

SUMMARY
When O2 is plentiful, the mitochondrial electron transport chain uses it as a terminal electron acceptor. However, the mammalian retina thrives in a hypoxic niche in the eye. We find that mitochondria in retinas adapt to their hypoxic environment by reversing the succinate dehydrogenase reaction to use fumarate to accept electrons instead of O2. Reverse succinate dehydrogenase activity produces succinate and is enhanced by hypoxia-induced downregulation of cytochrome oxidase. Retinas can export the succinate they produce to the neighboring O2-rich retinal pigment epithelium-choroid complex. There, succinate enhances O2 consumption by severalfold. Malate made from succinate in the pigment epithelium can then be imported into the retina, where it is converted to fumarate to again accept electrons in the reverse succinate dehydrogenase reaction. This malate-succinate shuttle can sustain these two tissues by transferring reducing power from an O2-poor tissue (retina) to an O2-rich one (retinal pigment epithelium-choroid).
Graphical Abstract
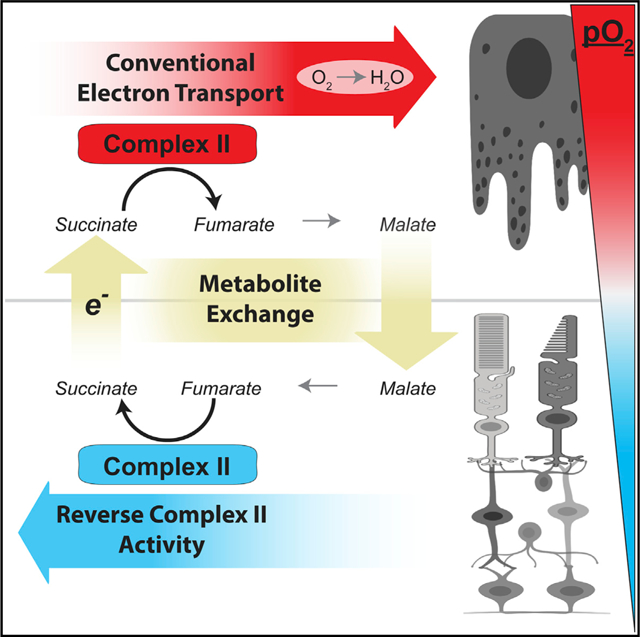
In Brief
Bisbach et al. describe a metabolite shuttle that can transfer electrons from a hypoxic tissue to an O2-rich one. In the retina, low O2 causes succinate dehydrogenase to operate in reverse, reducing fumarate to produce succinate. Retinas export this succinate, and the O2-rich RPE-choroid imports and oxidizes it.
INTRODUCTION
O2 is a key substrate in one of the most important and well-known reactions of energy metabolism. Normally, it is a terminal electron acceptor in the mitochondrial electron transport chain (ETC). The conventional model of the ETC suggests that, when O2 is limited, electrons from the ETC may be passed onto fumarate. In this “reverse” succinate dehydrogenase (SDH) reaction, SDH removes electrons from the ETC to reduce fumarate to succinate. This bypasses several steps in the ETC that drive ATP synthesis and the need for O2 (Chouchani et al., 2014; Hochachka et al., 1975). Succinate accumulates in muscle, heart, kidney, liver, brain, and blood during hypoxia (Cascarano et al., 1976; Chouchani et al., 2014; Hochachka et al., 1975). However, the degree to which the reverse SDH reaction contributes to succinate produced during hypoxia is debated, and the role of succinate in tissues that are in chronically hypoxic niches is largely unexplored (Chinopoulos, 2019; Chouchani et al., 2014; Zhang et al., 2018).
The unique architecture of the vertebrate eye places the retina in a chronically hypoxic niche. Choroidal vasculature in the sclera is the main source of O2 for the outer retina. A collagenous layer and a monolayer of cells, the retinal pigment epithelium (RPE), form a barrier that selectively regulates the flow of gases and nutrients from the choroid to the outer retina. This results in an O2-sufficient RPE but a steeply declining O2 gradient in the outer retina. The extent of hypoxia in the outer retina varies across species, but the partial pressure of O2 (pO2) in retinas can be as low as ~5 mm Hg in the mouse and can drop even lower in larger mammals (Linsenmeier and Zhang, 2017; Yu and Cringle, 2006).
To better understand the physiological consequences of this disparity in O2 tension, we investigated how the retina and RPE have adapted to their O2 environments in the eye. Retinas already are known to be very glycolytic (Chinchore et al., 2017; Kanow et al., 2017; Krebs, 1927; Winkler, 1981). We discovered that retinas also adapt to hypoxia by reducing fumarate to succinate and exporting the succinate. This form of reverse electron transport at SDH is a major pathway for succinate production in the retina. We found that retinas favor fumarate as an electron acceptor because the normal hypoxic state of the retina causes it to downregulate a subunit of mitochondrial complex IV, limiting its ability to use O2 to accept electrons.
These observations about retinal metabolism prompted us to explore the role of succinate in the overall metabolic ecosystem of the eye. The RPE relies on its mitochondria to oxidize diverse fuels, including lactate, fatty acids, glutamine, and proline, and some of these fuels can be supplied to the RPE by the retina (Adijanto et al., 2014; Du et al., 2016a; Kanow et al., 2017; Reyes-Reveles et al., 2017; Yam et al., 2019). In this report, we show that the RPE-choroid complex has an extraordinary capacity to oxidize succinate. When fueled with succinate, the RPE-choroid complex releases malate, which can be converted back into succinate in retinas by reverse SDH activity. Based on these findings, we propose that succinate shuttles unused reducing power from the hypoxic retina to the O2-rich RPE.
RESULTS
Retinas Release Succinate, which Can Fuel O2 Consumption in Eyecups
Retinas in an eye are in a chronically hypoxic environment (Linsenmeier and Zhang, 2017; Yu and Cringle, 2006). Exposure to hypoxia can induce tissues to release succinate, so we determined whether freshly isolated mouse retinas export succinate (Cascarano et al., 1976; Chouchani et al., 2014; Hochachka et al., 1975). We incubated freshly dissected retinas from C57BL/6J mice in 5 mM 12C-glucose at atmospheric (21%) O2 and 5% CO2 and measured the rate of tricarboxylic acid (TCA) cycle metabolite export by sampling the incubation medium at 30, 60, and 90 min (Figure 1A). Succinate was the most abundant TCA cycle metabolite found in the incubation medium and was exported by retinas at a rate of 0.24 (±0.04) pmol/μg/min.
Figure 1. Retinas Release Succinate, which Can Fuel O2 Consumption in Eyecups.
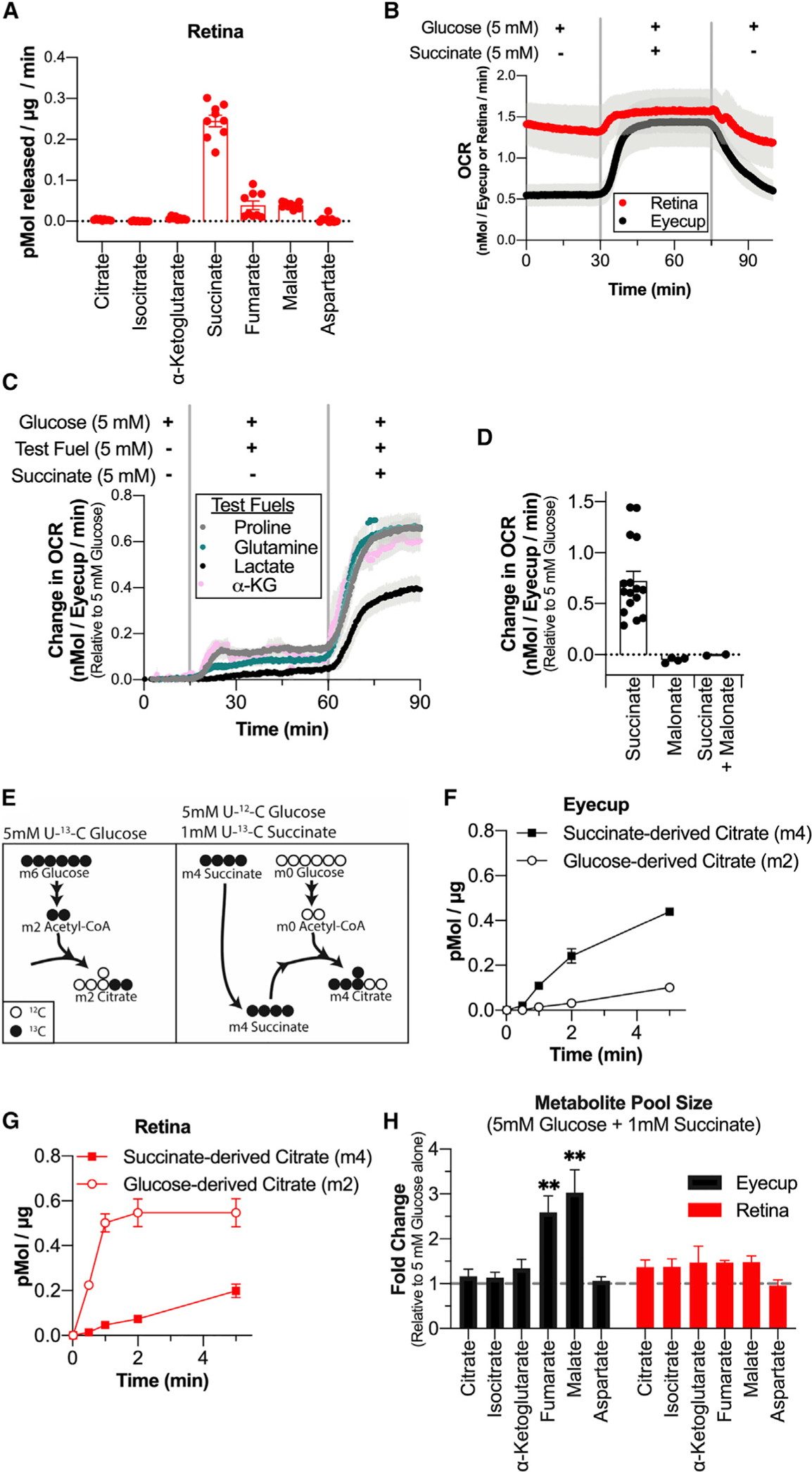
(A) Rate of TCA cycle metabolite release by retinas incubated in 5 mM 12C-glucose. Media samples were taken at 30, 60, and 90 min (n = 9 retinas, 3 per time point; error bars indicate SEM). (B) O2 consumption trace from retinas and eyecups perifused with media containing 5 mM glucose, then 5 mM glucose + 5 mM succinate, and then 5 mM glucose. Vertical gray bars indicate the approximate time when media containing the new metabolite condition reached the tissue (n = 4 retina and 4 eyecup chambers; error bars indicate SEM). (C) O2 consumption trace of eyecups supplied with 5 mM glucose, then 5 mM glucose + 5 mM of a test metabolite, and then 5 mM glucose + 5 mM test metabolite + 5 mM succinate. Test metabolites are proline (gray), glutamine (teal), lactate (black), and α-ketoglutarate (pink) (n = 3 eyecup chambers per condition; error bars indicate SEM). (D) Change in OCR (relative to 5 mM glucose alone) for eyecups supplied with 5 mM glucose + 5 mM succinate, 5 mM glucose + 20 mM malonate, or 5 mM glucose + 20 mM malonate + 5 mM succinate (n = 4 eyecup chambers for malonate and 2 eyecup chambers for malonate + succinate; error bars indicate SEM). (E) Labeling schematic showing isotopomers of citrate produced by U-13C-glucose alone (left) or 12C-glucose + U-13C-succinate (right). (F) Citrate production in eyecups supplied with 5 mM U-13C-glucose alone or 5 mM 12C-glucose + 1 mM U-13C-succinate (n = 2 eyecups per time point; error bars indicate SEM). (G) Citrate production in retinas supplied with 5 mM U-13C-glucose alone or 5 mM 12C-glucose + 1 mM U-13C-succinate (n = 2 retinas per time point; error bars indicate SEM). (H) Pool size of TCA cycle metabolites in eyecups and retinas supplied with 5 mM 12C-glucose + 1 mM U-13C-succinate for 10 min relative to tissue supplied with 5 mM 12C-glucose alone (n = 2 retinas or eyecups; error bars indicate SEM; ** indicates p < 0.005 using Welch’s t test).
The RPE consumes mitochondrial fuels. RPE cells are in direct contact with the retina, and metabolites exchange between these tissues in the eye (Adijanto et al., 2014; Du et al., 2016a; Kanow et al., 2017; Nickla and Wallman, 2010; Reyes-Reveles et al., 2017; Swarup et al., 2019; Yam et al., 2019; Young and Bok, 1969). Because we found that the retina secretes succinate, we tested the capacity of RPE cells to consume it. For these experiments, we used a custom-built perifusion oximeter loaded with intact retina or eyecup tissue (Sweet et al., 2002). Exposure of this tissue to 5 mM succinate caused a 0.7 (±0.3) nmol/eyecup/min increase in eyecup O2 consumption rate (OCR), which is an ~130% increase in OCR compared with eyecups supplied with 5 mM glucose alone (n = 17). In retinas, 5 mM succinate increased the OCR by only 0.2 (±0.1) nmol/retina/min, which is an ~15% increase in OCR compared with retinas supplied with 5 mM glucose alone (n = 7) (Figure 1B). The Michaelis-Menton constant Km for eyecup O2 consumption when fueled with succinate was 2.1 (±0.1) mM, and the maximal respiration rate was 1.0 (±0.4) nmol/eyecup/min (Figure S1A). No other glycolytic or TCA cycle metabolite we tested stimulated O2 consumption as much as succinate, and the metabolites we tested did not synergistically enhance succinate-induced OCR, although 5 mM lactate impaired the succinate-mediated increase in OCR (Figure 1C). Addition of the downstream TCA cycle metabolites fumarate and malate partially suppressed O2 consumption in eyecups stimulated by succinate (consistent with mass action) (Figure S1B). We confirmed the specificity of this effect to SDH, as succinate-stimulated increases in OCR were blocked by the SDH inhibitor malonate (Figure 1D; composite trace shown in Figure S1C). Compiled OCR measurements for all conditions tested are shown in Figure S1E.
We next used isotopic tracers to determine how succinate alters TCA cycle activity compared with glucose being the only fuel present. We supplied retinas and eyecups with either 5 mM U-13C-glucose or 5 mM 12C-glucose and 1 mM U-13C-succinate for times ranging from 0 to 5 min. We compared the formation of m2 citrate formed from glucose alone (Figure 1E, left) with the rate of m4 citrate formed from exogenous succinate (Figure 1E, right) in each tissue. Eyecups made citrate from succinate carbons (m4) ~5-fold faster than from glucose alone (m2) (Figure 1F). When supplied with U-13C-succinate, the eyecup fumarate pool was 54% (±0.8%) m4 labeled at 5 min, indicating that eyecups have a high capacity to import and metabolize succinate (Figure S1G). In striking contrast, retinas made citrate using carbons from glucose (m2) faster than with carbons from exogenous succinate (m4) (Figure 1G). When supplied with U-13C-succinate, the retinal fumarate pool was only 9% (±0.8%) m4 labeled at 5 min, suggesting that (1) retinas do not take up succinate well or (2) succinate is taken up well by retinas, but not easily converted to fumarate (Figure S1I). Other TCA cycle isotopomers and the full isotopic distribution for the 5-min time point are shown in Figure S1. Succinate did not significantly alter metabolite pool sizes in retinas, but it was anapleurotic in eyecups, acting to supplement fumarate and malate pools in eyecups ~3-fold relative to when glucose was the only fuel supplied (Figure 1H). Eyecup preparations were used for these experiments because removing the RPE (mechanically or enzymatically) from the eyecups can damage it and alter its metabolic features. Because eyecups contain multiple cell types (RPE, choroidal endothelium, and sclera), we repeated the flux analysis with cultured human fetal RPE (hfRPE) at 2 min to assess whether RPE uses succinate. These fully differentiated RPE cells made twice as much citrate from succinate carbons (m4) as citrate from glucose carbons (m2) (Figure S1D). This suggests that RPE cells are at least one of the types of cells in RPE-choroid preparations that oxidize succinate.
Succinate Enhances Export of Malate in Eyecups
The findings in Figure 1 show that retinas export succinate and that succinate fuels mitochondrial respiration in eyecups. We next tested the hypothesis that there is a reciprocal exchange of metabolites from the RPE-choroid to the retina. To determine whether eyecups release a metabolite that retinas can use to enhance succinate production, we supplied eyecups with 5 mM 12C-glucose and quantified the rate at which TCA cycle metabolites were released into the medium. Eyecups released citrate, α-ketoglutarate, malate, and aspartate, but not succinate (Figure 2A). Malate was the most abundant TCA cycle metabolite exported, released at a rate of 0.07 (±0.03) pmol/μg/min. We then determined whether succinate influences the export of any of these metabolites by supplying eyecups with U-13C-succinate in addition to 12C-glucose (Figure 2B). When we provided eyecups with 5 mM 12C-glucose and 2 mM U-13C-succinate for 60 and 90 min, export of fumarate and malate was enhanced. Malate was exported at a rate of 3.2 (±0.94) pmol/μg/min, and fumarate was exported at a rate of 4.1 (±1.1) pmol/μg/min. 93% (±2%) of the total fumarate exported was m4, and 94% (±2%) of the total malate exported was m4 (Figure 2C). Succinate present at 2 mM was consumed at a rate of 16.5 (±6.6) pmol/μg/min, and of the succinate carbons consumed, approximately 43% (±19%) were released as fumarate and malate.
Figure 2. Succinate Enhances Export of Malate in Eyecups.
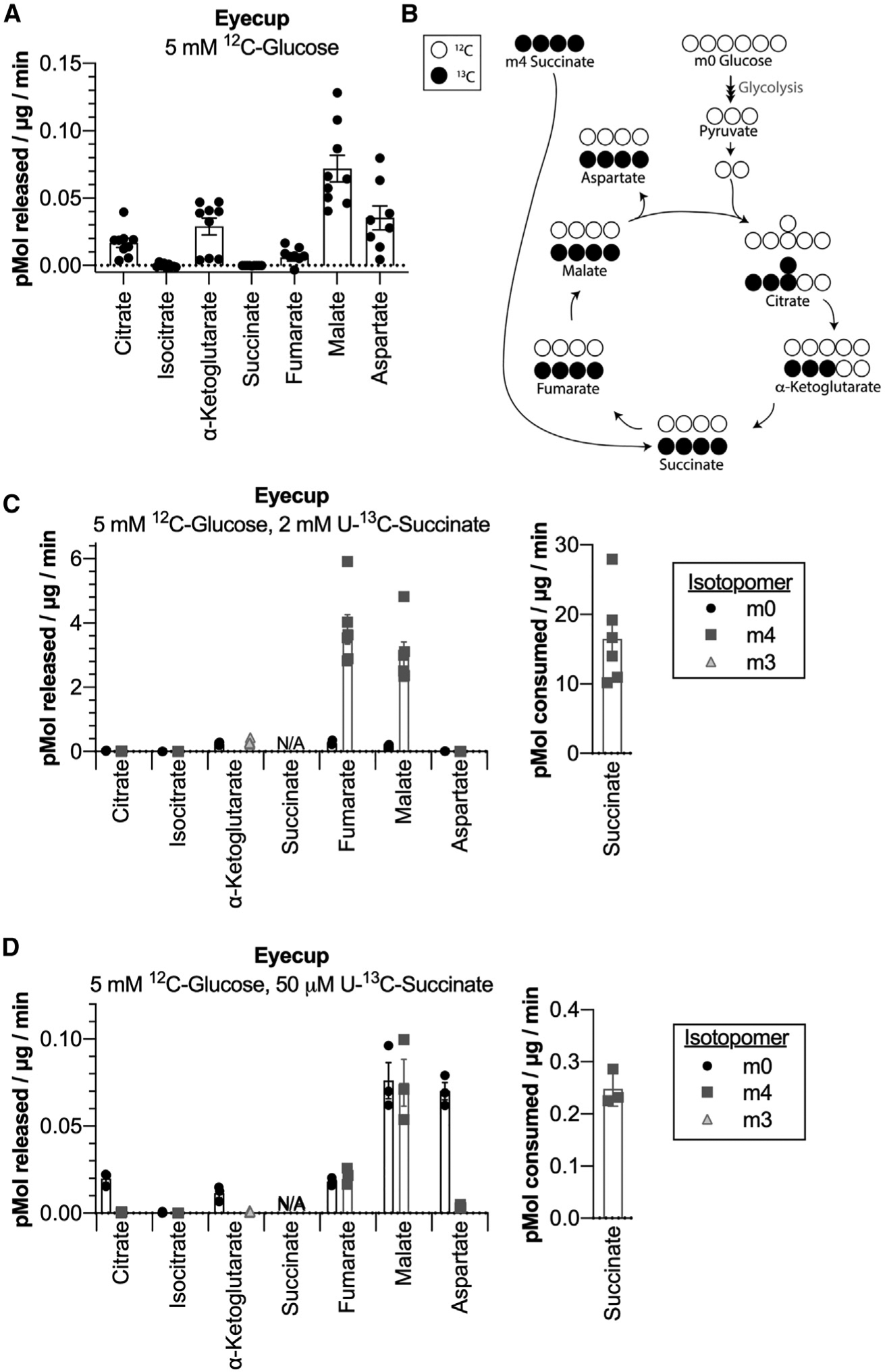
(A) Rate of TCA cycle metabolite release by eyecups incubated in 5 mM 12C-glucose. Media samples were taken at 30, 60, and 90 min (n = 9 eyecups, 3 per time point; error bars indicate SEM). (B) Labeling schematic showing TCA cycle isotopomers produced by eyecups supplied with 12C-glucose and U-13C-succinate. For simplicity, only isotopomers originating from either metabolite produced in a single turn of the TCA cycle are shown. (C) Rate of TCA cycle metabolite release by eyecups incubated in 5 mM 12C-glucose + 2 mM U-13C-succinate for 60 and 90 min. The rate at which succinate is consumed by eyecups during incubation is shown on the right (n = 6 eyecups, 3 per time point; error bars indicate SEM). (D) Rate of TCA cycle metabolite release by eyecups incubated in 5 mM 12C-glucose + 50 μM U-13C-succinate for 30 min. The rate at which succinate is consumed by eyecups during incubation is shown on the right (n = 3 eyecups; error bars indicate SEM).
Because the interface volume between the retina and RPE in a living eye is unknown, we could not determine the concentration of succinate RPE cells in an eye might receive from the retina. Plasma succinate in humans has been measured in the low micromolar range, so we also performed this experiment with a much lower concentration of succinate (Kushnir et al., 2001). When eyecups were supplied with 5 mM 12C-glucose and 50 μM U-13C-succinate for 30 min, malate was exported at a rate of 0.15 (±0.04) pmol/μg/min, and fumarate was exported at a rate of 0.04 (±0.01) pmol/μg/min. 54% (±3%) of the total fumarate exported was m4, and 49% (±2%) of the total malate exported was m4 (Figure 2D). The roughly equivalent amounts of m4 (produced from succinate) and m0 (produced from glucose) exported metabolites is remarkable, given that succinate was available at a 100-fold lower concentration than glucose in this experiment. Succinate present at 50 μM was consumed by eyecups at a rate of 0.25 (±0.03) pmol/μg/min. Similar to high succinate exposure, 39% (±11%) of succinate carbons consumed were released as fumarate and malate. Incubating retinas in 5 mM 12C-glucose and 50 μM U-13C-succinate for 30 min did not similarly enhance fumarate and malate export, and retinas did not measurably deplete succinate from the incubation medium (Figure S2A).
Retinas Can Use Malate to Produce Succinate via Reverse Electron Transport at SDH
Succinate enhances export of malate and fumarate from the RPE-choroid complex (Figure 2). We next tested whether malate exported by eyecups can influence retinal metabolism. Retinas in an eye are in a hypoxic environment, and hypoxic tissues can produce succinate by canonical oxidative TCA cycle activity as well as through reverse electron transport at SDH (Chouchani et al., 2014; Linsenmeier and Zhang, 2017; Yu and Cringle, 2006; Zhang et al., 2018). Because retinas export succinate (Figure 1), they require an anaplerotic source of metabolites to avoid running out of TCA cycle intermediates. Based on this premise, we tested whether retinas might use malate for anaplerosis to sustain succinate synthesis by reverse electron transport at SDH.
We used U-13C-malate to quantify reverse electron transport at SDH in retinas and eyecups (Figure 3A). We incubated retinas and eyecups for 5 min in U-13C-malate at a range of concentrations (5, 50, and 500 μM), all in the presence of 5 mM 12C-glucose. At all concentrations, retinas formed much more m4 succinate than eyecups (Figure 3B). None of these concentrations of malate increased the size of the fumarate pool in retinas (Figure 3C). This shows that the formation of m4 succinate we observed in retinas was due to a property inherent to retinas and not driven simply by an increase in concentration of the reactant of the reverse SDH reaction (fumarate). Total levels of other metabolites are reported in Figure S3A. To test whether the m4 succinate we observed was formed by the reverse activity of SDH or by an alternative metabolic pathway, we incubated retinas with 5 mM 12C-glucose, 50 μM U-13C-malate, and 20 mM of the SDH inhibitor malonate. Retinas still produced m4 fumarate but were unable to form m4 succinate in the presence of malonate, indicating that reversal of SDH is the pathway of m4 succinate formation (Figure 3D; other m4 metabolites shown in Figure S3B). To further validate SDH as the site of m4 succinate production, we tested another SDH inhibitor, Atpenin A5 (Miyadera et al., 2003). 2 μM Atpenin A5 also inhibited the formation of m4 succinate in retinas incubated with 5 mM 12C-glucose and 50 μM U-13C-malate (Figure S3C). However, we observed that incubating retinas in the same concentration of vehicle alone (0.2% DMSO) for 5 min caused a dramatic drop in total fumarate levels and a slight drop in total citrate and isocitrate levels compared with retinas without DMSO (Figure S3D). These unexpected changes in retinal metabolism because of DMSO led us to rely on malonate as an SDH inhibitor for our remaining experiments because it is water soluble.
Figure 3. Retinas Can Use Malate to Produce Succinate via Reverse Electron Transport at SDH.

(A) Labeling schematic showing isotopomers of TCA cycle metabolites produced by tissue incubated in U-13C-malate + 12C-glucose. (B) Isotopomers produced by retinas and eyecups incubated in 5 mM 12C-glucose and 5, 50, or 500 μM U-13C-malate for 5 min (n = 5 retinas or eyecups per concentration; error bars indicate SEM). (C) Total fumarate levels in retinas and eyecups incubated in 5 mM 12C-glucose and 5, 50, or 500 μM U-13C-malate for 5 min (n = 5 retinas or eyecups per concentration; error bars indicate SEM; * indicates p < 0.05 using Welch’s t test). (D) m4 fumarate and m4 succinate in retinas supplied with 5 mM 12C-glucose + 50 μM U-13C-malate in the presence or absence of 20 mM malonate for 5 min (n = 6 retinas; error bars indicate SEM; **** indicates p < 0.0001 using Welch’s t test). (E) m4 fumarate and m4 succinate in WT and AIPL1−/− retinas supplied with 5 mM 12C-glucose + 50 μM U-13C-malate for 5 min (n = 6 retinas; error bars indicate SEM; ** indicates p < 0.005 using Welch’s t test). (F) Labeling schematic showing isotopomers produced by 4-2H-glucose. (G) Accumulation of deuterated (m1) malate, fumarate, and succinate α-ketoglutarate in retinas and eyecups incubated in 5 mM 4-2H-glucose for 0.02, 0.5, 1, 2, and 5 min (n = 2 retinas per time point; error bars indicate SEM).
Retinas are composed of several types of neurons, so we next investigated whether a specific cell type in the retina favors reverse SDH activity. Photoreceptors are the neurons in the lowest pO2 layer of the mouse retina, and their outer segments are in direct contact with the RPE in an intact eye, so they are candidate sites of reverse SDH activity. To determine whether photoreceptors favor reverse SDH activity, we used retinas from Aipl1−/− mice, a genetic model of Leber’s congenital amaurosis, where photoreceptors degenerate before the first month of life is complete (Ramamurthy et al., 2004). We incubated Aipl1−/− retinas with 5 mM 12C-glucose and 50 μM U-13C-malate for 5 min. Aipl1−/− retinas produced equivalent amounts of m4 fumarate but less than half as much m4 succinate as WT retinas per microgram of protein (Figure 3E; other m4 metabolites shown in Figure S3E). This indicates that the contribution of photoreceptors to reverse SDH activity is greater than that of the inner retina.
We next determined whether succinate released by retinas might be in some part produced by reverse SDH activity. We incubated retinas in 5 mM 12C-glucose and 500 μM U-13C-malate for 60 and 90 min and calculated the rate of metabolites exported during that time (Figure S3F). We found that retinas exported m4 fumarate and m4 succinate, indicating that succinate produced via reverse SDH activity is exported from retinas. Succinate was exported at a rate of 0.47 (±0.04) pmol/μg/min, and 29% (±5%) of the succinate exported was found to be m4. Because it is impossible to determine what concentration of malate a retina might receive from the RPE in an intact eye, we also tested whether retinas would export succinate when incubated in lower concentrations of malate by incubating retinas in 5 mM 12C-glucose and 50 μM U-13C-malate for 30 min and measuring metabolites released into the incubation medium (Figure S3G). Retinas also exported m4 fumarate and m4 succinate when incubated in 50 μM U-13C-malate. Succinate was exported at a rate of 0.21 (±0.02) pmol/μg/min, and 5% (±0.04%) of the succinate exported was found to be m4. The approximately 10-fold decrease in fractional enrichment of m4 succinate exported from retinas supplied with 50 μM U-13C-malate versus 500 μM U-13C-malate corresponds to the 10-fold decrease in U-13C-malate with which we supplied the retinas in this experiment.
We also tested whether photoreceptors are a specific cell type in the retina that releases succinate by incubating wild-type (WT) and Aipl1−/− retinas in 5 mM 12C-glucose and 50 μM U-12C-malate for 30 min and measuring exported TCA cycle metabolites (Figure S3H). We found that retinas lacking photoreceptors released less succinate and more fumarate per microgram of protein. This indicates that, although photoreceptors are not the only retinal cell type that releases succinate, they are a major contributor because they release more succinate per microgram of cellular protein compared to the inner retina.
When supplying U-13C-malate to eyecups, we observed that sufficient concentrations (500 μM) of U-13C-malate can increase the total amount of fumarate and drive SDH to operate in reverse, consistent with Le Chatelier’s principle (Figures 3B and 3C). To confirm that the reverse SDH activity we observed in the retina was due to a characteristic inherent to them and not one we were driving artificially, we sought to also measure reverse SDH activity in a manner that did not perturb the size of the fumarate pool, i.e., in the presence of glucose alone. We used 4-2H-glucose to track reverse electron transport in the absence of added mitochondrial fuels. When cells metabolize 4-2H-glucose, the deuterium is transferred from the carbon skeleton to nicotinamide adenine dinucleotide (NAD+) during the glyceraldehyde 3-phosphate dehydrogenase (GAPDH) reaction in glycolysis, producing deuterated NADH (NAD2H) (Figure 3F; Lewis et al., 2014). This NAD2H can then be used in cytosolic reactions, including reduction of oxaloacetate to malate. Deuterated malate can then enter mitochondria. Because the fumarase reaction is readily reversible, deuterated malate will equilibrate with the fumarate pool. In the case of reverse SDH activity, deuterated fumarate will be converted to deuterated succinate. We incubated retinas and eyecups in 5 mM 4-2H-glucose for times ranging from 0 to 5 min and observed steady accumulation of m1 (deuterated) succinate in retinas, but not in eyecups (Figure 3G). This further indicates that retinas, but not eyecups, perform reverse SDH activity. To ensure that the deuterated succinate we observed was produced solely from reverse SDH activity, we confirmed that the deuterium was transferred off of the carbon skeleton during oxidative TCA cycle activity prior to formation of α-ketoglutarate (Figure 3G).
Reverse SDH Activity Maintains a Significant Portion of the Succinate Pool in Retinas
It has been estimated that reverse SDH activity produces only about ~6% of succinate during ischemia in the heart (Zhang et al., 2018). To determine whether reverse SDH activity produces significant amounts of succinate in the retina, we quantified the contributions of reverse SDH activity and oxidative (canonical forward) TCA cycle activity to the retinal succinate pool.
To compare the different modes of succinate production under the same conditions, we used U-13C-glucose to track oxidative TCA cycle activity and 4-2H-glucose to track reverse SDH activity. To assess the contribution of oxidative TCA cycle activity to succinate production, we incubated retinas in 5 mM U-13C-glucose for times ranging from 0 to 60 min and determined the fractional enrichment of m2 succinate at the steady state by fitting a curve assuming a first-order reaction (Figure 4A). At the steady state, 6.9% of the succinate pool was m2 in retinas. However, this did not mean that only 6.9% of the succinate pool was maintained by oxidative TCA cycle activity because its oxidative precursor α-ketoglutarate was only 22.8% m2-labeled at the steady state. To account for incomplete m2 labeling of the α-ketoglutarate pool, we scaled these values to determine what the fractional enrichment of m2 succinate would be if 100% of the α-ketoglutarate pool were m2 ((6:9% m2 succinate/22:8% m2 α - ketoglutarate) = (30:4% m2 succinate/100% m2 α - ketoglutarate)). This calculation predicts that the fraction of the succinate pool that is formed from α-ketoglutarate (via oxidative TCA cycle activity) is 30.4%.
Figure 4. Reverse SDH Activity Maintains a Significant Portion of the Succinate Pool in Retinas.
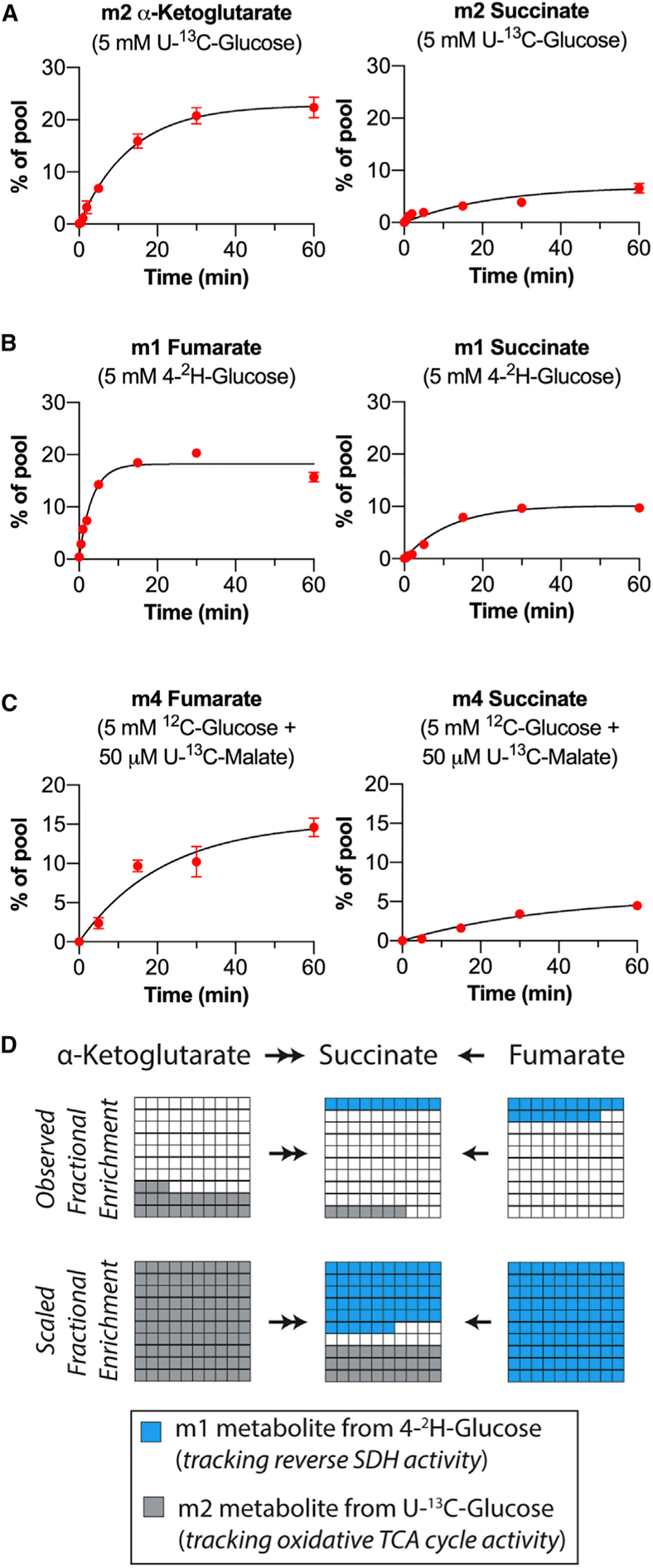
(A) Fractional enrichment of m2 α-ketoglutarate and m2 succinate in retinas supplied with 5 mM U-13C-glucose for 0.02, 0.5, 1, 2, 5, 15, 30, and 60 min (n = 3 to 7 retinas per time point; error bars indicate SEM). (B) Fractional enrichment of m1 fumarate and m1 succinate in retinas supplied with 5 mM 4-2H-glucose for 0.02, 0.5, 1, 2, 5, 15, 30, and 60 min (n = 3 retinas per time point; error bars indicate SEM). (C) Fractional enrichment of m4 fumarate and m4 succinate in retinas supplied with 5 mM 12C-glucose + 50 μM U-13C-malate for 0.02, 5, 15, 30, and 60 min (n = 3 to 5 retinas per time point; error bars indicate SEM). (D) Graphical description of observed (top) and maximal (bottom) contributions of oxidative TCA cycle activity and reverse SDH activity to maintaining the retinal succinate pool.
To directly measure the contribution of reverse SDH activity to succinate production when glucose is the only fuel source available, we repeated this experiment using 4-2H-glucose. At the steady state, the fractional enrichment of m1 fumarate was 18.2%, and that of m1 succinate was 10.2% (Figure 4B). Scaling these values for 100% m1 labeling of the fumarate pool predicts that reverse SDH activity maintains 55.8% of the succinate pool. m1 α-ketoglutarate was not detected at any time, indicating that m1 succinate does not form by oxidative TCA cycle activity (Figure S4A). To further confirm that reversal of SDH is responsible for producing all of the m1 succinate observed at the steady state, we incubated retinas in 5 mM 4-2H-glucose in the presence of malonate for 5 and 30 min (Figure S4B). Malonate inhibited formation of m1 succinate in retinas supplied with 4-2H-glucose at both time points, indicating that reversal of SDH is the pathway of m1 succinate formation. To further validate the use of 4-2H-glucose as a tracer that will lead to production of m1 succinate only through reverse SDH activity and not another pathway, even after 60 min, we performed an identical time course experiment using eyecups (which do not perform reverse SDH activity). If an alternative metabolic pathway was responsible for producing m1 succinate, then we might expect to see some accumulate in eyecups; however, m1 succinate was not observed in eyecups, even after 60 min in 5 mM 4-2H-glucose (Figure S4C).
We also determined the fraction of the retinal succinate pool that is maintained by reverse SDH activity from exogenous malate. We incubated retinas in 50 μM U-13C-malate and 5 mM 12C glucose for times ranging from 0 to 60 min (Figure 4C). The steady-state fractional enrichment was calculated as 15.3% for m4 fumarate and 5.7% for m4 succinate. Scaling these values for 100% m4 labeling of the fumarate pool predicts that 37.2% of the succinate pool is maintained from exogenous malate. This value is lower than the fraction of the succinate pool maintained by reverse SDH activity when glucose is the only fuel. Because the retina contains many cell types, a possible explanation for this difference is that there is a population of cells in the retina that consumes malate (producing m4 fumarate) but does not produce m4 succinate.
In summary, our analysis in retinas supplied with glucose alone (i.e., under identical conditions) predicts that 30.4% of retinal succinate is maintained by oxidative TCA cycle activity and 55.8% is maintained by reverse SDH activity (Figure 4D). All steady-state fractional enrichment values and reaction constants with confidence intervals are reported in Figure S4I. Fractional enrichment of all isotopomers for relevant metabolites at 60 min are reported in Figures S4D–S4F. Total levels of relevant metabolites for each time point are reported in Figures S4G and S4H.
Low Cytochrome c Oxidase Subunit IV (COXIV) Expression Drives Reversal of SDH in Retinas
Figures 3 and 4 show that reverse electron transport at SDH is a predominant pathway for succinate generation in retinas, but not in eyecups. Initially, we hypothesized that retinas utilize fumarate as an electron acceptor because they exist in a hypoxic niche in the eye, and O2 is limited. However, we observed, as shown in Figures 3 and 4, that fumarate may be converted to succinate even in retinal explants assayed at 21% O2. This suggests that lack of O2 alone does not directly drive reversal of SDH in the retina. Instead, chronic low O2 in the retina may drive a molecular adaptation that leads to reversal of SDH and persists at 21% O2. Because electrons from QH2 can reduce either O2 via complex IV or fumarate via SDH, we hypothesized that low O2 in retinal mitochondria might suppress complex IV expression, leading to an accumulation of QH2 that can drive reversal of SDH even during normoxia.
We evaluated expression of ETC component protein levels in retinal and eyecup homogenates by immunoblotting with antibodies that recognize representative subunits of the ETC complexes (Figure 5A). We found that freshly dissected retinas have a lower ratio of COXIV (a component of complex IV) to ATP5A (a component of ATP synthase) compared with eyecups (Figure 5A, lanes labeled “Fresh”). Hypoxia can decrease COXIV protein levels (Fukuda et al., 2007; Vijayasarathy et al., 2003). This motivated us to evaluate the influence of O2 levels on COXIV expression in retinas and eyecups. We incubated retinas at 0%, 1%, 5%, 21%, and 95% O2 for 2 h in 5 mM glucose and then analyzed ETC component expression by immunoblotting. The low COXIV/ATP5A ratio in freshly dissected retinas was similar to the ratio in retinas cultured in 0% O2. Increasing pO2 led to a higher COXIV/ATP5A ratio in retinas. Figure 5A shows a representative immunoblot, and quantification of multiple independent experiments is shown in Figure 5B. Altering pO2 did not have a substantial effect on the COXIV/ATP5A ratio in eyecups. These results indicate that the hypoxic niche where retinas reside could suppress COXIV expression and, thus, complex IV activity.
Figure 5. Low COXIV Expression Drives Reversal of SDH in Retinas.
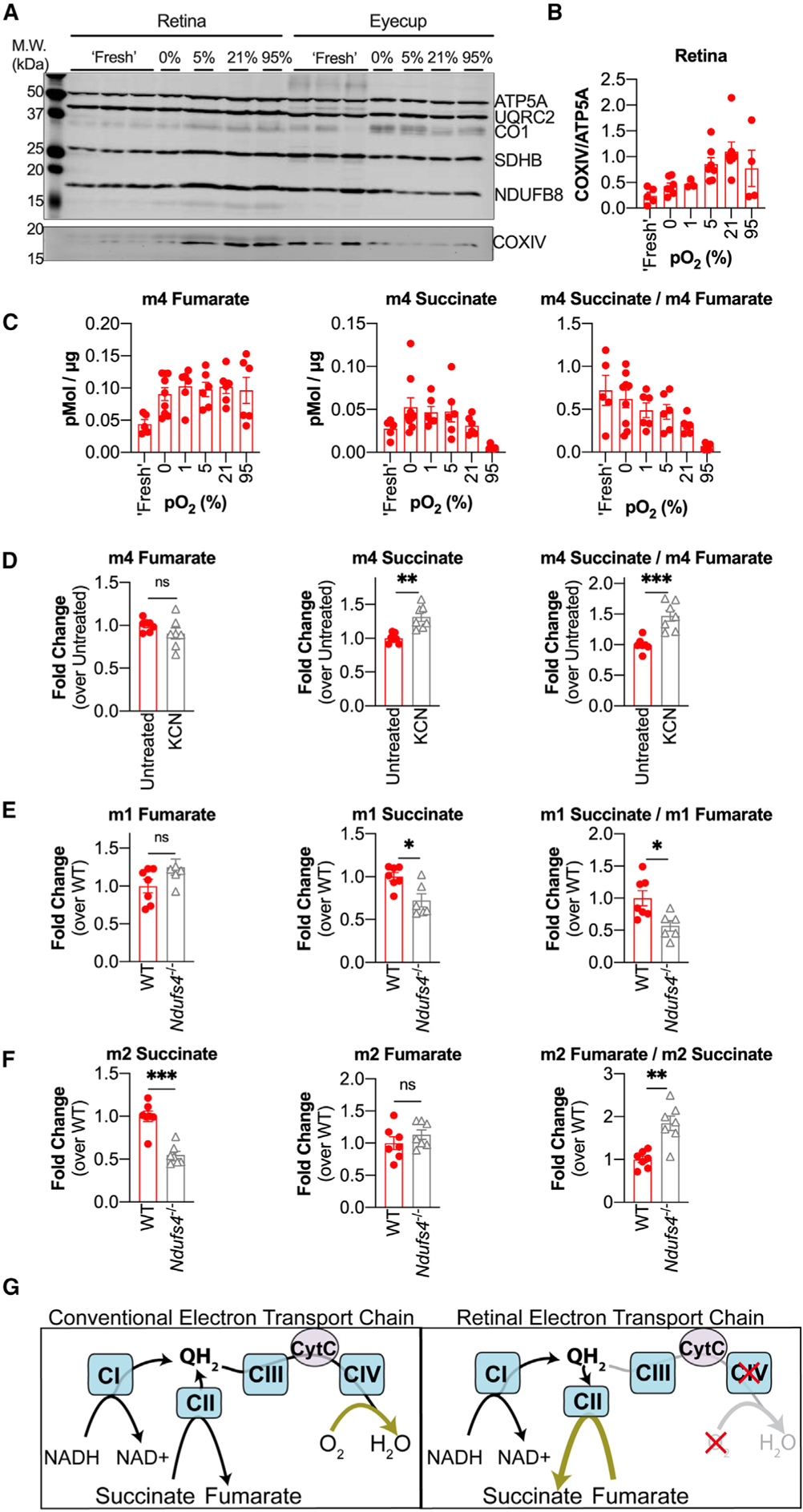
(A) Immunoblot of retinas and eyecups using an ETC component cocktail (top blot) and a single antibody against COXIV (bottom blot). ATP5A, ATP synthase subunit 5A; UQRC2, ubiquinol cytochrome c oxidoreductase subunit core 2; CO1, cytochrome c oxidase subunit 1; SDHB, succinate dehydrogenase B; NDUFB8, NADH ubiquinone oxidoreductase subunit B8; COXIV, cytochrome c oxidase subunit 4. (B) Quantification of multiple immunoblots probed with COXIV and ATP5A (each dot represents a biological replicate, n = 3 to 7 retinas per pO2 condition). (C) m4 fumarate, m4 succinate, and the m4 succinate/m4 fumarate ratio measured in retinas supplied with 5 mM 12C-glucose + 50 μM U-13C-malate for 5 min after pre-equilibration at specified pO2 for 2 h (“Fresh” data shown are duplicated from Figure 3B for ease of comparison) (n = 6–9 retinas per pO2 condition; error bars indicate SEM). (D) m4 fumarate, m4 succinate, and the m4 succinate/m4 fumarate ratio measured in retinas supplied with 5 mM 12C-glucose + 50 μM U-13C-malate in the presence of 3 mM KCN (n = 6 retinas; error bars indicate SEM; ** indicates p < 0.005 and *** indicates p < 0.001 using Welch’s t test). (E) m1 fumarate, m1 succinate, and the m1 succinate/m1 fumarate ratio in WT and Ndufs4−/− retinas supplied with 5 mM 4-2H-glucose for 5 min (n = 7 WT and 6 Ndufs4−/− retinas; error bars indicate SEM; * indicates p < 0.05 using Welch’s t test). (F) m2 fumarate, m2 succinate, and the m2 fumarate/m2 succinate ratio in WT and Ndufs4−/− retinas supplied with 5 mM U-13C-glucose for 5 min (n = 7 WT and 7 Ndufs4−/− retinas; error bars indicate SEM; ** indicates p < 0.005 and *** indicates p < 0.001 using Welch’s t test). (G) Diagram showing canonical ETC function (left) and proposed ETC function in retinas (right).
Based on these observations, we hypothesized that raising COXIV expression by pre-incubating retinas with higher O2 levels would diminish the conversion of fumarate to succinate. To test this, we preconditioned retinas and eyecups for 2 h in pre-equilibrated media containing 5 mM 12C-glucose at 0%, 1%, 5%, 21%, and 95% O2. After 2 h, reverse SDH activity was assessed by transferring retinas into fresh media containing 5 mM 12C-glucose and 50 μM U-13C-malate and incubating at the same pO2 as before for 5 min. Retinas incubated at increasing pO2 exhibited less reverse SDH activity, as measured by m4 succinate production and by the m4 succinate/m4 fumarate ratio (Figure 5C) However, we also noted that pre-equilibration for 2 h (at any pO2) drastically altered the total succinate pool size in retinas (Figure S5A).
Pre-incubating retinas at increasing pO2 increases COXIV levels and also decreases reverse SDH activity. However, changes in total metabolite levels suggest that pre-incubation also causes other metabolic changes that are independent of COXIV but could also influence reversal of SDH. For this reason, we also used other means to more directly interrogate the influence of complex IV on reverse SDH activity. We hypothesized that treating freshly dissected retinas with the complex IV inhibitor potassium cyanide (KCN) would further increase the reduction state of the retinal Q pool and drive more reverse SDH activity. We incubated retinas in 5 mM 12C-glucose and 50 μM U-12C-malate with 3 mM KCN for 5 min to allow QH2 to accumulate and then transferred the retinas to fresh media containing 5 mM 12C-glucose and 50 μM U-13C-malate with 3 mM KCN for 5 min to assay reverse SDH activity. We found that retinas treated with KCN produced more m4 succinate, which is consistent with a buildup of QH2 further driving reverse SDH activity (Figure 5D). We also observed that KCN treatment reduced the total fumarate pool in retinas (Figure S5B).
Next we tested whether in vivo disruption of the mitochondrial QH2 supply could influence the amount of reverse SDH activity we measured in our ex vivo assays. We used retinas from Ndufs4−/− mice, which lack a critical component of complex I (Kruse et al., 2008). We hypothesized that complex I deficiency would lead to retinal mitochondria from these mice accumulating less QH2, providing less driving force to reduce fumarate to succinate. We incubated retinas from post-natal day 21 (P21) and P30 Ndufs4−/− mice and WT littermates in 5 mM 4-2H-glucose for 5 min and found that Ndufs4−/− retinas still accumulated m1 fumarate but produced less m1 succinate, leading to a smaller m1 succinate/m1 fumarate ratio (Figure 5E). This indicates that retinas from Ndufs4−/− mice perform less reverse SDH activity. We also tested whether the decrease in the mitochondrial QH2 pool in Ndufs4−/− retinas would enhance forward SDH activity. We incubated retinas from P21 and P30 Ndufs4−/− mice and WT littermates in 5 mM U-13C-glucose for 5 min and found that Ndufs4−/− retinas accumulated less m2 succinate but equivalent amounts of m2 fumarate, leading to a higher m2 fumarate/m2 succinate ratio (Figure 5F). This indicates that the forward SDH reaction is enhanced in Ndufs4−/− retinas. Total fumarate and α-ketoglutarate levels were not different, but there was substantially less total succinate in Ndufs4−/− retinas incubated in 5 mM glucose for 5 min (Figure S5C). Enhanced forward SDH activity and diminished reverse SDH activity may contribute to the diminished size of the succinate pool in retinas from Ndufs4−/− mice.
DISCUSSION
We identified aspects of metabolic specialization in the retina and RPE-choroid that support the existence of a malate-succinate cycle between the two tissues. We found that the retina releases succinate and that the RPE-choroid imports succinate to fuel mitochondrial respiration and, in turn, exports malate, which is capable of refueling succinate production in the retina via reverse SDH activity. We found that pO2 influences expression of COXIV in retinal explants, suggesting that it is the hypoxic environment of the retina that keeps complex IV activity low and allows QH2 to build up to levels that can drive reversal of SDH (Figure 5G).
Previous studies have shown that reverse electron transport can produce succinate in the ischemic heart; however, it was calculated that only 5.7% of the succinate pool is made by reverse SDH activity (Chouchani et al., 2014; Zhang et al., 2018). In contrast, our data show that as much as 55% of the succinate pool in retinas derives from reverse SDH activity. Remarkably, this is greater than the amount of succinate produced from oxidative TCA cycle activity.
We specifically tested malate as a candidate metabolite used by retinas to resupply succinate pools because (1) eyecups release malate in the presence of glucose alone and (2) malate release from eyecups is enhanced by succinate. However, we also observed that, although eyecups do not release fumarate at a high rate in the presence of glucose alone, fumarate export is enhanced similarly to malate export in the presence of succinate. Thus, it is possible that retinas also use fumarate released by eyecups to resupply their succinate pools via reverse SDH activity.
The retina relies heavily on glycolysis to produce ATP (Kanow et al., 2017; Krebs, 1927). Because the RPE lies between the retina and the choroidal blood supply, glucose must pass through the RPE mostly unconsumed to fuel glycolysis in the retina. Export of succinate from the retina to the RPE could provide the RPE with an alternative fuel source to glucose. This would allow a greater fraction of glucose to pass through the RPE unconsumed so that it can reach the retina. This suggests that the RPE has specifically adapted to consume succinate; many other tissues (except brown fat) are thought to be impermeable to succinate (Ehinger et al., 2016; Hems et al., 1968; Mills et al., 2018).
Succinate exported from the retina is carrying reducing power that would be wasted if it were not used as a fuel by another tissue. The RPE is extremely well situated to use this succinate because it is positioned in an O2-rich niche where electrons from succinate can be donated to O2 via the ETC. This is not the first succinate-mediated “redox shuttle” to be proposed. During whole-body hypoxia in rats, succinate released from peripheral tissues has also been hypothesized to carry unused reducing power to the lungs, where O2 is relatively more accessible during hypoxia (Cascarano et al., 1976).
RPE consumption of succinate could protect the retina in more than one way. RPE consumption of succinate could prevent unwanted succinate accumulation in the retina. In an oxygen-induced retinopathy model, rats were exposed to 24-h cycles of hypoxia and hyperoxia from P0 to P21, which caused a 3-fold increase in retinal succinate. Accumulated succinate signaled through GPR91 in retinal ganglion cells to induce pathological extraretinal neovascularization (Sapieha et al., 2008). We have shown that retinas constitutively release succinate. If the RPE were not also constitutively consuming this succinate (as in the case of whole-body hypoxia), then it could accumulate in the retina and stimulate unwanted angiogenesis.
RPE consumption of succinate might also be protective by reducing oxidative stress in the retina. Mammalian retinas are composed of terminally differentiated neurons that cannot be replaced when damaged. Reactive oxygen species pose a great risk to these neurons because they can damage proteins, membranes, and nucleic acids. However, as long as photoreceptors release succinate, succinate can stimulate the RPE to consume a significant portion of O2 from the choroidal blood supply, preventing O2 from reaching the retina, where it could form reactive oxygen species. This is supported by the observation that, although mouse eyecups contain approximately 3-fold less cellular material than retinas, they are able to consume as much O2 as the entire retina when stimulated by succinate (Figure 1B).
The metabolic ecosystem formed by malate-succinate exchange between the retina and RPE illustrates another way in which photoreceptor degeneration can drastically affect eye health. In the absence of photoreceptors, retinal succinate export decreases (Figure S3H). We expect that, in an intact eye, this leads to a decrease in RPE O2 consumption. If succinate-stimulated RPE O2 consumption normally protects the retina, then loss of photoreceptors would exacerbate oxidative damage to the retina. This may occur in retinitis pigmentosa, where degeneration of rod photoreceptors causes an increase in photoreceptor layer O2 tension and leads to secondary cone photoreceptor death (Campochiaro and Mir, 2018; Yu et al., 2000). It may be possible to prevent secondary cone degeneration by supplying exogenous succinate to the RPE, which could recapitulate a critical aspect of this metabolic ecosystem for therapeutic benefits.
Overall, our findings suggest that the retina has adapted to its hypoxic niche in the eye by altering the stoichiometry of its respiratory complexes to favor a reversal of the SDH reaction. This adaptation can divert electrons out of the ETC to reduce fumarate to succinate. The succinate released from the retina can shuttle electrons to the RPE-choroid, where they reduce O2 to H2O. This exchange of metabolites facilitates transport of electrons away from a tissue that is not well poised to transfer them to O2 (the retina) to one that is better suited to use them to reduce O2 to H2O (the RPE-choroid). Succinate stimulates RPE cells to release malate as “empty” electron shuttles that retinas can then refill with electrons via reverse SDH activity. This malate-succinate exchange is another aspect of the metabolic ecosystem formed by the retina, RPE, and choroid, where tissue specialization of metabolic reactions is coordinated to generate the energy needed for visual function in the face of low O2 availability.
We rigorously demonstrated that these activities occur in isolated retinas and RPE-choroid complexes that are alive and functional. The next steps toward establishing the significance of this mechanism in vivo will be (1) to document SDH reversal and transfer of metabolites between these tissues in living animals; (2) to explore how malate-succinate exchange is influenced by genetic perturbations that interfere with the ability of the retina to produce succinate or the ability of the RPE-choroid to consume it; (3) to determine how disrupting this metabolite transfer in vivo influences the health of the retina and RPE-choroid; and (4) to identify how the exchange of succinate and malate is disrupted in disease.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
The main contact for this work is James B. Hurley (jbhhh@uw.ed).
Materials Availability
This work did not generate any new materials.
Data and Code Availability
This study did not generate any new datasets or codes.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mouse Models
Experimental procedures complied with NIH guidelines and were approved by the University of Washington IACUC. C57BL/6J mice (RRID:IMSR_JAX:000664) were purchased from The Jackson Laboratory at 6–8 weeks old and housed at the UW Medicine SLU 3.1 vivarium, where they experienced a 6AM–9PM (fall and winter) and 7AM–9PM (spring and summer) light/dark cycle and free access to food and water. Aipl1−/− mice were generated in house (Ramamurthy et al., 2004) and housed in the SLU 3.1 facility under the same conditions. Retinas from Ndufs4−/− mice were obtained from two sources: the Rong Tian lab at the University of Washington, and the Simon Johnson lab at Seattle Children’s Research Hospital (B6.129S4-Ndufs4tm1.1Rpa/J, RRID:IMSR_JAX:027058). C57BL/6J and Aipl1−/− mice were between 8–20 weeks old at the time of experiment. Ndufs4−/− mice were aged P21 (n = 4 mice) and P30 (n = 3 mice) at the time of experiment. Mice were euthanized by awake cervical dislocation for all experiments. Equal numbers of male and female mice were used.
RPE Cell Culture
RPE cell culture was performed in the lab of Jennifer Chao at the University of Washington. Human fetal RPE tissue with a gestational age of 115 days was obtained from the Birth Defects Research Laboratory (BDRL) at the University of Washington. Fetal RPE dissection and culture was performed according to (Sonoda et al., 2009). Fetal RPE sheets were cultured at 37°C with 5% CO2 in RPE media (MEM alpha (Life Technologies), 5% FBS (Atlanta Biologicals), N1-Supplement (Sigma-Aldrich), Nonessential Amino Acids (GIBCO), and Penicillin-Streptomycin (GIBCO)). 3–4 weeks after dissection, RPE sheets reached confluency and were strained using a 40 μm cell strainer to create a suspension of single cells. After counting, the RPE cells were plated onto a 12-well plate coated with matrigel (Corning) at 500,000 cells per insert.
METHOD DETAILS
Immunoblotting
Protein was extracted by homogenizing in RIPA buffer (150 mM NaCl, 1.0% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, 50 mM Tris, pH 8.0) and run on 12% polyacrylamide gels. After running, gels were transferred onto PVDF membranes (Millipore, IPFL00010) and blocked for 1 hr at room temperature in LI-COR Odyssey Blocking Buffer (LI-COR, 927–40000). Primary antibodies were diluted in blocking buffer at specified concentrations and incubated overnight at 4°C. Membranes were washed with 1x phosphate buffered saline (PBS) and PBS with 0.1% Tween-20 (PBS-T), then incubated with secondary antibody for 1 hr at 25°C and washed again before imaging. Membranes were imaged and bands were quantified using the LI-COR Odyssey CLx Imaging System (RRID: SCR_014579). Antibodies used are: [anti-Total Oxphos, 1:1000 dilution, RRID: AB_2629281, Lot# P3338], [anti-COXIV, 1:500 dilution, RRID: AB_2085424, Lot# 10], [anti-rabbit secondary, 1:2500, RRID: AB_2715510, Lot# C61012–02], [anti-mouse secondary, 1:2500, RRID: AB_2716622, Lot# C60217–15].
Ex vivo Isotopic Labeling of retinas and eyecups
Krebs-Ringer bicarbonate (KRB) buffer (98.5 mM NaCl, 4.9 mM KCl, 1.2 mM KH2PO4 1.2 mM MGSO4-7H2O, 20 mM HEPES, 2.6 mM CaCl-2H2O, 25.9 mM NaHCO3) optimized for isotopic labeling experiments in retinas was used in these experiments. Mice were euthanized by awake cervical dislocation and eyes were rapidly enucleated into a dish of Hank’s Buffered Salt Solution (HBSS; GIBCO, Cat#: 14025–076). Excess fat and connective tissue was trimmed from each eye, then retinas were dissected away from eyecups and the lens was removed. After dissection, retinas were placed in pre-warmed KRB containing labeled metabolites at concentrations in each experiment that was equilibrated at 21% O2 and 5% CO2 (unless otherwise specified in the text). Retinas were incubated for the specified time points at 37°C (at 5% CO2 and 21% oxygen, unless otherwise specified in the main text), then washed twice in ice-cold PBS and flash frozen in liquid nitrogen. Metabolites used were: D-[U-13C]-glucose (Cambridge Isotope Laboratories, CLM-1396), D-[U-13C]-malate (Cambridge Isotope Laboratories, CLM-8065), (D-[4-2H]-glucose, Omicron Biochemicals, GLC-035), ([U-13C]-succinic acid, Sigma #491985)
Metabolite Analysis by Gas-Chromatography Mass Spectrometry
Metabolites were extracted from both retinas and eyecups using ice-cold 80% MeOH. 150 μL extraction buffer was added to each sample and tissue was disrupted by sonication. Samples were incubated on dry ice for 45 minutes and centrifuged at maximum speed for 30 minutes. The resulting pellet was resuspended in RIPA buffer and the amount of protein in each retina or eyecup was determined by a BCA assay. The resulting supernatant was lyophilized at room-temperature until dry, then derivatized in 10 μL of 20 mg/mL Methoxyamine HCl (Sigma, Cat#: 226904) dissolved in pyridine (Sigma, Cat#: 270970) at 37°C for 90 minutes, and subsequently with 10 μL tert-butyldimethylsilyl-N-methyltrifluoroacetamide (Sigma, Cat#: 394882) at 70°C for 90 minutes. Metabolites were analyzed on an Agilent 7890/5975C GC-MS using selected-ion monitoring methods described extensively in previous work (Du et al., 2015). Peaks were manually integrated using MSD ChemStation software (Agilent), and correction for natural isotope abundance was performed using the software Isocor (Millard et al., 2012). Raw signals for each metabolite were converted to molar amounts using metabolite standard curves which were run alongside each experiment. Molar amounts were normalized to the total amount of protein (determined in a BCA assay) for each sample to determine the molar amount of metabolite per mg of cellular protein. To determine steady-state fractional enrichment values in Figure 4, a curve was fit to each time course using Graphpad Prism software (Version 8) assuming a first order reaction by using the equation: %metabolite = B *(1 −e−kt). B is the fractional enrichment at the steady state, k is the rate constant for the reaction, t is time, and %metabolite is the fractional enrichment at a given time.
Oxygen Consumption Measurement
Oxygen consumption measurements were performed using a perifusion flow-culture system adapted for analysis of retinal tissue (Du et al., 2016b; Sweet et al., 2002, 2004). KRB buffer modified for perifusion was prepared as follows: 98.5 mM NaCl, 4.9 mM KCl, 1.2 mM KH2PO4, 1.2 mM MgSO4-7H2O, 20 mM HEPES, and 2.6 mM CaCl2-2H2O, 25.9 mM NaHCO3, 1x GIBCO Antibiotic-Antimycotic, and 0.1 g/100 mL Fatty Acid Free Bovine Serum Albumin. Retinas and eyecups were dissected in KRB buffer supplemented with 5 mM glucose, then cut into quarters and loaded into chambers. Each chamber contained either tissue from 2 retinas or 4 eyecups. Retina and eyecup tissue were layered into chambers between Cytopore beads (Amersham Biosciences, Piscatawy, NJ) and kept in place by two porous frits. KRB buffer modified for perifusion was pumped through the system using a peristaltic pump. Media passed first through an artificial lung where it was oxygenated with a 21% O2, 5% CO2, 74% N2 mixture. Media then passed through a bubble trap before moving through the chambers containing retina or eyecup tissue. The glass of each chamber is coated with a thin layer of oxygen sensitive polymerized Pt(II) Meso-tetra(pentafluorophenyl)porphine dye (Frontier Scientific, Logan, UT). A fiber optic cable positioned to measure the oxygen-tension of the media after it has passed over the tissue detects emitted phosphorescent light from the oxygen-sensitive dye when pulsed with 405 nm ultraviolet LED. The lifetime of the phosphorescence signal decay is dependent on oxygen tension, and the quantitative relation was used to calibrate the signal.
QUANTIFICATION AND STATISTICAL ANALYSIS
Graphpad Prism software (Version 8) was used to perform statistical analysis. In all cases, comparisons between two conditions were made using Welch’s t test. In all figures, significance is defined as: * indicates p < 0.05; ** indicates p < 0.005, *** indicates p < 0.001, **** indicates p < 0.0001. The “n” used for each experiment is indicated in each figure legend. For oxygen consumption measurements, “n” indicates measurements made from a single chamber. Each retina chamber contained tissue from 2 retinas, and each eyecup chamber contained tissue from 4 eyecups. For isotopic labeling experiments, “n” indicates 1 retina or 1 eyecup. All values reported in the text are mean ± standard deviation, while error bars on graphs show standard error of the mean (SEM).
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-Total Oxphos (mouse) | Abcam | Cat# ab110413; RRID: AB_2629281 |
| anti-COXIV (rabbit) | Cell Signaling | Cat# 4850S; RRID: AB_2085424 |
| anti-rabbit secondary (donkey) | Li-Cor | RRID: AB_2715510 |
| anti-mouse secondary (donkey) | Li-Cor | RRID: AB_2716622 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| D-[U-13C]-glucose | Cambridge Isotope Laboratories | Cat# CLM-1396 |
| D-[U-13C]-malate | Cambridge Isotope Laboratories | Cat# CLM-8065 |
| D-[4-2H]-glucose | Omicron Biochemicals | Cat# GLC-035 |
| [U-13C]-succinic acid | Sigma | Cat# 491985 |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6J | Jackson Laboratory | RRID: IMSR_JAX:000664 |
| Mouse: AIPL1−/− | Ramamurthy et. al 2004 (Hurley Lab) | N/A |
| Mouse: B6.129S4-Ndufs4tm1.1Rpa/J | Jackson Laboratory | RRID: IMSR_JAX:027058 |
| Software and Algorithms | ||
| Isocor | Millard et. al 2012 | https://pypi.org/project/IsoCor/ |
| GraphPad Prism Version 8 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| Biological Samples | ||
| hfRPE tissue | Birth Defects Research Lab at University of Washington | N/A |
Highlights.
Succinate is produced by reversal of succinate dehydrogenase in the retina
Hypoxia decreases complex IV expression to drive retinal succinate production
The retina exports succinate, which is oxidized by the RPE-choroid
The RPE-choroid exports malate, which fuels retinal succinate production
ACKNOWLEDGMENTS
Work in the lab of J.B.H. was funded by NIH grants EY06641 and EY017863. Work in the lab of I.R.S. was funded by DK17047. C.M.B. was funded by NIH grant F31EY031165. D.T.H. was funded by NIH grant 5T32EY007031-42. We thank Whitney Cleghorn for assistance with maintaining the mouse colony in the lab of J.B.H. and Kristine Tsantilas for assisting with mass spectrometry method maintenance. We thank Abbi Engel and Jennifer Chao for supplying hfRPE cells. We also thank Rong Tian (University of Washington) and Simon C. Johnson and Rebecca Bornstein (Seattle Children’s Hospital) for sharing Ndufs4−/− mice.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.107606.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Adijanto J, Du J, Moffat C, Seifert EL, Hurle JB, and Philp NJ (2014). The retinal pigment epithelium utilizes fatty acids for ketogenesis. J. Biol. Chem 289, 20570–20582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campochiaro PA, and Mir TA (2018). The mechanism of cone cell death in Retinitis Pigmentosa. Prog. Retin. Eye Res 62, 24–37. [DOI] [PubMed] [Google Scholar]
- Cascarano J, Ades IZ, and O’Conner JD (1976). Hypoxia: a succinate-fumarate electron shuttle between peripheral cells and lung. J. Exp. Zool 198, 149–153. [DOI] [PubMed] [Google Scholar]
- Chinchore Y, Begaj T, Wu D, Drokhlyansky E, and Cepko CL (2017). Glycolytic reliance promotes anabolism in photoreceptors. eLife 6, 1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinopoulos C (2019). Succinate in ischemia: Where does it come from? Int. J. Biochem. Cell Biol 115, 105580. [DOI] [PubMed] [Google Scholar]
- Chouchani ET, Pell VR, Gaude E, Aksentijević D, Sundier SY, Robb EL, Logan A, Nadtochiy SM, Ord ENJ, Smith AC, et al. (2014). Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 515, 431–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J, Yanagida A, Knight K, Engel AL, Vo AH, Jankowski C, Sadilek M, Tran VTB, Manson MA, Ramakrishnan A, et al. (2016a). Reductive carboxylation is a major metabolic pathway in the retinal pigment epithelium. Proc. Natl. Acad. Sci. USA 113, 14710–14715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J, Linton JD, and Hurley JB (2015). Probing metabolism in the intact retina using stable isotope tracers. Methods Enzymol 561, 149–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J, Rountree A, Cleghorn WM, Contreras L, Lindsay KJ, Sadilek M, Gu H, Djukovic D, Raftery D, Satrústegui J, et al. (2016b). Phototransduction Influences Metabolic Flux and Nucleotide Metabolism in Mouse Retina. J. Biol. Chem 291, 4698–4710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehinger JK, Piel S, Ford R, Karlsson M, Sjövall F, Frostner EÅ, Morota S, Taylor RW, Turnbull DM, Cornell C, et al. (2016). Cell-permeable succinate prodrugs bypass mitochondrial complex I deficiency. Nat. Commun 7, 12317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda R, Zhang H, Kim JW, Shimoda L, Dang CV, and Semenza GL (2007). HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell 129, 111–122. [DOI] [PubMed] [Google Scholar]
- Hems R, Stubbs M, and Krebs HA (1968). Restricted permeability of rat liver for glutamate and succinate. Biochem. J 107, 807–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochachka PW, Owen TG, Allen JF, and Whittow GC (1975). Multiple end products of anaerobiosis in diving vertebrates. Comp. Biochem. Physiol. B 50, 17–22. [DOI] [PubMed] [Google Scholar]
- Kanow MA, Giarmarco MM, Jankowski CS, Tsantilas K, Engel AL, Du J, Linton JD, Farnsworth CC, Sloat SR, Rountree A, et al. (2017). Biochemical adaptations of the retina and retinal pigment epithelium support a metabolic ecosystem in the vertebrate eye. eLife 6, e28899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krebs H (1927). On the Metabolism of the Retina. Biochem. Z 189, 57–59. [Google Scholar]
- Kruse SE, Watt WC, Marcinek DJ, Kapur RP, Schenkman KA, and Palmiter RD (2008). Mice with mitochondrial complex I deficiency develop a fatal encephalomyopathy. Cell Metab. 7, 312–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kushnir MM, Komaromy-Hiller G, Shushan B, Urry FM, and Roberts WL (2001). Analysis of dicarboxylic acids by tandem mass spectrometry. High-throughput quantitative measurement of methylmalonic acid in serum, plasma, and urine. Clin. Chem 47, 1993–2002. [PubMed] [Google Scholar]
- Lewis CA, Parker SJ, Fiske BP, Mccloskey D, Gui DY, Green CR, Vokes NI, Feist AM, Heiden M.G., Vander, and Metallo CM (2014). Tracing Compartmentalized NADPH Metabolism in the Cytosol and Mitochondria of Mammalian Cells. Mol. Cell 55, 253–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linsenmeier RA, and Zhang HF (2017). Retinal oxygen: from animals to humans. Prog. Retin. Eye Res 58, 115–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millard P, Letisse F, Sokol S, and Portais JC (2012). IsoCor: correcting MS data in isotope labeling experiments. Bioinformatics 28, 1294–1296. [DOI] [PubMed] [Google Scholar]
- Mills EL, Pierce KA, Jedrychowski MP, Garrity R, Winther S, Vidoni S, Yoneshiro T, Spinelli JB, Lu GZ, Kazak L, et al. (2018). Accumulation of succinate controls activation of adipose tissue thermogenesis. Nature 560, 102–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyadera H, Shiomi K, Ui H, Yamaguchi Y, Masuma R, Tomoda H, Miyoshi H, Osanai A, Kita K, and Omura S (2003). Atpenins, potent and specific inhibitors of mitochondrial complex II (succinate-ubiquinone oxidoreductase). Proc. Natl. Acad. Sci. USA 100, 473–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickla DL, and Wallman J (2010). The multifunctional choroid. Prog. Retin. Eye Res 29, 144–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramamurthy V, Niemi GA, Reh TA, and Hurley JB (2004). Leber congenital amaurosis linked to AIPL1: a mouse model reveals destabilization of cGMP phosphodiesterase. Proc. Natl. Acad. Sci. USA 101, 13897–13902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes-Reveles J, Dhingra A, Alexander D, Bragin A, Philp NJ, and Boesze-Battaglia K (2017). Phagocytosis-dependent ketogenesis in retinal pigment epithelium. J. Biol. Chem 292, 8038–8047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapieha P, Sirinyan M, Hamel D, Zaniolo K, Joyal JS, Cho JH, Honoré JC, Kermorvant-Duchemin E, Varma DR, Tremblay S, et al. (2008). The succinate receptor GPR91 in neurons has a major role in retinal angiogenesis. Nat. Med 14, 1067–1076. [DOI] [PubMed] [Google Scholar]
- Sonoda S, Spee C, Barron E, Ryan SJ, Kannan R, and Hinton DR (2009). A protocol for the culture and differentiation of highly polarized human retinal pigment epithelial cells. Nat. Protoc 4, 662–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swarup A, Samuels IS, Bell BA, Han JYS, Du J, Massenzio E, Abel ED, Boesze-battaglia K, Peachey NS, Philp NJ, et al. (2019). Modulating GLUT1 expression in retinal pigment epithelium decreases glucose levels in the retina : impact on photoreceptors and Müller glial cells. Am. J. Physiol. Cell Physiol 316, C121–C133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweet IR, Khalil G, Wallen AR, Steedman M, Schenkman KA, Reems JA, Kahn SE, and Callis JB (2002). Continuous measurement of oxygen consumption by pancreatic islets. Diabetes Technol. Ther 4, 661–672. [DOI] [PubMed] [Google Scholar]
- Sweet IR, Cook DL, DeJulio E, Wallen AR, Khalil G, Callis J, and Reems J (2004). Regulation of ATP/ADP in pancreatic islets. Diabetes 53, 401–409. [DOI] [PubMed] [Google Scholar]
- Vijayasarathy C, Damle S, Prabu SK, Otto CM, and Avadhani NG (2003). Adaptive changes in the expression of nuclear and mitochondrial encoded subunits of cytochrome c oxidase and the catalytic activity during hypoxia. Eur. J. Biochem 270, 871–879. [DOI] [PubMed] [Google Scholar]
- Winkler BS (1981). Glycolytic and oxidative metabolism in relation to retinal function. J. Gen. Physiol 77, 667–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yam M, Engel AL, Wang Y, Zhu S, Hauer A, Zhang R, Lohner D, Huang J, Dinterman M, Zhao C, et al. (2019). Proline mediates metabolic communication between retinal pigment epithelial cells and the retina. J. Biol. Chem 294, 10278–10289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young RW, and Bok D (1969). Participation of the retinal pigment epithelium in the rod outer segment renewal process. J. Cell Biol 42, 392–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu DY, and Cringle SJ (2006). Oxygen distribution in the mouse retina. Invest. Ophthalmol. Vis. Sci 47, 1109–1112. [DOI] [PubMed] [Google Scholar]
- Yu DY, Cringle SJ, Su EN, and Yu PK (2000). Intraretinal oxygen levels before and after photoreceptor loss in the RCS rat. Invest. Ophthalmol. Vis. Sci 41, 3999–4006. [PubMed] [Google Scholar]
- Zhang J, Wang YT, Miller JH, Day MM, Munger JC, and Brookes PS (2018). Accumulation of Succinate in Cardiac Ischemia Primarily Occurs via Canonical Krebs Cycle Activity. Cell Rep. 23, 2617–2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate any new datasets or codes.