

Abstract
Members of genus Sphingopyxis are frequently found in diverse eco-environments worldwide and have been traditionally considered to play vital roles in the degradation of aromatic compounds. Over recent decades, many aromatic-degrading Sphingopyxis strains have been isolated and recorded, but little is known about their genetic nature related to aromatic compounds biodegradation. In this study, bacterial genomes of 19 Sphingopyxis strains were used for comparative analyses. Phylogeny showed an ambiguous relatedness between bacterial strains and their habitat specificity, while clustering based on Cluster of Orthologous Groups suggested the potential link of functional profile with substrate-specific traits. Pan-genome analysis revealed that 19 individuals were predicted to share 1,066 orthologous genes, indicating a high genetic homogeneity among Sphingopyxis strains. Notably, KEGG Automatic Annotation Server results suggested that most genes pertaining aromatic compounds biodegradation were predicted to be involved in benzoate, phenylalanine, and aminobenzoate metabolism. Among them, β-ketoadipate biodegradation might be the main pathway in Sphingopyxis strains. Further inspection showed that a number of mobile genetic elements varied in Sphingopyxis genomes, and plasmid-mediated gene transfer coupled with prophage- and transposon-mediated rearrangements might play prominent roles in the evolution of bacterial genomes. Collectively, our findings presented that Sphingopyxis isolates might be the promising candidates for biodegradation of aromatic compounds in pollution sites.
1. Introduction
Biodegradation of hazardous pollutants mediated by microorganisms is widely regarded as an effective strategy for reducing the risk of toxins [1]. Commonly, aromatic compounds are organic molecules that contain one or more aromatic rings, especially benzene ring, and are the most concerned environmental pollutants that severely threaten the environment and human health due to their prevalent and persistent characteristics and bioaccumulation via food web [2]. Over decades, various bacteria, such as Pseudomonas, Burkholderiales, and Rhodococcus [3–5], have been identified to harbor the ability of individually utilizing aromatic compounds as alternative carbon and energy source under the condition of nutrient-deficiency. On the basis of phylogenetic, chemotaxonomic, and physiological analyses, Takeuchi and Maruyama have stated that genus Sphingomonas were redivided into five independent genera (i.e., Sphingomonas, Sphingopyxis, Sphingobium, Novosphingobium, and Sphingosinicella) [6]. To our knowledge, strains of Sphingobium, Sphingomonas, and Novosphingobium have been extensively studied with respect to their potential for aromatic compounds degradation [7, 8].
In various habitats, members of Sphingopyxis isolates have been reported to efficiently degrade the aromatic compounds, such as microcystins (MCs) [9], tetralin [10], styrene [11], and triclosan [12], which generally cause environmental pollution and induce negative impact on human and ecosystem health [13]. For instance, Sphingopyxis sp. C-1 isolated from an eutrophic lake in China has been shown to harbor the ability of MCs degradation [9]. Sphingopyxis fribergensis Kp5.2, originally isolated from soils, was reported to degrade styrene [14]. In addition, Sphingopyxis granuli TFA isolated from the Rhine river was able to grow in organic solvent tetralin [15]. However, not all isolates exhibited the capability of aromatic compounds degradation. For example, Sphingopyxis ummariensis UI2, Sphingopyxis indica DS15, and Sphingopyxis flava R11H were isolated from different hexachlorocyclohexane (HCH)-contaminated soils, but there was no evidence to support their biodegradation capabilities. Furthermore, it was unclear whether Sphingopyxis sp. MWB1 had the ability of crude-oil-degradation, although it was isolated from crude oil-contaminated seashore [16]. There is a functional diversity among Sphingopyxis strains; it is thus of interest to explore the genetic potential for cleanup of pollutants.
Currently, a strain YF1 was isolated from the eutrophic Lake Taihu and phylogenetically affiliated to genus Sphingopyxis [17]. In our study, 18 reference genomes of other Sphingopyxis isolates were selected for comparison. Phylogeny of Sphingopyxis strains was inferred based on multiple methods to verify their evolutionary relationships. Comparative genomics was further performed to systematically analyze the functional profile and metabolic potential of Sphingopyxis isolates to investigate their genetic potential for aromatic compounds biodegradation.
2. Material and Methods
2.1. Strains Selection in This Study
In this study, 16S rRNA-based phylogeny showed that strain YF1 was phylogenetically affiliated to genus Sphingopyxis (Figure S1). Subsequently, 18 other Sphingopyxis strains that were originally isolated from various ecological niches, such as contaminated soil, marine water, and eutrophic lake, were used for further analysis. The general feature for each Sphingopyxis isolate was summarized in Table 1.
Table 1.
Summary for Sphingopyxis isolates used in this study.
| Stains | Genome size (Mbp) | Staus | No. of chromosomes (plasmid) | No. of 16S operons | G+C (%) | Genome completeness | Isolation source |
|---|---|---|---|---|---|---|---|
| Sphingopyxis alaskensis RB2256 | 3.37 | Complete | 1 (1) | 1 | 65.5 | 93.7% | Surface waters of Resurrection Bay, Alaska |
| Sphingopyxis baekryungensis DSM 16222 | 3.07 | Draft | N/A | 2 | 62.4 | 94.1% | Seawater of the Yellow Sea, Korea |
| Sphingopyxis bauzanensis DSM 22271 | 4.26 | Draft | N/A | 1 | 63.3 | 93.2% | Hydrocarbon-contaminated soil |
| Sphingopyxis flava R11H | 4.16 | Draft | N/A | 1 | 63.8 | 94.2% | Hexachlorocyclohexane dumpsite |
| Sphingopyxis fribergensis Kp52 | 5.2 | Complete | 1 (1) | 1 | 63.8 | 93.7% | Soil in Freiberg, Saxony, Germany |
| Sphingopyxis granuli NBRC 100800 | 4.26 | Draft | N/A | 1 | 66.4 | 93.2% | UASB bioreactor |
| Sphingopyxis indica DS15 | 4.15 | Draft | N/A | 1 | 65.7 | 94.2% | Hexachlorocyclohexane dumpsite in Lucknow |
| Sphingopyxis macrogoltabida 203 N | 5.95 | Complete | 1 (2) | 1 | 64.7 | 93.7% | Soil |
| Sphingopyxis sp. 113P3 | 4.66 | Complete | 1 (1) | 1 | 64.0 | 93.2% | Activated sludge |
| Sphingopyxis sp. C-1 | 4.58 | Draft | N/A | 1 | 63.7 | 93.2% | Blooms of cyanobacteria |
| Sphingopyxis sp. EG6 | 3.88 | Complete | 1 (1) | 1 | 64.6 | 93.7% | Industrial cooling water |
| Sphingopyxis sp. FD7 | 3.94 | Complete | 1 (1) | 1 | 65.2 | 93.7% | Industrial cooling water |
| Sphingopyxis sp. MC1 | 3.65 | Draft | N/A | 1 | 65.2 | 92.8% | Activated sludge |
| Sphingopyxis sp. MG | 4.22 | Complete | 1 (1) | 1 | 66.4 | 93.2% | Sewage and soil |
| Sphingopyxis sp. MWB1 | 3.12 | Draft | N/A | 1 | 62.8 | 92.3% | Crude oil contaminated seashore |
| Sphingopyxis terrae subsp. terrae 203-1 | 3.98 | Complete | 1 (1) | 1 | 64.6 | 93.2% | Activated sludge |
| Sphingopyxis terrae subsp. Ummariensis UI2 | 3.58 | Draft | N/A | 1 | 65.2 | 93.3% | HCH-contaminated dumpsite |
| Sphingopyxis witflariensis DSM14551 | 4.31 | Draft | N/A | 1 | 63.3 | 94.6% | Activated sludge |
| Sphingopyxis sp. YF1 | 4.37 | Complete | 1 (0) | 2 | 66.6 | 93.2% | Blooms of cyanobacteria |
2.2. Bacterial Phylogenetic Analysis
We employed four distinct methods for phylogenetic analysis to confirm the phylogenetic relationships among 19 Sphingopyxis strains. Multiple-sequence alignment of 16S rRNA genes was performed using ClustalW [18], and the phylogenetic tree was then constructed using MEGA version 7.0 [19] with the neighbor-joining method. Herein, the evolutionary distance was calculated with 1,000 bootstrap replicates. In order to ensure the reliability of the subsequent analysis, genome sequences of 19 Sphingopyxis strains were assessed using BUSCO v3 based on evolutionarily informed expectations of gene content with a proteobacteria_odb9 BUSCO lineage datasets containing 221 BUSCOs [20, 21]. Orthologous groups of proteins were identified amongst 19 Sphingopyxis strains using OrthoFinde [22] with diamond search and Markov Cluster Algorithm. All single-copy genes in 19 strains were aligned using MAFFT [23], and phylogenetic analysis was performed using MEGA version 7.0. Topology for species tree was constructed using CVTree3 [24] with K-tuple length of 6. Furthermore, values of average nucleotide identity (ANI) between pairs of Sphingopyxis genomes were calculated using web server JspeciesWS [25].
2.3. Orthologous Proteins Identification and Functional Annotation
All-versus-all BLASTP with an E value cut-off of 0.00001 was performed using the extracted protein sequences from each Sphingopyxis strain. The output results were used for further analysis, as described in previous studies [26–29]. Bacterial Pan Genome Analysis (sequence identity ≥50%; and E value ≤1e−5) was applied for the identification of orthologous genes shared by 19 Sphingopyxis strains [30]. The orthologous groups were classified as shared, distributed, and/or unique genes according to their distribution across bacterial genomes. Furthermore, the extracted sequences of shared and distributed genes were assigned into Clusters of Orthologous Groups (COG) categories by aligning against eggNOG v4.5.1 using the eggNOG-mapper tool [31]. Also, possible genes in all Sphingopyxis genomes were annotated using the KEGG Automatic Annotation Server [32]. Notably, the KEGG database [33, 34] was used for the identification of KEGG Orthology (KO), and genes potentially related to aromatic compounds metabolism were screened according to KO annotation. Finally, all results were manually checked.
2.4. Prediction of Mobile Genetic Elements (MGEs)
In order to investigate the evolution of Sphingopyxis strains, MGEs were detected in this study. Genomic islands (GIs) of Sphingopyxis spp. were predicted using the web server IslandViewer 4 [35] with methods SIGI-HMM [36] and IslandPath-DIMOB [37]. Insertion sequences (ISs) were identified by BLAST comparison (E value ≤1e−5) against the ISFinder database [38]. Online server CRISPRFinder [39] was employed to identify the Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) arrays via BLAST search against dbCRISPR (CRISPR database). In many bacterial and archaeal genomes, CRISPR were generally considered to be important for prokaryotic immunity to resist the phages and plasmids [26]. In addition, prophage sequences were detected using PHAST (PHAgeSearch Tool) [40] with default parameters.
3. Results and Discussion
3.1. General Features of 19 Sphingopyxis Gnomes
The general features of 19 Sphingopyxis strains are summarized in Table 1. Among these strains, S. macrogoltabida 203N and S. fribergensis Kp52 isolated from contaminated soil were identified to harbor a larger genome of over 5 Mbp, and S. baekryungensis DSM 16222 isolated from marine water had the smallest genome (3.07 Mbp). Except for YF1 and DSM 16222, all Sphingopyxis strains contained one copy of 16S rRNA gene. The small 16S rRNA copy number is a characteristic commonly associated with bacteria that inhabit the specialized environments and slowly respond to changing habitat conditions [41]. In addition, strain YF1 with a chromosome of 4.37 Mbp had the highest GC content of 66.6% compared to other Sphingopyxis strains.
3.2. Phylogenetic Analyses of Sphingopyxis Strains
Genome completeness of 19 Sphingopyxis strains was evaluated, and all values were higher than 92%, which ensured the reliability of the subsequent analysis. Phylogenetic relationships revealed that strain YF1 had an ambiguous status based on four different phylogenetic analyses (Figure 1 and Table S1). For instance, both 16S rRNA genes fragment and single-copy genes phylogenetic tree showed that strain YF1 was likely to be assigned into S. macrogoltabida species, while whole-genome-based phylogeny provided evidence that strain YF1 had a close relationship with S. witflariensis DSM 14551. In our study, ANI values between pairs of Sphingopyxis isolates were further calculated (Table S1), and the results (<95%) suggested that strain YF1 might be phylogenetically affiliated to a novel species of genus Sphingopyxis. Furthermore, phylogenetic analyses based on four different methods revealed that Sphingopyxis sp. MC1, Sphingopyxis terrae subsp. terrae 203-1, and Sphingopyxis terrae subsp. Ummariensis UI2 were grouped together in a separate clade, suggesting that strain MC1 might belong to Sphingopyxis terrae species. Similarly, Sphingopyxis sp. MG and S. granuli NBRC 100800 were clustered into a distinct group, and ANIb value (96.8%) further supports the phylogenetic relationship. In addition, Sphingopyxis sp. 113P3 and S. flava R11H, as well as Sphingopyxis sp. EG6 and S. bauzanensis DSM 22271 were grouped together, but ANI values further determined their taxonomy as separate species. Notably, strain DSM 16222 (formerly belonging to S. baekryungensis) was assigned into an outgroup, which suggested that this strain was likely to be phylogenetically affiliated to another genus instead of Sphingopyxis genus.
Figure 1.
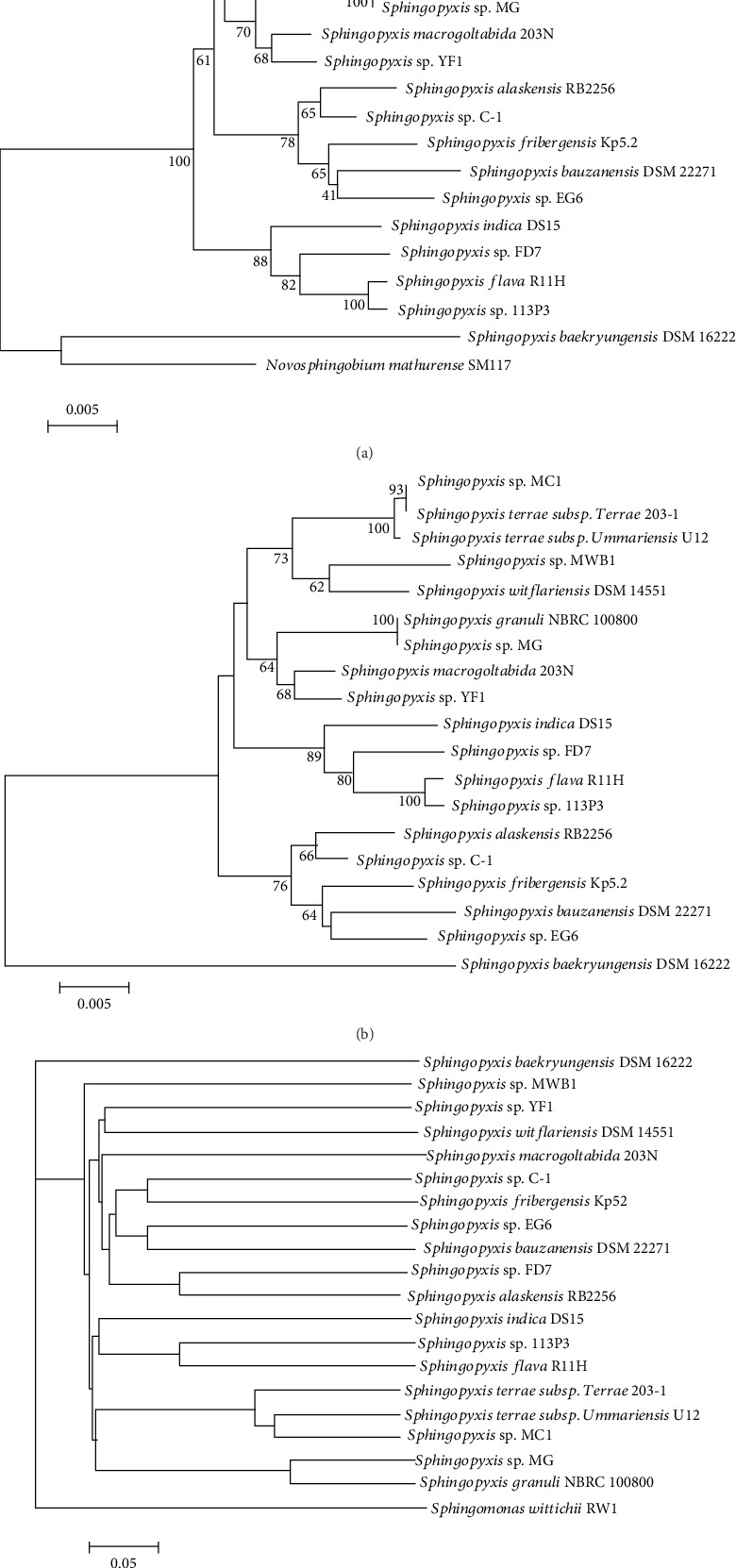
Phylogeny of 19 Sphingopyxis strains. Phylogenetic trees based on (a) 16S rRNA genes and (b) 1334 single-copy genes were constructed. The bars represent the number of substitutions per nucleotide position. Percentage bootstrap values (≥50%) were shown next to the nodes. Novosphingobium mathurense SM117 was used as an outgroup. (c) Whole-genome-based phylogenetic tree was generated using a composition vector approach with K-tuple length of 6. Sphingomonas wittichii RW1was set as an outgroup.
3.3. Pan-genome Analysis Reveals Difference in Gene Repertoire of Sphingopyxis
In this study, 68,244 protein-coding genes (CDS) from Sphingopyxis strains were clustered into 6,438 orthogroups. The number of orthogroups in these strains ranged from 2,477 to 5,003, indicating that most genes had no multiple copies. Comparative analysis further revealed that the percentage of shared genes (1,066) in each Sphingopyxis genome varied from 23.92% to 38.30% (Figure 2(a)-2(b)). This was similar to a previous study, in which S. granuli TFA harbored 1,371 shared genes in its gene repertoire [42]. In contrast, there was a relatively low percentage of core genome shared by Sphingomonas, Sphingobium, and Novosphingobium members. For example, comparative analyses based on 6, 22, and 27 Novosphingobium strains showed that 929, 674, and 220 shared genes were identified, respectively [43–45]. Another comparative genomics further suggested that 492 CDS were conserved in the complete genomes, and 268 CDS were universally conserved in all genomes of 26 bacterial strains, including 13 Sphingomonas spp., six Sphingobium spp., six Novosphingobium spp., and one Sphingopyxis sp. [7]. The findings indicated that genus Sphingopyxis was more conservative than its neighboring genera such as Novosphingobium and Sphingomonas with respect to their gene content [46]. In other words, core-genome and pan-genome had a pronounced heterogeneity in these neighboring genera, which suggested that caution should be taken when scaling from a single genus to the larger neighboring genera. Furthermore, the percentage of unique genes in S. baekryungensis DSM 16222 (44.99%) was much higher than that in other strains (3.97-20.23%). The result, to some extent, also supported that Sphingopyxis genus was a compact group, and the affiliation of strain DSM 16222 should be revised [42]. In addition, up to 20%, unique genes were predicted to be present in S. macrogoltabida 203N as its largest genome size contains a chromosome and two plasmids. With the exception of strain DSM 16222, the percentages of distributed genes in the 18 Sphingopyxis genomes varied from 52.17% (strain R11H) to 65.57% (strain YF1), and the proportions were relatively higher than that in previous studies [45]. In general, the high proportion of distributed genes were not essential to basic lifestyle, but they might confer bacteria with special features such as niche adaptation and reflect their variable metabolic profiles.
Figure 2.
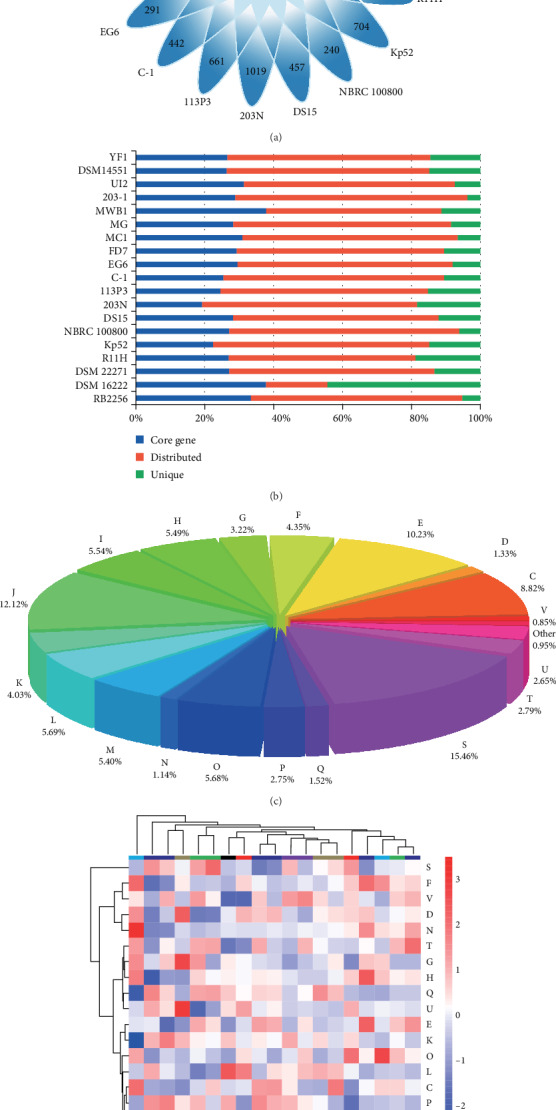
Comparison of orthologous groups in 19 Sphingopyxis genomes. (a) Venn diagram showing the numbers of shared genes and flexible genes in each Sphingopyxis strains. (b) Percentages of shared, distributed, and unique genes in each of the19 Sphingopyxis genomes. (c) Functional assignment of core-genome shared by 19 Sphingopyxis strains. (d) Functional profiling of the 19 Sphingopyxis genomes. Heatmap indicated the normalized relative abundance of COG categories of protein-coding genes in each Sphingopyxis genomes. Strains and COG categories were clustered using the Euclidean distance. The color scale represented the relative abundance of each COG category, normalized by sample mean. Abbreviations: C: energy production and conversion; D: cell cycle control; E: amino acid transport and metabolism; F: nucleotide transport and metabolism; G: carbohydrate transport and metabolism; H: coenzyme transport and metabolism; I: lipid transport and metabolism; J: translation, ribosomal structure, and biogenesis; K: transcription; L: replication, recombination, and repair; M: cell wall/membrane/envelope biogenesis; N: cell motility; O: posttranslational modification, protein turnover; P: inorganic ion transport and metabolism; Q: secondary metabolites biosynthesis and transport; S: function unknown; T: signal transduction mechanisms; U: intracellular trafficking, secretion, and vesicular transport; V: defense mechanisms.
COG assignment was performed to categorize the function of gene families in Sphingopyxis core-genome (Figure 2(c)). Results showed that a large number of CDSs were assigned into COG category [S] (function unknown, 16%). In addition, numerous genes were matched to COG categories [J] (ribosomal structure and biogenesis), [E] (amino acid transport and metabolism), and [C] (energy production and conversion), which accounted for 31.23% of the total shared genes. These essential genes were related to gain and loss of genetic information, uptake of nutrients from various environments, as well as the sustainment of basic lifestyle. Similarly, KO annotation showed that most of the shared genes in Sphingopyxis strains were involved in genetic information processing. Furthermore, there were many genes associated with carbohydrate and amino acid metabolism. With respect to pan-genome of 19 Sphingopyxis strains, the four most abundant CDSs were classified into COG categories [K] (transcription), [E], [P] (inorganic ion transport and metabolism), and [C]. In addition, functional analysis revealed that strain-specific genes were assigned into different COG categories in individuals, and their abundances were diverse.
Microorganisms could rearrange their metabolic profiles to better adapt to specific habitats and utilize the compounds to which they were exposed in eco-environments. In this study, COG clustering suggested a potential correlation between bacterial strains with their habitat specificity (Figure 2(d)). For instance, strains YF1 and C-1 originally isolated from cyanobacterial blooms were grouped together in the functional heat map. Similarly, S. bauzanensis DSM 22271 and S. flava R11H from hydrocarbon-contaminated soil were clustered into one subgroup, and S. fribergensis Kp52 from meadows, which were noncontaminated or contaminated with aliphatic and aromatic hydrocarbons, was clustered with S. indica DS15 that was from an HCH-contain dumpsite.
3.4. Pathways Prediction for Aromatic Compounds Degradation
In order to understand the metabolic diversity among 19 Sphingopyxis, we assigned CDSs into the KEGG database to identify the functional genes potentially involved in metabolisms of aromatic compounds. KAAS results suggested that there were many genes/gene clusters related to aromatic compounds degradation in Sphingopyxis strains (Figure 3, Figure S2, and Table S2). β-ketoadipate pathway, the most widely used aromatic compound-degrading pathway in microorganisms [4], was predicted in S. bauzanensis DSM 22271, S. flava R11H, and Sphingopyxis sp. MG. Genomic regions containing pca genes related to protocatechuate metabolism and cat genes were present in S. flava R11H and Sphingopyxis sp. MG, while genes for catechol degradation were identified in S. bauzanensis DSM 22271. In S. macrogoltabida 203N and S. bauzanensis DSM 22271, xyl genes associated with the catalytic reaction of benzoate to generate key intermediate of catechol were identified. Furthermore, bph genes involved in the degradation of polycyclic aromatic hydrocarbons were found in S. bauzanensis DSM 22271 [47]. More specifically, xyl genes were found to be clustered with bph genes in S. bauzanensis DSM 22271, but no cat genes were found in their flank. Likewise, cmt genes related to the cleavage of 2,3-dihydroxy-p-cumate to generate 2-hydroxypentadienoate [48] were identified to be clustered with xyl genes in S. macrogoltabida 203N. In addition, the two-component protocatechuate 4,5-dioxygenase (ligAB) was likely to be commonly involved in the cleavage of protocatechuate in Sphingopyxis. The nearly complete HCH-degrading pathway was only identified in S. fribergensis Kp52. In most Sphingopyxis strains, genes linA and linB encoding the dehydrochlorinase and haloalkane dehalogenase, respectively, were present, but the others have not been detected [49]. Among 19 Sphingopyxis isolates, it was suspected that four members isolated from the HCH-containing dumpsite had the potential to degrade HCH, but the absence of lin gene cluster suggested that these strains might undergo the loss event of genes related to HCH-degrading pathway which have been acquired at an early stage [50]. Notably, a complete pathway for phenylacetyl-CoA degradation was only present in Sphingopyxis sp. Kp5.2, while gene PaaK for phenylacetate-CoA ligase was absent in MC-degrading bacteria Sphingopyxis sp. YF1 and C-1 [51]. As previously reported, a sty gene cluster was present in Sphingopyxis sp. Kp5.2, which had the ability to convert styrene into phenylacetic acid [15, 52].
Figure 3.
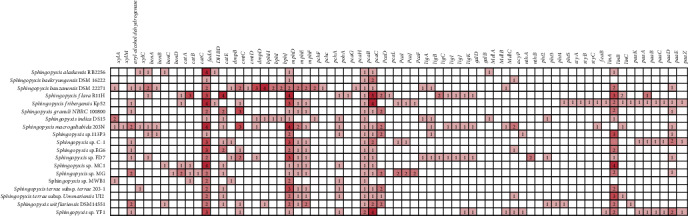
Comparison of main genes potentially involved in aromatic compounds degradation among Sphingopyxis strains. White box: absence of genes; red box: presence of genes with the number of copies in each strain. More details were listed in Table S2.
3.5. MGEs and CRISPRs Analysis
In general, MGEs such as phages, transposable, and IS elements are considered to be important for the evolution of microorganisms. Hence, MGEs were identified and compared among these Sphingopyxis genomes (Table 2 and Table S3). In our study, many MGEs existed in Sphingopyxis strains, suggesting their high plasticity and rapid adaptation in diverse environments. Some IS elements were observed in genomes of Sphingopyxis sp. YF1 (29), Sphingopyxis sp. MWB1 (35), and Sphingopyxis sp. C-1 (57), while these elements in S. bauzanensis DSM22271 (349), S. flava R11H (347), and S. witflariensis DSM 14551 (345) were predicted to outnumber that in the formers. Further analysis suggested that most of IS elements were classified into the IS3 families. Analysis of genomic regions showed that several IS- and transposase-coding genes were predicted to be located at the up- and downstream of genes related to aromatic compounds degradation, indicating that these functional genes might be acquired via horizontal gene transfer (HGT) rather than vertical inheritance. More especially, S. flava R11H was predicted to harbor the IS6100 elements, which were reported as mosaic distribution in the neighborhood of lin genes for the HCH-degrading pathway [53, 54] and genes for carbazole conversion enzymes involved in carbazole degradation pathway [55]. However, no IS element was identified at the up- and downstream of lin genes in S. fribergensis Kp52, which indicated that these functional genes were likely to be gained via vertically inheriting. Notably, gene cluster associated with MCs degradation was identified to be located at a genomic island with GC content of 59.1%, which suggested that HGT events might occur during the evolution of MC-degrading genes.
Table 2.
Statistics for predicted mobile genetic elements in Sphingopyxis genomes.
| Stains | IS element | Phage | CRISPR |
|---|---|---|---|
| Sphingopyxis alaskensis RB2256 | 121 | 2 | 0 |
| Sphingopyxis baekryungensis DSM 16222 | 167 | 2 | 0 |
| Sphingopyxis bauzanensis DSM 22271 | 119 | 2 | 0 |
| Sphingopyxis flava R11H | 196 | 3 | 0 |
| Sphingopyxis fribergensis Kp52 | 104 | 2 | 0 |
| Sphingopyxis granuli NBRC 100800 | 206 | 1 | 1 |
| Sphingopyxis indica DS15 | 186 | 2 | 1 |
| Sphingopyxis macrogoltabida 203 N | 29 | 14 | 0 |
| Sphingopyxis sp. 113P3 | 145 | 7 | 0 |
| Sphingopyxis sp. C-1 | 35 | 4 | 0 |
| Sphingopyxis sp. EG6 | 76 | 3 | 0 |
| Sphingopyxis sp. FD7 | 345 | 8 | 2 |
| Sphingopyxis sp. MC1 | 122 | 2 | 0 |
| Sphingopyxis sp. MG | 57 | 4 | 1 |
| Sphingopyxis sp. MWB1 | 118 | 1 | 0 |
| Sphingopyxis terrae subsp. terrae 203-1 | 125 | 1 | 0 |
| Sphingopyxis terrae subsp. Ummariensis UI2 | 347 | 2 | 0 |
| Sphingopyxis witflariensis DSM14551 | 349 | 3 | 2 |
| Sphingopyxis sp. YF1 | 90 | 2 | 0 |
Similar to Novosphingobium, Sphingopyxis strains were predicted to have few CRISPRs. In our study, two CRISPRs were identified in S. witflariensis DSM 14551 and Sphingopyxis sp. FD7. In addition, only one CRISPR was predicted to be present in S. granuli NBRC 100800 (with three spacers) and S. indica DS15 (with five spacers). As for other Sphingopyxis strains without CRISPRs, there might be a low frequency of viral attacks and vulnerability of the defense system.
4. Conclusions
This study has systematically investigated the genetic potential related to aromatic compounds bioremediation of Sphingopyxis strains. Phylogenetic analysis revealed that niche specificity had an insignificant influence on the evolutionary relationship. The high percentage of unique and distributed genes in each Sphingopyxis strain suggested that Sphingopyxis genome had a relatively high plasticity in response to environmental specificity. COG annotation showed that most of the core genes were predicted to be involved in ribosomal structure and biogenesis, amino acid transport, and metabolism, as well as energy production and conversion. Furthermore, COG clustering suggested a possible link between the functional profile and substrate-specific traits. Prediction of the metabolic profile was performed to identify the possible genes associated with aromatic compounds biodegradation (Figure 4). In Sphingopyxis strains, most aromatic compounds pathways were involved in benzoate degradation, phenylalanine metabolism, and aminobenzoate degradation. Our study showed that in Sphingopyxis strains partial bph and xyl genes were predicted, and β-ketoadipate pathway and peripheral phenylacetyl-CoA pathway were found to be the main pathway of aromatic compounds degradation. In addition, a large number of MGEs were present in the neighborhood of genes related to aromatic compounds metabolisms, which indicated that functional recruitment might be an efficient way to improve the environmental adaptation of bacterial strains in diverse ecosystems.
Figure 4.
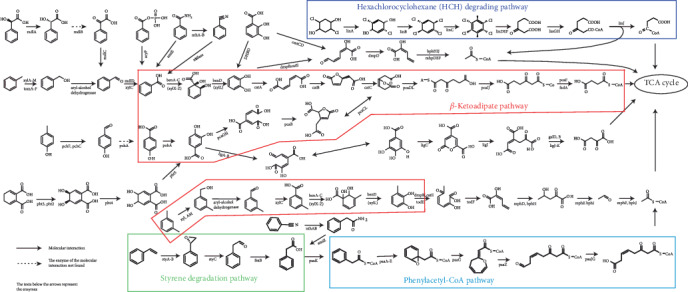
Prediction of aromatic compounds biodegradation in Sphingopyxis strains except for DSM 16222). Structure of aromatic compounds (such as benzoate, phenylalanine aminobenzoate, xylene, styrene, and HCH toluene) and their intermediates, and functional genes related to aromatic compounds degradation were shown.
Acknowledgments
The dataset used in this study was downloaded from the National Center for Biotechnology Information repository, including both genome sequences and 16S rRNA gene sequences. This work was supported by the National Natural Science Foundation of China (81502787, 81773393, and 81472972), National key research and development program of China (2016YFC0900800), Key Research and Development Projects in Hunan Province (2019SK2041), and Fundamental Research Funds for the Central Universities of Central South University (CX20190241).
Data Availability
The datasets used in this study are downloaded from the National Center for Biotechnology Information repository, including both 16S rRNA gene sequences and genomic sequences.
Conflicts of Interest
The authors declare that there is no conflict of interest regarding the publication of this paper.
Authors' Contributions
Fei Yang and Xian Zhang performed the conceptualization. Hai Feng worked the formal analysis. Fei Yang proposed the funding acquisition. Xian Zhang provided the software. Isaac Yaw Massey, Feiyu Huang, and Jian Guo were responsible for the supervision, and provided the validation. Hai Feng illustrated the visualization, and wrote the original draft. Xian Zhang reviewed and edited the manuscript.
Supplementary Materials
Figure S1: Phylogenetic analysis based on16S rRNA genes of Sphingopyxis strains. Novosphingobium mathurense SM117 was used as an outgroup. Figure S2: Schematic for main gene clusters potentially related to aromatic compounds metabolismsinSphingopyxis strains (except for S. baekryungensis DSM 16222).
Table S1: Genome-based phylogenetic indicators of Sphingopyxis strains. Table S2: Identification of gene clusters potentially involved in aromatic compounds degradation in Sphingopyxis strains. Table S3 Distribution of predicted IS elements in Sphingopyxis strains.
References
- 1.Bilal M., Adeel M., Rasheed T., Zhao Y., Iqbal H. M. N. Emerging contaminants of high concern and their enzyme-assisted biodegradation – a review. Environment International. 2019;124:336–353. doi: 10.1016/j.envint.2019.01.011. [DOI] [PubMed] [Google Scholar]
- 2.Seo J. S., Keum Y. S., Li Q. Bacterial degradation of aromatic compounds. International Journal of Environmental Research and Public Health. 2009;6(1):278–309. doi: 10.3390/ijerph6010278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pérez-Pantoja D., Donoso R., Agulló L., et al. Genomic analysis of the potential for aromatic compounds biodegradation in Burkholderiales. Environmental Microbiology. 2012;14(5):1091–1117. doi: 10.1111/j.1462-2920.2011.02613.x. [DOI] [PubMed] [Google Scholar]
- 4.Jiménez J. I., Nogales J., García J. L., Díaz E. Handbook of Hydrocarbon and Lipid Microbiology. Springer; 2010. A genomic view of the catabolism of aromatic compounds in Pseudomonas; pp. 1297–1325. [DOI] [Google Scholar]
- 5.Díaz E., Jiménez J. I., Nogales J. Aerobic degradation of aromatic compounds. Current Opinion in Biotechnology. 2013;24(3):431–442. doi: 10.1016/j.copbio.2012.10.010. [DOI] [PubMed] [Google Scholar]
- 6.Maruyama T., Park H. D., Ozawa K., et al. Sphingosinicella microcystinivorans gen. nov., sp. nov., a microcystin-degrading bacterium. International Journal of Systematic and Evolutionary Microbiology. 2006;56(1):85–89. doi: 10.1099/ijs.0.63789-0. [DOI] [PubMed] [Google Scholar]
- 7.Aylward F. O., McDonald B. R., Adams S. M., et al. Comparison of 26 Sphingomonad genomes reveals diverse environmental adaptations and biodegradative capabilities. Applied and Environmental Microbiology. 2013;79(12):3724–3733. doi: 10.1128/AEM.00518-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhao Q., Yue S., Bilal M., Hu H., Wang W., Zhang X. Comparative genomic analysis of 26 Sphingomonas and Sphingobium strains: dissemination of bioremediation capabilities, biodegradation potential and horizontal gene transfer. Science of the Total Environment. 2017;609:1238–1247. doi: 10.1016/j.scitotenv.2017.07.249. [DOI] [PubMed] [Google Scholar]
- 9.Massey I. Y., Zhang X., Yang F. Importance of bacterial biodegradation and detoxification processes of microcystins for environmental health. Journal of Toxicology and Environmental Health, Part B. 2018;21(6–8):357–369. doi: 10.1080/10937404.2018.1532701. [DOI] [PubMed] [Google Scholar]
- 10.Ledesma-García L., Sánchez-Azqueta A., Medina M., Reyes-Ramírez F., Santero E. Redox proteins of hydroxylating bacterial dioxygenases establish a regulatory cascade that prevents gratuitous induction of tetralin biodegradation genes. Scientific Reports. 2016;6(1, article 23848) doi: 10.1038/srep23848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schmidt G., Tischler D., Oelschlägel M., Kalinowski J., Rückert C., Schlömann M. Sphingopyxis fribergensis sp. nov., a soil bacterium with the ability to degrade styrene and phenylacetic acid. International Journal of Systematic and Evolutionary Microbiology. 2015;65(9):3008–3015. doi: 10.1099/ijs.0.000371. [DOI] [PubMed] [Google Scholar]
- 12.Lee D. G., Zhao F., Rezenom Y. H., Russell D. H., Chu K. H. Biodegradation of triclosan by a wastewater microorganism. Water Research. 2012;46(13):4226–4234. doi: 10.1016/j.watres.2012.05.025. [DOI] [PubMed] [Google Scholar]
- 13.Wei J., Xie X., Huang F., et al. Simultaneous Microcystis algicidal and microcystin synthesis inhibition by a red pigment prodigiosin. Environmental Pollution. 2020;256, article 113444 doi: 10.1016/j.envpol.2019.113444. [DOI] [PubMed] [Google Scholar]
- 14.Garcia-Romero I., Forstner K. U., Santero E., Floriano B. SuhB, a small non-coding RNA involved in catabolite repression of tetralin degradation genes in Sphingopyxis granuli strain TFA. Environmental Microbiology. 2018;20(10):3671–3683. doi: 10.1111/1462-2920.14360. [DOI] [PubMed] [Google Scholar]
- 15.Oelschlagel M., Zimmerling J., Schlomann M., Tischler D. Styrene oxide isomerase of Sphingopyxis sp. Kp5.2. Microbiology. 2014;160(11):2481–2491. doi: 10.1099/mic.0.080259-0. [DOI] [PubMed] [Google Scholar]
- 16.Kim J., Kim S. J., Kim S. H., et al. Draft genome sequence of Sphingopyxis sp. strain MWB1, a crude-oil-degrading marine bacterium. Genome Announcements. 2014;2(6, article e01256) doi: 10.1128/genomeA.01256-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang F., Huang F., Feng H., et al. A complete route for biodegradation of potentially carcinogenic cyanotoxin microcystin-LR in a novel indigenous bacterium. Water Research. 2020;174, article 115638 doi: 10.1016/j.watres.2020.115638. [DOI] [PubMed] [Google Scholar]
- 18.Thompson J. D., Higgins D. G., Gibson T. J. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Research. 1994;22(22):4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kumar S., Stecher G., Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Molecular Biology and Evolution. 2016;33(7):1870–1874. doi: 10.1093/molbev/msw054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Waterhouse R. M., Seppey M., Simão F. A., et al. BUSCO applications from quality assessments to gene prediction and phylogenomics. Molecular Biology and Evolution. 2018;35(3):543–548. doi: 10.1093/molbev/msx319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Simao F. A., Waterhouse R. M., Ioannidis P., Kriventseva E. V., Zdobnov E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 2015;31(19):3210–3212. doi: 10.1093/bioinformatics/btv351. [DOI] [PubMed] [Google Scholar]
- 22.Emms D. M., Kelly S. OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biology. 2015;16(1) doi: 10.1186/s13059-015-0721-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nakamura T., Yamada K. D., Tomii K., Katoh K. Parallelization of MAFFT for large-scale multiple sequence alignments. Bioinformatics. 2018;34(14):2490–2492. doi: 10.1093/bioinformatics/bty121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zuo G., Hao B. CVTree3 web server for whole-genome-based and alignment-free prokaryotic phylogeny and taxonomy. Genomics, Proteomics & Bioinformatics. 2015;13(5):321–331. doi: 10.1016/j.gpb.2015.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Richter M., Rosselló-Móra R., Oliver Glöckner F., Peplies J. JSpeciesWS: a web server for prokaryotic species circumscription based on pairwise genome comparison. Bioinformatics. 2016;32(6):929–931. doi: 10.1093/bioinformatics/btv681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang X., Liu X., Yang F., Chen L. Pan-genome analysis links the hereditary variation of Leptospirillum ferriphilum with its evolutionary adaptation. Frontiers in Microbiology. 2018;9:p. 577. doi: 10.3389/fmicb.2018.00577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang X., Liu Z., Wei G., Yang F., Liu X. In silico genome-wide analysis reveals the potential links between core genome of Acidithiobacillus thiooxidans and its autotrophic lifestyle. Frontiers in Microbiology. 2018;9:p. 1255. doi: 10.3389/fmicb.2018.01255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang X., Liu X., Li L., et al. Phylogeny, divergent evolution, and speciation of sulfur-oxidizing Acidithiobacillus populations. BMC Genomics. 2019;20(1):p. 438. doi: 10.1186/s12864-019-5827-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang X., Ye X., Chen L., et al. Functional role of bloom-forming cyanobacterium Planktothrix in ecologically shaping aquatic environments. Science of the Total Environment. 2020;710:p. 136314. doi: 10.1016/j.scitotenv.2019.136314. [DOI] [PubMed] [Google Scholar]
- 30.Chaudhari N. M., Gupta V. K., Dutta C. BPGA- an ultra-fast pan-genome analysis pipeline. Scientific Reports. 2016;6(1) doi: 10.1038/srep24373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huerta-Cepas J., Forslund K., Coelho L. P., et al. Fast genome-wide functional annotation through orthology assignment by eggNOG-mapper. Molecular Biology and Evolution. 2017;34(8):2115–2122. doi: 10.1093/molbev/msx148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moriya Y., Itoh M., Okuda S., Yoshizawa A. C., Kanehisa M. KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Research. 2007;35(Supplement 2):W182–W185. doi: 10.1093/nar/gkm321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kanehisa M., Araki M., Goto S., et al. KEGG for linking genomes to life and the environment. Nucleic Acids Research. 2008;36(Supplement 1):D480–D484. doi: 10.1093/nar/gkm882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kanehisa M., Goto S., Sato Y., Kawashima M., Furumichi M., Tanabe M. Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Research. 2013;42(D1):D199–D205. doi: 10.1093/nar/gkt1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bertelli C., Laird M. R., Williams K. P., et al. IslandViewer 4: expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Research. 2017;45(W1):W30–W35. doi: 10.1093/nar/gkx343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Waack S., Keller O., Asper R., et al. Score-based prediction of genomic islands in prokaryotic genomes using hidden Markov models. BMC Bioinformatics. 2006;7(1):p. 142. doi: 10.1186/1471-2105-7-142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hsiao W. W. L., Ung K., Aeschliman D., Bryan J., Finlay B. B., Brinkman F. S. L. Evidence of a large novel gene pool associated with prokaryotic genomic islands. PLoS Genetics. 2005;1(5):p. e62. doi: 10.1371/journal.pgen.0010062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Siguier P., Perochon J., Lestrade L., Mahillon J., Chandler M. ISfinder: the reference centre for bacterial insertion sequences. Nucleic Acids Research. 2006;34(90001) Supplement 1:D32–D36. doi: 10.1093/nar/gkj014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Grissa I., Vergnaud G., Pourcel C. CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Research. 2007;35(Supplement 2):W52–W57. doi: 10.1093/nar/gkm360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhou Y., Liang Y., Lynch K. H., Dennis J. J., Wishart D. S. PHAST: a fast phage search tool. Nucleic Acids Research. 2011;39(Supplement):W347–W352. doi: 10.1093/nar/gkr485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Klappenbach J. A., Dunbar J. M., Schmidt T. M. rRNA operon copy number reflects ecological strategies of bacteria. Applied and Environmental Microbiology. 2000;66(4):1328–1333. doi: 10.1128/aem.66.4.1328-1333.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.García-Romero I., Pérez-Pulido A. J., González-Flores Y. E., Reyes-Ramírez F., Santero E., Floriano B. Genomic analysis of the nitrate-respiring Sphingopyxis granuli (formerly Sphingomonas macrogoltabida) strain TFA. BMC Genomics. 2016;17(1) doi: 10.1186/s12864-016-2411-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gan H. M., Hudson A. O., Rahman A. Y. A., Chan K. G., Savka M. A. Comparative genomic analysis of six bacteria belonging to the genus Novosphingobium: insights into marine adaptation, cell-cell signaling and bioremediation. BMC Genomics. 2013;14(1):p. 431. doi: 10.1186/1471-2164-14-431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kumar R., Verma H., Haider S., et al. Comparative genomic analysis reveals habitat-specific genes and regulatory hubs within the genus Novosphingobium. mSystems. 2017;2(3, article e00020) doi: 10.1128/msystems.00020-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang J., Wang C., Li J., et al. Comparative genomics of degradative Novosphingobium strains with special reference to microcystin-degrading Novosphingobium sp. THN1. Frontiers in Microbiology. 2018;9:p. 2238. doi: 10.3389/fmicb.2018.02238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.van Tonder A. J., Bray J. E., Jolley K. A., et al. Genomic analyses of >3,100 nasopharyngeal pneumococci revealed significant differences between pneumococci recovered in four different geographical regions. Frontiers in Microbiology. 2019;10:p. 317. doi: 10.3389/fmicb.2019.00317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Velázquez F., de Lorenzo V., Valls M. The m-xylene biodegradation capacity of Pseudomonas putida mt-2 is submitted to adaptation to abiotic stresses: evidence from expression profiling of xyl genes. Environmental Microbiology. 2006;8(4):591–602. doi: 10.1111/j.1462-2920.2005.00936.x. [DOI] [PubMed] [Google Scholar]
- 48.Choi E. N., Cho M. C., Kim Y., Kim C. K., Lee K. Expansion of growth substrate range in Pseudomonas putida F1 by mutations in both cymR and todS, which recruit a ring-fission hydrolase CmtE and induce the tod catabolic operon, respectively. Microbiology. 2003;149(3):795–805. doi: 10.1099/mic.0.26046-0. [DOI] [PubMed] [Google Scholar]
- 49.Tabata M., Ohhata S., Nikawadori Y., et al. Comparison of the complete genome sequences of four γ-hexachlorocyclohexane-degrading bacterial strains: insights into the evolution of bacteria able to degrade a recalcitrant man-made pesticide. DNA Research. 2016;23(6):581–599. doi: 10.1093/dnares/dsw041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pearce S. L., Oakeshott J. G., Pandey G. Insights into ongoing evolution of the hexachlorocyclohexane catabolic pathway from comparative genomics of ten Sphingomonadaceae strains. G3: Genes, Genomes, Genetics. 2015;5(6):1081–1094. doi: 10.1534/g3.114.015933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang X., Yang F., Chen L., Feng H., Yin S., Chen M. Insights into ecological roles and potential evolution of Mlr-dependent microcystin-degrading bacteria. Science of the Total Environment. 2020;710, article 136401 doi: 10.1016/j.scitotenv.2019.136401. [DOI] [PubMed] [Google Scholar]
- 52.Oelschlägel M., Kaschabek S. R., Zimmerling J., Schlömann M., Tischler D. Co-metabolic formation of substituted phenylacetic acids by styrene-degrading bacteria. Biotechnology Reports. 2015;6:20–26. doi: 10.1016/j.btre.2015.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dogra C., Raina V., Pal R., et al. Organization of lin genes and IS6100 among different strains of hexachlorocyclohexane-degrading Sphingomonas paucimobilis: evidence for horizontal gene transfer. Journal of Bacteriology. 2004;186(8):2225–2235. doi: 10.1128/JB.186.8.2225-2235.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Malhotra S., Sharma P., Kumari H., Singh A., Lal R. Localization of HCH catabolic genes (lin genes) in Sphingobium indicum B90A. Indian Journal of Microbiology. 2007;47(3):271–275. doi: 10.1007/s12088-007-0050-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gai Z., Wang X., Liu X., et al. The genes coding for the conversion of carbazole to catechol are flanked by IS6100 elements in Sphingomonas sp. strain XLDN2-5. PLoS One. 2010;5(4, article e10018) doi: 10.1371/journal.pone.0010018. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: Phylogenetic analysis based on16S rRNA genes of Sphingopyxis strains. Novosphingobium mathurense SM117 was used as an outgroup. Figure S2: Schematic for main gene clusters potentially related to aromatic compounds metabolismsinSphingopyxis strains (except for S. baekryungensis DSM 16222).
Table S1: Genome-based phylogenetic indicators of Sphingopyxis strains. Table S2: Identification of gene clusters potentially involved in aromatic compounds degradation in Sphingopyxis strains. Table S3 Distribution of predicted IS elements in Sphingopyxis strains.
Data Availability Statement
The datasets used in this study are downloaded from the National Center for Biotechnology Information repository, including both 16S rRNA gene sequences and genomic sequences.