

Summary
Flowering of many plant species depends on interactions between basic leucine zipper (bZIP) transcription factors and systemically transported florigen proteins. Members of the genus Arabidopsis contain two of these bZIPs, FD and FDP, which we show have largely complementary expression patterns in shoot apices before and during flowering. CRISPR-Cas9-induced null mutants for FDP flower slightly earlier than wild-type, whereas fd mutants are late flowering. Identical G-box sequences are enriched at FD and FDP binding sites, but only FD binds to genes involved in flowering and only fd alters their transcription. However, both proteins bind to genes involved in responses to the phytohormone abscisic acid (ABA), which controls developmental and stress responses. Many of these genes are differentially expressed in both fd and fdp mutant seedlings, which also show reduced ABA sensitivity. Thus, florigen-interacting bZIPs have distinct functions in flowering dependent on their expression patterns and, at earlier stages in development, play common roles in phytohormone signaling.
Keywords: flowering, Arabidopsis, bZIPs, florigen, FD, FDP, FT, ChIP-seq, RNA-seq, ABA
Graphical Abstract
Highlights
-
•
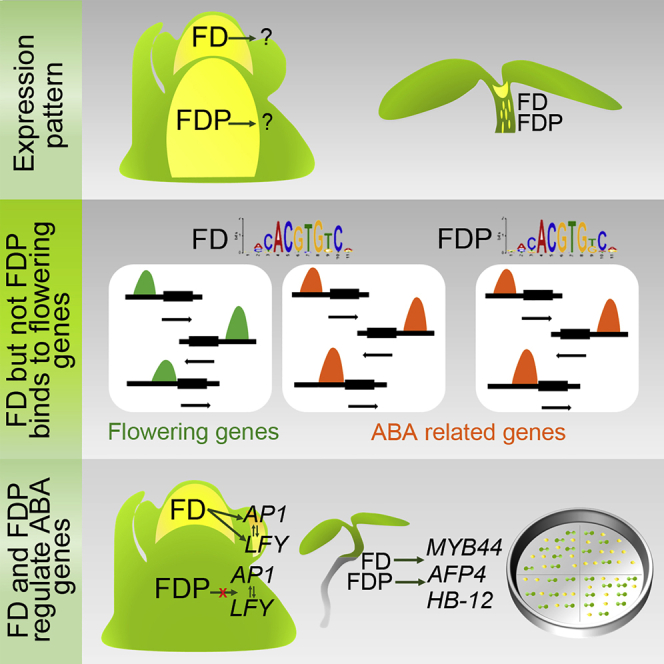
FDP is expressed at the shoot apex in a largely discrete pattern to FD
-
•
Flowering genes are bound only by FD, whereas other targets are bound by FD and FDP
-
•
FD and FDP bind to and regulate genes involved in ABA responses in seedlings
-
•
FT overexpressed in the phloem promotes flowering independently of FD and FDP
Florigen activating complex facilitates the response to day length in higher plants and contains a specific class of bZIP transcription factor. Romera-Branchat et al. analyze the two factors of this class in Arabidopsis and find they play distinct functions in flowering control and participate in ABA signaling in early development.
Introduction
The development of many plant species is controlled by day length, including seasonal control of floral development, regulation of bud dormancy in trees, and the initiation of tuberization in potato (Andrés and Coupland, 2012). In Arabidopsis thaliana, a genetic pathway and transcriptional cascade is activated by long days (LDs) typical of spring and early summer to induce flowering (Srikanth and Schmid, 2011, Turck et al., 2008). Strikingly, orthologs of several components of this pathway have conserved or related functions in photoperiodic responses of other plant species (Andrés and Coupland, 2012). This photoperiodic pathway activates floral induction in A. thaliana, the first step in reproduction during which the shoot apical meristem (SAM) transitions from vegetative growth and initiates flowers. In this pathway, FLOWERING LOCUS T (FT) is transcriptionally activated in the vascular tissue of the leaves under LDs, and the FT protein, which is related to phosphatidylethanolamine binding proteins (PEBPs) (Kardailsky et al., 1999, Kobayashi et al., 1999), moves systemically to the shoot apex by the phloem sieve elements (Chen et al., 2008, Corbesier et al., 2007, Jaeger and Wigge, 2007, Mathieu et al., 2007). Based on this systemic effect, FT proteins are often referred to as florigens. At the SAM, FT interacts with and forms a protein complex with the bZIP transcription factor FD (Abe et al., 2005, Abe et al., 2019, Wigge et al., 2005). This interaction is probably bridged by 14-3-3 proteins (Collani et al., 2019, Taoka et al., 2011). The FT/FD complex is proposed to mediate transcriptional reprogramming of the SAM by transcriptionally regulating flowering genes, such as SUPPRESSOR OF OVEREXPRESSION OF CO 1 (SOC1), and FRUITFULL (FUL), which encode MADS-box (MCM1, AGAMOUS, DEFICIENS, SRF) transcription factors and, thereby, initiate an inflorescence meristem (Torti et al., 2012, Searle et al., 2006, Schmid et al., 2003). In addition, in the floral primordia, the FT/FD module activates the floral meristem identity gene APETALA1 (AP1) (Abe et al., 2005, Wigge et al., 2005). Many early effects of FD on gene expression in the SAM and the floral primordium are caused by direct binding or recruitment of FD to the promoters of target genes, including AP1, SOC1, and FUL (Collani et al., 2019, Jung et al., 2016, Wigge et al., 2005). The FT/FD module is also required for the expression of later-acting genes in the inflorescence meristem, such as SQUAMOSA PROMOTER BINDING PROTEIN LIKE 3 (SPL3), SPL4, and SPL5 (Torti et al., 2012, Schmid et al., 2003). The encoded SPL proteins interact with FD and bind directly to AP1, LEAFY (LFY), and FUL, suggesting that they combinatorially activate gene expression (Jung et al., 2016, Yamaguchi et al., 2009, Yamaguchi et al., 2014, Wang et al., 2009).
Genetic analysis supports the relationship between FT and FD but is complicated by redundancy among family members. Mutants for FT retain a flowering response to LDs, but this is almost completely blocked in double mutants for FT and its closely related paralog TSF (Jang et al., 2009, Yamaguchi et al., 2005). Mutations in FD do not abolish the flowering response to LDs (Abe et al., 2005, Wigge et al., 2005, Koornneef et al., 1998), and these mutants are not as late flowering as ft-10 tsf-1 double mutants (Jang et al., 2009). Thus, FT and TSF might have independent functions to FD during floral transition, or genetic redundancy might exist between FD and genes encoding closely related group A bZIPs, which include the FD paralog FDP, as well as many transcription factors that confer responses to the phytohormone abscisic acid (ABA) (Dröge-Laser et al., 2018). FDP also interacts with FT and TSF (Jang et al., 2009, Abe et al., 2005, Wigge et al., 2005). Redundancy between FD and FDP has been difficult to test because no null alleles of FDP were available. One mutant allele of FDP was recovered and predicted to induce a single-amino acid change in the bZIP domain (Jaeger et al., 2013). This mutation enhances the late-flowering phenotype of fd, and the double mutant strongly suppressed the early-flowering phenotype caused by FT overexpression (Jaeger et al., 2013). These data suggested that FD and FDP have closely related functions in mediating the flowering function of FT TSF.
Here, we study FD and FDP byusing confocal microscopy and reverse genetic and genomic approaches. We find that these paralogous transcription factors have distinct expression patterns and surprisingly different functions in flowering control, as well as common functions in ABA responses of seedlings.
Results
fdp Null Alleles Induced with CRISPR-Cas9 Cause Early Flowering
To extend the genetic resources available for analyzing the function of FDP, we performed targeting induced local lesions in genomes (TILLING) and identified a second allele, fdp-2. This mutation resulted in a single-amino acid change (A182T) within the bZIP DNA binding domain at the adjacent residue to that in the previously described fdp-1 allele (R181K) (Jaeger et al., 2013; Figure 1A; Figure S1A). Both fdp-1 and fdp-2 expressed FDP mRNA (Figure S1B) and were significantly later flowering than wild-type (WT) plants under LDs but not under short days (SDs) (Figures 1C and 1D).
Figure 1.

Characterization and Flowering Time of fdp Mutants
(A) FDP mutant alleles. Red boxes: exons encoding the basic region of the bZIP domain. Blue boxes: exons encoding the leucine zipper domain. Light green box: exon encoding N-terminal coding sequence defined in this work. Green boxes: other exons. Dark gray lines: introns. Red lines: 5′ and 3′ UTRs. Red triangles: the TILLING alleles fdp-1 and fdp-2. The amino acid changes in fdp-1 and fdp-2 are shown. Orange triangles: the positions of single guide RNAs (sgRNAs) used to generate CRISPR alleles.
(B) Chromatogram of the nucleotide sequences of the CRISPR alleles. The PAM region is underlined in orange. In fdp-CRP1, the single-nucleotide deletion is marked by an orange arrow. In fdp-CRP2, one additional nucleotide is marked by an orange arrow. In fdp-CRP3, a 58-nt deletion exists between the two sgRNAs.
(C) Flowering time of WT, five fdp mutants, and fd-3 mutants under LDs.
(D) Flowering time of WT, five fdp mutants, and fd-3 mutants under SDs.
(E) Mutants and WT plants grown for 30 LDs.
(F) In situ hybridization of FUL mRNA in apices of 9-, 11-, and 14-day-old WT and fdp-CRP2 plants under LDs.
(G) In situ hybridization of AP1 mRNA in apices of 9-, 11-, 14-, and 17-day-old WT and fdp-CRP2 plants and 17-day-old fd-3 plants under LDs.
(H) Top: flowering time of fdp-1 homozygotes and heterozygotes compared to WT. Bottom: flowering time of fdp-2 homozygotes and heterozygotes compared to WT.
In (C), (D), and (H), one-way ANOVA followed by Turkey test was used for the statistical analysis. Letters shared in common between the genotypes in (C) and (D) indicate no significant differences in flowering time. In (C) and (H), groups were different with p ≤ 0.001, whereas in (D), p = 0201. In (H), ∗∗∗p ≤ 0.001, ∗∗p ≤ 0.01, ∗p ≤ 0.05; and ns, no significance. Scale bar, 2 cm (E); 50 μm (F and G). The whiskers are defined in the STAR Methods.
The fdp-1 and fdp-2 alleles might retain FDP activity; therefore, CRISPR-Cas9 was used to generate null alleles. Three mutations were recovered with independent guide RNAs. Two mutations caused frameshift mutations early in the coding sequence (fdp-CRP1, fdp-CRP2). In fdp-CRP1, translation could potentially reinitiate at an ATG codon (codon 40 in open reading frame [ORF]), whereas in fdp-CRP2, this could only occur at codon 66. In the third allele (fdp-CRP3) most of the region encoding the bZIP DNA-binding domain was deleted (Figures 1A and 1B; Figure S1C; STAR Methods). All three mutations expressed FDP mRNA, but it was consistently present at lower levels in fdp-CRP2 (Figure S1D). Thus, fdp-CRP2 was selected for most future genetic experiments because it was likely to be the strongest allele. Cas9 was segregated from each mutation, and all three mutant genomes were resequenced. No mutations at putative off-target sites were identified (Figure S1E). None of the CRISPR-induced mutants showed delayed flowering under LDs or SDs based on the number of leaves formed prior to flowering, in contrast to fdp-1 and fdp-2 (Figures 1C and 1D). However, all CRISPR-induced mutants reproducibly bolted to form an inflorescence 2–3 days earlier than WT plants under LDs (Figure 1E; Table S1). In situ hybridizations were therefore performed to determine whether genes expressed specifically in the inflorescence meristem are induced earlier in fdp-CRP mutants than in WT. FUL mRNA, which is expressed in the early inflorescence meristem during floral transition (Mandel and Yanofsky, 1995), was detected strongly throughout the SAM of 14-day-old fdp-CRP2 mutants, whereas its expression in WT was weaker and more restricted (Figure 1F). Similarly, AP1, which is induced in early floral primordia (Mandel et al., 1992), was expressed in apices of 14-day-old fdp-CRP2 mutants but was not detected in WT until 17 days (Figure 1G). These effects were confirmed by qRT-PCR (Figure S1F). Thus, under LDs, floral induction occurs approximately 2–3 days earlier in fdp-CRP mutants than in WT, in contrast to the late flowering of fdp-1 and fdp-2.
These data suggest that the amino acid changes in the DNA-binding domains of the proteins encoded by the fdp-1 and fdp-2 alleles might cause the mutant proteins to actively delay flowering. To test this, the fdp-1 mutant was crossed to WT and the flowering times of F1 plants were compared with each parent. F1 plants heterozygous for fdp-1 flowered later than WT, similar to fdp-1 homozygotes (Figure 1H), suggesting that fdp-1 is dominant. Similar results were obtained for the closely related fdp-2 allele (Figure 1H). Therefore, these appear to be dominant gain-of-function alleles that actively delay flowering.
Genetic Interactions between FD and FDP
The functional relationship between FD and FDP was studied using the newly generated FDP mutant alleles. The late-flowering phenotype of fd-2 is enhanced by fdp-1 (Jaeger et al., 2013). Consistent with this result, fdp-2 fd-3 flowered later than fd-3 under LDs (Figures 2A and 2B). Whether the loss-of-function alleles fdp-CRP2 and fdp-CRP3 also enhanced the late-flowering phenotype of fd-3 was tested. The double mutant fdp-CRP2 fd-3 was constructed and flowered slightly but significantly later than fd-3 but earlier than fdp-2 fd-3 (Figures 2A and 2B). The flowering time of fdp-CRP3 fd-3 was not statistically different from that of fd-3 (Figures 2A and 2B). As described above, fdp-CRP2 might be a stronger allele, and this might explain the stronger interaction with fd-3. In any case, we conclude that FDP only weakly promotes flowering in an fd-3 mutant background.
Figure 2.

Genetic Interactions between FD and FDP
(A) Flowering times of the illustrated genotypes under LDs.
(B) Plants of the illustrated genotypes grown under LDs for 36 days.
(C) Flowering times of 35S::HA:FDP and 35S::GFP:FDP transgenic plants compared to WT.
(D) Flowering times of the illustrated genotypes grown under LDs.
(E) Plants of the illustrated genotypes grown under LDs for 28 days.
(F) Rosette leaves series of 35S::HA:FD, 35S::HA:FD, and WT of 28-day-old plants.
In (A) and (F), one-way ANOVA followed by Turkey test was used. In (C), one-way ANOVA followed by the Holm-Sidak test was used. Shared letters between the genotypes in (A), (C), and (F) indicate no significant difference in flowering time. In (A) and (F), groups were considered statistically different when p ≤ 0.001, whereas in (C) when p ≤ 0.05. Scale bars, 1 cm (F) and 2.5 cm (B).
Phylogenetic analysis of FD and FDP protein sequences across Brassicaceae members demonstrated that after duplication, FDP and FD were retained in all species analyzed and that FDP has diverged from the ancestral sequence more than FD (Figure S2). To compare the functions of the proteins when expressed from the same promoters, FDP was expressed from the CaMV35S and FD promoters. Hemagglutinin (HA):FDP and GFP:FDP significantly promoted flowering of WT plants when expressed from the CaMV35S promoter (Figure 2C) but less strongly than 35S::HA:FD (Figure S2B). Also, introduction of an FD::FDP transgene into the fd-3 mutant background partially suppressed the late flowering of this mutant (Figure 2D). Therefore, FDP can partially confer the function of FD in flowering when expressed appropriately. Also, 35S::FDP formed rounder and lighter leaves instead of the more pointed leaves observed in 35S::FD plants (Figures 2E and 2F; Figures S2B and S2C) (Abe et al., 2005, Wigge et al., 2005), which are similar to those of 35S::FT plants (Teper-Bamnolker and Samach, 2005). Taken together, these data suggest that FD and FDP proteins have common and specific functions.
FT Strongly Promotes Flowering Independently of FD and FDP
The FT/TSF florigens interact with FDP in the yeast two-hybrid system (Jang et al., 2009, Abe et al., 2005, Wigge et al., 2005), and therefore, their genetic interactions were tested. The triple mutants ft-10 tsf-1 fdp-2 and ft-10 tsf-1 fdp-CRP2 flowered similarly to ft-10 tsf-1 double mutants (Figures 3A and 3B), supporting the idea that in LD, the effects of FDP on flowering time depend on FT TSF. Unexpectedly, however, the ft-10 tsf-1 fd-3 triple mutant and ft-10 tsf-1 fd-3 fdp-2 quadruple mutant flowered earlier after forming fewer leaves than ft-10 tsf-1, and in particular, the number of cauline leaves was reduced (Figure 3A; Figure S3A). Indeed, the number of cauline leaves was also lower in ft-10 tsf-1 fd-3 fdp-2 than in ft-10 tsf-1 fd-3 (Figure S3B), suggesting that FD and FDP redundantly delay floral development in the absence of FT and TSF.
Figure 3.

FT Promotes Flowering Independently of FD and FDP
(A) Flowering time of the illustrated genotypes incorporating fdp-2 grown under LDs.
(B) Flowering time of the illustrated genotypes incorporating fdp-CRP2 grown under LDs.
(C) Flowering time of the illustrated genotypes incorporating GAS1::FT into the allele fdp-2 grown under LDs.
(D) Flowering time of the illustrated genotypes incorporating GAS1:FT into allele fdp-CRP2 grown under LDs. In all panels, statistical analysis was performed as in Figure 1.
In (A) and (B), groups were considered statistically different when p ≤ 0.001, whereas in (C) and (D), when p ≤ 0.05.
Overexpression of FT in the companion cells of the phloem from the GAS1 promoter causes early flowering (Corbesier et al., 2007; Figures 3C and 3D). To further analyze functional redundancy among FD and FDP downstream of FT, the fd-3 fdp-2 GAS1::FT and fd-3 fdp-CRP2 GAS1::FT triple lines were constructed. The fdp-2 mutation slightly delayed flowering of fd-3 GAS1::FT, whereas fdp-CRP2 did not (Figures 3C and 3D). Both triple lines still flowered much earlier than the fd-3 mutant (Figures 3C and 3D), and the incorporation of fdp-2 or fdp-CRP2 into fd-3 GAS1::FT background had a weaker effect on flowering time than was previously described for the introduction of fdp-1 into fd-2 35S::FT (Jaeger et al., 2013). We conclude that overexpression of FT in companion cells can strongly promote flowering independently of FD and FDP.
FDP Is Expressed at the Shoot Apex in a Largely Complementary Pattern to FD
The genomic sequences of FDP and FD showed substantially divergent 5′ non-coding sequences (Figure 4A); therefore, their spatial and temporal expression patterns were compared. WT plants grown under SDs were transferred to LDs to induce floral transition, and FD and FDP mRNAs were monitored by in situ hybridization. Prior to exposure to LDs, FD mRNA was present on the flanks of the SAM and on the adaxial side of young leaves (Figure 4B), as expected (Abe et al., 2005, Wigge et al., 2005). At 3 LD and 5 LDs, FD mRNA was more highly expressed in the SAM and axillary meristems (Wigge et al., 2005). In contrast, FDP mRNA signals were weaker in SD-grown plants than those of FD but overlapped on the adaxial side of leaves (Figure S4A) and on the flanks of the SAM at the boundary between the SAM and leaf primordia (Figure 4B). After exposure to 3 or 5 LDs, FDP mRNA was also detected at the shoot apex (Figure 4A) but more basal within the apex than FD mRNA, in a region corresponding to the rib meristem. After 7 LDs, as the shoot elongated to form the inflorescence, the FDP mRNA signal in the rib meristem became stronger and extended into the vascular tissue. At this stage, FDP mRNA was also observed in the center of young floral primordia (stage 3–4) but in a more restricted pattern than FD mRNA (Figure S4B).
Figure 4.

Expression Patterns of FD and FDP
(A) VISTA plot generated from the pairwise alignment of the Arabidopsis thaliana FD and FDP loci. Sequence similarity (%) was calculated using a 100-bp sliding window; color indicates greater than 75% base identity.
(B) In situ hybridizations performed on apices of plants grown for 3 weeks under SD (0 LD) and then transferred to LDs for 3, 5, or 7 days. Sections were hybridized with FD (left panels) or FDP (right panels) probes. White arrow: adaxial leaf tissue. Red arrow: boundary region.
(C and E) Confocal analysis of dissected apices of pFD::VENUS:FD in fd-3 and pFDP-VENUS:FDP in fdp-2 grown for the indicated number of LDs. White arrow: adaxial leaf tissue. Red arrow: hypocotyl tissue.
(D) Cross sections of 10-day-old hypocotyls (two top panels) and stems (two bottom panels) below the apical meristems of 30-day-old plants grown under LDs. VENUS signal is shown in red color. White arrows point to signal from a single cell, whereas yellow arrows point to signal coming from several cells.
(E) Confocal images of seedlings of pFD::VENUS:FD and pFDP::VENUS:FDP 3DAS. Scale bars, 50 μm.
To analyze the distribution of FD and FDP proteins, translational fusions were constructed between FD or FDP and the VENUS chromophore in the context of full genomic sequences and transformed into fd-3 or fdp-2, respectively (STAR Methods; Figures S4C and S4D). Plants containing these fusions were analyzed by confocal microscopy. VENUS:FD was detected in the SAM and on the adaxial side of leaves prior to the floral transition at 6, 8, and 10 LDs after germination and throughout the domed inflorescence meristem at 12 LDs (Figure 4C, top panels). In SD-grown plants, VENUS:FD was detected in apices in a similar pattern to those of LDs (Figure S4F, left panel). VENUS:FD also accumulated in the vascular tissue of hypocotyls, as shown in cross sections (Figures 4D and S4G), longitudinal sections (Figure 4E), and in a 3D video (Video S1). The protein was also detected in the vascular tissue of inflorescence stems (Figure 4D) and at the root apical meristem (RAM) (Figures S4Ii and S4ii). VENUS:FDP was present in the apices of plants grown for 6 to 12 LDs but was expressed in a more restricted pattern at the SAM and was more associated with the lower rib meristem region (Figure 4C). In the inflorescence meristem at 12 LDs, VENUS:FDP was expressed more broadly in the rib-meristem region. Under SDs, VENUS:FDP was detected in a similar pattern to LDs (Figure S4F). VENUS:FDP was also present on the adaxial side of leaves (Figure 4C; Figures S4E and S4F), in the roots at the stele, and in the differentiation zone (Figures S4Iiii and S4iv), as well as in the vasculature of hypocotyl and stem (Figures 4D and 4E; Figure S4H; Video S2). Hence, the mRNA and protein localization reveal that FD and FDP are expressed broadly and are localized not only to the shoot apex but also strongly to the vascular tissue, young floral buds, floral organs, and roots.
Several Arabidopsis proteins transcriptionally regulate their paralogs (Nimchuk, 2017, Byrne et al., 2002). To test this, pFDP::VENUS:FDP was crossed into fd-3 and pFD::VENUS:FD into fdp-CRP2. In either background, no significant differences in the spatial expression of either FD:VENUS or FDP:VENUS were detected compared with their controls; therefore, these paralogs do not spatiotemporally regulate each other’s expression (Figures S4J and S4K).
FD but Not FDP Binds to Flowering and Floral Regulator Genes
To further compare the roles of FD and FDP, chromatin immunoprecipitation sequencing (ChIP-seq) was performed to compare their direct target genes. Inflorescences of 30-day-old VENUS:FD and VENUS:FDP plants were used because, at this developmental stage, VENUS accumulation was high in the shoot apices of both lines (Figures S5A and S5B). ChIP-seq experiments for each transcription factor were performed with biological replicates, and a high reproducibility between the replicates was found as described in the STAR Methods (R = 0.96 for VENUS:FDP and R = 0.88 for VENUS:FDP; Pearson correlation coefficient) (Figure S5C).
For ChIP-seq of VENUS:FD, 752 merged peaks were called (STAR Methods), which were assigned to 1,457 neighboring genes (Figure 5A; Figure S5E; Data S1). Most peaks were located within 500 bp upstream of the transcription start site (TSS) of a gene (Figure 5B). Furthermore, de novo motif analysis of the FD peak sequences revealed strong enrichment for G-boxes, consistent with the protein containing a bZIP domain, and these motifs were significantly enriched (p = 3.0e−59) in the center of the ChIP-seq peak, suggesting that they were directly bound by FD (Figure 5C). More than 50% of the peaks identified in FD ChIP-seq contain two G-box binding sites, and about 25% include three G-boxes (Figure 5D; Figure S5F). In addition, these motifs were mainly spaced 50–100 nucleotides (nt) apart (Figure 5E), a similar spacing to that observed in the promoters of non-FD/FDP target genes containing more than one G-box (Figure S5G). These data suggest that FD frequently binds to two or more closely spaced binding sites or that it acts in a combinatorial fashion with other transcription factors that recognize similar motifs.
Figure 5.

Genome-wide Target Sites of FD and FDP
(A–E) FD genome-wide targets. (A) The number of FD ChIP-seq peaks and associated genes. (B) Distribution of FD peaks in a region between 3 kb upstream of the transcription start site (TSS) and 1 kb downstream of the transcription termination site (TTS) of the closest gene. The solid line shows the positional distribution of observed FD peaks. The observed positional distribution was compared to those of 1,000 randomly generated peak sets. As the distributions were determined using bins, both the mean (dashed line) and the 95% confidence interval (light shaded color) for each bin are depicted. (C) Density plot of distances between the center of the G-boxes and the center of FDP peaks. The red line marks the shape of the distribution. The inset shows the logo of the enriched sequence motif identified by MEME motif analysis. The E-value indicates the statistical significance of the identified motif. (D) Number of FD ChIP-seq peaks containing the indicated number of G-box motifs. (E) Distance between neighboring G-box motifs in FD targets. The red line marks the shape of the distribution.
(F–J) FDP genome-wide targets. (F) Predicted number of FDP peaks and corresponding associated genes. (G) Distribution of FDP peaks as described for FD in (B). (H) Density plot of distances of G-boxes to the center of FDP peaks, as described for FD in (C). (I) Number of FD ChIP-seq peaks containing the indicated number of G-box motifs. (J) Distance between neighboring G-box motifs in FDP targets.
For ChIP-seq of VENUS:FDP, 291 merged peaks were identified, which were assigned to 552 neighboring genes (Figure 5F; Figure S5E; Data S1). Similar to FD, the highest frequency of FDP ChIP-seq peaks was found upstream of the TSS (Figure 5G). G-boxes were also significantly enriched in the center of the FDP peaks (p = 2.8e−54) (Figure 5H), and more than 50% of the peaks contained at least two closely spaced G-boxes (Figures 5I and 5J; Figures S5F and S5G), as observed for FD peaks.
The localization of these bZIP transcription factors was then compared. FD and FDP bound to 212 common regions (420 target genes; 80% of total FDP targets). In addition, FDP bound to 79 unique regions (representing 132 unique target genes) and FD bound to 540 unique regions (1,037 unique targets genes) (Figure 6A). The FDP unique target genes were of similar strength to the FDP common targets, whereas FD unique targets were, on average, slightly weaker than FD common targets, although they showed a wide distribution (Figure S5H). We also compiled unique target lists by using the more stringent criterion of identifying those regions bound by one factor in both replicates and not bound by the other factor in either of the two replicates. Using this criterion, 350 FD-bound regions were unique and 24 FDP-bound regions were unique. The unique target genes identified by both criteria are listed in Data S1. Afterward, we analyzed those unique genes identified by directly comparing the two original sets of target genes (132 for FDP and 1,037 for FD). Nine genes were identified as uniquely bound by FDP and related to ABA stimulus (p = 0.00026), and among them were CALCIUM-DEPENDENT PROTEIN KINASE 1 (CPK10), MOTHER OF FT (MFT), or SNF1-RELATED PROTEIN KINASE 2.7 (SNRK2.7) (Data S1; Fujii et al., 2011, Cutler et al., 2010, Xi et al., 2010). Uniquely bound by FD, were many flowering and floral regulator genes, such as SOC1, LFY, and AP1 (Figure 6B; Data S1), which were reduced in expression in fd mutants (Figure 6C; Collani et al., 2019, Searle et al., 2006, Abe et al., 2005, Wigge et al., 2005). Although FDP did not bind to these floral regulators, their mRNA levels increased earlier in fdp-CRP mutants (Figures 1F, 1G, and 6C; Figures S3A and S3B), suggesting that FDP indirectly regulates them. Strikingly, in an independent ChIP-seq dataset from 35S::GFP:FDP plants (Figure 2C), in which FDP is more broadly and highly expressed at the SAM, FDP was found to bind in the vicinity of most of these flowering genes, such as AP1, SOC1, and SEP3 (Data S1; Figure 6D). Furthermore, all except 11 of the binding sites (17 target genes) of FDP when expressed from its endogenous promoter were included in the 35S::GFP:FDP dataset (Figure S5I). Therefore, FDP can occupy the same sites as FD on flowering-related genes when expressed in a broader domain at the SAM or at higher levels. On the other hand, targets bound by both FD and FDP included genes involved in several biological processes, such as response to water deprivation (e.g., ABI5, ABF3, ARABIDOPSIS THALIANA HOMEOBOX 12 [HB-12]), and hormone pathways, including ABA (e.g., ABI FIVE BINDING PROTEIN 2 [AFP2], ABI FIVE BINDING PROTEIN 4 [AFP4], ABI5, ABF3, and HIGHLY ABA-INDUCED PP2C GENE 1 [HAI1]), gibberellin (GA) (e.g., GIBBERELLIN 2-OXIDASE 4 [GA2ox4]), and jasmonic acid (JA) (e.g., AUXIN-RESPONSIVE GH3 FAMILY PROTEIN [JAR1] and JASMONATE-ZIM-DOMAIN PROTEIN 6 [JAZ6]). We also identified genes encoding bZIP (e.g., ABF3 and ABI5) or SPL transcription factors (SPL8) and microRNAs (e.g., MIR156F). Strikingly, the bZIP genes include several associated with ABA responses within group A, suggesting that cross-regulation occurs within this group. However, FD did not bind to FDP or vice versa. Thus, despite both bZIP transcription factors containing identical DNA-binding regions (Figure S1A) and exhibiting identical enriched sequence motifs in ChIP-seq (Figures 5C and 5H), FDP binds to fewer sites and each of the proteins has unique binding sites.
Figure 6.

Characterization of FD and FDP Target Genes
(A) Venn diagram illustrating common peaks (top panel) and genes (bottom panel) in the FD and FDP ChIP-seq datasets.
(B) FD and FDP binding profiles to flowering genes. The three panels display FD and FDP and the control (INPUT) peaks at SOC1, LFY, and AP1 visualized with the Integrated Genome Browser (IGB).
(C) qRT-PCR analysis of SOC1, LFY, and AP1 mRNA abundance in apices of fd-3 and fdp-CRP2 mutants in 11- and 15-day-old plants under LDs.
(D) FDP-binding profiles to flowering genes when expressed from the 35S promoter.
(E) Proportion of DEGs (adj. p ≤ 0.05) that are up- or downregulated in the RNA-seq of 17 LD apices of fd-3 and fdp-CRP2. The number of DEGs directly bound by FD or FDP is illustrated in digits.
(F) GO terms enriched within the 109 genes differentially expressed and bound by FD using apices of 17-day-old plants. The color of the circles reflects the p value and the size the overrepresentation or underrepresentation of GO categories according a hypergeometric test (false discovery rate [FDR] < 0.05). The inset on the left shows an overview of all GO terms.
FD but Not FDP Directly Regulates Floral Transition
To further characterize FDP function, RNA sequencing (RNA-seq) was performed using apices of fdp-CRP2, fd-3, and WT grown under LDs for 17 days (Figure S6A), when both FDP and FD are expressed (Figures 4B and 4C; Figures S5A and S5B). At this stage, 153 and 1,458 differentially expressed genes (DEGs) (with adj. p ≤ 0.05) were identified in fdp-CRP2 and fd-3 mutants, respectively (Figure 6E; Data S2). The ChIP-seq data obtained in inflorescences were then compared with the DEGs in apices of each mutant. For FD, 109 DEGs were directly controlled by FD, a statistically significant overlap (one-sided Fisher’s exact test, p = 0.005094) (Figure 6E; Data S2). Of these 109 genes, 61 (56%) were upregulated in fd-3 and the others were downregulated (Figure 6E). Therefore, in inflorescences, FD transcriptionally activates or represses its direct targets. Gene Ontology (GO) analysis of FD directly regulated genes showed enrichment in biological processes, including maintenance of meristem identity, flower development, and response to abiotic stimulus (Figure 6F; Maere et al., 2005, Shannon et al., 2003). Genes within these clusters included the floral regulator genes SEPALLATA 1 (SEP1), SEP2, and SEP3; AP1; CAULIFLOWER (CAL); LFY; and SPL8; which were all downregulated in fd-3, whereas the GA catabolic enzyme ARABIDOPSIS THALIANA GIBBERELLIN 2-OXIDASE 6 (GA2ox6) as well as the negative regulator of ABA signaling ABI FIVE BINDING PROTEIN 3 (AFP3) were upregulated (Data S2). In contrast, a comparison of the DEGs in apices of fdp-CRP2 with the ChIP-seq of FDP identified only four genes in common in these two datasets (Figure 6E; Data S1), which was not a significant overlap (one-sided Fisher’s exact test, p = 0.5753) and suggested that most DEGs in the fdp-CRP2 mutant were indirectly regulated by FDP.
FD and FDP Act in Seedlings to Regulate ABA Responses and Cotyledon Greening
Many group A bZIP transcription factors related to FD and FDP are involved in ABA- and stress-signaling responses in seeds or young seedlings (Dröge-Laser et al., 2018). GO enrichment analysis of the overlapping set of FD and FDP target genes revealed a strong over-representation of genes involved in processes related to ABA (Figure 7A). Also, pFD::VENUS:FD and pFDP::VENUS:FDP expression was detected in apices of seedlings 2–3 days after sowing (DAS) (Figure 4E) in similar patterns to those observed in older plants (Figure 4C). RNA-seq was then performed on fd-3, fdp-CRP2, and WT seedlings 3 DAS (Figure S6B). Strikingly, more DEGs were identified in fdp-CRP2 and fd-3 mutants in seedlings, with 974 and 4,649 genes respectively (Figure 7B; Data S2), than in inflorescences (Figure 6E), where FD and FDP were believed to exert their main physiological function. A significant overlap between genotypes was detected, with almost 70% of the DEGs in fdp-CRP2 also found as DEGs in fd-3 (Figure 7C; Data S2).
Figure 7.

fd and fdp Are Less Sensitive to ABA in Greening Cotyledons
(A) GO analysis of the biological processes enriched in the common 420 genes bound by FD and FDP, analyzed and presented as in Figure 6F.
(B) Proportion of DEGs (adj. p ≤ 0.05) from the RNA-seq 3 DAS (of fd-3 and fdp-CRP2 mutants compared to WT), which are up- or downregulated, and within those, the subset of genes that are directly bound either by FD or FDP in inflorescences.
(C) Venn diagram showing the overlap between the DEGs in 3 DAS whole seedlings of fd-3 and fdp-CRP2 shown in (B).
(D) Venn diagram comparing the overlap of genes bound and regulated in fd-3 and fdp-CRP2 mutants in 3 DAS whole seedlings.
(E) FD- and FDP-binding profiles to ABA-related genes. The three panels display FD and FDP and the control (INPUT) peaks at the shown loci visualized with the IGB browser.
(F) Validation by ChIP-PCR of three common target genes involved in ABA responses. For each target, fold enrichment relative to its input is shown. Primers of TUBULIN (TUB) were used as negative controls. Data are shown as mean ± standard deviation.
(G) Greening cotyledon assay. The diagram represents the percentage of greening cotyledons (y axis) 7 DAS on plates containing different concentrations of ABA (x axes). The error bars represent standard errors (SEs) of two independent biological replicates.
The seedling DEGs were then compared with the direct target genes identified by ChIP-seq in inflorescences. For FD, 321 (25.7%) of the 1,250 expressed direct targets were DEGs between fd-3 and WT seedlings (Figure 7D), even though different tissues were used for the RNA-seq and the ChIP-seq analyses. The proportion of direct target genes misregulated in fd-3 seedlings is highly significant according to the one-sided Fisher’s exact test (p = 6.257 × 10−5). Similarly, a significant overlap (p = 0.000384) was found for FDP, where 38 (8%) of the 472 expressed direct targets were identified as DEGs in fdp-CRP2 (Figure 7D). Notably, 74% of FD DEGs that were bound by FD and 84% of FDP DEGs bound by FDP were more highly expressed in the corresponding mutant seedlings (Figure 7B), suggesting that FD and FDP mostly act as repressors of transcription at this stage, in contrast to the role of FD in inflorescences (Figure 6E). Furthermore, 50% (19 of 38) of the genes bound by FDP and differentially expressed in fdp-CRP2 were also found in the dataset of genes directly regulated by FD in seedlings (p = 1.36 10−27) (Figure 7D; Data S2). GO term analysis of this subset of 19 genes showed an enrichment in processes related to ABA, water deprivation, and JA (Figure S6A), which is similar to the GO term enrichments obtained within the DEGs in seedlings that are bound by FD or FDP (Figures S6C and S6D). We identified 20 genes (p = 2.5 × 10−7) within the 321 genes bound and DE in fd-3 that were related to ABA processes and mostly upregulated in expression in fd-3 seedlings. Among them, was CYP707A2 that encodes a cytochrome P450 enzyme involved in ABA catabolism (Kushiro et al., 2004), as well as ABA INSENSITIVE 1 (ABI1) and HYPERSENSITIVE TO ABA1 (HAB1), which encode phosphatases that inhibit ABA signaling (Saez et al., 2004, Leung et al., 1994, Meyer et al., 1994) and ABF3 (Data S1 and S2; Figure 7E). CYP707A2 and HAB1 were also upregulated in fdp-CRP2 but were not directly bound by FDP. In addition, CYP86A2, which encodes a fatty acid ω-hydroxylase involved in cutin biosynthesis, was bound by FD and upregulated in fd-3 seedlings (Data S1 and S2). Within the 38 genes bound by FDP and differentially expressed in fdp-CRP2, we identified 5 ABA-related genes (p = 0.0003), including the negative regulator of ABA signaling and stress response AFP2 (Garcia et al., 2008), which is also bound by FD (Data S1 and S2; Figure 7E). Within the 19 overlapping genes bound both by FD and FDP and upregulated in fd-3 and fdp-CRP2, we identified 4 ABA-related genes (21%, p value = 0.0002): AFP4/TMAC2, which belongs to the same gene family as AFP2 (Figure 7E); the ABA-inducible homeodomain transcription factor HB-12 that regulates stem growth (Son et al., 2010); and CIPK15/PKS3 (calcineurin B-like protein [CBL]-INTERACTING PROTEIN KINASE 15) and ARABIDOPSIS THALIANA MYB DOMAIN PROTEIN 44 (MYB44) that both mediate responses to abiotic stresses (Jaradat et al., 2013, Guo et al., 2002; Data S1 and S2). Also, binding of FD and FDP to the expected sites in ABI5, AFP2, and AFP4 was validated by ChIP-PCR (Figure 7F). Although FD and FDP are implicated in the regulation of ABA-related processes in seedlings, we detected no significant alteration in FD or FDP mRNA levels by application of ABA to seedlings.
Group A bZIPs, such as ABI5, are required during seedling establishment (Lopez-Molina et al., 2001). Hence, the sensitivity to ABA of fd and fdp mutants was determined in a cotyledon greening assay. The proportion of seedlings exhibiting green cotyledons 7 DAS was tested in the presence of several concentrations of ABA. Mutants that were less sensitive (abi5-7) or hypersensitive (hypersensitive germination3 [ahg3-2]) to ABA were used as controls (Figure 7G; Figure S7; Née et al., 2017, Nambara et al., 2002). In the absence of ABA, 100% of seedlings of all genotypes developed green cotyledons by 7 DAS (Figure 7F; Figures S7A and S7B). In the presence of 0.5 μM ABA, only 5% of WT seeds were able to establish green seedlings 7 DAS. In contrast, under the same conditions, approximately 80% of fd-3 and 40% of fdp-CRP2 seeds produced well-established autotrophic seedlings (Figure 7G; Figures S7A and S7B). Although exhibiting a weaker phenotype than abi5-7 mutants, fd-3 and each of the fdp-CRP mutants showed consistently reduced sensitivity to ABA along development compared to the WT (Figure S7B). Therefore, FD and FDP enhance the responsiveness of seedlings to ABA, consistent with them binding to and affecting the expression of many genes involved in ABA responses.
Discussion
Roles of FD and FDP in Flowering-Time Control and FT Activity
Only the gain-of-function missense mutations of FDP caused later flowering, whereas the deletion alleles flowered slightly earlier. The missense mutations fdp-1 and fdp-2 alter adjacent amino acids within the basic DNA-binding domain (Bensmihen et al., 2002; Figure S1A), without affecting the adjacent leucine zipper domain. An attractive explanation is that these mutant proteins heterodimerize with another basic leucine zipper transcription factor to form inactive heterodimers that cannot efficiently bind DNA, leading to late flowering. The proposed heterodimerization partner of FDP is unlikely to be FD because fdp-1 and fdp-2 enhanced the late-flowering phenotype of fd-3 mutants, in which the FD protein is likely to be absent, and FD and FDP are mainly expressed in different domains of the shoot apex. In contrast, the loss-of-function CRISPR alleles of FDP did not delay flowering of WT, but the allele predicted to cause the strongest reduction in FDP activity (fdp-CRP2) weakly delayed flowering in an fd-3 mutant background.
Surprisingly, the three FDP null alleles reproducibly caused earlier flowering under LDs. The early flowering of these mutants depends on the photoperiodic pathway, as it was not evident in SDs nor in the ft-10 tsf-1 mutant impaired in this pathway. FDP might, therefore, negatively modulate this pathway prior to floral induction. As FDP is expressed broadly in the plant, including in the vasculature of the stem and the rib meristem, the cells in which it acts to modulate flowering remain to be determined. In rice, bZIP proteins related to FD and FDP repress the transcription of FT-like genes in the vasculature (Brambilla et al., 2017), but we did not detect increased expression of FT or TSF in the fdp-CRP mutants. However, this might occur transiently or locally, making it difficult to detect by qRT-PCR, although such an effect would explain the epistatic genetic interaction between ft-10 tsf-1 and fdp-CRP.
The full effect of FT on floral induction cannot be explained by FD activity (Jaeger et al., 2013, Abe et al., 2005, Wigge et al., 2005). This residual effect of FT TSF at the SAM was assumed to be due to the activity of FDP (Jaeger et al., 2013, Jang et al., 2009, Abe et al., 2005). However, we found that FDP expression occurs in the rib meristem and did not detect binding of FDP to the flowering genes activated by FD. Similarly, neither fd-3 fdp-2, which was the latest flowering genotype generated by combining fd and fdp mutations, nor fd-3 fdp-CRP2, which included the strongest FDP loss-of-function allele, abolished the early flowering phenotype of plants overexpressing FT. Thus, FT can promote flowering independently of FD and FDP. Although, FD and FDP are present in a distinct branch of group A bZIPs (Dröge-Laser et al., 2018), FT might act through other members of the bZIP family or through unrelated classes of transcription factor,s as it does in axillary meristems (Niwa et al., 2013).
FD and FDP Bind to Common and Different Sets of Genes
Binding sites for both FD and FDP, as represented by peaks of ChIP-seq reads, are enriched at their center for a common motif related to a G-box (CACGTG). The 5′-C was less conserved than the other nucleotides, supporting variation in the precise recognition sequence (Figure 5C). Independently acquired ChIP-seq data for FD also detected enrichment for G-box motifs at FD-binding sites, but with slightly more conservation at the 5′ end of the G-box and less conservation at the 3′-G (Collani et al., 2019). We reanalyzed these data along with our data and detected an identical motif to the one described above (Figures S8A–S8D). Therefore, in vivo, FD and FDP bind to motifs with the central ACGT core, with some variation in the flanking bases but a preference for 5′-C and 3′-G. In flowering genes SEP3 and AP1, FD was also found to bind in vitro to a motif lacking the ACGT core (GTCGAC) (Collani et al., 2019). We did not detect this sequence by performing Multiple Em for Motif Elicitation (MEME) analysis on all FD targets or those misexpressed in apices or seedlings (Figures S8A, S8B, and S8D). However, this motif was overrepresented in the ChIP-seq peaks of FD and FDP compared to most random hexamer sequences but was much less frequent than the G-box (Figure S8F). The core ACGT sequence is recognized by most bZIPs, although some plant bZIPs do bind to non-ACGT sequences (Dröge-Laser et al., 2018). Many class A bZIPs closely related to FD and FDP, such as ABFs, bind to G-box-related ABA-responsive elements (ABREs) (PyACGTGG/TC) (Fujita et al., 2013). The bases 3′ to the core ACGT in the ABRE are also enriched in our MEME analysis of FD- and FDP-binding sites (ACGTGTC). Furthermore, on treatment with ABA, the G-box motif was over-represented among the binding sites of ABFs (Song et al., 2016). Thus, the binding sites of FD and FDP as defined in our ChIP-seq and the binding site of FD defined in the ChIP-seq of Collani et al. (2019) are very similar to those of other group A bZIPs and particularly of ABFs after ABA treatment.
Nevertheless, FD and FDP do not bind to identical gene sets. Generally, FD binds to more sites than FDP, including the promoters of genes involved in floral induction, such as AP1, SOC1, and LFY. The binding of FD to these flowering genes was also detected by Collani et al. (2019), and there was a statistically significant 50% overlap in the targets identified in that study with those identified here (Figure S8E), although different plant tissues were used. Furthermore, ABFs did not show significant binding to most of these flowering genes in ChIP-seq experiments after treatment of seedlings with ABA (Song et al., 2016). How the specificity of bZIPs for different target genes is determined, although they recognize related or identical motifs, is generally not clear (Dröge-Laser et al., 2018). However, in the case of FDP, expressing it from the FD or 35S promoters reduced its specificity, causing it to bind to established FD targets. Thus, the distinct patterns of transcription of FD and FDP are likely to, at least partially, explain the in vivo specificity of their protein products and allow FD to bind specifically to flowering genes. This proposal could be further tested by determining, for example, whether FD and FDP bind to the same sequences in vitro, and by performing ChIP-seq analysis on FDP expressed in the identical pattern to FD. As plant bZIP transcription factors bind to DNA as dimers or heterodimers (Vinson et al., 1989) and heterodimerization can determine their specificity (Mair et al., 2015, Ehlert et al., 2006), specific partners expressed in the SAM might determine FD target specificity. Similarly, combinatorial binding with transcription factors at adjacent motifs in promoters can also allow the recognition of specific targets (Zinzen et al., 2009). Alternatively, the open chromatin state or altered histone marks of target genes at the SAM during floral induction (You et al., 2017, Zhu et al., 2015) may allow FD to bind to flowering genes that are inaccessible to FDP in the cells in which it is expressed.
Relationship between FD/FDP Activity and ABA Signaling
ABA signaling by the group A bZIP ABI5 inhibits the acquisition of seedling autotrophy (Lopez-Molina et al., 2001). We found that FD and FDP are both expressed in young seedlings and participate in their ABA responses. Most ABA-related DEGs that were bound by FD or FDP were increased in expression in fd or fdp-CRP2 mutants, suggesting that they act as repressors of transcription in this process. Among the most highly upregulated genes in fd-3 were two related to ABA responses, namely, ATOEP16-2 and CYP707A2. Interestingly, mutations in these genes confer hypersensitivity to ABA, opposite to the phenotype observed in fd-3. Thus, further genetic studies will be required to establish the contribution of these genes to the fd phenotype. Collani et al. (2019) identified four ABA-related genes that were bound by FD and misexpressed in the apices of fd mutants during inflorescence development, but their function in ABA responses was not determined. Similarly, in hybrid aspen trees, a genetic analysis of FD-LIKE 1 (FDL1) and FD-LIKE 2 (FDL2) transcription factors showed they were involved in adaptive responses and bud dormancy (Tylewicz et al., 2015). Thus, in A. thaliana, as in poplar, these proteins have a conserved dual role in ABA signaling and reproductive development. Furthermore, in A. thaliana, the FT-like PEBP protein MFT negatively regulates ABA responses to repress seed germination (Vaistij et al., 2018, Xi et al., 2010), and FDP was found to bind to MFT in our ChIP-seq. MFT is a member of the most ancient clade within the PEBP proteins and is present in the moss Physcomitrella patens and the liverwort Marchantia polymorpha, whereas FT and TFL1-like proteins are not (Eklund et al., 2018, Hedman et al., 2009). Furthermore, in Marchantia sp., transcription of MFT is induced by application of ABA, suggesting an ancient role in ABA responses (Eklund et al., 2018). MFT proteins probably also interact with group A bZIPs related to FD (Hou and Yang, 2016). Thus, the relationship between the FT-interacting proteins FD and FDP with ABA responses that are described here might involve MFT and represent an evolutionarily ancient function. In this case, their roles in the flowering of higher plants are derived from an ancient role in ABA responses.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| GFP | Abcam | Cat#ab290 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| SCRI Renaissance 2200 | Musielak et al., 2015 | N/A |
| Propidium iodide | SIGMA | Cat#25535-16-4 |
| Protease Inhibitor Cocktail | SIGMA | Cat#P9599 |
| PMSF | SIGMA | Cat#329-98-6 |
| Clearsee | Kurihara et al., 2015 | N/A |
| Protein A Sepharose fast flow | GE Healthcare | GE17-1279-03 |
| Abscisic acid (ABA) | SIGMA | Cat#14375-45-2 |
| Bacterial and Virus Strains | ||
| Agrobacterium tumefaciens GV3101 | Intact Genomics | Cat #1282-12 |
| Critical Commercial Assays | ||
| DNA-free DNase | AMBION | Cat#AM1907M |
| RNeasy Plant Mini Kit | QIAGEN | Cat#74904 |
| iTaq Universal SYBR® Green Supermix | Bio-rad | Cat#1725121 |
| Ovation Ultralow System V2 (ChIP_seq) | NuGEN | Cat#0344NB-08 |
| NucleoSpin Gel and PCR Clean-up Kit | Macherey-Nagel | Cat#740609.10 |
| NTB Buffer | Macherey-Nagel | Cat#740595.1 |
| NEBNext® Ultra II Directional RNA Library Prep with Sample Purification Beads | NEBs | Cat#E7765 |
| Deposited Data | ||
| Raw RNA-seq | This paper | NCBI SRA: PRJNA560053 |
| whole genome sequencing | This paper | NCBI SRA:PRJNA560053 |
| ChiP-seq reads | This paper | NCBI SRA:PRJNA560053 |
| Software and Algorithms | ||
| CAS-OFFinder | Bae et al., 2014 | https://github.com/snugel/cas-offinder |
| BWA | Li and Durbin, 2009 | https://github.com/lh3/bwa |
| samtools | Li et al., 2009 | http://www.htslib.org/download/ |
| bcftools | Li, 2011 | http://www.htslib.org/download/ |
| cutadapt | Martin, 2011 | https://cutadapt.readthedocs.io/en/stable/installation.html |
| Trimmomatic | Bolger et al., 2014 | http://www.usadellab.org/cms/?page=trimmomatic |
| Bowtie2 | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| deepTools | Ramírez et al., 2014 | https://deeptools.readthedocs.io/en/develop/content/installation.html |
| R statistical language | R Development Core Team, 2009 | https://www.r-project.org |
| MACS 2 | Zhang et al., 2008 | https://pypi.org/project/MACS2/ |
| chipPeakAnno | Zhu et al., 2010 | https://www.bioconductor.org/packages/release/bioc/html/ChIPpeakAnno.html |
| MEME-ChIP | Ma et al., 2014 | http://meme-suite.org/meme-software/5.1.1/meme-5.1.1.tar.gz |
| TopHat2 | Kim et al., 2013 | https://ccb.jhu.edu/software/tophat/index.shtml |
| HTseq- count | Anders et al., 2015 | https://htseq.readthedocs.io/en/release_0.11.1/count.html |
| DESeq2 | Love et al., 2014 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| Cytoscape | Shannon et al., 2003 | https://cytoscape.org |
| BINGO | Maere et al., 2005 | Available through the cytoscape program |
| Exonerate | Slater and Birney, 2005 | https://www.ebi.ac.uk/about/vertebrate-genomics/software/exonerate |
| muscle | Edgar, 2004 | https://www.drive5.com/muscle/downloads.htm |
| trimAl | Capella-Gutiérrez et al., 2009 | http://trimal.cgenomics.org/downloads |
| MODELTEST | Posada and Crandall, 1998 | http://evomics.org/resources/software/molecular-evolution-software/modeltest/ |
| PhyML | Guindon et al., 2009 | http://www.atgc-montpellier.fr/phyml/binaries.php |
| Experimental Models: Organisms/Strains | ||
| Arabidopsis Col-0 | NASC | N1093 |
| Arabidopsis GAS1::FT | Jang et al., 2009 | N/A |
| Arabidopsis fd-3 | Jang et al., 2009 | N/A |
| Arabidopsis ft-10 | Torti et al., 2012 | N/A |
| Arabidopsis ft-10 tsf-1 | Torti et al., 2012 | N/A |
| Arabidopsis fdp-1 | Jaeger et al., 2013 | N/A |
| Arabidopsis tsf-1 | Torti et al., 2012 | N/A |
| Arabidopsis ahg3-2 | Née et al., 2017 | N/A |
| Arabidopsis abi5-7 | Née et al., 2017 | N/A |
| pFD::VENUS:FD in fd-3 | This study | N/A |
| pFDP::VENUS:FDP in fdp-2 | This study | N/A |
| pFD::FDP in fd-3 | This study | N/A |
| 35S::HA:FD in WT | This study | N/A |
| 35S::HA:FDP in WT | This study | N/A |
| 35S::GFP:FDP in WT | This study | N/A |
| Oligonucleotides | ||
| For cloning, please see Data S3 (sheet 1) | This study | N/A |
| For in situ hybridization, please see Data S3 (sheet 1) | This study | N/A |
| For ChIP-PCR, please see Data S3 (sheet 1) | This study | N/A |
| For qRT-PCR, please see Data S3 (sheet 1) | This study | N/A |
| For genotyping, please see Data S3 (sheet 1) | This study | N/A |
Resource Availability
Lead Contact
Further information and requests for resources should be directed to and will be fulfilled by the Lead Contact, George Coupland (coupland@mpipz.mpg.de).
Materials Availability
All materials generated in this study are available from the Lead Contact.
Data and Code Availability
The raw sequence read data can be obtained from the NCBI Sequence Read Archive under accession number PRJNA560053: Genome-wide analyses define distinct functions for the florigen interacting bZIP transcription factors FD and FDP in abscisic acid responses and flowering of Arabidopsis.
Experimental Model and Subject Details
Columbia-0 (Col) was used as WT in all experiments and for plant transformation.
The lines GAS1::FT, fd-3, ft-10, ft-10 tsf-1, tsf-1, ahg3-2 and abi5-7 were previously described (Née et al., 2017, Torti et al., 2012, Jang et al., 2009, Sohn et al., 2007, Nambara et al., 2002). The fdp-1 mutant was described in Jaeger et al. (2013) and backcrossed once to Col-0.
Seeds were stratified on soil for 2–3 days in the dark at 4°C. Plants were grown under controlled environmental conditions at 20°C, in LDs (16 h light/8 h dark) or in SDs (8 h light/16 h dark).
Method Details
Genotyping of TILLING lines
The fdp-2 TILLING line was obtained from BREECON GmbH (Potsdam). The fdp-2 mutation results in an amino-acid change at position 182 of the FDP protein. The original allele was backcrossed three times with Col-0 and homozygotes selected with a dCAPs detecting an AluI polymorphism (Data S3).
Generation of fdp-CRP mutants and sequencing
The fdp-CRP1 and fdp-CRP2 mutants were generated using the previously described protocol (Hyun et al., 2015; see Data S3 for primer list). The fdp-CRP3 line was obtained using the method described previously (Wang et al., 2015; see Data S3 for primer list). All three fdp-CRP mutants were backcrossed once to Col-0 to remove the Cas9 transgene and potential off-targets in the background. Whole-genome sequencing (20 × coverage) was performed on the backcrossed fdp-CRP mutants to confirm the expected mutations and detect any off-target mutations. Primers to genotype fdp-CRP2 and fdp-CRP3 are listed in Data S3.
Off-target analysis
The off-target analyses in this study were performed in three steps. First, potential off-targets were detected with maximum five mismatches using the dedicated off-target search program CAS-OFFinder (Bae et al., 2014). Second, whole-genome Illumina resequencing (single-end) was performed using the CRISPR mutant lines. The quality control of the resulting raw single-end reads was performed as for the ChIP-seq reads (see below). Reads were then mapped to the Arabidopsis thaliana TAIR10 genome using BWA (Li and Durbin, 2009) with default settings. BWA outputs both primary and, if present, secondary alignments. We only kept primary alignments with a mapping quality of at least 30. Samtools mpileup (Li et al., 2009) (default settings) was used in conjunction with bcftools (Li, 2011) for initial SNP calling. SNPs were filtered by minimum quality of 25, minimum depth of 5. Additionally, all regions of the genome devoid of any mapped reads were labeled as potential larger deletions. Finally, we searched for overlap between the predicted off-targets and the SNPs and larger deletions.
Construction of transgenic plants
The 35S::HA:FDP fusion was generated by introducing the FDP cDNA into pALLIGATOR (Bensmihen et al., 2004), whereas in the 35S::GFP:FDP construct (used for the ChIP-seq), the FDP cDNA was introduced into 35S::GFP:GW (gift J.Parker, MPIPZ, Cologne). For the pFD::FDP construct (used for the complementation analysis), the 35S promoter and the triple HA of the pAlligator2 vector were exchanged for the 3.1 kb FD promoter (see Data S3). The 35S:HA::FD construct was assembled in the same vector as used for FDP.
For the pFDP::VENUS:FDP and pFD::VENUS:FD constructs, the 8 kb and 5.5 kb genomic regions were cloned, respectively. Overlapping primers were designed at position −1 relative to the TSS, to insert the fluorescent VENUS protein with a poly 9-alanine linker attached to the first exon. Overlapping fragments were assembled by Polymerase Incomplete Primer Extension. All constructs were transferred to the binary vector pEarleyGate301 (Earley et al., 2006; See Data S3). At least 10 independent lines with similar flowering behavior were selected for each construct in the T1 generation and two to four lines showing a Mendelian segregation (3:1) were followed for further study. Most pFD::VENUS:FD fd-3 transformant lines flowered earlier than fd-3, confirming the functionality of the transgene, and a line (#3) that flowered at a similar time to WT plants was selected for further analysis (Figure S4C). The pFDP::VENUS:FDP transgene did not fully complement the late-flowering phenotype of fdp-2 (Figure S4D), probably because the fdp-2 allele is a dominant gain of function allele that partially impairs the complementation of the VENUS:FDP line. However, in four independent pFDP::VENUS:FDP lines, VENUS was expressed in the same pattern, which recapitulated the spatial and temporal patterns of FDP mRNA detected by in situ hybridization. Therefore, one of these lines (#10; Figure S4D) was selected for further analysis.
All constructs were introduced into Agrobacterium strain GV3101 (pMP90RK) and transformed into Col-0 by the floral dip method (Clough and Bent, 1998).
RNA extraction and qRT-PCR
Total RNA was extracted from apices collected at time Zeitgeber 3 (ZT3) using the RNAeasy extraction kit from QIAGEN and was treated with DNA-free DNase (Ambion). For reverse transcription, 2 μg RNA was used following manufacturer’s instructions (Invitrogen). RT-qPCR was performed with the LightCycler 480 instrument (Roche). The RT-qPCR primers are listed in Data S3. PEX4 was used as an internal control in all RT-qPCR experiments. All RT-qPCR assays were repeated at least twice for independent biological replicates.
In situ hybridization
Plants were grown for 3 weeks in SDs and then transferred to LDs for 3, 5 and 7 days. The samples were mainly apices collected at ZT3. In situ hybridization was performed as previously described (Torti et al., 2012). To amplify the FDP, FD, FUL and AP1 RNA probes, the primers listed in Data S3 were used.
Confocal microscopy analyses
The manually dissected shoot meristems were placed overnight in 4% PFA (Sigma-Aldrich) and then treated with ClearSee as described in Kurihara et al. (2015). Samples were kept in ClearSee solution for 4 days to one week. The samples were stained with SCRI Renaissance 2200 dye (Musielak et al., 2015) overnight and imaged using a confocal laser scanning microscope (Zeiss LSM780). The settings were optimized for visualizing Venus fluorescent proteins and DAPI (laser wavelength, 514 nm; detection wavelength, 517–569 nm).
Greening cotyledon assays
Seed batches were generated by growing all genotypes side by side in the green house under LDs and seeds were harvested when all siliques were dry and fully after-ripened. Between 30–60 seeds per genotype were sown on filter papers (Whatmann 3M grade 1, 85 mm) imbibed with 1.6 mL Mock or ABA solution. ABA stock solution was dissolved in DMSO in a final concentration: 0.05% v/v in distilled water. Seeds on Petri-dishes were stratified for 2 days in the dark and placed into a growth chamber (12 h light/12 h dark, 25/20°C cycle). After 7 days, the proportion of seedlings with open and green cotyledons was scored. Each assay was performed in technical triplicate, and was repeated after a 2-month interval. The data in Figure 6 combine results from both sets of assays.
ChIP experiments and RNA-seq materials
For ChIP experiments, pFD::VENUS:FD in fd-3 and pFDP::VENUS::FDP in fdp-2 were grown under LDs and 2 g inflorescence material (from plants approximately 10-cm tall) were harvested at ZT4. The 35S::GFP:FDP lines were grown in LDs for 2 weeks and above-ground tissue was collected at ZT4. Each ChiP experiment was performed twice following a previously described protocol (Mateos et al., 2015).
For transcriptome analysis, 3-day-old WT, CRP2-fdp and fd-3 seedlings were grown on plates containing filter paper moistened with demineralized water and incubated in a growth chamber (16 h light/8 h dark, 25/20°C cycle). For each genotype, three batches of 10 seedlings were harvested from three different plates growing simultaneously. The RNA pooled from the seedlings of each batch was considered as a biological replicate and used for RNA-seq library preparation.
ChIP-seq data analysis
Raw reads from ChIP-seq experiments and the corresponding control samples (INPUT) were preprocessed by removing potential adaptor sequences using cutadapt (Martin, 2011) and trimming low-quality ends using Trimmomatic (Bolger et al., 2014). Processed reads were mapped to the Arabidopsis thaliana genome version TAIR10 using Bowtie 2 (Langmead and Salzberg, 2012) with default settings that result in only the best alignment per read being reported (see Data S3). Only those alignments were kept that had a mapping quality of 30 or larger, which is indicative of a high probability that the read has been mapped to the correct region of the genome. To compare the ChIP-seq replicate samples, we determined the read coverage across the genome using the bamCoverage program of the deepTools suite (Ramírez et al., 2014) with the following parameters: binSize (-bs) 5000, extend reads (–extendReads) 250, RPKM normalization (–normalizeUsing RPKM). The RPKM values were transformed by adding 1 and taking the log2. Scatterplots and correlations between the values of replicate samples were obtained using the R statistical language (R Development Core Team, 2009).
MACS version 2.1.1.20160309 (Zhang et al., 2008) was used to identify peaks (FDR ≤ 0.01) on individual IP replicates together with their respective input samples. Peak length distributions were summarized using the density function available in R.
Peak annotation
The Bioconductor R package chipPeakAnno (Zhu et al., 2010) was used to relate peaks to Arabidopsis thaliana genes version TAIR10. Peaks were related to genes when they were located within 3 kb upstream and 1 kb downstream of the gene.
To determine the most favored peak-center position relative to gene bodies, peaks within 3 kb upstream and 1 kb downstream of the closest gene were classified based on whether the center resided in the (1) promotor, (2) downstream region and (3) open reading frame. For promoter regions, the distance was determined between the peak center to the transcription start site. For downstream regions, the distance was taken between the transcription termination site and the peak center. For those peaks with centers residing within the open reading frame, the distance was calculated between the transcription start site and the peak center and was then normalized by gene length. The peak position plots were generated in R according to the following method. First, the distances between the transcription start site and peak centers for those peaks in the promoter were converted to negative. The normalized distances between the transcription start site and peak-centers for the peaks within the open reading frame were multiplied arbitrarily by 2000 for plotting. Then, 2000 was added to the distances between the peak center and the transcription termination site for those peaks residing within the 1 kb downstream region of the gene to separate the peaks from the open reading frame. Finally, the converted positions were used to generate a histogram with the range −3,000 to 3,000 and bin-size of 200. The histogram range was divided as follows: positions −3,000 to 0, 1 to 2,000 and 2,001 to 3,000 corresponded to the 3 kb promoter region, open reading frame and 1 kb downstream region, respectively.
The observed peak position distributions were compared to those of 1000 random sets containing the same number of peaks and peak-length distributions as the observed peak sets (Figures 5B and 5G). A histogram similar to the one of the observed peaks was generated for each of the random peak set. Summarizing of the 1,000 histograms for the random peak sets was performed by calculating the mean and 95% confidence interval for each of the bins.
Overlapping peaks from the different replicates were merged when the overlap size was at least 80% of the smallest peak length (Figure S5D). A merged peak is at least as long or longer than the largest of the replicate peaks being merged. Only considering merged peaks ensured that each peak used in this study was present in all corresponding experimental replicates (Figure S5D).
Motif discovery
The sequences corresponding to identified peaks were extracted and analyzed for enriched motifs using MEME-ChIP (Ma et al., 2014) with the following adapted parameters: minimum motif width (-meme-minw 4);maximum motif width (-meme-minw) 12; motif database (-db ArabidopsisDAPv1.meme). The occurrences of predicted motifs were extracted from the corresponding FIMO output produced by the MEME-ChIP suite. Several motif positions reported by FIMO overlapped but were predicted on the opposite strand. In these specific cases the overlapping motif locations were merged. The positional distribution relative to the peak centers were derived by determining the distance between the peak center and the predicted motif centers.
RNA-seq data analysis
Raw RNA-seq single-end reads were pre-processed in the same way as the ChIP-seq and whole-genome resequencing reads. Processed RNA-seq reads were mapped to the genome of A. thaliana with TopHat2 (Kim et al., 2013) using the TAIR10 annotation as a guide. The following additional parameter settings were used for the mapping: maximum edit distance 5; maximum mismatches 5; minimum intron length 50; maximum intron length 10000; read gap length 2. Read counts per gene were summarized into an initial count table using HTseq- count (Anders et al., 2015). All genes in the count table with a rowSums of less than 10 were removed. The count table was used for further analysis with the DESeq2 (Love et al., 2014) R package using the simple design formula ~mutant where mutant indicated whether the samples were from WT, fd or fdp mutants. As a quality control, samples were clustered based on inter-sample distances defined as 0.5–0.5 ∗ cor(A,B), where cor(A,B) corresponds to the pairwise correlation between the regularized log expression of the 200 most frequently expressed genes in samples A and B. Clustering of the distances was performed using the hclust function in R. Differential expression analysis was performed using the DESeq function of DESeq2 and the contrasts between mutants versus the WT were extracted using the results function.
GO ontology analysis and data visualization
GO analysis was performed with the BINGO tool using the CYTOSCAPE platform for visualization. The cluster named “flowering and flower development” was generated using the list obtained from: https://www.mpipz.mpg.de/14637/Arabidopsis_flowering_genes, which contains related flowering genes, and the list generated using the website: https://bar.utoronto.ca/thalemine/template.do?name=Keyword_GO_genes&scope=all, which allows associated GO terms and genes to be queried by entering keywords: In this case,:“ flower development” was used. Both lists were combined and the subset of genes for FD and FDP were extracted when compared with the whole FD and FDP targets. To define the rest of GO defined in the Supplemental datasets, a similar procedure was followed using the ARAPORT tool and entering different keywords with the precise GO terms names obtained in the CYTOSCAPE tool.
Phylogenetic tree FDP-like sequences
The genomes of Arabidopsis halleri, A. lyrata, Boechera stricta, Capsella rubella C. grandiflora, Carica papaya and Eutrema salsugineum were downloaded from phytozome (Goodstein et al., 2012). The genome of Tarenaya hassleriana was downloaded from the CoGE webpage (Lyons and Freeling, 2008). The Arabis alpina genome was downloaded from http://www.arabis-alpina.org. Exonerate (Slater and Birney, 2005) was used to determine the gene structures of FD and FDP (if present) in the different genomes using the Arabidopsis AT4G35900 and AT2G17770 genes (from TAIR10) as the queries for searching for FD and FDP, respectively. The protein sequences corresponding to the identified gene (coding sequence) structures were aligned using muscle (Edgar, 2004). Noisy regions were removed from the alignment using trimAl (Capella-Gutiérrez et al., 2009) in the automated1 mode. The cleaned protein alignment was transformed into a CDS alignment by replacing amino acids by their corresponding codon triplet and each gap by three gaps. MODELTEST (Posada and Crandall, 1998) was used to select the best nucleotide substitution model that could be used for maximum likelihood tree estimation with the program PhyML (Guindon et al., 2009). Branch support for the maximum likelihood tree was derived from 100 bootstrap rounds.
Quantification and Statistical Analysis
All the standard statistical analyses were conducted using SigmaPlot software. When several data points were compared, an ANOVA and Kruskal-Wallis One Way Analysis of Variance on Ranks was used. For simpler two-way comparisons, t test. Most of the graphs were represented using the boxplots of SigmaPlot. The box depicts the distance between the 1st and 3rd quartiles, the Inter Quartile Range (IQR = Q3-Q1). The whiskers are 1.5∗IQR which corresponds to the highest / lowest point within the range. The dotted lines represent the outliers, which are the points greater than 1.5∗IQR.
Flowering time was quantified by counting total leaf number (including both cauline and rosette leaves) of at least 10 plants per genotype. Experiments for flowering time and qRT-PCR were performed at least twice independently. In general, data are represented by mean ± SD, except in the greening cotyledon assay, where the mean ± SE is represented.
The statistical analysis of hexamer overrepresentation was done as follows. There are in total 4096 (46) possible hexamers which can be reduced to 2080 accounting for palindromic reverse complement hexamers. For each of the 2080 remaining hexamers we determined the number of experimental peaks in which it occurs at least once. Next it was determined how many standard deviations (Z-score) this number was away from the mean number of peaks having at least one motif instance in 1000 random sets. Each of these random peak sets was generated as follows: Each experimental peak was re-assigned to a random gene and placed at the same distance from the transcription start site as the original associated gene. Finally, the Z-score of a particular hexamer was compared to that of all other hexamers. In the case of multiple occurrences of hexamers, the same approach was used but then the number of peaks with at least 2 or more motifs were considered.
To calculate motif spacing on peaks with multiple motifs, we recorded the distances between the centers of consecutive motifs. For comparison, we also analyzed the distance between consecutive motifs in the protomer (3kb upstream) of all genes that were not predicted to be targets of FD or FDP.
Acknowledgments
We thank John Chandler, Diarmuid O’Maoileidigh, Lucio Conti, Silvio Collani, and Alice Pajoro for their useful comments on the paper and on the interpretation of experiments. We thank BREECON GmbH (Potsdam) for carrying out the TILLING analysis. We thank Bruno Huettel from Max Planck-Genome Centre Cologne for all the technical support with the ChIP-seq and Samson Simon for helping with the phylogenetic analysis of FD and FDP. P.M. was supported by the EU Marie Curie Initial Training Network SYSFLO (agreement number 237909). G.C. was a recipient of the ERC Advanced Grant HyLife and his laboratory is supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany’s Excellence Strategy (EXC 2048/1 Project ID: 390686111) and a core grant from the Max Planck Society.
Author Contributions
Conceived and designed the experiments, M.R.B. and G.C.; Performed the experiments, M.R.B.; Analyzed the data, M.R.B., E.S., G.N., and G.C.; Participated in bioinformatics analysis, E.S., M.R.B., and P.M.; Interpreted the data, M.R.B., E.S., G.N., and G.C.; Contributed experiments and analysis tools, H.O., C.P., R.M.-G., C.V., G.N., S.J., and F.A.L.; Wrote the paper, M.R.B., E.S., G.N., and G.C.; All authors read and approved the final manuscript.
Declaration of Interests
The authors declare no competing interests.
Published: June 2, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.107717.
Supplemental Information
References
- Abe M., Kobayashi Y., Yamamoto S., Daimon Y., Yamaguchi A., Ikeda Y., Ichinoki H., Notaguchi M., Goto K., Araki T. FD, a bZIP protein mediating signals from the floral pathway integrator FT at the shoot apex. Science. 2005;309:1052–1056. doi: 10.1126/science.1115983. [DOI] [PubMed] [Google Scholar]
- Abe M., Kosaka S., Shibuta M., Nagata K., Uemura T., Nakano A., Kaya H. Transient activity of the florigen complex during the floral transition in Arabidopsis thaliana. Development. 2019;146:dev171504. doi: 10.1242/dev.171504. [DOI] [PubMed] [Google Scholar]
- Anders S., Pyl P.T., Huber W. HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrés F., Coupland G. The genetic basis of flowering responses to seasonal cues. Nat. Rev. Genet. 2012;13:627–639. doi: 10.1038/nrg3291. [DOI] [PubMed] [Google Scholar]
- Bae S., Park J., Kim J.S. Cas-OFFinder: a fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics. 2014;30:1473–1475. doi: 10.1093/bioinformatics/btu048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bensmihen S., Rippa S., Lambert G., Jublot D., Pautot V., Granier F., Giraudat J., Parcy F. The homologous ABI5 and EEL transcription factors function antagonistically to fine-tune gene expression during late embryogenesis. Plant Cell. 2002;14:1391–1403. doi: 10.1105/tpc.000869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bensmihen S., To A., Lambert G., Kroj T., Giraudat J., Parcy F. Analysis of an activated ABI5 allele using a new selection method for transgenic Arabidopsis seeds. FEBS Lett. 2004;561:127–131. doi: 10.1016/S0014-5793(04)00148-6. [DOI] [PubMed] [Google Scholar]
- Bolger A.M., Lohse M., Usadel B. Trimmomatic: A flexible trimmer for Illumina Sequence Data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brambilla V., Martignago D., Goretti D., Cerise M., Somssich M., de Rosa M., Galbiati F., Shrestha R., Lazzaro F., Simon R., Fornara F. Antagonistic Transcription Factor Complexes Modulate the Floral Transition in Rice. Plant Cell. 2017;29:2801–2816. doi: 10.1105/tpc.17.00645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrne M.E., Simorowski J., Martienssen R.A. ASYMMETRIC LEAVES1 reveals knox gene redundancy in Arabidopsis. Development. 2002;129:1957–1965. doi: 10.1242/dev.129.8.1957. [DOI] [PubMed] [Google Scholar]
- Capella-Gutiérrez S., Silla-Martínez J.M., Gabaldón T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics. 2009;25:1972–1973. doi: 10.1093/bioinformatics/btp348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z., Tan J.L., Ingouff M., Sundaresan V., Berger F. Chromatin assembly factor 1 regulates the cell cycle but not cell fate during male gametogenesis in Arabidopsis thaliana. Development. 2008;135:65–73. doi: 10.1242/dev.010108. [DOI] [PubMed] [Google Scholar]
- Clough S.J., Bent A.F. Floral dip: a simplified method for Agrobacterium-mediated transformation of Arabidopsis thaliana. Plant J. 1998;16:735–743. doi: 10.1046/j.1365-313x.1998.00343.x. [DOI] [PubMed] [Google Scholar]
- Collani S., Neumann M., Yant L., Schmid M. FT modulates genome-wide DNA-binding of the bZIP transcription factor FD. Plant Physiol. 2019;180:367–380. doi: 10.1104/pp.18.01505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbesier L., Vincent C., Jang S., Fornara F., Fan Q., Searle I., Giakountis A., Farrona S., Gissot L., Turnbull C., Coupland G. FT protein movement contributes to long-distance signaling in floral induction of Arabidopsis. Science. 2007;316:1030–1033. doi: 10.1126/science.1141752. [DOI] [PubMed] [Google Scholar]
- Cutler S.R., Rodriguez P.L., Finkelstein R.R., Abrams S.R. Abscisic acid: emergence of a core signaling network. Annu. Rev. Plant Biol. 2010;61:651–679. doi: 10.1146/annurev-arplant-042809-112122. [DOI] [PubMed] [Google Scholar]
- Dröge-Laser W., Snoek B.L., Snel B., Weiste C. The Arabidopsis bZIP transcription factor family-an update. Curr. Opin. Plant Biol. 2018;45:36–49. doi: 10.1016/j.pbi.2018.05.001. [DOI] [PubMed] [Google Scholar]
- Earley K.W., Haag J.R., Pontes O., Opper K., Juehne T., Song K., Pikaard C.S. Gateway-compatible vectors for plant functional genomics and proteomics. Plant J. 2006;45:616–629. doi: 10.1111/j.1365-313X.2005.02617.x. [DOI] [PubMed] [Google Scholar]
- Edgar R.C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehlert A., Weltmeier F., Wang X., Mayer C.S., Smeekens S., Vicente-Carbajosa J., Dröge-Laser W. Two-hybrid protein-protein interaction analysis in Arabidopsis protoplasts: establishment of a heterodimerization map of group C and group S bZIP transcription factors. Plant J. 2006;46:890–900. doi: 10.1111/j.1365-313X.2006.02731.x. [DOI] [PubMed] [Google Scholar]
- Eklund D.M., Kanei M., Flores-Sandoval E., Ishizaki K., Nishihama R., Kohchi T., Lagercrantz U., Bhalerao R.P., Sakata Y., Bowman J.L. An Evolutionarily Conserved Abscisic Acid Signaling Pathway Regulates Dormancy in the Liverwort Marchantia polymorpha. Curr. Biol. 2018;28:3691–3699.e3. doi: 10.1016/j.cub.2018.10.018. [DOI] [PubMed] [Google Scholar]
- Fujii H., Verslues P.E., Zhu J.K. Arabidopsis decuple mutant reveals the importance of SnRK2 kinases in osmotic stress responses in vivo. Proc. Natl. Acad. Sci. USA. 2011;108:1717–1722. doi: 10.1073/pnas.1018367108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita Y., Yoshida T., Yamaguchi-Shinozaki K. Pivotal role of the AREB/ABF-SnRK2 pathway in ABRE-mediated transcription in response to osmotic stress in plants. Physiol. Plant. 2013;147:15–27. doi: 10.1111/j.1399-3054.2012.01635.x. [DOI] [PubMed] [Google Scholar]
- Garcia M.E., Lynch T., Peeters J., Snowden C., Finkelstein R. A small plant-specific protein family of ABI five binding proteins (AFPs) regulates stress response in germinating Arabidopsis seeds and seedlings. Plant Mol. Biol. 2008;67:643–658. doi: 10.1007/s11103-008-9344-2. [DOI] [PubMed] [Google Scholar]
- Goodstein D.M., Shu S., Howson R., Neupane R., Hayes R.D., Fazo J., Mitros T., Dirks W., Hellsten U., Putnam N., Rokhsar D.S. Phytozome: a comparative platform for green plant genomics. Nucleic Acids Res. 2012;40:D1178–D1186. doi: 10.1093/nar/gkr944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon S., Delsuc F., Dufayard J.-F., Gascuel O. Estimating Maximum Likelihood Phylogenies with PhyML. In: Posada D., editor. Bioinformatics for DNA Sequence Analysis. Humana Press; 2009. pp. 113–137. [DOI] [PubMed] [Google Scholar]
- Guo Y., Xiong L., Song C.P., Gong D., Halfter U., Zhu J.K. A calcium sensor and its interacting protein kinase are global regulators of abscisic acid signaling in Arabidopsis. Dev. Cell. 2002;3:233–244. doi: 10.1016/s1534-5807(02)00229-0. [DOI] [PubMed] [Google Scholar]
- Hedman H., Källman T., Lagercrantz U. Early evolution of the MFT-like gene family in plants. Plant Mol. Biol. 2009;70:359–369. doi: 10.1007/s11103-009-9478-x. [DOI] [PubMed] [Google Scholar]
- Hou C.J., Yang C.H. Comparative analysis of the pteridophyte Adiantum MFT ortholog reveals the specificity of combined FT/MFT C and N terminal interaction with FD for the regulation of the downstream gene AP1. Plant Mol. Biol. 2016;91:563–579. doi: 10.1007/s11103-016-0489-0. [DOI] [PubMed] [Google Scholar]
- Hyun Y., Kim J., Cho S.W., Choi Y., Kim J.S., Coupland G. Site-directed mutagenesis in Arabidopsis thaliana using dividing tissue-targeted RGEN of the CRISPR/Cas system to generate heritable null alleles. Planta. 2015;241:271–284. doi: 10.1007/s00425-014-2180-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaeger K.E., Wigge P.A. FT protein acts as a long-range signal in Arabidopsis. Curr. Biol. 2007;17:1050–1054. doi: 10.1016/j.cub.2007.05.008. [DOI] [PubMed] [Google Scholar]
- Jaeger K.E., Pullen N., Lamzin S., Morris R.J., Wigge P.A. Interlocking feedback loops govern the dynamic behavior of the floral transition in Arabidopsis. Plant Cell. 2013;25:820–833. doi: 10.1105/tpc.113.109355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang S., Torti S., Coupland G. Genetic and spatial interactions between FT, TSF and SVP during the early stages of floral induction in Arabidopsis. Plant J. 2009;60:614–625. doi: 10.1111/j.1365-313X.2009.03986.x. [DOI] [PubMed] [Google Scholar]
- Jaradat M.R., Feurtado J.A., Huang D., Lu Y., Cutler A.J. Multiple roles of the transcription factor AtMYBR1/AtMYB44 in ABA signaling, stress responses, and leaf senescence. BMC Plant Biol. 2013;13:192. doi: 10.1186/1471-2229-13-192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung J.H., Lee H.J., Ryu J.Y., Park C.M. SPL3/4/5 Integrate Developmental Aging and Photoperiodic Signals into the FT-FD Module in Arabidopsis Flowering. Mol. Plant. 2016;9:1647–1659. doi: 10.1016/j.molp.2016.10.014. [DOI] [PubMed] [Google Scholar]
- Kardailsky I., Shukla V.K., Ahn J.H., Dagenais N., Christensen S.K., Nguyen J.T., Chory J., Harrison M.J., Weigel D. Activation tagging of the floral inducer FT. Science. 1999;286:1962–1965. doi: 10.1126/science.286.5446.1962. [DOI] [PubMed] [Google Scholar]
- Kim D., Pertea G., Trapnell C., Pimentel H., Kelley R., Salzberg S.L. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi Y., Kaya H., Goto K., Iwabuchi M., Araki T. A pair of related genes with antagonistic roles in mediating flowering signals. Science. 1999;286:1960–1962. doi: 10.1126/science.286.5446.1960. [DOI] [PubMed] [Google Scholar]
- Koornneef M., Alonso-Blanco C., Blankestijn-de Vries H., Hanhart C.J., Peeters A.J. Genetic interactions among late-flowering mutants of Arabidopsis. Genetics. 1998;148:885–892. doi: 10.1093/genetics/148.2.885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurihara D., Mizuta Y., Sato Y., Higashiyama T. ClearSee: a rapid optical clearing reagent for whole-plant fluorescence imaging. Development. 2015;142:4168–4179. doi: 10.1242/dev.127613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kushiro T., Okamoto M., Nakabayashi K., Yamagishi K., Kitamura S., Asami T., Hirai N., Koshiba T., Kamiya Y., Nambara E. The Arabidopsis cytochrome P450 CYP707A encodes ABA 8′-hydroxylases: key enzymes in ABA catabolism. EMBO J. 2004;23:1647–1656. doi: 10.1038/sj.emboj.7600121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung J., Bouvier-Durand M., Morris P.C., Guerrier D., Chefdor F., Giraudat J. Arabidopsis ABA response gene ABI1: features of a calcium-modulated protein phosphatase. Science. 1994;264:1448–1452. doi: 10.1126/science.7910981. [DOI] [PubMed] [Google Scholar]
- Li H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics. 2011;27:2987–2993. doi: 10.1093/bioinformatics/btr509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Molina L., Mongrand S., Chua N.H. A postgermination developmental arrest checkpoint is mediated by abscisic acid and requires the ABI5 transcription factor in Arabidopsis. Proc. Natl. Acad. Sci. USA. 2001;98:4782–4787. doi: 10.1073/pnas.081594298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons E., Freeling M. How to usefully compare homologous plant genes and chromosomes as DNA sequences. Plant J. 2008;53:661–673. doi: 10.1111/j.1365-313X.2007.03326.x. [DOI] [PubMed] [Google Scholar]
- Ma W., Noble W.S., Bailey T.L. Motif-based analysis of large nucleotide data sets using MEME-ChIP. Nat. Protoc. 2014;9:1428–1450. doi: 10.1038/nprot.2014.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maere S., Heymans K., Kuiper M. BiNGO: a Cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics. 2005;21:3448–3449. doi: 10.1093/bioinformatics/bti551. [DOI] [PubMed] [Google Scholar]
- Mair A., Pedrotti L., Wurzinger B., Anrather D., Simeunovic A., Weiste C., Valerio C., Dietrich K., Kirchler T., Nägele T. SnRK1-triggered switch of bZIP63 dimerization mediates the low-energy response in plants. eLife. 2015;4:e05828. doi: 10.7554/eLife.05828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandel M.A., Yanofsky M.F. The Arabidopsis AGL8 MADS box gene is expressed in inflorescence meristems and is negatively regulated by APETALA1. Plant Cell. 1995;7:1763–1771. doi: 10.1105/tpc.7.11.1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandel M.A., Gustafson-Brown C., Savidge B., Yanofsky M.F. Molecular characterization of the Arabidopsis floral homeotic gene APETALA1. Nature. 1992;360:273–277. doi: 10.1038/360273a0. [DOI] [PubMed] [Google Scholar]
- Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. 2011;17:10–12. [Google Scholar]
- Mateos J.L., Madrigal P., Tsuda K., Rawat V., Richter R., Romera-Branchat M., Fornara F., Schneeberger K., Krajewski P., Coupland G. Combinatorial activities of SHORT VEGETATIVE PHASE and FLOWERING LOCUS C define distinct modes of flowering regulation in Arabidopsis. Genome Biol. 2015;16:31. doi: 10.1186/s13059-015-0597-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathieu J., Warthmann N., Küttner F., Schmid M. Export of FT protein from phloem companion cells is sufficient for floral induction in Arabidopsis. Curr. Biol. 2007;17:1055–1060. doi: 10.1016/j.cub.2007.05.009. [DOI] [PubMed] [Google Scholar]
- Meyer K., Leube M.P., Grill E. A protein phosphatase 2C involved in ABA signal transduction in Arabidopsis thaliana. Science. 1994;264:1452–1455. doi: 10.1126/science.8197457. [DOI] [PubMed] [Google Scholar]
- Musielak T.J., Schenkel L., Kolb M., Henschen A., Bayer M. A simple and versatile cell wall staining protocol to study plant reproduction. Plant Reprod. 2015;28:161–169. doi: 10.1007/s00497-015-0267-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nambara E., Suzuki M., Abrams S., McCarty D.R., Kamiya Y., McCourt P. A screen for genes that function in abscisic acid signaling in Arabidopsis thaliana. Genetics. 2002;161:1247–1255. doi: 10.1093/genetics/161.3.1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Née G., Kramer K., Nakabayashi K., Yuan B., Xiang Y., Miatton E., Finkemeier I., Soppe W.J.J. DELAY OF GERMINATION1 requires PP2C phosphatases of the ABA signalling pathway to control seed dormancy. Nat. Commun. 2017;8:72. doi: 10.1038/s41467-017-00113-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimchuk Z.L. CLAVATA1 controls distinct signaling outputs that buffer shoot stem cell proliferation through a two-step transcriptional compensation loop. PLoS Genet. 2017;13:e1006681. doi: 10.1371/journal.pgen.1006681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niwa M., Daimon Y., Kurotani K., Higo A., Pruneda-Paz J.L., Breton G., Mitsuda N., Kay S.A., Ohme-Takagi M., Endo M., Araki T. BRANCHED1 interacts with FLOWERING LOCUS T to repress the floral transition of the axillary meristems in Arabidopsis. Plant Cell. 2013;25:1228–1242. doi: 10.1105/tpc.112.109090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posada D., Crandall K.A. MODELTEST: testing the model of DNA substitution. Bioinformatics. 1998;14:817–818. doi: 10.1093/bioinformatics/14.9.817. [DOI] [PubMed] [Google Scholar]
- R Development Core Team . R Foundation for Statistical Computing; 2009. R: A Language and Environment for Statistical Computing. [Google Scholar]
- Ramírez F., Dündar F., Diehl S., Grüning B.A., Manke T. deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res. 2014;42:W187–W191. doi: 10.1093/nar/gku365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saez A., Apostolova N., Gonzalez-Guzman M., Gonzalez-Garcia M.P., Nicolas C., Lorenzo O., Rodriguez P.L. Gain-of-function and loss-of-function phenotypes of the protein phosphatase 2C HAB1 reveal its role as a negative regulator of abscisic acid signalling. Plant J. 2004;37:354–369. doi: 10.1046/j.1365-313x.2003.01966.x. [DOI] [PubMed] [Google Scholar]
- Schmid M., Uhlenhaut N.H., Godard F., Demar M., Bressan R., Weigel D., Lohmann J.U. Dissection of floral induction pathways using global expression analysis. Development. 2003;130:6001–6012. doi: 10.1242/dev.00842. [DOI] [PubMed] [Google Scholar]
- Searle I., He Y., Turck F., Vincent C., Fornara F., Kröber S., Amasino R.A., Coupland G. The transcription factor FLC confers a flowering response to vernalization by repressing meristem competence and systemic signaling in Arabidopsis. Genes Dev. 2006;20:898–912. doi: 10.1101/gad.373506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P., Markiel A., Ozier O., Baliga N.S., Wang J.T., Ramage D., Amin N., Schwikowski B., Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slater G.S., Birney E. Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics. 2005;6:31. doi: 10.1186/1471-2105-6-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohn E.J., Rojas-Pierce M., Pan S., Carter C., Serrano-Mislata A., Madueño F., Rojo E., Surpin M., Raikhel N.V. The shoot meristem identity gene TFL1 is involved in flower development and trafficking to the protein storage vacuole. Proc. Natl. Acad. Sci. USA. 2007;104:18801–18806. doi: 10.1073/pnas.0708236104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son O., Hur Y.S., Kim Y.K., Lee H.J., Kim S., Kim M.R., Nam K.H., Lee M.S., Kim B.Y., Park J. ATHB12, an ABA-inducible homeodomain-leucine zipper (HD-Zip) protein of Arabidopsis, negatively regulates the growth of the inflorescence stem by decreasing the expression of a gibberellin 20-oxidase gene. Plant Cell Physiol. 2010;51:1537–1547. doi: 10.1093/pcp/pcq108. [DOI] [PubMed] [Google Scholar]
- Song L., Huang S.C., Wise A., Castanon R., Nery J.R., Chen H., Watanabe M., Thomas J., Bar-Joseph Z., Ecker J.R. A transcription factor hierarchy defines an environmental stress response network. Science. 2016;354:aag1550. doi: 10.1126/science.aag1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srikanth A., Schmid M. Regulation of flowering time: all roads lead to Rome. Cell. Mol. Life Sci. 2011;68:2013–2037. doi: 10.1007/s00018-011-0673-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taoka K., Ohki I., Tsuji H., Furuita K., Hayashi K., Yanase T., Yamaguchi M., Nakashima C., Purwestri Y.A., Tamaki S. 14-3-3 proteins act as intracellular receptors for rice Hd3a florigen. Nature. 2011;476:332–335. doi: 10.1038/nature10272. [DOI] [PubMed] [Google Scholar]
- Teper-Bamnolker P., Samach A. The flowering integrator FT regulates SEPALLATA3 and FRUITFULL accumulation in Arabidopsis leaves. Plant Cell. 2005;17:2661–2675. doi: 10.1105/tpc.105.035766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torti S., Fornara F., Vincent C., Andrés F., Nordström K., Göbel U., Knoll D., Schoof H., Coupland G. Analysis of the Arabidopsis shoot meristem transcriptome during floral transition identifies distinct regulatory patterns and a leucine-rich repeat protein that promotes flowering. Plant Cell. 2012;24:444–462. doi: 10.1105/tpc.111.092791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turck F., Fornara F., Coupland G. Regulation and identity of florigen: FLOWERING LOCUS T moves center stage. Annu. Rev. Plant Biol. 2008;59:573–594. doi: 10.1146/annurev.arplant.59.032607.092755. [DOI] [PubMed] [Google Scholar]
- Tylewicz S., Tsuji H., Miskolczi P., Petterle A., Azeez A., Jonsson K., Shimamoto K., Bhalerao R.P. Dual role of tree florigen activation complex component FD in photoperiodic growth control and adaptive response pathways. Proc. Natl. Acad. Sci. USA. 2015;112:3140–3145. doi: 10.1073/pnas.1423440112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaistij F.E., Barros-Galvão T., Cole A.F., Gilday A.D., He Z., Li Y., Harvey D., Larson T.R., Graham I.A. MOTHER-OF-FT-AND-TFL1 represses seed germination under far-red light by modulating phytohormone responses in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA. 2018;115:8442–8447. doi: 10.1073/pnas.1806460115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinson C.R., Sigler P.B., McKnight S.L. Scissors-grip model for DNA recognition by a family of leucine zipper proteins. Science. 1989;246:911–916. doi: 10.1126/science.2683088. [DOI] [PubMed] [Google Scholar]
- Wang J.W., Czech B., Weigel D. miR156-regulated SPL transcription factors define an endogenous flowering pathway in Arabidopsis thaliana. Cell. 2009;138:738–749. doi: 10.1016/j.cell.2009.06.014. [DOI] [PubMed] [Google Scholar]
- Wang Z.P., Xing H.L., Dong L., Zhang H.Y., Han C.Y., Wang X.C., Chen Q.J. Egg cell-specific promoter-controlled CRISPR/Cas9 efficiently generates homozygous mutants for multiple target genes in Arabidopsis in a single generation. Genome Biol. 2015;16:144. doi: 10.1186/s13059-015-0715-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wigge P.A., Kim M.C., Jaeger K.E., Busch W., Schmid M., Lohmann J.U., Weigel D. Integration of spatial and temporal information during floral induction in Arabidopsis. Science. 2005;309:1056–1059. doi: 10.1126/science.1114358. [DOI] [PubMed] [Google Scholar]
- Xi W., Liu C., Hou X., Yu H. MOTHER OF FT AND TFL1 regulates seed germination through a negative feedback loop modulating ABA signaling in Arabidopsis. Plant Cell. 2010;22:1733–1748. doi: 10.1105/tpc.109.073072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi A., Kobayashi Y., Goto K., Abe M., Araki T. TWIN SISTER OF FT (TSF) acts as a floral pathway integrator redundantly with FT. Plant Cell Physiol. 2005;46:1175–1189. doi: 10.1093/pcp/pci151. [DOI] [PubMed] [Google Scholar]
- Yamaguchi A., Wu M.F., Yang L., Wu G., Poethig R.S., Wagner D. The microRNA-regulated SBP-Box transcription factor SPL3 is a direct upstream activator of LEAFY, FRUITFULL, and APETALA1. Dev. Cell. 2009;17:268–278. doi: 10.1016/j.devcel.2009.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi N., Winter C.M., Wu M.F., Kanno Y., Yamaguchi A., Seo M., Wagner D. Gibberellin acts positively then negatively to control onset of flower formation in Arabidopsis. Science. 2014;344:638–641. doi: 10.1126/science.1250498. [DOI] [PubMed] [Google Scholar]
- You Y., Sawikowska A., Neumann M., Posé D., Capovilla G., Langenecker T., Neher R.A., Krajewski P., Schmid M. Temporal dynamics of gene expression and histone marks at the Arabidopsis shoot meristem during flowering. Nat. Commun. 2017;8:15120. doi: 10.1038/ncomms15120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nusbaum C., Myers R.M., Brown M., Li W., Liu X.S. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu L.J., Gazin C., Lawson N.D., Pagès H., Lin S.M., Lapointe D.S., Green M.R. ChIPpeakAnno: a Bioconductor package to annotate ChIP-seq and ChIP-chip data. BMC Bioinformatics. 2010;11:237. doi: 10.1186/1471-2105-11-237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu B., Zhang W., Zhang T., Liu B., Jiang J. Genome-Wide Prediction and Validation of Intergenic Enhancers in Arabidopsis Using Open Chromatin Signatures. Plant Cell. 2015;27:2415–2426. doi: 10.1105/tpc.15.00537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinzen R.P., Girardot C., Gagneur J., Braun M., Furlong E.E. Combinatorial binding predicts spatio-temporal cis-regulatory activity. Nature. 2009;462:65–70. doi: 10.1038/nature08531. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw sequence read data can be obtained from the NCBI Sequence Read Archive under accession number PRJNA560053: Genome-wide analyses define distinct functions for the florigen interacting bZIP transcription factors FD and FDP in abscisic acid responses and flowering of Arabidopsis.