

Abstract
After more than 4 months of the COVID‐19 pandemics with genomic information of SARS‐CoV‐2 around the globe, there are more than 1000 complete genomes of this virus. We used 691 genomes from the GISAID database. Several studies have been reporting mutations and hotspots according to viral evolution. Our work intends to show and compare positions that have variants in 30 complete viral genomes from South American countries. We classified strains according to point alterations and portray the source where strains came into this region. Most viruses entered South America from Europe, followed by Oceania. Only Chilean isolates demonstrated a relationship with Asian isolates. Some changes in South American genomes are near to specific domains related to viral replication or the S protein. Our work contributes to the global understanding of which sort of strains are spreading throughout South America, and the differences among them according to the first isolates introduced to this region.
Keywords: COVID‐19, genetic variants, phylogeny, SARS‐CoV‐2, South America
Highlights
This is the first overview of genomic comparison and source of introduction of SARS‐CoV‐2 in South America.
Most of SARS‐CoV‐2 strains in South America came from Europe, followed by Oceania.
South American isolates portrayed genetic variants on ORF1a (nsp1, nsp3 and nsp5 genes) region and the S protein.
1. INTRODUCTION
The current emergency of COVID‐19 arose after the end of 2019 in Wuhan (China) and was officially declared a pandemic by the World Health Organization (WHO) on 11 March 2020. Its causative agent was called novel coronavirus (nCoV), with a round or oval shape of this 60 to 140 nm enveloped structure. 1 Then, The Coronaviridae Study Group of the International Committee on Taxonomy of Viruses named it as SARS‐CoV‐2 2 based on genomic and phylogenetic analysis. SARS‐CoV‐2 is closely related to the bat coronavirus isolate RaTG13, due to its homology in phylogenetics analysis. 3 Hence, SARS‐CoV‐2 is a β‐coronavirus with more homology to RATG13 (around 96% sequence identity) and Pangolin‐CoV (91.02% identity) than to SARS‐CoV (79% identity) or MERS (51.8% identity) viruses. 1 , 3 , 4 , 5 , 6
SARS‐CoV‐2 genome sequences allowed us to understand the organization of this virus, which has nearly 29 890 base pairs (GenBank NC_045512.2), with genes that produce 29 proteins. These proteins are encoded by 10 open reading frames (ORFs), the most important viral proteins are Spike (S), Envelope (E), Membrane (M) and Nucleocapsid (N) proteins. In addition, the ORF1ab can translate 16 nonstructural proteins (nsp). 3
Genomic studies from China, allowed us to understand that the virus could accumulate mutations meanwhile spreading across the world, with a probable moderate mutation rate. 7 For instance, substitutions in positions 8750, 28 112 were the hotspots and were useful to define two groups of strains, and nt29063 was used by scientists to subdivide these groups. 8 Furthermore, another study evaluated 95 SARS‐CoV‐2 complete genomes and reported 13 variation hotspots in regions: ORF1ab, S, 3a, M, ORF8, and N regions. 7 SARS‐CoV‐2 sequences allowed classifying groups and subgroups according to fixed and cumulative mutations. Pachetti et al 9 demonstrated that European viral genomic mutation hotspots were located on positions 14 408, 23 403, and 3036; being the former first reported in Europe on the 9th February 2020, and in a position of RNA‐dependent RNA polymerase (RdRp or nsp12). These authors also found that positions 17 746, 17 857, and 18 060 were points of recurrent mutations in viruses isolated from American or Canadian patients.
The first report in the South American region of a patient with COVID‐19 was declared by Brazil, whereas Venezuela and Uruguay were the ultimate nations to confirm their patient zero. Different containment and mitigation strategies, and time‐points, have been implemented according to government's decisions. According to the situation report (No. 102) of the WHO, Brazil has the highest number of COVID‐19 confirmed cases (78 162) and deceases (5466) due to this disease, followed by Peru with 33 931 and 943, respectively. Countries with the fewest case fatality rates in the South American region are Chile followed by Colombia and Peru. Molecular or rapid immunological tests have been carried out mostly in Venezuela, Brazil, Peru, and Chile, in this descending order. 10 Conversely, little information was collected with next generation sequencing methodology in these countries.
A huge amount of SARS‐CoV‐2 genomes has been sequenced in a short time around the globe, and a lot of research was published. Nevertheless, South American countries have poor genomic information and lack of genomic analysis. To this date, there is only one official report of SARS‐CoV‐2 in South America, 11 and another online publication from Brazil. Thus, we aim to show a first overview of phylogenetics relationships and genetic variations of SARS‐CoV‐2 in South America.
2. MATERIALS AND METHODS
2.1. Genomic analysis
Analyses were performed to obtain an overview of genomic SARS‐CoV‐2 mutations of circulating strains in South America; we download a total of 30 complete genomes of South American countries from the GISAID (https://www.gisaid.org/) database. The alignment was carried out with Mauve 12 software, using the reference NC_045512.2 (from the initial report from Wuhan, China). The alignment was displayed in MEGA6 13 to extract nucleotide and amino acid mutations, for extracting the 10 ORFs regions the positions were assessed according to a previous study. 7
Additionally, we downloaded 688 genomes from the GISAID database, 14 the genomes were complete (>29 000 bp) and with high coverage according to this public resource. Furthermore, to obtain representative sequences of the seven different continents we chose 5 to 7 day intervals of the first strain isolated in each continent. The alignment of the genomes was performed using multiple alignment using fast fourier transform. 15 All gaps were replaced with N. The inference phylogenetics was used RaxML 16 with the model GTRCAT, with three rate categories and rapid hill‐climbing to accelerate computations. The Treetine 17 software was used for phylodynamic analysis using an approximate maximum likelihood approach with defaults parameters, and a time clock model was used. Finally the tree obtained from RaxML was analyzed with grapetree 18 using a minimum spanning algorithm to explore the fine‐grained population structure of South American genomes, related to continent expansion.
3. RESULTS
We have analyzed a total of 691 SARS‐Cov‐2 complete genomes from a wide variety of geographical sites. Only 30 genomes from South American countries (10 Brazil, 7 Chile, 3 Argentina, 2 Colombia, 1 Ecuador, and 1 Peru) were available (to date of this manuscript analysis, 12 April 2020). On the other hand, Colombia and Ecuador viral genomes have poor quality sequences, and were not included in the phylogenetic analyses (see Supporting information). Epidemiological data on the GISAID database indicated that the first genome reported in South America was on 28 February by a Brazilian research team. Afterward, Chile reported the 3rd of March the first SARS‐CoV‐2 genome sequence in this country. Colombian and Argentinian scientists uploaded viral genomic strains sequences 3 and 4 days later, respectively. Ecuador reported its proper isolate genome on March 9th whereas Peru did it 2 days after.
We aligned 30 SARS‐CoV‐2 genomes from South American isolates with the reference, demonstrating high homology with 29846 sites conserved, representing 99.98% of identity. Genomes have 57 SNPs sites in total (Table 1). Among them, 45.62% (26/57) represent amino acid substitutions in some proteins whereas 60.78% (31/51) corresponds to silent variations. The evaluation of the beginning (5′UTR) and end (3′ UTR) of the viral genome reported lots of ambiguous nucleotides in the analyzed sequences. We detected 11 positions in the ORF1a gene with amino acid variation. The G392D is a unique variation in fragment called nsp1, presenting in a Brazilian strain (EPI_ISL_416033). The region nsp2 has variations in T708I only presented in Brazilian strains (EPI_ISL_416033 and EPI_ISL_413016). Nevertheless, another Brazilian strain (EPI_ ISL_ 415128) has two amino acid changes I739V and P765S at same time. The Nsp3 gene presented two changes A876T and A1043V in strains from Chile (EPI_ISL_414580) and Argentina (EPI_ISL_420599), respectively. The change N2894D in nsp4 was found in the Peruvian strain; and the F3071Y is present in four Chilean (EPI_ISL 414579, EPI_ISL_415661, EPI_ISL_415660, and EPI_ISL_415658) and in one Brazilian isolates (EPI_ISL_417034). The fragment nsp5 has the amino acid change G3334S (Brazil ‐ EPI ISL 416034). Finally, one Brazilian (EPI_ISL_416034) strain has the change L3606F in the nsp6 region.
Table 1.
Alignment of 30 SARS‐CoV‐2 South American viral genomes and the reference: NC_045512.2

|
Note: On the top, the nucleotide changes with respect to the reference. At the bottom, genomic regions where changes occur. Amino acid alterations in red words.
The ORF1b has two positions with amino acid change, the first is P314L in the nsp12 region and is reported in 17 virus strains. The spike protein has two alterations, one is the position D614G present in 17 isolates of different countries and E1207V found in the analyzed Ecuadorian strain. ORF3a has three variants: Q57H for one Argentinian isolate (EPI_ISL_420599), the change G196V found in four Chilean samples (EPI_ISL_414579, EPI_ISL_415661, EPI_ISL_415660, EPI_ISL_415658), and the G251V amino acid alteration was found in a Brazilian strain (EPI_ISL_417034). The T175M substitution in the membrane gene (M) was only detected in three Brazilian isolates (EPI_ISL_414014, EPI_ISL_413016 and EPI_ISL_416028). Furthermore, we determined the L84S change in ORF8 in six of the Chilean strains (EPI_ISL_414578, EPI_ISL_414577, EPI_ISL_414579, EPI_ISL_415661, EPI_ISL_415660, EPI_ISL_415658), one Colombian (EPI_ISL_417924) and one Brazilian isolates (EPI_ISL_417034). Finally, the N gene depicts D103Y present in two Chilean (EPI_ISL_414577; EPI_ISL_414578), R191C in one Argentinian (EPI ISL 420598), S197L in four Chilean (EPI_ISL_414579, EPI_ISL_415661, EPI_ISL_415660, EPI_ISL_415658) including one Brazilian (EPI_ISL_417034) strains; and the alteration G238C is reported for a Colombian sample (EPI_ISL_417924). In addition the concomitant mutations R203K and G204R are present in 12 strains: 7 Brazil, 1 Peru, 1 Colombia, 1 Chile, and 1 Argentina.
Phylogenetics analysis of 688 genomes (Figure 1) was colored according to Tang et al, 19 the mutation a23403g permits to assign 17 genomes related to Clade G (light purple), and the nucleotide change t28144c classify eight genomes in the Clade S (light gray). On the other hand, five genomes did not belong to any clade. Figure 2 shows that clade G diverse and contains subtypes. At least one South American strain belongs to a subgroup of Clade G. It seems that South American isolates are more related to Western Europe and Oceania. Other virus samples from Colombia, Brazil, and Chile were classified in Clade S, closely related to Spain genomes. In addition, the information obtained from minimum spanning‐tree (Figure 2) is highly correlated to phylogenetic analysis, showing a star‐shaped distribution classical of rapid viral spreading among countries. SARS‐CoV‐2 origin is from Asia, with fast expansion to Europe and North America, then to Oceania. Our results portray circulating SARS‐CoV‐2 South American strains coming from Europe, North America, and Oceania.
Figure 1.
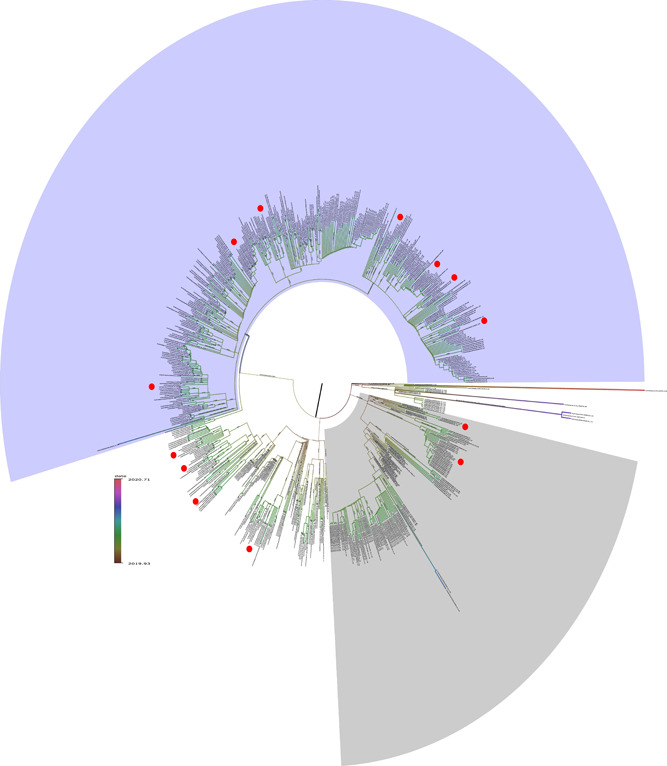
Phylogenetic tree using 688 genomes. The branch length reflect time rather than divergence and painted according to the heatmap bar. The South American SARS‐Cov‐2 isolates are highlighted with red circles inside Clade G (light purple, top) and Clade S (light gray, bottom). Five strains were unable to be assigned to any clade
Figure 2.
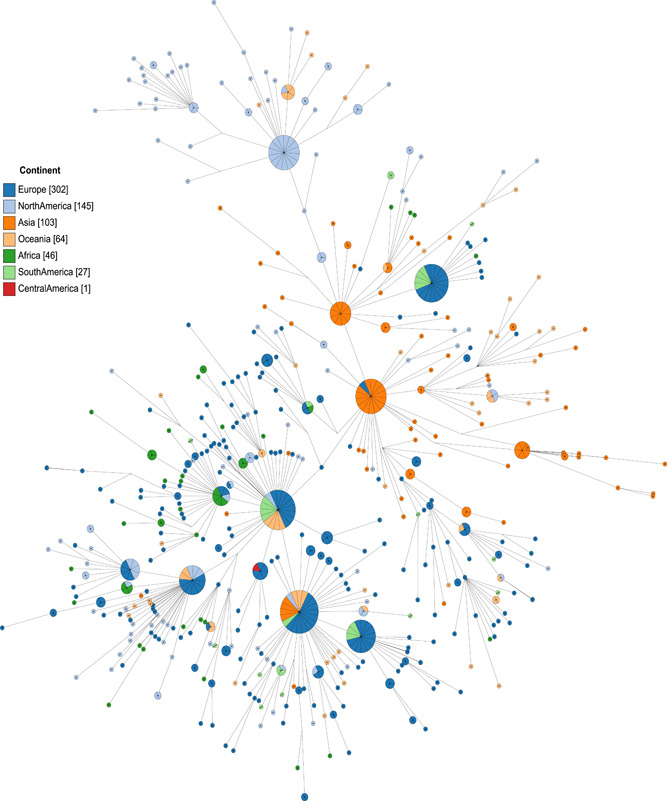
Minimum spanning‐tree to reconstruct and visualize the genomic relationships of South American SARS‐Cov‐2
4. DISCUSSION
Since the first officially reported patient in South America in Brazil, other countries have been informing COVID‐19 cases. Brazilian male patients were 61 and 32 years old, who weeks before had visited Italy (Lombardy and Milan, respectively) (http://virological.org/t/first-cases-of-coronavirus-disease-covid-19-in-brazil-south-america-2-genomes-3rd-march-2020/409). A recent study from Chile reported four patients, a couple who visited several European and Asian countries, on 21st and 24thFebruary they stayed in Madrid before returning to Santiago, Chile. Another patient visited London, Italy, and Spain (Madrid), the latter being the last city visited on 28th February to 3rd March. The fourth Chilean patient stayed in Italy (Milan) between 25th to 29th February, then returned to Chile. 11 Interestingly, our results portray to date four patients with the same SARS‐CoV‐2 strain, similar to the observed in the first confirmed Chilean case (EPI_ISL_414579). The first Peruvian patient was a 25‐year‐old male returning from the United Kingdom reported on 6th March, however the SARS‐CoV‐2 strain sequenced belonged to a woman of 65 years old who returned to Peru from Spain. Similar constraints occurred in other countries of the region, where only Chile and apparently Colombia (EPI_ISL_418262, sample collection date 3rd March 2020) succeeded to sequence the strain from the patient zero.
Our manuscript demonstrates variable sources of introduction of SARS‐CoV‐2 into South American countries. Phylogenetic analysis depicts that most of these strains are closely related to European viral isolates. Brazil had viruses from several parts of the globe mainly from Europe, including the United States and Africa. Only four Chilean strains were related to Asian isolates, corresponding to the same genome of the couple reported by Castillo et al, 11 and we assume the two other isolates could have been sampled from relatives or close people to patient zero. We hypothesize that our findings are related to the amount of samples of viral sequences which could not be done for other South American countries yet, or any other bias of sample selection. Furthermore, we were unable to include in our phylogenetic analysis viral genomes from Ecuador and Colombia, due to genome sequence quality. However, the GISAID SARS‐CoV‐2 portal (URL at: https://www.gisaid.org/epiflu-applications/next-hcov-19-app/) depicts the close relationship between an isolate from Oceania (EPI ISL 417211) and the strain sequenced in Ecuador. This online tool also shows that one of the strains sequenced from Colombian patients was related to an Australian origin (EPI_ISL_419834)—closely related to the Chilean strain EPI_ISL_414578—whereas the other closely related to European origin from Germany or Switzerland.
We were able to classify strains according to previous suggestions. 19 We demonstrate that strains from Clade G were the most common throughout South America; with 68.75%, 14.29%, 50%, 100% of strains in Brazil (11/16), Chile (1/7), Colombia (1/2), and the latter percentage for Peru (1/1) and Argentina (3/3), too. Up to this report, we only have an official publication from Chile and we were able to confirm analysis from this group, except the change of an amino acid (G196V in our analysis) reported in one strain of Clade S as G193V. 11 Currently, 85.71% of Chilean strains pertain to S Clade followed in frequency by Colombian and Brazilian isolates with 50% and 6.25%, respectively. All of S Clade Chilean strains were related to Asian origin, whereas Brazilian and Colombian isolates were related to viruses from Oceania.
Infectivity and pathogenicity of SARS‐CoV‐2 is related to S protein, mainly due to the human angiotensin‐converting enzyme 2 (h‐ACE2) binding ridge structural changes of the RBD domain, on residues 482 to 485: Gly, Val, Glu, and Gly. 20 Thus, novel mutations on S protein, especially on these residues or nearly of them could be of importance. Our report highlights two strains with novel variants on the S region, with no amino acid change in nt24022 (E1207E) whereas another nonsynonymous alteration in nt25182 (E1207V), for Peru (EPI_ISL_415787) and Ecuador (EPI_ISL_417482), respectively. However, these changes seem far away from the critical region of S protein for h‐ACE2 affinity.
Due to its prevalence across the world as in our sample of South American isolates, researchers are suggesting that Clade G strains could be more contagious than other subtypes; Zhang et al 8 suggested that it could be related to synonymous changes due to nucleotide changes in ORF1ab (nt8750) and N (nt29063) genes, which could enhance viral replication due to higher translational efficiencies compared to other clades. Furthermore, another study showed that there are some positions where mutations could arise more frequently in subsequent SARS‐CoV‐2 strains, corresponding to nt8782 of ORF1a, nt28144 of ORF8 and nt29095 of N region. 7 We highlight differences with this report because we found 8 (8/30) variations in both of nt8782 and nt28144 positions. Conversely, other regions seem to be hotspots in South American strains, with 11(36.67%) of these portraying changes at 5′UTR (nt241), nsp3 (nt3037), nsp12 (nt14408) and N/ORF9 (nt28881, nt28882, and nt28883) regions. This is paramount because changes in nsp1, nsp3, and nsp5 could be related to some functions of the viral incubation period and immune response evasion of SARS‐CoV‐2. 21
We found amino acid alterations in both of these regions, such as G392D (nsp1), A876T and A1043 (nsp3) and nsp5 (G3334S); and should be tested in further studies. Strikingly, we identified four changes—nt15324 in ORF1ab (RdRp), nt26144 in E gene and nt28580 and nt28657 in the nucleocapsid gene—in the suggested regions for primer annealing for SARS‐CoV‐2 specific fragments identification, according to real time RT‐PCR recommendations from the WHO. 22 Moreover, viral genomes with alterations on 14 408 and 23 403 positions have been correlated to more mutations (3‐4 per genome) than their counterparts without it. 9 All South American viruses of Clade G analyzed in this report have concomitantly mutations on 14 408 and 23 403 nucleotidic positions. Compared to the reference, we found an average of five mutations per genome among South American strains.
Our study represents the first overview of SARS‐CoV‐2 strains genomic comparison and phylogenetic analysis in South America. Surprisingly, five of the studied strains lack current classification, and we were not able to track all the global distribution of this virus due to our sampling methodology. However, we consider that our study highlights important findings such as two novel mutations in the S region, and novel hotspots positions. In addition, some external limitations such as primers design variations of the ORF1b 23 or N regions, could have influenced the sequencing process on some isolates from South America. 7 , 24 Some other limitations are the lack of epidemiological data for all patients (we mostly used media information or government's official websites), poor quality of some viral genome sequences; and specially the limited number of viral genomes reported in South America after almost 2 months of the arrival of SARS‐CoV‐2 to this part of the world. This must improve to identify mutations that could have an effect on the design of diagnostic and therapeutic measures, including vaccines or antiviral drugs. 25
We should take into account that this is a novel virus and could have higher mutation rates than currently expected, 7 and genetic drift and founder effect could influence specific SARS‐CoV‐2 subsequent strains and mutations which would be geographically constrained for a while. Moreover, South America should urgently strengthen the genomic epidemiology field for the current and further pandemics.
CONFLICT OF INTERESTS
The authors declare that there are no conflict of interests.
AUTHOR CONTRIBUTIONS
JAP and OM participated in conceptualization, study design, interpreting the data analysis, methodology design, visualization and wrote the whole manuscript. OM did the phylogenetics and genetic comparison of SARS‐CoV‐2 isolates in South America.
Supporting information
Supplementary information
Supplementary information
ACKNOWLEDGMENTS
The authors gratefully acknowledge the authors, originating and submitting laboratories of the SARS‐CoV‐2 sequences from GISAID's EpiFlu Database 14 (see S1). Moreover, we thank all people taking care of patients with COVID‐19 around the world. We all are part of this struggle against this virus, and we will succeed.
Poterico JA, Mestanza O. Genetic variants and source of introduction of SARS‐CoV‐2 in South America. J Med Virol. 2020;92:2139–2145. 10.1002/jmv.26001
Contributor Information
Julio A. Poterico, Email: jpoterico@insnsb.gob.pe.
Orson Mestanza, Email: orsomm@gmail.com.
REFERENCES
- 1. Zhu N, Zhang D, Wang W, et al. A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med. 2020;382(8):727‐733. 10.1056/NEJMoa2001017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Coronaviridae Study Group of the International Committee on Taxonomy of Viruses . The species severe acute respiratory syndrome‐related coronavirus: classifying 2019‐nCoV and naming it SARS‐CoV‐2. Nat Microbiol. 2020;5(4):536‐544. 10.1038/s41564-020-0695-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zhou P, Yang X‐L, Wang X‐G, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579(7798):270‐273. 10.1038/s41586-020-2012-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lu R, Zhao X, Li J, et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet. 2020;395(10224):565‐574. 10.1016/S0140-6736(20)30251-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ren Z, Lin L, Li Z, Sun XP, Yang K, Yang HX. Identification of a novel coronavirus causing severe pneumonia in human: a descriptive study. Chin Med J. 2020;133(9):1015‐1024. 10.1097/CM9.0000000000000722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhang T, Wu Q, Zhang Z. Probable pangolin origin of SARS‐CoV‐2 associated with the COVID‐19 outbreak. Curr Biol. 2020;30(7):1346‐1351. 10.1016/j.cub.2020.03.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wang C, Liu Z, Chen Z, et al. The establishment of reference sequence for SARS‐CoV‐2 and variation analysis, The establishment of reference sequence for SARS‐CoV‐2 and variation analysis. J Med Virol. 2020;92:667‐674. 10.1002/jmv.25762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhang L, Yang J‐R, Zhang Z, Lin Z. Genomic variations of SARS‐CoV‐2 suggest multiple outbreak sources of transmission. medRxiv. 2020. 10.1101/2020.02.25.20027953 [DOI] [Google Scholar]
- 9. Pachetti M, Marini B, Benedetti F, et al. Emerging SARS‐CoV‐2 mutation hot spots include a novel RNA‐dependent‐RNA polymerase variant [published online ahead of print March 29, 2020]. J Transl Med. 2020;18(179). 10.1186/s12967-020-02344-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. World Health Organization . Coronavirus disease (COVID‐19). Situation Report ‐ 102. World Health Organization; 2020:16. https://www.who.int/docs/default-source/coronaviruse/situation-reports/20200501-covid-19-sitrep.pdf?sfvrsn=742f4a18_2. Accessed on May 1, 2020.
- 11. Castillo AE, Parra B, Tapia P, et al. Phylogenetic analysis of the first four SARS‐CoV‐2 cases in Chile. J Med Virol. 2020;1–5. 10.1002/jmv.25797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Darling ACE. Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004;14(7):1394‐1403. 10.1101/gr.2289704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 2013;30(12):2725‐2729. 10.1093/molbev/mst197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Shu Y, McCauley J. GISAID: Global initiative on sharing all influenza data – from vision to reality. Euro Surveill. 2017;22(13):30494. 10.2807/1560-7917.ES.2017.22.13.30494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Katoh K. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002;30(14):3059‐3066. 10.1093/nar/gkf436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post‐analysis of large phylogenies. Bioinformatics. 2014;30(9):1312‐1313. 10.1093/bioinformatics/btu033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sagulenko P, Puller V, Neher RA. TreeTime: Maximum‐likelihood phylodynamic analysis. Virus Evol. 2018;4(1):vex042. 10.1093/ve/vex042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhou Z, Alikhan N‐F, Sergeant MJ, et al. GrapeTree: visualization of core genomic relationships among 100,000 bacterial pathogens. Genome Res. 2018;28(9):1395‐1404. 10.1101/gr.232397.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tang X, Wu C, Li X, et al. On the origin and continuing evolution of SARS‐CoV‐2 [published online ahead of print March 3, 2020]. Natl Sci Rev. 2020;0(0):1–12. 10.1093/nsr/nwaa036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Shang J, Ye G, Shi K, et al. Structural basis of receptor recognition by SARS‐CoV‐2. Nature. 2020;581(7807):221‐224. 10.1038/s41586-020-2179-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wen F, Yu H, Guo J, Li Y, Luo K, Huang S. Identification of the hyper‐variable genomic hotspot for the novel coronavirus SARS‐CoV‐2. J Infect. 2020;2020:671‐693. 10.1016/j.jinf.2020.02.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Corman VM, Landt O, Kaiser M, et al. Detection of 2019 novel coronavirus (2019‐nCoV) by real‐time RT‐PCR. Euro Surveill. 2020;25(3):2000045. 10.2807/1560-7917.ES.2020.25.3.2000045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chan JF‐W, Yuan S, Kok K‐H, et al. A familial cluster of pneumonia associated with the 2019 novel coronavirus indicating person‐to‐person transmission: a study of a family cluster. The Lancet. 2020;395(10223):514‐523. 10.1016/S0140-6736(20)30154-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chu DKW, Pan Y, Cheng SMS, et al. Molecular diagnosis of a novel coronavirus (2019‐nCoV) causing an outbreak of pneumonia. Clin Chem. 2020;66(4):549‐555. 10.1093/clinchem/hvaa029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gordon DE, Jang GM, Bouhaddou M, et al. A SARS‐CoV‐2 protein interaction map reveals targets for drug repurposing [published online ahead of print April 30, 2020]. Nature. 2020. 10.1038/s41586-020-2286-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary information
Supplementary information