugs, such as Taxol, may be a mechanism underlying therapeutic effects in cancer treatment (Weaver, 2014). Overexpression of kinases, polo like kinase 4 (PLK4) and Aurora A, during interphase cause centrosome amplification and multipolarity during subsequent mitosis (Holland et al., 2012; Coelho et al., 2015). Long delays at metaphase induce multipolar spindles in cells that have undergone chromatid separation in cohesion fatigue (Daum et al., 2011; Stevens et al., 2011).
In some cases, multipolarity in mitosis can occur without centrosome amplification caused by centriole disengagement or fragmentation of the PCM (reviewed by Maiato and Logarinho, 2014). The presence of damaged DNA may also generate multipolar spindles after cells initiate mitosis with a normal appearing bipolar spindle (Hut et al., 2003). Malignant cells with amplified centrosomes often cluster extra centrosomes into two poles to maintain the bipolar spindle and hence avoid massive chromosome instability (Quintyne et al., 2005; Gergely and Basto, 2008; Godinho et al., 2009).
Among the many proteins found within the PCM, some have clearly defined functions. For example, γ-tubulin is recruited to the PCM and aids in nucleating the microtubules that form the spindle during mitosis (Wiese and Zheng, 2006). The centrosome becomes enlarged and more defined as the cell approaches prophase of mitosis. How the PCM expands during the preparation for mitosis is poorly understood. The centrosome is not membrane bound and has been suggested to be an example of an organelle formed by phase separation or protein condensation (Zwicker et al., 2014; Woodruff et al., 2017; Boeynaems et al., 2018). Therefore, there are likely to be components of the centrosome central in maintaining its integrity. Fragmentation of PCM components during mitosis has been reported to occur in a manner that requires the presence of spindle microtubules (Asteriti et al., 2011).
In an effort to discover novel mitotic proteins, we used the global microarray meta-analysis (GAMMA) bioinformatics approach (Wren, 2009). Briefly, GAMMA processes over 80,000 publicly available high-throughput transcriptional experiments (i.e. microarray and RNA-sequencing) to identify highly correlated transcripts. Then, using a ‘guilt by association’ approach, even if nothing or little has been published on a candidate mitotic gene, if it does function in mitosis, the transcription of the candidate mitotic gene would be expected to be more strongly correlated with known mitotic genes than genes that function in other biological processes. In fact, we and others have successfully used GAMMA to prioritize potential mitotic proteins for further experimental characterization (Daum et al., 2009; Tipton et al., 2017; Fields et al., 2018). Using this approach, we identified a mitotic role for the poorly characterized chondrosarcoma-associated gene 1 (CSAG1). mRNA analyses indicate that the candidate gene CSAG1 is highly expressed in chondrosarcoma, other cancers and in certain normal tissues, such as testis and brain (https://www.proteinatlas.org/ENSG00000198930-CSAG1/tissue). Two transcript variants with different 5′ untranslated regions (UTRs) have been described, but both mRNAs encode the same 78 amino acid protein (Lin et al., 2002). Functions for CSAG1 have not yet been characterized and, generally, very little is known about the regulation of the CSAG1 gene. A closely related gene, CSAG2, which is also known as Taxol-resistant gene 3, is better studied and was shown through RNA analysis to be highly expressed in different cancers (Yao et al., 2004; Janjic et al., 2006; Ohta et al., 2006). Our laboratory has previously shown that depletion of CSAG1 significantly inhibits cell proliferation in a breast cancer stem cell model system (Fields et al., 2018).
In this study, we functionally characterize the CSAG1 gene product. We report that CSAG1 concentrates at centrosomes and its depletion by small interference RNA (siRNA) in HeLa cells results in a high level of multipolar anaphase. We found stretched and fragmented PCM in CSAG1-depleted cells, suggesting that CSAG1 functions in strengthening the integrity of the PCM during mitosis. In RPE1 and HCT116 cells, we found that cells that lack normal p53 function are more likely to undergo multipolar mitosis when depleted of CSAG1. Therefore, CSAG1 might be an example of mitotic addiction genes, i.e. genes whose expression is more indispensable in a p53-compromised background and, thus, are potential targets in cancer therapy.
RESULTS
CSAG1 depletion results in delayed mitotic progression and multipolar mitotic exit
Live cell imaging of HeLa cells stably expressing GFP-tagged histone H2B (GFP-histone H2B) revealed that CSAG1-depleted cells initiated mitosis with a bipolar spindle and, generally, advanced to a normal metaphase. Thereafter, in a large portion of cells, the metaphase plate became bent, which is indicative of multipolar mitosis, and then cells entered anaphase with the chromosomes segregating into 3-4 distinct DNA masses (Fig. 1A, middle and bottom panels; Movie 1). For comparison, see normal mitosis in cells treated with negative control siRNA (Fig. 1A, top panels; Movie 2). Depletion of CSAG1 in HeLa cells caused increased incidence (45%) of multipolar mitotic exit compared to those treated with negative control siRNA (NC siRNA) (Fig. 1B). In addition to the increased frequency of multipolar mitosis, CSAG1 depletion also caused chromosome alignment defects in a small number of cells (Fig. 1B). Other mitotic defects, such as premature exit from mitosis without alignment or lagging chromosomes, were rare. Additionally, depletion of CSAG1 increased the elapsed time from nuclear envelope breakdown (NEBD) to anaphase (Fig. 1C). Cells transfected with CSAG1 siRNA that did not exhibit the multipolar phenotype also showed delayed NEBD to anaphase compared to control cells (Fig. S1A). The longer duration of progression was attributable to both delays from NEBD to metaphase and from metaphase to anaphase.
Fig. 1.

Depletion of CSAG1 by RNAi causes multipolar mitosis. (A) Live-cell microscopy images of HeLa-H2B-GFP cells transfected with either negative control (NC) siRNA or CSAG1 siRNA (time is given in minutes). Top row: unperturbed mitosis in cells transfected with NC siRNA. Middle and bottom rows: cells transfected with CSAG1 siRNA exhibit multipolar mitosis, during which the metaphase plate bends shortly after onset of metaphase. (B) Cells as described in A were analyzed for multipolar spindles or other mitotic defects, such as delayed alignment, mitotic exit without alignment or lagging anaphase chromosomes. Sidak's multiple comparisons test was used for statistical analysis. Error bars represent +s.d.; ns, not significant. (C) Elapsed time from nuclear envelope breakdown (NEBD) to anaphase was determined in NC siRNA- or CSAG1 siRNA-treated cells. Mann–Whitney test was used for statistical analysis. A total of >200 cells from three independent experiments was analyzed. CSAG1-depleted cells exhibit multipolar mitosis and take longer to proceed through mitosis. Error bars represent +s.d.
CSAG1 accumulates at centrosomes in mitotic cells and expression of siRNA-resistant CSAG1 rescues the multipolar phenotype
Numerous attempts to detect the endogenous CSAG1 by using our in-house antibody and with a commercial antibody were unsuccessful. Therefore, to examine localization of CSAG1, we generated the stable HeLa-GFP-CSAG1 cell line with inducible GFP-tagged CSAG1 (GFP-CSAG1) using HeLa Flp-in TRex cells. In these cells, the region of cDNA insertion is predetermined by the flippase recognition target (FRT) sites, and the amount of protein expression can be regulated by the concentration of added doxycycline (Tighe et al., 2004). At lower magnification, >90% of HeLa-GFP-CSAG1 cells showed nuclear localization of the GFP signal in interphase cells when induced with 2 µg/ml doxycycline under live cell imaging conditions (Fig. S1B). Further examination of these cells at higher magnification by immunofluorescence analysis, with anti-GFP antibodies after doxycycline induction, showed clear localization of GFP-CSAG1 also at spindle poles and/or centrosomes in both mitotic and interphase cells. However, spindle pole localization of CSAG1 was more pronounced during mitosis. Centrosomal localization was confirmed by colocalization with antibodies against pericentrin. Centrosomal localization of CSAG1 peaked at prophase, similar to pericentrin (Fig. 2A). These data are consistent with a centrosomal function of CSAG1 in mitotic cells.
Fig. 2.

GFP-tagged CSAG1 localizes to spindle poles during mitosis. (A) HeLa cells stably expressing inducible GFP-CSAG1 were examined for localization of GFP signals (GFP-CSAG1) and the PCM component pericentrin during interphase and different stages of mitosis. First row: uninduced (parental) cells, showing no GFP signal. Second to fifth row: spindle pole and/or centrosome localization in cells expressing GFP-CSAG1 during late prophase, prometaphase, metaphase and interphase. Cell nuclei were stained with DAPI, merged images are shown in the last column. (B) Quantitative analysis of normal mitosis, multipolar mitosis or other defects (determined in cells as described in A and as indicated). Cells were transfected with CSAG1 siRNA in presence or absence of doxycycline to induce exogenous siRNA-resistant GFP-CSAG1. A total of >300 cells was analyzed from three independent experiments. Two-way ANOVA with Tukey's multiple comparisons test was used for statistical analysis. Error bars represent +s.d.; ns, not significant (C) Elapsed time (in minutes) from nuclear envelope breakdown (NEBD) to anaphase, determined in cells analyzed in B. A Kruskal–Wallis test was used for statistical analysis. Error bars represent +s.d.
We used these cells to validate the efficiency of CSAG1 depletion by using siRNA targeting the open reading frame of CSAG1 followed by western blotting with anti-CSAG1 antibody. In both soluble lysate and immune-precipitated samples, the GFP-CSAG1-specific band was reduced by >95% after siRNA treatment (Fig. S1C). To exclude non-specific effects of CSAG1 siRNA, we performed rescue experiments. CSAG1 depletion in uninduced GFP-CSAG1 HeLa Flp-in TRex cells caused the multipolar phenotype in ∼30% of cells. However, when cells were induced with doxycycline to express siRNA-resistant CSAG1 and then treated with the smart pool of siRNA targeting non-coding regions of endogenous CSAG1, only 6% of cells exhibited multipolar spindles (Fig. 2B). Furthermore, mitotic duration was also significantly reduced in GFP-CSAG1-induced cells compared to CSAG1-depleted cells (Fig. 2C). These results confirmed that the CSAG1-depletion phenotype was, indeed, due to loss of CSAG1 and not due to non-specific targeting of the siRNA. We also confirmed the siRNA specificity by using a different siRNA that targeted the open reading frame (ORF) of CSAG1, showing a similar induction of multipolar spindles but higher levels of chromosome alignment defects, perhaps owing to different levels of depletion (Fig. S1D).
Increasing mitotic duration and spindle microtubule stabilization exacerbates multipolarity caused by depletion of CSAG1
We noticed that depletion of CSAG1 in HeLa cells caused a significant delay in mitotic progression, even in cells that did not exhibit the multipolar phenotype (Fig. S1A). This observation led us to examine whether delaying mitotic progression experimentally might amplify the multipolar phenotype caused by CSAG1 depletion. To test this, we used low concentrations of Nocodazole (25 nM; a microtubule destabilizer), Taxol (1 nM; a microtubule stabilizer) or ProTAME [5 µM; an inhibitor of the anaphase-promoting complex/cyclosome (APC/C)] to delay mitotic progression, and Reversine (250 nM; an inhibitor of the MPS1 spindle checkpoint kinase) to accelerate mitosis (Sapkota et al., 2018). In control cells, Nocodazole, Taxol or ProTAME treatment alone increased the time between NEBD and anaphase (Fig. S2A) but did not cause multipolar mitosis (Fig. S2B). In contrast, in CSAG1-depleted cells, multipolarity increased to 52% with Nocodazole, 61% with Taxol and 80% with ProTAME, compared to 45% in control CSAG1-depleted cells (Fig. 3A). As expected, the drug treatments increased the mitotic duration in CSAG1-depleted cells (Fig. 3B). However, treatment of CSAG1-depleted cells with Reversine, abrogated the spindle checkpoint, accelerated mitosis, and eliminated the multipolar phenotype. Together, these data are consistent with the idea that longer mitotic duration enhances multipolarity induced by CSAG1 depletion.
Fig. 3.

Microtubule perturbation and prolonged mitotic duration increase multipolarity in CSAG1-depleted cells caused by spindle pole fragmentation. (A) The fraction of multipolar mitosis was determined in CSAG1-depleted cells treated with low concentrations of Nocodazole (5 ng/ml), Taxol (1 nM), ProTAME (5 µM) or Reversine (250 nM). A total of >300 cells was analyzed for each treatment. Error bars represent +s.e.m.; Kruskal–Wallis test was used for statistical analysis. ns, not significant. (B) Quantification of the elapsed time (in minutes) from nuclear envelope breakdown (NEBD) to anaphase was determined in cells shown in A. Increasing the duration of mitosis in cells with intact spindle pulling forces increases susceptibility for multipolarity in CSAG1-depleted cells. Error bars represent +s.e.m. (C) Immunofluorescence images of CSAG1-depleted HeLa cells labeled for the centriole markers γ-tubulin and pericentrin. The top row shows a CSAG1-depleted cell; the bottom row shows a negative control (NC) siRNA-transfected cell. The graph on the right shows the quantitative analysis of mitotic cells (as a percentage) containing three or more (≥3) γ-tubulin foci. (D) Cells as described in A were immuno-labeled for the centriole marker centrin 1 and pericentrin. The top set of panels shows a CSAG1-depleted cell and the bottom set shows a control siRNA-treated cell. The graph on the right shows the quantification of mitotic cells with three or more (≥3) pericentrin foci. In all cases, centrioles remained paired at two of the spindle poles. For both C and D, a total of >100 cells was analyzed; a Mann–Whitney test was used for statistical analysis. Labeling reveals fragmentation of pericentriolar material induced by CSAG1 depletion. Error bars represent +s.e.m.
Extra spindle poles induced by depletion of CSAG1 lack centrioles
Supernumerary centrosomes generate multipolar mitosis that may arise from centrosome reduplication, de-clustering of supernumerary centrosomes, premature centriole separation, or fragmentation of PCM (reviewed by Maiato and Logarinho, 2014). To determine whether CSAG1 depletion induced abnormal centrosome duplication of centrosomes and/or de-clustering of over duplicated centrosomes in HeLa cells, we used immunolabeling for γ-tubulin, the PCM component pericentrin and the centriole component centrin 1 in control and CSAG1-depleted HeLa cells. Control HeLa cells exhibited the normal number of centrosomes and centrioles during interphase and mitosis (Fig. S3A,B, top panels). Immunofluorescence images also showed that CSAG1-depleted interphase cells had normal numbers of centrosomes and centrioles (Fig. S3A,B, bottom panels). Thus, we found no evidence of preexisting centrosome or centriole overamplification, or of CSAG1 depletion-induced overamplification in HeLa cells. However, CSAG1-depleted mitotic cells with multiple spindle poles showed more than two γ-tubulin foci, and these additional pericentrin and γ-tubulin foci lacked centrioles (Fig. 3C). In contrast, the supernumerary spindle poles were positive for both of the PCM components γ-tubulin and pericentrin (Fig. 3C,D). These data suggest that CSAG1 aids to maintain PCM integrity during mitosis, and that depletion of CSAG1 promotes PCM fragmentation and multipolar mitosis.
Immunofluorescence data from fixed cell imaging led us to question whether pole fragmentation precedes or follows the bending of metaphase plate detected in our video analysis. We monitored the structure of spindle poles and the shape of the metaphase plate during mitosis in CSAG1-depleted cells. We used HeLa cells stably expressing GFP–tubulin to reveal spindle poles and applied the Far-Red DNA dye SiR-DNA, for labeling chromosomes. Whereas cells treated with control siRNA underwent normal bipolar division, in CSAG1-depleted cells the formation of extra poles directly preceded the bending of the metaphase plate (Movie 1). Thus, bending of the metaphase plate seems to be a consequence of microtubule reorganization in forming multipolar spindles. Taken together these data suggested that CSAG1-depleted cells have a normal number of centrosomes and enter mitosis with a normal bipolar spindle. However, extra spindle poles are generated during mitosis at metaphase.
Loss of CSAG1 alters PCM structure
To test whether CSAG1 depletion disrupts the PCM, which then fragments the poles, we compared distributions of pericentrin in interphase and mitotic HeLa cells after CSAG1 depletion. Interphase cells showed no differences in distribution or the apparent amount of the pericentrin associated with each centrosome (data not shown). However, in mitotic cells, particularly at metaphase, CSAG1 depletion disrupted normal pericentrin organization. Normally pericentrin occupied a well-defined oval shape at the spindle poles in metaphase. However, in some cells, this clear definition was not evident and the pericentrin spread to a stretched and/or non-oval distribution. We classified this type of unstructured and elongated pericentrin labeling as dispersed pericentrin. In control siRNA-transfected cells, 80% of bipolar metaphase cells showed defined, normal appearing pericentrin labeling and only 20% could be classified as dispersed. In contrast, in a population of CSAG1-depleted cells, ∼70% of bipolar cells showed dispersed pericentrin at poles at metaphase (Fig. 4A,B). To better quantify this effect, we measured the axes of PCM shape (as described in Fig. 4C, left) in CSAG1-depleted and control siRNA-transfected cells. In control metaphase cells, the average ratio of length to width of PCM structure was 1.5, whereas in CSAG1-depleted metaphase cells the average axis ratio was 2.7. By integrating the intensities of immuno-labeled pericentrin, we found that the total amount of pericentrin in each pole of CSAG1-depleted cells was comparable to that in control cells (Fig. 4D). Overall, CSAG1-depleted cells revealed a >80% increase in the axis ratio, revealed by labeling with pericentrin antibody (Fig. 4C). In extreme cases, CSAG1-depleted cells showed pericentrin labeling up to 5 μm from centrioles, something not seen in control cells. However, many of these cells retained bipolar spindles and metaphase plates that appeared normal. There was no indication of centriole dis-engagement as measured by distance between sister-centrioles (Fig. S3C). We also examined whether changes in PCM distribution and/or shape in CSAG1-depleted cells precede the formation of the spindle. However, quantification of the PCM axis ratio in prophase cells showed no difference between control and CSAG1-depleted cells (Fig. S3D).
Fig. 4.

Mitotic spindle microtubules are essential for CSAG1 depletion-induced changes in the distribution of pericentrin at metaphase. (A) Immunofluorescence images of HeLa cells labeled for centrin and pericentrin that were transfected with control siRNA (top set of panels) or CSAG1 siRNA (bottom set of panels). Cell nuclei were stained with DAPI (DNA), merged images are shown in the last column. (B) The fraction of bipolar metaphase cells with abnormal or dispersed, or normal (i.e. oval) distribution of pericentrin determined in cells described in A. A total of >100 cells was analyzed for each sample. (C) Schematic, showing the method to quantify the axis ratio (length/width) and the graph shows the ratios calculated. More than 50 cells were analyzed for each group. A Mann–Whitney test was used for statistical analysis. (D) Total amount of pericentrin was measured for the sum of z-stack images derived from cells as described in B. (E,F) Multipolarity (E), and pericentrin shape and/or distribution (F) was evaluated in CSAG1-depleted cells that had been treated with or without Nocodazole. CSAG1 depletion causes abnormal pericentrin shape and/or distribution in bipolar metaphase cells but does not alter the amount of pericentrin at poles. The redistribution requires intact microtubules. Mann–Whitney test was used for statistical analysis. Error bars represent +s.d.; ns, not significant.
The timing of the altered PCM shape and/or distribution coincided with formation of mitotic spindles in prometaphase and/or metaphase. We examined whether spindle microtubules are required for pole fragmentation and/or PCM disturbance. To test this, we disrupted microtubules by using a high concentration of Nocodazole (330 nM) for 6 h in control and CSAG1-depleted cells. This treatment abolished the formation of multiple poles, and any changes in PCM shape and distribution (Fig. 4E,F). Thus, PCM fragmentation induced by CSAG1 depletion is dependent on the presence of spindle microtubules.
To test the possibility that PCM shape change and PCM fragmentation was caused merely by metaphase delay, we examined the PCM distribution pattern in cells that had been arrested at metaphase in response to the proteasome inhibitor MG132 and found no change (Fig. S3E). These data suggest that CSAG1 depletion compromised PCM and caused it to become dispersed along the microtubules at the poles, thereby rendering it susceptible to fragmentation and formation of acentriolar supernumerary poles, which generated multipolar mitotic spindles at high frequency.
The multipolar phenotype is enhanced when p53 function or expression is compromised
A study of 56 different human cell lines shows large variations in CSAG1 transcript levels (Thul et al., 2017). In some cell types CSAG1 transcript levels are undetectable, while in others transcript levels are up to 80 reads per kilo base (RPKB) (Thul et al., 2017). This finding suggested that CSAG1 is not essential for mitosis in all cells but serves important roles in certain types of transformed cells. In particular, the RNAseq data revealed that CSAG1 transcript levels are substantially higher in malignant melanoma cell lines (Thul et al., 2017).
To examine whether CSAG1 is an essential protein for mitotic progression in non-transformed cells, we depleted CSAG1 in hTERT-RPE1 (hereafter referred to as RPE1) cells that are immortalized through expression of telomerase but are otherwise considered to reflect normal, non-transformed cells (Bodnar et al., 1998; Jiang et al., 1999). RPE1 cells depleted of CSAG1 did not show mitotic defects, although mitotic progression was marginally delayed (Fig. S4A,B) and a small fraction of CSAG1-depleted RPE1 cells exited mitosis without complete chromosome alignment (Fig. S4A). As a control for transfection efficiency, RPE1 cells were depleted of the essential mitotic regulator polo-like kinase 1 (PLK1), required for bipolar spindle formation. Here, nearly every PLK1-depleted cell arrested in mitosis due to spindle collapse (Fig. S4A).
Among many differences between RPE1 and HeLa cells is their p53 status. RPE1 cells have normal p53 function whereas in HeLa cells p53 is degraded via the papilloma virus E6 protein. We hypothesized that p53 status might be critical for unmasking the CSAG1 depletion phenotype. We compared phenotypes in RPE1 cell lines stably expressing control, non-specific shRNA or shRNA to p53 that were provided by Dr Tamara Potapova (Potapova et al., 2016). When CSAG1 was depleted in both lines, control RPE1 cells with intact p53 showed no multipolarity, whereas 19% of p53-depleted RPE1 cells exhibited multipolar mitosis (Fig. 5A). We further tested whether compromised p53 function promotes multipolarity in another cell line, HCT116. Parental HCT116 cells and a HCT116 derivative line, in which both copies of the gene encoding p53 were inactivated (HCT116 KO) were provided by Dr Ralf Janknecht (University of Oklahoma Health Sciences Center) and used with the permission of Dr. Bert Vogelstein (Johns Hopkins University), in whose laboratory the p53-knockout line originated (Bunz et al., 1998). Consistent with effects seen in RPE1 and HeLa cells, CSAG1 depletion in HCT116 KO cells caused multipolar mitotic exit in ∼40% of mitotic events, whereas only 5% of HCT116 parental cells showed the phenotype (Fig. 5B). Mitotic progression was delayed in both the parental and p53 KO HCT116 cells, but the delay was more severe in p53 KO cells. HCT116 parental cells were delayed by ∼8 min but p53 KO cells were delayed by 22 min (Fig. 5C).
Fig. 5.
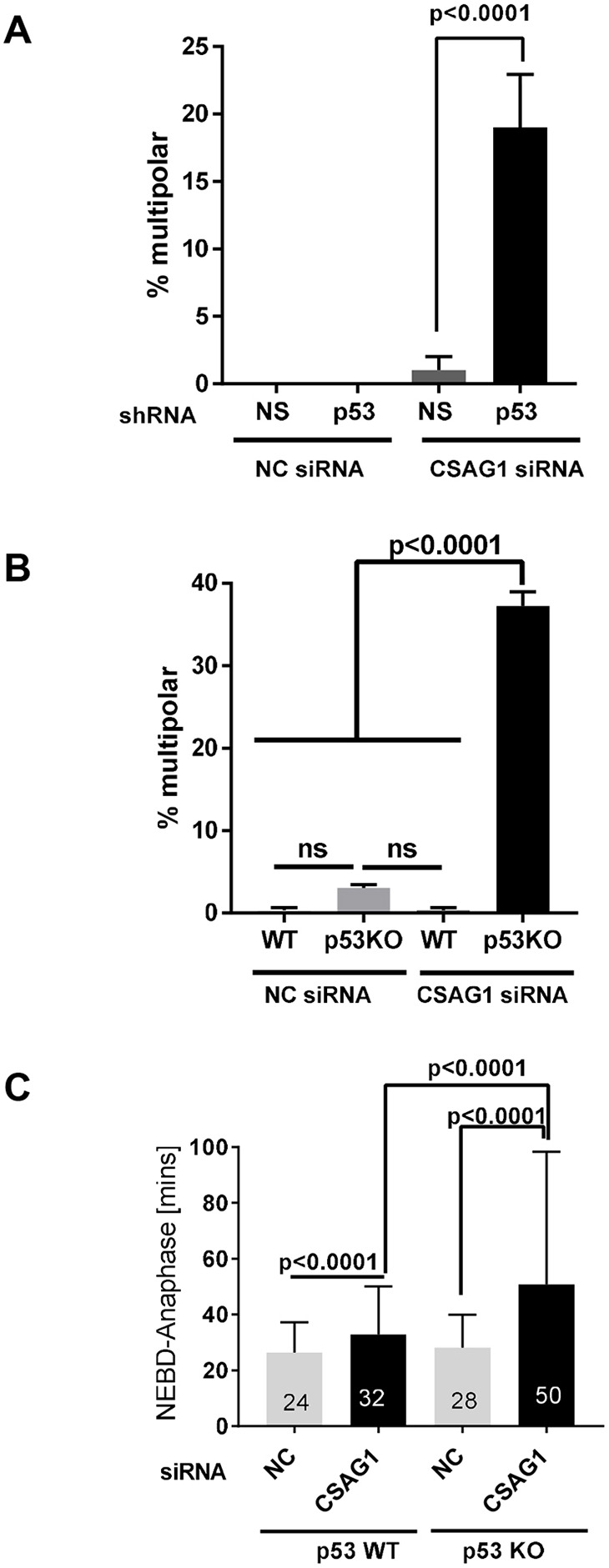
Multipolar mitosis due to CSAG1 depletion is affected by p53 expression. (A) Multipolar mitosis (in %) in RPE1 cells that had been depleted of p53 with shRNA and transfected with CSAG1 siRNA or negative (NC control siRNA). A total of 150 cells was analyzed from two independent experiments. A Kruskal–Wallis test was used for statistical analysis, error bars represent +s.e.m. (B) The number of multipolar mitotic cells (in %) were determined in HCT116 parental or p53 KO cells after treatment with NC or CSAG1 siRNA. A total of >300 cells was analyzed for mitotic defects from three independent experiments. Error bars represent +s.d. Two way ANOVA with Sidak's multiple comparisons test was used for statistical analysis. (C) Elapsed time from nuclear envelope breakdown (NEBD) to anaphase was determined for cells shown in A that did not show mitotic defects. A Kruskal-Wallis test was used for statistical analysis. A total of >200 cells from three independent experiments was analyzed. CSAG1 depletion caused pronounced multipolar mitosis in p53 KO cells, and increased mitotic duration in both HCT116 parental and p53 KO cells.
DISCUSSION
We have characterized mitotic functions for the previously uncharacterized gene CSAG1. Exogenously expressed CSAG1 localizes to the centrosome throughout the cell cycle, becoming more concentrated there during mitosis. By using RNA interference (RNAi) coupled with live cell imaging, we showed that CSAG1 plays an important role in maintaining mitotic spindle pole integrity, particularly in p53-deficient cells. Unfortunately, we were unsuccessful in detecting endogenous CSAG1 protein by western blotting or immunofluorescence using either our own and a commercial antibody. However, a study examining post-mortem brain tissue has used mass-spectrometry and detected a phospho-peptide specifically recognizing CSAG1 protein (Herskowitz et al., 2010). We speculate that the endogenous protein is expressed at very low levels.
Multipolarity has several potential sources, e.g. centrosome overamplification, failure to cluster extra centrosomes and premature centriole disengagement. None of these were evident in CSAG1-depleted HeLa cells. CSAG1-depleted cells enter mitosis with two normal spindle poles but these often exhibit extended areas of PCM compared to control cells. Immunofluorescence showed colocalization of pericentrin and GFP-CSAG1 (Fig. 2). Over time, in many CSAG1-depleted cells, supernumerary poles arose that lacked centrioles but contained PCM components pericentrin and γ-tubulin. Thus, new poles were formed by PCM fragmentation (Fig. 3C,D).
How the integrity of the centrosome is maintained and how it resists mechanical forces imposed by the mitotic spindle remains unclear. Several PCM components might contribute. One study suggested that protein phosphatase 4C promotes centrosome maturation and microtubule nucleation (Sumiyoshi et al., 2002), and a protein termed kizuna (Kiz), whose depletion induces a mitotic phenotype much like CSAG1 depletion, has been identified (Oshimori et al., 2006). Recent ideas suggest that centrosomes assemble through phase separation, which may be cell cycle regulated (Zwicker et al., 2014; Woodruff et al., 2017; Boeynaems et al., 2018). We speculate that CSAG1 contributes to mitotic centrosome PCM integrity, perhaps through a role in phase separation. In the absence of CSAG1, microtubule-dependent spindle forces cause disruption and formation of supernumerary poles. The exact molecular mechanisms through which CSAG1 stabilizes PCM are subjects of current and future inquiry. Although multipolarity was enhanced by treatments that delay anaphase onset in CSAG1-depleted cells, arrest at metaphase by itself does not induce spreading of PCM at poles. HeLa cells that were delayed at metaphase in response to MG132 did not show altered pericentrin distribution, indicating that centrosomes do not spontaneously fragment to form multiple poles after short mitotic delays (Fig. S3). An additional effect we detected in response to CSAG1 RNAi, particularly evident in RPE1 cells and HCT116 cells, was a 60–70% repression of mitosis entry. Because, unlike multipolarity, this effect could not be rescued by expression of wild-type CSAG1, we believe it is likely to reflect an off-target effect of RNAi used by us. siRNA targeting CSAG1 ORF does not reduce the mitotic entry, further strengthening the notion that mitotic entry phenotype is, indeed, a side effect of particular siRNAs that target the UTR of CSAG1.
Evidence from cells expressing uncompromised p53 suggest that CSAG1 is not strictly essential in normal cells, although it may increase the fidelity of chromosome segregation. Even in HeLa cells, which lack p53 function, only 45% of CSAG1-depleted mitotic cells show multipolarity. Some transformed cells take longer to proceed through mitosis, and those cells might be more likely to exhibit multipolarity when centrosome structure is compromised. The precise role that p53 plays in this process is uncertain, although other work has shown a correlation between p53 inactivation and sensitivity to depletion of mitotic regulators (McKinley and Cheeseman, 2017).
At first glance, multipolarity in CSAG1-depleted cells might be attributed to delays in mitotic progression. However, extended mitotic duration does not fully explain the extent of multipolarity observed with CSAG1 depletion. Cells treated with low concentrations of Nocodazole, Taxol or ProTAME exhibit mitotic delay that is equal to or longer than that induced by CSAG1 depletion but show no or low levels of multipolarity. Instead, we attribute the increased multipolarity in CSAG1-depleted cells to weakened PCM integrity, which fragments during mitotic delays. In CSAG1-depleted cells, spindle microtubules play a crucial role in PCM fragmentation and multipolar mitosis, as pole fragmentation is abolished in response to depolymerization of spindle microtubules with Nocodazole (Fig. 4F).
The connection between the multipolar phenotype and p53 is interesting from both a mechanistic level and as a lead in identifying cancer-specific targets. No correlation between p53 and CSAG1 gene expression has been detected, suggesting that one gene is not regulating the other gene at the transcriptional level (Thul et al., 2017). Cells depleted of centrosomal components, such as pericentrin, γ-tubulin and cNap1, appear to arrest at G1 at a p53-mediated G1 checkpoint (Mikule et al., 2007). This explanation, perhaps, explains why CSAG1-depleted HCT116 cells and RPE1 cells with normal p53 levels may exhibit defects in centrosome structure that cause arrest or delay in interphase, resulting in reduced mitotic entry.
Overall, our data establish CSAG1 as a centrosomal protein that aids in maintaining spindle pole integrity. Notably, defects are seen in transformed p53-deficient cells and enhanced by delayed mitotic progression. Identification of interacting partners and specific molecular functions of CSAG1 will provide a better understanding of how spindle pole integrity is maintained.
MATERIALS AND METHODS
Cell culture
HeLa, RPE1 and HCT116 cells were cultured in flasks in DMEM-based medium supplemented with 20 mM HEPES, non-essential amino acids (NEAA), sodium-pyruvate, 1× penicillin-streptomycin (P/S, Corning, 30-002-CI) and 10% FBS. Cells were maintained at 37°C under 5% CO2 in a water-jacketed incubator. Cells were subcultured by trypsinization every other day. Mycoplasma contamination was routinely tested cytologically. All cell lines used in this study were mycoplasma free.
RNAi, live-cell imaging and drug treatment
Cells grown in 4- or 8-well cover glass chambers were transfected with 100 nM siRNA using Lipofectamine 2000 (Thermofisher, cat. # 11668019). Either negative control siRNA (cat# D001810-10-20) or the CSAG1 siRNA pool from Dharmacon that targets both 5′- and 3′-UTRs (5′-GGCAGCCAGUAAUUCCCAG-3′, 5′-UUCCUUGGGCAGCCAGUAA-3′, 5′-GGCAGCCAGUAAUUCCCAG), or with siRNAs targeting CSAG1-ORF (5′-AGACAACCCAGAAGGGAAA-3′, 5′-GGAGUAGACUGUACAGAGA-3′) was used. At 30-36 h post transfection, medium was replaced with Fluorobright fluorescent imaging medium containing 500 nM of the dye SiR-DNA (Cytoskeleton). The chamber was then transferred to a Nikon Ti microscope fitted with an OKOlab environmental control chamber. Images were acquired every 7–10 min using a Nikon 20× objective and Nikon CMOS camera. After completion of imaging, files were exported as TIFF files and analyzed with Metamorph software. Elapsed time from nuclear envelope breakdown (NEBD) to metaphase and anaphase was determined for each cell that entered mitosis. Additionally, each cell entering mitosis was examined for mitotic defects, such as chromosome misalignment, mitotic exit without alignment, lagging chromosomes, anaphase bridges, multipolar mitotic exit and formation of micronuclei.
For drug treatment, HeLa cells transfected with control or CSAG1 siRNA were treated with either 25 nM Nocodazole, 1 nM Taxol, Reversine (250 nM) or 5 µM ProTAME at the beginning of live cell imaging.
Immunofluorescence
HeLa cells were grown on 22 mm2 coverslips in 6-well plates. 24 h after seeding, cells were transfected with 50-100 nM of CSAG1 siRNA. 36-48 h post siRNA transfection, cells were fixed with either −20°C methanol at −20°C for 20 min or co-fixed and extracted with 2% PFA+1% Triton X-100 in 1× PHEM buffer (60 mM PIPES, 25 mM HEPES, 10 mM EGTA and 4 mM MgCl2) for 15 min. Cells were then blocked with 20% boiled normal goat serum (BNGS) for 30 min followed by incubation with primary antibodies. Rabbit anti-pericentrin antibody (cat# Ab4448, Abcam) was used at 1:2500, mouse anti-Y-tubulin (cat# T5326, Sigma) was used at 1:300, mouse anti-centrin 1 (cat# 04-1624, EMD Millipore) was used at 1:2500 and anti-GFP Nano-body was used at 1:300 (Chromotek). Cells were washed 3× for 5 min with MBST (MOPS-buffered saline with 0.05% Tween 20) and then incubated with secondary antibodies; FITC conjugated to goat anti-rabbit IgG (1:800), Cy3 conjugated to goat-anti-mouse IgG (1:1500) for 2 h at room temperature. Cells were washed twice again then stained with 200 ng/ml of DAPI for 3 min. They were then washed and mounted in Vectashield, and coverslips were sealed with nail polish. Images were acquired using a Zeiss Axioplan II microscope with either 63× or 100× objectives, Hamamatsu ORCA II camera, and Metamorph software.
Generation of GFP-CSAG1 cell lines
The CSAG1 coding region was amplified from a Gateway entry vector plasmid obtained from DNASU to which a stop codon was added. The insert was cloned to a Topo D entry vector, then fused to a MYC-GFP- plasmid with pcDNA5/FRT/TO destination vector modified for gateway cloning. HeLa Flp-in TRex cells were transfected with 900 ng of flippase (pOGG44) plasmid and 100 ng of GFP-CSAG1 plasmid in a 6-well plate. After 48 h, cells were transferred to a 15-cm plate and selected with 200 µg/ml hygromycin over 20 days. Surviving clones were pooled, treated with doxycycline to induce CSAG1 for 48 h, then FACS sorted by fluorescence for GFP expression by using a FacsAriaIIIu. These cells were maintained in hygromycin for selection throughout the study.
For rescue experiments, GFP-CSAG1-HeLa Flp-in TRex cells were grown in chambered coverslips for 24 h with or without 2 µg/ml doxycycline, followed by transfection with either control siRNA or CSAG1 siRNA. 30-36 h later cells were imaged as described earlier, and quantified.
For CSAG1 localization, GFP-CSAG1-HeLa Flp-in TRex cells were grown on coverslips with or without doxycycline (2 µg/ml) for 48 h. Then, cells were co-fix extracted with 2% PFA+1% Triton X-100 in 1× PHEM buffer for 15 min at room temperature. Fixed cells were washed once, labeled with anti-pericentrin antibody and GFP-binding protein (nano-body) fused to Atto 488 (1:300). The protocol described earlier was followed for slide preparation and image acquisition.
Short metaphase arrest
HeLa cells were treated with 20 µM MG132 for 1.5 h and fixed with −20°C methanol as described earlier. Then cells were labeled with anti-pericentrin antibody followed by DAPI staining. Metaphase cells were randomly selected from untreated control cells and MG132-treated cells were then imaged. Distribution patterns of pericentrin were scored manually.
Antibody production and validation
GST-tev-CSAG1 fusion protein was produced in a bacterial expression system, purified and then sent to Cocalico (Stevens, PA) for antiserum production. Serum was tested periodically by western blotting for GST and CSAG1 protein. After confirming the presence of GST- and CSAG1-specific antibodies in the serum, the serum was collected and affinity purified using His-CSAG1 linked to the amino link plus coupling resins (Thermo Scientific, #20501) according to manufacturer's protocol. Serum was tested again by western blotting of CSAG1 recombinant protein for specificity and purity of purified antibodies. Serum specificity and reactivity was also tested in immunoprecipitated GFP-CSAG1 cell lysates. Clarified cell lysate was immunoprecipitated using GFP-Trap-A beads from Chromotek, then blotted with purified serum. Uninduced samples (no doxycycline treatment) of HeLa-GFP-CSAG1 cells were used as negative controls.
Immunoprecipitation
For IP using GFP-Trap-A beads (ChromoTek, # GTA-20), the manufacturer's protocol was followed with slight modifications. Briefly, inducible GFP-CSAG1 HeLa cells were grown in 10-cm tissue culture dishes for 48 h with or without 2 µg/ml of doxycycline. Cells were then harvested by trypsinization, washed in cold PBS and lysed in IP buffer (10 mM Tris-HCl pH 7.4, 150 mM NaCl, 1 mM EDTA, 0.5% NP-40 and 1:200 Protease inhibitor; Sigma, #P8340). Cells were lysed by vigorous pipetting every 10 min for a total of 30 min, followed by centrifugation at 16,000 g for 12 min. Clear supernatant was transferred to a new micro centrifuge tube, 25 µl of pre-equilibrated anti-GFP beads were added to the supernatants and incubated with mixing for 2 h at 4°C. Then, the beads were centrifuged at 200 g for 2 min and supernatant was discarded. Beads were washed 3× with 500 µl of IP buffer, then 30 µl of 3× LDS loading buffer with 150 mM DTT was added directly to the beads and samples were used for SDS-PAGE.
For western blots, 1× loading samples were prepared using 4× LDS (Thermofisher scientific, #NP008) sample buffer+100 mM DTT (final). The cell lysates (whole-cell lysate, supernatant or IPed samples) were treated at 95°C for 10 min. The samples were loaded onto a 4-12% Bis-Tris NuPage SDS gel and electrophoresed in MOPS SDS buffer at 120 V until the dye front reached the bottom of the gel. Samples were transferred to 45 μm Immobilon-FL PVDF membrane (Millipore, # IPFL00010), blocked with 1% fish gelatine in PBST (0.5% Tween-20) for 30 min, then incubated with in-house anti-CSAG1 antibody (1:100) at RT overnight and washed 3× with PBST, and incubated with Far-Red goat anti-rabbit IgG IR700 (1:10,000) from Azure (AC2128).
Statistical analysis
Statistical analysis for each experiment is stated in the corresponding figure legend. P-values are shown in the figures; ns is not significant. P>0.05 was classified as ns.
Supplementary Material
Acknowledgements
We thank Mr Evan Fields for technical assistance and for carrying out preliminary experiments. We thank Dr Andrew Holland (Johns Hopkins University, Baltimore, USA) for HeLa-flip-In TRex cells. We thank Dr Tamara Potapova (Stowers Institute, Kansas City, MO) for providing RPE1 cells expressing control and p53-directed shRNA. We thank Dr Bert Vogelstein (Johns Hopkins University, Sidney Kimmel Comprehensive Cancer Center, Baltimore, MD) for permission to use HCT116 53 KO cells, which were provided to us by Dr Ralf Janknecht (University of Oklahoma Health Sciences Center, Oklahoma City, OK). We also thank all members of Program in Cell Cycle and Cancer Biology at Oklahoma Medical Research Foundation (OMRF) for insightful discussion and comments.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization: H.S., G.J.G.; Methodology: H.S., J.D.W., G.J.G.; Software: J.D.W.; Validation: H.S., G.J.G.; Formal analysis: H.S., J.D.W., G.J.G.; Investigation: H.S., G.J.G.; Data curation: H.S.; Writing - original draft: H.S.; Writing - review & editing: G.J.G.; Visualization: H.S.; Supervision: G.J.G.; Project administration: G.J.G.; Funding acquisition: J.D.W., G.J.G.
Funding
This study was supported by the National Institute of General Medical Sciences (grant numbers: R35GM126980 to G.J.G. and P20GM103636 to J.D.W.) and the McCasland Foundation. Deposited in PMC for release after 12 months.
Supplementary information
Supplementary information available online at http://jcs.biologists.org/lookup/doi/10.1242/jcs.239723.supplemental
References
- Asteriti I. A., Giubettini M., Lavia P. and Guarguaglini G. (2011). Aurora-A inactivation causes mitotic spindle pole fragmentation by unbalancing microtubule-generated forces. Mol. Cancer 10, 131 10.1186/1476-4598-10-131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bharadwaj R. and Yu H. (2004). The spindle checkpoint, aneuploidy, and cancer. Oncogene 23, 2016-2027. 10.1038/sj.onc.1207374 [DOI] [PubMed] [Google Scholar]
- Bodnar A. G., Ouellette M., Frolkis M., Holt S. E., Chiu C. P., Morin G. B., Harley C. B., Shay J. W., Lichtsteiner S. and Wright W. E. (1998). Extension of life-span by introduction of telomerase into normal human cells. Science 279, 349-352. 10.1126/science.279.5349.349 [DOI] [PubMed] [Google Scholar]
- Boeynaems S., Alberti S., Fawzi N. L., Mittag T., Polymenidou M., Rousseau F., Schymkowitz J., Shorter J., Wolozin B., Van Den Bosch L. et al. (2018). Protein phase separation: a new phase in cell biology. Trends Cell Biol. 28, 420-435. 10.1016/j.tcb.2018.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunz F., Dutriaux A., Lengauer C., Waldman T., Zhou S., Brown J. P., Sedivy J. M., Kinzler K. W. and Vogelstein B. (1998). Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science 282, 1497-1501. 10.1126/science.282.5393.1497 [DOI] [PubMed] [Google Scholar]
- Coelho P. A., Bury L., Shahbazi M. N., Liakath-Ali K., Tate P. H., Wormald S., Hindley C. J., Huch M., Archer J., Skarnes W. C. et al. (2015). Over-expression of Plk4 induces centrosome amplification, loss of primary cilia and associated tissue hyperplasia in the mouse. Open Biol. 5, 150209 10.1098/rsob.150209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daum J. R., Wren J. D., Daniel J. J., Sivakumar S., McAvoy J. N., Potapova T. A. and Gorbsky G. J. (2009). Ska3 is required for spindle checkpoint silencing and the maintenance of chromosome cohesion in mitosis. Curr. Biol. 19, 1467-1472. 10.1016/j.cub.2009.07.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daum J. R., Potapova T. A., Sivakumar S., Daniel J. J., Flynn J. N., Rankin S. and Gorbsky G. J. (2011). Cohesion fatigue induces chromatid separation in cells delayed at metaphase. Curr. Biol. 21, 1018-1024. 10.1016/j.cub.2011.05.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drosopoulos K., Tang C., Chao W. C. and Linardopoulos S. (2014). APC/C is an essential regulator of centrosome clustering. Nat. Commun. 5, 3686 10.1038/ncomms4686 [DOI] [PubMed] [Google Scholar]
- Fields E., Wren J. D., Georgescu C., Daum J. R. and Gorbsky G. J. (2018). Predictive bioinformatics identifies novel regulators of proliferation in a cancer stem cell model. Stem. Cell Res. 26, 1-7. 10.1016/j.scr.2017.11.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gergely F. and Basto R. (2008). Multiple centrosomes: together they stand, divided they fall. Genes Dev. 22, 2291-2296. 10.1101/gad.1715208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godinho S. A., Kwon M. and Pellman D. (2009). Centrosomes and cancer: how cancer cells divide with too many centrosomes. Cancer Metastasis Rev. 28, 85-98. 10.1007/s10555-008-9163-6 [DOI] [PubMed] [Google Scholar]
- Herskowitz J. H., Seyfried N. T., Duong D. M., Xia Q., Rees H. D., Gearing M., Peng J., Lah J. J. and Levey A. I. (2010). Phosphoproteomic analysis reveals site-specific changes in GFAP and NDRG2 phosphorylation in frontotemporal lobar degeneration. J. Proteome Res. 9, 6368-6379. 10.1021/pr100666c [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland A. J., Fachinetti D., Da Cruz S., Zhu Q., Vitre B., Lince-Faria M., Chen D., Parish N., Verma I. M., Bettencourt-Dias M. et al. (2012). Polo-like kinase 4 controls centriole duplication but does not directly regulate cytokinesis. Mol. Biol. Cell 23, 1838-1845. 10.1091/mbc.e11-12-1043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hut H. M., Lemstra W., Blaauw E. H., Van Cappellen G. W., Kampinga H. H. and Sibon O. C. (2003). Centrosomes split in the presence of impaired DNA integrity during mitosis. Mol. Biol. Cell 14, 1993-2004. 10.1091/mbc.e02-08-0510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janjic B., Andrade P., Wang X. F., Fourcade J., Almunia C., Kudela P., Brufsky A., Jacobs S., Friedland D., Stoller R. et al. (2006). Spontaneous CD4+ T cell responses against TRAG-3 in patients with melanoma and breast cancers. J. Immunol. 177, 2717-2727. 10.4049/jimmunol.177.4.2717 [DOI] [PubMed] [Google Scholar]
- Jiang X. R., Jimenez G., Chang E., Frolkis M., Kusler B., Sage M., Beeche M., Bodnar A. G., Wahl G. M., Tlsty T. D. et al. (1999). Telomerase expression in human somatic cells does not induce changes associated with a transformed phenotype. Nat. Genet. 21, 111-114. 10.1038/5056 [DOI] [PubMed] [Google Scholar]
- Lin C., Mak S., Meitner P. A., Wolf J. M., Bluman E. M., Block J. A. and Terek R. M. (2002). Cancer/testis antigen CSAGE is concurrently expressed with MAGE in chondrosarcoma. Gene 285, 269-278. 10.1016/S0378-1119(02)00395-5 [DOI] [PubMed] [Google Scholar]
- Maiato H. and Logarinho E. (2014). Mitotic spindle multipolarity without centrosome amplification. Nat. Cell Biol. 16, 386-394. 10.1038/ncb2958 [DOI] [PubMed] [Google Scholar]
- McKinley K. L. and Cheeseman I. M. (2017). Large-scale analysis of CRISPR/Cas9 cell-cycle knockouts reveals the diversity of p53-dependent responses to cell-cycle defects. Dev. Cell 40, 405-420.e402. 10.1016/j.devcel.2017.01.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikule K., Delaval B., Kaldis P., Jurcyzk A., Hergert P. and Doxsey S. (2007). Loss of centrosome integrity induces p38-p53-p21-dependent G1-S arrest. Nat. Cell Biol. 9, 160-170. 10.1038/ncb1529 [DOI] [PubMed] [Google Scholar]
- Millband D. N., Campbell L. and Hardwick K. G. (2002). The awesome power of multiple model systems: interpreting the complex nature of spindle checkpoint signaling. Trends Cell Biol. 12, 205-209. 10.1016/S0962-8924(02)02276-6 [DOI] [PubMed] [Google Scholar]
- Mittal K., Choi D. H., Klimov S., Pawar S., Kaur R., Mitra A. K., Gupta M. V., Sams R., Cantuaria G., Rida P. C. G. et al. (2016). A centrosome clustering protein, KIFC1, predicts aggressive disease course in serous ovarian adenocarcinomas. J. Ovarian. Res. 9, 17 10.1186/s13048-016-0224-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta M., Tanaka F., Sadanaga N., Yamaguchi H., Inoue H. and Mori M. (2006). Expression of the TRAG-3 gene in human esophageal cancer: the frequent synchronous expression of MAGE-3 gene. Oncol. Rep. 15, 1529-1532. 10.3892/or.15.6.1529 [DOI] [PubMed] [Google Scholar]
- Oshimori N., Ohsugi M. and Yamamoto T. (2006). The Plk1 target Kizuna stabilizes mitotic centrosomes to ensure spindle bipolarity. Nat. Cell Biol. 8, 1095-1101. 10.1038/ncb1474 [DOI] [PubMed] [Google Scholar]
- Pannu V., Rida P. C., Ogden A., Turaga R. C., Donthamsetty S., Bowen N. J., Rudd K., Gupta M. V., Reid M. D., Cantuaria G. et al. (2015). HSET overexpression fuels tumor progression via centrosome clustering-independent mechanisms in breast cancer patients. Oncotarget 6, 6076-6091. 10.18632/oncotarget.3475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potapova T. A., Seidel C. W., Box A. C., Rancati G. and Li R. (2016). Transcriptome analysis of tetraploid cells identifies cyclin D2 as a facilitator of adaptation to genome doubling in the presence of p53. Mol. Biol. Cell 27, 3065-3084. 10.1091/mbc.e16-05-0268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintyne N. J., Reing J. E., Hoffelder D. R., Gollin S. M. and Saunders W. S. (2005). Spindle multipolarity is prevented by centrosomal clustering. Science 307, 127-129. 10.1126/science.1104905 [DOI] [PubMed] [Google Scholar]
- Sapkota H., Wasiak E., Daum J. R. and Gorbsky G. J. (2018). Multiple determinants and consequences of cohesion fatigue in mammalian cells. Mol. Biol. Cell 29, 1811-1824. 10.1091/mbc.E18-05-0315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern B. M. and Murray A. W. (2001). Lack of tension at kinetochores activates the spindle checkpoint in budding yeast. Curr. Biol. 11, 1462-1467. 10.1016/S0960-9822(01)00451-1 [DOI] [PubMed] [Google Scholar]
- Stevens D., Gassmann R., Oegema K. and Desai A. (2011). Uncoordinated loss of chromatid cohesion is a common outcome of extended metaphase arrest. PLoS ONE 6, e22969 10.1371/journal.pone.0022969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumiyoshi E., Sugimoto A. and Yamamoto M. (2002). Protein phosphatase 4 is required for centrosome maturation in mitosis and sperm meiosis in C. elegans. J. Cell Sci. 115, 1403-1410. [DOI] [PubMed] [Google Scholar]
- Thul P. J., Akesson L., Wiking M., Mahdessian D., Geladaki A., Ait Blal H., Alm T., Asplund A., Bjork L., Breckels L. M. et al. (2017). A subcellular map of the human proteome. Science 356, eaal3321 10.1126/science.aal3321 [DOI] [PubMed] [Google Scholar]
- Tighe A., Johnson V. L. and Taylor S. S. (2004). Truncating APC mutations have dominant effects on proliferation, spindle checkpoint control, survival and chromosome stability. J. Cell Sci. 117, 6339-6353. 10.1242/jcs.01556 [DOI] [PubMed] [Google Scholar]
- Tipton A. R., Wren J. D., Daum J. R., Siefert J. C. and Gorbsky G. J. (2017). GTSE1 regulates spindle microtubule dynamics to control Aurora B kinase and Kif4A chromokinesin on chromosome arms. J. Cell Biol. 216, 3117-3132. 10.1083/jcb.201610012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver B. A. (2014). How Taxol/paclitaxel kills cancer cells. Mol. Biol. Cell 25, 2677-2681. 10.1091/mbc.e14-04-0916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiese C. and Zheng Y. (2006). Microtubule nucleation: gamma-tubulin and beyond. J. Cell Sci. 119, 4143-4153. 10.1242/jcs.03226 [DOI] [PubMed] [Google Scholar]
- Woodruff J. B., Wueseke O. and Hyman A. A. (2014). Pericentriolar material structure and dynamics. Philos. Trans. R. Soc. Lond. B Biol. Sci. 369, 20130459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodruff J. B., Ferreira Gomes B., Widlund P. O., Mahamid J., Honigmann A. and Hyman A. A. (2017). The centrosome is a selective condensate that nucleates microtubules by concentrating tubulin. Cell 169, 1066-1077.e1010. 10.1016/j.cell.2017.05.028 [DOI] [PubMed] [Google Scholar]
- Wren J. D. (2009). A global meta-analysis of microarray expression data to predict unknown gene functions and estimate the literature-data divide. Bioinformatics 25, 1694-1701. 10.1093/bioinformatics/btp290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao X., Hu J. F., Li T., Yang Y., Sun Z., Ulaner G. A., Vu T. H. and Hoffman A. R. (2004). Epigenetic regulation of the taxol resistance-associated gene TRAG-3 in human tumors. Cancer Genet. Cytogenet. 151, 1-13. 10.1016/j.cancergencyto.2003.08.021 [DOI] [PubMed] [Google Scholar]
- Zwicker D., Decker M., Jaensch S., Hyman A. A. and Julicher F. (2014). Centrosomes are autocatalytic droplets of pericentriolar material organized by centrioles. Proc. Natl. Acad. Sci. USA 111, E2636-E2645. 10.1073/pnas.1404855111 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.