

Abstract
Sterol regulatory element binding protein (SREBP) cleavage activating protein (SCAP) is a key regulator of SREBP maturation. SCAP induces translocation of SREBP from the endoplasmic reticulum to the Golgi apparatus, allowing it to regulate cellular triglyceride and cholesterol levels. Previous studies have shown that suppression of SREBP activation in SCAP conditional knockout mice reduced the accumulation of intracellular triglycerides, which eventually causes the development of metabolic diseases such as atherosclerosis, diabetes, hepatic steatosis, and insulin resistance. However, despite the significance of SCAP as a regulator of SREBP, its function has not been thoroughly discussed. In this review, we have summarized the function of SCAP and its regulatory proteins. Furthermore, we discuss recent studies regarding SCAP as a possible therapeutic target for hypertriglyceridemia and hyperlipidemia.
Subject terms: Metabolic syndrome, Dyslipidaemias
Metabolic diseases: blocking fat and cholesterol production
The complex cellular functions of a membrane protein that influences fat and cholesterol levels have been reviewed by researchers in South Korea. The main role of this protein, SCAP, is to move certain transcription factors between organelles within cells, allowing them to regulate the synthesis of triglycerides (fat molecules) and cholesterol. Seung-Soon Im and co-workers at Keimyung University School of Medicine in Daegu reviewed studies of the numerous signalling pathways that influence SCAP activity, and of insulin-induced genes that bind to SCAP and inhibit its function. Genetic studies have shown that blocking SCAP in mice can reduce the development of diabetes, fatty liver disease and atherosclerosis. Several drugs for these metabolic conditions have been shown to indirectly inhibit SCAP activity, and therefore drugs that directly target SCAP should be a focus of future research.
Introduction
Sterol regulatory element binding protein (SREBP) cleavage-activating protein (SCAP) plays an important role in regulating triglyceride and cholesterol levels in the body1. SCAP is an endoplasmic reticulum (ER) sterol-sensing protein that chaperones SREBP-1 and SREBP-2 from the ER to the Golgi apparatus2. In the Golgi, two proteases, site-1 protease (S1P) and site-2 protease (S2P) release the N-terminus of SREBP in a two-step proteolytic process, thereby allowing its entry into the nucleus3. However, cholesterol buildup in ER membranes prevents the exit of SCAP/SREBP complexes, subsequently aborting the proteolytic processing of SREBPs and leading to a decrease in the transcription of target genes4. Although SCAP plays an important role in the regulation of SREBP activity, intracellular fatty acid homeostasis and cholesterol synthesis, studies on SCAP are insufficient, and few review articles are available. Therefore, this review will discuss the various roles of SCAP in lipogenesis and the inflammatory response as well as newly discovered antagonists of SCAP as putative therapeutic targets for hypertriglyceridemia and hypercholesterolemia.
Molecular features of SCAP
SCAP (≈140 kDa) is a polytopic membrane protein composed of 1276 amino acids and can be divided into two functional regions5: the transmembrane N-terminal region and a soluble C-terminal domain that consists of multiple copies of a WD40 repeat motif to aid protein–protein interactions (Fig. 1a)6. The former region is composed of approximately 735 amino acids and functions to mediate membrane attachment5. It contains eight transmembrane helicases (TMs) organized into eight α-helices separated by hydrophilic loops7,8. These TMs are linked by four small and three large hydrophilic loops9. Two large rings (loops 1 and 7) are in the ER lumen, while the other large rings (loop 6) face the cytosol to combine with the coat protein II (COPII) protein to move towards the Golgi10. Cholesterol binding to loop 1 changes the composition of loop 6 to exclude COPII binding and prevent the exit of SCAP from the ER9. The latter domain, containing approximately 540 amino acids, extends into the cytosol and includes at least four WD repeat sequences that mediate its binding to SREBPs7. The SCAP protein forms a homotetramer with its membrane region to form a stable complex with SREBF1/SREBP1 or SREBF2/SREBP2 through its C-terminal cytoplasmic domain11. The translocation machinery of SCAP containing SREBP is regulated by the intracellular sterol concentration.
Fig. 1. Schematic model of the domain organization and processing mechanism of SCAP.
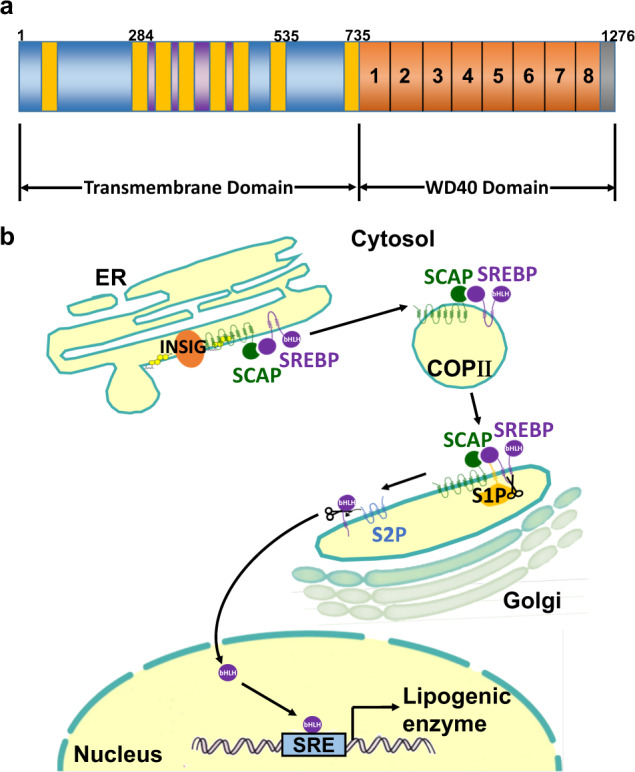
a SCAP proteins consist of an amino-terminal domain of eight transmembrane helices and a carboxyl-terminal WD40 domain. Transmembrane helices 2-6 of SCAP constitute a sterol-sensing domain. b When mammalian cells are deprived of cholesterol, SCAP escorts SREBPs in COPII vesicles from the ER to the Golgi. Two Golgi proteases (S1P and S2P) then sequentially cleave SREBPs. The NH2-terminal region of SREBPs moves to the nucleus and activates the transcription of target genes. To date, SREBPs are known to directly enhance the transcription of more than 30 genes needed for the uptake and synthesis of cholesterol, fatty acids, triglycerides, and phospholipids. Despite acting in diverse biosynthetic pathways, the activity of each SREBP isoform is regulated by sterols and SCAP. COPII, coat protein II; ER, endoplasmic reticulum; INSIG, insulin-induced gene; S1P, site-1 protease; S2P, site-2 protease; SCAP, SREBP cleavage-activating protein; SRE, sterol regulatory element; SREBP, Sterol regulatory element-binding protein.
At high sterol concentrations, SCAP forms a ternary complex with insulin-induced gene (INSIG) via its transmembrane domains and interacts with the Sec23/24 complex in a SAR1-GTP-dependent manner through an ER export signal in its third cytoplasmic loop. Cholesterol buildup in ER membranes exceeding a threshold of 4–5% of the total lipid levels causes sterol binding to SCAP, which triggers a conformational change that, in turn, causes SCAP to bind to insulin-induced gene (INSIG) proteins (Fig. 1b)12,13. The addition of sterols to either intact cells or isolated membranes triggers SCAP binding to INSIGs3. The importance and role of INSIG were first discovered when the membrane domain of SCAP was overexpressed in cells via transfection8. Under these conditions, endogenous INSIGs became saturated, and sterols no longer prevented transport from the ER to Golgi14. When INSIGs bind SCAP, which is mediated by helices 2-6, binding of the Sec23/24-Sar1 complex is prohibited, consequently preventing SCAP from binding SREBP, resulting in suppression of movement from the ER3. Loop 6 of the N-terminal regions of SCAP facing the cytosol contains the hexapeptide sequence methionine-glutamic acid-leucine-alanine-aspartic acid-leucine (MELADL), which acts as the binding site for COPII proteins. The basic functional units of COPII coat proteins are Sar1, Sec23/24 and Sec13/3115. When sterols such as cholesterol and 25-hydroxycholesterol are used to treat cells, the lateral movement of SREBPs into COPII-coated vesicles is obstructed on ER membranes, thereby preventing SREBP maturation to suppress cholesterol synthesis16. To understand the molecular mechanisms by which sterols block the binding of COPII proteins to the SCAP–SREBP complex, however, it is necessary to establish an in vitro system in which this binding can be blocked by the addition of sterols to isolated membranes rather than to pre-incubated cells16. The feasibility of this assay is reinforced by findings that demonstrate the requirement of INSIGs, resident proteins of the ER that function as anchors, for sterol-mediated inhibition of SCAP/SREBP transport17. Otherwise, under sterol-depleted conditions, the SCAP/SREBP complex exits the ER by budding from the ER membranes18. SCAP mediates this exit using the general mechanisms defined for yeast and mammalian membrane proteins that move from the ER to the Golgi19.
Roles of SCAP in lipid metabolism
SREBPs are transcription factors involved in regulating the synthesis and uptake of fatty acids and cholesterol through activating their processing mechanism by SCAP in mammalian cells (Fig. 2)20. In these cells, the synthesis of cholesterol and other lipids is governed by the lateral transfer of a membrane-embedded protein complex into coated vesicles, which then move from the ER to the Golgi21. Upon entering the nucleus, the NH2-terminal domains of SREBPs activate the transcription of several genes that encode proteins involved in cholesterol synthesis (e.g., 3-hydroxy-3-methyl-glutaryl-CoA synthase [HMG-CoA synthase], HMG-CoA reductase, farnesyl diphosphate synthase, squalene synthase, and others), cholesterol uptake (low-density lipoprotein receptor [LDLR]), fatty acid synthesis (acetyl-CoA carboxylase, fatty acid synthase, and stearoyl-CoA desaturase), and triglyceride synthesis (glycerol-3-phosphate acyltransferase)2. Sterols hinder the proteolytic cleavage of SREBP precursors, resulting in downregulated transcription of these genes7. Then, it allows cells to maintain a constant membrane composition despite large changes in cholesterol demand22.
Fig. 2. The function of SREBP-SCAP in lipid metabolism.
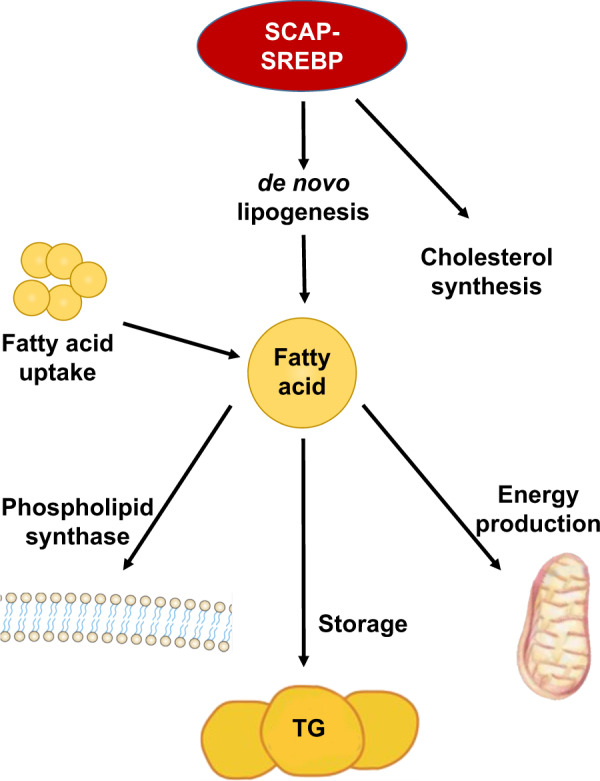
Activation of SCAP–SREBP as the master regulator of lipid metabolism stimulates the transcription of enzymes required for de novo lipogenesis and receptors that mediate the uptake of fatty acids released by lipolysis.
INSIG1 and INSIG2 mediate the feedback control of lipid synthesis by sterol-dependent binding to SCAP23,24. The role of SCAP and INSIGs in activating SREBPs has been demonstrated in previous studies25. Briefly, the SCAP pathway plays a crucial role in feedback regulation of lipid metabolism and may be involved in the development of obesity. In a previous study, liver-specific loss of SCAP in high-fat diet-fed obese mice inhibited hepatic de novo lipogenesis and prevented hepatosteatosis, demonstrating the singularly important role for SREBPs relative to other nutritionally stimulated lipogenic factors26. An abnormal increase in de novo lipogenesis has been suggested to contribute to the pathogenesis of non‐alcoholic fatty liver disease27, a highly prevalent metabolic disease that is linked to the development of type 2 diabetes mellitus28.
The roles of SCAP in inflammation
Cholesterol is an essential lipid in various biological processes, and the pivotal role of SCAP as a cholesterol sensor in the regulation of intracellular cholesterol homeostasis is well established8,29. Cholesterol deposition in dendritic cells stimulates the development of autoimmunity, possibly at the transcriptional level, through the nucleotide-binding oligomerization domain, leucine rich repeat and pyrin domain containing protein 3 (NLRP3) inflammasome30,31. The SCAP-SREBP2 complex promotes NLRP3 inflammasome activation, which is mainly dependent on its ER-to-Golgi translocation rather than an effect on cholesterol homeostasis. Mechanistically, NLRP3 associates with the SCAP-SREBP2 complex to form a ternary complex, which then translocates to the Golgi apparatus adjacent to a mitochondrial cluster for optimal inflammasome assembly32. In addition, SCAP-SREBP2 plays a role as a signaling center that integrates the inflammatory response and cholesterol metabolism in macrophages32. Ouyang et al. reported that the overexpression of SCAP induced cholesterol synthesis, while its knockdown reduced lipid accumulation in THP-1 human macrophages. Moreover, the overexpression of SCAP increased the levels of tumor necrosis factor α (TNFα), interleukin (IL)-1β and Monocyte Chemoattractant Protein-1 (MCP-1) production33. SCAP dysfunction in THP-1 human macrophages was found to affect the expression of inflammatory cytokines and lipid metabolism when the loss of SCAP significantly reduced expression levels of the TNFα, IL-1β and MCP-1 genes.
However, a controversial study demonstrated that dysfunctional SCAP stimulates an inflammatory response in THP-1 cells34. Suppression of the intracellular cholesterol content in THP-1 macrophages did not affect the expression of inflammatory cytokines, suggesting that the SCAP-mediated inflammatory response was independent of the regulation of cholesterol synthesis33. Furthermore, in a recent study regarding the molecular mechanisms of crosstalk between the inflammatory response and dyslipidemia, the loss of SCAP attenuated lipopolysaccharide-stimulated IκB phosphorylation in human macrophages and decreased the level of p65 in the nucleus, suggesting that SCAP dysfunction stimulates the inflammatory response by activating the NF-κB signaling pathway33. These results indicated that the function of SCAP in inflammation is independent of its role in lipid metabolism.
Inflammation disrupts the feedback regulation of LDLR to stimulate foam cell formation in macrophages35. SCAP recycling is a key process in the feedback regulation of LDLR and HMG-CoA reductase by controlling SCAP glycosylation in the Golgi36. In macrophages treated with inflammatory cytokines, mRNA and protein expression levels of mannosidase II were increased by accumulating lipid droplets and induced activation of the SREBP2/LDLR pathway37. SCAP glycosylation may prevent the degradation of SCAP and prolong its half-life, thereby facilitating increased SCAP recycling and activating the SREBP2/LDLR pathway.
The role of SCAP/SREBPs in the innate immune system in the context of viral, but not bacterial stimuli, was previously studied using lysozyme 2-Cre-mediated selective deletion of SCAP in macrophages (LysM-SCAP−/− mice)38. LysM-SCAP−/− mice were reported to be resistant to respiratory infection with MHV-68 and demonstrated increased expression of interferon-stimulated genes (ISGs) in the lung. As expected, cholesterol and fatty acid synthesis were reduced in SCAP-deficient macrophages. In cultured cells in which cholesterol synthesis was inhibited due to SCAP deficiency, interferon signaling was improved, and the levels of the ISGs interferon β (IFNβ) 1, myxovirus resistance (Mx)1, Mx2, and chemokine (C-C motif) ligand 2 (Ccl2) were dramatically increased38. Conversely, IFNβ downregulated the intracellular synthesis of cholesterol in macrophages39. The connection between IFNβ signaling and SCAP was further investigated using in vivo siRNAs targeting SCAP to reduce its level in the liver and leukocyte cells40. Compared to LysM-SCAP−/− mice, an animal model treated with SCAP siRNAs was more vulnerable to viral infection and contained decreased plasma levels of IFNβ38. SCAP interference suppressed the expression of the ISGs, IFNα4, IFNβ1, and C-X-C motif chemokine 10 in virus-exposed macrophages41.
In the study on SREBP function in the adaptive immune system, SCAP was selectively deleted in T cells using Cd4-Cre (Cd4-SCAP−/− mice)42. T cells derived from Cd4-SCAP−/− mice failed to increase their levels of cholesterol and FAs after mitogen stimulation compared to wild-type mice41. Mitogen-stimulated SCAP-deficient T cells failed to enter the S phase of the cell cycle and thus could not proliferate. This defect was prevented by the addition of exogenous cholesterol41,43.
Synthetic antagonists of SCAP
To date, several drugs that inhibit lipid and cholesterol synthesis have been developed44–46. Among them, fatostatin, betulin and xanthohumol suppress SCAP/SREBP translocation47. The small synthetic molecule 125B11, named fatostatin, is an organic antagonist of SCAP (Fig. 3a). By binding to SCAP48, fatostatin inhibits the dissociation of SCAP from INSIGs, thereby restricting the translocation of SREBPs to the ER and subsequently reducing lipogenesis and fat accumulation in obese mice48–50. In a previous study, fatostatin was one of the compounds that prevented insulin-induced fat production in the library of 10,000 compounds51. To obtain information about the specific molecular pathways affected by fatostatin, DU145 cells were treated with fatostatin, and extracted mRNA samples were analyzed by Affymetrix DNA microarray mapping of 33,000 genes48. Among the reduced genes, the ratios of genes associated with SCAP at high levels were analyzed. Using modified fatostatin derivatives, fatostatin was found to interact with ER proteins, and the direct interaction between SCAP and fatostatin was confirmed through binding analysis. Kamisuki et al. demonstrated that fatostatin directly interacts with an NH2-terminal fragment of SCAP (amino acids 1-448) including its sterol-binding domain48. Furthermore, previous studies have suggested that fatostatin prevents insulin-induced adipogenesis of 3T3-L1 cells51 and inhibits glucose-mediated activation of TGF-β in primary rat mesangial cells by inhibiting SCAP activity34.
Fig. 3. Small-molecule inhibitors of the SCAP-SREBP complex.
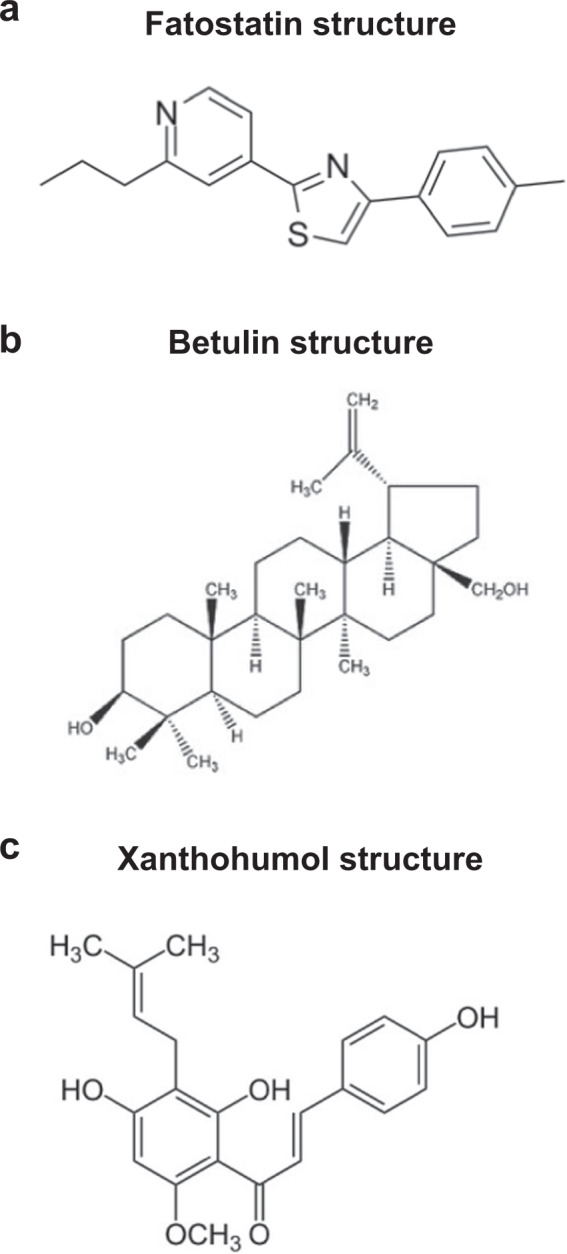
a Fatostatin is a chemical inhibitor of the SREBP pathway that directly binds SCAP and blocks its ER-to-Golgi transport. b Betulin enhances the binding of SCAP with INSIGs, thereby promoting the retention of SREBPs in the ER. c Xanthohumol, a prenylated flavonoid in hops, is an antagonist of SREBP.
Similarly, betulin binds SCAP and enhances its interaction with INSIGs to suppress SCAP/SREBP translocation (Fig. 3b)44. To identify small-molecule inhibitors of the SREBP pathway, various compounds were used to treat Huh7 cells44. Interestingly, betulin efficiently decreased the promoter activity of target genes. Furthermore, betulin stimulated the interaction between SCAP and INSIG-1, demonstrating its mechanism of action. Betulin reduces the activity of genes related to cholesterol and fatty acid biosynthesis and prevents the build-up of intracellular lipids. In diet-induced obese mice, betulin increased insulin sensitivity and reduced cholesterol and triglyceride levels44.
Another novel SCAP/SREBP inhibitor is xanthohumol, a prenylated flavonoid extracted from hops (Fig. 3c)49,52. To identify xanthohumol as a new SCAP/SREBP-inactive agent, numerous food ingredients were screened46. Xanthohumol was found to inhibit SREBP activity in Huh7 cells. In addition, in diet-induced obese mice, dietary xanthohumol reduced SCAP/SREBP target gene expression in the liver, thereby reducing the mature form of liver SREBP-1, which inhibited the development of obesity and hepatic steatosis. Xanthohumol inhibits triglyceride synthesis and apolipoprotein B secretion associated with the inactivation of diacylglycerol acyltransferase 1 and microsomal triglyceride transfer protein45. Xanthohumol interacts with Sec23/24 and blocks sorting of the SCAP/SREBP complex into COPII vesicles, thereby suppressing the ER-to-Golgi translocation of the complex46. Dietary xanthohumol inhibits the maturation of SREBP and transcription of its target genes46. Xanthohumol contributes to the amelioration of diet-induced obesity and fatty liver53. However, putative drugs that directly target SCAP have been still discovered.
Concluding remarks
SCAP, a regulator of SREBP, controls the intracellular biosynthesis of cholesterol, fatty acids and triglycerides. SCAP-mediated hyperlipidemia and hypertriglyceridemia are directly related to metabolic diseases such as arteriosclerosis, obesity, and type II diabetes. These metabolic diseases secondary to high-calorie diets and hyperlipidemia are becoming a global problem. Therefore, SCAP presents a promising target for pharmacologic suppression in the treatment of metabolic diseases.
Acknowledgements
This study was supported by grants from the Korea Research Foundation and an NRF grant funded by the Korean Government (MSIP) (2019R1A2C2085302) and Medical Research Center (2014R1A5A2010008).
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Shimano H, Sato R. SREBP-regulated lipid metabolism: convergent physiology-divergent pathophysiology. Nat. Rev. Endocrinol. 2017;13:710–730. doi: 10.1038/nrendo.2017.91. [DOI] [PubMed] [Google Scholar]
- 2.Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Invest. 2002;109:1125–1131. doi: 10.1172/JCI15593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goldstein JL, DeBose-Boyd RA, Brown MS. Protein sensors for membrane sterols. Cell. 2006;124:35–46. doi: 10.1016/j.cell.2005.12.022. [DOI] [PubMed] [Google Scholar]
- 4.Goldstein JL, Rawson RB, Brown MS. Mutant mammalian cells as tools to delineate the sterol regulatory element-binding protein pathway for feedback regulation of lipid synthesis. Arch. Biochem. Biophys. 2002;397:139–148. doi: 10.1006/abbi.2001.2615. [DOI] [PubMed] [Google Scholar]
- 5.Motamed M, et al. Identification of luminal Loop 1 of Scap protein as the sterol sensor that maintains cholesterol homeostasis. J. Biol. Chem. 2011;286:18002–18012. doi: 10.1074/jbc.M111.238311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Radhakrishnan A, Sun LP, Kwon HJ, Brown MS, Goldstein JL. Direct binding of cholesterol to the purified membrane region of SCAP: mechanism for a sterol-sensing domain. Mol. Cell. 2004;15:259–268. doi: 10.1016/j.molcel.2004.06.019. [DOI] [PubMed] [Google Scholar]
- 7.Sakai J, Nohturfft A, Goldstein JL, Brown MS. Cleavage of sterol regulatory element-binding proteins (SREBPs) at site-1 requires interaction with SREBP cleavage-activating protein. Evidence from in vivo competition studies. J. Biol. Chem. 1998;273:5785–5793. doi: 10.1074/jbc.273.10.5785. [DOI] [PubMed] [Google Scholar]
- 8.Yang T, et al. Crucial step in cholesterol homeostasis: sterols promote binding of SCAP to INSIG-1, a membrane protein that facilitates retention of SREBPs in ER. Cell. 2002;110:489–500. doi: 10.1016/s0092-8674(02)00872-3. [DOI] [PubMed] [Google Scholar]
- 9.Brown MS, Radhakrishnan A, Goldstein JL. Retrospective on cholesterol homeostasis: the central role of scap. Annu. Rev. Biochem. 2018;87:783–807. doi: 10.1146/annurev-biochem-062917-011852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang Y, Motamed M, Seemann J, Brown MS, Goldstein JL. Point mutation in luminal loop 7 of Scap protein blocks interaction with loop 1 and abolishes movement to Golgi. J. Biol. Chem. 2013;288:14059–14067. doi: 10.1074/jbc.M113.469528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Espenshade PJ, Hughes AL. Regulation of sterol synthesis in eukaryotes. Annu. Rev. Genet. 2007;41:401–427. doi: 10.1146/annurev.genet.41.110306.130315. [DOI] [PubMed] [Google Scholar]
- 12.Sokolov A, Radhakrishnan A. Accessibility of cholesterol in endoplasmic reticulum membranes and activation of SREBP-2 switch abruptly at a common cholesterol threshold. J. Biol. Chem. 2010;285:29480–29490. doi: 10.1074/jbc.M110.148254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Afonso MS, et al. Molecular pathways underlying cholesterol homeostasis. Nutrients. 2018;10:760. doi: 10.3390/nu10060760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dong XY, Tang SQ, Chen JD. Dual functions of Insig proteins in cholesterol homeostasis. Lipids Health Dis. 2012;11:173. doi: 10.1186/1476-511X-11-173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bi X, Corpina RA, Goldberg J. Structure of the Sec23/24-Sar1 pre-budding complex of the COPII vesicle coat. Nature. 2002;419:271–277. doi: 10.1038/nature01040. [DOI] [PubMed] [Google Scholar]
- 16.Sun LP, Li L, Goldstein JL, Brown MS. Insig required for sterol-mediated inhibition of Scap/SREBP binding to COPII proteins in vitro. J. Biol. Chem. 2005;280:26483–26490. doi: 10.1074/jbc.M504041200. [DOI] [PubMed] [Google Scholar]
- 17.Radhakrishnan A, Ikeda Y, Kwon HJ, Brown MS, Goldstein JL. Sterol-regulated transport of SREBPs from endoplasmic reticulum to Golgi: oxysterols block transport by binding to Insig. Proc. Natl Acad. Sci. USA. 2007;104:6511–6518. doi: 10.1073/pnas.0700899104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nohturfft A, Yabe D, Goldstein JL, Brown MS, Espenshade PJ. Regulated step in cholesterol feedback localized to budding of SCAP from ER membranes. Cell. 2000;102:315–323. doi: 10.1016/s0092-8674(00)00037-4. [DOI] [PubMed] [Google Scholar]
- 19.Espenshade PJ, Li WP, Yabe D. Sterols block binding of COPII proteins to SCAP, thereby controlling SCAP sorting in ER. Proc. Natl Acad. Sci. USA. 2002;99:11694–11699. doi: 10.1073/pnas.182412799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ye J, DeBose-Boyd RA. Regulation of cholesterol and fatty acid synthesis. Cold Spring Harb. Perspect. Biol. 2011;3:a004754. doi: 10.1101/cshperspect.a004754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.van Meer G, Voelker DR, Feigenson GW. Membrane lipids: where they are and how they behave. Nat. Rev. Mol. Cell Biol. 2008;9:112–124. doi: 10.1038/nrm2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Breslow DK, Weissman JS. Membranes in balance: mechanisms of sphingolipid homeostasis. Mol. Cell. 2010;40:267–279. doi: 10.1016/j.molcel.2010.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gong Y, et al. Sterol-regulated ubiquitination and degradation of Insig-1 creates a convergent mechanism for feedback control of cholesterol synthesis and uptake. Cell Metab. 2006;3:15–24. doi: 10.1016/j.cmet.2005.11.014. [DOI] [PubMed] [Google Scholar]
- 24.McFarlane MR, Liang G, Engelking LJ. Insig proteins mediate feedback inhibition of cholesterol synthesis in the intestine. J. Biol. Chem. 2014;289:2148–2156. doi: 10.1074/jbc.M113.524041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.DeBose-Boyd RA, Ye J. SREBPs in lipid metabolism, insulin signaling, and beyond. Trends Biochem. Sci. 2018;43:358–368. doi: 10.1016/j.tibs.2018.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moon YA, et al. The Scap/SREBP pathway is essential for developing diabetic fatty liver and carbohydrate-induced hypertriglyceridemia in animals. Cell Metab. 2012;15:240–246. doi: 10.1016/j.cmet.2011.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang X, Ji X, Wang Q, Li JZ. New insight into inter-organ crosstalk contributing to the pathogenesis of non-alcoholic fatty liver disease (NAFLD) Protein Cell. 2018;9:164–177. doi: 10.1007/s13238-017-0436-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mu W, et al. Potential nexus of non-alcoholic fatty liver disease and type 2 diabetes mellitus: insulin resistance between hepatic and peripheral tissues. Front. Pharm. 2018;9:1566. doi: 10.3389/fphar.2018.01566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tall AR, Yvan-Charvet L. Cholesterol, inflammation and innate immunity. Nat. Rev. Immunol. 2015;15:104–116. doi: 10.1038/nri3793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ito A, et al. Cholesterol accumulation in CD11c(+) Immune cells is a causal and targetable factor in autoimmune disease. Immunity. 2016;45:1311–1326. doi: 10.1016/j.immuni.2016.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Westerterp M, et al. Cholesterol accumulation in dendritic cells links the inflammasome to acquired immunity. Cell Metab. 2017;25:1294–1304 e1296. doi: 10.1016/j.cmet.2017.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guo C, et al. Cholesterol homeostatic regulator SCAP-SREBP2 Integrates NLRP3 inflammasome activation and cholesterol biosynthetic signaling in macrophages. Immunity. 2018;49:842–856 e847. doi: 10.1016/j.immuni.2018.08.021. [DOI] [PubMed] [Google Scholar]
- 33.Ouyang N, et al. Dysfunction of cholesterol sensor SCAP promotes inflammation activation in THP-1 macrophages. Exp. Cell Res. 2018;367:162–169. doi: 10.1016/j.yexcr.2018.03.032. [DOI] [PubMed] [Google Scholar]
- 34.Uttarwar L, Gao B, Ingram AJ, Krepinsky JC. SREBP-1 activation by glucose mediates TGF-beta upregulation in mesangial cells. Am. J. Physiol. Ren. Physiol. 2012;302:F329–F341. doi: 10.1152/ajprenal.00136.2011. [DOI] [PubMed] [Google Scholar]
- 35.Rosenfeld ME, Ross R. Macrophage and smooth muscle cell proliferation in atherosclerotic lesions of WHHL and comparably hypercholesterolemic fat-fed rabbits. Arteriosclerosis. 1990;10:680–687. doi: 10.1161/01.atv.10.5.680. [DOI] [PubMed] [Google Scholar]
- 36.Yuan Y, et al. Advanced glycation end products (AGEs) increase human mesangial foam cell formation by increasing Golgi SCAP glycosylation in vitro. Am. J. Physiol. Ren. Physiol. 2011;301:F236–F243. doi: 10.1152/ajprenal.00646.2010. [DOI] [PubMed] [Google Scholar]
- 37.Zhou C, et al. Enhanced SCAP glycosylation by inflammation induces macrophage foam cell formation. PLoS ONE. 2013;8:e75650. doi: 10.1371/journal.pone.0075650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.York AG, et al. Limiting cholesterol biosynthetic flux spontaneously engages Type I IFN signaling. Cell. 2015;163:1716–1729. doi: 10.1016/j.cell.2015.11.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Boshuizen MC, et al. Interferon-beta promotes macrophage foam cell formation by altering both cholesterol influx and efflux mechanisms. Cytokine. 2016;77:220–226. doi: 10.1016/j.cyto.2015.09.016. [DOI] [PubMed] [Google Scholar]
- 40.Chen W, et al. ER adaptor SCAP translocates and recruits IRF3 to perinuclear microsome induced by cytosolic microbial DNAs. PLoS Pathog. 2016;12:e1005462. doi: 10.1371/journal.ppat.1005462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Engelking LJ, Cantoria MJ, Xu Y, Liang G. Developmental and extrahepatic physiological functions of SREBP pathway genes in mice. Semin. Cell Dev. Biol. 2018;81:98–109. doi: 10.1016/j.semcdb.2017.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kidani Y, et al. Sterol regulatory element-binding proteins are essential for the metabolic programming of effector T cells and adaptive immunity. Nat. Immunol. 2013;14:489–499. doi: 10.1038/ni.2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McFarlane MR, et al. Scap is required for sterol synthesis and crypt growth in intestinal mucosa. J. Lipid Res. 2015;56:1560–1571. doi: 10.1194/jlr.M059709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tang JJ, et al. Inhibition of SREBP by a small molecule, betulin, improves hyperlipidemia and insulin resistance and reduces atherosclerotic plaques. Cell Metab. 2011;13:44–56. doi: 10.1016/j.cmet.2010.12.004. [DOI] [PubMed] [Google Scholar]
- 45.Casaschi A, et al. The chalcone xanthohumol inhibits triglyceride and apolipoprotein B secretion in HepG2 cells. J. Nutr. 2004;134:1340–1346. doi: 10.1093/jn/134.6.1340. [DOI] [PubMed] [Google Scholar]
- 46.Miyata S, Inoue J, Shimizu M, Sato R. Xanthohumol improves diet-induced obesity and fatty liver by suppressing sterol regulatory element-binding protein (SREBP) Activation. J. Biol. Chem. 2015;290:20565–20579. doi: 10.1074/jbc.M115.656975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mustafa M, Wang TN, Chen X, Gao B, Krepinsky JC. SREBP inhibition ameliorates renal injury after unilateral ureteral obstruction. Am. J. Physiol. Ren. Physiol. 2016;311:F614–F625. doi: 10.1152/ajprenal.00140.2016. [DOI] [PubMed] [Google Scholar]
- 48.Kamisuki S, et al. A small molecule that blocks fat synthesis by inhibiting the activation of SREBP. Chem. Biol. 2009;16:882–892. doi: 10.1016/j.chembiol.2009.07.007. [DOI] [PubMed] [Google Scholar]
- 49.Cheng X, Li J, Guo D. SCAP/SREBPs are central players in lipid metabolism and novel metabolic targets in cancer therapy. Curr. Top. Med. Chem. 2018;18:484–493. doi: 10.2174/1568026618666180523104541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pharr PN, Ogawa M. Enhancement of the proliferation of murine fetal liver erythroid progenitors by infection with Harvey sarcoma virus. Leukemia. 1990;4:210–215. [PubMed] [Google Scholar]
- 51.Choi Y, Kawazoe Y, Murakami K, Misawa H, Uesugi M. Identification of bioactive molecules by adipogenesis profiling of organic compounds. J. Biol. Chem. 2003;278:7320–7324. doi: 10.1074/jbc.M210283200. [DOI] [PubMed] [Google Scholar]
- 52.Liu M, et al. Xanthohumol, a prenylated chalcone from beer hops, acts as an alpha-glucosidase inhibitor in vitro. J. Agric. Food Chem. 2014;62:5548–5554. doi: 10.1021/jf500426z. [DOI] [PubMed] [Google Scholar]
- 53.Mahli A, et al. Therapeutic application of micellar solubilized xanthohumol in a western-type diet-induced mouse model of obesity, diabetes and non-alcoholic Fatty liver disease. Cells. 2019;8:359. doi: 10.3390/cells8040359. [DOI] [PMC free article] [PubMed] [Google Scholar]