

Keywords: Clostridioides difficile, enteritis, goblet cells, mucin 2, mucus, actin, inflammatory bowel disease (IBD), live imaging, cell rounding, enteroids, organoids, toxins
Abstract
Clostridioides difficile is an important nosocomial pathogen that produces toxins to cause life-threatening diarrhea and colitis. Toxins bind to epithelial receptors and promote the collapse of the actin cytoskeleton. C. difficile toxin activity is commonly studied in cancer-derived and immortalized cell lines. However, the biological relevance of these models is limited. Moreover, no model is available for examining C. difficile-induced enteritis, an understudied health problem. We hypothesized that human intestinal enteroids (HIEs) express toxin receptors and provide a new model to dissect C. difficile cytotoxicity in the small intestine. We generated biopsy-derived jejunal HIE and Vero cells, which stably express LifeAct-Ruby, a fluorescent label of F-actin, to monitor actin cytoskeleton rearrangement by live-cell microscopy. Imaging analysis revealed that toxins from pathogenic C. difficile strains elicited cell rounding in a strain-dependent manner, and HIEs were tenfold more sensitive to toxin A (TcdA) than toxin B (TcdB). By quantitative PCR, we paradoxically found that HIEs expressed greater quantities of toxin receptor mRNA and yet exhibited decreased sensitivity to toxins when compared with traditionally used cell lines. We reasoned that these differences may be explained by components, such as mucins, that are present in HIEs cultures, that are absent in immortalized cell lines. Addition of human-derived mucin 2 (MUC2) to Vero cells delayed cell rounding, indicating that mucus serves as a barrier to toxin-receptor binding. This work highlights that investigation of C. difficile infection in that HIEs can provide important insights into the intricate interactions between toxins and the human intestinal epithelium.
NEW & NOTEWORTHY In this article, we developed a novel model of Clostridioides difficile-induced enteritis using jejunal-derived human intestinal enteroids (HIEs) transduced with fluorescently tagged F-actin. Using live-imaging, we identified that jejunal HIEs express high levels of TcdA and CDT receptors, are more sensitive to TcdA than TcdB, and secrete mucus, which delays toxin-epithelial interactions. This work also optimizes optically clear C. difficile-conditioned media suitable for live-cell imaging.
INTRODUCTION
Clostridioides difficile infection, also known as CDI, is a prevalent complication of long-term hospitalization. Risk factors for disease include antibiotic usage that alters gut microbiota, immunocompromised status, and advanced age (9, 18, 37, 160). CDI causes a range of clinical manifestations, including mild to severe watery diarrhea, pseudomembranous colitis, toxic megacolon, and death. The pathogenesis of infection involves ingestion of C. difficile spores and germination of the spores in the presence of unconjugated primary bile acids, which are abundant in the small intestine, followed by vegetative cell colonization of the intestinal mucus layer (45, 58, 75, 101, 135, 139). C. difficile toxins are responsible for clinically significant CDI disease manifestations (5, 7, 15–17, 27, 28, 32, 33, 40, 56, 59, 61, 69, 72, 73, 94, 98, 104, 106, 108, 111, 113).
CDI is most frequently documented in the colon, prompting a substantial effort devoted to understanding pathogenesis in this intestinal segment (17, 18, 23, 27, 30, 44, 45, 66). Although CDI in the colon is well recognized, CDI in the small intestine, termed CDI-enteritis, has also been reported (1, 3, 4, 6, 8, 12–14, 22, 35, 43, 50, 51, 54, 55, 60, 62, 65, 71, 86–89, 92, 95, 97, 99, 100, 103, 105, 107, 109, 114–116, 121, 122, 127, 136–138, 142, 143, 145, 146, 148, 152, 154–158, 161). These CDI cases include infection of the jejunum and ileum. CDI-enteritis frequently occurs in individuals with inflammatory bowel disease (IBD), a condition that is increasing in prevalence (6, 87, 103, 114). Although C. difficile-associated enteritis is observed at a lower incidence than colonic CDI, it has been speculated that small bowel enteritis is more prevalent than previously suspected (35).
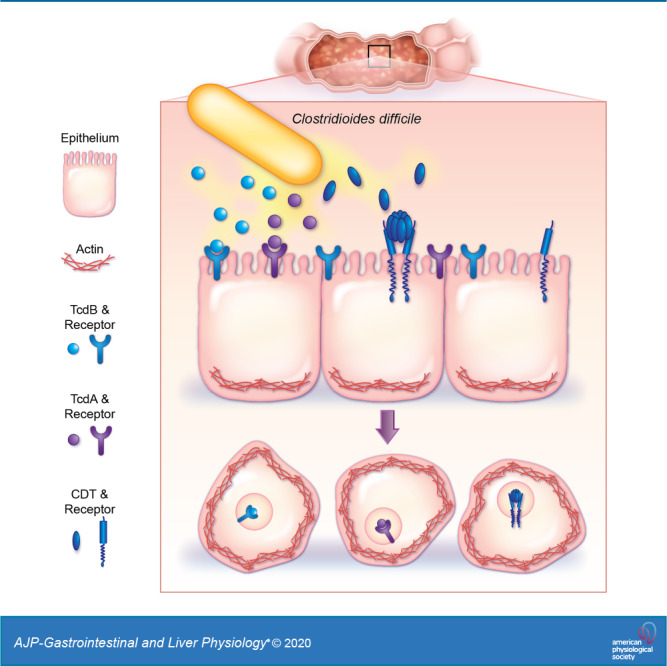
Most C. difficile strains produce two major toxins, TcdA and TcdB, which belong to the family of large clostridial toxins (23). In addition, hypervirulent strains of C. difficile can produce an additional binary toxin called CDT (57). These C. difficile toxins enter epithelial cells by binding to one or more receptors present on the cell surface (2, 26, 30, 61, 68, 98, 112, 119, 124–126, 141, 149). Receptor binding is followed by endocytosis, which rapidly initiates multiple signaling events (48, 49, 66, 86). For TcdA and TcdB, toxin endocytosis leads to the inactivation of Rho GTPases (77, 78), which results in disassembly of actin stress fibers and collapse of the actin cytoskeleton. This actin cytoskeleton collapse can be visualized as the rounding of cells, decreased cell volume, loss of cell-cell contacts, and formation of neurite-like retraction fibers (33, 67, 130). For CDT, toxin endocytosis results in ADP-ribose caps in the actin protein, which prevents actin filament elongation, resulting in the collapse of the actin cytoskeleton. Thus, all C. difficile toxins result in cell rounding.
Because toxins are necessary for symptomatic infection, toxin activity has become a hallmark of the study of C. difficile pathogenesis. Traditionally, cancer-derived cell lines, including HeLa, HT29, Caco-2, and T84 cells, or immortalized monkey fibroblast Vero cells have been used to examine toxin activity. However, multiple studies have noted variations in the cytotoxic potencies of the same toxin across different cell lines. Another challenge in studying CDI-enteritis comes from the cell type the traditional model systems originated from; HeLa and Vero cells are of non-intestinal origin, whereas HT29, Caco-2, and T84 cells are derived from colonic adenocarcinomas. Thus, the ability of these cell lines to recapitulate normal human intestinal epithelial cell biology and responses to CDI is limited. Moreover, there are no immortalized human small intestine epithelium cell lines available, making it difficult to decipher the mechanism of CDI-induced enteritis.
The ability to propagate primary human intestinal epithelial cells has circumvented the need for immortalization. These primary cells, known as human intestinal enteroids (HIEs), have revolutionized the study of the intestinal epithelium (52, 53, 91, 132, 133, 159). HIEs are a complex epithelial culture system produced from nontransformed stem cells isolated from intestinal tissue or biopsy samples. Instead of the single cell type represented in traditional cell lines, HIEs harbor all cell types of the native intestinal epithelium, including enterocytes, goblet cells, stem cells, Paneth cells, and enteroendocrine cells (118). As a result, HIEs recapitulate human intestinal physiology and can be maintained in culture, thereby enabling the establishment of long-term in vitro model systems. Thus, HIEs offer a promising new way to study toxin-epithelial interactions in an in vitro culture system. In these studies, we investigated the impact of C. difficile toxins on the small intestinal cytoskeleton dynamics using HIEs with a fluorescently labeled actin. Our work reveals that, when compared with Vero cells, HIEs are significantly more resistant to cytoskeletal rearrangement. We further show that this resistance is attributable to the presence of mucin in the intestinal epithelium and alterations in toxin receptor expression. These results provide a foundation for future studies to elucidate the mechanisms of CDI-enteritis.
METHODS
Bacterial strains and culture conditions.
Clostridioides (previously Clostridium) difficile strains R20291 and 630 were obtained from American Type Culture Collection (ATCC); M68 was isolated in 2004 from an outbreak in Dublin, Ireland (36), and nontoxic CD37 was obtained from a Texas Medical Center Hospital. All C. difficile cultures were grown in brain-heart-infusion medium supplemented with 2% yeast extract (BHIS) at 37°C in an Anaerobe Systems AS-580 anaerobic chamber supplied with a mixture of 10% CO2, 5% H2, and 85% N2. For cell treatment, cultures of C. difficile were grown in BHIS for 24 h, adjusted to an optical density (OD600nm) = 1.5, and pelleted by centrifugation at 4,000 g. Cell pellets were washed three times in sterile anaerobic phosphate-buffered saline (PBS), suspended in an equal volume of anaerobic prewarmed Fluorobrite DMEM (cat. no. A1896701; ThermoFisher), and incubated for an additional 24 h. C. difficile cells were pelleted by centrifugation at 7,000 g for 5 min, Fluorobrite DMEM supernatant was collected, and 0.22 µM was filtered for sterility. Filtered supernatant was adjusted for HIE treatment by adding 1× Glutamax (Invitrogen), 1× HEPES (Invitrogen), and 100 U/mL penicillin-streptomycin (Invitrogen).
Human intestinal enteroid cultures.
Human intestinal enteroid (HIE) culture procedures were approved by the Baylor College of Medicine Institutional Review Board (IRB) committee, and all cultures were deidentified. HIEs were produced from stem cells isolated from surgical specimens of adult jejunum obtained at Baylor College of Medicine via the Texas Medical Center Digestive Diseases Center Gastrointestinal Experimental Model Systems (GEMS) Core, as previously described (24, 132, 133). Jejunal HIE culture J3 was used for all these studies. Complete media with and without growth factors (CMGF+ and CMGF−, respectively), differentiation media, and high Wnt3a CMGF+ (hW-CMGF+) were prepared as previously described (24, 46, 132, 133), with the modification that the differentiation media was created without any R-spondin.
The LifeActRuby (plasmid no. 51009; Addgene) was cloned into pLVX-IRES-Hygro. Lentivirus vectors for the construct were packaged in human embryonic kidney (HEK)-293T cells as described previously (123). Jejunum human intestinal enteroids from patient J3 stably expressing LifeActRuby were created using lentivirus transduction as described previously and grown in high Wnt3a CMGF+ with 50 µg/mL hygromycin B for selection (24). The jHIE-LifeActRuby enteroids were propagated and maintained in hW-CMGF+ with the selection drug. HIEs were seeded in phenol red-free, growth factor-reduced Matrigel (Corning) on 24-well plates (Corning) and passaged every 7–10 days.
HIE monolayers (2D) were prepared from 3D cultures, as previously described (24, 46, 151), in flat 96-well cultures slides that were pretreated with Matrigel diluted in 1× PBS and incubated in CMGF+ supplemented with 10 µM Y-27632 Rock inhibitor at 37°C with 5% CO2. After 24 h of growth in CMGF+, HIE media was replaced with differentiation media, and monolayers were differentiated for 4–5 days. For live-cell imaging assays, HIEs were washed twice with sterile DMEM Fluorobrite supplemented with 15 mM HEPES, 1× sodium pyruvate, 1× Glutamax, and 1× nonessential amino acids (Invitrogen). Then HIEs were treated with DMEM Fluorobrite (control) or C. difficile-conditioned DMEM Fluorobrite supplemented with 1× GlutaMAX (Invitrogen), 10 mM HEPES buffer (Invitrogen), and 100 U/mL penicillin-streptomycin (Invitrogen).
RNA-sequencing and transcriptional analysis of HIEs.
HIEs were prepared and differentiated in transwell inserts. Transwells were washed with ice-cold Dulbecco’s PBS (lacking Ca2+ and Mg2+), and RNA was isolated with TRIzol reagent (Invitrogen) according the manufacturer’s details and purified using a Qiagen RNeasy Isolation Kit (Qiagen). RNA integrity was examined by Agilent Bioanalyzer analysis (Agilent 2100 Bioanalyzer) from six biological replicates. Novogene was used to examine paired-end Illumina sequencing of mRNA-enriched samples. STAR software mapped reads using the human reference genome with an output of counts per gene and transcript per million reads.
Mammalian culture conditions.
HeLa (ATCC CCL-2; epithelial), Vero (ATCC CCL-81; epithelial), T84 (ATCC CCL-248; epithelial), HT29 (ATCC HTB-38; epithelial), and LS174T (ATCC CL-188; goblet like) cells were obtained from ATCC. Cells were regularly maintained in a complete growth medium (CGM) of Gibco Dulbecco’s modified Eagle’s medium (ThermoFisher) supplemented with 10% fetal bovine serum (FBS). All cultures were maintained in a humidified atmosphere at 37°C and 5% CO2 and routinely tested for Mycoplasma using the Mycoplasma Detection Kit (cat. no. LT07-518; Lonza). For actin imaging studies, cells were cultured in 12-well plates to 50–60% confluency, transduced using lentivirus pLVX-LifeActRuby with 10 µg/mL polybrene (cat. no. TR-1003-G; Millipore), and incubated for 48 h. After transduction, stably expressing cells were selected using hygromycin (30 μg/mL).
Epifluorescence live-cell microscopy.
For live-cell imaging, LifeActRuby-expressing cells were grown to confluence on 10-well CELLview chamber slides (GreinerBio) and then changed to an optically clear FluoroBrite DMEM media (Invitrogen) supplemented with 15 mM HEPES (Invitrogen), 1× sodium pyruvate (Invitrogen), 1× GlutaMax (Invitrogen), and 1× nonessential amino acids (Invitrogen) (Fluorobrite-Plus) C. difficile conditioned FluoroBrite DMEM was added in uninoculated FluoroBrite DMEM (50% final concentration). HIEs, HeLa, and Vero cells were placed in an Okolabs stage-top incubation chamber with CO2 and humidity control. The stage was then placed on a Nikon TiE inverted wide-field epifluorescence microscope (Nikon) with a motorized X, Y, and Z stage for software-controlled multi-position imaging. Cell images were obtained with wide-field epifluorescence using a 20× Plan Apo (NA 0.75) differential interference contrast objective, using a SPECTRA X LED light source (Lumencor). Videos were recorded using an ORCA-Flash 4.0 sCMOS camera (Hamamatsu). Data acquisition and image analysis were performed using Nikon Elements version 4.5 software. For analysis, FIJI (Formerly ImageJ; National Institutes of Health) was used to define cell membranes (as denoted by actin), and cell diameter was recorded in four regions per well, two wells per slide.
After the experiment, cell viability was examined by addition of resazurin as previously described (42). Briefly, 100 µM of resazurin (7-hydroxy-3H-phenoxazin-3-one 10-oxide; Sigma Aldrich) in Fluorobrite DMEM was added to each well at a final concentration of 44 µM, and plates were incubated for 2 h at 37°C, 5% CO2. After the incubation, the contents of the wells were transferred to a 96-well plate, and the reduction of resazurin to resorufin was measured using a microplate spectrofluorometer at an excitation wavelength of 570 nm and an emission wavelength of 600 nm. Cell viability was assessed by comparing readings against those obtained for wells incubated only in Fluorobrite DMEM medium and is represented as a percentage of cell viability relative to this control.
Human mucin 2 purification.
Mucin 2 (MUC2) was purified from human goblet-like LS179T cells, as previously described with minor modifications (76, 118). Briefly, LS174T cells were grown in large T300 flasks (VWR) with 10 µM of DAPT (Sigma-Aldrich) to promote goblet cell differentiation. Cells were incubated rocking for 5 days to stimulate maximal mucus release. Supernatants were collected and mucin proteins precipitated by ETOH precipitation, followed by guanidinium chloride and cesium chloride gradient ultracentrifugation in a Beckman Coulter Ultra-Centrifuge (30.2 Ti rotor), followed by dialysis, as previously described (125). Fractions containing MUC2 were selected by dot blot analysis using a MUC2 antibody (cat. no. NBP1-31231, 1:100 dilution; Novus) with the LI-COR Odyssey Blotting reagents, Odyssey imaging system and Image Studio software (LI-CORE Biosciences). MUC2 protein was quantified by BCA assay. For tissue culture toxin inhibition assays, human MUC2 was added to Vero cells at a concentration of 1 mg/ml in the presence of toxins.
Scanning electron microscopy.
After imaging, the wells of the slides were washed gently with PBS containing Mg2+ and Ca2+ (2×) and fixed in 2.5% glutaraldehyde in PBS for 1 h at room temperature. The black compartment of the CELLview slide was detached, and the slide was dehydrated with ethanol and coated in 20 nm of gold using a desktop sputtering system (Denton Desk II). All slides were viewed in a scanning electron microscope (FEI XL-30FEG) at 12 kV.
Quantitative real-time PCR.
Untreated HIEs, Vero, HeLa, T84, and HT29 cells were grown as monolayers on 96-well plates, and cells were collected in TRIzol. RNA was extracted according to the manufacturer’s details, with genomic DNA removal performed by the Ambion Turbo DNA-free kit, following the manufacturer’s instructions (ThermoFisher, Waltham, MA). RNA (0.5 µg) was converted to cDNA using the SensiFAST cDNA synthesis kit (Bioline USA Inc.). cDNA was examined by quantitative real-time PCR (qPCR) using the listed primers (Table 1) and FastSYBR Green (Thermo-Fisher) on a QuantStudio 3 qPCR machine (Applied Biosystems). Relative fold changes were examined using the ΔΔCT method with the housekeeping gene glyceraldehyde-3-phosphate dehydrogenase (GAPDH).
Table 1.
qPCR Primers for human toxin receptors
| Toxin | Receptor | Forward Primer | Reverse Primer |
|---|---|---|---|
| CDT | LSR | CAGGAGAATCACCATCACAGGAA | AGTAATACACTCCACTGTCTCCCCAG |
| TcdA | GP96 | TGTTTCCCGCGAGACTCTTC | CACACCAGCACTGCTGTGCACCACAGTGG |
| TcdB | CSPG4 | CTTCACTCAGGCAGAGGTCTACG | GAGGACAGCTGGAGCTCTAGGGT |
| TcdB | PVRL3 | ATGCAAAGCCGTTACATTCC | TTAAAGAATCCGGCCCTTTT |
| TcdB | FZD1 | CATCGTCATCGCCTGCTACT | TAGCGTAGCTCTTGCAGCTC |
| TcdB | FZD5 | CGCGAGCACAACCACATC | AGAAGTAGACCAGGAGGAAGACGAT |
| TcdB | FZD7 | CGCCTCTGTTCGTCTACCTC | TCATGATGGTGCGGATACGG |
CSPG4, chondroitin sulfate proteoglycan 4; FZD1, -5, and -7, Frizzled 1, 5, and 7, respectively; GP96, glycoprotein 96; LSR, lipolysis-stimulated lipoprotein receptor; qPCR, quantitative PCR; PVRL3, poliovirus receptor-related 3; TcdA, toxin A; TcdB, toxin B.
Immunostaining.
HIE two-dimensional monolayers on CELLview chamber slides (GreinerBio) were fixed in Clarke’s solution for 30 min at room temperature, and cells were permeabilized by incubation with 0.1% Triton-X for 30 min at room temperature. Slides were then blocked for 1 h at room temperature in 10% donkey serum. A MUC2 antibody (1:200 dilution; cat. no. sc-515032, Santa Cruz Biotechnology) was added overnight at 4°C. After PBS washes, cells were incubated with donkey-anti-mouse AlexaFluor 555 (1:1,000 dilution; cat. no. A11004; Life Technologies) for 1 h at room temperature. Nuclei were visualized with DAPI (cat. no. R37606; Thermo Fisher Scientific) at room temperature for 5 min. Slides were imaged on the Nikon TiE.
C. difficile strains (Cd37, M68, 630, and R20291) were grown in BHIS overnight, transferred to Fluorobrite DMEM overnight, and stained with an FM4-64 Lipophilic Styryl Dye (ThermoFisher cat. no. T13320), as previously described (5). Briefly, bacterial cells were stained with 500 μg/mL FM4–6 for 1 h, washed twice with 1× PBS, and then mounted on slides. The slides were imaged on a Zeiss AxioImager Z1 fluorescent microscope at 100× with a Hamamatsu Electron Multiplier charge-coupled device (CCD) camera. For live/dead cell staining, C. difficile strains were incubated with LIVE/DEAD BacLight Bacterial Viability Stains (cat. no. L7012; ThermoFisher) according to the manufacturer’s details. Briefly, 1 × 108 C. difficile bacteria were mixed with a 2× LIVE/DEAD BacLight staining reagent mixture and incubated for 15 min in the dark at 37°C anaerobically. Then 100 µL of each of the bacterial cell suspension was added into separate wells of a 96-well flat-bottom black-walled microplate. Plates were read at excitation: 485 nm/emission: 530 nm (green) and excitation: 485 nm/emission: 630 nm (red) on a Synergy H1 Microplate Reader (Bio-Tek Instruments). Viabilities were calculated by dividing the fluorescence intensity of the stained bacterial suspensions at 485/530 by the fluorescence intensity at 485/630 × 100% (ratio green/red × 100%).
Toxin ELISAs.
Toxins A and B were evaluated by enzyme-linked immunosorbent assay (ELISA: cat. no. DFA35-K01, DFB35-K01; Eagle Biosciences). A common method for producing C. difficile toxin-containing media is to grow the bacteria for 48 h in BHIS. However, this solution contains multiple autofluorescent compounds and is not conducive to live-cell imaging. To generate optically clear media that contains C. difficile toxins, we grew up C. difficile for 24 h in BHIS and then transferred to anaerobic Fluorobrite DMEM and incubated for an additional 24 h (for a total of 48 h). To ensure that our optically clear “conditioned media” contained toxins and to compare it with the standard BHIS method, we performed toxin ELISAs. Supernatants from these two methods (BHIS for 48 h vs. BHIS for 24 h and Fluorobrite DMEM for 24 h) were collected, sterile filtered, and examined by ELISA. ELISAs were performed according to the manufacturers’ protocols. Plates were read at 450 nm in a Synergy H1 Microplate Reader (Bio-Tek Instruments). The optical densities (ODs) were used to calculate concentrations according to the manufacturers’ instructions.
Whole genome alignments of distinct C. difficile genomes.
Publicly available genome sequences of four C. difficile strains (3 complete and 1 draft sequence) used in this study were downloaded from NCBI and whole genome fasta files aligned and visualized with progressive Mauve (31) using default settings. The Newick guide tree output was visualized using iTOL (Letunic I and Bork P. 2019. NAR doi:10.1093/nar/gkz239). GenBank assembly accession numbers for each strain are R20291 (GCA_000027105.1), CD630 (GCA_000009205.2), M68 (GCA_000210395.1), and cd37 (GCA_900166435.1).
Data availability statement.
The raw data supporting the conclusions of this manuscript will be made available by the authors upon request.
Statistical analysis.
All graphs were generated using GraphPad Prism software (version 8; GraphPad). Comparisons were made with either one-way ANOVA or repeated-measures ANOVA with the Holm-Sidak post hoc test. The data are presented as means ± SD, with P < 0.05.
RESULTS
Optimization of C. difficile toxins for live-cell imaging.
The virulence of C. difficile strains is primarily attributed to the presence of clostridial toxins. To examine the effects of C. difficile-produced toxin on epithelial cells, we selected C. difficile strains with unique toxin profiles: 1) C. difficile M68 (ribotype 017), which produces only toxin B; 2) C. difficile 630 (ribotype 012), which produces toxin A and B; 3) C. difficile R20291 (ribotype 027), which produces toxin A, toxin B, and binary toxin; and 4) C. difficile 37, which is nontoxigenic (Fig. 1, A and B). In traditional toxin studies, C. difficile is cultured for 48 h in a rich medium such as brain-heart-infusion broth supplemented with yeast extract and cysteine (BHIS). However, this rich medium contains numerous autofluorescent compounds that are not suitable for live-cell fluorescence imaging. Therefore, we modified C. difficile toxin production in an optically clear medium by a two-step culturing process. Strains were initially cultured anaerobically for 24 h in BHIS broth, and then cells were resuspended in an equal volume of anaerobic prewarmed Fluorobrite DMEM and incubated for additional 24 h to secrete toxins into the optically clear medium (see methods). The sterile supernatant was termed “C. difficile conditioned Fluorobrite.” We observed similar quantities of toxin production by ELISA in C. difficile 630, R20291, and M68 in BHIS (48 h) and Fluorobrite (24 h BHIS, 24 h Fluorobrite), with no toxin production by the nontoxigenic control strain 37 (Fig. 2A). Importantly, Fluorobrite-conditioned medium exhibited low autofluorescence (data not shown). We also examined cell viability by membrane staining and live/dead cell staining (Fig. 2, B and C). We found that all C. difficile strains maintained their membrane integrity, as denoted by FM Lipophilic Styryl dye. All strains exhibited >40% viability by fluorescence measurements using live/dead cell staining of C. difficile in BHIS (48 h) and Fluorobrite DMEM (24 h BHIS, 24 h Fluorobrite). These data establish Fluorobrite DMEM cultures as a suitable method to generate optically clear C. difficile-derived toxin solutions for downstream analysis.
Fig. 1.

Clostridioides difficile strains exhibit differences in their genome and toxin genes. A, left: phylogenetic tree generated from a concatenated nucleotide sequence alignment of C. difficile strains R20291 (ribotype 027), 630 (ribotype 012), M68 (ribotype 017), and nontoxic Cd37 core genes. Horizontal bar at the base (scale tree) represents 0.001 substitutions per nucleotide site. A, right: genome alignment of C. difficile strains complete genomes (single assembled contig) was performed using progressive Mauve. B, left: phylogenetic tree. B, right: toxin genes were examined in each strain and graphically represented by arrows.
Fig. 2.

Clostridioides difficile strains secrete toxins, maintain shape and viability after incubation in Fluorobrite DMEM. A: toxin A and B ELISAs with C. difficile strains 37, M28, 630, and R20291 after 48 h in BHIS or 24-h incubation in BHIS and an additional 24 h incubation in Fluorobrite DMEM. ELISAs were examined on a plate reader at optical density (OD) of 450 nm. B: FM 4–64 membrane staining in C. difficile 37, M28, 630, and R20291 after 24-h incubation with BHIS and an additional 24 h incubation in Fluorobrite DMEM. Scale bar = 50 μm. C: cell viability as determined by BacLight live/dead cell staining. Cell staining was examined at excitation: 485 nm/emission: 530 nm (live) and excitation: 485 nm/emission: 630 nm (dead) on a Synergy H1 Microplate Reader, and viabilities were calculated by dividing the fluorescence intensity of 485/530 by the fluorescence intensity at 485/630 × 100%.
Human intestinal enteroids derived from the jejunum respond to C. difficile toxins over time.
There is a gap in knowledge regarding the interaction of C. difficile toxins and the small intestinal epithelium. Therefore, we sought to characterize the cytoskeletal response of jejunal HIEs to C. difficile toxins, an infection site that has been previously documented (12, 92, 110, 142). To visualize cytoskeletal actin rearrangements, we created HIEs stably expressing LifeAct-Ruby, which marks F-actin with the red fluorescent protein mRuby, using lentivirus transduction (129). We then monitored the responses of HIE monolayers to C. difficile-conditioned Fluorobrite DMEM by live-cell, time-lapse imaging (Fig. 3, A–C). Cell rounding was defined as a 50% decrease in cell diameter. As expected, no cell rounding was observed in media controls or Fluorobrite-conditioned media from nontoxigenic CD37. Cell rounding was observed for all toxigenic strains, with significant rounding occurring 6–10 h posttoxin addition. Incubation with conditioned media from hypervirulent (TcdA-, TcdB-, and CDT-producing) R20291 induced cell rounding at 6.75 h. Interestingly, incubation with conditioned media from TcdB-producing M68 and TcdA- and TcdB-producing 630 induced cell rounding at ∼10 h (Fig. 3, A and B). To better capture the cellular responses over the entire 16 h of treatment, the area under the curve of the cell diameter was calculated (Fig. 3C), indicating that HIEs from the small intestine are sensitive to C. difficile-derived toxins.
Fig. 3.

Human intestinal enteroids (HIEs) exhibited delayed cell rounding in response to toxigenic Clostridioides difficile strains. HIEs derived from the jejunum were transduced with the LifeAct-Ruby sensor, which labels F-actin with red fluorescent protein. Cell rounding was visualized by live cell microscopy on a Nikon TiE with a ×20 Plan Apo (NA 0.75) differential interference contrast objective, using a SPECTRA X LED light source and ORCA-Flash 4.0 sCMOS camera (scale bar, 50 µm). A: representative images of LifeAct-Ruby HIEs over time (1–16 h) after exposure to Fluorobrite DMEM medium alone or conditioned Fluorobrite from C. difficile strains CD37, M68, 630, or R20291. Insets indicate significant rounding occurs with toxigenic C. difficile strains (M68, 630, and R20291), but this cell rounding occurs at later time points. B: FIJI (formerly ImageJ) software was used to define cell membranes (as denoted by actin labeling) and cell diameter over time. C: resulting curves were assessed for the area under the curve (AUC), which demonstrates decreased cell diameter with M68, 630, and R20291. *P < 0.05, 1-way ANOVA; n = 3 independent experiments, n = 6/experiment. NS, not significant.
Next, we queried HIE response to a range of concentrations of purified toxins (Fig. 4). Cell rounding was observed within 4 h after the addition of 1 μg/mL TcdA (Fig. 4, A–C). However, lower doses of TcdA required longer incubation times to induce similar rounding. For example, 0.1 μg/mL TcdA achieved cell rounding at 8 h, and 0.001 μg/mL TcdA required 10 h for cell rounding. No cell rounding was observed at 1 ng/mL TcdA or below. We also examined the responsiveness of HIEs to purified TcdB and observed a similar dose-dependent response at the higher concentrations (Fig. 5, A–C). Similar to the HIE response to TcdA, cell rounding was observed at 4 h with 1 μg/mL TcdB. However, no cell rounding was observed with 0.01 μg/mL TcdB, indicating that the HIEs are more sensitive to TcdA than TcdB at low concentrations.
Fig. 4.

Human intestinal enteroids (HIEs) are less responsive than Vero cells to purified Clostridioides difficile toxin A: HIEs derived from jejunum were transduced with the LifeAct-Ruby sensor, which labels F-actin with red fluorescent protein. Cell rounding was visualized by live-cell microscopy on a Nikon TiE with a ×20 Plan Apo (NA 0.75) differential interference contrast objective, using a SPECTRA X LED light source and ORCA-Flash 4.0 sCMOS camera (scale bar, 50 µm). A: representative images of LifeAct-Ruby HIEs over time (1–16 h) after exposure to Fluorobrite DMEM medium alone or purified TcdA (List Biologicals) in Fluorobrite DMEM. Insets indicate that significant rounding occurs with TcdA as low as 0.01 μg/mL (10 ng/mL). Similar to the C. difficile strains, cell rounding in response to purified toxins occurs at later time points. B: FIJI (formerly ImageJ) software was used to define cell membranes (as denoted by actin labeling) and cell diameter over time. C: resulting curves were assessed for the area under the curve (AUC), which demonstrates decreased cell diameter with TcdA. *P < 0.05, 1-way ANOVA; n = 3 independent experiments, n = 6/experiment.
Fig. 5.

Human intestinal enteroids (HIEs) are less responsive than Vero cells to purified Clostridium difficile toxin B. HIEs derived from jejunum were transduced with the LifeAct-Ruby sensor, which labels F-actin with red fluorescent protein. Cell rounding was visualized by live-cell microscopy on a Nikon TiE with a ×20 Plan Apo (NA 0.75) differential interference contrast objective, using a SPECTRA X LED light source and ORCA-Flash 4.0 sCMOS camera (scale bar, 50 µm). A: representative images of LifeAct-Ruby HIEs cell over time (1–16 h) after exposure to Fluorobrite DMEM medium alone or purified TcdB (List Biologicals) in Fluorobrite DMEM. Insets indicate significant rounding occurs with TcdB as low as 0.1 μg/mL (100 ng/mL). Similar to the C. difficile strains, cell rounding in response to purified toxins occurs at later time points. B: FIJI (formerly ImageJ) software was used to define cell membranes (as denoted by actin labeling) and cell diameter over time. C: resulting curves were assessed for the area under the curve (AUC), which demonstrates decreased cell diameter with TcdB treatment. *P < 0.05, 1-way ANOVA; n = 3 independent experiments, n = 6/experiment.
Vero cells respond rapidly to C. difficile toxins.
The immortalized monkey kidney Vero cell line is a classic model for C. difficile toxin activity. To directly compare the cell-rounding results in jejunal HIEs and confirm toxin activity, we generated Vero expressing with LifeAct-Ruby and examined changes in cell morphology by live-cell time lapse imaging. Consistent with our HIE model, we detected cell rounding of LifeAct-labeled Vero cells with C. difficile 630-, R20291-, and M68-conditioned Fluorobrite DMEM and no cell rounding with the nontoxigenic C. difficile 37-conditioned Fluorobrite DMEM (Fig. 6, A and B). In contrast to HIEs, cell rounding was observed by 1 h of incubation with toxigenic strains. Area under the curve calculations revealed similar cell rounding rates for the three toxigenic C. difficile strains (Fig. 6C). To quantitatively assess toxin activity and to compare sensitivity, a range of concentrations of purified TcdA and TcdB were applied to Vero cells (Fig. 7). The threshold concentration to produce cell rounding was 100 ng/mL for TcdA, whereas TcdB produced cell rounding with concentrations as low as 10 pg/mL within 4 h (Fig. 7, A–C). Overall, Vero cells exhibited 1,000 times greater sensitivity to TcdB than TcdA, whereas HIEs were 10 times more sensitive to TcdA than TcdB. Furthermore, differences in were found in cell morphology between HIEs and Vero cells after exposure to C. difficile R20291-conditioned Fluorobrite DMEM, as seen by scanning electron microscopy (SEM) (Fig. 8). These findings highlight key differences between our small intestinal HIEs and the classically used Vero cell model, suggesting that HIEs may serve as an important tool in the investigation of CDI-enteritis.
Fig. 6.

Toxigenic Clostridioides difficile strains cause rapid cell rounding in monkey kidney fibroblast Vero cells. Vero cells were transduced with the LifeAct-Ruby sensor, which labels F-actin with red fluorescent protein. Cell rounding was visualized by live cell microscopy on a Nikon TiE with a ×20 Plan Apo (NA 0.75) differential interference contrast objective, using a SPECTRA X LED light source and ORCA-Flash 4.0 sCMOS camera (scale bar, 50 µm). A: representative images of LifeAct-Ruby Vero cells over time (1–4 h) after exposure to Fluorobrite DMEM medium alone or conditioned Fluorobrite from C. difficile strains CD37, M68, 630, or R20291. Insets indicate significant rounding occurs with toxigenic C. difficile strains (M68, 630, and R20291). B: FIJI (formerly ImageJ) software was used to define cell membranes (as denoted by actin staining) and cell diameter over time. C: resulting curves were assessed for the area under the curve, which demonstrates decreased cell diameter with M68, 630, and R20291. *P < 0.05, 1-way ANOVA; n = 3 independent experiments, n = 6/experiment.
Fig. 7.

Purified toxin A (TcdA) and B (TcdB) cause rapid cell rounding in monkey kidney fibroblast Vero cells. LifeAct-Ruby-transduced Vero cells were incubated with varying concentrations of purified TcdA or TcdB (List Biologicals). Cell rounding was visualized by live-cell microscopy on a Nikon TiE with ×20 Plan Apo (NA 0.75) differential interference contrast objective, using a SPECTRA X LED light source and ORCA-Flash 4.0 sCMOS camera (scale bar = 50 µm). A: representative images of LifeAct-Ruby Vero cells 4 h after exposure to Fluorobrite DMEM medium alone or purified toxins in Fluorobrite DMEM. FIJI (formerly ImageJ) software was used to define cell membranes (as denoted by actin staining) and cell diameter over time for TcdA (B) and TcdB (C). Area under the curve (AUC) analysis of TcdA (D) and TcdB (E). One-way ANOVA; n = 3 independent experiments, n = 6/experiment.
Fig. 8.

Vero cell and human intestinal enteroids (HIEs) exhibit distinct morphologies following Clostridioides difficile R20291 toxin exposure by scanning electron microscopy (SEM). SEM was performed on Vero and HIE monolayers exposed to C. difficile R20291 Fluorobrite DMEM for 16 h. Images depict increasing magnification for both cell types, ending with a 10-μm scale.
HIEs have increased expression of TcdA receptor genes.
To investigate the cause of the phenotypic differences between HIEs and Vero cells, we examined the expression of toxin receptors in HIEs by RNAseq. C. difficile toxin receptor binding has been characterized by multiple groups. TcdA binds to sucrase-isomaltase (SI) (125, 126) and glycoprotein 96 (GP96) (112). TcdB binds to the glycoproteins Frizzled receptors (FZD), CSPG4, and PVRL3 (26, 61, 98, 141), and binary toxin (CDT) binds to LSR (68, 119, 120). Transcriptional analysis revealed the most abundantly expressed receptors in the jejunum were GP96, SI, and LSR (Fig. 9A). In contrast, TcdB-associated receptor transcripts were expressed at lower levels in jejunal HIEs, with the notable exception of FZD5 and PVRL3 (Fig. 9A). We speculate that the elevated levels of TcdA receptor expression may explain in part why jejunal HIEs are more sensitive to TcdA than TcdB.
Fig. 9.

Human intestinal enteroids (HIEs) express higher levels of toxin receptor mRNA than cell lines. A: RNA-seq of HIEs (untreated) revealed a unique profile of toxin receptors. We examined binary toxin (CDT) receptor LSR, TcdA receptor Gp96, sucrase isomaltase (SI), and TcdB receptors [CSPG4, PVRL3, and Frizzled genes (FZD)]. Heat maps were generated from FPKM (fragments per kilobase of transcript per million mapped reads). B: quantitative PCR (qPCR) analysis of HIEs, Vero, and cancer-derived HeLa, T84, and HT29 cells revealed high expression of toxin receptors in HIEs. Data represented as ΔΔCT normalized to 18S. *P < 0.05, 1-way ANOVA; n = 3 independent experiments, n = 6/experiment.
To confirm our HIE RNA-seq data and directly compare toxin receptor expression in commonly used in vitro models, we performed quantitative PCR on a subset of toxin receptors in HIE, Vero, HeLa, T84, and HT29 cells (Fig. 9B). Surprisingly, we found that HIEs expressed significantly greater quantities of FZD1, FZD7, PVRL3, GP96, and LSR compared with all cell lines. No differences were observed between HIEs and cell lines in the expression of TcdB receptor CSPG4. The qPCR and cell-rounding experiment results indicate that HIEs paradoxically express greater quantities of toxin receptor transcripts compared with cell lines, but exhibit reduced sensitivity to toxins, suggesting that another component present in HIE culture may be responsible for the delayed cell-rounding phenotype.
Epithelial mucin, MUC2, delays toxin-induced cell rounding.
One important difference between HIEs and other cell culture models is the heterogeneity of cell types present. HIEs derived from the jejunum are known to express enteroendocrine cells, Paneth cells, stem cells, enterocytes, and goblet cells (24, 46, 132). Notably, goblet cells synthesize and secrete MUC2, a mucin protein that is heavily covered with glycan structures that resemble epithelial surface receptors. TcdA and TcdB have been shown to bind extensively to glycan structures, particularly ones that terminate in galactose, fucose, and sialic acid (63). These structures are also commonly found in mucin glycans. These findings suggest that mucin glycans could act as decoys and delay the adherence of toxins to cell surface receptors. Previous in vitro studies have found results consistent with this mechanism. For example, the presence of mucus in mucus-producing HT29-MTX cells correlated with decreased markers of C. difficile toxin-induced inflammation and necrosis compared with non-mucus-producing parental HT29 cells (34, 47, 80, 117). Therefore, we hypothesized that goblet cells within the HIE culture produce, potentially delaying adherence of toxins to cell surface receptors.
To investigate our hypothesis, we examined transcript levels of known goblet cell markers in jejunal HIEs. RNA-seq analysis revealed significant expression of many goblet cell markers (AGR2, CDX2, MEP1B, FCGBP, KLK1) and the mucin protein MUC2 (Fig. 10A). We validated mucin production through immunostaining, which revealed MUC2-positive goblet cells present in HIE monolayers in differentiated culture. These mucins may interfere with C. difficile toxin binding to epithelial cell receptors (Fig. 10B). To test whether mucins could delay the onset of cell rounding, we preincubated TcdA (0.01 μg/mL) or TcdB (0.001 μg/mL) for 30 min with human-derived MUC2 before adding to Vero cells. We observed delayed onset of cell rounding in Vero cells, with no visible rounding at 30 min, with the MUC2-incubated toxin treatments. This is in contrast to the distinct cell rounding observed in Vero cells treated with media control or methylcellulose, a polysaccharide that mirrors the viscosity of mucus (83). These results support that MUC2 has a critical role in limiting C. difficile toxin-epithelial interactions and highlights key differences between HIEs and Vero cells and traditional cell lines. Based on these studies, we propose that HIEs are a robust system in which to investigate the interactions of C. difficile toxins with the small intestinal epithelium.
Fig. 10.

Human intestinal enteroids (HIEs) secrete mucin 2 (MUC2), which acts as a decoy for toxin A (TcdA) and B (TcdB). A: RNA-seq analysis of goblet cell markers anterior gradient 2 (AGR2), caudal-related homeobox protein 2 (CDX2), meprin β (MEP1B), Fc-γ binding protein (FCGBP), kallikrein-1 (KLK-1), and MUC2 in HIEs (untreated). Data expressed as FPKM (fragments per kilobase of transcript per million mapped reads). B: MUC2 immunostaining in HIE monolayers demonstrates MUC2-positive goblet cells. C: decoy assays in Vero cells. Purified TcdA (10 ng) and TcdB (1 ng) were preincubated with 1 mg/mL methylcellulose or 1 mg/mL human MUC2 derived from LS174T goblet-like cells. Cell rounding was observed by differential interference contrast (DIC) microscopy at 30 min on a Nikon TiE with ×20 Plan Apo (NA 0.75) differential interference contrast objective, using a SPECTRA X LED light source and ORCA-Flash 4.0 sCMOS camera. Scale bar, 100 μm; n = 3 independent experiments, n = 3/experiment.
DISCUSSION
In this article, we report the use of jejunal human intestinal enteroids (HIEs) and live-cell time-lapse imaging to visualize the cytotoxicity kinetics of C. difficile toxins on the small intestinal epithelium as a model of CDI-enteritis. This characteristic cellular rounding morphology has been used as a proxy for C. difficile toxin activity in numerous cell culture models, with Vero cells being the most widely adopted in pathogenicity studies (108, 140, 150). We observed time-dependent cell rounding of HIEs, a typical morphological event associated with the actin cytoskeleton disruption key for C. difficile-induced disease. We demonstrate that cell rounding in HIEs requires both increased concentrations of toxins and longer incubation time when compared with Vero cells in the same conditions despite having higher mRNA levels of several known toxin receptors. This study establishes a physiologically relevant small intestinal model to interrogate epithelial-toxin interactions and understand CDI-induced enteritis pathophysiology.
Although the pathophysiology of C. difficile enteritis is not well understood, CDI enteritis in patients appears to mirror their colonic disease. Similar to colonic infection, antibiotic use, advanced age, immunocompromised states, and IBD predispose patients to CDI of the small intestine (54, 65, 71, 92, 110, 148, 152, 158). Additionally, the histological findings of CDI enteritis resemble that of the colon, with loss of epithelial integrity, areas of necrosis, neutrophil infiltration, and inflammation (54, 65, 71, 92, 148, 152, 157). Similar to the colon, infection of the small intestine may also be facilitated the presence of bile acids (128). Studies have shown taurocholate and cholate, bile acids that serve as germinants for C. difficile spores, are present at high concentrations in the small intestine under normal conditions (144). Antibiotic use, a common prerequisite of CDI, is known to profoundly alter both the gut microbiota and increase the concentration of bile acids in the small intestine (96, 134, 144, 153). Importantly, Theriot et al. (144) demonstrated that antibiotics that promote CDI significantly elevate unconjugated bile acids in the small intestine above the already high baseline level. In principle, C. difficile could use primary bile acids in the small intestine, germinate, and cause small intestinal disease.
Although infection of the small and large bowel has been documented, the sensitivity of the small and large intestine to C. difficile toxins appears to be different. Although both TcdA and TcdB are potent cytotoxins, TcdA possesses greater enterotoxic effects in rodent and rabbit small intestine than TcdB (147). Injection of TcdA into rat and rabbit intestinal loops causes mucosal damage, increased mucosal permeability, fluid secretion, and release of inflammatory mediators (19–21, 85, 147). In pigs, TcdA was observed to have more robust binding in the villi of the ileum than in the colon (84). These data suggest that the small intestine is highly sensitive and responsive to TcdA. This is consistent with our observations that jejunal HIEs are more sensitive to TcdA than TcdB. We also observed that the jejunal HIEs express higher levels of known TcdA receptors than TcdB receptors, which might explain why the small intestine is more sensitive to TcdA. Interestingly, human intestinal organoids (HIOs) generated from pluripotent stem cells, which appear to resemble the human small intestine more than the colon, also exhibit higher sensitivity to TcdA than TcdB (102).
In lieu of colonic epithelial cells, several studies have implemented human colon explants to dissect the sensitivity of human colonic cells to C. difficile toxins. In human colonic explants, it has been observed that both TcdA and TcdB impair tight-junction function and increase paracellular permeability (90, 130). However, TcdB elicited greater epithelial damage in colon explants than TcdA (90, 130, 131). These findings may represent key differences between the intestinal regions. Based on these studies, we hypothesize that the human colonic enteroids will express more TcdB receptors and will be more sensitive to TcdB than TcdA. In the future, studies using colonic HIEs would help shed light on the differences between CDI-induced enteritis and colitis. Our data suggest that enteritis may be influenced by expression of TcdA in C. difficile strains. In the future, it would be beneficial to note the toxin type of the C. difficile strains that are observed in CDI enteritis. Additionally, it would be interesting to determine which receptors the toxins preferentially bind. Based on our RNA-seq data, we would predict that TcdA would bind highly to GP96 and TcdB would bind highly to FZD5, FZD7, and PVRL3 in the small intestine. However, additional studies are needed to understand toxin binding in the small intestine.
Importantly, these studies have identified the major nonanchored intestinal mucin MUC2 as playing a critical protective role in the inhibition of C. difficile toxin interactions with the small intestinal cell surface. Mucus is composed primarily of mucin proteins that anchor high molecular weight glycoproteins forming an entangled network. In the intestine, MUC2 is the major gel-forming mucin (74), although goblet cells can also express MUC5AC during nematode infection (64). The mucin protein backbone is rich in repeats of serine and threonine, and these domains are heavily glycosylated with oligosaccharides. These oligosaccharides contain N‐acetylglucosamine (GlcNAc), N‐acetylgalactosamine (GalNAc), galactose, fucose, and sialic acid (10). These oligosaccharide chains can contain up to 19 sugar molecules and account for 80% of the mucin molecular weight (10). The sialic acid residues and carbohydrate‐bound ester sulphates provide mucus with a net negative charge (29). Consequently, mucus forms a steric barrier to luminal contents, limiting the free diffusion of components within and through the mucus. Furthermore, mucus represents a dynamic barrier due to its continuous secretion from goblet cells.
Previous studies have identified that TcdA and TcdB bind to glycoproteins and recognize several glycan structures (26, 63, 93, 112, 141). C. difficile toxin A and B both possess carbohydrate-binding sites that can bind to human milk oligosaccharides, compounds which mimic select mucin glycans (41, 42). In fact, the presence of mucus has been associated with decreased inflammation and necrosis in the presence of C. difficile and its toxins (34, 47, 117), indicating the mucus could prevent toxin injury to some degree. Studies have also shown that compounds that increase mucus production or bolster the mucus layer, for example, exogenous phosphatidylcholine (PC) administration, decrease epithelial TcdA uptake and lessen proinflammatory cytokine production (34). Consistent with these studies, our data confirm that human MUC2 can act as a toxin decoy and delay cell rounding when added to Vero cells, indicating a critical role of MUC2 glycans in limiting toxin effects. Although we have not examined supplementing HIES with exogenous MUC2, we speculate that there would be a delay in rounding similar to Vero cells.
Our immunostaining data reveal MUC2-positive goblet cells in enteroid monolayers, which we believe is critical for delaying toxin effects. It should be noted that the multiple washes required for immunostaining and that lack of stool, which normally compresses the mucus layer for optimal staining, likely removes the secreted mucus layer since it is not attached firmly to the epithelium. We believe that in the undisturbed state, the mucus covers the monolayers and provides a barrier which limits toxin-epithelial interactions. Although our HIE live-imaging system was static (i.e., no flow), peristalsis in the intestine or even CDI-induced diarrhea may serve to wash away toxins bound to mucin glycans, thereby limiting the severity of infection. This might represent a possible host mechanism for protection.
In addition to using the novel LifeAct-Ruby HIE as a small intestinal model, this work also optimized a system for examining C. difficile produced toxins suitable for live-cell imaging. We demonstrate that C. difficile can secret toxins in Fluorobrite DMEM, generating an optically clear solution for live-cell imaging applications. Culture media are known to influence the composition of C. difficile toxin production (11). When C. difficile is grown in rich medium, toxin gene expression occurs when the cells enter stationary phase and nutrient limitation, or accumulation of growth inhibiting substances occurs (38, 70, 81). It has been demonstrated that glucose and biotin starvation drive toxin production (38, 70, 81). In contrast, the presence of metabolizable carbon sources or certain amino acids inhibits toxin production (79, 81, 82). By transferring C. difficile from BHIS to Fluorobrite, we are still supporting C. difficile viability but not providing enough nutrients to inhibit toxin production. Our data demonstrate that C. difficile secretes toxins A and B in Fluorobrite and maintains >40% cell viability for all tested C. difficile strains. Of the strains we tested, we found that C. difficile 630 had the highest viability (∼60%) after incubation in BHIS, then Fluorobrite DMEM. C. difficile 630 has been shown to be more resistant to cell stress (ethanol, atmospheric oxygen, and hydrogen peroxide) when compared with other C. difficile strains (39). We speculate that the ability of C. difficile 630 to deal with cell stress results in an increased survival in our Fluorobrite DMEM compared with other C. difficile strains.
Although our use of nontoxigenic C. difficile-conditioned media and purified toxins points to the role of toxins in mediating cell rounding, it would be useful in the future to include antitoxin antibodies as controls. The use of antitoxin antibodies would confirm that toxigenic C. difficile strains are not producing other metabolites that may also modulate the actin cytoskeleton. Using our conditioned media system, we can address these important questions by fluorescent imaging. In total, we believe these studies provide foundational insight into the potential of HIEs generated from the small intestine as a model to study the mechanism of CDI-enteritis pathogenicity and that future studies may provide insights to inform future therapeutic interventions of this deadly condition.
GRANTS
This work was supported by grants from the National Institute of Diabetes and Digestive and Kidney Diseases (Grant P30-DK-56338 to Texas Medical Center Digestive Disease Center, Gastrointestinal Experimental Model Systems), a Young Investigator Pilot Award (to M. A. Engevik) and NIH P30-DK-56338 (to M. A. Engevik), National Institutes of Health (NIH) Grant F32AI136404 (to H. A. Danhof), NIH Grant F30-DK-112563 (to A. L. Chang-Graham), the BCM Medical Scientist Training Program (to A. L. Chang-Graham), NIH Grant T32-DK-07644 (to K. A. Engevik), Pediatric GI T32-DK-007664, NIH NIAID Grant U01-AI-124290 (to K. W. Garey and R. A. Britton), NIH Grants R01-DK-103759 and R01-AI-123278 (to R. A. Britton), and unrestricted research support from BioGaia AB (Stockholm, Sweden) (to R. A. Britton and J. Versalovic).
DISCLOSURES
J. Versalovic serves on the scientific advisory boards of Biomica, Plexus Worldwide and Seed Health, and J. Versalovic and R.A. Britton receive unrestricted research support from BioGaia, AB. R.A. Britton serves on the scientific advisory board of Tenza, is a cofounder of Mikrovia, and consults for Takeda and Probiotech. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
AUTHOR CONTRIBUTIONS
M.A.E. and H.A.D. conceived and designed research; M.A.E., A.L.C.-G., J.K.S., K.A.E., B.H., and B.T.E. performed experiments; M.A.E., J.K.S., and K.A.E. analyzed data; M.A.E., K.W.G., J.M.H., R.A.B., and J.V. interpreted results of experiments; M.A.E. prepared figures; M.A.E. drafted manuscript; M.A.E., H.A.D., A.L.C.-G., J.K.S., K.A.E., B.H., B.T.E., K.W.G., J.M.H., R.A.B., and J.V. edited and revised manuscript; J.V. approved final version of manuscript.
REFERENCES
- 1.Abid H, Bischof E. An unusual presentation of severe sepsis due to Clostridium difficile enteritis. Cureus 11: e4162, 2019. doi: 10.7759/cureus.4162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anderson DM, Sheedlo MJ, Jensen JL, Lacy DB. Structural insights into the transition of Clostridioides difficile binary toxin from prepore to pore. Nat Microbiol 5: 102–107, 2020. doi: 10.1038/s41564-019-0601-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Atkinson MH, McLeod BD. Reactive arthritis associated with Clostridium difficile enteritis. J Rheumatol 15: 520–522, 1988. [PubMed] [Google Scholar]
- 4.Aujla AK, Averbukh LD, Potashinsky A, Rossi L. A rare case of Clostridium difficile enteritis: a common bug in an uncommon place. Cureus 11: e4519, 2019. doi: 10.7759/cureus.4519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barbieri JT, Riese MJ, Aktories K. Bacterial toxins that modify the actin cytoskeleton. Annu Rev Cell Dev Biol 18: 315–344, 2002. doi: 10.1146/annurev.cellbio.18.012502.134748. [DOI] [PubMed] [Google Scholar]
- 6.Beal EW, Bass R, Harzman AE. Two patients with fulminant Clostridium difficile enteritis who had not undergone total colectomy: a case series and review of the literature. Case Rep Surg 2015: 1–5, 2015. doi: 10.1155/2015/957257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bhargava A, Clifton MS, Mhaske P, Liao M, Pothoulakis C, Leeman SE, Grady EF. Local injection of dsRNA targeting calcitonin receptor-like receptor (CLR) ameliorates Clostridium difficile toxin A-induced ileitis. Proc Natl Acad Sci USA 110: 731–736, 2013. [Erratum in: Proc Natl Acad Sci USA 110: 3651, 2013.] 10.1073/pnas.1219733110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bhattacharya MK, Niyogi SK, Rasaily R, Bhattacharya SK, Dutta P, Nag A, Pal SC. Clinical manifestation of Clostridium difficile enteritis in Calcutta. J Assoc Physicians India 39: 683–684, 1991. [PubMed] [Google Scholar]
- 9.Bignardi GE. Risk factors for Clostridium difficile infection. J Hosp Infect 40: 1–15, 1998. doi: 10.1016/S0195-6701(98)90019-6. [DOI] [PubMed] [Google Scholar]
- 10.Boegh M, Nielsen HM. Mucus as a barrier to drug delivery–understanding and mimicking the barrier properties. Basic Clin Pharmacol Toxicol 116: 179–186, 2015. doi: 10.1111/bcpt.12342. [DOI] [PubMed] [Google Scholar]
- 11.Boetzkes A, Felkel KW, Zeiser J, Jochim N, Just I, Pich A. Secretome analysis of Clostridium difficile strains. Arch Microbiol 194: 675–687, 2012. doi: 10.1007/s00203-012-0802-5. [DOI] [PubMed] [Google Scholar]
- 12.Boey CC, Ramanujam TM, Looi LM. Clostridium difficile-related necrotizing pseudomembranous enteritis in association with Henoch-Schonlein purpura. J Pediatr Gastroenterol Nutr 24: 426–429, 1997. doi: 10.1097/00005176-199704000-00012. [DOI] [PubMed] [Google Scholar]
- 13.Boku S. [A role of platelet activating factor in experimental hemorrhagic enteritis induced by Clostridium difficile toxin]. Nihon Shokakibyo Gakkai Zasshi 91: 1407–1414, 1994. [PubMed] [Google Scholar]
- 14.Boland E, Thompson JS. Fulminant Clostridium difficile enteritis after proctocolectomy and ileal pouch-anal anastamosis. Gastroenterol Res Pract 2008: 1–5, 2008. doi: 10.1155/2008/985658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Borriello SP. Pathogenesis of Clostridium difficile infection. J Antimicrob Chemother 41, Suppl C: 13–19, doi: 10.1093/jac/41.suppl_3.13. 1998. [DOI] [PubMed] [Google Scholar]
- 16.Borriello SP, Ketley JM, Mitchell TJ, Barclay FE, Welch AR, Price AB, Stephen J. Clostridium difficile—a spectrum of virulence and analysis of putative virulence determinants in the hamster model of antibiotic-associated colitis. J Med Microbiol 24: 53–64, 1987. doi: 10.1099/00222615-24-1-53. [DOI] [PubMed] [Google Scholar]
- 17.Branka JE, Vallette G, Jarry A, Bou-Hanna C, Lemarre P, Van PN, Laboisse CL. Early functional effects of Clostridium difficile toxin A on human colonocytes. Gastroenterology 112: 1887–1894, 1997. doi: 10.1053/gast.1997.v112.pm9178681. [DOI] [PubMed] [Google Scholar]
- 18.Britton RA, Young VB. Interaction between the intestinal microbiota and host in Clostridium difficile colonization resistance. Trends Microbiol 20: 313–319, 2012. doi: 10.1016/j.tim.2012.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Castagliuolo I, Keates AC, Qiu B, Kelly CP, Nikulasson S, Leeman SE, Pothoulakis C. Increased substance P responses in dorsal root ganglia and intestinal macrophages during Clostridium difficile toxin A enteritis in rats. Proc Natl Acad Sci USA 94: 4788–4793, 1997. doi: 10.1073/pnas.94.9.4788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Castagliuolo I, Kelly CP, Qiu BS, Nikulasson ST, LaMont JT, Pothoulakis C. IL-11 inhibits Clostridium difficile toxin A enterotoxicity in rat ileum. Am J Physiol Gastrointest Liver Physiol 273: G333–G341, 1997. doi: 10.1152/ajpgi.1997.273.2.G333. [DOI] [PubMed] [Google Scholar]
- 21.Castagliuolo I, LaMont JT, Nikulasson ST, Pothoulakis C. Saccharomyces boulardii protease inhibits Clostridium difficile toxin A effects in the rat ileum. Infect Immun 64: 5225–5232, 1996. doi: 10.1128/IAI.64.12.5225-5232.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Causey MW, Spencer MP, Steele SR. Clostridium difficile enteritis after colectomy. Am Surg 75: 1203–1206, 2009. [PubMed] [Google Scholar]
- 23.Chandrasekaran R, Lacy DB. The role of toxins in Clostridium difficile infection. FEMS Microbiol Rev 41: 723–750, 2017. doi: 10.1093/femsre/fux048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chang-Graham AL, Danhof HA, Engevik MA, Tomaro-Duchesneau C, Karandikar UC, Estes MK, Versalovic J, Britton RA, Hyser JM. Human intestinal enteroids with inducible neurogenin-3 expression as a novel model of gut hormone secretion. Cell Mol Gastroenterol Hepatol 8: 209–229, 2019. doi: 10.1016/j.jcmgh.2019.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen P, Tao L, Wang T, Zhang J, He A, Lam KH, Liu Z, He X, Perry K, Dong M, Jin R. Structural basis for recognition of frizzled proteins by Clostridium difficile toxin B. Science 360: 664–669, 2018. doi: 10.1126/science.aar1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chumbler NM, Farrow MA, Lapierre LA, Franklin JL, Lacy DB. Clostridium difficile toxins TcdA and TcdB cause colonic tissue damage by distinct mechanisms. Infect Immun 84: 2871–2877, 2016. doi: 10.1128/IAI.00583-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Clark GF, Krivan HC, Wilkins TD, Smith DF. Toxin A from Clostridium difficile binds to rabbit erythrocyte glycolipids with terminal Gal alpha 1-3Gal beta 1-4GlcNAc sequences. Arch Biochem Biophys 257: 217–229, 1987. doi: 10.1016/0003-9861(87)90561-3. [DOI] [PubMed] [Google Scholar]
- 29.Cone RA. Barrier properties of mucus. Adv Drug Deliv Rev 61: 75–85, 2009. doi: 10.1016/j.addr.2008.09.008. [DOI] [PubMed] [Google Scholar]
- 30.Cowardin CA, Buonomo EL, Saleh MM, Wilson MG, Burgess SL, Kuehne SA, Schwan C, Eichhoff AM, Koch-Nolte F, Lyras D, Aktories K, Minton NP, Petri WA Jr. The binary toxin CDT enhances Clostridium difficile virulence by suppressing protective colonic eosinophilia. Nat Microbiol 1: 16108, 2016. doi: 10.1038/nmicrobiol.2016.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Darling AE, Mau B, Perna NT. progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One 5: e11147, 2010. doi: 10.1371/journal.pone.0011147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Denève C, Janoir C, Poilane I, Fantinato C, Collignon A. New trends in Clostridium difficile virulence and pathogenesis. Int J Antimicrob Agents 33, Suppl 1: S24–S28, 2009. doi: 10.1016/S0924-8579(09)70012-3. [DOI] [PubMed] [Google Scholar]
- 33.Di Bella S, Ascenzi P, Siarakas S, Petrosillo N, di Masi A. Clostridium difficile toxins A and B: insights into pathogenic properties and extraintestinal effects. Toxins (Basel) 8: 134, 2016. doi: 10.3390/toxins8050134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Diebel LN, Liberati DM. Reinforcement of the intestinal mucus layer protects against Clostridium difficile intestinal injury in vitro. J Am Coll Surg 219: 460–468, 2014. doi: 10.1016/j.jamcollsurg.2014.05.005. [DOI] [PubMed] [Google Scholar]
- 35.Dineen SP, Bailey SH, Pham TH, Huerta S. Clostridium difficile enteritis: a report of two cases and systematic literature review. World J Gastrointest Surg 5: 37–42, 2013. doi: 10.4240/wjgs.v5.i3.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Drudy D, Harnedy N, Fanning S, O’Mahony R, Kyne L. Isolation and characterisation of toxin A-negative, toxin B-positive Clostridium difficile in Dublin, Ireland. Clin Microbiol Infect 13: 298–304, 2007. doi: 10.1111/j.1469-0691.2006.01634.x. [DOI] [PubMed] [Google Scholar]
- 37.Dubberke ER, Carling P, Carrico R, Donskey CJ, Loo VG, McDonald LC, Maragakis LL, Sandora TJ, Weber DJ, Yokoe DS, Gerding DN. Strategies to prevent Clostridium difficile infections in acute care hospitals: 2014 update. Infect Control Hosp Epidemiol 35, Suppl 2: S48–S65, 2014. doi: 10.1017/S0899823X00193857. [DOI] [PubMed] [Google Scholar]
- 38.Dupuy B, Sonenshein AL. Regulated transcription of Clostridium difficile toxin genes. Mol Microbiol 27: 107–120, 1998. doi: 10.1046/j.1365-2958.1998.00663.x. [DOI] [PubMed] [Google Scholar]
- 39.Edwards AN, Karim ST, Pascual RA, Jowhar LM, Anderson SE, McBride SM. Chemical and stress resistances of Clostridium difficile spores and vegetative cells. Front Microbiol 7: 1698, 2016. doi: 10.3389/fmicb.2016.01698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Eglow R, Pothoulakis C, Itzkowitz S, Israel EJ, O’Keane CJ, Gong D, Gao N, Xu YL, Walker WA, LaMont JT. Diminished Clostridium difficile toxin A sensitivity in newborn rabbit ileum is associated with decreased toxin A receptor. J Clin Invest 90: 822–829, 1992. doi: 10.1172/JCI115957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.El-Hawiet A, Kitova EN, Kitov PI, Eugenio L, Ng KK, Mulvey GL, Dingle TC, Szpacenko A, Armstrong GD, Klassen JS. Binding of Clostridium difficile toxins to human milk oligosaccharides. Glycobiology 21: 1217–1227, 2011. doi: 10.1093/glycob/cwr055. [DOI] [PubMed] [Google Scholar]
- 42.El-Hawiet A, Kitova EN, Klassen JS. Recognition of human milk oligosaccharides by bacterial exotoxins. Glycobiology 25: 845–854, 2015. doi: 10.1093/glycob/cwv025. [DOI] [PubMed] [Google Scholar]
- 43.El Muhtaseb MS, Apollos JK, Dreyer JS. Clostridium difficile enteritis: a cause for high ileostomy output. ANZ J Surg 78: 416, 2008. doi: 10.1111/j.1445-2197.2008.04494.x. [DOI] [PubMed] [Google Scholar]
- 44.Engevik MA, Engevik KA, Yacyshyn MB, Wang J, Hassett DJ, Darien B, Yacyshyn BR, Worrell RT. Human Clostridium difficile infection: inhibition of NHE3 and microbiota profile. Am J Physiol Gastrointest Liver Physiol 308: G497–G509, 2015. doi: 10.1152/ajpgi.00090.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Engevik MA, Yacyshyn MB, Engevik KA, Wang J, Darien B, Hassett DJ, Yacyshyn BR, Worrell RT. Human Clostridium difficile infection: altered mucus production and composition. Am J Physiol Gastrointest Liver Physiol 308: G510–G524, 2015. doi: 10.1152/ajpgi.00091.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ettayebi K, Crawford SE, Murakami K, Broughman JR, Karandikar U, Tenge VR, Neill FH, Blutt SE, Zeng XL, Qu L, Kou B, Opekun AR, Burrin D, Graham DY, Ramani S, Atmar RL, Estes MK. Replication of human noroviruses in stem cell-derived human enteroids. Science 353: 1387–1393, 2016. doi: 10.1126/science.aaf5211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Eveillard M, Fourel V, Bare MC, Kernéis S, Coconnier MH, Karjalainen T, Bourlioux P, Servin AL. Identification and characterization of adhesive factors of Clostridium difficile involved in adhesion to human colonic enterocyte-like Caco-2 and mucus-secreting HT29 cells in culture. Mol Microbiol 7: 371–381, 1993. doi: 10.1111/j.1365-2958.1993.tb01129.x. [DOI] [PubMed] [Google Scholar]
- 48.Fiorentini C, Donelli G, Nicotera P, Thelestam M. Clostridium difficile toxin A elicits Ca(2+)-independent cytotoxic effects in cultured normal rat intestinal crypt cells. Infect Immun 61: 3988–3993, 1993. doi: 10.1128/IAI.61.9.3988-3993.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fiorentini C, Thelestam M. Clostridium difficile toxin A and its effects on cells. Toxicon 29: 543–567, 1991. doi: 10.1016/0041-0101(91)90050-2. [DOI] [PubMed] [Google Scholar]
- 50.Fleming F, Khursigara N, O’Connell N, Darby S, Waldron D. Fulminant small bowel enteritis: a rare complication of Clostridium difficile-associated disease. Inflamm Bowel Dis 15: 801–802, 2009. doi: 10.1002/ibd.20758. [DOI] [PubMed] [Google Scholar]
- 51.Follmar KE, Condron SA, Turner II, Nathan JD, Ludwig KA. Treatment of metronidazole-refractory Clostridium difficile enteritis with vancomycin. Surg Infect (Larchmt) 9: 195–200, 2008. doi: 10.1089/sur.2006.089. [DOI] [PubMed] [Google Scholar]
- 52.Foulke-Abel J, In J, Kovbasnjuk O, Zachos NC, Ettayebi K, Blutt SE, Hyser JM, Zeng XL, Crawford SE, Broughman JR, Estes MK, Donowitz M. Human enteroids as an ex-vivo model of host-pathogen interactions in the gastrointestinal tract. Exp Biol Med (Maywood) 239: 1124–1134, 2014. doi: 10.1177/1535370214529398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Foulke-Abel J, In J, Yin J, Zachos NC, Kovbasnjuk O, Estes MK, de Jonge H, Donowitz M. Human enteroids as a model of upper small intestinal ion transport physiology and pathophysiology. Gastroenterology 150: 638–649.e8, 2016. doi: 10.1053/j.gastro.2015.11.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Freiler JF, Durning SJ, Ender PT. Clostridium difficile small bowel enteritis occurring after total colectomy. Clin Infect Dis 33: 1429–1431, 2001. doi: 10.1086/322675. [DOI] [PubMed] [Google Scholar]
- 55.Gagandeep D, Ira S. Clostridium difficile enteritis 9 years after total proctocolectomy: a rare case report. Am J Gastroenterol 105: 962–963, 2010. doi: 10.1038/ajg.2009.680. [DOI] [PubMed] [Google Scholar]
- 56.Genth H, Dreger SC, Huelsenbeck J, Just I. Clostridium difficile toxins: more than mere inhibitors of Rho proteins. Int J Biochem Cell Biol 40: 592–597, 2008. doi: 10.1016/j.biocel.2007.12.014. [DOI] [PubMed] [Google Scholar]
- 57.Gerding DN, Johnson S, Rupnik M, Aktories K. Clostridium difficile binary toxin CDT: mechanism, epidemiology, and potential clinical importance. Gut Microbes 5: 15–27, 2014. doi: 10.4161/gmic.26854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gomez-Trevino M, Boureau H, Karjalainen T, Bourlioux P. Clostridium difficile Adherence to Mucus: Results of an in vivo and ex vivo Assay. Microb Ecol Health Dis 9: 329–334, 1996. doi:. [DOI] [Google Scholar]
- 59.Greco A, Ho JG, Lin SJ, Palcic MM, Rupnik M, Ng KK. Carbohydrate recognition by Clostridium difficile toxin A. Nat Struct Mol Biol 13: 460–461, 2006. doi: 10.1038/nsmb1084. [DOI] [PubMed] [Google Scholar]
- 60.Guillemin P, Gerster JC. [Reactive arthritis induced by Clostridium difficile enteritis]. Praxis (Bern 1994) 94: 471–474, 2005. doi: 10.1024/0369-8394.94.12.471. [DOI] [PubMed] [Google Scholar]
- 61.Gupta P, Zhang Z, Sugiman-Marangos SN, Tam J, Raman S, Julien JP, Kroh HK, Lacy DB, Murgolo N, Bekkari K, Therien AG, Hernandez LD, Melnyk RA. Functional defects in Clostridium difficile TcdB toxin uptake identify CSPG4 receptor-binding determinants. J Biol Chem 292: 17290–17301, 2017. doi: 10.1074/jbc.M117.806687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hariri S, Gouin P, Tuech JJ, Veber B, Dureuil B. Clostridium difficile infection causing multiple organ failure and small-bowel enteritis. Clin Res Hepatol Gastroenterol 35: 142–144, 2011. doi: 10.1016/j.clinre.2010.10.004. [DOI] [PubMed] [Google Scholar]
- 63.Hartley-Tassell LE, Awad MM, Seib KL, Scarselli M, Savino S, Tiralongo J, Lyras D, Day CJ, Jennings MP. Lectin activity of the TcdA and TcdB toxins of Clostridium difficile. Infect Immun 87: e00676-18, 2019. doi: 10.1128/IAI.00676-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hasnain SZ, Evans CM, Roy M, Gallagher AL, Kindrachuk KN, Barron L, Dickey BF, Wilson MS, Wynn TA, Grencis RK, Thornton DJ. Muc5ac: a critical component mediating the rejection of enteric nematodes. J Exp Med 208: 893–900, 2011. doi: 10.1084/jem.20102057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hayetian FD, Read TE, Brozovich M, Garvin RP, Caushaj PF. Ileal perforation secondary to Clostridium difficile enteritis: report of 2 cases. Arch Surg 141: 97–99, 2006. doi: 10.1001/archsurg.141.1.97. [DOI] [PubMed] [Google Scholar]
- 66.He D, Sougioultzis S, Hagen S, Liu J, Keates S, Keates AC, Pothoulakis C, Lamont JT. Clostridium difficile toxin A triggers human colonocyte IL-8 release via mitochondrial oxygen radical generation. Gastroenterology 122: 1048–1057, 2002. doi: 10.1053/gast.2002.32386. [DOI] [PubMed] [Google Scholar]
- 67.Hecht G, Pothoulakis C, LaMont JT, Madara JL. Clostridium difficile toxin A perturbs cytoskeletal structure and tight junction permeability of cultured human intestinal epithelial monolayers. J Clin Invest 82: 1516–1524, 1988. doi: 10.1172/JCI113760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hemmasi S, Czulkies BA, Schorch B, Veit A, Aktories K, Papatheodorou P. Interaction of the Clostridium difficile binary toxin CDT and its host cell receptor, lipolysis-stimulated lipoprotein receptor (LSR). J Biol Chem 290: 14031–14044, 2015. doi: 10.1074/jbc.M115.650523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ho JG, Greco A, Rupnik M, Ng KK. Crystal structure of receptor-binding C-terminal repeats from Clostridium difficile toxin A. Proc Natl Acad Sci USA 102: 18373–18378, 2005. doi: 10.1073/pnas.0506391102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hundsberger T, Braun V, Weidmann M, Leukel P, Sauerborn M, von Eichel-Streiber C. Transcription analysis of the genes tcdA-E of the pathogenicity locus of Clostridium difficile. Eur J Biochem 244: 735–742, 1997. doi: 10.1111/j.1432-1033.1997.t01-1-00735.x. [DOI] [PubMed] [Google Scholar]
- 71.Jacobs A, Barnard K, Fishel R, Gradon JD. Extracolonic manifestations of Clostridium difficile infections. Presentation of 2 cases and review of the literature. Medicine (Baltimore) 80: 88–101, 2001. doi: 10.1097/00005792-200103000-00002. [DOI] [PubMed] [Google Scholar]
- 72.Jank T, Aktories K. Structure and mode of action of clostridial glucosylating toxins: the ABCD model. Trends Microbiol 16: 222–229, 2008. doi: 10.1016/j.tim.2008.01.011. [DOI] [PubMed] [Google Scholar]
- 73.Jank T, Giesemann T, Aktories K. Rho-glucosylating Clostridium difficile toxins A and B: new insights into structure and function. Glycobiology 17: 15R–22R, 2007. doi: 10.1093/glycob/cwm004. [DOI] [PubMed] [Google Scholar]
- 74.Johansson ME, Sjövall H, Hansson GC. The gastrointestinal mucus system in health and disease. Nat Rev Gastroenterol Hepatol 10: 352–361, 2013. doi: 10.1038/nrgastro.2013.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jump RL, Pultz MJ, Donskey CJ. Vegetative Clostridium difficile survives in room air on moist surfaces and in gastric contents with reduced acidity: a potential mechanism to explain the association between proton pump inhibitors and C. difficile-associated diarrhea? Antimicrob Agents Chemother 51: 2883–2887, 2007. doi: 10.1128/AAC.01443-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Juntunen M, Kirjavainen PV, Ouwehand AC, Salminen SJ, Isolauri E. Adherence of probiotic bacteria to human intestinal mucus in healthy infants and during rotavirus infection. Clin Diagn Lab Immunol 8: 293–296, 2001. doi: 10.1128/CDLI.8.2.293-296.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Just I, Selzer J, von Eichel-Streiber C, Aktories K. The low molecular mass GTP-binding protein Rho is affected by toxin A from Clostridium difficile. J Clin Invest 95: 1026–1031, 1995. doi: 10.1172/JCI117747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Just I, Wilm M, Selzer J, Rex G, von Eichel-Streiber C, Mann M, Aktories K. The enterotoxin from Clostridium difficile (ToxA) monoglucosylates the Rho proteins. J Biol Chem 270: 13932–13936, 1995. doi: 10.1074/jbc.270.23.13932. [DOI] [PubMed] [Google Scholar]
- 79.Karasawa T, Maegawa T, Nojiri T, Yamakawa K, Nakamura S. Effect of arginine on toxin production by Clostridium difficile in defined medium. Microbiol Immunol 41: 581–585, 1997. doi: 10.1111/j.1348-0421.1997.tb01895.x. [DOI] [PubMed] [Google Scholar]
- 80.Karjalainen T, Barc MC, Collignon A, Trollé S, Boureau H, Cotte-Laffitte J, Bourlioux P. Cloning of a genetic determinant from Clostridium difficile involved in adherence to tissue culture cells and mucus. Infect Immun 62: 4347–4355, 1994. doi: 10.1128/IAI.62.10.4347-4355.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Karlsson S, Burman LG, Åkerlund T. Suppression of toxin production in Clostridium difficile VPI 10463 by amino acids. Microbiology 145: 1683–1693, 1999. doi: 10.1099/13500872-145-7-1683. [DOI] [PubMed] [Google Scholar]
- 82.Karlsson S, Lindberg A, Norin E, Burman LG, Akerlund T. Toxins, butyric acid, and other short-chain fatty acids are coordinately expressed and down-regulated by cysteine in Clostridium difficile. Infect Immun 68: 5881–5888, 2000. doi: 10.1128/IAI.68.10.5881-5888.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kavanaugh NL, Zhang AQ, Nobile CJ, Johnson AD, Ribbeck K. Mucins suppress virulence traits of Candida albicans. MBio 5: e01911-14, 2014. doi: 10.1128/mBio.01911-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Keel MK, Songer JG. The distribution and density of Clostridium difficile toxin receptors on the intestinal mucosa of neonatal pigs. Vet Pathol 44: 814–822, 2007. doi: 10.1354/vp.44-6-814. [DOI] [PubMed] [Google Scholar]
- 85.Kelly CP, Becker S, Linevsky JK, Joshi MA, O’Keane JC, Dickey BF, LaMont JT, Pothoulakis C. Neutrophil recruitment in Clostridium difficile toxin A enteritis in the rabbit. J Clin Invest 93: 1257–1265, 1994. doi: 10.1172/JCI117080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kim H, Rhee SH, Kokkotou E, Na X, Savidge T, Moyer MP, Pothoulakis C, LaMont JT. Clostridium difficile toxin A regulates inducible cyclooxygenase-2 and prostaglandin E2 synthesis in colonocytes via reactive oxygen species and activation of p38 MAPK. J Biol Chem 280: 21237–21245, 2005. doi: 10.1074/jbc.M413842200. [DOI] [PubMed] [Google Scholar]
- 87.Kim JH, Muder RR. Clostridium difficile enteritis: a review and pooled analysis of the cases. Anaerobe 17: 52–55, 2011. doi: 10.1016/j.anaerobe.2011.02.002. [DOI] [PubMed] [Google Scholar]
- 88.Kim KA, Wry P, Hughes E Jr, Butcher J, Barbot D. Clostridium difficile small-bowel enteritis after total proctocolectomy: a rare but fatal, easily missed diagnosis. Report of a case. Dis Colon Rectum 50: 920–923, 2007. doi: 10.1007/s10350-006-0784-y. [DOI] [PubMed] [Google Scholar]
- 89.Köksal F. [The role of Clostridium difficile in antibiotic-associated enteritis]. Mikrobiyol Bul 19: 109–111, 1985. [PubMed] [Google Scholar]
- 90.Koon HW, Shih DQ, Hing TC, Yoo JH, Ho S, Chen X, Kelly CP, Targan SR, Pothoulakis C. Human monoclonal antibodies against Clostridium difficile toxins A and B inhibit inflammatory and histologic responses to the toxins in human colon and peripheral blood monocytes. Antimicrob Agents Chemother 57: 3214–3223, 2013. doi: 10.1128/AAC.02633-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kovbasnjuk O, Zachos NC, In J, Foulke-Abel J, Ettayebi K, Hyser JM, Broughman JR, Zeng XL, Middendorp S, de Jonge HR, Estes MK, Donowitz M. Human enteroids: preclinical models of non-inflammatory diarrhea. Stem Cell Res Ther 4, Suppl 1: S3, 2013. doi: 10.1186/scrt364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kralovich KA, Sacksner J, Karmy-Jones RA, Eggenberger JC. Pseudomembranous colitis with associated fulminant ileitis in the defunctionalized limb of a jejunal-ileal bypass. Report of a case. Dis Colon Rectum 40: 622–624, 1997. doi: 10.1007/BF02055391. [DOI] [PubMed] [Google Scholar]
- 93.Krivan HC, Clark GF, Smith DF, Wilkins TD. Cell surface binding site for Clostridium difficile enterotoxin: evidence for a glycoconjugate containing the sequence Gal alpha 1-3Gal beta 1-4GlcNAc. Infect Immun 53: 573–581, 1986. doi: 10.1128/IAI.53.3.573-581.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kuehne SA, Cartman ST, Heap JT, Kelly ML, Cockayne A, Minton NP. The role of toxin A and toxin B in Clostridium difficile infection. Nature 467: 711–713, 2010. doi: 10.1038/nature09397. [DOI] [PubMed] [Google Scholar]
- 95.Kuntz DP, Shortsleeve MJ, Kantrowitz PA, Gauvin GP. Clostridium difficile enteritis. A cause of intramural gas. Dig Dis Sci 38: 1942–1944, 1993. doi: 10.1007/BF01296124. [DOI] [PubMed] [Google Scholar]
- 96.Kuribayashi H, Miyata M, Yamakawa H, Yoshinari K, Yamazoe Y. Enterobacteria-mediated deconjugation of taurocholic acid enhances ileal farnesoid X receptor signaling. Eur J Pharmacol 697: 132–138, 2012. doi: 10.1016/j.ejphar.2012.09.048. [DOI] [PubMed] [Google Scholar]
- 97.Kurtz LE, Yang SS, Bank S. Clostridium difficile-associated small bowel enteritis after total proctocolectomy in a Crohn’s disease patient. J Clin Gastroenterol 44: 76–77, 2010. doi: 10.1097/MCG.0b013e3181a7481b. [DOI] [PubMed] [Google Scholar]
- 98.LaFrance ME, Farrow MA, Chandrasekaran R, Sheng J, Rubin DH, Lacy DB. Identification of an epithelial cell receptor responsible for Clostridium difficile TcdB-induced cytotoxicity. Proc Natl Acad Sci USA 112: 7073–7078, 2015. doi: 10.1073/pnas.1500791112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lamont JT, Trnka YM, Lamont JT, Trnka YM. Therapeutic implications of Clostridium difficile toxin during relapse of chronic inflammatory bowel disease. Lancet 315: 381–383, 1980. doi: 10.1016/S0140-6736(80)90939-3. [DOI] [PubMed] [Google Scholar]
- 100.Lavallée C, Laufer B, Pépin J, Mitchell A, Dubé S, Labbé AC. Fatal Clostridium difficile enteritis caused by the BI/NAP1/027 strain: a case series of ileal C. difficile infections. Clin Microbiol Infect 15: 1093–1099, 2009. doi: 10.1111/j.1469-0691.2009.03004.x. [DOI] [PubMed] [Google Scholar]
- 101.Lawley TD, Croucher NJ, Yu L, Clare S, Sebaihia M, Goulding D, Pickard DJ, Parkhill J, Choudhary J, Dougan G. Proteomic and genomic characterization of highly infectious Clostridium difficile 630 spores. J Bacteriol 191: 5377–5386, 2009. doi: 10.1128/JB.00597-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Leslie JL, Huang S, Opp JS, Nagy MS, Kobayashi M, Young VB, Spence JR. Persistence and toxin production by Clostridium difficile within human intestinal organoids result in disruption of epithelial paracellular barrier function. Infect Immun 83: 138–145, 2015. doi: 10.1128/IAI.02561-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Lundeen SJ, Otterson MF, Binion DG, Carman ET, Peppard WJ. Clostridium difficile enteritis: an early postoperative complication in inflammatory bowel disease patients after colectomy. J Gastrointest Surg 11: 138–142, 2007. doi: 10.1007/s11605-006-0022-x. [DOI] [PubMed] [Google Scholar]
- 104.Lyras D, O’Connor JR, Howarth PM, Sambol SP, Carter GP, Phumoonna T, Poon R, Adams V, Vedantam G, Johnson S, Gerding DN, Rood JI. Toxin B is essential for virulence of Clostridium difficile. Nature 458: 1176–1179, 2009. doi: 10.1038/nature07822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.MacDonald KS, McLeod J, Nicolle L. Clostridium difficile enteritis in a Canadian tertiary care hospital. Can J Infect Control 8: 37–40, 1993. [PubMed] [Google Scholar]
- 106.Mahida YR, Galvin A, Makh S, Hyde S, Sanfilippo L, Borriello SP, Sewell HF. Effect of Clostridium difficile toxin A on human colonic lamina propria cells: early loss of macrophages followed by T-cell apoptosis. Infect Immun 66: 5462–5469, 1998. doi: 10.1128/IAI.66.11.5462-5469.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Mann SD, Pitt J, Springall RG, Thillainayagam AV. Clostridium difficile infection–an unusual cause of refractory pouchitis: report of a case. Dis Colon Rectum 46: 267–270, 2003. doi: 10.1007/s10350-004-6533-1. [DOI] [PubMed] [Google Scholar]
- 108.May M, Wang T, Müller M, Genth H. Difference in F-actin depolymerization induced by toxin B from the Clostridium difficile strain VPI 10463 and toxin B from the variant Clostridium difficile serotype F strain 1470. Toxins (Basel) 5: 106–119, 2013. doi: 10.3390/toxins5010106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.McBride M. Antibiotic-associated pseudomembranous enteritis due to Clostridium difficile. Clin Infect Dis 21: 455, 1995. doi: 10.1093/clinids/21.2.455. [DOI] [PubMed] [Google Scholar]
- 110.Miller DL, Sedlack JD, Holt RW. Perforation complicating rifampin-associated pseudomembranous enteritis. Arch Surg 124: 1082, 1989. doi: 10.1001/archsurg.1989.01410090092020. [DOI] [PubMed] [Google Scholar]
- 111.Mitchell MJ, Laughon BE, Lin S. Biochemical studies on the effect of Clostridium difficile toxin B on actin in vivo and in vitro. Infect Immun 55: 1610–1615, 1987. doi: 10.1128/IAI.55.7.1610-1615.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Na X, Kim H, Moyer MP, Pothoulakis C, LaMont JT. gp96 is a human colonocyte plasma membrane binding protein for Clostridium difficile toxin A. Infect Immun 76: 2862–2871, 2008. doi: 10.1128/IAI.00326-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Na X, Zhao D, Koon HW, Kim H, Husmark J, Moyer MP, Pothoulakis C, LaMont JT. Clostridium difficile toxin B activates the EGF receptor and the ERK/MAP kinase pathway in human colonocytes. Gastroenterology 128: 1002–1011, 2005. doi: 10.1053/j.gastro.2005.01.053. [DOI] [PubMed] [Google Scholar]
- 114.Nasser H, Munie S, Shakaroun D, Ivanics T, Nalamati S, Killu K. Clostridium difficile Enteritis after total abdominal colectomy for ulcerative colitis. Case Rep Crit Care 2019: 1–4, 2019. doi: 10.1155/2019/2987682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Navaneethan U, Giannella RA. Thinking beyond the colon-small bowel involvement in Clostridium difficile infection. Gut Pathog 1: 7, 2009. doi: 10.1186/1757-4749-1-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Oka S, Inamatsu T, Urayama K, Shimada K. [Adverse effects of antimicrobiological agents and countermeasures--Clostridium difficile enteritis: with special reference to early diagnosis]. Nihon Rinsho 44: 951–954, 1986. [PubMed] [Google Scholar]
- 117.Olson A, Diebel LN, Liberati DM. Effect of host defenses on Clostridium difficile toxin-induced intestinal barrier injury. J Trauma Acute Care Surg 74: 983–990, 2013. doi: 10.1097/TA.0b013e3182858477. [DOI] [PubMed] [Google Scholar]
- 118.Ouwehand AC, Isolauri E, Kirjavainen PV, Salminen SJ. Adhesion of four Bifidobacterium strains to human intestinal mucus from subjects in different age groups. FEMS Microbiol Lett 172: 61–64, 1999. doi: 10.1111/j.1574-6968.1999.tb13450.x. [DOI] [PubMed] [Google Scholar]
- 119.Papatheodorou P, Carette JE, Bell GW, Schwan C, Guttenberg G, Brummelkamp TR, Aktories K. Lipolysis-stimulated lipoprotein receptor (LSR) is the host receptor for the binary toxin Clostridium difficile transferase (CDT). Proc Natl Acad Sci USA 108: 16422–16427, 2011. doi: 10.1073/pnas.1109772108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Papatheodorou P, Hornuss D, Nölke T, Hemmasi S, Castonguay J, Picchianti M, Aktories K. Clostridium difficile binary toxin CDT induces clustering of the lipolysis-stimulated lipoprotein receptor into lipid rafts. MBio 4: e00244-13, 2013. doi: 10.1128/mBio.00244-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Parikh VA, Edlund JW. Fatal Clostridium difficile enteritis after total abdominal colectomy. J Clin Gastroenterol 22: 329, 1996. doi: 10.1097/00004836-199606000-00022. [DOI] [PubMed] [Google Scholar]
- 122.Peacock O, Speake W, Shaw A, Goddard A. Clostridium difficile enteritis in a patient after total proctocolectomy. BMJ Case Rep 2009: bcr10.20008.1165, 2009. doi: 10.1136/bcr.10.2008.1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Perry JL, Ramachandran NK, Utama B, Hyser JM. Use of genetically-encoded calcium indicators for live cell calcium imaging and localization in virus-infected cells. Methods 90: 28–38, 2015. doi: 10.1016/j.ymeth.2015.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Popoff MR, Rubin EJ, Gill DM, Boquet P. Actin-specific ADP-ribosyltransferase produced by a Clostridium difficile strain. Infect Immun 56: 2299–2306, 1988. doi: 10.1128/IAI.56.9.2299-2306.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Pothoulakis C, Galili U, Castagliuolo I, Kelly CP, Nikulasson S, Dudeja PK, Brasitus TA, LaMont JT. A human antibody binds to alpha-galactose receptors and mimics the effects of Clostridium difficile toxin A in rat colon. Gastroenterology 110: 1704–1712, 1996. doi: 10.1053/gast.1996.v110.pm8964394. [DOI] [PubMed] [Google Scholar]
- 126.Pothoulakis C, Gilbert RJ, Cladaras C, Castagliuolo I, Semenza G, Hitti Y, Montcrief JS, Linevsky J, Kelly CP, Nikulasson S, Desai HP, Wilkins TD, LaMont JT. Rabbit sucrase-isomaltase contains a functional intestinal receptor for Clostridium difficile toxin A. J Clin Invest 98: 641–649, 1996. doi: 10.1172/JCI118835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Ramos Martínez A, Romero Pizarro Y, Martínez Arrieta F, Balandín Moreno B, Múñez Rubio E, Cuiñas León K, Sánchez Romero I, Cantos López de Ibargüen B, Asensio Vegas A. [Clostridium difficile enteritis]. Gastroenterol Hepatol 34: 539–545, 2011. doi: 10.1016/j.gastrohep.2011.03.005. [DOI] [PubMed] [Google Scholar]
- 128.Ridlon JM, Kang DJ, Hylemon PB. Bile salt biotransformations by human intestinal bacteria. J Lipid Res 47: 241–259, 2006. doi: 10.1194/jlr.R500013-JLR200. [DOI] [PubMed] [Google Scholar]
- 129.Riedl J, Crevenna AH, Kessenbrock K, Yu JH, Neukirchen D, Bista M, Bradke F, Jenne D, Holak TA, Werb Z, Sixt M, Wedlich-Soldner R. Lifeact: a versatile marker to visualize F-actin. Nat Methods 5: 605–607, 2008. doi: 10.1038/nmeth.1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Riegler M, Sedivy R, Pothoulakis C, Hamilton G, Zacherl J, Bischof G, Cosentini E, Feil W, Schiessel R, LaMont JT. Clostridium difficile toxin B is more potent than toxin A in damaging human colonic epithelium in vitro. J Clin Invest 95: 2004–2011, 1995. doi: 10.1172/JCI117885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Riegler M, Sedivy R, Sogukoglu T, Castagliuolo I, Pothoulakis C, Cosentini E, Bischof G, Hamilton G, Teleky B, Feil W, Lamont JT, Wenzl E. Epidermal growth factor attenuates Clostridium difficile toxin A- and B-induced damage of human colonic mucosa. Am J Physiol Gastrointest Liver Physiol 273: G1014–G1022, 1997. doi: 10.1152/ajpgi.1997.273.5.G1014. [DOI] [PubMed] [Google Scholar]
- 132.Sato T, Stange DE, Ferrante M, Vries RG, Van Es JH, Van den Brink S, Van Houdt WJ, Pronk A, Van Gorp J, Siersema PD, Clevers H. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterology 141: 1762–1772, 2011. doi: 10.1053/j.gastro.2011.07.050. [DOI] [PubMed] [Google Scholar]
- 133.Saxena K, Blutt SE, Ettayebi K, Zeng XL, Broughman JR, Crawford SE, Karandikar UC, Sastri NP, Conner ME, Opekun AR, Graham DY, Qureshi W, Sherman V, Foulke-Abel J, In J, Kovbasnjuk O, Zachos NC, Donowitz M, Estes MK. Human intestinal enteroids: a new model to study human rotavirus infection, host restriction, and pathophysiology. J Virol 90: 43–56, 2016. doi: 10.1128/JVI.01930-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Sayin SI, Wahlström A, Felin J, Jäntti S, Marschall HU, Bamberg K, Angelin B, Hyötyläinen T, Orešič M, Bäckhed F. Gut microbiota regulates bile acid metabolism by reducing the levels of tauro-beta-muricholic acid, a naturally occurring FXR antagonist. Cell Metab 17: 225–235, 2013. doi: 10.1016/j.cmet.2013.01.003. [DOI] [PubMed] [Google Scholar]
- 135.Semenyuk EG, Poroyko VA, Johnston PF, Jones SE, Knight KL, Gerding DN, Driks A. Analysis of bacterial communities during Clostridium difficile infection in the mouse. Infect Immun 83: 4383–4391, 2015. doi: 10.1128/IAI.00145-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Shortland JR, Spencer RC, Williams JL. Pseudomembranous colitis associated with changes in an ileal conduit. J Clin Pathol 36: 1184–1187, 1983. doi: 10.1136/jcp.36.10.1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Siddiqui J, Campion T, Wei R, Kuzmich S. Clostridium difficile enteritis: diffuse small bowel radiological changes in a patient with abdominal sepsis. BMJ Case Rep 2018: bcr-2017-222209, 2018. doi: 10.1136/bcr-2017-222209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Söderlin MK, Alasaarela E, Hakala M. Reactive arthritis induced by Clostridium difficile enteritis as a complication of Helicobacter pylori eradication. Clin Rheumatol 18: 337–338, 1999. doi: 10.1007/s100670050113. [DOI] [PubMed] [Google Scholar]
- 139.Sorg JA, Sonenshein AL. Inhibiting the initiation of Clostridium difficile spore germination using analogs of chenodeoxycholic acid, a bile acid. J Bacteriol 192: 4983–4990, 2010. doi: 10.1128/JB.00610-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Steele J, Chen K, Sun X, Zhang Y, Wang H, Tzipori S, Feng H. Systemic dissemination of Clostridium difficile toxins A and B is associated with severe, fatal disease in animal models. J Infect Dis 205: 384–391, 2012. doi: 10.1093/infdis/jir748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Tao L, Zhang J, Meraner P, Tovaglieri A, Wu X, Gerhard R, Zhang X, Stallcup WB, Miao J, He X, Hurdle JG, Breault DT, Brass AL, Dong M. Frizzled proteins are colonic epithelial receptors for C. difficile toxin B. Nature 538: 350–355, 2016. doi: 10.1038/nature19799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Testore GP, Nardi F, Babudieri S, Giuliano M, Di Rosa R, Panichi G. Isolation of Clostridium difficile from human jejunum: identification of a reservoir for disease? J Clin Pathol 39: 861–862, 1986. doi: 10.1136/jcp.39.8.861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Thai H, Guerron AD, Bencsath KP, Liu X, Loor M. Fulminant Clostridium difficile enteritis causing abdominal compartment syndrome. Surg Infect (Larchmt) 15: 821–825, 2014. doi: 10.1089/sur.2013.026. [DOI] [PubMed] [Google Scholar]
- 144.Theriot CM, Bowman AA, Young VB. Antibiotic-induced alterations of the gut microbiota alter secondary bile acid production and allow for Clostridium difficile spore germination and outgrowth in the large intestine. MSphere 1: e00045-15, 2016. doi: 10.1128/mSphere.00045-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Tjandra JJ, Street A, Thomas RJ, Gibson R, Eng P, Cade J. Fatal Clostridium difficile infection of the small bowel after complex colorectal surgery. ANZ J Surg 71: 500–503, 2001. doi: 10.1046/j.1440-1622.2001.02083.x. [DOI] [PubMed] [Google Scholar]
- 146.Tjellström B, Stenhammar L, Eriksson S, Magnusson KE. Oral immunoglobulin A supplement in treatment of Clostridium difficile enteritis. Lancet 341: 701–702, 1993. doi: 10.1016/0140-6736(93)90477-X. [DOI] [PubMed] [Google Scholar]
- 147.Triadafilopoulos G, Pothoulakis C, O’Brien MJ, LaMont JT. Differential effects of Clostridium difficile toxins A and B on rabbit ileum. Gastroenterology 93: 273–279, 1987. doi: 10.1016/0016-5085(87)91014-6. [DOI] [PubMed] [Google Scholar]
- 148.Tsutaoka B, Hansen J, Johnson D, Holodniy M. Antibiotic-associated pseudomembranous enteritis due to Clostridium difficile. Clin Infect Dis 18: 982–984, 1994. doi: 10.1093/clinids/18.6.982. [DOI] [PubMed] [Google Scholar]
- 149.Tucker KD, Wilkins TD. Toxin A of Clostridium difficile binds to the human carbohydrate antigens I, X, and Y. Infect Immun 59: 73–78, 1991. doi: 10.1128/IAI.59.1.73-78.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Valdés L, Gueimonde M, Ruas-Madiedo P. Monitoring in real time the cytotoxic effect of Clostridium difficile upon the intestinal epithelial cell line HT29. J Microbiol Methods 119: 66–73, 2015. doi: 10.1016/j.mimet.2015.09.022. [DOI] [PubMed] [Google Scholar]
- 151.VanDussen KL, Marinshaw JM, Shaikh N, Miyoshi H, Moon C, Tarr PI, Ciorba MA, Stappenbeck TS. Development of an enhanced human gastrointestinal epithelial culture system to facilitate patient-based assays. Gut 64: 911–920, 2015. doi: 10.1136/gutjnl-2013-306651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Vesoulis Z, Williams G, Matthews B. Pseudomembranous enteritis after proctocolectomy: report of a case. Dis Colon Rectum 43: 551–554, 2000. doi: 10.1007/BF02237205. [DOI] [PubMed] [Google Scholar]
- 153.Vrieze A, Out C, Fuentes S, Jonker L, Reuling I, Kootte RS, van Nood E, Holleman F, Knaapen M, Romijn JA, Soeters MR, Blaak EE, Dallinga-Thie GM, Reijnders D, Ackermans MT, Serlie MJ, Knop FK, Holst JJ, van der Ley C, Kema IP, Zoetendal EG, de Vos WM, Hoekstra JB, Stroes ES, Groen AK, Nieuwdorp M. Impact of oral vancomycin on gut microbiota, bile acid metabolism, and insulin sensitivity. J Hepatol 60: 824–831, 2014. doi: 10.1016/j.jhep.2013.11.034. [DOI] [PubMed] [Google Scholar]
- 154.Wada Y, Satoh T, Kubo M. [5 cases of Clostridium difficile enteritis in infants]. Kansenshogaku Zasshi 69: 608–614, 1995. doi: 10.11150/kansenshogakuzasshi1970.69.608. [DOI] [PubMed] [Google Scholar]
- 155.Wee B, Poels JA, McCafferty IJ, Taniere P, Olliff J. A description of CT features of Clostridium difficile infection of the small bowel in four patients and a review of literature. Br J Radiol 82: 890–895, 2009. doi: 10.1259/bjr/57970083. [DOI] [PubMed] [Google Scholar]
- 156.Wood MJ, Hyman N, Hebert JC, Blaszyk H. Catastrophic Clostridium difficile enteritis in a pelvic pouch patient: report of a case. J Gastrointest Surg 12: 350–352, 2008. doi: 10.1007/s11605-007-0440-4. [DOI] [PubMed] [Google Scholar]
- 157.Yafi FA, Selvasekar CR, Cima RR. Clostridium difficile enteritis following total colectomy. Tech Coloproctol 12: 73–74, 2008. [PubMed] [Google Scholar]
- 158.Yee HF Jr, Brown RS Jr, Ostroff JW. Fatal Clostridium difficile enteritis after total abdominal colectomy. J Clin Gastroenterol 22: 45–47, 1996. doi: 10.1097/00004836-199601000-00013. [DOI] [PubMed] [Google Scholar]
- 159.Zachos NC, Kovbasnjuk O, Foulke-Abel J, In J, Blutt SE, de Jonge HR, Estes MK, Donowitz M. Human enteroids/colonoids and intestinal organoids functionally recapitulate normal intestinal physiology and pathophysiology. J Biol Chem 291: 3759–3766, 2016. doi: 10.1074/jbc.R114.635995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Zhang S, Palazuelos-Munoz S, Balsells EM, Nair H, Chit A, Kyaw MH. Cost of hospital management of Clostridium difficile infection in United States-a meta-analysis and modelling study. BMC Infect Dis 16: 447, 2016. doi: 10.1186/s12879-016-1786-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Zoppi G, Vinco A, Cinquetti M, Barbato G, Luciano A. [A case of slowly resolving enteritis caused by Clostridium difficile]. Pediatr Med Chir 17: 459–460, 1995. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The raw data supporting the conclusions of this manuscript will be made available by the authors upon request.