

ABSTRACT
Frontotemporal dementia and parkinsonism (FTDP-17) caused by the 10+16 splice-site mutation in the gene encoding microtubule-associated protein tau (MAPT) provides an established platform to model tau-related dementia in vitro. Neurons derived from human induced pluripotent stem cells (iPSCs) have been shown to recapitulate the neurodevelopmental profile of tau pathology during in vitro corticogenesis, as in the adult human brain. However, the neurophysiological phenotype of these cells has remained unknown, leaving unanswered questions regarding the functional relevance and the gnostic power of this disease model. In this study, we used electrophysiology to explore the membrane properties and intrinsic excitability of the generated neurons and found that human cells mature by ∼150 days of neurogenesis to become compatible with matured cortical neurons. In earlier FTDP-17, however, neurons exhibited a depolarized resting membrane potential associated with increased resistance and reduced voltage-gated Na+- and K+-channel-mediated conductance. Expression of the Nav1.6 protein was reduced in FTDP-17. These effects led to reduced cell capability of induced firing and changed the action potential waveform in FTDP-17. The revealed neuropathology might thus contribute to the clinicopathological profile of the disease. This sheds new light on the significance of human in vitro models of dementia.
KEY WORDS: Tau pathology, Human cells, Maturation of iPSC-derived neurons, Neuronal excitability, Neuropathological phenotype, Frontotemporal dementia and parkinsonism
Summary: Pathophysiological excitability was revealed for human cells in dementia. This provides a major advance in understanding of tau-induced brain neuropathology and sheds new light on human models of dementia.
INTRODUCTION
A recurrent problem in dementia research is that the generated animal models do not show neuropathological profiles that match pathological changes found in the human brain (Van Dam and De Deyn, 2006; Irwin et al., 2018; Rohrer et al., 2011; Götz and Ittner, 2008). Although the wide use of animal models (∼180 models of Alzheimer's disease are currently available) remains under scrutiny, the latest advances in generating patient-specific nerve cells from induced pluripotent stem cells (iPSCs) have enabled studies in live human cells (Livesey, 2014; Shi et al., 2012b). Because such model systems are ideally suited for exploring the mechanistic basis of cellular pathogenesis in live cells, they could provide an essential tool for exploring the cell and molecular biology of dementia (Iovino et al., 2015; Ortiz-Virumbrales et al., 2017; Sposito et al., 2015).
Various types of dementia share common functional impairments that occur in nerve cells as the disease progresses. This suggests that different dementia forms have a similar cellular basis. As an example, a class of tauopathies that includes, among others, Alzheimer's disease, Pick's disease and inherited frontotemporal dementia and parkinsonism linked to chromosome 17 (FTDP-17) features the abnormal tau protein, which appears to underlie the pathology at the neuronal level. The primary molecular mechanism involves self-aggregation of hyperphosphorylated tau, toxicity of the tau deposits and, eventually, neuronal death in Alzheimer's disease (Goedert et al., 1989; Matsuo et al., 1994) and FTDP-17 (Hutton et al., 1998; Pickering-Brown et al., 2002; Poorkaj et al., 1998). However, exactly how tau pathology affects human nerve cell function leading to cell death remains unknown for the whole class of tauopathies.
We therefore sought to understand the changes, if any, in the neurophysiological properties of human cells in tau-related dementia, in particular, in a human iPSC model of FTDP-17 caused by the 10+16 intronic mutation in the gene coding the microtubule-associated protein tau (MAPT). This experimental model recapitulates the developmental changes in tau-splicing pathology (increased splicing of 4R tau isoforms) as found in the adult human brain (Sposito et al., 2015; Verheyen et al., 2018; Paonessa et al., 2019). Because very little is known about the neurophysiology of human cells with tau pathology at any stage of earlier or overt symptoms, we sought first to establish the timeline of functional neuronal maturation in vitro. Having established the timing of well-developed neurophysiological properties of human iPSC-derived neurons in controls (healthy donor iPSC lines), we next examined the biophysical properties of neurons with the 10+16 MAPT mutation to document the pathophysiological phenotype of intrinsic excitability for human cells in FTDP-17.
RESULTS
Time-dependent maturation of intrinsic excitability of human iPSC-derived cortical neurons
Human iPSC-derived cells were positively stained with neuronal markers for several cortical layers, detectable following 80 days of in vitro neurogenesis in control lines (healthy donors) and those obtained from patients with FTDP-17 caused by the 10+16 intronic mutation in MAPT (Sposito et al., 2015). Consistently, cells displayed a clear neuronal morphology at 100 days of corticogenesis in these cell lines (Fig. 1A). Hence, patch-clamp recordings were performed on iPSC-derived neurons at 100 and 150 days in vitro (DIV) to trace the maturation of their biophysical properties over an extended period of neurogenesis for the anticipated neurophysiological maturation of the differentiated neurons. At this developmental stage, iPSC-derived neurons with the mutation express both 3R and 4R tau isoforms, whereas control cells express only the 3R tau isoform (Sposito et al., 2015).
Fig. 1.

Time-dependent development of intrinsic excitability of human iPSC-derived cortical neurons in control (non-demented) cell lines. (A) Immunofluorescence staining of human iPSC-derived cells for β-tubulin III (red) and Hoechst 33342 (blue), showing a clear neuronal morphology of the cells at 100 days of corticogenesis in vitro. (B) Left, transmitted light image of human iPSC-derived neurons at 100 DIV for electrophysiological testing; right, membrane capacitance (Cm) of the cells recorded at 100 DIV (n=17) and 150 DIV (n=37). (C) Resting membrane potential (Vrest) of generated neurons at 100 (n=10) and 150 days (n=23) of neurogenesis in vitro. (D) Representative examples of induced firing (upper row) by iPSC-derived neurons at 100 and 150 DIV in response to injecting a series of square current pulses (indicated on the bottom). Note the high-frequency discharge induced by a current of lower stimulating intensity in a neuron at 150 DIV compared to the single AP spike elicited at 100 DIV (red traces). Data are mean±s.e.m. ***P<0.001 (ANOVA with Bonferroni post-hoc test).
Intrinsic membrane properties determine key aspects of neuronal function and behaviour. We, therefore, started by comparing the passive membrane properties of the generated cells at different time points in control lines. First, there was a substantial increase (almost twofold) in the membrane capacitance (Cm) between 100 and 150 DIV (from 29.6±2.6 pF, n=17 to 61.0±6.9 pF; n=37, P<0.001; Fig. 1B). Second, we measured the resting membrane potential (Vrest, immediately after membrane breakthrough). At 100 DIV, Vrest was relatively depolarized, as observed in all tested cells (a range from −17 mV to −32 mV, an average of −25.3±1.8 mV, n=10; Fig. 1C). This suggests that the generated neurons at this developmental age remain underdeveloped. Indeed, only a proportion of iPSC-derived neurons at the age of 100 DIV (6 out of 18) enabled generation of a single action potential (AP) in response to current injection (Vrest held at −60 mV to −70 mV; Fig. 1D). We never observed a cell capable of generating a train of induced APs at 100 DIV. The Vrest markedly developed by ∼150 DIV (a range from −43 mV to −70 mV, average −54.7±2.5 mV, n=23; Fig. 1C). Consistent with the ‘maturation’ of Vrest, neurons developed their capability for a high-frequency discharge (Fig. 1D).
Other passive properties of the membrane, such as the input resistance (Rin, indicating membrane conductance, i.e. a pool of readily available functional ion channels) and the time constant (τm, the time for the neuron membrane potential to reach 63% of its resting value) were also markedly improved by ∼150 DIV (see next section).
Next, we assessed the voltage-dependent membrane properties underlying the intrinsic excitability of differentiated cells. Because the hyperpolarization-activated cation current (Ih), a mixed current carried by both K+ and Na+, makes an important contribution to setting of the Vrest (Biel et al., 2009; Ludwig et al., 1998) and hence reflects a maturation level, we measured cell responses to hyperpolarizing current steps. For quantitative comparisons, we calculated the drop in resting potential (Vdrop) and the sag response (ratio of the peak to steady-state) for a hyperpolarizing current pulse of −150 pA (Fig. 2A). The iPSC-derived neurons showed a more prominent Vdrop at 150 DIV than at 100 DIV (120.9±14.0 mV, n=18 versus 46.3±12.6 mV, n=17; P<0.001; Fig. 2B). In line with this, neurons at the age of 150 DIV exhibited a larger voltage sag, which was followed in some cells by a rebound spike on termination of the hyperpolarization step (Fig. 2A); cells at 100 DIV had a minor sag response or none (0.31±0.05 versus 0.10±0.04, P<0.01; Fig. 2B). Because hyperpolarization-activated and cyclic nucleotide-gated (HCN) channels largely mediate Ih (Bennett et al., 2000; Biel et al., 2009), the increased Ih (threefold increase in the sag ratio) indicates increased HCN channel density by ∼150 DIV (i.e. improved neuronal maturation).
Fig. 2.
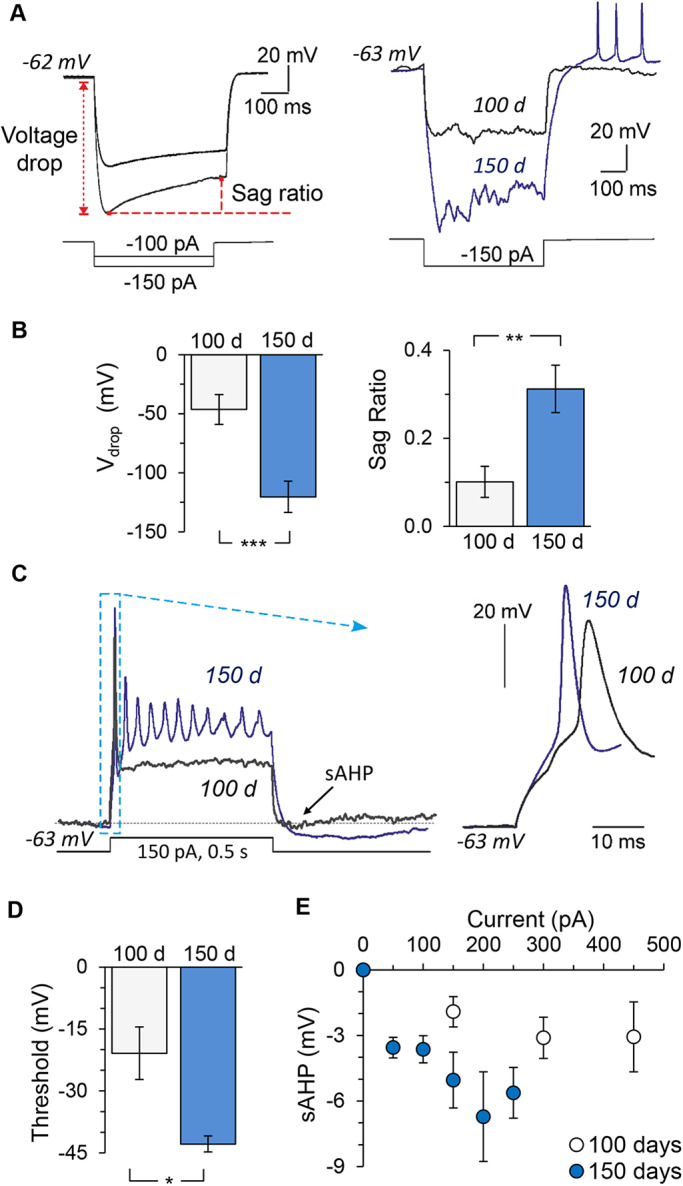
Time-dependent maturation of neurophysiological properties of human cells in control cell lines. (A) Left, examples of patch-clamp recordings made from iPSC-derived neurons in response to a hyperpolarizing current (indicated on the bottom), depicting the method used to measure the voltage drop (Vdrop) and the sag ratio. Right, overlay of hyperpolarizing currents in response to −150 pA by iPSC-derived neurons at the ages of 100 DIV (black line) and 150 DIV (blue line). (B) The Vdrop and sag ratio in iPSC-derived neurons at 100 (n=18) and 150 days (n=17) of neurogenesis. (C) Examples of induced firing by iPSC-derived neurons at 100 DIV (black trace) and 150 DIV (blue trace), showing the sAHP measured at the end of a prolonged (500 ms duration) square current pulse (indicated on the bottom). Box depicts the first AP spikes illustrated on an expanded scale (right) to compare different time points of neuronal maturation. (D) AP threshold in iPSC-derived neurons at 100 and 150 days of neurogenesis (n=9 and 4, respectively). Analysis was carried out for the first AP spike elicited by injecting a slow-ramp current. (E) sAHP (measured as indicated in C) in response to varied depolarizing current pulses recorded in iPSC-derived neurons at 100 DIV (n=17 cells) and 150 DIV (n=17). Data are mean±s.e.m. *P<0.05, **P<0.01, ***P<0.001 (ANOVA with Bonferroni post-hoc test).
To assess the functional expression of fast-activating Na+ channels underlying cell firing activity, we next examined the AP upstroke in the generated neurons (Fig. 2C). In line with a cell's capability of generating a train of induced APs at 150 DIV (as compared with a generated single AP spike by iPSC-derived neurons at 100 DIV; Fig. 1D and Fig. 2C, left traces), the AP spike was of a larger amplitude at 150 DIV (Fig. 2C, right). Importantly, the AP threshold had further developed, as revealed by analysis of the first AP in a train induced by a slow-injecting ramp current (−20.9±6.4 mV at 100 DIV versus −42.9±2.0 mV at 150 DIV; n=9, P<0.05; Fig. 2D). The threshold thus became comparable to that of primary brain neurons (Kopach et al., 2018b; Kopach et al., 2018a). The slow after-hyperpolarization (sAHP) mediated by K+ channels, which largely contribute to AP repolarization and spike shaping (Wilson and Goldberg, 2006), was also developed. The sAHP measured at the end of a square depolarizing pulse train (Fig. 2C) markedly increased by ∼150 DIV, with a clear difference between ∼100 and ∼150 DIV detectable for all depolarizing currents tested, including relatively small currents (0-450 pA; Fig. 2E). This reflects a time-dependent increase in the K+ channel density, especially Ca2+-activated K+ channel subtypes, as an ionic basis of the sAHP component in pyramidal neurons (Andrade et al., 2012; King et al., 2015).
Together, the results show that human iPSC-derived cortical neurons require ∼150 DIV to mature intrinsic excitability up to a common neurophysiological level.
Human iPSC-derived neurons with the MAPT mutation display normal development but changed intrinsic excitability
Having established the time of maturation of neurophysiological excitability for control human cells (non-demented group) as ∼150 DIV, we next investigated the intrinsic excitability of cells with the 10+16 intronic mutation in MAPT (FTDP-17 group).
First, we assessed iPSC-derived neurons with the mutation for their morphological development at ∼100 DIV, an earlier stage of corticogenesis. For analysis of neuronal morphology, cells were immunostained for β-tubulin III and GFAP, together with nuclear staining, and two parameters of the area and the diameter of neuronal somata were quantified in both control and FTDP-17 lines (Fig. 3A). We analysed a total of 367 neurons derived from the two control lines and 431 neurons derived from iPSC lines obtained from two FTDP-17 patients with the 10+16 MAPT mutation (at least six independent preparations per group tested). Quantitative analysis of the neuronal somata area revealed a similar distribution in the density (number of cells) of pooled control and FTDP-17 groups (Fig. 3B). Similarly, the soma diameter did not differ between the groups (median, ∼8.8 µm in control and ∼8.3 µm in FTDP-17; P=0.359 Mood's median test; Fig. 3C). These results demonstrate that human cells with the mutation develop neuronal morphology to a similar extent as control cells at the age of ∼100 DIV.
Fig. 3.

Similar morphological development of human iPSC-derived neurons in control and FTDP-17 cell lines. (A) Immunofluorescence staining of iPSC-derived cells for β-tubulin III (red), GFAP (green) and nuclear marker Hoechst 33342 (blue) at 100 DIV, showing neuronal morphology in a control culture (left and middle images) and one derived from an FTDP-17 patient (right image). Dashed rectangle depicts the area shown on an expanded scale; dotted circles show the method used for measuring neuronal somata size. (B) Distribution profiles of the neuronal somata area in control and FTDP-17 cultures at the age of 100 DIV (n=367 neurons quantified in two control lines and n=431 neurons in two cell lines from FTDP patients). (C) Neuronal soma diameter, with scatter plots showing the parameter distribution across control neurons and those with the mutation at the age of 100 DIV. Boxes show median values; the numbers of cells analysed are same as for B (nonparametric Moods median test value indicated).
Second, we made patch-clamp recordings from iPSC-derived neurons with the mutation. In full agreement with morphological assessment, electrophysiological studies showed no significant difference in the Cm of iPSC-derived neurons in FTDP-17 compared with age-matched controls at 100 DIV, but a trend to increase in FTDP-17 (29.6±2.6 pF, n=17 in control versus 44.6±9.6 pF, n=7 in FTDP-17; P=0.175; Fig. 4A). Neither was a difference observed over an extended period of neurogenesis (at 150 DIV), regardless of comparison between pooled groups (61.0±6.9 pF, n=37 in control versus 60.3±5.1 pF, n=66 in FTDP-17; P=0.935; Fig. 4A, left) or between individual cases (median, ∼43.3 pF, n=37 in control neurons versus ∼45.6 pF, n=31 in neurons from patient 1, P=0.667 versus ∼52.73, n=34 in neurons from patient 2, P=0.797, Mann–Whitney test; Fig. 4A, right).
Fig. 4.

Human iPSC-derived neurons with tau pathology display changed passive membrane properties during extended neurogenesis. (A) Membrane capacitance (Cm) of iPSC-derived neurons in control cell lines (non-demented group) and in cell lines obtained from two patients (FTDP-17 group) at 100 and 150 DIV. Left plots show pooled data at 100 DIV (n=17 in control, n=7 in FTDP-17) and 150 DIV (n=37 in control, n=66 in FTDP-17). Scatter plots demonstrate the parameter distribution for each individual case (i and ii) at 150 DIV (FTDP-i, n=32; FTDP-ii, n=34). Lines represent median values (nonparametric Mann–Whitney test values are indicated). (B) Membrane constant (τm) in control neurons and neurons with the mutation at 100 DIV (n=17 in control, n=7 in FTDP-17) and 150 DIV (n=37 in control, n=66 in FTDP-17). (C) Same as for B, but for the input resistance (Rin); notations as in B. (D) Same as for A, but for the resting membrane potential (Vrest). Right, pooled data at 100 DIV (n=10 in control, n=5 in FTDP-17) and 150 DIV (n=24 in control, n=51 in FTDP-17). Scatter plots show the parameter distribution per case at 150 DIV (FTDP-i, n=22; FTDP-ii, n=24). Lines represent median values. ***P<0.001 (nonparametric Mann–Whitney test). Data are mean±s.e.m. unless indicated otherwise. *P<0.05; ***P<0.001 (ANOVA with Bonferroni post-hoc test); ###P<0.001 (150 DIV versus 100 DIV for control); §§§P<0.001 (150 DIV versus 100 DIV for FTDP-17 groups).
Third, one of the other passive properties of membranes, τm, was not different in neurons from FTDP-17 lines and age-matched controls at any time point tested. The τm was 5.6±1.0 ms (n=17) in control versus 13.9±4.5 ms (n=7, P=0.120) in FTDP-17 at 100 DIV and 43.4±5.5 ms (n=37) versus 46.2±3.7 ms (n=66, P=0.665) at 150 DIV (Fig. 4B). This suggests ‘normal’ development of iPSC-derived neurons with the 10+16 mutation in MAPT (i.e. similar to that in age-matched controls) over the examined period of neurogenesis.
However, neurons with the mutation exhibited changed membrane excitability by ∼150 DIV. In comparison with the control group, neurons with the mutation had an increased Rin (690.2±90.9 MΩ, n=37 in control versus 1000.6±112.7 MΩ, n=66 in FTDP-17; P<0.05; Fig. 4C), although no difference was found at the age of 100 DIV (196.5±33.4 MΩ, n=17 in control versus 270.7±42.3 MΩ, n=7 in FTDP-17; P=0.190; Fig. 4C). Furthermore, neurons with the mutation displayed a depolarized Vrest at 150 DIV compared with the age-matched control (Fig. 4D), which was significant for each individual case (median value, −53 mV, n=23 in control versus −39.0 mV, n=22 in patient 1, P<0.001 and versus −41.5 mV, n=24 in patient 2, P<0.001; Mann–Whitney test; Fig. 4D). Again, such a depolarized Vrest could not be a result of slower Vrest maturation in neurons with the mutation because at 100 DIV the Vrest developed by these cells was even more hyperpolarized than for control cells (−25.3±1.8 mV, n=10 in control versus −45.9±7.0 mV, n=5 in FTDP-17; P<0.05; Fig. 4D, left).
To assess whether such an increase in membrane excitability of human cells in FTDP-17 could be a result of impairments in the voltage-dependent conductance mediated by HCN channels, we recorded the Ih. Surprisingly, there was no significant difference in the Ih between control and FTDP-17 neurons at 150 DIV (Vdrop, n=17 in control and n=32 in FTDP-17, P=0.190; sag ratio, n=16 and 32, respectively, P=0.798, Mann–Whitney test; Fig. 5A), nor at 100 DIV (Vdrop, n=17 in control and n=6 in FTDP-17, P=0.403; sag ratio, n=17 in control and n=6 in FTDP-17, P=0.806; Fig. 5A). Western blot analysis further confirmed a similar level of HCN1 protein expression at ∼150 DIV in control neurons and neurons with the MAPT mutation (Fig. 5B). This suggests that other voltage-gated conductance, instead of HCN channels, is involved in the increased membrane excitability of neurons with the MAPT mutation by the age of ∼150 DIV.
Fig. 5.
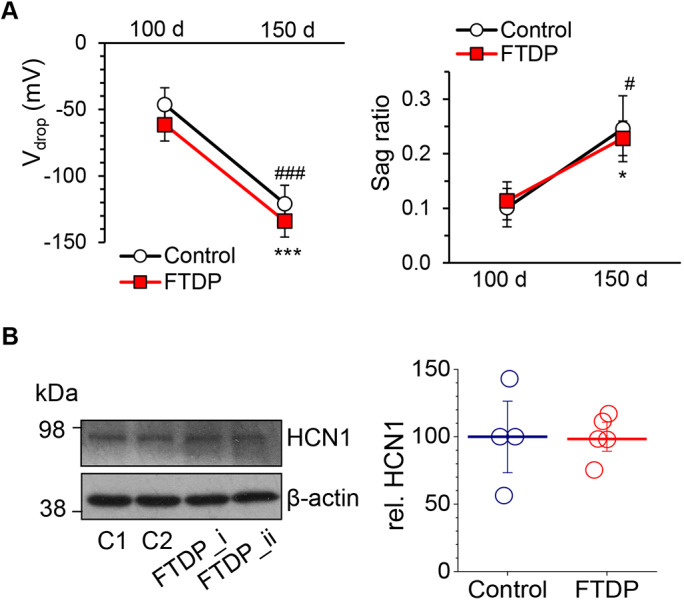
Time-dependent maturation of the neurophysiological properties of human cells in FTDP-17: preserved HCN-channel function during extended neurogenesis. (A) Voltage drop and sag ratio for a hyperpolarizing current pulse of −100 pA in control (non-demented) and FTDP-17 groups at 100 and 150 DIV (Vdrop, n=17 in control, n=6 in FTDP-17 at 100 DIV and n=17, n=32 at 150 DIV; sag ratio, n=17 in control, n=6 in FTDP-17 at 100 DIV and n=16, n=32 at 150 DIV). (B) Left, representative western blot for HCN1 in iPSC-derived neurons in two control lines (C1 and C2) and two FTDP-17 lines (i and ii) at 150-165 DIV. β-actin was used as a loading control. Right, HCN1 protein level (relative for β-actin) in control and FTDP-17 samples (n=4 samples in control and n=5 in FTDP-17; three independent experiments performed). Lines depict median values. Data are mean±s.e.m. unless indicated otherwise. *P<0.05; ***P<0.001 (150 DIV versus 100 DIV for control, ANOVA with Bonferroni post-hoc test); #P<0.05; ###P<0.001 (150 DIV versus 100 DIV for FTDP-17).
Functional downregulation of voltage-gated Na+ and K+ channels in human cells with tau pathology: implication of the neuronal Nav1.6 channel in FTDP-17
We next looked at whether the voltage-gated membrane conductance mediated by Na+ and K+ channels could be affected in human cells by the mutation in MAPT. To assess this, we recorded Na+ (INa) and K+ (IK) currents in differentiated neurons at 150 DIV at different membrane potentials and plotted the current–voltage (I–V) relationships for both channels to compare control and FTDP-17 cells. Most recorded cells displayed robust voltage-activated INa and IK (Fig. 6A). The I–V relationship showed that INa activation occurs at a range of membrane potentials more positive than approximately −50 mV, with no bias in the I–V curve shape, but a clear reduction in the INa density in neurons with the mutation (n=12 in control and n=36 in FTDP-17; Fig. 6B). The peak INa density was dramatically decreased in cells with the mutation (median, ∼7.39 pA/pF in control versus ∼1.9 pA/pF in FTDP-17; P<0.05 Mann–Whitney test; Fig. 6B). Also, there was a decrease in the density of IK (consisting of fast and slow non-inactivating components) in FTDP-17; this reduction appeared at membrane potentials more positive than −30 mV (n=7 in control and n=30 in FTDP-17; Fig. 6C). The results indicate the functional downregulation of Na+ and K+ channels in FTDP-17.
Fig. 6.

Tau-related downregulation of Na+- and K+-channel-mediated conductance in human cells with the 10+16 MAPT mutation: loss of the Nav1.6 channel. (A) Example voltage-clamp recordings from a control iPSC-derived neuron at 150 DIV; box depicts the fast-activating INa shown beneath on an expanded scale. The protocol consisted of voltage steps from −70 mV to +50 mV. (B) Left, the I–V relationship for Na+-channel-mediated conductance (INa) in control neurons (blue) and neurons with the mutation (red) at 150 DIV; right, scatter plots of the peak INa density in control (n=12) and FTDP-17 (n=36) neurons. Red and blue lines represent median values. *P<0.05 (Mann–Whitney test). (C) The I-V relationship for K+-channel-mediated conductance (IK) in human cells in control and FTDP groups at 150 DIV. (D) Expression of the Nav1.6 channel in iPSC-derived neurons at 130-165 DIV in control and FTDP-17 groups, as analysed by western blot. Left, representative western blot including two control cell lines (C1 and C2) and two FTDP-17 lines (i and ii), with β-actin as loading control. Right, Nav1.6 protein levels (relative to β-actin) in control (n=6) and FTDP-17 (n=8) cell cultures, obtained in four independent experiments. Lines depict median values. *P<0.05 (Mann–Whitney test).
To evaluate the mechanism for tau-related functional downregulation of Na+ channels, we examined the protein expression in iPSC-derived neurons in control and FTDP-17 cell lines at 130-165 DIV. Western blot experiments demonstrated reduced expression of the Nav1.6 channel in neurons with the mutation (n=6 samples in control and n=8 in FTDP-17, 4 independent experiments performed; P<0.05, Mann–Whitney test; Fig. 6D). This indicates loss of neuronal Nav1.6 channel as a mechanism contributing to changed intrinsic excitability of human cells in FTDP-17.
Because voltage-gated Na+ and K+ channels are largely responsible for neuronal firing, functional downregulation of these channels would lead to impaired cell function (i.e. changes in firing activity). Therefore, we examined changes in AP discharge at 150 DIV by neurons with the mutation, using two different experimental protocols for eliciting firing: injecting a slow ramp current (200 pA/s; Fig. 7A) or injecting a series of short, square depolarizing pulses (Fig. 7C). First, we found that neurons with the mutation exhibited a depolarized AP threshold (measured for the first AP spike in a train elicited by a ramp protocol) compared with that in age-matched control neurons (−42.4±2.3 mV, n=4 in control versus −31.1±2.6 mV, n=9 in FTDP-17; P<0.05; Fig. 7A). Thus, there was a shift in the threshold of ∼11 mV (depolarizing shift). Second, there was a reduction in amplitude of the induced AP spike (from 46.9±2.9 mV, n=14 in control to 35.5±2.5 mV, n=36 in FTDP-17; P<0.05; Fig. 7B). Third, neurons with the mutation exhibited an increased rheobase, which is the magnitude of depolarizing current required to shift membrane potential to the spike threshold to drive firing (Fig. 7C). A twofold stronger current was needed to bring neurons with tau pathology to firing compared with control neurons (48.2±8.2 pA, n=11 in control versus 96.1±11.5 pA, n=41 in FTDP; P<0.05; Fig. 7C). The latter indicates that neurons with tau pathology need a stimulus almost twice as strong to drive firing. In addition, neurons with the mutation exhibited decreased sAHP, as observed at different depolarizing current pulses (50-250 pA; Fig. 7D), consistent with a reduced K+ channel density in these neurons. Overall, these changes characterize the phenotypic spectrum of pathological neuronal excitability of human cells in FTDP-17.
Fig. 7.

Phenotypic pathophysiological firing by human cells with the 10+16 MAPT mutation. (A) Left, example recordings of AP discharge by a human iPSC-derived neuron bearing the mutation (red trace) and by a control neuron (blue trace on an expanded scale) in response to injection of a slow-ramp current (indicated on bottom); dotted box illustrates the first AP spike for the analysis; insert shows overlay of expanded first AP spikes in FTDP-17 versus control neurons; arrows indicate the AP threshold. Right, AP threshold at 150 DIV in control neurons and those with the mutation (analysis performed for the first AP spike as noted on the right; n=9). (B) AP spike amplitude in control neurons (n=14) and neurons with the mutation (n=36). (C) Left, examples of firing discharge induced by square depolarizing currents (indicated on the bottom) in a control neuron (black) and neuron with the mutation (red). Right, rheobase in control (n=13) and FTDP-17 (n=41). (D) The sAHP in iPSC-derived neurons in control (non-demented) and FTDP-17 groups (n=15 and 32, respectively). Data are mean±s.e.m. *P<0.05 (ANOVA with Bonferroni post-hoc test).
DISCUSSION
Development of human stem cell models that employ patient-specific cell types was a milestone in dementia research. However, physiological studies of iPSC-derived neurons are challenging and sparse. Here, we show that human iPSC-derived cortical neurons require ∼150 days of neurogenesis to match the physiological traits of mature cortical neurons. For the first time, we reveal the phenotypic pathophysiological intrinsic excitability of human cells in frontotemporal dementia and parkinsonism related to tau protein pathology.
Maturation of neurophysiological properties of human cells in vitro
Despite the fact that dementia is a disorder that gradually progresses over a considerably prolonged time, at present, there is no precise treatment to cease or modify the disease. The major challenge in developing therapies has been the lack of a clear understanding of the mechanistic basis underlying the neuropathology. Because animal models appear to not fully replicate neuronal loss as it occurs in the human brain, being associated with a clear clinicopathological profile of memory loss and cognitive decline (somewhat debatable for animal species), human stem cell models have been used for modelling genetic forms of Alzheimer and non-Alzheimer types of dementia for the direct study of patient-specific cells. This study demonstrates the electrophysiological expertise of human iPSC-derived cortical neurons to establish, in the first instance, neuronal maturation of human cells in control and FTDP-17 with tau pathology.
Determining the time when iPSC-derived neurons mature and set neurophysiological activity has always been a challenge that most studies overcome via routine use of various antigens and markers. Although immunocyto(histo)chemistry can document firm expression of receptors and proteins in generated nerve cells, it cannot prove the cell function, with regard to a constitutive level of neurophysiological activity (Kopach, 2019). From the neurophysiological point of view, the biophysical properties of human iPSC-derived cortical neurons at 150 days of neurogenesis in control (non-demented) cell lines confirm that these cells are physiologically credible. The matured intrinsic excitability of generated neurons was confirmed cumulatively by their Vrest and AP threshold – both comparable to that of primary brain neurons (Kopach et al., 2018b) – and a cell capability of high-frequency firing by the age of ∼150 DIV. In comparison with control neurons, neurons with the mutation in MAPT (demented group) did not appear to have underdeveloped biophysical properties at the age of 100 DIV, nor at 150 DIV. For instance, the Ih (reflecting the functional HCN channel expression responsible for recovery from hyperpolarization) was comparable to that in control neurons, consistently with a similar level of HCN1 protein expression (Fig. 5). Likewise, some of the passive membrane properties, such as the capacitance and the time constant were also similar between the groups (Fig. 4). Quantification of neuronal morphology (the somata size) further confirmed similar neuronal development between groups, using an approach other than electrophysiology (Fig. 3).
Overall, varied tests (quantitative comparison of morphological and electrophysiological parameters) indicate that human iPSC-derived neurons mature intrinsic excitability to a constitutive neurophysiological level by ∼150 days of corticogenesis, in both control and FTDP-17 cell lines.
Pathophysiological neuronal excitability of human cells in FTDP-17
The established biophysical properties of the generated neurons suggest that the model is reliable for use in functional studies of genetic forms of tauopathy. In this stem cell model, the developmental changes in tau splicing pathology have been already confirmed (Paonessa et al., 2019; Sposito et al., 2015; Iovino et al., 2015), detecting both 3R and 4R adult brain tau isoforms after a few months of in vitro corticogenesis in FTDP-17 but only 3R in control (non-demented) cell lines. This study documents the tau-related neuropathological phenotype of human cell function through detecting the pathophysiological intrinsic excitability of neurons with the mutation at the development stage between ∼100 and 150 DIV. The phenotype can be described by (i) a depolarized Vrest, (ii) an increased Rin, (iii) a downregulated Na+- and K+-channel-mediated conductance, leading to (iv) functional impairments in firing activity and (v) the changed waveform of APs in FTDP-17. At this earlier stage of the pathology, cells exhibited a depolarized AP threshold (depolarizing shift in ∼11 mV), generated APs of a smaller amplitude and altered shape, and displayed a reduced capability of induced firing (a stimulus nearly twice as strong was needed to drive firing).
Numerous experimental studies have shown the tau-related neuronal dysfunction using animal models, at either earlier or advanced stages of the pathology. Results include (but are not limited to) reduced firing of neocortical neurons in a transgenic model of tauopathy in vivo (Menkes-Caspi et al., 2015), frontotemporal dementia and amyotrophic lateral sclerosis (Radzicki et al., 2016), Alzheimer models (Busche et al., 2019) and parkinsonism (Fieblinger et al., 2014) that opposes neuronal hyperexcitability in aged animals (Crimins et al., 2012). The emerged discrepancy could relate to severe morphological impairment (regression of the brain structure) at later stages, namely loss of synapses and cell death (Crimins et al., 2011). Given the important physiological role of tau in neuronal excitability (DeVos et al., 2013) and synaptic plasticity (Kimura et al., 2014), careful interpolation of obtained phenotypes in transgenic models is needed, because tau overexpression would rapidly shift neurons to hyperexcitability.
The revealed phenotype of pathological neuronal excitability of human cells in FTDP-17 has emerged, at least partially, due to the loss of function of voltage-gated Na+ channels, in particular the neuronal Nav1.6 channel as one of the predominant candidates. The role of the Nav1.6 channel in AP initiation and propagation in cortical excitatory pyramidal neurons (Hu et al., 2009) implies impaired AP parameters and firing discharge by neurons with the MAPT mutation. Dysfunction of Nav1.6-mediated currents is associated with altered learning and memory, being linked to a number of neurological and psychiatric brain disorders, such as epilepsy, intellectual disability and sudden unexpected death in epilepsy (Estacion et al., 2014; Lopez-Santiago et al., 2017; Wagnon et al., 2017). Thus far, we have only revealed a reduced Nav1.6 channel expression in FTDP-17, although other voltage-gated channels could also be involved in this neuropathology. For example, our data indicate that K+ channels are very likely to contribute to the phenotypic pattern of human cell excitability in FTDP-17. Because the slow component of AHP, lasting hundreds of milliseconds to seconds, is Ca2+ dependent (Azouz et al., 1996; Power et al., 2002), the reduced sAHP in FTDP-17 suggests functional downregulation of Ca2+-dependent K+ channels (Andrade et al., 2012; King et al., 2015), which mediate AP repolarization (Azouz et al., 1996; King et al., 2015) and are essential for the generation of rhythmic bursts (Wilson and Goldberg, 2006). Taking into account that slowly inactivating, voltage-gated K+ conductance could be mediated by several channel types, not limited to the Kv1, Kv2, Kv3 and Kv7 subfamilies, numerous candidates could be considered as potential contributors to the pathophysiological excitability of human cells with the MAPT mutation. The most promising candidates apparently refer to the Kv1 subfamily (containing Kv1.1, Kv1.2 and Kv1.6), whose expression contributes to changes in rheobase, AP threshold and the spike waveform (Wykes et al., 2012; Brew et al., 2003).
The tau-induced impairments in intrinsic neuronal excitability can be accompanied by changes in synaptic excitability. The abnormal synaptic function was observed in transgenic animal models before tau aggregates appeared (Van der Jeugd et al., 2012) and prior to neurodegeneration (Menkes-Caspi et al., 2015). Whether this phenotype of intrinsic neuronal excitability of human cells in FTDP-17 is associated with altered synaptic excitation (transmission) remains to be determined. Further studies are needed to decipher in detail the tau-related changes in human cell function at the neuronal network level.
This study provides evidence for the time-dependent maturation of the neurophysiological properties of human iPSC-derived cortical neurons during in vitro neurogenesis, an essential factor for reliable model use in studies of tau-related dementia in vitro. The pathophysiological phenotype of intrinsic neuronal excitability of human cells with the 10+16 splice-site mutation in MAPT has been revealed at 150 DIV, providing a major advance in our understanding of human brain cell dysfunctions at earlier stages of tau-induced dementia. These findings shed new light on human stem cell models of dementia.
MATERIALS AND METHODS
Human stem cell model of FTDP-17
This study included four human cell lines: two control iPSC lines (non-demented group) and two obtained from patients bearing the 10+16 MAPT mutation (FTDP-17 group). Human iPSC lines were as reported previously in detail (Esteras et al., 2017; Sposito et al., 2015). In particular, two 10+16 MAPT iPSC lines were generated by retroviral-transduction reprogramming of fibroblasts obtained from the National Hospital for Neurology and Neurosurgery (London, UK). One control iPSC line, also generated using retroviral transduction from fibroblasts, was obtained from the laboratory of Dr Kunath (Edinburgh University, UK). Another control was purchased from ThermoFisher Scientific. Human iPSC-derived neurons were generated and characterized as previously described (Shi et al., 2012a; Sposito et al., 2015; Esteras et al., 2017). Briefly, differentiation of the pluripotent stem cells into cortical neurons was performed via dual SMAD inhibition for 10 days followed by in vitro neurogenesis. At 46 days after induction, cells were plated on polyornithine/laminin-coated glass coverslips and maintained until use in neural maintenance medium composed of a mixture of N2 and B27 media, as described elsewhere (Shi et al., 2012a), with changes of medium twice a week.
For each group, at least six different preparations were examined. Patch-clamp experiments were performed by the experimenter in a blind to experimental group manner.
Immunocytochemistry
Immunocytochemistry was performed using a common immunostaining protocol by fixing neuronal cultures with 4% paraformaldehyde (15 min at room temperature), followed by permeabilization/blocking with 0.2% Triton X-100 and 10% bovine albumin serum (BSA) for 1 h. Cells were then incubated with Alexa-Fluor-conjugated primary antibodies (anti-β-tubulin III and anti-GFAP, 1:200; Abcam, UK) overnight at 4°C. Hoechst 33342 (10 μM) was used to stain nuclei. Confocal imaging was carried out using a Zeiss 710 VIS CLMS confocal microscope.
Western blot analysis
Cells were washed with PBS and lysed in an ice-cold RIPA lysis buffer supplemented with protease and phosphatase inhibitors (ThermoFisher, Paisley, UK). Samples were snap frozen, sonicated and centrifuged at 14,000 rpm; the protein content was determined using the Pierce BCA protein assay (ThermoFisher). Protein (20 μg) was fractionated on SDS polyacrylamide gel (4-12%) (ThermoFisher), transferred to a PVDF membrane (Bio-Rad, Richmond, CA) and blocked with 5% non-fat milk. Membranes were incubated overnight with primary antibodies (HCN1 and Nav1.6, 1:500, Alomone Labs, Jerusalem, Israel; β-actin, 1:5000, Cell Signaling Technologies, Danvers, MA) diluted in 5% BSA and then with horseradish peroxidase (HRP)-conjugated secondary antibodies. The luminol-based Pierce ECL Western Blotting Substrate (ThermoFisher) was used to detect HRP activity. Protein band densities were quantified using ImageJ software (NIH, Bethesda, MD) after scanning of the X-ray films, and normalized to the control.
Whole-cell electrophysiology
Whole-cell recordings were made from iPSC-derived neurons (Fig. 1A) using a Multipatch 700B amplifier controlled by pClamp 10.2 software package (Molecular Devices, San Jose, CA). For the recordings, a neuronal culture was placed in a recording chamber mounted on the stage of Olympus BX51WI upright microscope (Olympus, Japan) equipped with a LUMPlanFI/IR 40×0.8 objective coupled to an infrared DIC imaging system. Recordings were performed in a bicarbonate-buffered solution containing 126 mM NaCl, 3 mM KCl, 2 mM MgSO4, 2 mM CaCl2, 26 mM NaHCO3, 1.25 mM NaH2PO4 and 10 mM D-glucose (95% O2 and 5% CO2; pH 7.4; osmolarity 300–310 mOsmol) at 31–33°C. Recording electrodes had a resistance of 2.5-5 MΩ when filled with a potassium gluconate-based intracellular solution containing 126 mM potassium gluconate, 10 mM HEPES, 4 mM KCl, 4 mM MgCl2, 2 mM BAPTA, 4 mM Mg-ATP and 0.4 mM GTP-Na, adjusted to pH 7.2 with KOH (osmolarity ∼290 mOsmol). Once in the whole-cell, iPSC-derived neurons were recorded for passive membrane properties, including the resting membrane potential (Vrest), which was detected immediately after membrane breakthrough, membrane capacitance (Cm), input resistance (Rin) and the membrane time constant (τm), measured from the hyperpolarizing square current pulse steps in current mode. The constant, measured as a function of two membrane properties, Rm and Cm, was calculated using the equation τm=Rm×Cm.
Experimental protocols and data analysis
For the assessment of intrinsic excitability, iPSC-derived neurons were tested in the first instance for their capability to generate induced AP. Neuronal firing was elicited using either of two experimental protocols. The first consisted of a series of sub- and supra-threshold rectangular current pulses of 500 ms duration applied with stepwise increased stimulus intensity (an increment of 30–150 pA). The second protocol was a slow-ramp current injection, ramped up with a slope of 100–200 pA/s. For the recordings, Vrest was set at −60 mV to −70 mV by injecting a hyperpolarizing bias current, if needed. Analysis of the parameters of individual AP spikes was performed for the first AP only, in any experimental protocol applied. For the I–V relationship for Na+- and K+-channel-mediated conductance, voltage-clamp recordings consisted of voltage steps of 250 ms duration applied from −100 mV with an increment of 5-10 mV (Kopach et al., 2018a). The voltage drop (Vdrop) during hyperpolarization was measured as the difference between the peak for a hyperpolarizing current (Vmin) and Vrest (Fig. 2A). The hyperpolarization-activated cationic current (Ih) was calculated as the sag ratio by measuring the ratio between the Vmin and its steady-state (Vst) in cell responses to −150 pA or −100 pA pulse injection as (Vmin–Vst)/Vst. Quantitative comparisons of the slow after-hyperpolarization (sAHP) were performed at the end of a spike train in response to a square depolarizing pulse of 500 ms duration (Fig. 2B). Clampfit 10.3 software (Molecular Devices) and Origin Pro (OriginLab, Northampton, MA) were used for analysis.
Statistical analysis
Data are presented as mean±s.e.m., with n referring to the number of cells analysed. The data sets not normally distributed are presented as median values. The Shapiro–Wilk test was used for testing whether experimental data sets were distributed normally. For determining the statistical differences between experimental groups, one-way analysis of variance (ANOVA) with Bonferroni or Tukey multiple comparisons post-hoc test was used as appropriate. The nonparametric Mann–Whitney test or Moods median test was used to compare the medians from two or more populations. P<0.05 was considered as a statistically significant difference between the groups for either test used.
Supplementary Material
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization: O.K., A.Y.A.; Methodology: O.K., N.E., S.W.; Validation: O.K., N.E.; Formal analysis: O.K., N.E.; Investigation: O.K., N.E.; Resources: D.A.R., A.Y.A.; Data curation: D.A.R., A.Y.A.; Writing - original draft: O.K.; Writing - review & editing: O.K., N.E., D.A.R., A.Y.A.; Visualization: O.K., N.E.; Supervision: D.A.R., A.Y.A.; Project administration: A.Y.A.; Funding acquisition: D.A.R., A.Y.A.
Funding
This work was supported by an Engineering and Physical Sciences Research Council grant (EP/R024898/1) to A.Y.A. and a Wellcome Trust Principal Fellowship (212251_Z_18_Z) and European Commission NEUROTWIN grant (857562) to D.A.R. Deposited in PMC for release after 6 months.
Peer review history
The peer review history is available online at https://jcs.biologists.org/lookup/doi/10.1242/jcs.241687.reviewer-comments.pdf
References
- Andrade R., Foehring R. C. and Tzingounis A. V. (2012). The calcium-activated slow AHP: cutting through the Gordian knot. Front. Cell Neurosci. 6, 47 10.3389/fncel.2012.00047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azouz R., Jensen M. S. and Yaari Y. (1996). Ionic basis of spike after-depolarization and burst generation in adult rat hippocampal CA1 pyramidal cells. J. Physiol. 492, 211-223. 10.1113/jphysiol.1996.sp021302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett B. D., Callaway J. C. and Wilson C. J. (2000). Intrinsic membrane properties underlying spontaneous tonic firing in neostriatal cholinergic interneurons. J. Neurosci. 20, 8493-8503. 10.1523/JNEUROSCI.20-22-08493.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biel M., Wahl-Schott C., Michalakis S. and Zong X. (2009). Hyperpolarization-activated cation channels: from genes to function. Physiol. Rev. 89, 847-885. 10.1152/physrev.00029.2008 [DOI] [PubMed] [Google Scholar]
- Brew H. M., Hallows J. L. and Tempel B. L. (2003). Hyperexcitability and reduced low threshold potassium currents in auditory neurons of mice lacking the channel subunit Kv1.1. J. Physiol. 548, 1-20. 10.1113/jphysiol.2002.035568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busche M. A., Wegmann S., Dujardin S., Commins C., Schiantarelli J., Klickstein N., Kamath T. V., Carlson G. A., Nelken I. and Hyman B. T. (2019). Tau impairs neural circuits, dominating amyloid-β effects, in Alzheimer models in vivo. Nat. Neurosci. 22, 57-64. 10.1038/s41593-018-0289-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crimins J. L., Rocher A. B., Peters A., Shultz P., Lewis J. and Luebke J. I. (2011). Homeostatic responses by surviving cortical pyramidal cells in neurodegenerative tauopathy. Acta Neuropathol. 122, 551-564. 10.1007/s00401-011-0877-0 [DOI] [PubMed] [Google Scholar]
- Crimins J. L., Rocher A. B. and Luebke J. I. (2012). Electrophysiological changes precede morphological changes to frontal cortical pyramidal neurons in the rTg4510 mouse model of progressive tauopathy. Acta Neuropathol. 124, 777-795. 10.1007/s00401-012-1038-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devos S. L., Goncharoff D. K., Chen G., Kebodeaux C. S., Yamada K., Stewart F. R., Schuler D. R., Maloney S. E., Wozniak D. F., Rigo F. et al. (2013). Antisense reduction of tau in adult mice protects against seizures. J. Neurosci. 33, 12887-12897. 10.1523/JNEUROSCI.2107-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estacion M., O'brien J. E., Conravey A., Hammer M. F., Waxman S. G., Dib-Hajj S. D. and Meisler M. H. (2014). A novel de novo mutation of SCN8A (Nav1.6) with enhanced channel activation in a child with epileptic encephalopathy. Neurobiol. Dis. 69, 117-123. 10.1016/j.nbd.2014.05.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteras N., Rohrer J. D., Hardy J., Wray S. and Abramov A. Y. (2017). Mitochondrial hyperpolarization in iPSC-derived neurons from patients of FTDP-17 with 10+16 MAPT mutation leads to oxidative stress and neurodegeneration. Redox Biol. 12, 410-422. 10.1016/j.redox.2017.03.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fieblinger T., Graves S. M., Sebel L. E., Alcacer C., Plotkin J. L., Gertler T. S., Chan C. S., Heiman M., Greengard P., Cenci M. A. et al. (2014). Cell type-specific plasticity of striatal projection neurons in parkinsonism and L-DOPA-induced dyskinesia. Nat. Commun. 5, 5316 10.1038/ncomms6316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedert M., Spillantini M. G., Jakes R., Rutherford D. and Crowther R. A. (1989). Multiple isoforms of human microtubule-associated protein tau: sequences and localization in neurofibrillary tangles of Alzheimer's disease. Neuron 3, 519-526. 10.1016/0896-6273(89)90210-9 [DOI] [PubMed] [Google Scholar]
- Götz J. and Ittner L. M. (2008). Animal models of Alzheimer's disease and frontotemporal dementia. Nat. Rev. Neurosci. 9, 532-544. 10.1038/nrn2420 [DOI] [PubMed] [Google Scholar]
- Hu W., Tian C., Li T., Yang M., Hou H. and Shu Y. (2009). Distinct contributions of Na(v)1.6 and Na(v)1.2 in action potential initiation and backpropagation. Nat. Neurosci. 12, 996-1002. 10.1038/nn.2359 [DOI] [PubMed] [Google Scholar]
- Hutton M., Lendon C. L., Rizzu P., Baker M., Froelich S., Houlden H., Pickering-Brown S., Chakraverty S., Isaacs A., Grover A. et al. (1998). Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 393, 702-705. 10.1038/31508 [DOI] [PubMed] [Google Scholar]
- Iovino M., Agathou S., Gonzalez-Rueda A., Del Castillo Velasco-Herrera M., Borroni B., Alberici A., Lynch T., O'dowd S., Geti I., Gaffney D. et al. (2015). Early maturation and distinct tau pathology in induced pluripotent stem cell-derived neurons from patients with MAPT mutations. Brain 138, 3345-3359. 10.1093/brain/awv222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin D. J., Mcmillan C. T., Xie S. X., Rascovsky K., Van Deerlin V. M., Coslett H. B., Hamilton R., Aguirre G. K., Lee E. B., Lee V. M. Y. et al. (2018). Asymmetry of post-mortem neuropathology in behavioural-variant frontotemporal dementia. Brain 141, 288-301. 10.1093/brain/awx319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura T., Whitcomb D. J., Jo J., Regan P., Piers T., Heo S., Brown C., Hashikawa T., Murayama M., Seok H. et al. (2014). Microtubule-associated protein tau is essential for long-term depression in the hippocampus. Philos. Trans. R. Soc. Lond. B Biol. Sci. 369, 20130144 10.1098/rstb.2013.0144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- King B., Rizwan A. P., Asmara H., Heath N. C., Engbers J. D., Dykstra S., Bartoletti T. M., Hameed S., Zamponi G. W. and Turner R. W. (2015). IKCa channels are a critical determinant of the slow AHP in CA1 pyramidal neurons. Cell Rep. 11, 175-182. 10.1016/j.celrep.2015.03.026 [DOI] [PubMed] [Google Scholar]
- Kopach O. (2019). Monitoring maturation of neural stem cell grafts within a host microenvironment. World J. Stem. Cells 11, 982-989. 10.4252/wjsc.v11.i11.982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopach O., Rybachuk O., Krotov V., Kyryk V., Voitenko N. and Pivneva T. (2018a). Maturation of neural stem cells and integration into hippocampal circuits - a functional study in an in situ model of cerebral ischemia. J. Cell Sci. 131, jcs210989 10.1242/jcs.210989 [DOI] [PubMed] [Google Scholar]
- Kopach O., Zheng K., Dong L., Sapelkin A., Voitenko N., Sukhorukov G. B. and Rusakov D. A. (2018b). Nano-engineered microcapsules boost the treatment of persistent pain. Drug Deliv. 25, 435-447. 10.1080/10717544.2018.1431981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livesey F. J. (2014). Human stem cell models of dementia. Hum. Mol. Genet. 23, R35-R39. 10.1093/hmg/ddu302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Santiago L. F., Yuan Y., Wagnon J. L., Hull J. M., Frasier C. R., O'malley H. A., Meisler M. H. and Isom L. L. (2017). Neuronal hyperexcitability in a mouse model of SCN8A epileptic encephalopathy. Proc. Natl. Acad. Sci. USA 114, 2383-2388. 10.1073/pnas.1616821114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig A., Zong X., Jeglitsch M., Hofmann F. and Biel M. (1998). A family of hyperpolarization-activated mammalian cation channels. Nature 393, 587-591. 10.1038/31255 [DOI] [PubMed] [Google Scholar]
- Matsuo E. S., Shin R. W., Billingsley M. L., Van Devoorde A., O'connor M., Trojanowski J. Q. and Lee V. M. (1994). Biopsy-derived adult human brain tau is phosphorylated at many of the same sites as Alzheimer's disease paired helical filament tau. Neuron 13, 989-1002. 10.1016/0896-6273(94)90264-X [DOI] [PubMed] [Google Scholar]
- Menkes-Caspi N., Yamin H. G., Kellner V., Spires-Jones T. L., Cohen D. and Stern E. A. (2015). Pathological tau disrupts ongoing network activity. Neuron 85, 959-966. 10.1016/j.neuron.2015.01.025 [DOI] [PubMed] [Google Scholar]
- Ortiz-Virumbrales M., Moreno C. L., Kruglikov I., Marazuela P., Sproul A., Jacob S., Zimmer M., Paull D., Zhang B., Schadt E. E. et al. (2017). CRISPR/Cas9-Correctable mutation-related molecular and physiological phenotypes in iPSC-derived Alzheimer's PSEN2 (N141I) neurons. Acta Neuropathol. Commun. 5, 77 10.1186/s40478-017-0475-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paonessa F., Evans L. D., Solanki R., Larrieu D., Wray S., Hardy J., Jackson S. P. and Livesey F. J. (2019). Microtubules deform the nuclear membrane and disrupt nucleocytoplasmic transport in tau-mediated frontotemporal dementia. Cell Rep. 26, 582-593.e5. 10.1016/j.celrep.2018.12.085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickering-Brown S. M., Richardson A. M. T., Snowden J. S., McDonagh A. M., Burns A., Braude W., Baker M., Liu W.-K., Yen S.-H., Hardy J. et al. (2002). Inherited frontotemporal dementia in nine British families associated with intronic mutations in the tau gene. Brain 125, 732-751. 10.1093/brain/awf069 [DOI] [PubMed] [Google Scholar]
- Poorkaj P., Bird T. D., Wijsman E., Nemens E., Garruto R. M., Anderson L., Andreadis A., Wiederholt W. C., Raskind M. and Schellenberg G. D. (1998). Tau is a candidate gene for chromosome 17 frontotemporal dementia. Ann. Neurol. 43, 815-825. 10.1002/ana.410430617 [DOI] [PubMed] [Google Scholar]
- Power J. M., Wu W. W., Sametsky E., Oh M. M. and Disterhoft J. F. (2002). Age-related enhancement of the slow outward calcium-activated potassium current in hippocampal CA1 pyramidal neurons in vitro. J. Neurosci. 22, 7234-7243. 10.1523/JNEUROSCI.22-16-07234.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radzicki D., Liu E., Deng H. X., Siddique T. and Martina M. (2016). Early impairment of synaptic and intrinsic excitability in mice expressing ALS/dementia-linked mutant UBQLN2. Front. Cell Neurosci. 10, 216 10.3389/fncel.2016.00216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohrer J. D., Lashley T., Schott J. M., Warren J. E., Mead S., Isaacs A. M., Beck J., Hardy J., De Silva R., Warrington E. et al. (2011). Clinical and neuroanatomical signatures of tissue pathology in frontotemporal lobar degeneration. Brain 134, 2565-2581. 10.1093/brain/awr198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y., Kirwan P. and Livesey F. J. (2012a). Directed differentiation of human pluripotent stem cells to cerebral cortex neurons and neural networks. Nat. Protoc. 7, 1836-1846. 10.1038/nprot.2012.116 [DOI] [PubMed] [Google Scholar]
- Shi Y., Kirwan P., Smith J., Robinson H. P. and Livesey F. J. (2012b). Human cerebral cortex development from pluripotent stem cells to functional excitatory synapses. Nat. Neurosci. 15, 477-486. 10.1038/nn.3041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sposito T., Preza E., Mahoney C. J., Setó-Salvia N., Ryan N. S., Morris H. R., Arber C., Devine M. J., Houlden H., Warner T. T. et al. (2015). Developmental regulation of tau splicing is disrupted in stem cell-derived neurons from frontotemporal dementia patients with the 10+16 splice-site mutation in MAPT. Hum. Mol. Genet. 24, 5260-5269. 10.1093/hmg/ddv246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Dam D. and De Deyn P. P. (2006). Drug discovery in dementia: the role of rodent models. Nat. Rev. Drug Discov. 5, 956-970. 10.1038/nrd2075 [DOI] [PubMed] [Google Scholar]
- Van der Jeugd A., Hochgräfe K., Ahmed T., Decker J. M., Sydow A., Hofmann A., Wu D., Messing L., Balschun D., D′hooge R. et al. (2012). Cognitive defects are reversible in inducible mice expressing pro-aggregant full-length human Tau. Acta Neuropathol. 123, 787-805. 10.1007/s00401-012-0987-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verheyen A., Diels A., Reumers J., Van Hoorde K., Van Den Wyngaert I., Van Outryve D'ydewalle C., De Bondt A., Kuijlaars J., De Muynck L., De Hoogt R. et al. (2018). Genetically engineered iPSC-derived FTDP-17 MAPT neurons display mutation-specific neurodegenerative and neurodevelopmental phenotypes. Stem Cell Reports 11, 363-379. 10.1016/j.stemcr.2018.06.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagnon J. L., Barker B. S., Ottolini M., Park Y., Volkheimer A., Valdez P., Swinkels M. E. M., Patel M. K. and Meisler M. H. (2017). Loss-of-function variants of SCN8A in intellectual disability without seizures. Neurol. Genet. 3, e170 10.1212/NXG.0000000000000170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson C. J. and Goldberg J. A. (2006). Origin of the slow afterhyperpolarization and slow rhythmic bursting in striatal cholinergic interneurons. J. Neurophysiol. 95, 196-204. 10.1152/jn.00630.2005 [DOI] [PubMed] [Google Scholar]
- Wykes R. C., Heeroma J. H., Mantoan L., Zheng K., Macdonald D. C., Deisseroth K., Hashemi K. S., Walker M. C., Schorge S. and Kullmann D. M. (2012). Optogenetic and potassium channel gene therapy in a rodent model of focal neocortical epilepsy. Sci. Transl. Med. 4, 161ra152 10.1126/scitranslmed.3004190 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.