

Abstract
N6-methyladenosine (m6A), the most prevalent chemical modification found on eukaryotic mRNA, is associated with almost all stages of mRNA metabolism and influences various human diseases. Recent research has implicated the aberrant regulation of m6A mRNA modification in many human cancers. An increasing number of studies have revealed that dysregulation of m6A-containing gene expression via the abnormal expression of m6A methyltransferases, demethylases, or reader proteins is closely associated with tumorigenicity. Notably, the molecular functions and cellular consequences of m6A mRNA modification often show opposite results depending on the degree of m6A modification in specific mRNA. In this review, we highlight the current progress on the underlying mechanisms of m6A modification in mRNA metabolism, particularly the functions of m6A writers, erasers, and readers in the context of tumorigenesis.
Subject terms: RNA metabolism, Translation, Transcriptomics, Cancer
Cancer: monitoring messenger RNA modifications
A thorough investigation into the role and function of RNA modifications in cancers could yield novel therapies. The chemical modification of messenger RNA (mRNA), the molecule that carries code from DNA to synthesize proteins, is thought to play a role in influencing genetically inherited traits and diseases. A common modification found in mRNA is N6-methyladenosine (m6A). Disruption to the regulation of m6A modification has been linked with human cancers. Junho Choe and Seung Hun Han at Hanyang University in Seoul, South Korea, reviewed current understanding of the molecular mechanisms behind m6A modification, with particular reference to tumor formation. The researchers point out that abnormal expression of proteins associated with m6A may lead to heightened expression of cancer-related genes. More extensive m6A modification levels are also linked to tumor formation.
Introduction
Since the discovery of the DNA double-helix structure in the 1950s, how genetic information is controlled and inherited has been a fundamental question. The discovery that alteration of the chromatin structure and DNA modifications affect heritable phenotypes in addition to the DNA sequence itself opened up a new field of epigenetics1. Similarly, many recent studies have proposed various chemical modifications of RNA as another layer of post-transcriptional gene expression regulation termed “epitranscriptomics”2–4. Post-transcriptional regulation is critical for the control of gene expression programs that dictate a variety of cellular functions and cell fate decisions. To date, at least 160 different chemical modifications have been identified in multiple RNA species, including messenger RNAs (mRNAs), transfer RNAs (tRNAs), ribosomal RNAs (rRNAs), noncoding RNAs (ncRNAs), and viral RNA genomes4,5. Although the majority of these modifications map to noncoding RNAs, increasing evidence implicates multiple mRNA modifications as components of another layer of gene expression regulation2,6.
Discovered in the 1970s, N6-methyladenosine (m6A) is the best-characterized RNA modification and particularly is involved in almost all stages of the mRNA life cycle, including splicing, export, translation, and stability7–11. It is the most prevalent mRNA modification, with approximately one-fourth of the eukaryotic mRNAs harboring at least one m6A-modified base3,12. The m6A modification is found in multiple organisms and associated with various cell functions, including meiosis in yeast13,14, plant development15, mouse spermatogenesis16, mouse embryogenesis17, and various cancers18–22.
In this review, we discuss the current understanding of m6A mRNA modification regulation at the molecular level and its various cellular effects. In particular, we highlight the emerging understanding of m6A mRNA modification in cancer.
Mechanism of dynamic m6A modification
The discovery of methyltransferases (also known as m6A writers) and demethylases (also known as m6A erasers) provided evidence that m6A modification is a dynamic and reversible event23. In addition to the combined action of m6A writers and erasers on m6A modification regulation, m6A reader proteins contribute to the regulation of the fate of m6A-containing RNAs. The m6A modification is the methylation of the sixth position of nitrogen atom of adenosine, with the cellular methyltransferase substrate S‑adenosylmethionine serving as the methyl donor for m6A formation24,25. Methyltransferase-like protein 3 (METTL3, also known as MT-A70) and METTL14 form a heterodimer at a ratio of 1:1, and they functions as a catalytic core complex recognizing the DRACH motif (D = A, G, or U; R = G or A; and H = A, C, or U) and inducing m6A modification of mRNA12,24. Growing evidence has revealed that METTL3 plays a central role in introducing m6A onto nascent transcripts cotranscriptionally, while METTL14 supports binding of the METTL3 protein to the target mRNA (Fig. 1)26,27. In addition, at least five other proteins are involved in the regulation of m6A mRNA modification, although it often shows a slightly different composition of the protein complex in each study. While they lack methyltransferase activity, they stabilize the METTL3/14 complex and facilitate its localization to the specific RNA sites for m6A modification28,29. Wilms tumor 1-associated protein [WTAP, also known as female-lethal(2)d] recruits other proteins to the METTL3/14 complex, thereby affecting the overall levels of m6A modification30. RNA-binding motif 15 (RBM15) protein and its paralog RBM15B have been shown to interact with METTL3 in a WTAP-dependent manner28,31. It has been suggested that they preferentially bind to U-rich regions in RNA and recruit the METTL3/14-WTAP complex to sites proximal to the m6A consensus motifs31. Vir-like m6A methyltransferase associated protein (VIRMA, also known as Virilizer or KIAA1429) was recently found to mediate mRNA methylation near the stop codon in the 3′ untranslated region (UTR), where it plays a role in alternative polyadenylation32. In mouse embryonic stem cells (mESCs), Cbl proto-oncogene like 1 (CBLL1, also known as Hakai) protein and zinc finger CCCH-type containing 13 (ZC3H13) proteins have been shown to be required for the nuclear localization of ZC3H13-WTAP-VIRMA-CBLL1, which promotes m6A mRNA modification29. In Drosophila, ZC3H13 has also been shown to act as an adapter protein between WTAP and RBM15 in the methyltransferase complex to support efficient methylation28. On the other hand, methyltransferase-like protein 16 (METTL16) was recently found to be critical for m6A modification in several pre-mRNAs, U6 small nuclear RNAs (U6 snRNAs), and noncoding RNAs containing a specific stem-loop structure33–35. Interestingly, METTL16 has been shown to control S-adenosylmethionine levels by regulating the expression of a S-adenosylmethionine synthetase methionine adenosyltransferase 2 A (MAT2A) by the enhanced splicing of a retained intron33,34. When METTL16 is depleted, the level of m6A in a cell decreases by ~20%33.
Fig. 1. An overview of cotranscriptional m6A mRNA modification.
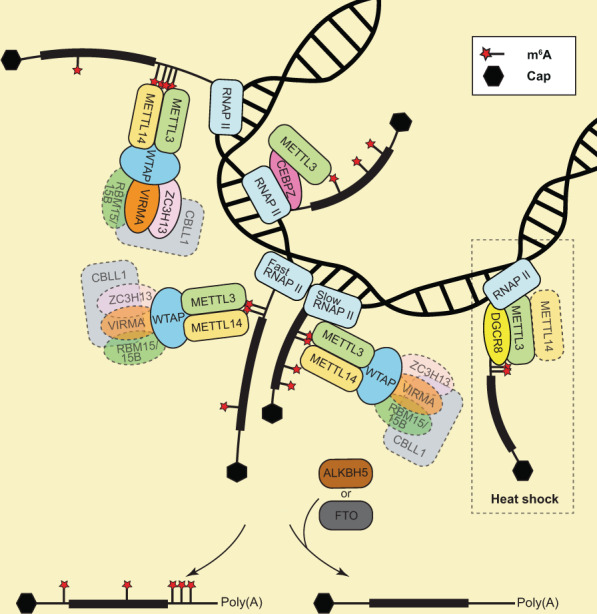
Introduction of m6A modification is currently suggested to occur cotranscriptionally in the nucleus. Individually transcribing mRNAs illustrate the different modes of cotranscriptional m6A modification, including different compositions of associated DNA- and RNA-binding proteins with distinct methylation sites. The thick line represents the coding sequence, and the thin line represents the UTR. The dashed box indicates the heat shock condition.
To date, two mammalian m6A demethylating enzymes have been identified, namely, the fat mass and obesity-associated protein (FTO) and a-ketoglutarate-dependent dioxygenase alk B homolog 5 (ALKBH5) protein36,37. FTO was the first identified m6A demethylase originally found to be associated with increased body mass and obesity in humans36,38. Demethylation of m6A by FTO generates an intermediate product, N6-hydroxymethyladenosine (hm6A), which is then further oxidized to N6-formyladenosine (f6A)39. However, the potential functions of these intermediate products remain unclear. While several studies have provided evidence that depletion of FTO increases the level of total m6A, another recent report suggested FTO preferentially demethylates 2′-O-dimethyladenosine (m6Am), which is found adjacent to the 7-methylguanosine (m7G) cap in mRNA, thereby influencing mRNA stability40. Most recently, FTO was also shown to demethylate N1-methyladenosine (m1A) in tRNAs41. ALKBH5, the second identified m6A demethylase, preferentially recognizes the m6A mark for demethylation in a consensus sequence-dependent manner; thus, it is considered as a better candidate for global m6A demethylation37.
Molecular functions of m6A in mRNA metabolism
Gene expression is the result of orchestrated transcriptional and post-transcriptional regulation. Recently, an increasing number of studies have suggested m6A mRNA modification as a layer of gene expression regulation previously unrecognized. Various m6A reader proteins are involved in many processes of overall mRNA metabolism (Fig. 2).
Fig. 2. Molecular details for m6A-mediated mRNA metabolism.

Multiple m6A reader proteins dynamically regulate m6A-containing mRNA metabolism, including alternative splicing, mRNA export, structural switch, translation, and mRNA stability, depending on the specific m6A-bound reader protein. The thick line represents the coding sequence, and the thin line represents the UTR. The dashed box indicates the heat shock condition.
Cotranscriptional m6A modification
In general, m6A modifications of mRNAs are enriched near translation stop codons in the 3′ UTR3,12,42. However, this characteristic varies among different mRNAs and depends on the tissue. There are several lines of evidence indicating that the m6A modification is a cotranscriptional event (Fig. 1)18,26,27. One report showed that METTL3 binds to chromatin in a transcription-dependent manner and cotranscriptionally methylates nascent transcripts26. In a case of acute myeloid leukemia (AML), METTL3 can be recruited to the promoter region independent of METTL14 by binding to CCAAT/enhancer-binding protein zeta (CEBPZ)18. METTL3 can induce m6A modification cotranscriptionally within the coding region of the associated transcripts, ultimately resulting in translation enhancement18. Moreover, it has been shown that cotranscriptional modification of m6A is dependent on the activity of RNA polymerase II (RNAP II)27. A low rate of transcriptional activity induces increased levels of m6A modification throughout the gene body, resulting in reduced levels of translation27. On the other hand, in the case of heat shock stress, METTL3 can be recruited with DGCR8 to the chromatin of heat shock responsive genes in the region of the transcription ending site, where it subsequently methylates nascent mRNAs, leading to the degradation of the target mRNAs as a consequence26. Considering the accumulating evidence that the m6A modification is mainly found around the translation stop codon in mRNAs3,12,42 and that VIRMA preferentially mediates mRNA methylation near the stop codon in the 3′ UTR32, further studies are required to clarify such discrepancies in the methylation mechanism. Moreover, despite consistent results showing that m6A modification is a cotranscriptional event, the molecular consequences of this modification vary among different studies. Therefore, further research is required to determine the regulating factors that lead to these discrepancies.
m6A promotes alternative splicing
Multiple model organism studies have shown that dynamic m6A modification alters mRNA splicing. In Drosophila, mutation of IME4 (a METTL3 homolog) influences sex determination by modulating female-specific splicing of the Sex-lethal (Sxl) gene43,44. In addition, the Drosophila orthologs of VIRMA and/or ZC3H13 have been shown to regulate alternative splicing of pre-mRNAs involved in sex determination28. m6A demethylases were also reported to be involved in splicing machinery37,45,46. FTO regulates mouse pre-adipocyte differentiation by regulating the alternative splicing of the genes involved in adipogenesis45. ALKBH5 regulates splicing by removing m6A from pre-mRNAs and allows the production of a subset of mRNAs containing relatively long 3′ UTRs in mouse germ cells37,46. While m6A writers and erasers regulate alternative splicing by modulating the levels of m6A modification, m6A reader proteins directly regulate splicing8,47. The m6A-bound YTHDC1 associating with splicing factor SRSF3 has been shown to block the binding of SRSF10 to m6A-modified RNA, promoting exon inclusion in the selected transcripts8,48. Moreover, m6A modification influences mRNA structural changes, which allows heterogeneous nuclear ribonucleoprotein C (hnRNPC) and hnRNPG binding9,47. While hnRNPC binds opposite strand U-rich sequences after the disruption of RNA base pairing by m6A modification9, hnRNPG preferentially binds to purine-rich motifs, including m6A sites47. Binding of either hnRNPC or hnRNPG influences the alternative splicing of m6A-modified transcripts9,47. Finally, METTL16 induces the m6A modification of U6 snRNA, which base pairs with 5′ splice sites of pre-mRNAs during splicing, suggesting that METTL16 plays an important role in mRNA splicing34,35.
m6A facilitates mRNA export
mRNA export is also influenced by m6A modification. ALKBH5-deficient cells exhibit increased levels of cytoplasmic m6A-containing mRNA, suggesting that the m6A modification accelerates mRNA export37. Another report showed that YTHDC1 facilitates the export of m6A-modified mRNAs via its interaction with nuclear RNA export factor 1 (NXF1)10.
m6A alters RNA structure
It has been well established that gene expression is largely affected by the secondary and tertiary structures of mRNA49. Introduction of m6A modification promotes the destabilization of A/U pairings, resulting in alterations to the thermostability of RNA duplexes and changes in the RNA secondary structure50. Another study demonstrated that RNA structural changes caused by the introduction of m6A also alter the interaction between RNAs and proteins9,47.
m6A regulates translation efficiency
Many m6A reader proteins are reported to be crucial for the efficient translation of methylated mRNAs. Members of the YT521-B homology (YTH) domain-containing protein family have been identified as direct m6A readers, including YTHDF1, YTHDF2, YTHDF3, YTHDC1, and YTHDC27,11,51–56. Among these proteins, YTHDF1, YTHDF3, and YTHDC2 have been shown to promote target mRNA translation11,51–53. YTHDF1 selectively binds to m6A sites near the stop codon and cooperates with translation initiation factors to promote the translation of the target mRNAs51. YTHDF3 cooperates with YTHDF1 in the regulation of translation by interacting with a common set of ribosomal proteins52. YTHDC2 has been suggested to play a role in enhanced translation levels while reducing target mRNA abundance53. Furthermore, increased levels of YTHDF2 translocate to the nucleus under heat shock stress and bind m6A in the 5′ UTR of a subset of stress-induced mRNAs, protecting them from FTO-mediated demethylation and promoting their cap-independent translation57. Eukaryotic translation initiation factor 3 (eIF3) is also considered an m6A-binding protein. mRNAs containing m6A modification in the 5′ UTR can be recognized by direct binding of eIF3 to the methylated region, which in turn recruits the 43 S complex to initiate translation in a cap-independent manner in the absence of the cap-binding protein eIF4E58. However, the mechanism of eIF3 in the recognition of m6A is not yet clearly understood. Interestingly, most recent studies have suggested that the m6A writer protein METTL3 also functions as a reader protein in the cytoplasm, promoting the translation of a large subset of target mRNAs21,22. These studies revealed that 3′ UTR m6A modification near the stop codon significantly increases translation through mRNA looping, governed by the interaction between METTL3 at the 3′ UTR and the translation initiation factor eIF3 subunit h (eIF3h) at the 5′ end21,22.
m6A regulates mRNA stability
An increasing number of studies have demonstrated that m6A modification influences mRNA stability. Various structural and functional studies suggest that all three YTHDF reader proteins (YTHDF1, YTHDF2, and YTHDF3) may share the same subset of target mRNAs51,52. However, accumulating evidence suggests that YTHDF2 is the major factor involved in the degradation of m6A-containing mRNA either through exoribonucleolytic decay or the endoribonucleolytic cleavage pathway55,56. YTHDF2 has been shown to selectively recognize m6A sites and recruit the CCR4-NOT deadenylase complex directly, which in turn recruits exosomes (3′-to-5′ exoribonuclease) to initiate mRNA decay56. Other recent studies revealed that YTHDF2 promotes the translocation of m6A-containing mRNA from the translation machinery to processing bodies (P bodies), where cellular proteins participating in mRNA degradation are enriched7,59. In addition, a very recent study revealed the YTHDF2-mediated endoribonucleolytic cleavage of m6A-containing mRNAs55. Mechanistically, heat-responsive protein 12 (HRSP12, also known as reactive intermediate imine deaminase A homolog, UK114 antigen homolog, and 14.5 kDa translational inhibitor protein) bridges m6A-bound YTHDF2 to an endoribonuclease, RNase P/MRP, triggering the endoribonucleolytic cleavage of an m6A-containing mRNA55. Another study suggested that YTHDC2 recruits the 5′ to 3′ exoribonuclease XRN1 for subsequent m6A-containing mRNA degradation54. In addition to the YTH proteins, a variety of other RNA-binding proteins are involved in the regulation of m6A-containing mRNA stability. Fragile X mental retardation protein (FMRP) can bind to the sequence motifs YGGA (Y = C or U) and GAC, which likely overlap with the DRACH motif involved in m6A modification, resulting in stabilization of the m6A-containing mRNA through the competition of FMRP with YTHDF260. In another case, stress granule protein (G3BP1) has binding affinity for m6A-methylated transcripts, promoting their demethylation and resulting in stabilization of the target mRNAs61. Insulin-like growth factor 2 mRNA-binding protein (IGF2BP) 1, 2, and 3 or human antigen R (HuR, also known as ELAVL1) have also been reported to stabilize m6A-containing mRNAs62,63.
Molecular functions of m6A in various cancers
Interest in m6A modification has been extended to many human diseases as well as to its molecular function. In particular, an increasing number of studies are examining the role of m6A-mediated gene expression regulation in cancers. In general, many different signaling pathways converge onto translation machinery to satisfy the increased anabolic demands of cancers. Given the crucial function of m6A modification in regulating mRNA metabolism, it is reasonable to speculate that m6A modification plays an important role in human carcinogenesis. Nonetheless, the molecular details of how m6A modification affects the cellular phenotype of cancer are still being investigated. The physiological effects of m6A mRNA modification in cancer often lead to opposite results (Table 1); thus, further understanding of a balanced m6A modification is required for the treatment of cancer. Here, we highlight recent insights into the biological functions of m6A mRNA modification and the underlying molecular mechanisms of m6A regulatory proteins in various cancers (Fig. 3 and Table 1).
Table 1.
Cellular effects of m6A mRNA modification in cancer.
| Positive regulation of m6A in cancer | Negative regulation of m6A in cancer | |||||
|---|---|---|---|---|---|---|
| Molecular function | Target | Reference | Molecular function | Target | Reference | |
| Lung cancer | Translation | A subset of mRNAs | 21,22 | mRNA level change | A subset of mRNAs | 68 |
| EGFR, TAZ, MAPKAPK2, DNMT3A mRNA | 67 | |||||
| Protein level change | BAX, BCL-2 | 66 | mRNA stabilization | MZF1 mRNA | 69 | |
| Acute myeloid leukemia | Translation | A subset of mRNAs | 18 | mRNA stabilization | ASB2, RARA mRNA | 19 |
| MYC, BCL2, PTEN mRNA | 71 | |||||
| Translation | MYB, MYC mRNA | 73 | mRNA decay | A subset of mRNAs | 72 | |
| mRNA stabilization | 73 | |||||
| Hepatocellular carcinoma | mRNA level change | SOCS2 mRNA | 75 | NR | NR | |
| mRNA decay | EGFR mRNA | 76 | ||||
| mRNA level change | SON, SREBBP mRNA | 77 | ||||
| Breast cancer | mRNA level change | HBXIP mRNA | 79 | mRNA level change | NANOG, KFL4 mRNA | 80,81 |
| Gastric cancer | mRNA stabilization | SEC62 mRNA | 83 | Unknown | Unknown | 85,86 |
| HDGF mRNA | 84 | |||||
| Bladder cancer | mRNA level change | AFF4, MYC mRNA | 20 | NR | NR | |
| Protein level change | AFF4, MYC, IKBKB, RELA | 20 | ||||
| Translation | ITGA6 mRNA | 87 | ||||
| Glioblastoma | mRNA stabilization | SOX2 mRNA | 92 | mRNA level change | Nascent FOXM1 transcript | 89 |
| mRNA level change | A subset of mRNAs | 91 | ||||
| Colorectal cancer | mRNA stabilization | SOX2 mRNA | 93 | NR | NR | |
| Renal cell carcinoma | NR | NR | Unknown | Unknown | 95 | |
| Endometerial Cancer | NR | NR | Translation | PHLPP2 mRNA | 96 | |
| mRNA decay | PRR5, PRR5L, mTOR mRNA | 96 | ||||
| Cervical cancer | NR | NR | Unknown | Unknown | 97 | |
| Pancreatic cancer | NR | NR | Protein level change | YAP | 98 | |
Some studies did not identify molecular mechanisms or targets, but only measured m6A levels and their effects on cancer, which are marked as “unknown”. “Translation” indicates the m6A-mediated translation enhancement. “Protein level change” and “mRNA level change” indicate their steady-state levels without specifying the translation efficiency or mRNA stability.
NR not reported.
Fig. 3. m6A-mediated mRNA regulation in tumorigenesis.

A number of studies have identified the molecular mechanism of m6A-mediated mRNA regulation and their effects on tumorigenesis. To date, m6A-mediated regulation of translation or mRNA stability has been demonstrated, while the relevance of pre-mRNA splicing or mRNA export remains unclear for specific cancer types.
Lung cancer
Lung cancer causes the greatest number of cancer-related deaths worldwide. There are two main histological types of lung cancer: small-cell lung cancer and non-small-cell lung cancer (NSCLC). Approximately 85% are classified as NSCLC, which statistically shows just a 15.9% 5-year survival rate64,65. Nevertheless, therapeutic efforts have improved only slightly over the last few decades. Therefore, it is urgent to explore new treatments and deepen our understanding of the underlying mechanisms of lung cancer occurrence and development. The relevance of m6A modification in lung cancer has been extensively studied, and several lines of evidence show that METTL3 is highly expressed in NSCLC cells and is associated with cell proliferation, invasion, and viability21,22,66–68. Two recent studies from the same group revealed intriguing effects of METTL3 in lung cancer progression. These studies showed that cytoplasm-localized METTL3 functions as an m6A reader protein that enhances translation of a large subset of oncogenic mRNAs without affecting mRNA abundance21,22. Mechanistically, the 3′ UTR near the stop codon-bound METTL3 directly interacts with eIF3h. This interaction mediates mRNA looping to facilitate the recycling of ribosomes at the termination codon in a similar way to canonical eukaryotic mRNA looping mediated by the interactions between eIF4E (a cap-binding protein), eIF4G (a translation initiation factor), and PABP (a poly(A)-binding protein)21,22. Indeed, ectopic expression of METTL3, but not a mutant that fails to interact with eIF3h, promotes cell proliferation, invasion, and oncogenic transformation21. Other studies have shown that METTL3 mRNA can be targeted by microRNAs (miRNAs)66,67. Exogenously expressed miR-600 targets the 3′ UTR of METTL3 mRNA, resulting in the inhibition of METTL3 expression66. Depletion of METTL3 inhibits the survival and proliferation of A549 and H1299 cells and leads to increased levels of the pro-apoptotic regulator BAX and decreased levels of the anti-apoptotic regulator BCL-2, suggesting that the altered expression ratio of BAX/BCL-2 triggers the mitochondrial apoptotic pathway66. In addition, knocking down METTL3 decreases the phosphorylation of AKT, thus affecting cell growth and apoptosis via the alteration of the PI3K/AKT/mTOR pathway66. Another miRNA, miR-33a, has also been shown to reduce METTL3 expression and, as a result, inhibits NSCLC cell proliferation67. On the other hand, METTL3 is SUMOylated by small ubiquitin-related modifier 1 (SUMO1), which modifies METTL3 at lysine residues and represses its methyltransferase activity without altering its stability, localization, or interaction with two other writer proteins, METTL14 and WTAP68. The SUMOylation of METTL3 reduces m6A levels and subsequently changes the mRNA expression profiles, ultimately promoting the development of NSCLC68. Besides, the m6A demethylase FTO has also been shown to play a critical role in lung squamous cell carcinoma (LUSC), one of the most common NSCLCs. FTO knockdown effectively inhibits cell proliferation and invasion while promoting apoptosis of L78 and NCI-H520 cells69. In contrast, overexpression of FTO, but not its mutant form, facilitates the acquisition of malignant phenotypes69. Mechanistically, FTO increases the stability of myeloid zinc finger 1 (MZF1) mRNA by reducing its m6A level, leading to high levels of protein expression, which has an oncogenic function69. MZF1 is a member of the SCAN-zinc finger transcription factor family, which contributes to cell proliferation, migration, and metastasis through the regulation of diverse target genes.
Acute myeloid leukemia (AML)
AML is one of the most prevalent hematopoietic malignancies. It is often derived from genetic mutations and aberrant regulation of epigenetic modification, including DNA methylation and histone modification70. Recently, many studies have pointed to m6A mRNA modification as a new role for a gene expression regulator associated with AML18,19,71,72. As previously described, promoter-bound METTL3 induces m6A modification within coding regions of a subset of nascent transcripts independent of METTL1418. In this way, the genes necessary for AML growth enhance their translation efficiency by relieving ribosome stalling at the GAN (GAG, GAT, GAC, and GAA) codons during translation elongation18. Another study revealed that increased expression levels of METTL3 promote the translation of MYC proto-oncogene (c-MYC), B-cell lymphoma 2 (BCL2), and phosphatase and tensin homolog (PTEN) mRNAs by increasing the levels of m6A modification, thereby altering phosphoinositide 3-kinase (PI3K) and protein kinase B (PKB, also known as AKT) signaling, an intracellular signaling pathway important in regulating the cell cycle, to control cell differentiation and self-renewal71. METTL14 was also shown to function in a similar way by promoting translation of its target mRNAs, the proto-oncogenes MYB and MYC, through m6A modifications, which in turn leads to block the myeloid differentiation73. Notably, in addition to m6A writers, differentially expressed eraser or reader proteins seem to contribute to various AML subtypes through the modulation of m6A modification in a target mRNA-specific manner. Elevated expression of FTO enhances cell transformation and leukemogenesis by downregulating both the mRNA and protein expression of targets, such as ASB2 and RARA mRNAs, by reducing the m6A levels in their UTRs19. On the other hand, YTHDF2 overexpression plays a crucial role in disease initiation and propagation in human and mouse AML by destabilizing a subset of mRNAs, including tumor necrosis factor receptor TNFRSF2 mRNA72.
Hepatocellular carcinoma (HCC)
HCC is a major type of primary liver cancer and is a highly progressive malignant tumor associated with a low survival rate74. It was recently reported that METTL3 levels are increased in human HCC, leading to increased m6A modification of the tumor suppressor SOCS2 mRNA75. Increased levels of m6A in SOCS2 mRNA can be targeted by YTHDF2, leading to its rapid degradation, which is associated with the efficient proliferation of HCC cells75. Besides, overexpression of YTHDF2 has been shown to suppress cell proliferation and tumor growth in HCC cells76. Mechanistically, the m6A-modified 3′ UTR of epidermal growth factor receptor (EGFR) mRNA is recognized by YTHDF2 and undergoes degradation, which in turn impairs mitogen-activated protein kinase kinase (MEK) and extracellular signal-regulated kinases (ERK)76. Similarly, another report showed that YTHDF2 mRNA can be targeted by miR145, leading to an increase in overall m6A levels in HCC cells, which is associated with HCC malignancy77.
Breast cancer (BrC)
Of all malignant tumors in women, BrC is highly metastatic and has the highest cancer-related mortality78. One interesting report suggested a potential positive feedback loop between mammalian hepatitis B X-interacting protein (HBXIP) and METTL379. High expression levels of HBXIP elevate METTL3 expression through the suppression of let-7g, and increased METTL3 upregulates HBXIP expression through m6A modifications of mRNA. This positive feedback loop leads to the acceleration of cell proliferation in BrC. On the other hand, a decrease in m6A modification also promotes BrC tumorigenesis. In BrC stem cells, hypoxic stress induces overexpression of ALKBH5 and/or ZNF217, leading to inhibition of the methylation of pluripotency markers NANOG and KLF4 mRNAs80,81. Increasing the expression of NANOG and KLF4 mRNA by inhibiting m6A modification promotes the specification of BrC stem cells80,81. Another report also showed that m6A levels increased by METTL14 overexpression or ALKBH5 knockdown inhibited BrC growth and metastasis82.
Gastric cancer (GC)
GC is a prevalent tumor occurring in the digestive system. One clear mechanism showed that the preprotein translocation factor SEC62 mRNA can undergo m6A modification by METTL383. In turn, IGF2BP1 recognizes m6A and facilitates the stabilization of SEC62 mRNA. Moreover, miR4429 has been suggested to target METTL3 and prevent the m6A modification of SEC62 mRNA, thus destabilizing SEC62 mRNA83. Downregulated SEC62 inhibits GC cell proliferation and promotes apoptosis83. Another report showed that METTL3 transcription is elevated in GC by a histone acetyltransferase, P300, which mediates H3K27 acetylation at the METTL3 promoter region, which in turn induces the methylation of hepatoma-derived growth factor (HDGF) mRNA84. The methylated HDGF mRNA is then recognized and stabilized by IGF2BP3. Overexpressed HDGF protein can be secreted and promotes tumor angiogenesis, while nuclear HDGF stimulates the expression of glucose transporter type 4 (GLUT-4) and enolase 2 (ENO2) mRNAs, resulting in increased levels of glycolysis and subsequently causing tumor growth and liver metastasis84. On the other hand, it has been suggested that FTO and ALKBH1 play crucial roles in GC progression and metastasis, although the relevance of m6A in these processes is unclear85. It has been shown statistically that lower ALKBH1 protein expression correlates with larger tumor size, while lower FTO protein expression is associated with shorter overall survival in patients with GC85. Another report revealed that the downregulation of m6A modification by METTL14 knockdown leads to the acquisition of oncogenic phenotypes through the alteration of Wnt and PI3K-AKT signaling pathways, although the exact upstream regulatory mechanism is unclear86.
Bladder cancer (BlC)
BlC is the most prevalent urogenital cancer. Recent studies suggest that increased levels of m6A modification are correlated with BlC20,87. One study identified the mRNAs of AF4/FMR2 family member 4 (AFF4), two key regulators of the NF-κB pathway (IKBKB and RELA), and MYC as direct METTL3 targets for m6A modification20. METTL3 depletion led to a reduction in AFF4 and MYC mRNA and protein expression, while only the protein expression was reduced for IKBKB and RELA. METTL3 downregulation in BlC drastically reduced cell proliferation, invasion, and survival in vitro and tumorigenicity in vivo20. Considering the results indicating that (1) MYC is a well-known oncogene that triggers the expression of target genes to benefit cell proliferation, cell survival, and stemness maintenance and (2) AFF4 and NF-κB are known to regulate MYC expression, through which NF-κB signaling enhances the proliferation and survival of cancer cells during the development and recurrence of BlC, it can be speculated that m6A modification by METTL3 affects the AFF4/NF-κB/MYC signaling network to regulate BlC progression20. In addition, upregulated METTL3 promotes the translation of integrin alpha-6 (ITGA6) mRNA via the recognition of m6A in the 3′ UTR by the m6A reader proteins YTHDF1 and YTHDF387. As a result, the upregulated ITGA6 protein promotes BlC cell adhesion, migration, and invasion, similar to multiple other types of cancer, in which ITGA6 overexpression promotes tumorigenesis and metastasis87.
Glioblastoma (GBM)
GBM is a primary malignant brain tumor prevalent in adults88. GBMs have heterogeneous characteristics and contain cells with stem-like properties89. These self-renewing GBM stem-like cells (GSCs) contribute to tumor initiation and therapeutic resistance90. Intriguingly, the expression levels of both METTL3 and ALKBH5 are elevated in GSCs, with opposite results on m6A-mediated tumor formation in a target-specific manner89,91,92. High METTL3 expression levels exhibit oncogenic function through efficient m6A modification in the 3′ UTR of sex-determining region Y (SRY)-box 2 (SOX2) mRNA, which is stabilized by binding of HuR92. Silencing METTL3 expression reduces SOX2 expression and, as a result, inhibits GBM tumor growth and prolongs the survival of mice92. In contrast, ALKBH5 is highly expressed in GSCs and demethylates FOXM1 nascent transcripts, leading to FOXM1 overexpression, stem-like cell proliferation, and tumorigenesis89. The elevated levels of the transcription factor FOXM1 play critical roles in regulating GSC proliferation, self-renewal, and tumorigenicity89. Similarly, another study suggested a tumor-suppressive function for the m6A modification in GSCs91. Reduction of m6A modification by the depletion of METTL3 or METTL14 or the chemical inhibition of FTO upregulates the mRNA expression of critical oncogenes such as ADAM19, EPHA3, and KLF4 and downregulates the mRNA expression of many tumor suppressors, including CDKN2A, BRCA2, and TP53I11 mRNAs, resulting in overall enhanced GBM stem cell growth, self-renewal, and tumorigenesis91.
Colorectal cancer (CrC)
In CrC, METTL3 and YTHDF1 expression is significantly upregulated93,94. High levels of METTL3 expression have been shown to significantly upregulate m6A methylation in the coding sequences of SOX2 mRNA, a well-known CrC marker that is involved in maintaining the properties of tumor-initiating cells93. Methylated SOX2 mRNA is subsequently recognized by IGF2BP2, preventing mRNA degradation. Indeed, knocking down METTL3 reduces the SOX2 expression level, inhibiting CrC development and metastasis93. On the other hand, c-MYC has been suggested to promote YTHDF1 transcription94. A statistical analysis suggests that patients with high YTHDF1 expression have significantly poorer overall survival94. Moreover, knocking down YTHDF1 results in the inhibition of cell proliferation and sensitization of cells to anticancer drugs such as fluorouracil and oxaliplatin94.
Other cancers
Similar to the cancers discussed above, modulation of m6A modification plays a critical role in renal cell carcinoma, endometrial cancer, and cervical cancer95–97. In renal cell carcinoma, depletion of METTL3 promotes cell proliferation, cell invasion, and migration, and induces G0/G1 arrest95. Conversely, upregulation of METTL3 results in significant suppression of tumor growth95. Moreover, knocking down METTL3 promotes the acquisition of an epithelial phenotype and represses the manifestation of a mesenchymal phenotype, while overexpression of METTL3 reverses epithelial–mesenchymal transition progression95. Furthermore, the observation that increased phosphorylation levels of PI3K/AKT/mTOR due to METTL3 knockdown suggests that these METTL3-mediated pathways may also be involved in renal cell carcinoma progression95. A report revealed that METTL14 is frequently mutated and METTL3 expression is significantly reduced in endometrial cancer96. Mechanistically, m6A mRNA modification affects the YTHDF1-dependent translation enhancement of the negative AKT regulator PHLPP2 and YTHDF2-dependent destabilization of the mRNAs of positive AKT regulators PRR5, PRR5L, and mTOR. Thus, either METTL14 mutation or decreased METTL3 expression leads to m6A reduction in these target mRNAs and, as a result, promotes cell proliferation and tumorigenicity of endometrial cancer through AKT activation96. In cervical cancer, downregulation of m6A modification enhances cell proliferation, while upregulation inhibits tumor development97. However, the exact mechanism remains unknown. Last, YTHDF2 is upregulated in pancreatic cancer and has two roles in cancer development: 1) YTHDF2 promotes cell proliferation, since it was observed that knocking down YTHDF2 results in the activation of the AKT/GSK3β/Cyclin D1 pathway, leading to G1 arrest, and 2) the YTHDF2-mediated decay of yes-associated protein (YAP) may influence the epithelial–mesenchymal transition, since overexpression of YAP results in decreased expression of epithelial markers and increases in mesenchymal markers98.
Concluding remarks and future perspectives
Considering the increasing number of studies revealing that m6A modification plays a critical role in almost all stages of mRNA metabolism10,48,56,62, we can easily speculate that aberrant regulation of these modifications affects many cellular phenotypes. Nevertheless, the molecular mechanisms and cellular effects of m6A mRNA modifications are not yet fully understood, since they do not always function in the same way. For instance, although it is well known that the m6A modification sites in mRNAs are mainly enriched in the 3′ UTR near the stop codon3,12,42, several recent findings showed that cotranscriptional methylation occurs in coding sequences (Fig. 1)18,27. In addition, it is still unclear why some mRNAs are not methylated. Considering that the m6A modification is reversible, the demethylases FTO and/or ALKBH5 may play critical roles in balancing the methylation of specific mRNAs in a cell type-dependent manner.
In recent years, m6A modification studies in various cancers have been conducted. Remarkably, an increasing number of studies have revealed that altered expression levels of m6A methyltransferases, demethylases, and reader proteins aberrantly regulate m6A modification on target mRNAs, resulting in abnormal expression of cancer-associated genes. In particular, increased methyltransferase expression levels were detected in most cancers, suggesting that higher m6A modification levels are closely related to tumorigenesis. However, the molecular functions and cellular consequences of m6A modification differed in each study, depending on the degree of methylation in the specific target mRNAs (Table 1). For instance, increased levels of m6A modification by higher levels of METTL3 or METTL14 expression promoted the translation or stabilization of c-MYC, BCL2, PTEN, or MYB mRNAs in AML71,73. In contrast, FTO also showed an elevated level of expression, which downregulated both the translation and abundance of ASB2 and RARA mRNAs through demethylation19. Taken together, the coordinated functions of methylation and demethylation of specific targets seem to be critical for tumorigenesis.
Interest in m6A modification resurged quite recently. To date, most of the m6A studies in cancer have been demonstrated based on the discovery of the m6A modification itself rather than the underlying mechanisms with reader proteins (Fig. 3 and Table 1) because efforts to define the molecular mechanism and the biological relevance have been carried out in parallel. To date, only a single m6A reader-dependent molecular mechanism has been demonstrated in most cancer types (Fig. 3). In addition, cancer-related studies on other outcomes of m6A-dependent mRNA regulation, such as pre-mRNA splicing or mRNA export, remain insufficient. Considering that multiple reader proteins recognize m6A, it might be possible to crosstalk between readers on a single or a multiple m6A modification in an mRNA for the tight gene expression regulation. Therefore, to develop novel tumor therapies based on the regulation of m6A modifications, more thorough mechanistic and functional studies are required for each cancer type.
Acknowledgements
This work was supported by the research fund of Hanyang University (HY-2019).
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Heard E, Martienssen RA. Transgenerational epigenetic inheritance: myths and mechanisms. Cell. 2014;157:95–109. doi: 10.1016/j.cell.2014.02.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Esteller M, Pandolfi PP. The epitranscriptome of noncoding RNAs in cancer. Cancer Discov. 2017;7:359–368. doi: 10.1158/2159-8290.CD-16-1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Meyer KD, et al. Comprehensive analysis of mRNA methylation reveals enrichment in 3’ UTRs and near stop codons. Cell. 2012;149:1635–1646. doi: 10.1016/j.cell.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Helm M, Motorin Y. Detecting RNA modifications in the epitranscriptome: predict and validate. Nat. Rev. Genet. 2017;18:275–291. doi: 10.1038/nrg.2016.169. [DOI] [PubMed] [Google Scholar]
- 5.Nachtergaele S, He C. Chemical modifications in the life of an mRNA transcript. Annu. Rev. Genet. 2018;52:349–372. doi: 10.1146/annurev-genet-120417-031522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Meyer KD, Jaffrey SR. The dynamic epitranscriptome: N6-methyladenosine and gene expression control. Nat. Rev. Mol. Cell Biol. 2014;15:313–326. doi: 10.1038/nrm3785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang X, et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature. 2014;505:117–120. doi: 10.1038/nature12730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kasowitz SD, et al. Nuclear m6A reader YTHDC1 regulates alternative polyadenylation and splicing during mouse oocyte development. PLoS Genet. 2018;14:e1007412. doi: 10.1371/journal.pgen.1007412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu N, et al. N(6)-methyladenosine-dependent RNA structural switches regulate RNA-protein interactions. Nature. 2015;518:560–564. doi: 10.1038/nature14234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Roundtree IA, et al. YTHDC1 mediates nuclear export of N(6)-methyladenosine methylated mRNAs. Elife. 2017;6:1–28. doi: 10.7554/eLife.31311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li A, et al. Cytoplasmic m(6)A reader YTHDF3 promotes mRNA translation. Cell Res. 2017;27:444–447. doi: 10.1038/cr.2017.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dominissini D, et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature. 2012;485:201–206. doi: 10.1038/nature11112. [DOI] [PubMed] [Google Scholar]
- 13.Schwartz S, et al. High-resolution mapping reveals a conserved, widespread, dynamic mRNA methylation program in yeast meiosis. Cell. 2013;155:1409–1421. doi: 10.1016/j.cell.2013.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Clancy MJ, Shambaugh ME, Timpte CS, Bokar JA. Induction of sporulation in Saccharomyces cerevisiae leads to the formation of N6-methyladenosine in mRNA: a potential mechanism for the activity of the IME4 gene. Nucleic Acids Res. 2002;30:4509–4518. doi: 10.1093/nar/gkf573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhong S, et al. MTA is an Arabidopsis messenger RNA adenosine methylase and interacts with a homolog of a sex-specific splicing factor. Plant Cell. 2008;20:1278–1288. doi: 10.1105/tpc.108.058883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lin Z, et al. Mettl3-/Mettl14-mediated mRNA N(6)-methyladenosine modulates murine spermatogenesis. Cell Res. 2017;27:1216–1230. doi: 10.1038/cr.2017.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang Y, et al. Publisher correction: N(6)-methyladenosine RNA modification regulates embryonic neural stem cell self-renewal through histone modifications. Nat. Neurosci. 2018;21:1139. doi: 10.1038/s41593-018-0169-2. [DOI] [PubMed] [Google Scholar]
- 18.Barbieri I, et al. Promoter-bound METTL3 maintains myeloid leukaemia by m(6)A-dependent translation control. Nature. 2017;552:126–131. doi: 10.1038/nature24678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li Z, et al. FTO plays an oncogenic role in acute myeloid leukemia as a N(6)-methyladenosine RNA demethylase. Cancer Cell. 2017;31:127–141. doi: 10.1016/j.ccell.2016.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cheng M, et al. The m(6)A methyltransferase METTL3 promotes bladder cancer progression via AFF4/NF-kappaB/MYC signaling network. Oncogene. 2019;38:3667–3680. doi: 10.1038/s41388-019-0683-z. [DOI] [PubMed] [Google Scholar]
- 21.Choe J, et al. mRNA circularization by METTL3-eIF3h enhances translation and promotes oncogenesis. Nature. 2018;561:556–560. doi: 10.1038/s41586-018-0538-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lin S, Choe J, Du P, Triboulet R, Gregory RI. The m(6)A methyltransferase METTL3 promotes translation in human cancer cells. Mol. Cell. 2016;62:335–345. doi: 10.1016/j.molcel.2016.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wu R, Jiang D, Wang Y, Wang X. N (6)-Methyladenosine (m(6)A) methylation in mRNA with a dynamic and reversible epigenetic modification. Mol. Biotechnol. 2016;58:450–459. doi: 10.1007/s12033-016-9947-9. [DOI] [PubMed] [Google Scholar]
- 24.Wang P, Doxtader KA, Nam Y. Structural basis for cooperative function of Mettl3 and Mettl14 methyltransferases. Mol. Cell. 2016;63:306–317. doi: 10.1016/j.molcel.2016.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jia G, Fu Y, He C. Reversible RNA adenosine methylation in biological regulation. Trends Genet. 2013;29:108–115. doi: 10.1016/j.tig.2012.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Knuckles P, et al. RNA fate determination through cotranscriptional adenosine methylation and microprocessor binding. Nat. Struct. Mol. Biol. 2017;24:561–569. doi: 10.1038/nsmb.3419. [DOI] [PubMed] [Google Scholar]
- 27.Slobodin B, et al. Transcription impacts the efficiency of mRNA translation via co-transcriptional N6-adenosine Methylation. Cell. 2017;169:326–337 e312. doi: 10.1016/j.cell.2017.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Knuckles P, et al. Zc3h13/Flacc is required for adenosine methylation by bridging the mRNA-binding factor Rbm15/Spenito to the m(6)A machinery component Wtap/Fl(2)d. Genes Dev. 2018;32:415–429. doi: 10.1101/gad.309146.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wen J, et al. Zc3h13 regulates nuclear RNA m(6)A methylation and mouse embryonic stem cell self-renewal. Mol. Cell. 2018;69:1028–1038 e1026. doi: 10.1016/j.molcel.2018.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ping XL, et al. Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res. 2014;24:177–189. doi: 10.1038/cr.2014.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Patil DP, et al. m(6)A RNA methylation promotes XIST-mediated transcriptional repression. Nature. 2016;537:369–373. doi: 10.1038/nature19342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yue Y, et al. VIRMA mediates preferential m(6)A mRNA methylation in 3’UTR and near stop codon and associates with alternative polyadenylation. Cell Discov. 2018;4:10. doi: 10.1038/s41421-018-0019-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pendleton KE, et al. The U6 snRNA m(6)A methyltransferase METTL16 regulates SAM synthetase intron retention. Cell. 2017;169:824–835 e814. doi: 10.1016/j.cell.2017.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Warda AS, et al. Human METTL16 is a N(6)-methyladenosine (m(6)A) methyltransferase that targets pre-mRNAs and various non-coding RNAs. EMBO Rep. 2017;18:2004–2014. doi: 10.15252/embr.201744940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shima H, et al. S-Adenosylmethionine synthesis is regulated by selective N(6)-adenosine methylation and mRNA degradation involving METTL16 and YTHDC1. Cell Rep. 2017;21:3354–3363. doi: 10.1016/j.celrep.2017.11.092. [DOI] [PubMed] [Google Scholar]
- 36.Jia G, et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat. Chem. Biol. 2011;7:885–887. doi: 10.1038/nchembio.687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zheng G, et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol. Cell. 2013;49:18–29. doi: 10.1016/j.molcel.2012.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dina C, et al. Variation in FTO contributes to childhood obesity and severe adult obesity. Nat. Genet. 2007;39:724–726. doi: 10.1038/ng2048. [DOI] [PubMed] [Google Scholar]
- 39.Fu Y, et al. FTO-mediated formation of N6-hydroxymethyladenosine and N6-formyladenosine in mammalian RNA. Nat. Commun. 2013;4:1798. doi: 10.1038/ncomms2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mauer J, et al. Reversible methylation of m(6)Am in the 5’ cap controls mRNA stability. Nature. 2017;541:371–375. doi: 10.1038/nature21022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wei J, et al. Differential m(6)A, m(6)Am, and m(1)A demethylation mediated by FTO in the cell nucleus and cytoplasm. Mol. Cell. 2018;71:973–985 e975. doi: 10.1016/j.molcel.2018.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Linder B, et al. Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome. Nat. Methods. 2015;12:767–772. doi: 10.1038/nmeth.3453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lence T, et al. m(6)A modulates neuronal functions and sex determination in Drosophila. Nature. 2016;540:242–247. doi: 10.1038/nature20568. [DOI] [PubMed] [Google Scholar]
- 44.Haussmann IU, et al. m(6)A potentiates Sxl alternative pre-mRNA splicing for robust Drosophila sex determination. Nature. 2016;540:301–304. doi: 10.1038/nature20577. [DOI] [PubMed] [Google Scholar]
- 45.Zhao X, et al. FTO-dependent demethylation of N6-methyladenosine regulates mRNA splicing and is required for adipogenesis. Cell Res. 2014;24:1403–1419. doi: 10.1038/cr.2014.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tang C, et al. ALKBH5-dependent m6A demethylation controls splicing and stability of long 3’-UTR mRNAs in male germ cells. Proc. Natl Acad. Sci. USA. 2018;115:E325–E333. doi: 10.1073/pnas.1717794115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu N, et al. N6-methyladenosine alters RNA structure to regulate binding of a low-complexity protein. Nucleic Acids Res. 2017;45:6051–6063. doi: 10.1093/nar/gkx141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xiao W, et al. Nuclear m(6)A reader YTHDC1 regulates mRNA splicing. Mol. Cell. 2016;61:507–519. doi: 10.1016/j.molcel.2016.01.012. [DOI] [PubMed] [Google Scholar]
- 49.Jacobs E, Mills JD, Janitz M. The role of RNA structure in posttranscriptional regulation of gene expression. J. Genet Genomics. 2012;39:535–543. doi: 10.1016/j.jgg.2012.08.002. [DOI] [PubMed] [Google Scholar]
- 50.Roost C, et al. Structure and thermodynamics of N6-methyladenosine in RNA: a spring-loaded base modification. J. Am. Chem. Soc. 2015;137:2107–2115. doi: 10.1021/ja513080v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang X, et al. N(6)-methyladenosine modulates messenger RNA translation efficiency. Cell. 2015;161:1388–1399. doi: 10.1016/j.cell.2015.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shi H, et al. YTHDF3 facilitates translation and decay of N(6)-methyladenosine-modified RNA. Cell Res. 2017;27:315–328. doi: 10.1038/cr.2017.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hsu PJ, et al. Ythdc2 is an N(6)-methyladenosine binding protein that regulates mammalian spermatogenesis. Cell Res. 2017;27:1115–1127. doi: 10.1038/cr.2017.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wojtas MN, et al. Regulation of m(6)A transcripts by the 3’->5’ RNA helicase YTHDC2 is essential for a successful meiotic program in the mammalian germline. Mol. Cell. 2017;68:374–387 e312. doi: 10.1016/j.molcel.2017.09.021. [DOI] [PubMed] [Google Scholar]
- 55.Park OH, et al. Endoribonucleolytic cleavage of m(6)A-containing RNAs by RNase P/MRP complex. Mol. Cell. 2019;74:494–507 e498. doi: 10.1016/j.molcel.2019.02.034. [DOI] [PubMed] [Google Scholar]
- 56.Du H, et al. YTHDF2 destabilizes m(6)A-containing RNA through direct recruitment of the CCR4-NOT deadenylase complex. Nat. Commun. 2016;7:12626. doi: 10.1038/ncomms12626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhou J, et al. Dynamic m(6)A mRNA methylation directs translational control of heat shock response. Nature. 2015;526:591–594. doi: 10.1038/nature15377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Meyer KD, et al. 5’ UTR m(6)A promotes cap-independent translation. Cell. 2015;163:999–1010. doi: 10.1016/j.cell.2015.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ries RJ, et al. m(6)A enhances the phase separation potential of mRNA. Nature. 2019;571:424–428. doi: 10.1038/s41586-019-1374-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang F, et al. Fragile X mental retardation protein modulates the stability of its m6A-marked messenger RNA targets. Hum. Mol. Genet. 2018;27:3936–3950. doi: 10.1093/hmg/ddy292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Edupuganti RR, et al. N(6)-methyladenosine (m(6)A) recruits and repels proteins to regulate mRNA homeostasis. Nat. Struct. Mol. Biol. 2017;24:870–878. doi: 10.1038/nsmb.3462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Huang H, et al. Recognition of RNA N(6)-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat. Cell Biol. 2018;20:285–295. doi: 10.1038/s41556-018-0045-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang Y, et al. N6-methyladenosine modification destabilizes developmental regulators in embryonic stem cells. Nat. Cell Biol. 2014;16:191–198. doi: 10.1038/ncb2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chen Z, Fillmore CM, Hammerman PS, Kim CF, Wong KK. Non-small-cell lung cancers: a heterogeneous set of diseases. Nat. Rev. Cancer. 2014;14:535–546. doi: 10.1038/nrc3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Molina JR, Yang P, Cassivi SD, Schild SE, Adjei AA. Non-small cell lung cancer: epidemiology, risk factors, treatment, and survivorship. Mayo Clin. Proc. 2008;83:584–594. doi: 10.4065/83.5.584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wei W, Huo B, Shi X. miR-600 inhibits lung cancer via downregulating the expression of METTL3. Cancer Manag. Res. 2019;11:1177–1187. doi: 10.2147/CMAR.S181058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Du M, et al. MiR-33a suppresses proliferation of NSCLC cells via targeting METTL3 mRNA. Biochem Biophys. Res Commun. 2017;482:582–589. doi: 10.1016/j.bbrc.2016.11.077. [DOI] [PubMed] [Google Scholar]
- 68.Du Y, et al. SUMOylation of the m6A-RNA methyltransferase METTL3 modulates its function. Nucleic Acids Res. 2018;46:5195–5208. doi: 10.1093/nar/gky156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Liu J, et al. m(6)A demethylase FTO facilitates tumor progression in lung squamous cell carcinoma by regulating MZF1 expression. Biochem. Biophys. Res. Commun. 2018;502:456–464. doi: 10.1016/j.bbrc.2018.05.175. [DOI] [PubMed] [Google Scholar]
- 70.Chen J, Odenike O, Rowley JD. Leukaemogenesis: more than mutant genes. Nat. Rev. Cancer. 2010;10:23–36. doi: 10.1038/nrc2765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Vu LP, et al. The N(6)-methyladenosine (m(6)A)-forming enzyme METTL3 controls myeloid differentiation of normal hematopoietic and leukemia cells. Nat. Med. 2017;23:1369–1376. doi: 10.1038/nm.4416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Paris J, et al. Targeting the RNA m(6)A reader ythdf2 selectively compromises cancer stem cells in acute myeloid leukemia. Cell Stem Cell. 2019;25:137–148 e136. doi: 10.1016/j.stem.2019.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Weng H, et al. METTL14 inhibits hematopoietic stem/progenitor differentiation and promotes leukemogenesis via mRNA m(6)A modification. Cell Stem Cell. 2018;22:191–205 e199. doi: 10.1016/j.stem.2017.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sia D, Villanueva A, Friedman SL, Llovet JM. Liver cancer cell of origin, molecular class, and effects on patient prognosis. Gastroenterology. 2017;152:745–761. doi: 10.1053/j.gastro.2016.11.048. [DOI] [PubMed] [Google Scholar]
- 75.Chen M, et al. RNA N6-methyladenosine methyltransferase-like 3 promotes liver cancer progression through YTHDF2-dependent posttranscriptional silencing of SOCS2. Hepatology. 2018;67:2254–2270. doi: 10.1002/hep.29683. [DOI] [PubMed] [Google Scholar]
- 76.Zhong L, et al. YTHDF2 suppresses cell proliferation and growth via destabilizing the EGFR mRNA in hepatocellular carcinoma. Cancer Lett. 2019;442:252–261. doi: 10.1016/j.canlet.2018.11.006. [DOI] [PubMed] [Google Scholar]
- 77.Yang Z, et al. MicroRNA-145 modulates N(6)-methyladenosine levels by targeting the 3’-untranslated mRNA region of the N(6)-methyladenosine binding YTH domain family 2 protein. J. Biol. Chem. 2017;292:3614–3623. doi: 10.1074/jbc.M116.749689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.DeSantis CE, Ma J, Goding Sauer A, Newman LA, Jemal A. Breast cancer statistics, 2017, racial disparity in mortality by state. CA Cancer J. Clin. 2017;67:439–448. doi: 10.3322/caac.21412. [DOI] [PubMed] [Google Scholar]
- 79.Cai X, et al. HBXIP-elevated methyltransferase METTL3 promotes the progression of breast cancer via inhibiting tumor suppressor let-7g. Cancer Lett. 2018;415:11–19. doi: 10.1016/j.canlet.2017.11.018. [DOI] [PubMed] [Google Scholar]
- 80.Zhang C, et al. Hypoxia-inducible factors regulate pluripotency factor expression by ZNF217- and ALKBH5-mediated modulation of RNA methylation in breast cancer cells. Oncotarget. 2016;7:16. doi: 10.18632/oncotarget.11743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zhang C, et al. Hypoxia induces the breast cancer stem cell phenotype by HIF-dependent and ALKBH5-mediated m(6)A-demethylation of NANOG mRNA. Proc. Natl Acad. Sci. USA. 2016;113:E2047–E2056. doi: 10.1073/pnas.1602883113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wu L, Wu D, Ning J, Liu W, Zhang D. Changes of N6-methyladenosine modulators promote breast cancer progression. BMC Cancer. 2019;19:326. doi: 10.1186/s12885-019-5538-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.He H, Wu W, Sun Z, Chai L. MiR-4429 prevented gastric cancer progression through targeting METTL3 to inhibit m(6)A-caused stabilization of SEC62. Biochem. Biophys. Res Commun. 2019;517:581–587. doi: 10.1016/j.bbrc.2019.07.058. [DOI] [PubMed] [Google Scholar]
- 84.Wang, Q. et al. METTL3-mediated m(6)A modification of HDGF mRNA promotes gastric cancer progression and has prognostic significance. Gut (2019). [Epub ahead of print] [DOI] [PubMed]
- 85.Li Y, et al. Expression of demethylase genes, FTO and ALKBH1, is associated with prognosis of gastric cancer. Dig. Dis. Sci. 2019;64:1503–1513. doi: 10.1007/s10620-018-5452-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zhang C, et al. Reduced m6A modification predicts malignant phenotypes and augmented Wnt/PI3K-Akt signaling in gastric cancer. Cancer Med. 2019;8:4766–4781. doi: 10.1002/cam4.2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Jin H, et al. N(6)-methyladenosine modification of ITGA6 mRNA promotes the development and progression of bladder cancer. EBioMedicine. 2019;47:195–207. doi: 10.1016/j.ebiom.2019.07.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Thakkar JP, et al. Epidemiologic and molecular prognostic review of glioblastoma. Cancer Epidemiol. Biomark. Prev. 2014;23:1985–1996. doi: 10.1158/1055-9965.EPI-14-0275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zhang S, et al. m(6)A demethylase ALKBH5 maintains tumorigenicity of glioblastoma stem-like cells by sustaining FOXM1 expression and cell proliferation program. Cancer Cell. 2017;31:591–606 e596. doi: 10.1016/j.ccell.2017.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lathia JD, Mack SC, Mulkearns-Hubert EE, Valentim CL, Rich JN. Cancer stem cells in glioblastoma. Genes Dev. 2015;29:1203–1217. doi: 10.1101/gad.261982.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Cui Q, et al. m(6)A RNA methylation regulates the self-renewal and tumorigenesis of glioblastoma stem cells. Cell Rep. 2017;18:2622–2634. doi: 10.1016/j.celrep.2017.02.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Visvanathan A, et al. Essential role of METTL3-mediated m(6)A modification in glioma stem-like cells maintenance and radioresistance. Oncogene. 2018;37:522–533. doi: 10.1038/onc.2017.351. [DOI] [PubMed] [Google Scholar]
- 93.Li T, et al. METTL3 facilitates tumor progression via an m(6)A-IGF2BP2-dependent mechanism in colorectal carcinoma. Mol. Cancer. 2019;18:112. doi: 10.1186/s12943-019-1038-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Nishizawa Y, et al. Oncogene c-Myc promotes epitranscriptome m6A reader YTHDF1 expression in colorectal cancer. Oncotarget. 2017;9:11. doi: 10.18632/oncotarget.23554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Li X, et al. The M6A methyltransferase METTL3: acting as a tumor suppressor in renal cell carcinoma. Oncotarget. 2017;8:14. doi: 10.18632/oncotarget.21726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Liu J, et al. m(6)A mRNA methylation regulates AKT activity to promote the proliferation and tumorigenicity of endometrial cancer. Nat. Cell Biol. 2018;20:1074–1083. doi: 10.1038/s41556-018-0174-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wang X, et al. Reduced m6A mRNA methylation is correlated with the progression of human cervical cancer. Oncotarget. 2017;8:13. doi: 10.18632/oncotarget.22041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Chen J, et al. YTH domain family 2 orchestrates epithelial-mesenchymal transition/proliferation dichotomy in pancreatic cancer cells. Cell Cycle. 2017;16:2259–2271. doi: 10.1080/15384101.2017.1380125. [DOI] [PMC free article] [PubMed] [Google Scholar]