

Abstract
Acute lung injury is a major complication of hemorrhagic shock and the required resuscitation with large volumes of crystalloid fluids and blood products. We previously identified a role of macrophage-derived chemokine (CCL22/MDC) pulmonary inflammation following hemorrhage and resuscitation. However, further details regarding the induction of CCL22/MDC and its precise role in pulmonary inflammation after trauma remain unknown. In the current study we used in vitro experiments with a murine alveolar macrophage cell line, as well as an in vivo mouse model of hemorrhage and resuscitation, to identify key regulators in CCL22/MDC production. We show that trauma induces expression of IFNγ, which leads to production of CCL22/MDC through a signaling mechanism involving p38 MAPK, NF-κB, JAK, and STAT-1. IFNγ also activates TNFα production by alveolar macrophages, potentiating CCL22/MDC production via an autocrine mechanism. Neutralization of IFNγ or TNFα with specific antibodies reduced histological signs of pulmonary injury after hemorrhage and reduced inflammatory cell infiltration into the lungs.
Keywords: alveolar macrophages, JAK, NF-κB, p38 MAPK, pulmonary inflammation, STAT-1
INTRODUCTION
Although traumatic injury is an individual event on the patient level, it can be considered a pandemic disease. Worldwide, 5.8 million people die each year as a result of injuries (52b); road injuries alone are the eighth-most-common cause of death worldwide (52a). In the United States, one person dies from traumatic injury every 3 min (5). Hemorrhage following traumatic injury accounts for 30–40% of traumatic deaths (43). Hemorrhage is not exclusively an acute problem, as hemorrhage followed by resuscitation (H/R) initiates a cascade of systemic inflammatory events that can lead to multiorgan damage, significant morbidity, and even delayed mortality (11). One particular complication is acute lung injury. Experimental models have shown that pulmonary injury after resuscitation manifests as hypoxia, pulmonary edema, and immune cell infiltration into the lungs (14, 30, 31, 41) and identified interleukin (IL)-8, macrophage inflammatory protein-2, IL-1, TNF-α, and IL-6 as the inflammatory mediators (1, 2, 13, 26, 29, 48, 51). Recently, our group also identified a role of macrophage-derived C-C motif chemokine ligand 22 (CCL22/MDC) (41).
CCL22/MDC is a member of the CC chemokine family that binds to CC chemokine receptor 4 (CCR4) (20). It was originally known as stimulated T cell chemoattractant protein-1 because of its action on CD4 T cells (3). CCL22/MDC is a strong chemoattractant for CCR4-expressing T helper type 2 and cytotoxic T type 2 cells and amplifies type II immune responses (32). Monocytes, dendritic cells, and natural killer cells have also been shown to migrate toward CCL22/MDC in a dose-dependent manner (12, 21). Previous publications suggest that CCL22/MDC contributes to diseases such as allergic reactions, Crohn’s disease, HIV infection, and neoplasia (22, 32, 38). It has also been implicated in the pathogenesis of several lung diseases, including asthma- and cigarette-induced lung inflammation (42). Our own previous work demonstrates that CCL22/MDC levels are elevated following H/R in a mouse model. Neutralization of CCL22/MDC through antibody injection reduced inflammatory cell recruitment to the lungs, whereas administration of recombinant CCL22/MDC augmented pulmonary inflammation (41).
CCL22/MDC is known to be produced by dendritic cells, epithelial cells, macrophages, and basophils (10, 13). However, the cell type responsible for CCL22/MDC production following H/R has yet to be determined. Given the key role of alveolar macrophages in the pulmonary inflammatory response to trauma (14), we hypothesized that alveolar macrophages are likely producers of CCL22/MDC in response to proinflammatory mediators released early after H/R.
To test this hypothesis, we determined levels of interferon (IFN)-γ in the serum and lungs of mice subjected to H/R and the ability of IFNγ to stimulate CCL22/MDC production in vitro. By using specific inhibitors to different signaling pathways, we identified TNFα as another likely driver of CCL22/MDC production by alveolar macrophages and confirmed that neutralizing either IFNγ or TNFα or both through injection of specific antibodies abrogated CCL22/MDC production after H/R and ameliorated pulmonary inflammation.
METHODS
Ethical statement.
All murine experiments were approved by the Institutional Animal Care and Use Committee of the University of Cincinnati (protocol no. 10-05-10-01).
Mouse housing.
C57BL/6 male mice were obtained from Harlan Laboratories (Indianapolis, IN) at 9–14 wk of age. Mice were housed in groups of four in standard environmental conditions with corn-cob bedding. A standard pellet diet and water were provided ab libitum. Mice were acclimated for ≥1 wk before experimentation.
H/R model.
Mice were initially anesthetized with 0.1 mg/g body wt ip pentobarbital and redosed when necessary to maintain the proper surgical plane of anesthesia throughout the experiment. To maintain a constant body temperature and avoid hypothermia, mice were kept on a 41°C heating pad throughout the procedure. Anesthetized mice underwent femoral artery cannulation, as previously described (4, 31). Briefly, the fur above the left femur was clipped, and the skin was sterilized with povidone-iodine solution and alcohol. The left femoral artery was exposed, isolated, and cannulated with a polyethylene catheter. Catheters were connected to pressure transducers (Harvard Apparatus, Holliston, MA) for continuous real-time hemodynamic monitoring. Mice were allowed to equilibrate for 10 min. Subsequently, blood was withdrawn through the femoral artery cannula until a systolic blood pressure of 25 ± 5 mmHg was reached. Mice were maintained at this hypotensive pressure for 60 min through removal or administration of additional autologous blood as necessary. At the end of the hemorrhage period, mice were resuscitated to a systolic blood pressure of 75 ± 5 mmHg with Ringer’s lactate (LR) for 20 min. Mice were then decannulated and euthanized for analysis at 30 min or 4 h after surgery. Sham mice underwent identical femoral artery cannulation but were neither hemorrhaged nor resuscitated.
Identification of CCL22/MDC-producing cells by flow cytometry.
Mice were injected with brefeldin A (250 µg ip; Sigma Aldrich, St. Louis, MO) at 30 min before hemorrhage to inhibit protein export. Mice were euthanized 4 h postresuscitation, and lungs were harvested and homogenized using a lung dissociation kit (Miltenyi Biotec, Bergisch Gladbach, Germany) and a gentleMACS dissociator (Miltenyi Biotec) according to the manufacturer’s instructions. Red blood cells were depleted using ammonium-chloride-potassium lysing buffer (155 mM ammonium chloride, 10 mM potassium bicarbonate, and 0.1 mM EDTA, pH 7.2–7.4). The following fluorescent-labeled antibodies were obtained from BioLegend (San Diego, CA): anti-CD11b (clone M1/70), anti-CD11c (clone N418), anti-CD24 (clone M1/69), anti-CD64/FcγRI (clone X54-9/7.1), anti-CD170/Siglec-F (clone S17007L), anti-Ly6C (clone HK1.4), and anti-Ly6G (clone 1A8). Identification of different immune cell subsets in the lung was based on cell surface antigen expression, as previously described (33). Briefly, alveolar macrophages were defined as CD11b−CD11c+CD24−CD64+, interstitial macrophages as CD11b+CD11c+CD24−CD64+, Ly6C− monocytes/undifferentiated macrophages as CD11b+Siglec-F−Ly6G−CD24−Ly6C−, Ly6C+ monocytes as CD11b+Siglec-F−Ly6G−CD24−Ly6C+, granulocytes as CD11b+CD24+, and CD11b+ dendritic cells as CD11b−CD11c+CD24+CD64−. For intracellular CCL22/MDC staining, cells were fixed with 1% paraformaldehyde, permeabilized with 0.1% saponin, exposed to an anti-CCL22/MDC antibody (clone EPR1362, Abcam, Cambridge, UK), and then stained with fluorescent-labeled goat anti-rabbit IgG (polyclonal; Invitrogen, Carlsbad, CA).
Cytokine quantification.
Mice were euthanized 4 h postresuscitation, and blood was drawn via cardiac puncture and collected into serum separator tubes. After the blood clotted, serum was cleared by centrifugation and stored at −80°C until further analysis. The right lung was removed, snap-frozen in liquid nitrogen, and stored at −80°C until further analysis. For protein extraction, lungs were homogenized in tissue extraction buffer [PBS supplemented with 10 mM EDTA, 2 mM phenylmethylsulfonyl fluoride (Sigma Aldrich), 1× complete protease inhibitor cocktail (Roche, Indianapolis, IN), 0.1 mg/mL soybean trypsin inhibitor (Sigma Aldrich), and 1 mg/mL each aprotinin, leupeptin, and pepstatin (Sigma Aldrich)] and incubated on ice for 2 h. Debris was cleared by centrifugation with 12,500 g at 4°C for 10 min.
For in vitro cytokine analysis, cells were removed by centrifugation, and the cell-free supernatant was frozen at −80°C until further analysis.
CCL22/MDC and TNFα levels were quantified by enzyme-linked immunosorbent assays (ELISA) according to the manufacturer’s instructions (R & D Systems, Minneapolis, MN). Because of the short half-life of IFNγ in vivo, a specialized in vivo capture assay was employed for IFNγ quantification (BD Biosciences, San Diego, CA), as previously described (10, 50). Mice received an anti-mouse IFNγ antibody (0.5 mg ip) 2 h before H/R. This antibody forms a soluble complex with IFNγ, preventing its degradation and excretion in vivo, thereby allowing IFNγ to accumulate and increasing the sensitivity of the subsequent cytokine quantification by ELISA. The anti-IFNγ antibody used in the ELISA is directed against an epitope different from the capture antibody used in vivo.
Alveolar macrophage cell culture and in vitro treatments.
The AMJ2-C11 cell line (American Type Culture Collection, Manassas, VA) was used for all in vitro experiments. This alveolar macrophage cell line has been described previously and undergoes activation upon stimulation with IFNγ (36, 37). Cells were maintained in Dulbecco’s modified Eagle’s medium with 4 mM l-glutamine, 1.5 g/L sodium bicarbonate, 4.5 g/L glucose, 5 mM HEPES, and 5% fetal bovine serum and kept at 37°C with 5% CO2.
On the day before an experiment, cells (50,000/mL) were plated in tissue culture plates. Alveolar macrophages were activated with recombinant mouse IFNγ (R & D Systems). Unless otherwise stated, 1 ng/mL IFNγ was used, and cells were stimulated for 24 h until supernatants were collected for cytokine analysis. Cells were harvested and stained with fluorescent-labeled anti-CD11b (clone M1/70, BioLegend), annexin V (BioLegend), and propidium iodide in annexin V binding buffer (BD Biosciences) to assess cellular viability by flow cytometry.
To identify signal transduction pathways involved in CCL22/MDC production in response to IFNγ, a literature search was conducted to compile a list of potentially involved transcription factors and signaling molecules and their chemical inhibitors. The following candidates were tested: nuclear factor (NF)-κB, inhibited by ammonium pyrrolidinedithiocarbamate (PDTC; Sigma Aldrich); signal transducer and activator of transcription (STAT)-1, inhibited by l-sulforaphane (Sigma Aldrich); p38 mitogen-activated protein kinase (MAPK), inhibited by SB203580 (Cell Signaling Technology, Danvers, MA); activator protein (AP)-1, inhibited by SR11302 (R & D Systems); and Janus-like kinase (JAK)-1, inhibited by ruxolitinib (Selleck Chem, Houston, TX). Previous studies reported that PDTC is a potent inhibitor of NF-κB (27, 34, 46) and also blocks IκBα phosphorylation, preventing the dissociation of NF-κB from IκBα and subsequent NF-κB translocation from the nucleus in response to inflammatory stimulation (28). PDTC has also been suggested to downregulate chemokine receptor expression (47, 55). l-Sulforaphane has been reported to block the IFNγ-induced induction of STAT-1 phosphorylation (39), as well as IFNγ-induced protein kinase B phosphorylation (39), decrease IFN regulatory factor-1 mRNA expression (39), and activate the transcription factor nuclear factor erythroid 2-related factor 2 (18). SB203580 is a widely used small-molecule inhibitor of p38 MAPK (7) but has also been reported to inhibit protein kinase B phosphorylation (25). SR11302 is a selective inhibitor of AP-1 that does not activate retinoic acid response element (9, 19). Ruxolitinib is a selective JAK1/2 inhibitor, with weaker inhibition of the other JAK family kinases Tyk2 and JAK3 (52). In an in vitro near-kinome-wide survey, ruxolitinib also showed inhibitory capacity toward a number of other kinases and kinase mutants (57). Alveolar macrophages were treated with these inhibitors at the indicated concentrations for 24 h in the presence of 1 ng/mL IFNγ before CCL22/MDC quantification.
To test the influence of TNFα on CCL22/MDC production, alveolar macrophages were treated with polyclonal goat IgG antibodies against TNFα, TNFα receptor subtype 1, or TNFα receptor subtype 2 (all from R & D Systems). Cells were treated in the presence of 1 ng/mL IFNγ for 24 h before CCL22/MDC quantification. Control groups were treated with an IgG isotype control, which had no effect (data not shown).
In vivo antibody treatments.
To test the roles of IFNγ and TNFα on CCL22/MDC production in vivo, mice were injected intraperitoneally with either 100 µg/mouse polyclonal rat anti-mouse IFNγ IgG (Affymetrix eBioscience, San Diego, CA) or 100 µg/mouse polyclonal goat anti-mouse TNFα IgG (R & D Systems) 2 h before hemorrhage.
Lung histology and immunohistochemistry.
Mice were euthanized 4 h after H/R. The left lung was inflated with 10% neutral buffered formalin (Thermo Fisher Scientific, Waltham, MA) through the left main stem bronchus for fixation and then harvested. The tissue was fixed further in 10% neutral buffered formalin for 48 h and then dehydrated with ethanol, embedded in paraffin, and sectioned at 5–10 µm. Sections were dewaxed and rehydrated. For general histological assessment, sections were stained with hematoxylin and eosin. For assessment and quantification of inflammatory cell infiltration, sections were subjected to antigen retrieval and stained with rat anti-mouse Ly-6B.2 antibody (clone 7/4, AbD Serotec, Raleigh, NC), followed by detection with a horseradish peroxidase-coupled anti-rat secondary antibody (BD PharMingen, San Diego, CA) and an UltraVision Detection System-DAB Plus Substrate System (Thermo Fisher Scientific, Fremont, CA). Stained sections were analyzed with a light microscope (Zeiss, Thornwood, NY). A combined histological score for pulmonary inflammation (0–4) and pulmonary edema (0–3) was calculated by a blinded investigator based on the average score of 10 vision fields per mouse, as previously described (6, 53).
Statistics.
GraphPad Prism 6.0 (GraphPad Software, La Jolla, CA) was used for statistical testing. A two-tailed Student’s t test was used for comparisons between two groups or one-way ANOVA with Tukey’s post hoc analysis for comparisons between more than two groups. Individual data points from two independently repeated experiments, as well as means ± SE, are shown. Representative images were chosen for histology.
RESULTS
IFNγ is elevated after H/R and induces CCL22/MDC production by alveolar macrophages.
We previously demonstrated that CCL22/MDC levels are elevated following H/R in a mouse model. To identify the cellular source of CCL22/MDC production in the lung following H/R, we analyzed CCL22/MDC production by a number of different myeloid cells by flow cytometry. Protein export was blocked with brefeldin A, and mice were subjected to a pressure-clamped model of hemorrhage for 60 min and resuscitation with LR for 20 min, as previously described (4, 31). Among the analyzed cellular subsets, only CCL22/MDC+ alveolar macrophages and CCL22/MDC+CD103+ dendritic cells showed a significant expansion by 4 h after H/R (Fig. 1A). The fold increase was more pronounced in CCL22/MDC+ alveolar macrophages than in CCL22/MDC+CD103+ dendritic cells (4.0 ± 0.6 vs 2.7 ± 1.6; Fig. 1A) and the number of CCL22/MDC+ alveolar macrophages was ~16 times greater than the number of CCL22/MDC+CD103+ dendritic cells (0.7 ± 0.1 × 106 vs. 0.04 ± 0.02 × 106/lung; data not shown). These data indicate that alveolar macrophages are most likely the predominant source of CCL22/MDC production in response to H/R.
Fig. 1.

Serum and pulmonary levels of IFNγ are elevated after hemorrhage and resuscitation (H/R), and IFNγ induces macrophage-derived C-C motif chemokine ligand 22 (CCL22/MDC) production by alveolar macrophages in vitro. Male C57BL/6 mice were subjected to a pressure-clamped model of H/R with Ringer’s lactate (LR) or a sham procedure. A: flow cytometry analysis of different immune cell subsets for CCL22/MDC production. The experiment was conducted in the presence of the protein export inhibitor brefeldin A, which was administered before the H/R procedure. Thus the data show cumulative CCL22/MDC production until the mice were euthanized 4 h after the hypotensive episode. Alveolar macrophages (MΦ) were defined as CD11b−CD11c+CD24−CD64+, interstitial macrophages as CD11b+CD11c+CD24−CD64+,Ly6C−, monocytes (Mo)/undifferentiated macrophages as CD11b+Siglec-F−Ly6G−CD24−Ly6C−, Ly6C+ monocytes as CD11b+Siglec-F−Ly6G−CD24−Ly6C+, granulocytes as CD11b+CD24+, and CD11b+ dendritic cells (DC) as CD11b−CD11c+CD24+CD64− (n = 5 mice/group). *P < 0.05 vs. respective population in sham mice (by ANOVA with Sidak’s post test). B and C: quantitation of cumulative IFNγ levels in serum and lungs by an in vivo capture assay 4 h after the hypotensive episode (n = 5 mice/group). *P < 0.05 vs. sham (by Student’s t test). D: alveolar macrophages were stimulated in vitro with escalating doses of murine recombinant IFNγ, and CCL22/MDC levels in the supernatant were quantified by ELISA after 24 h (n = 6 replicates/group). *P < 0.05 vs. 0 µM IFNγ (by ANOVA with Tukey’s post test). E: alveolar macrophages were treated with 1 ng/mL IFNγ, supernatants were collected at 0–24 h, and CCL22/MDC levels were quantified by ELISA (n = 6 replicates/group). *P < 0.05 vs. 0 h (by ANOVA with Tukey’s post test).
IFNγ has been reported to drive CCL22/MDC production in keratinocytes (17, 56). To determine if IFNγ could also be a potential driver of CCL2/MDC production after H/R, we next determined if IFNγ is elevated after H/R. Because of the short half-life of IFNγ in vivo, a specialized in vivo capture assay was employed for cumulative IFNγ quantification, as previously described (10, 50). By 4 h after H/R, significantly elevated levels of IFNγ were noted in the serum (Fig. 1B), as well as in the lungs (Fig. 1C).
We next treated AMJ2-C11 cells with escalating doses of IFNγ to see if IFNγ could induce CCL22/MDC production specifically by alveolar macrophages. We did indeed observe a dose-dependent increase in CCL22/MDC production upon IFNγ stimulation (Fig. 1D). The smallest effective IFNγ dose for the 24-h stimulation was 0.05 ng/mL, and a dose of 1 ng/mL had the greatest effect, approximately tripling CCL22/MDC production by alveolar macrophages. To determine the kinetics of CCL22/MDC production, we used the highest-effective dose and assessed cytokine levels at different time points. While slight elevations were seen as early as 1 h post-IFNγ stimulation, CCL22/MDC levels were not significantly elevated until 24 h after stimulation (Fig. 1E). Thus, for subsequent experiments, we chose 1 ng/mL IFNγ and 24 h of stimulation as the test parameters.
IFNγ-induced production of CCL22/MDC involves JAK, STAT-1, NF-κB, and p38 MAPK signaling pathways.
To determine potential signaling pathways involved in IFNγ-driven CCL22/MDC production, we treated IFNγ-stimulated AMJ2-C11 cells with specific inhibitors to different signaling molecules and assessed CCL22/MDC production. No decreases or only mild decreases in cell viability of AMJ2-C11 cells were observed with inhibitor treatments (data not shown). Inhibition of JAK-1 with ruxolitinib significantly reduced IFNγ-induced CCL22/MDC production by alveolar macrophages in a dose-dependent manner. The best effect was a ~50% reduction with 5 µM ruxolitinib (Fig. 2A). STAT-1 inhibition with higher doses (20 µM) of l-sulforaphane also reduced CCL22/MDC production to levels equivalent to baseline (Fig. 2B). AP-1 inhibition with SR11302 had no effect (Fig. 2C). The NF-κB inhibitor PDTC significantly reduced (~50%) CCL22/MDC production at higher doses (50 and 100 µM; Fig. 2D). Finally, inhibition of p38 MAPK with SB203580 also reduced CCL22/MDC production by AMJ2-C11 cells in a dose-dependent manner, achieving a ~30% reduction (Fig. 2E). Collectively, these findings indicate that IFNγ signals through JAK-1/STAT-1, NF-κB, and p38 MAPK pathways to induce CCL22/MDC production by alveolar macrophages.
Fig. 2.

IFNγ-induced production of macrophage-derived C-C motif chemokine ligand 22 (CCL22/MDC) involves JAK, STAT-1, NF-κB, and p38 MAPK signaling pathways. Alveolar macrophages were treated with inhibitors in the presence of 1 ng/mL IFNγ. After 24 h, supernatants were collected, and CCL22/MDC levels were quantified by ELISA (n = 4 replicates/group). A: JAK-1 inhibition with ruxolitinib. B: STAT-1 inhibition with l-sulforaphane. C: AP-1 inhibition with SR11302. D: NF-κB inhibition with ammonium pyrrolidinedithiocarbamate (PDTC). E: p38 MAPK inhibition with SB203580. *P < 0.05 vs. 0 µM (by ANOVA with Tukey’s post test).
TNFα is elevated after H/R and produced by alveolar macrophages upon IFNγ stimulation.
NF-κB and p38 MAPK are two well-known factors within the TNFα signaling pathway. To test if IFNγ stimulation does indeed induce TNFα production in alveolar macrophages, we quantified TNFα levels in the supernatants of AMJ2-C11 cells treated with IFNγ. We noted a significant increase in TNFα production at 12 and 24 h. At 24 h, TNFα production had approximately doubled from baseline values (Fig. 3A).
Fig. 3.
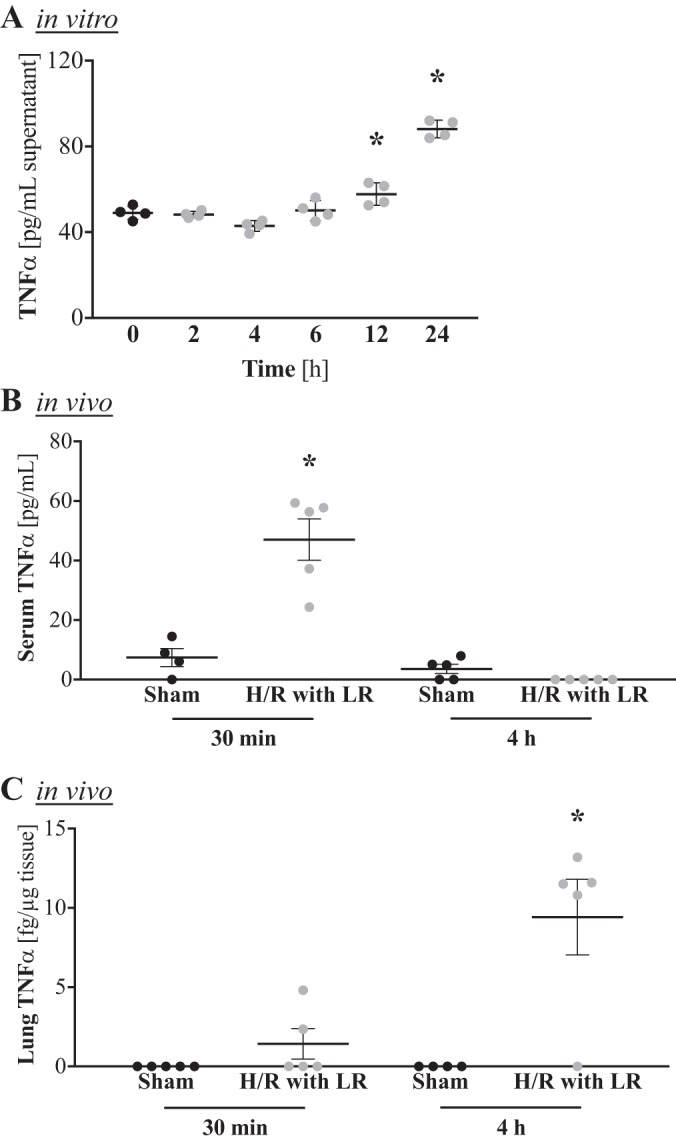
IFNγ induces TNFα production by alveolar macrophages in vitro, and TNFα is elevated after hemorrhage and resuscitation (H/R) in vivo. A: alveolar macrophages were stimulated in vitro with 1 ng/mL IFNγ, and macrophage-derived C-C motif chemokine ligand 22 (CCL22/MDC) levels in the supernatant were quantified by ELISA at 0–24 h (n = 4 replicates/group). *P < 0.05 vs. 0 h (by ANOVA with Tukey’s post test). B and C: male C57BL/6 mice were subjected to a pressure-clamped model of H/R with Ringer’s lactate (LR) or a sham procedure, and TNFα levels were quantified by ELISA in serum and lung 30 min and 4 h after the hypotensive episode (n = 5 mice/group). *P < 0.05 vs. respective sham (by ANOVA with Sidak’s post test).
To translate these in vitro data back to our in vivo model of H/R, we measured TNFα levels in injured mice. At 30 min after the H/R procedure, serum TNFα levels in hemorrhaged mice were nearly six times higher than in control animals (Fig. 3B). At 4 h, however, no serum TNFα was detectable, indicating consumption (Fig. 3B). Pulmonary TNFα levels were not detectable in control animals but were increased in two of five injured mice as early as 30 min after H/R and in all but one injured animal at 4 h after H/R (Fig. 3C), showing that TNFα does increase in response to H/R.
Neutralization of TNFα signaling partially blocks CCL22/MDC production by alveolar macrophages in vitro and prevents elevation of serum CCL22/MDC levels after H/R in vivo.
To associate the elevated TNFα levels upon H/R to CCL22/MDC production by alveolar macrophages, we stimulated AMJ2-C11 cells with IFNγ in the presence of antibodies against TNFα or TNFα receptor subtype 1 or 2 to block TNFα signaling. Direct neutralization of TNFα significantly reduced IFNγ-driven CCL22/MDC production, whereas blockade of TNFα signaling through its receptor had a significant effect only when both receptor subtypes were blocked simultaneously (Fig. 4A).
Fig. 4.

Neutralization of TNFα signaling partially blocks production of macrophage-derived C-C motif chemokine ligand 22 (CCL22/MDC) by alveolar macrophages in vitro and prevents elevation of serum CCL22/MDC levels after hemorrhage and resuscitation (H/R) in vivo. A: alveolar macrophages were treated with antibodies against TNFα (α-TNFα), TNFα receptor subtype 1 or 2 (α-TNFR1 or α-TNFR2), or a combination of the anti-TNFα receptor antibodies (α-TNFR1 + R2) in vitro in the presence of 1 ng/mL IFNγ, and CCL22/MDC levels in the supernatant were quantified by ELISA after 24 h (n = 4 replicates/group). *P < 0.05 vs. IFNγ only (by ANOVA with Tukey’s post test). B: male C57BL/6 mice were injected intraperitoneally with antibodies against IFNγ and/or TNFα. After 2 h, mice were subjected to a pressure-clamped model of H/R with Ringer’s lactate (LR) or a sham procedure. CCL22/MDC levels were quantified by ELISA in the serum 4 h after the hypotensive episode (n = 5 mice/group). *P < 0.05 vs. H/R only (by ANOVA with Tukey’s post test).
To test if CCL22/MDC production could also be blocked in vivo, we pretreated mice with antibodies against either IFNγ or TNFα or both antibodies and then subjected mice to H/R 2 h later. Both antibodies completely blocked CCL22/MDC induction after H/R. Blockade of both IFNγ and TNFα had a synergistic effect, reducing CCL22/MDC levels below those in control mice (Fig. 4B).
Neutralization of IFNγ and/or TNFα signaling ameliorates pulmonary inflammation following H/R.
We previously reported that increased CCL22/MDC levels correlate with increased myeloid cell recruitment to the lungs after H/R. Now knowing that IFNγ and TNFα are the drivers of alveolar macrophage activation after H/R, we tested whether IFNγ and/or TNFα neutralization could ameliorate acute lung injury after H/R. We analyzed the lungs of antibody-treated injured mice histologically 4 h after H/R. Staining with hematoxylin and eosin demonstrated decreased alveolar wall thickening and proteinaceous deposition within all antibody-treated groups (Fig. 5A). Staining of inflammatory myeloid cells with a Ly-6B.2-specific antibody showed reduced cell infiltration within the pulmonary parenchyma in the antibody-treated groups (Fig. 5B). Pulmonary injury and inflammation were also quantified using a histological scoring system, as previously reported (6, 53), which confirmed a significant reduction of lung damage and inflammation in the antibody-treated groups (Fig. 5C).
Fig. 5.
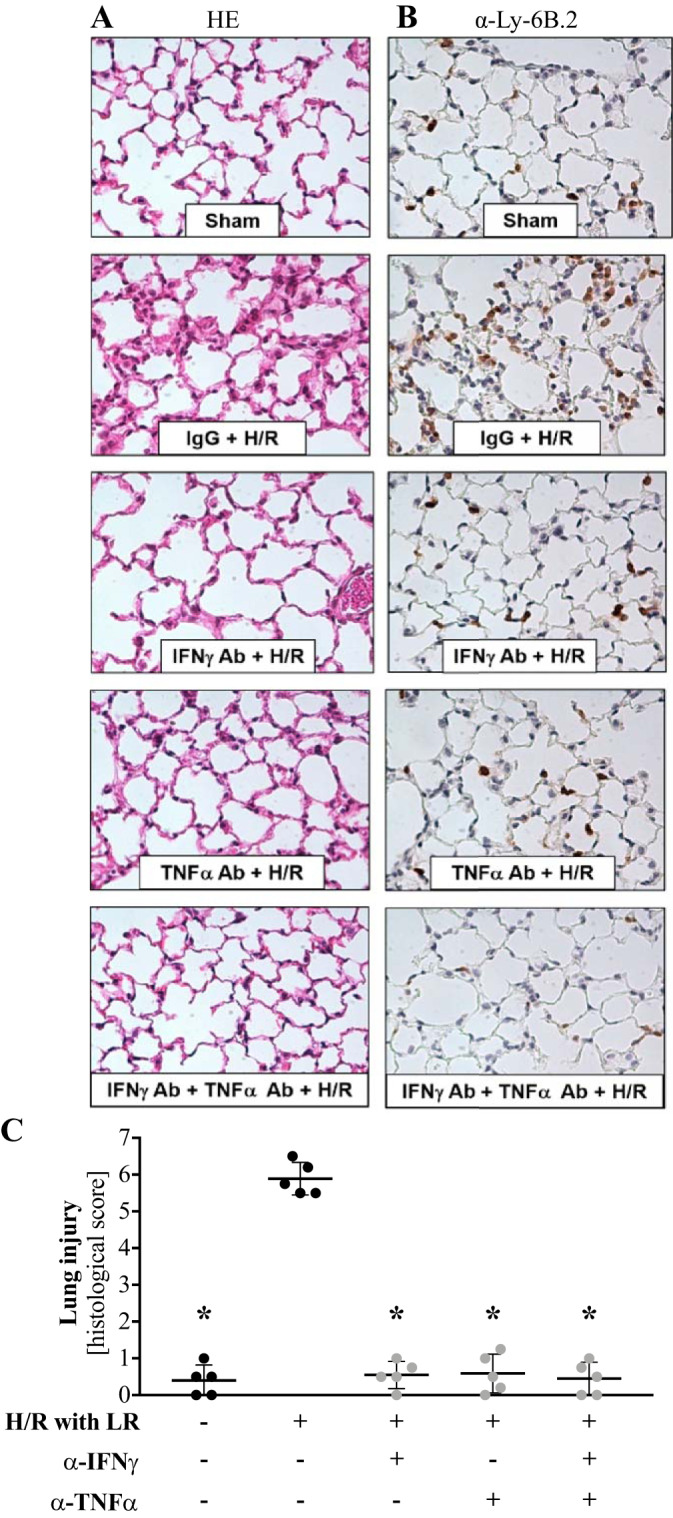
Neutralization of IFNγ and/or TNFα signaling ameliorates pulmonary inflammation following hemorrhage and resuscitation (H/R). At 2 h after intraperitoneal injection with antibodies against IFNγ (α-IFNγ) and/or TNFα (α-TNFα), mice were subjected to a pressure-clamped model of H/R with Ringer’s lactate (LR) or a sham procedure. After 4 h, mice were euthanized, and the left lung was harvested for histology. A and B: lung sections stained with hematoxylin and eosin (HE) and anti-Ly-6B.2 antibody. C: quantification of immune cell infiltration by a blinded investigator in 10 vision fields per mouse (n = 5 mice/group). *P < 0.05 vs. H/R only (by ANOVA with Tukey’s post test).
DISCUSSION
We previously reported that CCL22/MDC levels are elevated after H/R (41). We now show that, among the tested immune subsets in the lung, only CCL22/MDC+ alveolar macrophages and CCL22/MDC+CD103+ dendritic cells undergo significant expansion in response to H/R, with alveolar macrophages being 16 times more abundant than CD103+ dendritic cells after H/R. Thus, alveolar macrophages are most likely the predominant cellular source of CCL22/MDC in the lungs after H/R.
Given that IFNγ is known to drive CCL22/MDC production in keratinocytes (17, 56), we determined if IFNγ levels are also elevated in the context of H/R. To our knowledge, our study is the first to describe increases in systemic and pulmonary IFNγ levels after hemorrhage. IFNγ is typically produced by natural killer cells, natural killer T cells, T helper type 1 CD4 cells, and CD8 cytotoxic T lymphocytes (44). While we did not investigate which cell type produces IFNγ upon H/R, we did confirm that IFNγ stimulated CCL22/MDC production by alveolar macrophages. Subsequently, this was shown to involve JAK, STAT-1, NF-κB, and p38 MAPK, but not AP-1, signaling pathways. This is congruent with previous reports: IFNγ led to p38 MAPK-dependent nuclear translocation of NF-κB and phosphorylation of STAT-1 and subsequent CCL22/MDC production in keratinocytes (24), MDC production in response to LPS stimulation of monocytes also involved p38 MAPK and NF-κB (49), and MDC production in bladder cancer involved NF-κB, but not AP-1 (54). One limitation of these data is that all inhibitors, except ruxolitinib, resulted in some reduction in cell viability, which could also account for a decrease in CCL22/MDC production. However, at the chosen concentrations, the inhibitor’s effect on cell viability was mild (<10%; data not shown), whereas the effect on CCL22/MDC production was much more pronounced (30–50%), suggesting that the effect on CCL22/MDC production was indeed specific to the respective pathway inhibition.
The involvement of NF-κB and p38 MAPK prompted us to investigate the role of TNFα as well, since both factors are well-known components of the TNFα signaling pathway. We could indeed confirm that IFNγ stimulation enhances TNFα production by alveolar macrophages. Given the short half-life of IFNγ in vivo and, thus, the necessity for cumulative IFNγ quantification, we could not confirm the kinetics of IFNγ and TNFα production in our in vivo model. We did, however, confirm that TNFα levels are elevated systemically and in the lungs after H/R. Systemic TNFα levels rose earlier than pulmonary TNFα levels, indicating that early TNFα production in vivo is driven by nonlung cells. We further showed that blockage of TNFα signaling abrogated CCL22/MDC production by alveolar macrophages in vitro, suggesting an autocrine signaling mechanism. Blockage of TNFα signaling also successfully prevented CCL22/MDC elevation in vivo.
Hemorrhagic shock and its sequelae are a leading cause of morbidity and mortality among trauma patients (23). Hemorrhagic patients require resuscitation with crystalloid fluids, as well as human blood products, to restore circulatory volume and maintain end-organ perfusion (8, 15, 16, 45). This can cause substantial lung injury due to the induction of several inflammatory cytokines and chemokines, with subsequent pulmonary edema, neutrophil infiltration, and tissue hypoxia (51). Our laboratory previously showed that CCL22/MDC is elevated after H/R and that this event was associated with enhanced cell recruitment to the lungs. We now offer a more detailed understanding of this pathway, showing that CCL22/MDC is produced by alveolar macrophages in response to IFNγ and TNFα. In other contexts, CCL22/MDC is known to predominantly promote type II immune responses (54). This could indicate that its production in response to proinflammatory and cytotoxic cytokines, such as IFNγ and TNFα, is actually a mechanism to promote tissue healing after injury. Consistent with this idea, we did observe ameliorated signs of lung injury and reduced pulmonary inflammatory cell infiltration in hemorrhagic mice when IFNγ and/or TNFα signaling was blocked with neutralizing antibodies. Further studies are necessary to determine the mechanisms of pulmonary injury upon hemorrhage and the precise role of CCL22/MDC, IFNγ, and TNFα.
In summary, we have outlined a novel mechanism mediating lung inflammation after H/R: IFNγ is elevated after H/R and drives TNFα and MDC production by alveolar macrophages via a signaling mechanism involving p38 MAPK, NF-κB, JAK, and STAT-1. Additionally, TNFα is also induced systemically early after H/R in alveolar macrophages, and TNFα potentiates CCL22/MDC production in an autocrine manner. Neutralization of IFNγ or TNFα signaling with specific antibodies works synergistically to block CCL22/MDC production in vivo and ameliorate pulmonary injury upon H/R. Thorough understanding of the mediators of pulmonary injury offers an opportunity to identify new therapeutic targets and ameliorate morbidity and mortality associated with acute lung injury after trauma.
GRANTS
This work was supported by National Institute of General Medical Sciences Grants R01 GM-107625 (to T. A. Pritts) and T32 GM-08478 (to J. M. Sutton).
DISCLAIMERS
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health and National Institute of General Medical Sciences.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
N.B., J.M.S., R.S.H., P.L.J., T.A.J., A.B.L., C.C.C., and T.A.P. conceived and designed research; N.B., J.M.S., R.S.H., P.L.J., L.A.F., T.A.J., and R.M.S. performed experiments; N.B., J.M.S., R.S.H., P.L.J., T.A.J., A.B.L., C.C.C., and T.A.P. analyzed data; N.B., J.M.S., R.S.H., P.L.J., T.A.J., A.B.L., C.C.C., and T.A.P. interpreted results of experiments; N.B. prepared figures; N.B. drafted manuscript; N.B., J.M.S., R.S.H., P.L.J., L.A.F., T.A.J., R.M.S., A.B.L., C.C.C., and T.A.P. edited and revised manuscript; N.B., J.M.S., R.S.H., P.L.J., L.A.F., T.A.J., R.M.S., A.B.L., C.C.C., and T.A.P. approved final version of manuscript.
REFERENCES
- 1.Abraham E, Carmody A, Shenkar R, Arcaroli J. Neutrophils as early immunologic effectors in hemorrhage- or endotoxemia-induced acute lung injury. Am J Physiol Lung Cell Mol Physiol 279: L1137–L1145, 2000. doi: 10.1152/ajplung.2000.279.6.L1137. [DOI] [PubMed] [Google Scholar]
- 2.Abraham E, Jesmok G, Tuder R, Allbee J, Chang YH. Contribution of tumor necrosis factor-α to pulmonary cytokine expression and lung injury after hemorrhage and resuscitation. Crit Care Med 23: 1319–1326, 1995. doi: 10.1097/00003246-199508000-00004. [DOI] [PubMed] [Google Scholar]
- 3.Andrew DP, Chang MS, McNinch J, Wathen ST, Rihanek M, Tseng J, Spellberg JP, Elias CG III. STCP-1 (MDC) CC chemokine acts specifically on chronically activated Th2 lymphocytes and is produced by monocytes on stimulation with Th2 cytokines IL-4 and IL-13. J Immunol 161: 5027–5038, 1998. [PubMed] [Google Scholar]
- 4.Belizaire RM, Prakash PS, Richter JR, Robinson BR, Edwards MJ, Caldwell CC, Lentsch AB, Pritts TA. Microparticles from stored red blood cells activate neutrophils and cause lung injury after hemorrhage and resuscitation. J Am Coll Surg 214: 648–655, 2012. doi: 10.1016/j.jamcollsurg.2011.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Centers for Disease Control and. Prevention Fatal Injury Data. WISQARS, 2015. [Google Scholar]
- 6.Cimolai N, Taylor GP, Mah D, Morrison BJ. Definition and application of a histopathological scoring scheme for an animal model of acute Mycoplasma pneumoniae pulmonary infection. Microbiol Immunol 36: 465–478, 1992. doi: 10.1111/j.1348-0421.1992.tb02045.x. [DOI] [PubMed] [Google Scholar]
- 7.Cuenda A, Rouse J, Doza YN, Meier R, Cohen P, Gallagher TF, Young PR, Lee JC. SB 203580 is a specific inhibitor of a MAP kinase homologue which is stimulated by cellular stresses and interleukin-1. FEBS Lett 364: 229–233, 1995. doi: 10.1016/0014-5793(95)00357-F. [DOI] [PubMed] [Google Scholar]
- 8.Duchesne JC, Barbeau JM, Islam TM, Wahl G, Greiffenstein P, McSwain NE Jr. Damage control resuscitation: from emergency department to the operating room. Am Surg 77: 201–206, 2011. [DOI] [PubMed] [Google Scholar]
- 9.Fanjul A, Dawson MI, Hobbs PD, Jong L, Cameron JF, Harlev E, Graupner G, Lu XP, Pfahl M. A new class of retinoids with selective inhibition of AP-1 inhibits proliferation. Nature 372: 107–111, 1994. doi: 10.1038/372107a0. [DOI] [PubMed] [Google Scholar]
- 10.Finkelman FD, Morris SC. Development of an assay to measure in vivo cytokine production in the mouse. Int Immunol 11: 1811–1818, 1999. doi: 10.1093/intimm/11.11.1811. [DOI] [PubMed] [Google Scholar]
- 11.Giannoudis PV. Current concepts of the inflammatory response after major trauma: an update. Injury 34: 397–404, 2003. doi: 10.1016/S0020-1383(02)00416-3. [DOI] [PubMed] [Google Scholar]
- 12.Godiska R, Chantry D, Raport CJ, Sozzani S, Allavena P, Leviten D, Mantovani A, Gray PW. Human macrophage-derived chemokine (MDC), a novel chemoattractant for monocytes, monocyte-derived dendritic cells, and natural killer cells. J Exp Med 185: 1595–1604, 1997. doi: 10.1084/jem.185.9.1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hierholzer C, Kalff JC, Omert L, Tsukada K, Loeffert JE, Watkins SC, Billiar TR, Tweardy DJ. Interleukin-6 production in hemorrhagic shock is accompanied by neutrophil recruitment and lung injury. Am J Physiol Lung Cell Mol Physiol 275: L611–L621, 1998. doi: 10.1152/ajplung.1998.275.3.L611. [DOI] [PubMed] [Google Scholar]
- 14.Hierholzer C, Kelly E, Tsukada K, Loeffert E, Watkins S, Billiar TR, Tweardy DJ. Hemorrhagic shock induces G-CSF expression in bronchial epithelium. Am J Physiol Lung Cell Mol Physiol 273: L1058–L1064, 1997. doi: 10.1152/ajplung.1997.273.5.L1058. [DOI] [PubMed] [Google Scholar]
- 15.Holcomb JB, Jenkins D, Rhee P, Johannigman J, Mahoney P, Mehta S, Cox ED, Gehrke MJ, Beilman GJ, Schreiber M, Flaherty SF, Grathwohl KW, Spinella PC, Perkins JG, Beekley AC, McMullin NR, Park MS, Gonzalez EA, Wade CE, Dubick MA, Schwab CW, Moore FA, Champion HR, Hoyt DB, Hess JR. Damage control resuscitation: directly addressing the early coagulopathy of trauma. J Trauma 62: 307–310, 2007. doi: 10.1097/TA.0b013e3180324124. [DOI] [PubMed] [Google Scholar]
- 16.Holcomb JB, Pati S. Optimal trauma resuscitation with plasma as the primary resuscitative fluid: the surgeon’s perspective. Hematology (Am Soc Hematol Educ Program) 2013: 656–659, 2013. doi: 10.1182/asheducation-2013.1.656. [DOI] [PubMed] [Google Scholar]
- 17.Horikawa T, Nakayama T, Hikita I, Yamada H, Fujisawa R, Bito T, Harada S, Fukunaga A, Chantry D, Gray PW, Morita A, Suzuki R, Tezuka T, Ichihashi M, Yoshie O. IFN-γ-inducible expression of thymus and activation-regulated chemokine/CCL17 and macrophage-derived chemokine/CCL22 in epidermal keratinocytes and their roles in atopic dermatitis. Int Immunol 14: 767–773, 2002. doi: 10.1093/intimm/dxf044. [DOI] [PubMed] [Google Scholar]
- 18.Houghton CA, Fassett RG, Coombes JS. Sulforaphane and other nutrigenomic Nrf2 activators: can the clinician’s expectation be matched by the reality? Oxid Med Cell Longev 2016: 1, 2016. doi: 10.1155/2016/7857186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang C, Ma WY, Dawson MI, Rincon M, Flavell RA, Dong Z. Blocking activator protein-1 activity, but not activating retinoic acid response element, is required for the antitumor promotion effect of retinoic acid. Proc Natl Acad Sci USA 94: 5826–5830, 1997. doi: 10.1073/pnas.94.11.5826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Imai T, Chantry D, Raport CJ, Wood CL, Nishimura M, Godiska R, Yoshie O, Gray PW. Macrophage-derived chemokine is a functional ligand for the CC chemokine receptor 4. J Biol Chem 273: 1764–1768, 1998. doi: 10.1074/jbc.273.3.1764. [DOI] [PubMed] [Google Scholar]
- 21.Imai T, Nagira M, Takagi S, Kakizaki M, Nishimura M, Wang J, Gray PW, Matsushima K, Yoshie O. Selective recruitment of CCR4-bearing Th2 cells toward antigen-presenting cells by the CC chemokines thymus and activation-regulated chemokine and macrophage-derived chemokine. Int Immunol 11: 81–88, 1999. doi: 10.1093/intimm/11.1.81. [DOI] [PubMed] [Google Scholar]
- 22.Jugde F, Alizadeh M, Boissier C, Chantry D, Siproudhis L, Corbinais S, Quelvennec E, Dyard F, Campion JP, Gosselin M, Bretagne JF, Sémana G, Heresbach D. Quantitation of chemokines (MDC, TARC) expression in mucosa from Crohn’s disease and ulcerative colitis. Eur Cytokine Netw 12: 468–477, 2001. [PubMed] [Google Scholar]
- 23.Kauvar DS, Lefering R, Wade CE. Impact of hemorrhage on trauma outcome: an overview of epidemiology, clinical presentations, and therapeutic considerations. J Trauma 60, Suppl: S3–S11, 2006. doi: 10.1097/01.ta.0000199961.02677.19. [DOI] [PubMed] [Google Scholar]
- 24.Kwon DJ, Bae YS, Ju SM, Goh AR, Youn GS, Choi SY, Park J. Casuarinin suppresses TARC/CCL17 and MDC/CCL22 production via blockade of NF-κB and STAT1 activation in HaCaT cells. Biochem Biophys Res Commun 417: 1254–1259, 2012. doi: 10.1016/j.bbrc.2011.12.119. [DOI] [PubMed] [Google Scholar]
- 25.Lali FV, Hunt AE, Turner SJ, Foxwell BM. The pyridinyl imidazole inhibitor SB203580 blocks phosphoinositide-dependent protein kinase activity, protein kinase B phosphorylation, and retinoblastoma hyperphosphorylation in interleukin-2-stimulated T cells independently of p38 mitogen-activated protein kinase. J Biol Chem 275: 7395–7402, 2000. doi: 10.1074/jbc.275.10.7395. [DOI] [PubMed] [Google Scholar]
- 26.Lee BH, Lee TJ, Jung JW, Oh DJ, Choi JC, Shin JW, Park IW, Choi BW, Kim JY. The role of keratinocyte-derived chemokine in hemorrhage-induced acute lung injury in mice. J Korean Med Sci 24: 775–781, 2009. doi: 10.3346/jkms.2009.24.5.775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu SF, Ye X, Malik AB. Inhibition of NF-κB activation by pyrrolidine dithiocarbamate prevents in vivo expression of proinflammatory genes. Circulation 100: 1330–1337, 1999. doi: 10.1161/01.CIR.100.12.1330. [DOI] [PubMed] [Google Scholar]
- 28.Liu SF, Ye X, Malik AB. Pyrrolidine dithiocarbamate prevents IκB degradation and reduces microvascular injury induced by lipopolysaccharide in multiple organs. Mol Pharmacol 55: 658–667, 1999. [PubMed] [Google Scholar]
- 29.Lomas JL, Chung CS, Grutkoski PS, LeBlanc BW, Lavigne L, Reichner J, Gregory SH, Doughty LA, Cioffi WG, Ayala A. Differential effects of macrophage inflammatory chemokine-2 and keratinocyte-derived chemokine on hemorrhage-induced neutrophil priming for lung inflammation: assessment by adoptive cells transfer in mice. Shock 19: 358–365, 2003. doi: 10.1097/00024382-200304000-00011. [DOI] [PubMed] [Google Scholar]
- 30.Makley AT, Goodman MD, Belizaire RM, Friend LA, Johannigman JA, Dorlac WC, Lentsch AB, Pritts TA. Damage control resuscitation decreases systemic inflammation after hemorrhage. J Surg Res 175: e75–e82, 2012. doi: 10.1016/j.jss.2011.11.1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Makley AT, Goodman MD, Friend LA, Deters JS, Johannigman JA, Dorlac WC, Lentsch AB, Pritts TA. Resuscitation with fresh whole blood ameliorates the inflammatory response after hemorrhagic shock. J Trauma 68: 305–311, 2010. doi: 10.1097/TA.0b013e3181cb4472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mantovani A, Gray PA, Van Damme J, Sozzani S. Macrophage-derived chemokine (MDC). J Leukoc Biol 68: 400–404, 2000. [PubMed] [Google Scholar]
- 33.Misharin AV, Morales-Nebreda L, Mutlu GM, Budinger GR, Perlman H. Flow cytometric analysis of macrophages and dendritic cell subsets in the mouse lung. Am J Respir Cell Mol Biol 49: 503–510, 2013. doi: 10.1165/rcmb.2013-0086MA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ohta K, Nakayama K, Kurokawa T, Kikuchi T, Yoshimura N. Inhibitory effects of pyrrolidine dithiocarbamate on endotoxin-induced uveitis in Lewis rats. Invest Ophthalmol Vis Sci 43: 744–750, 2002. [PubMed] [Google Scholar]
- 36.Palleroni AV, Hajos S, Wright RB, Palleroni NJ. Nitric oxide synthase induction in lines of macrophages from different anatomical sites. Cell Mol Biol (Noisy-le-grand) 44: 527–535, 1998. [PubMed] [Google Scholar]
- 37.Palleroni AV, Varesio L, Wright RB, Brunda MJ. Tumoricidal alveolar macrophage and tumor infiltrating macrophage cell lines. Int J Cancer 49: 296–302, 1991. doi: 10.1002/ijc.2910490226. [DOI] [PubMed] [Google Scholar]
- 38.Panina-Bordignon P, Papi A, Mariani M, Di Lucia P, Casoni G, Bellettato C, Buonsanti C, Miotto D, Mapp C, Villa A, Arrigoni G, Fabbri LM, Sinigaglia F. The C-C chemokine receptors CCR4 and CCR8 identify airway T cells of allergen-challenged atopic asthmatics. J Clin Invest 107: 1357–1364, 2001. doi: 10.1172/JCI12655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Park YK, Ramalingam M, Kim S, Jang BC, Park JW. Sulforaphane inhibits the interferon-γ-induced expression of MIG, IP-10 and I-TAC in INS-1 pancreatic β-cells through the downregulation of IRF-1, STAT-1 and PKB. Int J Mol Med 40: 907–912, 2017. doi: 10.3892/ijmm.2017.3054. [DOI] [PubMed] [Google Scholar]
- 41.Richter JR, Sutton JM, Belizaire RM, Friend LA, Schuster RM, Johannigman TA, Miller SG, Lentsch AB, Pritts TA. Macrophage-derived chemokine (CCL22) is a novel mediator of lung inflammation following hemorrhage and resuscitation. Shock 42: 525–531, 2014. doi: 10.1097/SHK.0000000000000253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ritter M, Göggel R, Chaudhary N, Wiedenmann A, Jung B, Weith A, Seither P. Elevated expression of TARC (CCL17) and MDC (CCL22) in models of cigarette smoke-induced pulmonary inflammation. Biochem Biophys Res Commun 334: 254–262, 2005. doi: 10.1016/j.bbrc.2005.06.084. [DOI] [PubMed] [Google Scholar]
- 43.Sauaia A, Moore FA, Moore EE, Moser KS, Brennan R, Read RA, Pons PT. Epidemiology of trauma deaths: a reassessment. J Trauma 38: 185–193, 1995. doi: 10.1097/00005373-199502000-00006. [DOI] [PubMed] [Google Scholar]
- 44.Schoenborn JR, Wilson CB. Regulation of interferon-γ during innate and adaptive immune responses. Adv Immunol 96: 41–101, 2007. doi: 10.1016/S0065-2776(07)96002-2. [DOI] [PubMed] [Google Scholar]
- 45.Schorn MN, Phillippi JC. Volume replacement following severe postpartum hemorrhage. J Midwifery Womens Health 59: 336–343, 2014. doi: 10.1111/jmwh.12186. [DOI] [PubMed] [Google Scholar]
- 46.Schreck R, Meier B, Männel DN, Dröge W, Baeuerle PA. Dithiocarbamates as potent inhibitors of nuclear factor-κB activation in intact cells. J Exp Med 175: 1181–1194, 1992. doi: 10.1084/jem.175.5.1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sozzani S, Ghezzi S, Iannolo G, Luini W, Borsatti A, Polentarutti N, Sica A, Locati M, Mackay C, Wells TN, Biswas P, Vicenzi E, Poli G, Mantovani A. Interleukin 10 increases CCR5 expression and HIV infection in human monocytes. J Exp Med 187: 439–444, 1998. doi: 10.1084/jem.187.3.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Strieter RM, Kunkel SL. Acute lung injury: the role of cytokines in the elicitation of neutrophils. J Investig Med 42: 640–651, 1994. [PubMed] [Google Scholar]
- 49.Tsai YC, Chang HW, Chang TT, Lee MS, Chu YT, Hung CH. Effects of all-trans retinoic acid on Th1- and Th2-related chemokines production in monocytes. Inflammation 31: 428–433, 2008. doi: 10.1007/s10753-008-9095-x. [DOI] [PubMed] [Google Scholar]
- 50.Tschöp J, Martignoni A, Goetzman HS, Choi LG, Wang Q, Noel JG, Ogle CK, Pritts TA, Johannigman JA, Lentsch AB, Caldwell CC. γδT cells mitigate the organ injury and mortality of sepsis. J Leukoc Biol 83: 581–588, 2008. doi: 10.1189/jlb.0707507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.van Meurs M, Wulfert FM, Knol AJ, De Haes A, Houwertjes M, Aarts LP, Molema G. Early organ-specific endothelial activation during hemorrhagic shock and resuscitation. Shock 29: 291–299, 2008. doi: 10.1097/SHK.0b013e318145a7c1. [DOI] [PubMed] [Google Scholar]
- 52.Wernig G, Kharas MG, Okabe R, Moore SA, Leeman DS, Cullen DE, Gozo M, McDowell EP, Levine RL, Doukas J, Mak CC, Noronha G, Martin M, Ko YD, Lee BH, Soll RM, Tefferi A, Hood JD, Gilliland DG. Efficacy of TG101348, a selective JAK2 inhibitor, in treatment of a murine model of JAK2V617F-induced polycythemia vera. Cancer Cell 13: 311–320, 2008. doi: 10.1016/j.ccr.2008.02.009. [DOI] [PubMed] [Google Scholar]
- 52a.World Health Organization The 10 Leading Causes of Death in the World, 2000–2016. https://www.who.int/news-room/fact-sheets/detail/the-top-10-causes-of-death [November 2018].
- 52b.World Health Organization Injuries and Violence: The Facts. https://www.who.int/violence_injury_prevention/key_facts/en/. [November 2018].
- 53.Xia BT, Beckmann N, Winer LK, Pugh AM, Pritts TA, Nomellini V, Gulbins E, Caldwell CC. Amitriptyline reduces inflammation and mortality in a murine model of sepsis. Cell Physiol Biochem 52: 565–579, 2019. [DOI] [PubMed] [Google Scholar]
- 54.Yamada H, Luo Y, Matsumoto T, O’Donnell MA. A novel expression of macrophage derived chemokine in human bladder cancer. J Urol 173: 990–995, 2005. doi: 10.1097/01.ju.0000155188.04120.f8. [DOI] [PubMed] [Google Scholar]
- 55.Yang CH, Fang IM, Lin CP, Yang CM, Chen MS. Effects of the NF-κB inhibitor pyrrolidine dithiocarbamate on experimentally induced autoimmune anterior uveitis. Invest Ophthalmol Vis Sci 46: 1339–1347, 2005. doi: 10.1167/iovs.04-0640. [DOI] [PubMed] [Google Scholar]
- 56.Yano C, Saeki H, Komine M, Kagami S, Tsunemi Y, Ohtsuki M, Nakagawa H. Mechanism of macrophage-derived chemokine/CCL22 production by HaCaT keratinocytes. Ann Dermatol 27: 152–156, 2015. doi: 10.5021/ad.2015.27.2.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhou T, Georgeon S, Moser R, Moore DJ, Caflisch A, Hantschel O. Specificity and mechanism-of-action of the JAK2 tyrosine kinase inhibitors ruxolitinib and SAR302503 (TG101348). Leukemia 28: 404–407, 2014. [Erratum in Leukemia 28: 471-472, 2014.] doi: 10.1038/leu.2013.205. [DOI] [PubMed] [Google Scholar]