

The global pandemic of severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) has resulted in a critical need to rapidly develop new pharmacologic interventions and disseminate information. This has led to confusing and conflicting information on drug efficacy. Remdesivir has emerged as a promising treatment for SARS‐CoV‐2 infection yet published clinical pharmacology and drug interaction studies are limited. Additional studies of the disposition of remdesivir, its active metabolite (GS‐441524), and its triphosphate metabolite (GS‐443902) are needed.
As of April 30, 2020, the global pandemic of SARS‐CoV‐2 infections has resulted in > 3.4 million cases of coronavirus disease 2019 (COVID‐19) and almost 240,000 deaths worldwide. 1 This has led to an urgency to develop new pharmacologic treatment options. Since the start of the pandemic, information on potential therapeutic options have been rapidly disseminated on an almost daily basis, often with incomplete or conflicting results and without adequate peer review. Understandably, while the focus of these investigations has centered on therapeutic efficacy, attention should also be paid to the clinical pharmacology and potential for drug‐drug interactions, which is often lacking published data. For the front‐line clinician, this information is vital to providing optimal care for patients with COVID‐19 with complex underlying medical conditions receiving multiple medications.
Remdesivir (GS‐5734), developed by Gilead Sciences, has recently emerged as a promising new antiviral agent against SARS‐CoV‐2. Initially developed to combat Ebola, remdesivir has been administered to over 1,800 patients worldwide since January 2020 through clinical trials, individual compassionate use, and expanded access. Published reports of remdesivir have shown mixed efficacy. 2 , 3 A case series of 53 hospitalized patients who received remdesivir via the compassionate use program showed an overall mortality of 13%, with higher mortality (18%) in patients who were mechanically ventilated. A randomized, double‐blind placebo‐controlled trial from China found no benefit of remdesivir compared with placebo. However, the study was terminated early due to failure to achieve its target enrollment and was, therefore, underpowered to show clinical benefit. Adverse events were similar in both studies, ranging from 60–66% of patient receiving remdesivir (compared with 64% patients receiving placebo). Serious adverse events occurred in 20–23% of patients. Notable adverse events include rash (8%) and abnormal liver function tests (5–23%). More promising preliminary results are emerging from the National Institute of Health Adaptive COVID‐19 Treatment Trial (ACTT), which showed a statistically significant decrease in time to recovery, defined as return to normal activity or hospital discharge. The median time to recovery was 11 days for patients treated with remdesivir, compared with 15 days for patients receiving placebo (P < 0.001). Although not statistically significant, results did suggest a survival benefit with a mortality of 8.0% of patients in the remdesivir group compared with 11.6% of patients in the placebo group (P = 0.059). 4 Topline results from the open‐label phase III SIMPLE trial showed that patients with severe COVID‐19 manifestations taking a 10‐day treatment course of remdesivir achieved similar improvement in clinical status compared with patients taking a 5‐day course of therapy (odds ratio 0.75; 95% confidence interval 0.51–1.12). 5 Based upon these data, the US Food and Drug Administration (FDA) issued an Emergency Use Authorization (EUA) for use of remdesivir for the treatment of hospitalized patients with COVID‐19 on May 1, 2020. 6
Remdesivir (GS‐5734) is the monophosphoramidate prodrug of the adenosine analog, GS‐441524 (Figure 1 ). Remdesivir has broad antiviral activity against RNA viruses, including filoviruses (Ebola and Marburg) and coronaviruses, such as severe acute respiratory syndrome coronavirus (SARS‐CoV), Middle East respiratory syndrome coronavirus (MERS‐CoV), and SARS‐CoV‐2. 7 Remdesivir exhibits potent in vitro activity against SARS‐CoV‐2 with an half‐maximal effective concentration of 0.77 μM. 8 The majority of pharmacokinetic studies of remdesivir stem from in vitro, in vivo, and limited clinical studies for the treatment of Ebola. For the treatment of COVID‐19, remdesivir is given as a 200 mg i.v. injection on day 1, followed by 100 mg i.v. on days 2–10. Following i.v. administration, remdesivir is rapidly converted in plasma to the intermediate metabolite, GS‐704277, and to the nucleoside analog, GS‐441524. Due to the high first‐pass hepatic extraction of phosphoramidates and expected low bioavailability, oral administration of remdesivir was not explored. 9 In respiratory epithelial cells, GS‐441524 is further converted by intracellular esterases into the pharmacologically active nucleoside triphosphate (GS‐443902), which competes with naturally occurring adenosine phosphate and functions as a delayed RNA‐dependent RNA polymerase inhibitor. 7 Although the plasma half‐lives of remdesivir and the intermediate metabolite are short (~ 0.5–1 hour), the plasma and intracellular half‐lives of the GS‐441524 are long (~ 24 and 40 hours, respectively) allowing for once‐daily dosing. Remdesivir has wide distribution to most tissues, including the kidneys, liver, prostate gland, and salivary gland, but does not cross the blood‐brain barrier. Metabolism is mediated predominantly by hydrolases with 74% and 18% of remdesivir recovered in urine and feces. 10 , 11 The majority of remdesivir recovered in urine was metabolite GS‐441524; 10% was recovered as remdesivir. It is worth noting that remdesivir is formulated in sulfobutylether‐β‐cyclodextrin (SBECD), an excipient to enhance solubility. SBECD has been shown to accumulate in moderate to severe renal dysfunction and, in preclinical studies, has resulted in renal vacuolation. 12 Caution should be exercised when co‐administered with other parenteral medications formulated in SBECD (e.g., voriconazole and amiodarone), particularly in patients with creatinine clearance < 30 mL/minute.
Figure 1.
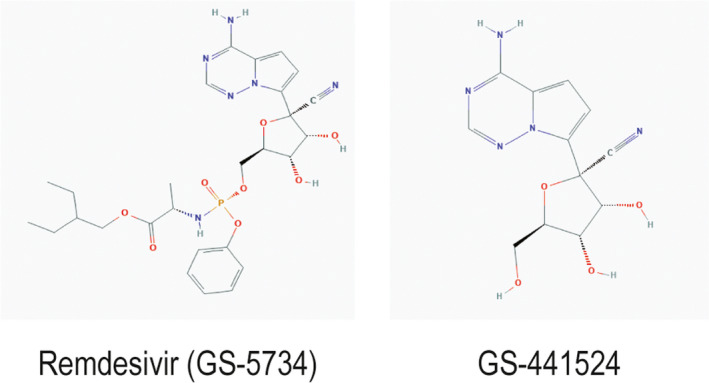
Chemical structures of remdesivir (GS‐5734) and the nucleoside metabolite, GS‐441524. Images sourced from PubChem Database, National Center for Biotechnology Information.
To date, there are only limited in vitro transporter and enzyme‐mediated drug interaction studies on remdesivir. Remdesivir is a substrate of multiple cytochrome P450 enzymes, including CYP2C8, CYP2D6, and CYP3A4, and is also a substrate of the organic anion transporting polypeptide OATP1B1, and P‐glycoprotein (P‐gp). Remdesivir is also a weak inhibitor of CYP3A4, OATP1B1, OATP1B3, bile acid export pump, multidrug resistance‐associated protein 4, and sodium‐taurocholate cotransporter protein. However, metabolism of remdesivir is believed to be mediated predominantly via hydrolases and not CYP enzymes, as noted above. 11 Due to the high extraction ratio of remdesivir, hepatic clearance is likely driven by hepatic blood flow and not metabolic enzyme activity. This, in addition to the short plasma half‐life and need for i.v. administration that avoids hepatic first‐pass metabolism, likely results in a low potential for drug‐drug interactions with the administered drug. Although there was no evidence for CYP induction by either the intermediate metabolite, GS‐7042277, or the active nucleoside metabolite, GS‐441524, it is unclear if either of these metabolites are substrates or inhibitors of P450s or affected by transporters. This will require further study to determine the full clinical impact of drug‐drug interactions with remdesivir therapy.
Although there are no published drug‐drug interaction studies with remdesivir, there are published guidance on the management of potential drug interactions (Table 1 ). Unfortunately, the guidance can be confusing or even contradictory. The Liverpool Drug Interaction Group, well‐known for providing detailed drug interactions for cancer therapeutics as well as drugs for the treatment of hepatitis B, hepatitis C, and HIV, recently launched a comprehensive website detailing drug interactions with experimental COVID‐19 therapies. 13 Listed drugs include remdesivir, lopinavir/ritonavir, chloroquine, hydroxychloroquine, tocilizumab, and favipiravir among others. According to the website (accessed May 4, 2020), co‐administration of strong CYP3A4 inhibitors, such as voriconazole, is labeled as “no clinically significant interaction expected.” In contrast, co‐administration with strong CYP3A4 inducers, such as rifampicin, can lead to decreased remdesivir levels and it is recommended that “these drugs should not be coadministered.” To add to the confusion, three recent published reviews or guidelines of COVID‐19 therapeutics resulted in discordant conclusions regarding the significance and clinical management of potential CYP3A4 interactions. McCreary et al. correctly noted that the initial ACTT protocol erroneously stated that “remdesivir is a prodrug that is metabolized to its active form as a substrate of CYP3A4” when, in fact, remdesivir is activated to the active metabolite GG‐441524 via plasma carboxylesterases and metabolized by hydrolases. 14 The authors concluded that, although remdesivir is a substrate of multiple CYP isoforms, drug interactions with CYP3A4 inhibitors or inducers were unlikely. This is in contrast to a recent review of COVID‐19 pharmacologic therapeutics published in JAMA, which states that while “not a significant inducer/inhibitor of CYP enzymes, monitor with strong inducers/inhibitors.” 15 On April 21, 2020, the National Institutes of Health (NIH) released their own COVID‐19 treatment guidelines. 16 In their review, the authors note that although remdesivir is unlikely to be affected by CYP enzymes or P‐gp transporters, remdesivir should not be administered with strong CYP or P‐gp inducers but may be co‐administered with strong CYP inhibitors. The basis for this recommendation is unclear but is clearly aimed at minimizing the potential for subtherapeutic remdesivir levels. Whereas the impact of strong CYP inhibitors, such as voriconazole, is typically limited to the major isoenzyme, CYP3A4, CYP inducers can affect multiple CYP isoenzymes and transporters leading to a larger cumulative decrease in substrate concentration when metabolized and transported via multiple pathways. At present, the contribution of each CYP isoenzyme and transporter to the overall metabolism of remdesivir and its metabolites is unclear. This approach also assumes that increased remdesivir levels, and potentially increased levels of the active metabolite, GS‐441524, will not lead to increased toxicity, which may or may not be warranted given the short plasma half‐life and safety profile of remdesivir and clinical studies are needed to validate this approach. The Full FDA EUA Prescribing Information released on May 1, 2020, notes that drug‐drug interaction trials of remdesivir and other concomitant medication have not been conducted in humans and the clinical relevance of the in vitro studies have not been established, further underscoring the need for additional clinical studies. 10
Table 1.
Published guidance regarding remdesivir metabolism and drug‐drug interactions
| Source | Website | Date published or updated | Conclusions and recommendations |
|---|---|---|---|
|
McCreary et al. 14 SIDP |
https://doi.org/10.1093/ofid/ofaa105 | March 23, 2020 | “There is no reason to believe that any significant drug interactions between remdesivir and CYP3A4 inhibitors or inducers are likely” |
| Liverpool Drug Interaction Group 13 | https://www.covid19‐druginteractions.org | April 9, 2020 |
Voriconazole: no clinically significant interaction expected. Rifampicin, carbamazepine, phenytoin: Potential decreased exposure of [remdesivir]. These drugs should not be co‐administered. |
| Sanders et al. 15 | https://doi.org/10.1001/jama.2020.6019 | April 13, 2020 | Not a significant inducer/inhibitor of CYP enzymes, monitor with strong inducers/inhibitors |
| NIH COVID‐19 Treatment Guidelines 16 | https://covid19treatmentguidelines.nih.gov | April 21, 2020 |
RDV levels are unlikely to be markedly altered by CYP2C8, CYP2D6, or CYP3A4 enzymes, or by P‐gp or OATP drug transporters. It may be administered with weak to moderate inducers or with strong inhibitors of CYP450, OATP, or P‐gp. Strong induction of P‐gp is expected to modestly reduce RDV levels. The clinical relevance of lower RDV levels is unknown. The use of RDV with known inducers of P‐gp (e.g., rifampin) is not recommended. |
| Fact Sheet for Health Care Providers Emergency Use Authorization of Remdesivir (GS‐5734) 10 | https://www.fda.gov/media/137566/download | May 3, 2020 | Drug‐drug interaction trials of remdesivir and other concomitant medications have not been conducted in humans. In vitro, remdesivir is a substrate for drug metabolizing enzymes CYP2C8, CYP2D6, and CYP3A4, and is a substrate for OAPT1B1 and P‐gp transporters. In vitro, remdesivir is an inhibitor of CYP3A4, OATP1B1, OATP1B3, BSEP, MRP4, and NTCP. The clinical relevance of these in vitro assessments has not been established. |
BSEP, bile acid export pump; COVID‐19, coronavirus disease 2019; CYP, cytochrome; EUA, Emergency Use Authorization; MRP4, multidrug resistance‐associated protein 4; NIH, National Institutes of Health; NTCP, Na+‐taurocholate co‐transporting polypeptide; OATP, organic anion transporting polypeptide; P‐gp, P‐glycoprotein; RDV, remdesivir; SIDP, Society of Infectious Diseases Pharmacists.
With the recent announcement of the FDA EUA for remdesivir, Gilead Sciences has pledged to donate its existing supply of remdesivir of 1.5 million doses—enough to treat ~ 140,000 patients worldwide. With the focus on therapeutic efficacy, clinical data on transporter and enzyme‐mediated drug interactions will likely not be available in the foreseeable future. In the absence of human pharmacokinetic interaction studies, the use of mechanistic physiologically‐based pharmacokinetic modeling studies will be particularly important to quantify the magnitude of drug‐drug interactions with both remdesivir and its intermediate and active metabolites. A clearer understanding of the clinical pharmacology and significance of potential drug interactions of remdesivir are needed to provide safe and effective care in the ongoing pandemic.
Funding
No funding was received for this work.
Conflict of Interest
The author declared no competing interests for this work.
References
- 1. Johns Hopkins University & Medicine . COVID‐19 map. Johns Hopkins Coronavirus Resource Center. <https://coronavirus.jhu.edu/map.html>. Accessed May 1, 2020. [Google Scholar]
- 2. Grein, J. et al Compassionate use of remdesivir for patients with severe Covid‐19. N. Engl. J. Med. (2020). 10.1056/NEJMoa2007016. [e‐pub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wang, Y. et al Remdesivir in adults with severe COVID‐19: a randomised, double‐blind, placebo‐controlled, multicentre trial. Lancet 395, 1569–1578 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gilead Sciences . Gilead sciences statement on positive data emerging From National Institute of Allergy and Infectious Diseases’ study of investigational antiviral remdesivir for COVID‐19. <https://www.gilead.com/news‐and‐press/press‐room/press‐releases/2020/4/gilead‐sciences‐statement‐on‐positive‐data‐emerging‐from‐national‐institute‐of‐allergy‐and‐infectious‐diseases‐study‐of‐investigational‐antiviral‐rem>. Accessed April 29, 2020.
- 5. Gilead Sciences . Gilead announces results from phase 3 trial of investigational antiviral remdesivir in patients with severe COVID‐19. <https://www.gilead.com/news‐and‐press/press‐room/press‐releases/2020/4/gilead‐announces‐results‐from‐phase‐3‐trial‐of‐investigational‐antiviral‐remdesivir‐in‐patients‐with‐severe‐covid‐19>. Accessed April 29, 2020.
- 6. US Food & Drug Administration . Authorization for emergency use of remdesivir for the treatment of COVID‐19 (letter). <https://www.fda.gov/media/137564/download>. Accessed May 3, 2020.
- 7. Gordon, C.J. , Tchesnokov, E.P. , Feng, J.Y. , Porter, D.P. & Gotte, M. The antiviral compound remdesivir potently inhibits RNA‐dependent RNA polymerase from Middle East respiratory syndrome coronavirus. J. Biol. Chem. 295, 4773–4779 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wang, M. et al Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019‐nCoV) in vitro. Cell Res. 30, 269–271 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Siegel, D. et al Discovery and synthesis of a phosphoramidate prodrug of a Pyrrolo[2,1‐f][triazin‐4‐amino] adenine C‐Nucleoside (GS‐5734) for the treatment of Ebola and emerging viruses. J. Med. Chem. 60, 1648–1661 (2017). [DOI] [PubMed] [Google Scholar]
- 10. US Food & Drug Administration . Fact Sheet for Health Care Providers Emergency Use Authorization (EUA) of Remdesivir (GS‐5734). <https://www.fda.gov/media/137566/download>. Accessed May 3, 2020.
- 11. European Medicines Agency . Summary on compassionate use Remdesivir Gilead. <https://www.ema.europa.eu/en/documents/other/summary‐compassionate‐use‐remdesivir‐gilead_en.pdf>. Accessed April 19, 2020.
- 12. Luke, D.R. , Tomaszewski, K. , Damle, B. & Schlamm, H.T. Review of the basic and clinical pharmacology of sulfobutylether‐β‐cydodextrin (SBECD). J. Pharmaceut. Sci. 99, 3291–3301 (2010). [DOI] [PubMed] [Google Scholar]
- 13. Liverpool Drug Interactions Group University of Liverpool . COVID‐19 drug interactions. <https://www.covid19‐druginteractions.org>. Accessed May 4, 2020.
- 14. McCreary, E.K. & Pogue, J.M. Coronavirus disease 2019 treatment: a review of early and emerging options. Open Forum Infect. Dis. 7, ofaa105 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sanders, J.M. , Monogue, M.L. , Jodlowski, T.Z. & Cutrell, J.B. Pharmacologic treatments for coronavirus disease 2019 (COVID‐19): a review. JAMA. 10.1001/jama.2020.6019. [e‐pub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 16. National Institutes of Health . Coronavirus disease 2019 (COVID‐19) treatment guidelines. <https://www.covid19treatmentguidelines.nih.gov>. Accessed April 21, 2020. [PubMed]