

Abstract
We examined the effect of acute intermittent hypoxia (IH) on sympathetic neural firing patterns and the role of the carotid chemoreceptors. We hypothesized exposure to acute IH would increase muscle sympathetic nerve activity (MSNA) via an increase in action potential (AP) discharge rates and within-burst firing. We further hypothesized any change in discharge patterns would be attenuated during acute chemoreceptor deactivation (hyperoxia). MSNA (microneurography) was assessed in 17 healthy adults (11 male/6 female; 31 ± 1 yr) during normoxic rest before and after 30 min of experimental IH. Prior to and following IH, participants were exposed to 2 min of 100% oxygen (hyperoxia). AP patterns were studied from the filtered raw MSNA signal using wavelet-based methodology. Compared with baseline, multiunit MSNA burst incidence (P < 0.01), AP incidence (P = 0.01), and AP content per burst (P = 0.01) were increased following IH. There was an increase in the probability of a particular AP cluster firing once (P < 0.01) and more than once (P = 0.03) per burst following IH. There was no effect of hyperoxia on multiunit MSNA at baseline or following IH (P > 0.05); however, hyperoxia following IH attenuated the probability of particular AP clusters firing more than once per burst (P < 0.01). Acute IH increases MSNA by increasing AP discharge rates and within-burst firing. A portion of the increase in within-burst firing following IH can be attributed to the carotid chemoreceptors. These data advance the mechanistic understanding of sympathetic activation following acute IH in humans.
Keywords: carotid bodies, carotid chemoreceptors, hypoxia, intermittent hypoxia, muscle sympathetic nerve activity, sympathetic nervous system
INTRODUCTION
Sleep apnea is the most common form of sleep-disordered breathing. Patients with sleep apnea experience transient, repetitive reductions in arterial oxygen saturation [intermittent hypoxia (IH)], which lead to persistent activation of the sympathetic nervous system (44, 51, 66, 67). IH resulting from recurrent apneas has been implicated as the primary stimulus for increases in sympathetic nervous system activity (6, 42, 53). In animal models, IH-mediated sympathoexcitation occurs as a result of altered carotid body chemoreceptor function (12, 38, 48, 56). Along these lines, carotid body denervation has emerged as a potential treatment for hypertension and other sympathoexcitatory conditions (47, 57). Although a link between IH, sympathetic activation, and hypertension is established in animal models, the pathogenic mechanisms responsible in humans are not completely understood.
The technique of microneurography in humans makes it possible to evaluate sympathetic outflow directed toward the peripheral vasculature (77). Sympathetic neural recordings are often rectified and integrated, resulting in multiunit “bursts” that represent simultaneous firing of several axons in close proximity to the electrode tip (63). Unfortunately, it is not clear whether increases in the incidence of multiunit integrated muscle sympathetic nerve activity (MSNA) bursts are due to increases in the firing rate of already active neurons and/or the recruitment of additional neurons (61). The frequency of neural discharge determines neurotransmitter release patterns (27), and the number of active neurons may vary with each multiunit burst of activity (34). Work in humans has shown IH may have differential effects on the occurrence versus the amplitude of multiunit bursts of MSNA (32); with this, no information is available regarding firing or recruitment of postganglionic sympathetic neurons or mechanisms driving these responses in human IH. This is likely important in the context of hypertension, given that the size or strength of neural discharge can predict the magnitude of skeletal muscle vasoconstriction (14). Consistent with this, recent observations in patients with hypertension have uncovered augmented neural firing/recruitment patterns that were otherwise undetected within the traditional integrated neurogram (3).
Data suggest changes in multiunit burst occurrence and size may reflect distinct sites involved in the control of MSNA (21, 22). Consistent with this concept, previous work in humans has shown that larger amplitude axons [which fire with low probability (16)] primarily contribute to larger amplitude multiunit bursts (61). These larger axons are relatively silent under baseline conditions and are recruited with chemoreflex activation (61–63, 69). In this way, the presence of certain postganglionic action potentials (APs) may vary depending on the influence of chemoreflex inputs. Preclinical work suggests the carotid chemoreceptors are not obligatory in the sympathetic response to acute IH and are instead more important after longer-term exposure (48). In contrast, data from Querido and colleagues (55) suggest the carotid chemoreceptors are obligatory for the increase in MSNA following acute hypoxia in humans. Interestingly, the carotid chemoreceptors were shown to mediate changes in MSNA burst occurrence following hypoxia but not total MSNA (which takes into account burst amplitude) (55).
With this information in mind, we aimed to characterize sympathetic neuronal discharge patterns in response to acute IH in humans and identify the contribution of the carotid body chemoreceptors to sympathetic discharge patterns following acute IH in humans. We hypothesized IH increases the discharge rate and number of postganglionic sympathetic axons recruited following return to room-air breathing. We further hypothesized that the carotid body chemoreceptors contribute to MSNA neural firing and recruitment patterns after acute IH exposure in healthy adults.
METHODS
Participants.
All experiments and procedures were approved by the Institutional Review Board (16-004563) and conformed to the Declaration of Helsinki. Preliminary results from the present investigation were published previously in abstract form (17). All participants were young (<40 yr of age), healthy, nonobese (body mass index <30 kg/m2), and nonsmokers. Participants were without chronic disease, and women were not pregnant (negative pregnancy urine test). One male participant was prescribed Sumatriptan for migraines, which he took on an as-needed basis. Importantly, this individual did not take the medication for a minimum 48 h before study participation. No other individuals reported medication use other than oral contraceptives. Women were studied in the placebo phase of oral contraceptive use (n = 6). Participants were asked to refrain from alcohol, caffeine, and exercise for 24 h and fast for 12 h before the screen and study visits. Written informed consent was obtained from all participants on a screen visit, followed by medical history and fasting blood chemistries. Participants were given an overnight pulse oximeter to wear during sleep. Participants were instructed to sleep for a standard 8–9 h with oxygen saturation and heart rate monitored continuously (Wrist Ox, model 3150, Nonin Medical, Plymouth, MN). The device provided data for an automated scoring algorithm, which calculated an oxygen desaturation event index (4% drop in for a minimum duration of 10 s) adjusted for artifacts. All participants were required to have an adjusted index of <10 per hour to avoid potential effects of undiagnosed sleep apnea.
Instrumentation.
On the study day, participants were admitted to the laboratory at 0800 h. Participants rested supine during instrumentation, which included a 3-lead electrocardiogram and pulse oximetry (GE Datex-Ohmeda Cardiocap/5, GE Healthcare, Chicago, IL). A 20-gauge, 5-cm catheter was placed in the brachial artery under aseptic conditions after local anesthesia (2% lidocaine) for periodic blood sampling (i.e., norepinephrine, epinephrine) and beat-by-beat arterial blood pressure measurement. Of note, the use of an arterial catheter has the potential for underdamping/resonance of the blood pressure tracing, which can cause blood pressure to read artificially high (58, 59). Blood samples were placed on ice and centrifuged at 4°C. Plasma was removed and samples were stored at −80°C for later analysis. Plasma catecholamines were measured with reverse-phase high-performance liquid chromatography with electrochemical detection after extraction with activated alumina.
Muscle sympathetic nerve activity.
Resting MSNA was recorded using the technique of microneurography (60, 77). Multiunit MSNA was recorded using 2.0 ± 0.4 MΩ impedance tungsten microelectrode with a 200-µm shaft diameter tapering to 1–5 µm at the uninsulated tip (FHC, Bowdoin, ME). The microelectrode was placed percutaneously into the peroneal nerve, posterior to the fibular head under direct two-dimensional ultrasound guidance (8). A muscle sympathetic fascicle was identified when taps on the muscle belly or passive muscle stretch evoked mechanoreceptive impulses, and no afferent neural response was evoked by skin stimuli. A reference electrode was positioned subcutaneously ~4 cm from the recording electrode. The recorded signal was amplified 80,000-fold, band-pass filtered (700–2,000 Hz), rectified, and integrated (resistance-capacitance integrator circuit, time constant 0.1 s) by a nerve-traffic analyzer (662C-3; Bioengineering of University of Iowa). Sympathetic neurograms were analyzed by computer-assisted inspection of the mean voltage neurogram based on an expected burst latency of 1.3 s from the preceding R-wave. A voltage deviation was identified as a burst if it exceeded a noise “threshold” (typically 20% of the maximal deviation from zero). A trained individual (J. K. Limberg) reviewed the record and manually added or removed bursts as appropriate using visual appraisal of the raw neurogram for confirmation (29).
AP patterns were detected and extracted using wavelet-based methodology on the raw, band-pass-filtered neurogram using techniques developed in the laboratory of Dr. J. Kevin Shoemaker (60). As described previously (30, 46), a continuous wavelet transform using a “mother wavelet” with the same morphology as a physiological postganglionic sympathetic AP was used for AP detection. APs were only extracted if they occurred within an integrated burst (i.e., 0.8 s around the peak of the burst). A continuous wavelet transform with the matched mother wavelet was applied to the filtered MSNA signal to provide a wavelet coefficient (i.e., resemblance index) between the AP of interest and the mother wavelet such that the resemblance index was largest in the presence of APs and negligible when applied to noise. Wavelet coefficients related to APs and noise were separated based on threshold analysis.
Nonoverlapping APs were grouped into separate clusters based on their morphology using a 32-point matching model (32-point K-means). Extracted APs were ordered based on peak-to-peak amplitude, and histogram analysis was performed to separate APs into amplitude-based clusters. Cluster bin widths were automatically defined based on Scott’s rule, which balanced bin width, data bias, and variance to minimize the integrated mean square root error (64). The number of total clusters (i.e., bins or groups of APs with similar peak-to-peak amplitudes) in the neurogram varied by intervention (i.e., pre- and post-IH). Therefore, bin characteristics (minimum histogram bin width, maximum bin center, and total number of bins) were normalized within each individual to the condition with the largest detected AP to ensure corresponding clusters at baseline and following IH contained AP with identical peak-to-peak amplitudes.
AP cluster firing probability distributions were constructed to evaluate the firing probability of each sympathetic AP cluster at baseline and following IH. To do so, we counted the number of times a given cluster fired only once per burst, divided that number by the total number of bursts that occurred within the condition of interest, and multiplied by 100 to give a percentage (23). The probability of 100% indicates clusters within the bin fired once in every integrated burst, whereas a probability of less than 100% indicates the cluster was not active in every burst. We also calculated the probability of a given cluster firing more than once per burst (number of times a given cluster fired more than once per burst, divided by the total number of bursts, multiplied by 100). The probability of 100% indicates clusters within the bin fired more than once in every integrated burst, whereas a probability less than 100% indicates the cluster did not fire multiple times in every burst. Because the number of total clusters varies by individual, normalization procedures were completed using methods published previously (5, 30, 46). Briefly, APs were normalized to the largest detected cluster (given a value of 100%). Each cluster was placed into 1 of 10 equally sized bins containing a 10% range of the largest detected cluster (i.e., 0–9.9% of largest detected cluster).
Acute intermittent hypoxia.
Participants were instrumented with a mask connected to a nonrebreathing valve. Breath-by-breath tidal volume (Universal Ventilation meter, VacuMed, Ventura, CA), respiratory rate, and inspired/expired gasses (GE Datex-Ohmeda Cardiocap/5, GE Healthcare, Chicago, IL) were monitored and used to calculate minute ventilation. IH was achieved by alternating between a hypercapnic hypoxic (5% oxygen, 3% carbon dioxide, balance nitrogen) gas cylinder and room air (21% oxygen, normoxia) to target ~15 events over 30 min (31). A 50-Liter meteorological balloon served as a volume reservoir. This technique results in arterial desaturation events similar to those observed in sleep apnea [5–10% desaturations; 15–30 events per hour (31, 67)]. A ventilatory response test was conducted immediately before and following acute IH. Tests took ~15 min to complete, at which time hypoxia was achieved using 2–6 breaths of 5% oxygen/3% carbon dioxide followed by room air (7, 49). This was repeated up to 5 times per test. Chemosensitivity was assessed as the slope of the regression line for minute ventilation versus oxygen saturation ().
Modified Dejour’s test.
Before and after acute IH, participants breathed room air (21% oxygen, normoxia) freely followed by acute exposure to 100% oxygen (hyperoxia) (11, 80). During hyperoxia, participants were quietly switched to 100% oxygen for 2 min to experimentally “turn off” the carotid chemoreceptors (11, 71). A 50-Liter meteorological balloon served as a volume reservoir. The modified Dejour’s test was followed by a minimum 10-min washout period to avoid any potential carry over on subsequent testing.
Statistical analysis.
Data were recorded at 10,000 Hz using a computer data acquisition system (PowerLab, ADInstruments) and stored for offline analysis (60). Normoxic resting data were analyzed over a 5-min baseline period before and following acute IH. Data during acute hyperoxia were analyzed from the first 2 min of hyperoxia exposure and, for consistency, compared with the last 2-min of normoxia baseline immediately preceding the exposure (72).
Multiunit MSNA was then expressed as burst frequency (bursts/min), burst incidence (bursts per 100 heartbeats), burst amplitude [arbitrary units (AU)], and mean burst area (AU per min). AP data are reported as AP frequency (number of APs per minute), AP incidence (number of APs per 100 heartbeats), and mean AP content per integrated burst, which are indicative of the number of efferent postganglionic sympathetic neurons active within a specific frame of time. Cluster data are reported as the average number of active clusters per integrated burst (mean clusters/burst) and the maximum number of active clusters per integrated burst (max clusters/burst). With this, the presence of a new cluster represents recruitment of additional populations of APs following IH, which were not present at baseline.
Statistical analysis was completed using SigmaPlot 14.0 (Systat Software, Inc). An αof P < 0.05 was considered statistically significant. All data are reported as means ± SE. The effect of IH (pre-IH, post-IH) on main outcome variables was assessed using a one-way repeated measures analysis of variance (ANOVA). Normality was assessed using the Shapiro–Wilk test. If not normally distributed, a Friedman repeated measures ANOVA on Ranks was conducted. Data presented in Fig. 3 were analyzed with a two-way repeated measures ANOVA to determine the main effect of relative cluster size (10–100% of total clusters), condition (pre-IH, post-IH), and the interaction of cluster and condition. The effect of IH (pre-IH, post-IH) and chemoreceptor desensitization (normoxia, hyperoxia) on main outcome variables was assessed using a two-way repeated measures ANOVA. Pairwise comparisons were done with the Holm–Sidak method.
Fig. 3.

Effect of acute intermittent hypoxia (IH) on sympathetic nervous system activity during quiet normoxic rest. A: firing once per burst. B: firing more than once per burst. Data are reported as means ± SE from n = 17 (11 male/6 female) participants. The effect of relative cluster size (10–100% of total clusters) and IH (pre-IH, post-IH) and the interaction of cluster and IH were assessed using a two-way repeated measures ANOVA. *P < 0.05 vs. pre-IH.
RESULTS
Participant demographics.
Data from 17 (11 male/6 female) young (31 ± 1 yr), healthy individuals are included in the present investigation, and participant characteristics are reported in Table 1.
Table 1.
Participant demographics
| Sex (male/female) | 11/6 |
| Age, yr | 31 ± 1 |
| Height, cm | 176 ± 2 |
| Weight, kg | 81 ± 3 |
| Body mass index, kg/m2 | 26 ± 1 |
| Oxygen desaturation index, events/h | 2 ± 1 |
| Glucose, mg/dL | 88 ± 2 |
| Total cholesterol, mg/dL | 185 ± 7 |
| Triglycerides, mg/dL | 87 ± 9 |
| HDL cholesterol, mg/dL | 61 ± 4 |
Data are reported as means ± SE from n = 17 participants unless otherwise noted (Glucose n = 16).
Intermittent hypoxia.
Acute IH resulted in 15 events where inspired oxygen was reduced (inspired oxygen: 12.8 ± 0.7%). This exposure resulted in significant reductions in oxygen saturation (: baseline 99 ± 1% vs. average nadir 92 ± 1%, P < 0.01), whereas end-tidal carbon dioxide during the 30-min protocol was maintained at pre-IH levels (5.6 ± 0.1% vs. 5.6 ± 0.1%, P = 0.33) (Fig. 1). After IH and return to steady-state room-air breathing, participants continued to exhibit greater diastolic blood pressure, breathing frequency, and minute ventilation compared with baseline (P < 0.05 for all). See Table 2. An increase in chemosensitivity following acute IH was also observed (Table 2).
Fig. 1.

Acute intermittent hypoxia protocol. Representative data from male participant [27 yr, 21.9 kg/m2; muscle sympathetic nerve activity (MSNA) signal-to-noise ratio = 4.2]. BP, blood pressure; HR, heart rate; MSNAint, integrated MSNA signal; , oxygen saturation; Vt, tidal volume.
Table 2.
Effect of acute intermittent hypoxia on hemodynamics during quiet normoxic rest
| Pre-IH | Post-IH | P Value | |
|---|---|---|---|
| Heart rate, beats/min | 60 ± 2 | 62 ± 2 | 0.06 |
| Systolic blood pressure, mmHg | 139 ± 2 | 140 ± 2 | 0.77 |
| Diastolic blood pressure, mmHg | 77 ± 2 | 78 ± 2 | 0.04 |
| Mean blood pressure, mmHg | 98 ± 2 | 100 ± 2 | 0.09 |
| Breathing frequency, breaths/min | 12 ± 1 | 13 ± 1 | 0.14 |
| Tidal volume, mL/breath | 606 ± 51 | 598 ± 45 | 0.74 |
| Minute ventilation, L/min | 6.8 ± 0.4 | 7.6 ± 0.4 | <0.01 |
| End-tidal carbon dioxide, % | 5.6 ± 0.1 | 5.2 ± 0.1 | <0.01 |
| Oxygen saturation () | 99 ± 0.3 | 99 ± 0.2 | 0.63 |
| Hypoxic ventilatory response, L/min/% | −0.64 ± 0.08 | −0.85 ± 0.09 | 0.03 |
Data are reported as means ± SE from n = 17 (11 male/6 female) participants. The effect of intermittent hypoxia (IH) (pre-IH, post-IH) was assessed using a one-way repeated measures ANOVA.
Neural firing and recruitment.
Compared with baseline, an increase in multiunit MSNA burst frequency (bursts/min) and burst incidence (bursts/100 heartbeats) was observed, in addition to an increase in mean burst area (P < 0.01 for all) (Fig. 2, Table 3). Consistent with this, there was an increase in plasma norepinephrine (P = 0.03) and epinephrine (P = 0.01) following acute IH. The increase in multiunit MSNA following acute IH was due to an increase in the frequency and incidence of AP spikes (AP/min, AP/burst, AP/100 heartbeats; all P < 0.01) (Table 3). Additionally, there was an increase in the probability of APs from the same cluster firing once per integrated MSNA burst (single firing, Fig. 3A; main effect of IH, P < 0.01) and more than once per burst (multiple firing, Fig. 3B; main effect of IH, P = 0.03) following IH. Specifically, there was an increase in the probability of multiple within-burst firing in the smallest two cluster bins (i.e., populations of APs with the smallest peak-to-peak amplitudes) following acute IH (Fig. 3B, interaction of cluster and IH, P < 0.01). A significant increase in cluster incidence was also observed (P ≤ 0.01, Table 3), signifying recruitment of latent axons. The AP cluster size-latency profile was unaffected by acute IH (main effect of IH, P = 0.97) (Fig. 4).
Fig. 2.

Steady-state normoxia before and after acute intermittent hypoxia. Representative data from male participant (35 yr, 25.5 kg/m2; muscle sympathetic nerve activity (MSNA) signal-to-noise ratio pre = 5.6, post = 6.0). BP, blood pressure; HR, heart rate; MSNAint, integrated MSNA signal; Vt, tidal volume.
Table 3.
Effect of acute intermittent hypoxia on sympathetic nervous system activity during quiet normoxic rest
| Pre-IH | Post-IH | P Value | |
|---|---|---|---|
| Burst frequency, bursts/min | 19 ± 2 | 25 ± 2 | <0.01 |
| Burst incidence, bursts/100 heartbeats | 33 ± 4 | 43 ± 5 | <0.01 |
| Mean burst amplitude, AU | 0.15 ± 0.01 | 0.17 ± 0.01 | 0.14 |
| Mean burst area, AU/min | 0.9 ± 0.1 | 1.4 ± 0.2 | <0.01 |
| Action potential frequency, AP/min | 155 ± 26 | 286 ± 56 | <0.01 |
| Action potential incidence, AP/100 heartbeats | 272 ± 50 | 501 ± 112 | 0.01 |
| Action potential incidence, AP/burst | 7.4 ± 0.8 | 9.9 ± 1.4 | 0.01 |
| Mean cluster incidence, clusters/burst | 4.3 ± 0.4 | 5.4 ± 0.5 | <0.01 |
| Max cluster incidence, clusters/burst | 8.5 ± 0.7 | 10.4 ± 1.0 | 0.01 |
| MSNA signal-to-noise ratio | 4.4 ± 0.2 | 4.8 ± 0.2 | 0.02 |
| Norepinephrine, pg/mL | 185 ± 17 | 210 ± 17 | 0.03 |
| Epinephrine, pg/mL | 52 ± 6 | 72 ± 11 | 0.01 |
Data are reported as means ± SE from n = 17 (11 male/6 female) participants. The effect of intermittent hypoxia (IH) (pre-IH, post-IH) was assessed using a one-way repeated measures ANOVA. AP, action potential; AU, arbitrary units; MSNA, muscle sympathetic nerve activity.
Fig. 4.
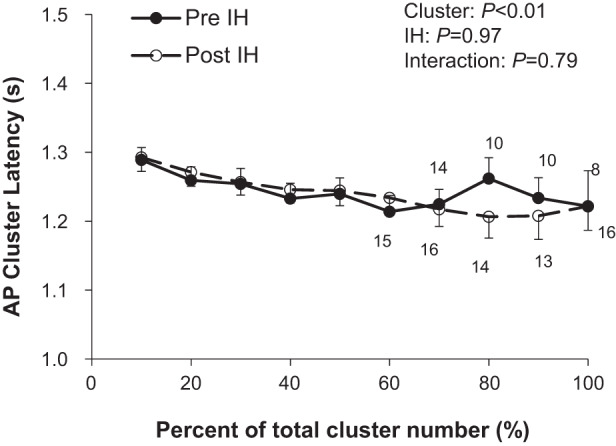
Effect of acute intermittent hypoxia (IH) on action potential (AP) cluster latency. Data are reported as means ± SE from n = 17 (11 male/6 female) participants unless otherwise noted. Numbers above data points indicate number of participants. The effect of relative cluster size (10–100% of total clusters) and IH (pre-IH, post-IH) and the interaction of cluster and IH were assessed using a two-way repeated measures ANOVA.
Acute hyperoxia.
Of the 17 individuals who completed the IH protocol, stable MSNA signals were available during the modified Dejour’s test before and following IH from 11 individuals (6 male/5 female). Acute hyperoxia resulted in a fall in heart rate and blood pressure (main effect of hyperoxia, P < 0.01) that did not differ from pre- to post-IH (interaction of IH and hyperoxia, P value range 0.19–0.49; Table 4). No changes in multiunit MSNA were observed during acute hyperoxia, nor were any changes in the frequency or incidence of AP spikes observed (main effect of hyperoxia, P > 0.05 for all) (Table 4). No changes in cluster incidence (clusters/burst) were observed under hyperoxic conditions (P > 0.05, Table 4). Consistent with this, there were no changes with hyperoxia in the probability of APs from the same cluster firing once (Fig. 5A) or more than once (Fig. 5B) per integrated MSNA burst before IH (main effect of hyperoxia, P > 0.05; interaction of cluster and hyperoxia, P > 0.05). In contrast, following acute IH, there was a main effect of hyperoxia on the probability of APs from the same cluster firing once (Fig. 5C; main effect of hyperoxia, P = 0.02) and more than once (Fig. 5D; interaction of cluster and hyperoxia, P < 0.01) per burst. Specifically, there was a decrease in the probability of multiple firing in the smallest two cluster bins with exposure to acute hyperoxia (Fig. 5D). The AP cluster size-latency profile was unaffected by acute hyperoxia (Fig. 6, P value range 0.48–1.00).
Table 4.
Effect of acute hyperoxia on the sympathetic and hemodynamic response to intermittent hypoxia
| Pre-IH |
Post-IH |
P Value |
|||||
|---|---|---|---|---|---|---|---|
| Normoxia | Hyperoxia | Normoxia | Hyperoxia | IH | Hyperoxia | Interaction | |
| Heart rate, beats/min | 58 ± 3 | 56 ± 3 | 63 ± 3 | 59 ± 3 | 0.02 | <0.01 | 0.22 |
| Systolic blood pressure, mmHg | 141 ± 4 | 140 ± 3 | 143 ± 3 | 142 ± 3 | 0.37 | <0.01 | 0.19 |
| Diastolic blood pressure, mmHg | 77 ± 2 | 76 ± 2 | 80 ± 2 | 79 ± 2 | 0.04 | <0.01 | 0.49 |
| Mean blood pressure, mmHg | 99 ± 3 | 98 ± 3 | 102 ± 3 | 101 ± 3 | 0.09 | <0.01 | 0.37 |
| Breathing frequency, breaths/min | 12 ± 1 | 12 ± 1 | 13 ± 1 | 14 ± 2 | 0.14 | 0.82 | 0.52 |
| Tidal volume, mL/breath | 548 ± 58 | 493 ± 57 | 591 ± 55 | 598 ± 59 | 0.08 | 0.56 | 0.36 |
| Minute ventilation, L/min | 6.6 ± 0.6 | 5.9 ± 0.7 | 7.5 ± 0.5 | 8.3 ± 1.3 | 0.01 | 0.95 | 0.03 |
| End-tidal carbon dioxide, % | 5.4 ± 0.1 | 5.5 ± 0.1 | 5.2 ± 0.1 | 5.1 ± 0.1 | <0.01 | 0.38 | 0.20 |
| Oxygen saturation (), % | 99 ± 0.3 | 100 ± 0.2 | 100 ± 0.2 | 100 ± 0.2 | 0.13 | <0.01 | 0.36 |
| Burst frequency, bursts/min | 17 ± 1 | 18 ± 2 | 23 ± 2 | 23 ± 2 | <0.01 | 0.82 | 0.91 |
| Burst incidence, bursts/100 heartbeats | 30 ± 3 | 33 ± 4 | 38 ± 5 | 41 ± 6 | 0.04 | 0.13 | 0.64 |
| Mean burst amplitude, AU | 0.15 ± 0.01 | 0.15 ± 0.01 | 0.17 ± 0.01 | 0.18 ± 0.01 | <0.01 | 0.30 | 0.85 |
| Mean burst area, AU/min | 0.8 ± 0.1 | 0.8 ± 0.1 | 1.2 ± 0.1 | 1.3 ± 0.1 | <0.01 | 0.09 | 0.10 |
| Action potential frequency, AP/min | 142 ± 18 | 146 ± 17 | 245 ± 36 | 254 ± 40 | <0.01 | 0.36 | 0.64 |
| Action potential incidence, AP/100 heartbeats | 258 ± 39 | 275 ± 41 | 431 ± 89 | 482 ± 113 | 0.02 | 0.09 | 0.28 |
| Action potential incidence, AP/burst | 8.3 ± 0.9 | 8.5 ± 0.8 | 10.5 ± 1.2 | 10.9 ± 1.3 | 0.01 | 0.30 | 0.91 |
| Mean cluster incidence, clusters/burst | 4.4 ± 0.3 | 4.5 ± 0.3 | 5.2 ± 0.3 | 5.4 ± 0.3 | <0.01 | 0.19 | 0.68 |
| Max cluster incidence, clusters/burst | 8.1 ± 0.6 | 7.8 ± 0.4 | 9.5 ± 0.6 | 9.4 ± 0.4 | <0.01 | 0.38 | 0.82 |
| MSNA signal-to-noise ratio | 4.9 ± 0.3 | 5.0 ± 0.3 | 5.4 ± 0.3 | 5.4 ± 0.3 | 0.03 | 0.07 | 0.99 |
Data are reported as means ± SE from n = 11 (6 male/5 female) participants. The effect of intermittent hypoxia (IH) (pre-IH, post-IH), hyperoxia (normoxia, hyperoxia), and the interaction of IH and hyperoxia were assessed using a two-way repeated measures ANOVA. AP, action potential; AU, arbitrary units; MSNA, muscle sympathetic nerve activity.
Fig. 5.

Effect of acute hyperoxia on the sympathetic response to intermittent hypoxia (IH). Probability of action potentials from the cluster firing once per burst (A) and more than once per burst (B) before IH and firing once per burst (C) and more than once per burst (D) after acute IH. Data are reported as means ± SE from n = 11 (6 male/5 female) participants. The effect of relative cluster size (10–100% of total clusters) and hyperoxia (normoxia, hyperoxia) and the interaction of cluster and hyperoxia were assessed using a two-way repeated measures ANOVA. *P < 0.05 vs. normoxia.
Fig. 6.
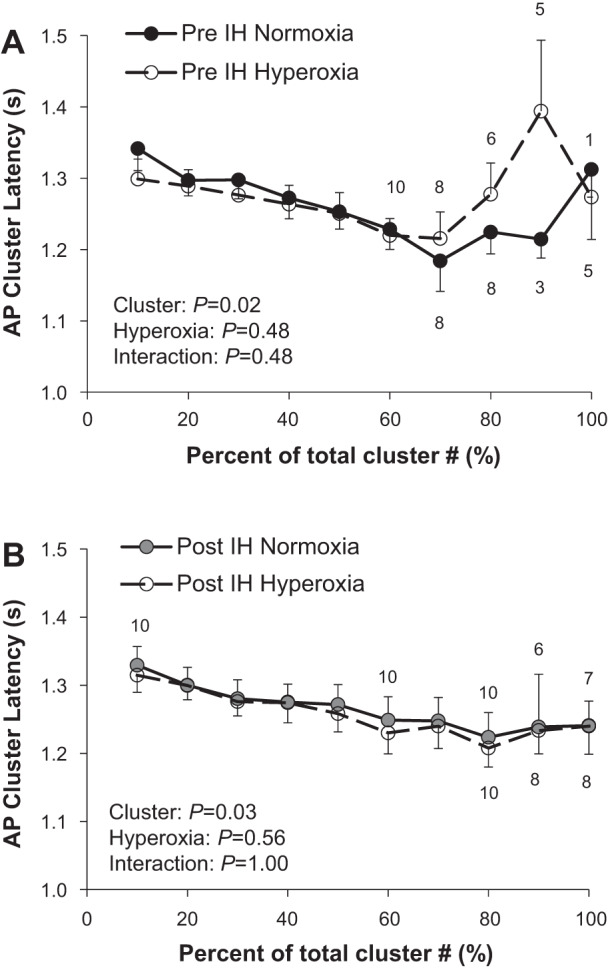
A and B: effect of acute hyperoxia on action potential (AP) cluster latency. Data are reported as means ± SE from n = 11 (6 male/5 female) participants unless otherwise noted. The effect of relative cluster size (10–100% of total clusters) and hyperoxia (normoxia, hyperoxia) and the interaction of cluster and hyperoxia were assessed using a two-way ANOVA. Numbers above data points indicate number of participants. IH, intermittent hypoxia.
DISCUSSION
Main findings from the present study are twofold: 1) following acute IH, sympathetic neuronal discharge is increased via an increase in within-burst firing of smaller APs and recruitment of previously latent, larger amplitude APs and 2) following acute IH, attenuation of carotid body chemoreceptor activity (i.e., hyperoxia) attenuates within-burst firing of smaller amplitude APs with no effect on recruitment. These data have important implications for the mechanistic understanding of sympathetic activation following IH in humans as well as potential downstream effects.
Intermittent hypoxia-induced sympathoexcitation.
Repeated, intermittent exposure to hypoxia in humans results in consistent increases in MSNA that persist beyond the duration of the hypoxic exposure; this phenomenon has been repeatedly observed in both clinical (13, 37, 66, 75) and laboratory settings (9, 10, 19, 28, 42, 73, 74, 81). Along these lines, we observed an increase in integrated multiunit MSNA burst frequency and burst incidence following IH, which persisted upon return to room air (Table 3). We also observed an increase in burst area following acute IH, which is evident in our representative tracing (Fig. 2). An increase in burst amplitude and/or area following exposure to hypoxia is consistent with some (9, 10, 28, 41, 42, 70, 81) but not all (32) previous recordings in humans, and in a number of instances, burst amplitude and/or area is not reported (15, 19, 20, 40, 73). In contrast to present data, Lusina and colleagues (32) found acute hypoxic exposures over 10 days in healthy adults increased multiunit MSNA burst frequency, but not burst amplitude, following subsequent hypoxic exposure. Differences in hypoxic stimulus (steady-state vs. acute intermittent) may have contributed to discrepancies between results.
Changes in burst area likely reflect a change in total number of neurons recruited and/or recruitment of different populations of neurons (36, 45). However, prior work examining the integrated raw MSNA neurogram is limited in its ability to explore this phenomenon. In the present investigation, we show for the first time that an acute bout of IH facilitates an increase in firing probability and mean firing rates of individual groups of sympathetic neurons (Table 3), as well as an increase in multiple within-burst firing (Fig. 3). Patients with sleep apnea exhibit an increase in firing probability and mean firing rates of individual neurons when compared with control (13, 33). Similarly, patients with sleep apnea also exhibit greater multiple within-burst firing [neurons firing more than once within a cardiac interval (13)] compared with healthy controls. This pattern of multiple within-burst firing in patients with sleep apnea is consistent with other respiratory diseases (e.g., chronic obstructive pulmonary disease) but is unlike what is observed in individuals with heart failure (13, 33). Together, these data support patterns of sympathetic activation that differ between pathophysiological conditions (i.e., cardiovascular vs. respiratory) (33, 35).
Multiple within-burst firing in itself may cause greater norepinephrine release. Specifically, Lambert and colleagues (26) found that greater incidence of multiple within-burst firing from sympathetic neurons in the peroneal nerve was associated with greater cardiac norepinephrine spillover. The observed increase in the incidence of multiple within-burst firing following IH in the present investigation may be associated with preferential increases in sympathetic outflow. Thus, an increase in multiple firing will have significant physiological consequences that have important clinical implications for conditions prone to repeated hypoxic exposures (e.g., sleep apnea). Of note, although we observed significant increases in circulating norepinephrine and epinephrine following acute IH (Table 3), there are multiple factors that influence plasma levels (including release, binding, and reuptake). With this, a post hoc analysis did not identify any direct relationships between changes in norepinephrine and key MSNA variables (data not shown).
In addition to an increase in within-burst firing, an increase in multiunit burst amplitude may be attributed to the presence of larger-amplitude, higher-threshold axons (i.e., larger amplitude AP clusters that fire with low probability) (61–63, 69). Consistent with this, we observed recruitment of latent (i.e., not present at baseline) sympathetic neurons following exposure to IH (Table 3). In this setting, recruitment of latent, larger-amplitude clusters is likely a normal physiological response to acute IH. In contrast, augmented sympathetic recruitment has been postulated to contribute to elevated blood pressures in disease states (3). Larger sympathetic bursts containing larger APs likely elicit greater vasoconstriction and, in the setting of IH, provide a potential mechanism by which the progression of hypertension in individuals with sleep apnea may occur (14, 61). Recent work from Stuckless et al. (73) showed greater neurovascular transduction following IH; based on present data, it is likely that alterations in neural firing and/or recruitment play an important role in this response. Unfortunately, whether the increase in firing probability and mean firing rates following acute IH is due to augmented descending drive and/or a reduction in the firing threshold of the neurons themselves cannot be explained with the present data.
An inverse relationship exists between burst latency (delay of an integrated MSNA burst from a representative ECG R-wave) and burst size, such that larger bursts exhibit shorter reflex latencies (78). Consistent with this, we observed an inverse relationship between AP cluster amplitude and conduction velocity (Figs. 4 and 6). However, we did not observe any effect of acute IH on the AP cluster size-latency profile (Fig. 4). Although we did not have participants quantify how stressful they perceived the IH exposure, anecdotally it is markedly less stressful than the breathhold apnea or Valsalva maneuvers, which are typically held to volitional tolerance. Therefore, it is unlikely that acute IH alters central processing times; rather, this appears to occur only in situations of perceived stress (1, 2, 4, 5).
Acute intermittent hypoxia and the carotid body chemoreceptors.
Preclinical animal models have shown that an intact carotid body and neural chemoreflex arc are required to elicit IH-mediated increases in sympathetic nervous system activity (25, 51, 52). In humans, Lusina and colleagues (32) uncovered a strong positive relationship between the increase in chemosensitivity and MSNA following acute hypoxia. Along these lines, acute inhibition of the carotid body chemoreceptors (hyperoxia) results in a transient decrease in integrated multiunit MSNA burst area following acute isocapnic hypoxia exposure in humans (55). Together, these data support long-lasting facilitation of sympathetic outflow following acute hypoxic exposure that is dependent on input from the carotid body chemoreceptors. In contrast, there are data that show at least 3 days of IH exposure are required to induce carotid body sensory plasticity in preclinical models (48, 50) and rather only central components of the chemoreflex mediate sympathetic responses to acute IH (39, 54).
With this information in mind, we highlight 3 key observations from the present study: 1) acute hyperoxia has no effect on MSNA before exposure to acute IH (Table 4, Fig. 5, A and B), 2) acute hyperoxia has no effect on integrated multiunit MSNA following exposure to acute IH (Table 4), and 3) attenuation of carotid body chemoreceptor activity (i.e., hyperoxia) following IH attenuates within-burst firing of smaller APs (Fig. 5D) with no effect on recruitment (Table 4). These data agree with previous studies showing little-to-no contribution of the carotid body chemoreceptors to basal MSNA in healthy adults (42, 65, 72), as well as data from humans supporting a role for the carotid body chemoreceptors in the increase in MSNA following acute hypoxia exposure (42, 55).
Interestingly, using a novel analytical approach we see the effect of acute hyperoxia on MSNA following IH is highly variable across clusters (i.e., populations of APs with similar peak-to-peak amplitudes). Lower amplitude APs (related to smaller neurons with a lower recruitment threshold) tend to increase firing probability following IH (Fig. 3B), and these same axons decrease their probability of firing with acute exposure to hyperoxia post-IH exposure (Fig. 5D). This effect of hyperoxia is not observed at rest and thus it is unlikely an independent effect of high oxygen air. Rather, these data appear to support a role for augmented carotid body chemoreceptor activation in multiple within-burst firing of low-threshold axons following IH in healthy humans. This intriguing result is consistent with data showing that continuous positive airway pressure (CPAP) treatment, which reduces hypoxic exposures in patients with sleep apnea, is effective at attenuating MSNA firing probabilities (43, 79).
In contrast to their role in increased firing of low-threshold axons, the carotid chemoreceptors do not play a measurable role in recruitment of previously latent axons following acute IH in the individuals studied. Specifically, there was no effect of acute hyperoxia on firing of high-threshold, higher-amplitude axons (Table 4). This is in contrast to what has been observed in healthy adults using other “derecruitment” protocols [e.g., phenylephrine, trimethaphan (24, 30)], whereby the firing of larger, low-probability APs is abolished first. With this, the mechanisms by which IH leads to sustained MSNA in humans is likely multifactorial.
Experimental considerations.
Strengths of the study design include high-quality, stable neurograms with good signal-to-noise ratios from a relatively large number of human participants. Based on previous work (60), the present level of signal to noise (Tables 3 and 4) would produce a correct AP detection rate of ~90% and false positive detection rate of <3%. Thus, although the detection of APs may vary depending on the signal-to-noise ratio of the neurogram, the quality of MSNA signals (average signal to noise >4.4) was such that this is a minor concern. Another strength is the addition of carbon dioxide during the IH protocol to maintain isocapnia (Fig. 1). The addition of carbon dioxide is important to our IH paradigm to replicate changes that may accompany sleep apnea and prevent hypocapnia, which occurs with acute hyperventilation. End-tidal carbon dioxide levels returned quickly to baseline following the gas exposure and, if anything, participants were mildly hypocapnic following IH due to the slightly elevated resting ventilation (Table 2). Importantly, hypocapnia during post-IH measurements would, if anything, reduce chemoreceptor activation and lower MSNA following IH, leading us to underestimate (rather than overestimate) the effect of IH and the chemoreceptors on MSNA.
With these strengths in mind, there are also three important considerations. First, both male and female participants were included in the present investigation. Very little is known regarding sex differences in the neural response to IH; however, there are data to support (76) and refute (18, 68) sex differences in the peak MSNA response to chemoreflex activation. Although we are likely underpowered, we did not observe any noticeable sex differences in the effect of acute IH on MSNA neural firing patterns (data not shown). Second, we did not include a time control. There are recent data that show integrated MSNA is relatively stable across a similar time period (73); however, the effect of time on neural recruitment patterns has yet to be examined. Last, the “intensity” of the IH stimulus was relatively mild. The goal of the present investigation was to achieve a rate of hypoxic events consistent with moderate-to-severe sleep apnea (25–30 events per hour) with an oxygen desaturation index consistent with clinical cutoffs (≥4% oxygen desaturation from pre-event baseline). With this, much lower saturations are often observed in patients with sleep apnea, and other experimental IH models have elicited much lower saturations during IH cycles. Thus, it is likely that a more severe stimulus would exaggerate the observed responses.
Perspectives and Significance
These data provide new evidence regarding the effect of IH on sympathetic neural firing and recruitment strategies, as well as the role of the carotid chemoreceptors in human sympathetic control. We show that the carotid chemoreceptors contribute, in part, to the firing probabilities of small, low-threshold APs following IH. However, the recruitment of previously silent, larger amplitude and faster-conducting sympathetic neurons following IH is unaffected by acute carotid chemoreceptor deactivation. Augmented sympathetic recruitment has been postulated to contribute to elevated blood pressures in disease states (3). With this, present findings provide a deeper understanding of potential mechanisms by which the progression of cardiovascular disease in individuals exposed to IH (e.g., sleep apnea) may occur (14, 61).
GRANTS
This work was supported by the American Heart Association Grant AHA15SDG25080095 (to J. K. Limberg), National Institutes of Health Grant HL130339 (to J. K. Limberg), and Mayo Clinic Center for Biomedical Discovery (J. K. Limberg).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
E.P.O., J.K.S., and J.K.L. conceived and designed research; E.P.O., D.W.J., S.E.B., W.W.H., Z.M.S., J.K.S., and J.K.L. performed experiments; E.P.O., Z.M.S., and J.K.L. analyzed data; E.P.O., D.W.J., S.E.B., W.W.H., Z.M.S., J.K.S., and J.K.L. interpreted results of experiments; E.P.O. and J.K.L. prepared figures; J.K.L. drafted manuscript; E.P.O., D.W.J., S.E.B., W.W.H., Z.M.S., J.K.S., and J.K.L. edited and revised manuscript; E.P.O., D.W.J., S.E.B., W.W.H., Z.M.S., J.K.S., and J.K.L. approved final version of manuscript.
REFERENCES
- 1.Badrov MB, Barak OF, Mijacika T, Shoemaker LN, Borrell LJ, Lojpur M, Drvis I, Dujic Z, Shoemaker JK. Ventilation inhibits sympathetic action potential recruitment even during severe chemoreflex stress. J Neurophysiol 118: 2914–2924, 2017. doi: 10.1152/jn.00381.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Badrov MB, Lalande S, Olver TD, Suskin N, Shoemaker JK. Effects of aging and coronary artery disease on sympathetic neural recruitment strategies during end-inspiratory and end-expiratory apnea. Am J Physiol Heart Circ Physiol 311: H1040–H1050, 2016. doi: 10.1152/ajpheart.00334.2016. [DOI] [PubMed] [Google Scholar]
- 3.Badrov MB, Okada Y, Yoo JK, Vongpatanasin W, Shoemaker JK, Levine BD, Fu Q. Sex differences in the sympathetic neural recruitment and hemodynamic response to head-up tilt in older hypertensives. Hypertension 75: 458–467, 2020. doi: 10.1161/HYPERTENSIONAHA.119.14009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Badrov MB, Olver TD, Shoemaker JK. Central vs. peripheral determinants of sympathetic neural recruitment: insights from static handgrip exercise and postexercise circulatory occlusion. Am J Physiol Regul Integr Comp Physiol 311: R1013–R1021, 2016. doi: 10.1152/ajpregu.00360.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Badrov MB, Usselman CW, Shoemaker JK. Sympathetic neural recruitment strategies: responses to severe chemoreflex and baroreflex stress. Am J Physiol Regul Integr Comp Physiol 309: R160–R168, 2015. doi: 10.1152/ajpregu.00077.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brooks D, Horner RL, Kozar LF, Render-Teixeira CL, Phillipson EA. Obstructive sleep apnea as a cause of systemic hypertension. Evidence from a canine model. J Clin Invest 99: 106–109, 1997. doi: 10.1172/JCI119120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chua TP, Ponikowski PP, Harrington D, Chambers J, Coats AJ. Contribution of peripheral chemoreceptors to ventilation and the effects of their suppression on exercise tolerance in chronic heart failure. Heart 76: 483–489, 1996. doi: 10.1136/hrt.76.6.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Curry TB, Charkoudian N. The use of real-time ultrasound in microneurography. Auton Neurosci 162: 89–93, 2011. doi: 10.1016/j.autneu.2011.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cutler MJ, Swift NM, Keller DM, Wasmund WL, Burk JR, Smith ML. Periods of intermittent hypoxic apnea can alter chemoreflex control of sympathetic nerve activity in humans. Am J Physiol Heart Circ Physiol 287: H2054–H2060, 2004. doi: 10.1152/ajpheart.00377.2004. [DOI] [PubMed] [Google Scholar]
- 10.Cutler MJ, Swift NM, Keller DM, Wasmund WL, Smith ML. Hypoxia-mediated prolonged elevation of sympathetic nerve activity after periods of intermittent hypoxic apnea. J Appl Physiol (1985) 96: 754–761, 2004. doi: 10.1152/japplphysiol.00506.2003. [DOI] [PubMed] [Google Scholar]
- 11.Dejours P. Control of respiration by arterial chemoreceptors. Ann NY Acad Sci 109: 682–695, 1963. doi: 10.1111/j.1749-6632.1963.tb13497.x. [DOI] [PubMed] [Google Scholar]
- 12.Dick TE, Hsieh YH, Wang N, Prabhakar N. Acute intermittent hypoxia increases both phrenic and sympathetic nerve activities in the rat. Exp Physiol 92: 87–97, 2007. doi: 10.1113/expphysiol.2006.035758. [DOI] [PubMed] [Google Scholar]
- 13.Elam M, McKenzie D, Macefield V. Mechanisms of sympathoexcitation: single-unit analysis of muscle vasoconstrictor neurons in awake OSAS subjects. J Appl Physiol (1985) 93: 297–303, 2002. doi: 10.1152/japplphysiol.00899.2001. [DOI] [PubMed] [Google Scholar]
- 14.Fairfax ST, Padilla J, Vianna LC, Davis MJ, Fadel PJ. Spontaneous bursts of muscle sympathetic nerve activity decrease leg vascular conductance in resting humans. Am J Physiol Heart Circ Physiol 304: H759–H766, 2013. doi: 10.1152/ajpheart.00842.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gilmartin GS, Tamisier R, Curley M, Weiss JW. Ventilatory, hemodynamic, sympathetic nervous system, and vascular reactivity changes after recurrent nocturnal sustained hypoxia in humans. Am J Physiol Heart Circ Physiol 295: H778–H785, 2008. doi: 10.1152/ajpheart.00653.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Henneman E. Relation between size of neurons and their susceptibility to discharge. Science 126: 1345–1347, 1957. doi: 10.1126/science.126.3287.1345. [DOI] [PubMed] [Google Scholar]
- 17.Jacob DW, Baker SE, Scruggs ZM, Ott EP, Harper JL, Manrique C, Shoemaker JK, Limberg JK. Sympathetic discharge patterns and neurovascular transduction following acute intermittent hypoxia. FASEB J 33, Suppl 1: 562.8, 2019. [Google Scholar]
- 18.Jones PP, Davy KP, Seals DR. Influence of gender on the sympathetic neural adjustments to alterations in systemic oxygen levels in humans. Clin Physiol 19: 153–160, 1999. doi: 10.1046/j.1365-2281.1999.00158.x. [DOI] [PubMed] [Google Scholar]
- 19.Jouett NP, Moralez G, Raven PB, Smith ML. Losartan reduces the immediate and sustained increases in muscle sympathetic nerve activity after hyperacute intermittent hypoxia. J Appl Physiol (1985) 122: 884–892, 2017. doi: 10.1152/japplphysiol.00683.2016. [DOI] [PubMed] [Google Scholar]
- 20.Jouett NP, Moralez G, White DW, Eubank WL, Chen S, Tian J, Smith ML, Zimmerman MC, Raven PB. N-Acetylcysteine reduces hyperacute intermittent hypoxia-induced sympathoexcitation in human subjects. Exp Physiol 101: 387–396, 2016. doi: 10.1113/EP085546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Keller DM, Cui J, Davis SL, Low DA, Crandall CG. Heat stress enhances arterial baroreflex control of muscle sympathetic nerve activity via increased sensitivity of burst gating, not burst area, in humans. J Physiol 573: 445–451, 2006. doi: 10.1113/jphysiol.2006.108662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kienbaum P, Karlsson T, Sverrisdottir YB, Elam M, Wallin BG. Two sites for modulation of human sympathetic activity by arterial baroreceptors? J Physiol 531: 861–869, 2001. doi: 10.1111/j.1469-7793.2001.0861h.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Klassen SA, Limberg JK, Baker SE, Nicholson WT, Curry TB, Joyner MJ, Shoemaker JK. The role of the paravertebral ganglia in human sympathetic neural discharge patterns. J Physiol 596: 4497–4510, 2018. doi: 10.1113/JP276440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Klassen SA, Moir ME, Limberg JK, Baker SE, Nicholson WT, Curry TB, Joyner MJ, Shoemaker JK. Asynchronous action potential discharge in human muscle sympathetic nerve activity. Am J Physiol Heart Circ Physiol 317: H754–H764, 2019. doi: 10.1152/ajpheart.00258.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kumar P, Prabhakar N. Sensing hypoxia: carotid body mechanisms and reflexes in health and disease. Respir Physiol Neurobiol 157: 1–3, 2007. doi: 10.1016/j.resp.2007.02.003. [DOI] [PubMed] [Google Scholar]
- 26.Lambert E, Dawood T, Schlaich M, Straznicky N, Esler M, Lambert G. Single-unit sympathetic discharge pattern in pathological conditions associated with elevated cardiovascular risk. Clin Exp Pharmacol Physiol 35: 503–507, 2008. doi: 10.1111/j.1440-1681.2008.04905.x. [DOI] [PubMed] [Google Scholar]
- 27.Lambert EA, Schlaich MP, Dawood T, Sari C, Chopra R, Barton DA, Kaye DM, Elam M, Esler MD, Lambert GW. Single-unit muscle sympathetic nervous activity and its relation to cardiac noradrenaline spillover. J Physiol 589: 2597–2605, 2011. doi: 10.1113/jphysiol.2011.205351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Leuenberger UA, Brubaker D, Quraishi SA, Hogeman CS, Imadojemu VA, Gray KS. Effects of intermittent hypoxia on sympathetic activity and blood pressure in humans. Auton Neurosci 121: 87–93, 2005. [Erratum in Auton Neurosci 183: 120, 2014]. doi: 10.1016/j.autneu.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 29.Limberg JK, Morgan BJ, Schrage WG, Dempsey JA. Respiratory influences on muscle sympathetic nerve activity and vascular conductance in the steady state. Am J Physiol Heart Circ Physiol 304: H1615–H1623, 2013. doi: 10.1152/ajpheart.00112.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Limberg JK, Ott EP, Holbein WW, Baker SE, Curry TB, Nicholson WT, Joyner MJ, Shoemaker JK. Pharmacological assessment of the contribution of the arterial baroreflex to sympathetic discharge patterns in healthy humans. J Neurophysiol 119: 2166–2175, 2018. doi: 10.1152/jn.00935.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Louis M, Punjabi NM. Effects of acute intermittent hypoxia on glucose metabolism in awake healthy volunteers. J Appl Physiol (1985) 106: 1538–1544, 2009. doi: 10.1152/japplphysiol.91523.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lusina SJ, Kennedy PM, Inglis JT, McKenzie DC, Ayas NT, Sheel AW. Long-term intermittent hypoxia increases sympathetic activity and chemosensitivity during acute hypoxia in humans. J Physiol 575: 961–970, 2006. doi: 10.1113/jphysiol.2006.114660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Macefield VG. Firing patterns of muscle vasoconstrictor neurons in respiratory disease. Front Physiol 3: 153, 2012. doi: 10.3389/fphys.2012.00153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Macefield VG, Elam M. Why do human postganglionic neurones primarily only fire once during a sympathetic burst? Acta Physiol Scand 177: 247–253, 2003. doi: 10.1046/j.1365-201X.2003.01078.x. [DOI] [PubMed] [Google Scholar]
- 35.Macefield VG, Wallin BG. Physiological and pathophysiological firing properties of single postganglionic sympathetic neurons in humans. J Neurophysiol 119: 944–956, 2018. doi: 10.1152/jn.00004.2017. [DOI] [PubMed] [Google Scholar]
- 36.Malpas SC. The rhythmicity of sympathetic nerve activity. Prog Neurobiol 56: 65–96, 1998. doi: 10.1016/S0301-0082(98)00030-6. [DOI] [PubMed] [Google Scholar]
- 37.Malpas SC. Sympathetic nervous system overactivity and its role in the development of cardiovascular disease. Physiol Rev 90: 513–557, 2010. doi: 10.1152/physrev.00007.2009. [DOI] [PubMed] [Google Scholar]
- 38.Marcus NJ, Li YL, Bird CE, Schultz HD, Morgan BJ. Chronic intermittent hypoxia augments chemoreflex control of sympathetic activity: role of the angiotensin II type 1 receptor. Respir Physiol Neurobiol 171: 36–45, 2010. doi: 10.1016/j.resp.2010.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mifflin S, Cunningham JT, Toney GM. Neurogenic mechanisms underlying the rapid onset of sympathetic responses to intermittent hypoxia. J Appl Physiol (1985) 119: 1441–1448, 2015. doi: 10.1152/japplphysiol.00198.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Miller AJ, Sauder CL, Cauffman AE, Blaha CA, Leuenberger UA. Endurance training attenuates the increase in peripheral chemoreflex sensitivity with intermittent hypoxia. Am J Physiol Regul Integr Comp Physiol 312: R223–R228, 2017. doi: 10.1152/ajpregu.00105.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Monahan KD, Leuenberger UA, Ray CA. Effect of repetitive hypoxic apnoeas on baroreflex function in humans. J Physiol 574: 605–613, 2006. doi: 10.1113/jphysiol.2006.108977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Morgan BJ, Crabtree DC, Palta M, Skatrud JB. Combined hypoxia and hypercapnia evokes long-lasting sympathetic activation in humans. J Appl Physiol (1985) 79: 205–213, 1995. doi: 10.1152/jappl.1995.79.1.205. [DOI] [PubMed] [Google Scholar]
- 43.Narkiewicz K, Kato M, Phillips BG, Pesek CA, Davison DE, Somers VK. Nocturnal continuous positive airway pressure decreases daytime sympathetic traffic in obstructive sleep apnea. Circulation 100: 2332–2335, 1999. doi: 10.1161/01.CIR.100.23.2332. [DOI] [PubMed] [Google Scholar]
- 44.Narkiewicz K, Somers VK. The sympathetic nervous system and obstructive sleep apnea: implications for hypertension. J Hypertens 15: 1613–1619, 1997. doi: 10.1097/00004872-199715120-00062. [DOI] [PubMed] [Google Scholar]
- 45.Ninomiya I, Malpas SC, Matsukawa K, Shindo T, Akiyama T. The amplitude of synchronized cardiac sympathetic nerve activity reflects the number of activated pre- and postganglionic fibers in anesthetized cats. J Auton Nerv Syst 45: 139–147, 1993. doi: 10.1016/0165-1838(93)90125-E. [DOI] [PubMed] [Google Scholar]
- 46.Ott EP, Baker SE, Holbein WW, Shoemaker JK, Limberg JK. Effect of varying chemoreflex stress on sympathetic neural recruitment strategies during apnea. J Neurophysiol 122: 1386–1396, 2019. doi: 10.1152/jn.00319.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Paton JF, Sobotka PA, Fudim M, Engelman ZJ, Hart EC, McBryde FD, Abdala AP, Marina N, Gourine AV, Lobo M, Patel N, Burchell A, Ratcliffe L, Nightingale A. The carotid body as a therapeutic target for the treatment of sympathetically mediated diseases. Hypertension 61: 5–13, 2013. [Erratum in Hypertension 61: e26, 2013.] doi: 10.1161/HYPERTENSIONAHA.111.00064. [DOI] [PubMed] [Google Scholar]
- 48.Peng YJ, Overholt JL, Kline D, Kumar GK, Prabhakar NR. Induction of sensory long-term facilitation in the carotid body by intermittent hypoxia: implications for recurrent apneas. Proc Natl Acad Sci USA 100: 10073–10078, 2003. doi: 10.1073/pnas.1734109100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ponikowski P, Chua TP, Amadi AA, Piepoli M, Harrington D, Volterrani M, Colombo R, Mazzuero G, Giordano A, Coats AJ. Detection and significance of a discrete very low frequency rhythm in RR interval variability in chronic congestive heart failure. Am J Cardiol 77: 1320–1326, 1996. doi: 10.1016/S0002-9149(96)00199-3. [DOI] [PubMed] [Google Scholar]
- 50.Prabhakar NR, Dick TE, Nanduri J, Kumar GK. Systemic, cellular and molecular analysis of chemoreflex-mediated sympathoexcitation by chronic intermittent hypoxia. Exp Physiol 92: 39–44, 2007. doi: 10.1113/expphysiol.2006.036434. [DOI] [PubMed] [Google Scholar]
- 51.Prabhakar NR, Kumar GK. Mechanisms of sympathetic activation and blood pressure elevation by intermittent hypoxia. Respir Physiol Neurobiol 174: 156–161, 2010. doi: 10.1016/j.resp.2010.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Prabhakar NR, Peng YJ, Jacono FJ, Kumar GK, Dick TE. Cardiovascular alterations by chronic intermittent hypoxia: importance of carotid body chemoreflexes. Clin Exp Pharmacol Physiol 32: 447–449, 2005. doi: 10.1111/j.1440-1681.2005.04209.x. [DOI] [PubMed] [Google Scholar]
- 53.Prabhakar NR, Peng YJ, Kumar GK, Pawar A. Altered carotid body function by intermittent hypoxia in neonates and adults: relevance to recurrent apneas. Respir Physiol Neurobiol 157: 148–153, 2007. doi: 10.1016/j.resp.2006.12.009. [DOI] [PubMed] [Google Scholar]
- 54.Prabhakar NR, Semenza GL. Oxygen sensing and homeostasis. Physiology (Bethesda) 30: 340–348, 2015. doi: 10.1152/physiol.00022.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Querido JS, Kennedy PM, Sheel AW. Hyperoxia attenuates muscle sympathetic nerve activity following isocapnic hypoxia in humans. J Appl Physiol (1985) 108: 906–912, 2010. doi: 10.1152/japplphysiol.01228.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rey S, Del Rio R, Alcayaga J, Iturriaga R. Chronic intermittent hypoxia enhances cat chemosensory and ventilatory responses to hypoxia. J Physiol 560: 577–586, 2004. doi: 10.1113/jphysiol.2004.072033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ribeiro MJ, Sacramento JF, Gonzalez C, Guarino MP, Monteiro EC, Conde SV. Carotid body denervation prevents the development of insulin resistance and hypertension induced by hypercaloric diets. Diabetes 62: 2905–2916, 2013. doi: 10.2337/db12-1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Romagnoli S, Ricci Z, Quattrone D, Tofani L, Tujjar O, Villa G, Romano SM, De Gaudio AR. Accuracy of invasive arterial pressure monitoring in cardiovascular patients: an observational study. Crit Care 18: 644, 2014. doi: 10.1186/s13054-014-0644-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Romagnoli S, Romano SM, Bevilacqua S, Lazzeri C, Gensini GF, Pratesi C, Quattrone D, Dini D, De Gaudio AR. Dynamic response of liquid-filled catheter systems for measurement of blood pressure: precision of measurements and reliability of the Pressure Recording Analytical Method with different disposable systems. J Crit Care 26: 415–422, 2011. doi: 10.1016/j.jcrc.2010.08.010. [DOI] [PubMed] [Google Scholar]
- 60.Salmanpour A, Brown LJ, Shoemaker JK. Spike detection in human muscle sympathetic nerve activity using a matched wavelet approach. J Neurosci Methods 193: 343–355, 2010. doi: 10.1016/j.jneumeth.2010.08.035. [DOI] [PubMed] [Google Scholar]
- 61.Salmanpour A, Brown LJ, Steinback CD, Usselman CW, Goswami R, Shoemaker JK. Relationship between size and latency of action potentials in human muscle sympathetic nerve activity. J Neurophysiol 105: 2830–2842, 2011. doi: 10.1152/jn.00814.2010. [DOI] [PubMed] [Google Scholar]
- 62.Salmanpour A, Frances MF, Goswami R, Shoemaker JK. Sympathetic neural recruitment patterns during the Valsalva maneuver. 2011 Annual International Conference of the IEEE Engineering in Medicine and Biology Society. 2011: 6951–6954, 2011. doi: 10.1109/IEMBS.2011.6091757 [DOI] [PubMed] [Google Scholar]
- 63.Salmanpour A, Shoemaker JK. Baroreflex mechanisms regulating the occurrence of neural spikes in human muscle sympathetic nerve activity. J Neurophysiol 107: 3409–3416, 2012. doi: 10.1152/jn.00925.2011. [DOI] [PubMed] [Google Scholar]
- 64.Scott D. On optimal and data-based histograms. Biometrika 66: 605–610, 1979. doi: 10.1093/biomet/66.3.605. [DOI] [Google Scholar]
- 65.Smorschok MP, Sobierajski FM, Purdy GM, Riske LA, Busch SA, Skow RJ, Matenchuk BA, Pfoh JR, Vanden Berg ER, Linares A, Borle K, Lavoie L, Saran G, Dyck R, Funk DR, Day TA, Boulé NG, Davenport MH, Steinback CD. Peripheral chemoreceptor deactivation attenuates the sympathetic response to glucose ingestion. Appl Physiol Nutr Metab 44: 389–396, 2019. doi: 10.1139/apnm-2018-0062. [DOI] [PubMed] [Google Scholar]
- 66.Somers VK, Dyken ME, Clary MP, Abboud FM. Sympathetic neural mechanisms in obstructive sleep apnea. J Clin Invest 96: 1897–1904, 1995. doi: 10.1172/JCI118235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Somers VK, White DP, Amin R, Abraham WT, Costa F, Culebras A, Daniels S, Floras JS, Hunt CE, Olson LJ, Pickering TG, Russell R, Woo M, Young T; American Heart Association Council for High Blood Pressure Research Professional Education Committee, Council on Clinical Cardiology; American Heart Association Stroke Council; American Heart Association Council on Cardiovascular Nursing; American College of Cardiology Foundation . Sleep apnea and cardiovascular disease: an American Heart Association/American College Of Cardiology Foundation Scientific Statement from the American Heart Association Council for High Blood Pressure Research Professional Education Committee, Council on Clinical Cardiology, Stroke Council, and Council On Cardiovascular Nursing. In collaboration with the National Heart, Lung, and Blood Institute National Center on Sleep Disorders Research (National Institutes of Health). Circulation 118: 1080–1111, 2008. doi: 10.1161/CIRCULATIONAHA.107.189420. [DOI] [PubMed] [Google Scholar]
- 68.Souza GM, Bonagamba LG, Amorim MR, Moraes DJ, Machado BH. Inspiratory modulation of sympathetic activity is increased in female rats exposed to chronic intermittent hypoxia. Exp Physiol 101: 1345–1358, 2016. doi: 10.1113/EP085850. [DOI] [PubMed] [Google Scholar]
- 69.Steinback CD, Breskovic T, Banic I, Dujic Z, Shoemaker JK. Autonomic and cardiovascular responses to chemoreflex stress in apnoea divers. Auton Neurosci 156: 138–143, 2010. doi: 10.1016/j.autneu.2010.05.002. [DOI] [PubMed] [Google Scholar]
- 70.Steinback CD, Kevin Shoemaker J. Differential regulation of sympathetic burst frequency and amplitude following acute hypoxia in humans. Am J Physiol Regul Integr Comp Physiol 303: R633–R638, 2012. doi: 10.1152/ajpregu.00130.2012. [DOI] [PubMed] [Google Scholar]
- 71.Stickland MK, Fuhr DP, Haykowsky MJ, Jones KE, Paterson DI, Ezekowitz JA, McMurtry MS. Carotid chemoreceptor modulation of blood flow during exercise in healthy humans. J Physiol 589: 6219–6230, 2011. doi: 10.1113/jphysiol.2011.218099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Stickland MK, Morgan BJ, Dempsey JA. Carotid chemoreceptor modulation of sympathetic vasoconstrictor outflow during exercise in healthy humans. J Physiol 586: 1743–1754, 2008. doi: 10.1113/jphysiol.2007.147421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Stuckless TJ, Vermeulen TD, Brown CV, Boulet LM, Shafer BM, Wakeham DJ, Steinback CD, Ayas NT, Floras JS, Foster GE. Acute intermittent hypercapnic hypoxia and sympathetic neurovascular transduction in men. J Physiol 598: 473–487, 2020. doi: 10.1113/JP278941. [DOI] [PubMed] [Google Scholar]
- 74.Tamisier R, Pépin JL, Rémy J, Baguet JP, Taylor JA, Weiss JW, Lévy P. 14 nights of intermittent hypoxia elevate daytime blood pressure and sympathetic activity in healthy humans. Eur Respir J 37: 119–128, 2011. doi: 10.1183/09031936.00204209. [DOI] [PubMed] [Google Scholar]
- 75.Taylor KS, Millar PJ, Murai H, Haruki N, Kimmerly DS, Bradley TD, Floras JS. Cortical autonomic network gray matter and sympathetic nerve activity in obstructive sleep apnea. Sleep (Basel) 41: zsx208, 2018. doi: 10.1093/sleep/zsx208. [DOI] [PubMed] [Google Scholar]
- 76.Usselman CW, Steinback CD, Shoemaker JK. Effects of one’s sex and sex hormones on sympathetic responses to chemoreflex activation. Exp Physiol 101: 362–367, 2016. doi: 10.1113/EP085147. [DOI] [PubMed] [Google Scholar]
- 77.Vallbo AB, Hagbarth KE, Torebjörk HE, Wallin BG. Somatosensory, proprioceptive, and sympathetic activity in human peripheral nerves. Physiol Rev 59: 919–957, 1979. doi: 10.1152/physrev.1979.59.4.919. [DOI] [PubMed] [Google Scholar]
- 78.Wallin BG, Burke D, Gandevia S. Coupling between variations in strength and baroreflex latency of sympathetic discharges in human muscle nerves. J Physiol 474: 331–338, 1994. doi: 10.1113/jphysiol.1994.sp020025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Waradekar NV, Sinoway LI, Zwillich CW, Leuenberger UA. Influence of treatment on muscle sympathetic nerve activity in sleep apnea. Am J Respir Crit Care Med 153: 1333–1338, 1996. doi: 10.1164/ajrccm.153.4.8616563. [DOI] [PubMed] [Google Scholar]
- 80.Whipp BJ, Wasserman K. Carotid bodies and ventilatory control dynamics in man. Fed Proc 39: 2668–2673, 1980. [PubMed] [Google Scholar]
- 81.Xie A, Skatrud JB, Crabtree DC, Puleo DS, Goodman BM, Morgan BJ. Neurocirculatory consequences of intermittent asphyxia in humans. J Appl Physiol (1985) 89: 1333–1339, 2000. doi: 10.1152/jappl.2000.89.4.1333. [DOI] [PubMed] [Google Scholar]