

Abstract
Preclinical and clinical proof-of-concept studies have suggested the effectiveness of pharmacological modulation of inflammatory cytokines in ischemic stroke. Experimental evidence shows that targeting tumor necrosis factor (TNF) and interleukin (IL)-1 holds promise, and these cytokines are considered prime targets in the development of new stroke therapies. So far, however, information on the cellular expression of TNF and IL-1 in the human ischemic brain is sparse.
We studied 14 cases of human post-mortem ischemic stroke, representing 21 specimens of infarcts aged 1 to > 8 days. We characterized glial and leukocyte reactions in the infarct/peri-infarct (I/PI) and normal-appearing tissue (NAT) and the cellular location of TNF, TNF receptor (TNFR)1 and TNFR2, IL-1α, IL-1β, and IL-1 receptor antagonist (IL-1Ra). The immunohistochemically stained tissue sections received a score reflecting the number of immunoreactive cells and the intensity of the immunoreactivity (IR) in individual cells where 0 = no immunoreactive cells, 1 = many intermediately to strongly immunoreactive cells, and 2 = numerous and intensively immunoreactive cells. Additionally, we measured blood TNF, TNFR, and IL-1 levels in surviving ischemic stroke patients within the first 8 h and again at 72 h after symptom onset and compared levels to healthy controls.
We observed IL-1α and IL-1β IR in neurons, glia, and macrophages in all specimens. IL-1Ra IR was found in glia, in addition to macrophages. TNF IR was initially found in neurons located in I/PI and NAT but increased in glia in older infarcts. TNF IR increased in macrophages in all specimens. TNFR1 IR was found in neurons and glia and macrophages, while TNFR2 was expressed only by glia in I/PI and NAT, and by macrophages in I/PI. Our results suggest that TNF and IL-1 are expressed by subsets of cells and that TNFR2 is expressed in areas with increased astrocytic reactivity. In ischemic stroke patients, we demonstrate that plasma TNFR1 and TNFR2 levels increased in the acute phase after symptom onset compared to healthy controls, whereas TNF, IL-1α, IL-1β, and IL-1Ra did not change.
Our findings of increased brain cytokines and plasma TNFR1 and TNFR2 support the hypothesis that targeting post-stroke inflammation could be a promising add-on therapy in ischemic stroke patients.
Keywords: Apoplexy, Microglia, Leukocytes, Cytokines
Introduction
Inflammation is integral to the pathophysiology of ischemic stroke and a prime target for the development of new stroke therapies. The inflammatory cytokines interleukin (IL)-1 and tumor necrosis factor (TNF) are pivotal in regulating immune responses following ischemic stroke (reviewed in [31]) and are potential targets in stroke therapy. IL-1 comprises two agonists, IL-1α and IL-1β, both signaling through IL-1 receptor type I that is expressed by neurons, glia, and endothelial cells [1, 36]. In response to stroke, IL-1β is rapidly upregulated in the blood [41, 55, 69] and brain [45] and exacerbates injury in experimental animal models (reviewed in [31]). The IL-1 receptor antagonist (IL-1Ra) is a natural competitive inhibitor of IL-1 signaling [23] and modulates the size of ischemic infarcts in experimental animal models [12, 37, 44, 49]. An IL-1β haplotype in postmenopausal women and hypertensive persons [3], and an IL-1α haplotype in Koreans [64] are associated with increased stroke risk.
TNF is an inflammatory cytokine with both beneficial and detrimental properties for the CNS (reviewed in [28, 31]). It is expressed at constitutively low levels in the healthy CNS and, along with its two known receptors TNFR1 and TNFR2, increases after both experimental and human ischemic stroke [10, 17, 30, 45, 52, 61]. Plasma TNF levels are increased in stroke patients compared with controls [5], and common polymorphisms of the TNF gene promoter leading to increased circulating TNF levels appear to be associated with cardiovascular risk factors and ischemic stroke in Asians [14, 63]. TNF exists as both membrane-bound TNF (mTNF) and soluble TNF (solTNF), and emerging experimental data suggest that solTNF is detrimental whereas mTNF has protective properties and is necessary for the maintenance of innate immunity [6, 35, 38, 47, 58, 70].
The brain is extremely susceptible to both extrinsic and intrinsic challenges, and ischemic stroke initiates an acute and long-lasting inflammatory response characterized by glial activation and leukocyte infiltration. The inflammatory response is accompanied by increased cytokine production, both centrally in the brain but also in the periphery (reviewed in [7, 28]). In the present study, we determined the cellular expression of IL-1α, IL-1β, IL-1Ra, TNF, TNFR1, and TNFR2 in post-mortem ischemic brain tissue and, in parallel, the expression of the microglial/macrophage markers CD68, CD45 and Iba1 and the astrocytic marker glial fibrillary acidic protein (GFAP). In addition, we compared blood levels of IL-1α, IL-1β, IL-1Ra, TNF, TNFR1, and TNFR2 in samples from ischemic stroke survivors and healthy controls.
Materials and methods
Participants and recruitment
The prospective part of this study was conducted at the Department of Neurology and the Department of Orthopaedic Surgery, Odense University Hospital (OUH), from September 2017 to April 2018 and from May 2016 to February 2019, respectively.
Neurological patients presenting with classic clinical symptoms of stroke were eligible if they were over 18 years old and admitted within 48 h of symptom onset (n = 34). Both men and women were included, as were patients who underwent treatment with thrombolysis or thrombectomy. Exclusion criteria were the presence of a space-occupying lesion, sinus thrombosis, pregnancy, and the inability to write and understand Danish. A standard non-contrast head CT scan was taken at admission on all patients suspected of stroke. Some had MRI taken for further diagnostic clarification. All scans were assessed by radiologists at OUH for ischemic or hemorrhagic changes in the neural tissue. Patients were diagnosed by a combination of clinical history, physical examination, and CT scans, and only patients diagnosed with ischemic stroke were included. Patients received treatment according to the national standards in Denmark.
Controls (n = 7) were recruited through the relatives. Further, orthopedic patients over 18 years old undergoing surgery for rotator cuff tendon tear (n = 21) were included. Exclusion criteria included diabetes, autoimmune diseases, previous shoulder surgery, a fractured or dislocated shoulder, and the inability to write and understand Danish.
Outcome measures
Stroke patients were assessed on admission by a clinician using the Scandinavian Stroke Scale (SSS), which measures stroke severity based on physical examination on a scale from 0 to 58, with milder symptoms giving a higher score [22]. Modified Rankin Scale (mRS), a 7-step scale from 0 (no functional disabilities) to 6 (death), was used to assess functional outcome 3 months after discharge via a telephone interview.
Other data collection
Data were collected on age, sex, weight, height, smoking and drinking habits, pre-existing diabetes, and use of anti-inflammatory medication. In ischemic stroke patients, we further collected data on first time of clinical symptoms, symptoms at time of admission, and type and location of the lesion. In orthopedic patients, we further collected data on pre-existing rheumatic disease. None displayed any signs of rheumatic disease. As our healthy controls and orthopedic patients were comparable in sex, age, body mass index (BMI), drinking habits, and anti-inflammatory medication, although not smoking habits (Suppl. Table 1), we combined them into one control group for further analysis.
Procedures
Venous blood was drawn in neurological patients at admission (n = 34) and 72 h later (non-discharged patients, n = 9) and in orthopedic patients just prior to surgery for rotator cuff tendon tear (n = 21, only plasma). Blood was collected in 4 ml vacutainer and EDTA tubes and then centrifuged. Samples were aliquoted and kept at − 80 °C until analysis. Differential leukocyte counts (total leukocyte, neutrophil, lymphocyte, and monocyte counts) in orthopedic patients were all within normal range (Suppl. Table 1).
Cytokine and cytokine receptor analysis
TNF, TNFR1, TNFR2 concentrations in plasma, and IL-1α, IL-1β, and IL-1Ra concentrations in serum and plasma were analyzed in duplicate using commercially available V-Plex Plus human kits (Mesoscale, Rockville, USA) according to the manufacturer’s instructions. Prior to measurement, the samples were diluted two-fold in Diluent 41. MSD Discovery Workbench software was used for analysis [8]. Samples with a coefficient of variation (CV) values > 25% in individual analyses were excluded. Only samples without repeated freeze-thaw cycles were used.
Post-mortem brain tissue
The retrospective part of this study included 21 autopsy specimens from 14 patients admitted to OUH in 2000–2005. These were nine males (median age 61 years, interquartile range (IQR) 38–76 years) and five females (median age 78 years, IQR 67–83 years). Table 1 shows age, sex, infarcted brain area, age of infarct, and cause of death. Mixed organ tissue blocks were used as controls for immunohistochemical staining, as was brain tissue free of brain-specific disease (n = 3). Parallel tissue sections were used in previous studies [10, 12, 31, 32, 46].
Table 1.
Clinical data from post-mortem brain tissue
| Case | Sex/Age at death | Infarcted brain area | Infarct age | Cause of death |
|---|---|---|---|---|
| 1 | M/76 | Left temporal lobe | < 7 days | Multiorgan failure due to sepsis |
| 2 | F/73 | Right temporal lobe | < 7 days | Rupture of thoracic aortic aneurysm |
| 3 | F/78 | Right hemisphere | 2 days | Perforated gastric ulcer with peritonitis and infarct |
| 4 | M/67 | Left temporal lobe | < 7 days | Cardiac arrest |
| 5 | M/61 | Right parietal lobe |
< 1 day > 7 days |
Multiorgan failure due to sepsis, cardiac infarct and endocarditis |
| 6 | F/83 |
Right hippocampus Cerebellum |
1 day > 7 days |
Pulmonary embolism |
| 7 | F/80 |
Right frontal lobe Medulla Pons Striatum |
1 day < 5 days 1 day < 7 days |
Bronchopneumonia |
| 8 | M/68 | Left parietal lobe | < 1 day | Cerebral infarct, pneumonia |
| 9 | M/68 |
Caudate nucleus Insula |
> 7 days > 7 days |
Bronchopneumonia |
| 10 | M/59 | Right parietal lobe | > 7 days | Pneumonia |
| 11 | M/57 | Left internal capsule | > 7 days | Pneumonia |
| 12 | F/67 |
Right occipital lobe Right occipital lobe |
1 day > 2 days |
Pulmonary infarcts Respiratory distress syndrome |
| 13 | M/38 | Right parietal lobe | < 5 days | Cerebral herniation, subdural empyema, ARDS |
| 14 | M/48 | Right temporal lobe | > 7 days | Pneumonia, hypoglycemia |
ARDS acute respiratory distress syndrome, F female, M male
Preparation of tissue
Human post-mortem tissue encompassing infarcted brain tissue was formalin-fixed, embedded in paraffin, and cut into 2 μm thick, serial sections on a microtome. Tissue sections were then dewaxed in xylene and rehydrated in ethanol. For immunohistochemical staining, endogenous peroxidase activity was quenched using 1.5% hydrogen peroxide in Tris-buffered saline (TBS). For optimal staining protocols, heat-induced epitope retrieval was performed using T-EG buffer (10 mM Tris, 0.5 mM EGTA, pH 9) for chromogen staining and citrate buffer (10 mM citrate, pH 6) for fluorescence staining.
Hematoxylin and eosin (HE) staining
For visualization of nuclei and cytoplasmic inclusions, one section from each specimen was stained using HE according to standard protocols at the Department of Pathology, OUH. HE-stained tissue sections were evaluated by two independent neuropathologists.
Immunohistochemistry
Immunohistochemical staining was performed using the Dako autostainer platform (Dako, Denmark) as previously described [24]. Sections were stained using the following primary antibodies: mouse anti-CD68 (clone PG-M1, 1:100, Dako), mouse anti-CD45 (clone 2B11, 1:200, Dako), rabbit anti-Iba1 (ionized calcium binding adaptor molecule 1, 1:1000, Wako), rabbit anti-GFAP (1:2000, Dako), mouse anti-neurofilament (NF) (phosphorylated and non-phosphorylated NF-heavy chain; clone N52, 1:1000, Sigma-Aldrich), mouse anti-IL-1α (clone 4414, 1:1200, R&D Systems), mouse anti-IL-1β (clone 2E8, 1:50, BioRad), rabbit anti-TNF (1:100, ThermoFisher Scientific), rabbit anti-TNFR1 (clone H-271, 1:50, Santa Cruz), rabbit anti-TNFR2 (1:50, Sigma-Aldrich), and rat anti-IL-1Ra (clone 40,007, 1:1500, R&D Systems). The antigen-antibody complex was visualized using EnVision+System horse-radish peroxidase-labelled Polymer (Dako), PowerVision+Poly-HRP IHC (AH Diagnostics), or CSAII (Dako) detection systems.
Control reactions
Controls for antibody specificity and non-specific staining were performed by substituting the primary antibodies with rabbit IgG (TNF, TNFR1, and TNFR2), mouse IgG2a (IL-1α), mouse IgG1 (IL-1β), or rat IgG2a (IL-1Ra) in the same IgG concentrations or by omitting the primary antibody in the protocol. Immunoabsorption was performed using a mixture of the primary antibody and a 100-fold excess of a recombinant human (rh) protein (rhTNF (210-TA); rhTNFR1/TNFRSF1A (636-R1); rhTNFR2/TNFRSF1B (aa 24–206; all from R&D Systems); and rhIL-1β (rcyec-hil1b, InvivoGen)). Controls were devoid of staining or showed a reduced signal (Suppl. Fig. 1).
Immunofluorescent and immunohistochemical double staining
Sections were bleached in Autofluorescence Eliminator Reagent (Millipore) according to the manufacturer’s guidelines. This treatment completely removed autofluorescence in the tissue (Suppl. Fig. 2). Sections were then pre-incubated with 5% normal serum from secondary antibody species diluted in phosphate-buffered saline (PBS) containing 0.25% Triton (PBS-T). Sections were incubated overnight with primary antibodies diluted in PBS-T as described above for IL-1α, IL-1β, TNF, TNFR1, and TNFR2. Next day, sections were rinsed 2 × 5 min in PBS, followed by 3 × 10 min in PBS-T, and incubated overnight with specific antibodies directed against Iba1 (1:600), NeuN-488 (clone EPR12763, 1:250, Abcam), CD68-FITC (clone KP1, 1:500, Dako), GFAP-Cy3 (clone G-A-5, 1:500, Sigma-Aldrich), or NF-heavy chain (clone TA51, 1:50, Millipore). The following day, sections were rinsed in PBS-T for 10 min and incubated with Alexa-488-conjugated donkey anti-rat, Alexa-488-conjugated goat anti-rabbit, Alexa-488-conjugated donkey anti-rabbit, Alexa-555-conjugated donkey anti-mouse, or Alexa-594-conjugated donkey anti-rabbit (all 1:500 and from Invitrogen) for 2 h at room temperature. Finally, sections were rinsed 3x in PBS and 1x in TBS before being mounted in ProLong Gold Antifade Reagent with 4′,6-diamidine-2′-phenylindole dihydrochloride (DAPI) (Thermo Fisher Scientific). Immunohistochemical double staining for IL-1Ra and CD45 was performed as previously described [12].
Scoring of cell-specific staining intensity
The sections were scored independently by a neuropathologist and a neurobiologist who were blinded to the age and the location of infarct, and cause of death. Three categories were used to generate a score reflecting the number of immunoreactive cells and the intensity of the immunoreactivity (IR) of individual cells: 0 = no immunoreactive cells, 1 = many intermediately to strongly immunoreactive cells, 2 = numerous and intensively immunoreactive cells [12, 43] (for representative images of the scoring system, please refer to Suppl. Fig. 3). A low sensitivity 0–2 scoring scale was applied because of the multiple variations in post-mortem autolysis and fixation time this autopsy tissue has been exposed to, and which could not be controlled for prior to the actual immunohistochemical staining. Variation in post-mortem autolysis and fixation time could have more impact on the data and introduce bias in the results with the use of more precise quantification methods, such as computerized pixel count. Based on our previous findings of a microglial/macrophage expression of TNF, TNFR2, IL-1α, IL-1β, and IL-1Ra in experimental stroke models [12, 29, 30], cells of particular interest were glia and infiltrating macrophages, although neuronal expression was also evaluated. Cells were scored based on their morphology. Microglia were characterized by a ramified morphology (representative images are shown in Fig. 1a,e,i) and macrophages by a round morphology (representative images are shown in Fig. 1g,k). The intensity was evaluated in normal-appearing tissue (NAT) and in infarct/peri-infarct (I/PI) tissue. Scoring was performed using a conventional Leica bright field microscope.
Fig. 1.

Characterization of glial reactivity in post-mortem ischemic stroke brain tissue. a-c Iba1 staining demonstrating ramified microglia and round macrophage-like cells in ≤2-day-old (a), 3–7-day-old (b), and ≥ 8-day-old (c) infarcts. d Scoring of Iba1 staining intensity demonstrating higher Iba1 expression in I/PI than in NAT in both microglia and macrophages. e-h CD45 staining demonstrating ramified microglia in ≤2-day-old infarcts (e) and round macrophage-like cells in 3–7-day-old (f) and ≥ 8-day-old (g) infarcts. h Scoring of CD45 staining intensity demonstrating higher CD45 expression in I/PI than in NAT in both microglia and macrophages. CD45+ macrophages were rarely detected in NAT. i-k CD68 staining demonstrating ramified microglia in ≤2-day-old infarcts (i) and round macrophage-like cells in 3–7-day-old (j), and ≥ 8-day-old (k) infarcts. (l) Scoring of CD68 staining intensity demonstrating higher CD68 expression in I/PI than in NAT in both microglia and macrophages. CD68+ macrophages were rarely detected in NAT. m-o GFAP staining demonstrating gemistocytic astrocytes in a ≤ 2-day-old infarct (m), shrunken astrocytes with absence of astrocytic processes in a 3–7-day-old infarct (n), and gemistocytic astrocytes in a ≥ 8-day-old infarct (o). p Scoring of GFAP staining intensity demonstrating comparable staining intensity in I/PI and NAT at all timepoints. GFAP, glial fibrillary acidic protein; Iba1, ionized calcium-binding adapter molecule 1; I/PI, infarct/peri-infarct; NAT, normal appearing tissue. Results are presented as mean ± SD. *p < 0.05, **p < 0.01, ***p < 0.001; one-way ANOVA followed by Sidak’s multiple comparisons test (n = 3–9/group). Scale bar = 40 μm
Statistical analysis
To compare stroke patients and controls, we used chi-square test for smoking, alcohol, and anti-inflammatory medication, Fisher’s exact test for sex, and Mann-Whitney test for age and BMI. For analysis of blood markers, the Shapiro-Wilk normality test was used to test for normal distribution of data, ROUT’s test to detect outliers, and Kruskal-Wallis test followed by Dunn’s multiple comparison to compare groups. One-way ANOVA test followed by Sidak’s multiple comparison was used to compare NAT and I/PI tissue.
Data are presented as mean ± SD or as median with IQR [25;75], and p ≤ 0.05 was considered statistically significant. Statistical analyses were performed using the Prism 5 software for Mac (GraphPad).
Results
Glial and macrophage reactivity in post-mortem ischemic brain tissue
We scored glial and macrophage reactivity in parallel I/PI and NAT brain tissue sections using the microglial/macrophage markers Iba1 (Fig. 1a-d), CD45 (Fig. 1e-h), and CD68 (Fig. 1i-l), and the astrocytic marker GFAP (Fig. 1m-p) in ≤2-day-old, 3–7-day-old, and ≥ 8-day-old infarcts (Fig. 1d,h,l,p). At all timepoints, intensely Iba1 immunoreactive (Iba1+) microglia and macrophages were present in I/PI and to a lesser extent Iba1+ microglia in NAT (Fig. 1d). In ≤2-day-old and ≥ 8-day-old infarcts, the Iba1 score was significantly higher in I/PI than in NAT for both microglia and macrophages. The Iba1 score for macrophages was very low in NAT (Fig. 1d, right graph). Similar findings were observed for CD45 (Fig. 1h) and CD68 (Fig. 1k), despite a tendency to increased IR in I/PI over time.
In ≤2-day-old lesions, microglial CD68 IR and infiltration of CD68+ macrophages were minimal. In one cortical infarct (Case #3, Table 1), a few CD68+ macrophages appeared to have infiltrated from meningeal vessels into the cortical layer, and CD68+ perivascular cells were present. Microglial CD68 IR, however, was minimal. At this early stage, subtle microglial reactivity and macrophage infiltration were observed in several cases. One case of infarction in the pyramids in the medulla oblongata (Case #7, Table 1), however, displayed pronounced infiltration of CD68+ macrophages in a pattern almost encircling both pyramids. In another case with infiltration of CD68+ meningeal and perivascular macrophages (Case #12, Table 1), parenchymal microglial CD68 IR was sparse.
In up to 7-day-old lesions, CD68+ microglia and infiltration of CD68+ macrophages were observed in the peri-infarct, while the infarcted area per se appeared less infiltrated by macrophages at this early stage. In one infarct, affecting the striatum and internal capsule (Case #11, Table 1), CD68+ microglia were located among the myelinated axons as an indication of Wallerian degeneration. Infarcts up to 7-days-old had pronounced infiltration of CD68+ macrophages, and microglial CD68 IR concomitantly increased in peri-infarct areas. In one case (Case #10, Table 1), CD68+ microglial rod cells appeared in the cerebral cortex adjacent to the infarct.
In older infarcts, CD68+ microglia were observed at the border of the infarct, but most microglial activity had subsided. CD68+ macrophages were present in the parenchyma particularly perivascularly. One old cortical infarct with laminar necrosis (Case #4, Table 1) showed extensive CD68+ macrophage infiltration in the laminar infarct and pronounced adjacent microglial reactivity.
The score for Iba1 generally paralleled the score for CD68 but showed different intensity. As microglia typically labeled more intensely with the anti-Iba1 antibody than with the anti-CD68 antibody, the ramified morphology of microglia was most clearly visible in the Iba1 staining. In contrast, macrophages labeled more intensely when stained for CD68. In the older infarcts, microglia generally displayed a ramified morphology though microglial Iba1 IR was still enhanced.
Regarding the score for CD45 in early infarcts, CD45+ microglia occasionally appeared intensified around lesions. In 3–7-day-old infarcts and in older lesions, microglial CD45 IR was less pronounced in peri-infarct areas than in the corresponding Iba1 staining, consistent with the CD45low phenotype of microglia. CD45+ macrophages were not as intensely labeled as when stained for CD68.
Similar to previous findings [25, 33], the GFAP score, reflecting the number of GFAP+ astrocytes and the GFAP IR of individual astrocytes, was decreased within the infarct, creating an outline of the infarcted area compared to the surrounding GFAP+ vital peri-infarct (Fig. 1p). At all timepoints investigated, GFAP IR was enhanced in the cortical peri-infarct areas compared to the infarct.
IL-1β IR is present in neurons, glia, and infiltrating macrophages
In tissue sections from ≤2-day-old infarcts, IL-1β IR was located in the neuronal cytoplasm, extending out to the proximal dendrites (Fig. 2a) and in glial cells (Fig. 2a, insert) in both I/PI and NAT, whereas IL-1β IR was only located in macrophage-like cells in I/PI.
Fig. 2.

Characterization of cellular IL-1 expression and serum IL-1 levels in ischemic stroke. a IL-1β immunohistochemical staining demonstrating IL-1β+ neurons (arrowheads) and IL-1β+ glial cells (insert) in a ≤ 2-day-old infarct. b IL-1β+ glial cell (arrow) located in a 3–7-day-old infarct. c IL-1β+ neurons (arrowheads) in a ≥ 8-day-old infarct. d Scoring of IL-1β staining intensity showing comparable IL-1β expression in neurons and glial cells in I/PI and NAT, whereas IL-1β was only expressed in macrophages located in I/PI. e IL-1Ra immunohistochemical staining demonstrating IL-1Ra+ glial cells (arrows) and IL-1Ra+ macrophages extravasating from capillaries (insert). f IL-1Ra+ macrophages (arrows) in I/PI in a 3–7-day-old infarct. g IL-1Ra+ glia (arrows) in a ≥ 8-day-old infarct. h Scoring of staining intensity showed that IL-1Ra expression was low in neurons in I/PI and NAT early after stroke and was absent in older infarcts. IL-1Ra was mainly expressed in glia located in NAT and I/PI, and in IL-1Ra+ macrophages located in I/PI. IL-1Ra+ macrophages were absent from NAT. i IL-1α immunohistochemical staining demonstrating IL-1α+ glial cell (arrow) and IL-1α+ macrophage (insert) in a ≤ 2-day-old infarct. j IL-1α+ macrophages in a 3–7-day-old infarct. k IL-1α+ macrophages in a ≥ 8-day-old infarct. l Scoring of IL-1α staining intensity showing comparable IL-1α expression in neurons and glia in I/PI and NAT, whereas IL-1α was primarily expressed in macrophages in I/PI and to a lesser extent in macrophages in NAT. m Immunofluorescent staining demonstrating co-localization of IL-1β (red) with Iba1+ (green) microglia (arrows) and Iba1+ (green) macrophages (insert). Note that not all IL-1β+ cells (arrowheads) co-localized to Iba1+ cells. n Immunofluorescent staining demonstrating co-localization of IL-1β (red) with NF+ (green) neurons (arrows). o Immunohistochemical double staining demonstrating co-localization of IL-1Ra+ (red) and CD45+ (brown) microglia. p Immunofluorescent staining demonstrating co-localization of IL-1α (red) with Iba1+ (green) microglia (arrows) and CD68+ (green) macrophages (insert). q-s V-Plex analysis demonstrating comparable plasma levels of IL-1β (q), IL-1Ra (r), and IL-1α (s) between controls (n = 7) and ischemic stroke patients at < 8 h (n = 34) and 72 h (n = 9) after symptom onset. For IL-1β, CV > 25% in one 72-h sample, for IL-1RA CV > 25% in one < 8-h sample and in one 72-h sample. IL-1α, interleukin-1alpha; IL-1β, interleukin-1beta; IL-1Ra, interleukin-1 receptor antagonist; I/PI, infarct/peri-infarct; NF, neurofilament; NAT, normal appearing tissue. Results are presented as mean ± SD (staining intensity) or mean with IQR (V-plex analysis). *p < 0.05, **p < 0.01, ***p < 0.001; one-way ANOVA followed by Sidak’s multiple comparisons test (scoring of staining intensity, n = 3–9/group) or Kruskal Wallis test followed by Dunn’s multiple comparison test (V-plex analysis). Scale bars: a-c, e-g, i-k = 40 μm, m-n, p = 100 μm, and o = 10 μm
In tissue sections from 3 to 7-day-old (Fig. 2b) and ≥ 8-day-old (Fig. 2c) infarcts, the cellular distribution of IL-1β IR was comparable to that of ≤2-day-old infarcts. IL-1β IR was observed in glial processes, infiltrating macrophages, intraparenchymal microglia and macrophages, and macrophages located inside blood vessels.
Scoring of the number of IL-1β+ cells and their IR yielded the highest score in neurons and glial cells in I/PI and NAT tissue and in macrophages located in I/PI at all timepoints (Fig. 2d). In ≥8-day-old infarcts, the score for IL-1β was highest in I/PI, with no IL-1β+ cells in NAT (Fig. 2d, lower graph). Information on IL-1β IR in control tissues is provided in Suppl. Fig. 4.
IL-1Ra IR is mainly present in glia and infiltrating macrophages
In tissue sections from ≤2-day-old infarcts, IL-1Ra IR was confined to glial cells (Fig. 2e) and neurons in the NAT and I/IP, whereas IL-1Ra IR was located in macrophages only in I/IP. IL-1Ra+ macrophages appeared to extravasate from capillaries located in I/IP (Fig. 2e, insert). IL-1Ra IR was located in the cytoplasm and in microglia, also to cellular processes. In tissue sections from 3 to 7-day-old infarcts, IL-1Ra IR was only located in glia in NAT, though still present in neurons, glial cells, and in macrophages in I/PI (Fig. 2f). At ≥8 days, no neuronal IL-1Ra IR was observed, and IL-1Ra IR was only present in glia in NAT and glia and macrophages in I/PI (Fig. 2g).
Scoring of IL-1Ra staining showed that although IL-1Ra IR was present in neurons (Fig. 2h, upper graph), glia scored higher in I/PI and NAT tissue in ≤2-day-old infarcts, and additionally increased in 3–7-day-old and ≥ 8-day-old infarcts (Fig. 2h, middle graph). Finally, scoring of macrophage IL-1Ra staining intensity showed that IL-1Ra+ macrophages were only observed in the I/PI, not in NAT (Fig. 2d, lower graph). Information on IL-1Ra IR in control tissues is provided in Suppl. Fig. 5.
IL-1α IR is present in neurons, glia, and infiltrating macrophages
In tissue sections from ≤2-day-old infarcts, IL-1α IR was located in the cytoplasm of neurons and in glia (Fig. 2i) both in NAT and I/IP. The same was true for 3–7-day-old and ≥ 8-day-old infarcts (Fig. 2j,k), though with decreasing number and IR of both neurons and glia in NAT (Fig. 2l, upper and middle graphs). Scoring showed that the number of IL-1α+ macrophages and their IR were low in macrophages in NAT at all timepoints and significantly increased in I/PI tissue from ≤2-day- and 3–7-day-old infarcts (Fig. 2l, lower graph). At ≥8 days, the level of IR was higher in macrophages in I/PI than in NAT, though not statistically significant (Fig. 2l, lower graph). Information on IL-1α IR in control tissues is provided in Suppl. Fig. 6.
TNF IR is present in ischemic neurons, glia, and infiltrating macrophages
In tissue sections from ≤2-day-old infarcts, TNF IR was located to small vesicles in the cytoplasm or in association with the cellular surface of neurons (Fig. 3a), glia, and macrophages. In 3–7-day-old infarcts, TNF IR was mainly located to glia and macrophages located within the infarct (Fig. 3b). At ≥8 days, TNF+ cells were primarily macrophages located in I/PI and glia located in I/PI and NAT (Fig. 3c), however, neuronal TNF IR was also observed. TNF IR was both observed in glial processes, infiltrating macrophages, and macrophages within blood vessels.
Fig. 3.

Characterization of cellular localization and plasma levels of TNF, TNFR1, and TNFR2 in ischemic stroke. a TNF immunohistochemical staining showing a TNF+ neuron (arrowhead) in a ≤ 2-day-old infarct. b TNF+ glial- and macrophage-like cells (arrows) located in a 3–7-day-old infarct. c TNF+ cells (arrows) and TNF+ macrophages extravasating from capillaries (arrows in insert) in a ≥ 8-day-old infarct. d Scoring of TNF staining intensity showing significantly higher TNF expression in neurons located in I/PI in ≤2-day-old infarcts compared to ≥8-day-old infarcts and in neurons located in NAT in ≤2-day-old infarcts compared to both 3–7-day-old and ≥ 8-day-old infarcts (upper graph). TNF expression was comparable in glial cells located in I/PI and NAT in ≤2-day-old and 3–7-day-old infarcts, with significantly increased expression in I/PI in ≥8-day-old infarcts compared to ≤2-day- and 3–7-day-old infarcts and compared to NAT (middle graph). At all timepoints, TNF expression in macrophages was only observed in I/PI (lower graph). e TNFR1 immunohistochemical staining demonstrating a TNFR1+ glial cell (arrow) and TNFR1+ neuron (arrowhead) in a ≤ 2-day-old infarct. f TNFR1+ neurons (arrowheads) in a 3–7-day-old infarct. g TNFR1+ neuron (arrowhead) in a ≥ 8-day-old infarct. h Scoring of staining intensity showed that TNFR1 expression was comparable in neurons and glia in I/PI and NAT at all timepoints. TNFR1 was mainly expressed in macrophages located in I/PI and absent from NAT. i TNFR2 immunohistochemical staining demonstrating TNFR2+ glial end-feet encircling the blood vessels (arrow) and a TNFR2+ glia cell (insert) in ≤2-day-old infarcts. j TNFR2+ cell (arrow) in a 3–7-day-old infarct. k TNFR2+ glia in a ≥ 8-day-old infarct. l Scoring of TNFR2 staining intensity showing comparable TNFR2 expression in glia located in I/PI and NAT in ≤2-day-old and 3–7-day-old infarcts and with a significant increase in TNFR2 expression in glia located in I/PI compared to NAT in ≥8-day-old infarcts. TNFR2 expression was only found in macrophages located in I/PI. No neurons were found to express TNFR2. m Immunofluorescent staining demonstrating co-localization of TNF (red) with NeuN+ (green) neurons (arrows). n Immunofluorescent staining demonstrating co-localization of TNFR1 (red) with NF+ (green) neurons and their proximal dentrites (arrows). o Immunofluorescent staining demonstrating co-localization of TNFR2 (green) with GFAP+ astrocytes (red). p High magnification of (o). q V-Plex analysis demonstrating comparable plasma TNF levels between controls and ischemic stroke patients at < 8 and 72 h after symptom onset. r-s V-Plex analysis demonstrating significantly increased plasma TNFR1 (r) and TNFR2 (s) levels in ischemic stroke patients < 8 h after symptom onset compared to controls. For TNF, CV > 25% in three control samples and one < 8-h sample. For TNFR1, CV > 25% in four < 8-h samples, 12 control samples, and one 72-h sample. For TNFR2, CV > 25% in three < 8-h samples, 11 control samples, and three 72-h samples. GFAP, glial fibrillary acidic protein; I/PI, infarct/peri-infarct; NF, neurofilament; NAT, normal appearing tissue; TNF, tumor necrosis factor; TNFR1, tumor necrosis factor receptor 1; TNFR2, tumor necrosis factor receptor 2. Results are presented as mean ± SD (staining intensity) or mean with IQR (V-plex analysis). *p < 0.05, **p < 0.01, ***p < 0.001; one-way ANOVA followed by Sidak’s multiple comparisons test (scoring of staining intensity, n = 3–9/group) or Kruskal Wallis test followed by Dunn’s multiple comparison test (V-plex analysis). Scale bars: a-c, e-g, i-k, p, inserts = 40 μm, and m-o = 100 μm
Scoring of TNF staining intensity showed that the number of TNF+ cells and their IR were highest in I/PI and NAT neurons in ≤2-day-old infarcts, with decreasing score in tissue sections from patients with 3–7-day-old and ≥ 8-day-old infarcts (Fig. 3d, upper graph). Scoring of TNF staining in glia showed a significantly increased TNF score in I/PI in ≥8-day-old infarcts compared to ≤2-day-old and 3–7-day-old infarcts (Fig. 3d, middle graph). Glial TNF IR was also observed in NAT (Fig. 3d, middle graph). Scoring of macrophage TNF staining yielded a high TNF score in I/PI, with no TNF IR in NAT (Fig. 3d, lower graph). Information on TNF IR in control tissues is provided in Suppl. Fig. 7.
TNFR1 IR is primarily in neurons but is also upregulated in macrophages and glia
In general, TNFR1 IR was mainly located to neurons, extending into their proximal dendrites (Fig. 3e,g). At ≤2 days, TNFR1 IR was primarily located to neurons and their proximal dendrites (Fig. 3e) but also to glial cells and macrophages (insert in Fig. 3e). At 3–7 days, TNFR1 IR was still observed in neurons but also in infiltrating macrophages and glia (Fig. 3f). At ≥8 days, TNFR1 IR was primarily observed in neurons (Fig. 3g), but a few macrophages and glia also upregulated their TNFR1 IR. Scoring of TNFR1 staining in neurons showed that TNFR1 IR was mainly found in I/PI tissue at all timepoints (Fig. 3h, upper graph). Glia located in both I/PI and NAT showed TNFR1 IR (Fig. 3 h, middle graph), and macrophages in I/PI tissue showed TNFR1 IR (Fig. 3h, lower graph). Information on TNFR1 IR in control tissues is provided in Suppl. Fig. 8.
TNFR2 IR is primarily in astrocytes, but also in microglia and macrophages
Astrocytes (Fig. 3i) were the most predominant cell type showing TNFR2 IR in ≤2-day-old infarcts. At 3–7 days, also macrophages in I/PI tissue (Fig. 3j) in addition to astrocytes showed TNFR2 IR. At ≥8 days, TNFR2 IR was also observed in glia (Fig. 3k). At all timepoints, TNFR2 IR was associated to the cellular processes. Scoring of TNFR2 staining intensity demonstrated that glia in I/PI and NAT showed TNFR2 IR at all timepoints (Fig. 3l, upper graph). In addition, TNFR2 IR was located to macrophages in the I/PI area at all timepoints (Fig. 3l, lower graph). No neuronal TNFR2 IR was observed. Information on TNFR2 IR in control tissues is provided in Suppl. Fig. 9.
IL-1α, IL-1β, IL-1Ra, and TNF IR is located to subsets of cells
We showed previously that TNF and IL-1β [11] and IL-1α, IL-1β, IL-1Ra [12] are produced by subsets of microglia in experimental stroke. We therefore compared parallel tissue sections from two stroke cases stained for IL-1α, IL-1β, IL-1Ra, TNF, TNFR1, and TNFR2 (Figs. 4 and 5). The first (case #7, Table 1) was a < 1-day-old right frontal lobe infarct in an 80-year-old female (Fig. 4). In line with our findings for all infarcts ≤2-day-old, we observed increased Iba1 (Fig. 4a), CD45 (Fig. 4b), and CD68 (Fig. 4c) IR in microglia/macrophages located within the infarct. Similar to our experimental stroke studies, IL-1β (Fig. 4d), IL-1α (Fig. 4c), IL-1Ra (Fig. 4f), and TNF (Fig. 4g) IR was located to subsets of cells (please compare Fig. 4d-g). TNFR1 IR was sparse (Fig. 4h), and TNFR2 IR (Fig. 4i) appeared to co-localize with areas of increased GFAP IR (Fig. 4j), suggesting that TNFR2 is expressed primarily by astrocytes. The second case (case #9, Table 1) was a > 7-day-old caudate nucleus infarct in a 68-year-old male (Fig. 5). Comparing parallel tissue sections, we observed that at this timepoint, IL-1β (Fig. 5a), IL-1α (Fig. 5b), IL-Ra (Fig. 5c), and TNF (Fig. 5d) IR was located to subsets of cells within the infarct. TNFR1 IR appeared to be increased on microglial-like cells and macrophages located within the infarct (Fig. 5e), whereas TNFR2 IR appeared to be increased on astrocyte-like cells (Fig. 5f).
Fig. 4.

Characterization of glial markers, IL-1, TNF, and TNF receptors in post-mortem tissue sections. a-j Immunohistochemical staining of parallel tissue sections from a < 1-day-old infarct for glial markers Iba1 (a), CD45 (b), and CD68 (c), and for cytokines IL-1β (d), IL-1α (e), IL-1Ra (f), and TNF (g), and for TNFR1 (h), TNFR2 (i), and the astrocyte marker GFAP (j). GFAP, glial fibrillary acidic protein; IL-1α, interleukin-1alpha; IL-1β, interleukin-1beta; IL-1Ra, interleukin-1 receptor antagonist; TNF, tumor necrosis factor; TNFR1, tumor necrosis factor receptor 1; TNFR2, tumor necrosis factor receptor 2. Scale bar = 40 μm
Fig. 5.
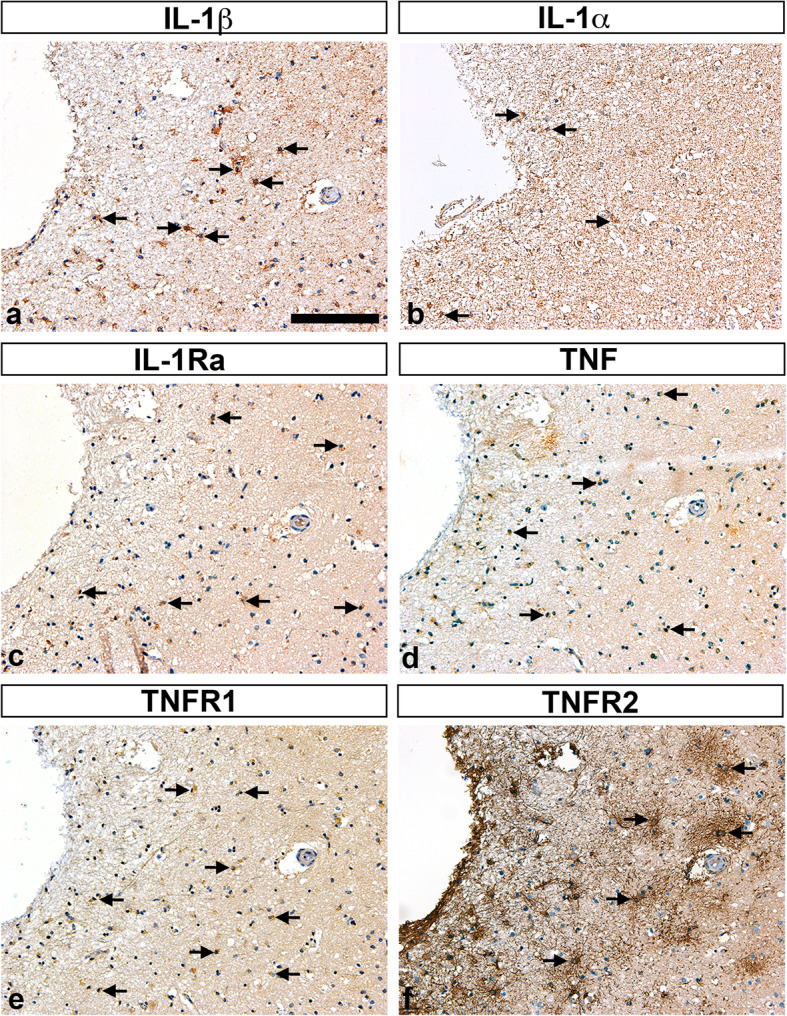
Characterization of IL-1, TNF, and TNF receptors TNFR1, and TNFR2 in post-mortem tissue sections. a-f Immunohistochemical staining of parallel tissue sections from a > 7-day-old infarct for cytokines IL-1β (arrows in a), IL-1α (arrows in b), IL-1Ra (arrows in c), and TNF (arrows in d), and for TNFR1 (arrows in e), and TNFR2 (arrows in f). IL-1α, interleukin-1alpha; IL-1β, interleukin-1beta; IL-1Ra, interleukin-1 receptor antagonist; TNF, tumor necrosis factor; TNFR1, tumor necrosis factor receptor 1; TNFR2, tumor necrosis factor receptor 2. Scale bar = 40 μm
These studies prompted us to perform co-localization studies, trying to identify the cellular sources of IL-1α, IL-1β, IL-1Ra (Fig. 2m-0), TNF, TNFR1, and TNFR2 (Fig. 3m-p). We found that IL-1β co-localized both to Iba1+ microglia (Fig. 2m), Iba1+ macrophage-like cells (insert in Fig. 2 m), and NF+ neurons (Fig. 2n). IL-1Ra IR co-localized to CD45+ cells (Fig. 2o), and IL-1α to Iba1+ (Fig. 2p) and CD68+ (insert in Fig. 2p) cells. TNF IR co-localized to NeuN+ neurons (Fig. 3m) and CD45+ cells (not shown), and TNFR1 to NF+ neurons and their proximal dendrites (Fig. 3n), while TNFR2 IR co-localized to GFAP+ astrocytes (Fig. 3o-p). The double immunofluorescent staining experiments thereby support the immunohistochemical staining and the qualitative scores.
Serum IL-1α, IL-1β, and IL-1Ra levels do not change in ischemic stroke patients
Baseline characteristics of ischemic stroke patients and controls are presented in Table 2. Importantly, we observed no difference in the use of anti-inflammatory medicine (p = 0.21) between ischemic stroke patients and controls.
Table 2.
Characteristics of study participants
| Variable | Controls | Ischemic stroke | p-value |
|---|---|---|---|
| Men, n (%) | 19 (68) | 19 (56) | 0.34 |
| Age, years (mean ± SD) | 59.3 ± 12.8 | 64.5 ± 12.8 | *0.03 |
| BMI, kg/m2 (mean ± SD) | 26.1 ± 3.8 | 26.6 ± 5.5 | 0.84 |
| SSS, points (mean ± SD) | 49.5 ± 9.2 | ||
| mRS, points (mean ± SD) | 1.8 ± 1.8 | ||
| Smoking | **0.003 | ||
| - Current smokers, n (%) | 4 (14.2) | 9 (26.4) | |
| - Previous smokers, n (%) | 5 (17.9) | 17 (50.0) | |
| - Non-smokers, n (%) | 14 (50) | 4 (11.8) | |
| - Not known, n (%) | 5 (17.9) | 4 (11.8) | |
| Alcohol consumption a | *0.04 | ||
| < health authorities’ recommendations, n (%) | 18 (64.3) | 25 (73.5) | |
| > health authorities’ recommendations, n (%) | 2 (7.1) | 9 (26.5) | |
| Not known, n (%) | 8 (28.6) | ||
| Anti-inflammatory medication, n (%) | 4 (14.2) | 14 (41.2) | 0.21 |
| - Topical steroids | 1 (3.6) | 8 (23.5) | |
| - NSAID | 3 (10.7) | 5 (14.7) | |
| - Systemic glucocorticoid | 3 (10.7) | 2 (5.9) | |
| - Antibodies | 1 (2.9) | ||
| Arterial supply area affected | |||
| - Anterior | 16 (52.9) | ||
| - Posterior (PCA & basilar/vertebral) | 6 (17.7) | ||
| - Nuclei | 5 (14.7) | ||
| - Brain stem & spinal cord | 3 (8.8) | ||
| - Fiber tracts | 3 (8.8) | ||
| - Not identified | 1 (2.9) | ||
| Treatment | |||
| - Thrombolysis | 5 (14.7) | ||
| - Thrombectomy | 5 (14.7) | ||
| Time to first sample, hours (mean ± SD) | 7.5 (5.6) | ||
BMI body mass index, mRS modified Rankin Scale, NSAID non-steroidal anti-inflammatory drug, PCA posterior communicating artery, SSS Scandinavian Stroke Scale
aThe Danish Health authority recommends < 7 units per week for women (1 unit equals 1 glass of wine) and < 14 units per week for men
Serum IL-1β was almost undetectable, and we observed no significant difference in IL-1β levels between controls (< 0.000001 pg/ml [< 0.000001;< 0.000001]) and ischemic stroke patients when serum was sampled either < 8 h (< 0.000001 pg/ml [< 0.000001;0.00016]) or 72 h after the vascular event (0.0013 pg/ml [< 0.000001;0.005]) (p = 0.47) (Fig. 2q).
No significant differences were observed in serum IL-1Ra levels between controls (237.7 pg/ml [232.2;320.6]) and ischemic stroke patients when serum was sampled either < 8 h (234.2 pg/ml [192.9;336.3]) or 72 h after the vascular event (202.6 pg/ml [169.2;239.2]) (p = 0.90) (Fig. 2r).
Finally, IL-1α was only detected in two ischemic stroke patients (Fig. 2s) and thus did not differ between groups.
Similar findings were obtained when analyzing plasma samples for IL-1α, IL-1β, and IL-1Ra (data not shown).
Plasma TNFR1 and TNFR2 are upregulated in the acute phase after ischemic stroke
We observed no significant differences in plasma TNF levels between controls (1.9 pg/ml [1.6;2.6]) and ischemic stroke patients when plasma was sampled < 8 h (2.0 pg/ml [0.7;2.6]) or 72 h (2.0 pg/ml [1.8;3.1], n = 9) after the vascular event (p = 0.57) (Fig. 3q).
In contrast, TNFR1 plasma levels were significantly upregulated within 8 h of the vascular event in ischemic stroke patients (376.4 pg/ml [146.8;3202]) compared to controls (131.9 pg/ml [118.5;158.7]) (**p = 0.006) (Fig. 3r). At 72 h after ischemic stroke, TNFR1 levels were not significantly changed (303.3 pg/ml [141.;1936]) compared to controls (Fig. 3r).
Similarly, TNFR2 levels were significantly upregulated within 8 h of the vascular event in ischemic stroke patients (413.8 pg/ml [251.3;7090]) compared to controls (276.7 pg/ml [236.7;357.8]) (*p = 0.04) (Fig. 3s). Although TNFR2 levels were higher at 72 h (518.7 pg/ml [266.7;6605]) compared to < 8 h, we observed no significant difference at this timepoint.
Discussion
To our knowledge, this study determines for the first time the detailed cellular localization of IL-1α, IL-1β, and IL-1Ra IR in human ischemic stroke. Furthermore, the study adds to the scarce literature on the cellular localization of TNF, TNFR1, and TNFR2 IR. We immunohistochemically characterized neuronal, glial and leukocyte reactions in 14 cases of human post-mortem ischemic stroke, using 21 specimens of infarcts aged 1 to > 8 days. At all timepoints, IL-1α and IL-1β IR was located to neurons and glia in I/PI and NAT, and to macrophages in I/PI. IL-1Ra IR was primarily located to glia in I/PI and NAT, and to macrophages in I/PI. TNF IR was located to neurons in I/PI and NAT ≤2 days after stroke onset, to glia in I/PI and NAT with increased intensity in ≥8-day-old infarcts, and to macrophages in I/PI at all timepoints. TNFR1 IR was located to neurons and glia in I/PI and NAT and to macrophages in I/PI, whereas TNFR2 IR was observed only in glia in I/PI and NAT, and in macrophages in I/PI, being absent in neurons.
The distribution of IL-1α, IL-1β, IL-1Ra, and TNF in parallel sections suggest that these cytokines are expressed by subsets of cells in I/IP and NAT and that TNFR2 is expressed in areas with increased GFAP IR. Supporting this, immunofluorescent double labeling demonstrated that TNFR2 IR co-localized to GFAP+ astrocytes and TNFR1 IR to NF+ neurons. TNF IR was found in NeuN+ neurons and CD45+ cells. Furthermore, IL-1α IR co-localized to Iba1+ and CD68+ cells, IL-1β IR to Iba1+ cells and neurons, and IL-1Ra IR to CD45+ cells. Finally, plasma TNFR1 and TNFR2 levels increased significantly in the acute phase after symptom onset in ischemic stroke patients compared to healthy controls whereas TNF, IL-1α, IL-1β, and IL-1Ra did not change.
To our knowledge, no other papers besides those published by our laboratory [12, 31] have described the cellular localization of IL-1α, IL-1β, or IL-1Ra in the human ischemic brain. From animal studies we know that IL-1α is primarily expressed by CD11b+ microglia and CD41+ platelets following focal cerebral ischemia [12, 16]. IL-1β is expressed primarily by CD11b+ microglia and macrophages in the acute phase [12, 15] and by astrocytes at more chronic phases [68]. IL-1Ra is preferentially produced by resident microglia [12]. In the present study, we also observed IL-1α, IL-1β, and IL-1Ra IR in glial cells and infiltrating macrophages, however, but we observed IL-1α and IL-1β IR in neurons as well.
Few studies have investigated cellular TNF expression in human post-mortem ischemic brain tissue. Sairanen et al. [51] found TNF to be expressed in neurons 1–6 days post-stroke, peaking at days 2–3, and gone by day 6. Microglial TNF expression was apparent in the acute phase, astrocytic TNF expression dominated later (17–18 days), and leukocytes expressed TNF around day 3. In contrast, two other studies [17, 61] found the most intense TNF IR in microglia and concluded that these cells were the major source. Also in animal models, TNF is primarily expressed by microglia and infiltrating macrophages, although neurons have been reported to express TNF (reviewed in [28]).
Several studies have investigated blood IL-1β levels in ischemic stroke. While some report increased blood IL-1β [55, 62, 69] compared to controls, other studies are in line with our findings and report no changes [18, 20, 48, 59] or even reduced [72] blood IL-1β levels in ischemic stroke patients. Most of these studies were performed in the first 72 h after symptom onset, and the discrepancy in results can be explained by the different timings of blood samples and stroke etiologies. In the study by Tuttolomondo et al. [62] and Licata et al. [34], patients with cardioembolic subtype of stroke had significantly higher plasma IL-1β levels than patients with other stroke subtypes, suggesting that plasma IL-1β changes depend on stroke etiology.
In line with our studies, plasma IL-1α did not change after stroke [72], suggesting that peripheral IL-1α production does not contribute to post-stroke neuroinflammation.
Several studies have demonstrated increased IL-1Ra plasma levels in acute stroke patients [2, 48] and some with an association to functional outcome at 6 months [67]. We observed no significant increase in blood IL-1Ra levels. Given the protective effects of IL-1Ra in experimental animal stroke models (reviewed in [31]), rhIL-1Ra has been administered to ischemic stroke patients within 6 h of symptom onset [19]. Treatment with rhIL-1Ra reduced neutrophil and total white cell counts, C-reactive protein, and IL-6 levels, while 3-month clinical outcomes were better than in placebo-treated. Together with findings of a detrimental role of IL-1β in experimental stroke models, the use of rhIL-1Ra, such as anakinra, holds promise as a neuroprotective treatment in ischemic stroke patients.
In line with our studies, serum TNF levels were unchanged in ischemic stroke patients [18, 20, 48, 69]. Despite the lack of healthy controls, Fassbender et al. [20] found no change in serum TNF levels sampled at 4, 6, 8, 10, and 14 h and 1, 3, 5, and 7 days after symptom onset. Some studies, however, reported on increased blood TNF levels sampled within 12 h [34, 62], 24 h [55, 66, 71], or 72 h [72] after symptom onset in ischemic stroke patients compared to healthy controls, though not in patients with lacunar stroke [34, 62]. In one study, serum TNF levels did not increase until 4–10 days after symptom onset compared to controls [26], suggesting that TNF levels only increase in the chronic phase after ischemic stroke. Adding to the discrepancy in the literature on peripheral TNF levels in ischemic stroke patients, increased plasma TNF correlated with infarct volume in some studies [71] but not in others [26, 55]; with stroke severity at admission in some studies [34, 41, 62] but not in others [72]; and with functional outcome in some studies [41, 71], but not in others [26, 66].
TNFR1 and TNFR2 are associated with arterial stiffness [13, 27] that itself is associated with an increased risk of stroke [39, 65]. One study reported that serum TNFR1 levels predict the risk of recurrent vascular events in patients with lacunar stroke [4]. In another study, high plasma levels of TNFR1 and TNFR2 were associated with incident intracerebral hemorrhage and with poor functional outcome [57], just as increased venous TNFR1 levels were associated with poor outcome in subarachnoid hemorrhage [21]. TNF-mediated inflammation could thus be associated with vascular changes preceding intracerebral hemorrhage. Supporting this, the present study found that plasma TNFR1 and TNFR2 levels increased in acute stroke patients compared to healthy controls. Plasma TNFR1, but not TNFR2, levels were also significantly upregulated in a study [18], where plasma TNFR1 was strongly correlated with CT infarct volume at 5–7 days, and mRS and Barthel Index at 3 and 6 months. Our data differ, however, to findings by Fassbender et al. [20], who found no changes in serum TNFR1 and TNFR2 levels over time in acute ischemic stroke patients. The weakness of this study, however, was the lack of healthy controls. Altogether, this could suggest that the body’s response to ischemic stroke is to increase soluble TNFR1 levels capable of binding soluble TNF, which we know exerts detrimental effects in experimental stroke [70].
In our preclinical stroke model, we have previously shown that IL-1β and TNF are produced by largely segregated populations of microglia and macrophages after experimental stroke in mice [11], providing evidence of the functional heterogeneity among microglia and macrophages post-stroke. This, along with similar findings in the present study, may inform the design of successful anti-inflammatory therapies in stroke. Multiple laboratories have examined IL-1Ra as a therapy in preclinical stroke models in mice (reviewed in [31]). A meta-analysis of the efficacy of IL-1Ra in rodent models of stroke reported on an overall decrease in infarct volumes and an improvement in functional outcomes [42]. In addition, we recently demonstrated, that cell-based delivery of IL-1Ra was neuroprotective and improved functional outcome in mice subjected to ischemia [12] and a multicenter preclinical study examining the short- and long-term effects of subcutaneous IL-1Ra therapy demonstrated consistent decreases in infarct volumes and improvements in neurological deficits for up to 28 days post-treatment [40]. These preclinical data, along with clinical studies demonstrating beneficial effects of IL-1Ra treatment in ischemic stroke patients [19, 54], support IL-1Ra as a candidate therapy for ischemic stroke.
Also, TNF inhibitors hold promise as candidate therapies in ischemic stroke. Peri-spinal administration of the non-selective TNF antagonist etanercept provided benefits for post-stroke pain and improved cognitive dysfunction in chronic stroke patients [50, 60], demonstrating the potential of anti-TNF inhibitors as anti-inflammatory treatment in ischemic stroke. A major barrier to the efficacy of etanercept as a TNF inhibitor in ischemic stroke, however, is its poor blood-brain barrier (BBB) permeability due to its large size. Furthermore, because etanercept targets both solTNF and mTNF, this can predispose patients to an increased risk of systemic infections and demyelinating and cardiovascular diseases [53]. Therefore, formulations of TNF inhibitors with better BBB penetrance and selective inhibition of solTNF have been designed and tested in preclinical stroke models (reviewed in [31]). The use of intravenous injection of cTfRMAb-TNFR, a drug designed to ferry the TNFR2 fusion protein [56] across the BBB using the transferrin receptor (TfR), resulted in reduced infarct volumes and reduced neural deficits 1 and 7 days post-stroke, whereas etanercept had no effect [56]. We previously examined the effect of a selective solTNF inhibitor, XPro1595, versus etanercept in our preclinical stroke model [9, 70]. When XPro1595 and etanercept were given systemically, both improved functional outcome and decreased the acute phase response in the liver, indicating an important effect of tmTNF on the peripheral immune response; however, neither reduced infarct volumes [9]. When XPro1595 or etanercept were delivered directly to the infarcted area for three consecutive days, only XPro1595 significantly reduced infarct volumes, modified microglial activation, and the post-stroke inflammatory response [70], suggesting that targeting solTNF alone may be efficient for the treatment of post-stroke inflammation. Even though more preclinical studies are needed, the use of selective solTNF inhibitors, sparing mTNF, continue to hold promise in stroke therapy.
A limitation of our study is the lack of estimation of blood IL-1 receptor type I and II levels. Plasma levels of IL-1 receptor type II were previously unchanged in ischemic stroke patients within 12 h and up to 1 year after the stroke [18]. Immunohistochemical characterization of the cellular expression of the IL-1 receptors in the brain would have strengthened this paper. Another limitation is the low number of acute ischemic stroke patients and controls included in plasma analyses of peripheral cytokine and receptor levels.
The strengths of the present study are the use of parallel tissue sections from the same post-mortem ischemic stroke brains and the range of controls for specific staining using absorption controls, serum and isotype controls, combined with control tissue from various organs. The thorough scoring of the staining intensity by two independent raters strengthens the conclusions of this paper, as does the inclusion of double immunofluorescent staining (a very tricky protocol to perform on post-mortem human tissue) to verify cellular localization.
Conclusions
In conclusion, the present study determines the cellular sources of IL-1α, IL-1β, and IL-1Ra in human post-mortem ischemic stroke tissue, and the cellular sources of TNF, TNFR1, and TNFR2 in parallel tissue sections. The results support findings from animal studies [11, 12] that IL-1α, IL-1β, IL-1Ra, and TNF are produced by subsets of primarily microglia and leukocytes in ischemic stroke tissue. Finally, our findings of increased brain cytokines and plasma TNFR1 and TNFR2 support hypotheses that targeting post-stroke inflammation is a promising add-on therapy in ischemic stroke patients.
Supplementary information
Additional file 1: Supplementary Figs. 1–9 and Supplementary Table 1.
Acknowledgements
The excellent work of technicians Helle Wohlleben, Ulla Damsgaard Munk, and Karen Rich is greatly acknowledged. We thank senior consultants Charlotte Madsen, Alex Alban Christensen, Søren Bak, and Anne-Mette Homborg for assistance with the recruitment of ischemic stroke patients and research nurse Annie Gam-Petersen for assistance with recruitment of rotator cuff patients. Claire Gudex is acknowledged for proofreading.
Abbreviations
- BBB
Blood-brain barrier
- BMI
Body mass index
- DAPI
4′,6′-diamidine-2′-phenylindole dihydrochloride
- GFAP
Glial fibrillary acidic protein
- HE
Hematoxylin and eosin
- Iba1
Ionized calcium-binding adaptor molecule 1
- IL-1
Interleukin-1
- IL-1Ra
Interleukin-1 receptor antagonist
- IR
Immunoreactivity
- I/PI
Infarct/peri-infarct
- IQR
Interquartile range
- mRS
Modified Rankin Scale
- NAT
Normal-appearing tissue
- NIHSS
National Institute of Stroke Severity Scale
- NF
Neurofilament
- OUH
Odense University Hospital
- PBS
Phosphate-buffered saline
- PBS-T
Phosphate-buffered saline containing 0.25% Triton
- rh
Recombinant human
- SD
Standard deviation
- SSS
Scandinavian Stroke Scale
- TBS
Tris-buffered saline
- TNF
Tumor necrosis factor
- TNFR1
Tumor necrosis factor receptor 1
- TNFR2
Tumor necrosis factor receptor 2
Authors’ contributions
KØ identified eligible post-mortem brain tissue for histological evaluation. HDS and BWK made the initial neuropathological evaluation of HE-stained post-mortem brain tissue. BHC and MW evaluated the staining intensity in post-mortem brain tissue and helped draft the manuscript. SSH recruited and collected patient data and blood samples from ischemic stroke patients and control relatives with the assistance of HHN. LHF recruited and collected patient data and blood samples from rotator cuff patients. BF and KLL conceived the study. MW, SSH, BHC, and KLL analyzed data and interpreted results. KLL performed statistical analysis and wrote the manuscript. All authors read and approved the final manuscript.
Funding
This work was supported by research grants from the Danish MRC (BF, 271–05-0737), the Danish Council for Independent Research – Medical Sciences (KLL, DFF-4183-00033), P.A. Messerschmidt og Hustrus Fond (KLL), Overlægerådets Legatudvalg at OUH, (KLL), Aase og Ejnar Danielsens Fond (BHC & KLL), the Lundbeck Foundation, and the Danish Neurological Society (KLL & SSH).
Availability of data and materials
All data are hosted at Open Patient data Explorative Network (OPEN; https://open.rsyd.dk/), and requests to access datasets should be directed to klambertsen@health.sdu.dk.
Ethics approval and consent to participate
Sampling of patients’ blood and the use of human post-mortem brain tissue were approved by the Regional Committees on Health Research Ethics for Southern Denmark (Journal numbers S-20160152G, S-20160037, and S-20080042) and the projects were reported to the Danish Data Protection Agency. Eligible patients and controls donating blood provided written informed consent prior to study participation according to the Declaration of Helsinki. Patient information was gathered from patient journals after consent had been given.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Bettina H. Clausen, Email: bclausen@health.sdu.dk
Martin Wirenfeldt, Email: martin.nielsen@rsyd.dk.
Sofie S. Høgedal, Email: sofie.sander.hogedal@rsyd.dk
Lars H. Frich, Email: lars.henrik.frich@rsyd.dk
Helle H. Nielsen, Email: helle.hvilsted.nielsen@rsyd.dk
Henrik D. Schrøder, Email: henrik.daa.schroeder@rsyd.dk
Kamilla Østergaard, Email: kamillaoe@gmail.com.
Bente Finsen, Email: bfinsen@health.sdu.dk.
Bjarne W. Kristensen, Email: bwk@rsyd.dk
Kate L. Lambertsen, Email: klambertsen@health.sdu.dk
Supplementary information
Supplementary information accompanies this paper at 10.1186/s40478-020-00957-y.
References
- 1.Ban EM. Interleukin-1 receptors in the brain: characterization by quantitative in situ autoradiography. Immunomethods. 1994;5:31–40. doi: 10.1006/immu.1994.1035. [DOI] [PubMed] [Google Scholar]
- 2.Beamer NB, Coull BM, Clark WM, Hazel JS, Silberger JR. Interleukin-6 and interleukin-1 receptor antagonist in acute stroke. Ann Neurol. 1995;37:800–805. doi: 10.1002/ana.410370614. [DOI] [PubMed] [Google Scholar]
- 3.Bis JC, Heckbert SR, Smith NL, Reiner AP, Rice K, Lumley T, Hindorff LA, Marciante KD, Enquobahrie DA, Monks SA, et al. Variation in inflammation-related genes and risk of incident nonfatal myocardial infarction or ischemic stroke. Atherosclerosis. 2008;198:166–173. doi: 10.1016/j.atherosclerosis.2007.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boehme AK, McClure LA, Zhang Y, Luna JM, Del Brutto OH, Benavente OR, Elkind MS. Inflammatory markers and outcomes after lacunar Stroke: levels of inflammatory markers in treatment of Stroke study. Stroke. 2016;47:659–667. doi: 10.1161/STROKEAHA.115.012166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bokhari FA, Shakoori TA, Butt A, Ghafoor F. TNF-alpha: a risk factor for ischemic stroke. J Ayub Med Coll Abbottabad. 2014;26:111–114. [PubMed] [Google Scholar]
- 6.Brambilla R, Ashbaugh JJ, Magliozzi R, Dellarole A, Karmally S, Szymkowski DE, Bethea JR. Inhibition of soluble tumour necrosis factor is therapeutic in experimental autoimmune encephalomyelitis and promotes axon preservation and remyelination. Brain. 2011;134:2736–2754. doi: 10.1093/brain/awr199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brambilla R, Couch Y, Lambertsen KL. The effect of stroke on immune function. Mol Cell Neurosci. 2013;53:26–33. doi: 10.1016/j.mcn.2012.08.011. [DOI] [PubMed] [Google Scholar]
- 8.Butt JH, Rostrup E, Hansen AS, Lambertsen KL, Kruuse C. Induction of migraine-like headache, but not aura, by cilostazol in patients with migraine with aura. Brain. 2018;141:2943–2951. doi: 10.1093/brain/awy228. [DOI] [PubMed] [Google Scholar]
- 9.Clausen B, Degn M, Martin N, Couch Y, Karimi L, Ormhoj M, Mortensen ML, Gredal H, Gardiner C, Sargent II, et al. Systemically administered anti-TNF therapy ameliorates functional outcomes after focal cerebral ischemia. J Neuroinflammation. 2014;11:203. doi: 10.1186/PREACCEPT-2982253041347736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Clausen BH, Degn M, Sivasaravanaparan M, Fogtmann T, Andersen MG, Trojanowsky MD, Gao H, Hvidsten S, Baun C, Deierborg T, et al. Conditional ablation of myeloid TNF increases lesion volume after experimental stroke in mice, possibly via altered ERK1/2 signaling. Sci Rep. 2016;6:29291. doi: 10.1038/srep29291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Clausen BH, Lambertsen KL, Babcock AA, Holm TH, Dagnaes-Hansen F, Finsen B. Interleukin-1beta and tumor necrosis factor-alpha are expressed by different subsets of microglia and macrophages after ischemic stroke in mice. J Neuroinflammation. 2008;5:46. doi: 10.1186/1742-2094-5-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Clausen BH, Lambertsen KL, Dagnaes-Hansen F, Babcock AA, von Linstow CU, Meldgaard M, Kristensen BW, Deierborg T, Finsen B (2016) Cell therapy centered on IL-1Ra is neuroprotective in experimental stroke. Acta Neuropathol. 10.1007/s00401-016-1541-5 [DOI] [PMC free article] [PubMed]
- 13.Cortez-Cooper M, Meaders E, Stallings J, Haddow S, Kraj B, Sloan G, McCully KK, Cannon JG. Soluble TNF and IL-6 receptors: indicators of vascular health in women without cardiovascular disease. Vasc Med. 2013;18:282–289. doi: 10.1177/1358863X13508336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cui G, Wang H, Li R, Zhang L, Li Z, Wang Y, Hui R, Ding H, Wang DW. Polymorphism of tumor necrosis factor alpha (TNF-alpha) gene promoter, circulating TNF-alpha level, and cardiovascular risk factor for ischemic stroke. J Neuroinflammation. 2012;9:235. doi: 10.1186/1742-2094-9-235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Davies CA, Loddick SA, Toulmond S, Stroemer RP, Hunt J, Rothwell NJ. The progression and topographic distribution of interleukin-1beta expression after permanent middle cerebral artery occlusion in the rat. J Cereb Blood Flow Metab. 1999;19:87–98. doi: 10.1097/00004647-199901000-00010. [DOI] [PubMed] [Google Scholar]
- 16.Denes A, Ferenczi S, Kovacs KJ. Systemic inflammatory challenges compromise survival after experimental stroke via augmenting brain inflammation, blood- brain barrier damage and brain oedema independently of infarct size. J Neuroinflammation. 2011;8:164. doi: 10.1186/1742-2094-8-164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dziewulska D, Mossakowski MJ. Cellular expression of tumor necrosis factor a and its receptors in human ischemic stroke. Clin Neuropathol. 2003;22:35–40. [PubMed] [Google Scholar]
- 18.Emsley HC, Smith CJ, Gavin CM, Georgiou RF, Vail A, Barberan EM, Illingworth K, Scarth S, Wickramasinghe V, Hoadley ME, et al. Clinical outcome following acute ischaemic stroke relates to both activation and autoregulatory inhibition of cytokine production. BMC Neurol. 2007;7:5. doi: 10.1186/1471-2377-7-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Emsley HC, Smith CJ, Georgiou RF, Vail A, Hopkins SJ, Rothwell NJ, Tyrrell PJ, Acute Stroke I. A randomised phase II study of interleukin-1 receptor antagonist in acute stroke patients. J Neurol Neurosurg Psychiatry. 2005;76:1366–1372. doi: 10.1136/jnnp.2004.054882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fassbender K, Rossol S, Kammer T, Daffertshofer M, Wirth S, Dollman M, Hennerici M. Proinflammatory cytokines in serum of patients with acute cerebral ischemia: kinetics of secretion and relation to the extent of brain damage and outcome of disease. J Neurol Sci. 1994;122:135–139. doi: 10.1016/0022-510x(94)90289-5. [DOI] [PubMed] [Google Scholar]
- 21.Fragata I, Bustamante A, Penalba A, Ferreira P, Nunes AP, Canhao P, Montaner J. Venous and arterial TNF-R1 predicts outcome and complications in acute subarachnoid hemorrhage. Neurocrit Care. 2019;31:107–115. doi: 10.1007/s12028-019-00669-9. [DOI] [PubMed] [Google Scholar]
- 22.Group SSS Multicenter trial of hemodilution in ischemic stroke--background and study protocol. Scandinavian Stroke Study Group. Stroke. 1985;16:885–890. doi: 10.1161/01.STR.16.5.885. [DOI] [PubMed] [Google Scholar]
- 23.Hannum CH, Wilcox CJ, Arend WP, Joslin FG, Dripps DJ, Heimdal PL, Armes LG, Sommer A, Eisenberg SP, Thompson RC. Interleukin-1 receptor antagonist activity of a human interleukin-1 inhibitor. Nature. 1990;343:336–340. doi: 10.1038/343336a0. [DOI] [PubMed] [Google Scholar]
- 24.Hermansen SK, Christensen KG, Jensen SS, Kristensen BW. Inconsistent immunohistochemical expression patterns of four different CD133 antibody clones in glioblastoma. J Histochem Cytochem. 2011;59:391–407. doi: 10.1369/0022155411400867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huang L, Wu ZB, Zhuge Q, Zheng W, Shao B, Wang B, Sun F, Jin K. Glial scar formation occurs in the human brain after ischemic stroke. Int J Med Sci. 2014;11:344–348. doi: 10.7150/ijms.8140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Intiso D, Zarrelli MM, Lagioia G, Di Rienzo F, Checchia De Ambrosio C, Simone P, Tonali P, Cioffi RP. Tumor necrosis factor alpha serum levels and inflammatory response in acute ischemic stroke patinets. Neurol Sci. 2003;24:390–396. doi: 10.1007/s10072-003-0194-z. [DOI] [PubMed] [Google Scholar]
- 27.Kim HL, Lee JP, An JN, Kim JH, Lim WH, Seo JB, Chung WY, Oh YK, Kim YS, Lim CS, et al. Soluble tumor necrosis factor receptors and arterial stiffness in patients with coronary atherosclerosis. Am J Hypertens. 2017;30:313–318. doi: 10.1093/ajh/hpw134. [DOI] [PubMed] [Google Scholar]
- 28.Lambertsen KL, Biber K, Finsen B. Inflammatory cytokines in experimental and human stroke. J Cereb Blood Flow Metab. 2012;32:1677–1698. doi: 10.1038/jcbfm.2012.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lambertsen KL, Clausen BH, Babcock AA, Gregersen R, Fenger C, Nielsen HH, Haugaard LS, Wirenfeldt M, Nielsen M, Dagnaes-Hansen F, et al. Microglia protect neurons against ischemia by synthesis of tumor necrosis factor. J Neurosci. 2009;29:1319–1330. doi: 10.1523/JNEUROSCI.5505-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lambertsen KL, Clausen BH, Fenger C, Wulf H, Owens T, Dagnaes-Hansen F, Meldgaard M, Finsen B. Microglia and macrophages express tumor necrosis factor receptor p75 following middle cerebral artery occlusion in mice. Neuroscience. 2007;144:934–949. doi: 10.1016/j.neuroscience.2006.10.046. [DOI] [PubMed] [Google Scholar]
- 31.Lambertsen KL, Finsen B, Clausen BH (2018) Post-stroke inflammation-target or tool for therapy? Acta Neuropathol. 10.1007/s00401-018-1930-z [DOI] [PMC free article] [PubMed]
- 32.Lambertsen KL, Ostergaard K, Clausen BH, Hansen S, Stenvang J, Thorsen SB, Meldgaard M, Kristensen BW, Hansen PB, Sorensen GL, et al. No effect of ablation of surfactant protein-D on acute cerebral infarction in mice. J Neuroinflammation. 2014;11:123. doi: 10.1186/1742-2094-11-123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Leifer D, Kowall NW. Immunohistochemical patterns of selective cellular vulnerability in human cerebral ischemia. J Neurol Sci. 1993;119:217–228. doi: 10.1016/0022-510X(93)90137-N. [DOI] [PubMed] [Google Scholar]
- 34.Licata G, Tuttolomondo A, Di Raimondo D, Corrao S, Di Sciacca R, Pinto A. Immuno-inflammatory activation in acute cardio-embolic strokes in comparison with other subtypes of ischaemic stroke. Thromb Haemost. 2009;101:929–937. doi: 10.1160/TH08-06-0375. [DOI] [PubMed] [Google Scholar]
- 35.Liguz-Lecznar M, Zakrzewska R, Kossut M. Inhibition of Tnf-alpha R1 signaling can rescue functional cortical plasticity impaired in early post-stroke period. Neurobiol Aging. 2015;36:2877–2884. doi: 10.1016/j.neurobiolaging.2015.06.015. [DOI] [PubMed] [Google Scholar]
- 36.Liu X, Nemeth DP, McKim DB, Zhu L, DiSabato DJ, Berdysz O, Gorantla G, Oliver B, Witcher KG, Wang Y, et al. Cell-type-specific interleukin 1 receptor 1 signaling in the brain regulates distinct Neuroimmune activities. Immunity. 2019;50:764–766. doi: 10.1016/j.immuni.2019.02.012. [DOI] [PubMed] [Google Scholar]
- 37.Loddick SA, Rothwell NJ. Neuroprotective effects of human recombinant interleukin-1 receptor antagonist in focal cerebral ischaemia in the rat. J Cereb Blood Flow Metab. 1996;16:932–940. doi: 10.1097/00004647-199609000-00017. [DOI] [PubMed] [Google Scholar]
- 38.Madsen PM, Clausen BH, Degn M, Thyssen S, Kristensen LK, Svensson M, Ditzel N, Finsen B, Deierborg T, Brambilla R, et al. Genetic ablation of soluble tumor necrosis factor with preservation of membrane tumor necrosis factor is associated with neuroprotection after focal cerebral ischemia. J Cereb Blood Flow Metab. 2016;36:1553–1569. doi: 10.1177/0271678X15610339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mattace-Raso FU, van der Cammen TJ, Hofman A, van Popele NM, Bos ML, Schalekamp MA, Asmar R, Reneman RS, Hoeks AP, Breteler MM, et al. Arterial stiffness and risk of coronary heart disease and stroke: the Rotterdam study. Circulation. 2006;113:657–663. doi: 10.1161/CIRCULATIONAHA.105.555235. [DOI] [PubMed] [Google Scholar]
- 40.Maysami S, Wong R, Pradillo JM, Denes A, Dhungana H, Malm T, Koistinaho J, Orset C, Rahman M, Rubio M, et al. A cross-laboratory preclinical study on the effectiveness of interleukin-1 receptor antagonist in stroke. J Cereb Blood Flow Metab. 2016;36:596–605. doi: 10.1177/0271678X15606714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mazzotta G, Sarchielli P, Caso V, Paciaroni M, Floridi A, Floridi A, Gallai V. Different cytokine levels in thrombolysis patients as predictors for clinical outcome. Eur J Neurol. 2004;11:377–381. doi: 10.1111/j.1468-1331.2004.00798.x. [DOI] [PubMed] [Google Scholar]
- 42.McCann SK, Cramond F, Macleod MR, Sena ES. Systematic review and meta-analysis of the efficacy of Interleukin-1 receptor antagonist in animal models of Stroke: an update. Transl Stroke Res. 2016;7:395–406. doi: 10.1007/s12975-016-0489-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Morris CS, Esiri MM. The expression of cytokines and their receptors in normal and mildly reactive human brain. J Neuroimmunol. 1998;92:85–97. doi: 10.1016/S0165-5728(98)00181-7. [DOI] [PubMed] [Google Scholar]
- 44.Mulcahy NJ, Ross J, Rothwell NJ, Loddick SA. Delayed administration of interleukin-1 receptor antagonist protects against transient cerebral ischaemia in the rat. Br J Pharmacol. 2003;140:471–476. doi: 10.1038/sj.bjp.0705462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nguyen TV, Frye JB, Zbesko JC, Stepanovic K, Hayes M, Urzua A, Serrano G, Beach TG, Doyle KP. Multiplex immunoassay characterization and species comparison of inflammation in acute and non-acute ischemic infarcts in human and mouse brain tissue. Acta Neuropathol Commun. 2016;4:100. doi: 10.1186/s40478-016-0371-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nielsen HH, Soares CB, Høgedahl SS, Madsen JS, Hansen RB, Christensen AA, Madsen C, Clausen BH, Frich LH, Degn M et al (2020) Acute neurofilament light chain plasma levels correlate with stroke severity and clinical outcome in ischemic stroke patients. Frontiers in Neurology In press. 10.3389/fneur.2020.00448 [DOI] [PMC free article] [PubMed]
- 47.Olleros ML, Vesin D, Lambou AF, Janssens JP, Ryffel B, Rose S, Fremond C, Quesniaux VF, Szymkowski DE, Garcia I. Dominant-negative tumor necrosis factor protects from Mycobacterium bovis Bacillus Calmette Guerin (BCG) and endotoxin-induced liver injury without compromising host immunity to BCG and Mycobacterium tuberculosis. J Infect Dis. 2009;199:1053–1063. doi: 10.1086/597204. [DOI] [PubMed] [Google Scholar]
- 48.Ormstad H, Aass HC, Lund-Sorensen N, Amthor KF, Sandvik L. Serum levels of cytokines and C-reactive protein in acute ischemic stroke patients, and their relationship to stroke lateralization, type, and infarct volume. J Neurol. 2011;258:677–685. doi: 10.1007/s00415-011-6006-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pinteaux E, Rothwell NJ, Boutin H. Neuroprotective actions of endogenous interleukin-1 receptor antagonist (IL-1ra) are mediated by glia. Glia. 2006;53:551–556. doi: 10.1002/glia.20308. [DOI] [PubMed] [Google Scholar]
- 50.Ralph SJ, Weissenberger A, Bonev V, King LD, Bonham MD, Ferguson S, Smith AD, Goodman-Jones AA, Espinet AJ. Phase I/II parallel double-blind randomized controlled clinical trial of perispinal etanercept for chronic stroke: improved mobility and pain alleviation. Expert Opin Investig Drugs. 2020;29:311–326. doi: 10.1080/13543784.2020.1709822. [DOI] [PubMed] [Google Scholar]
- 51.Sairanen T, Carpen O, Karjalainen-Lindsberg ML, Paetau A, Turpeinen U, Kaste M, Lindsberg PJ. Evolution of cerebral tumor necrosis factor-alpha production during human ischemic stroke. Stroke. 2001;32:1750–1758. doi: 10.1161/01.STR.32.8.1750. [DOI] [PubMed] [Google Scholar]
- 52.Sairanen TR, Lindsberg PJ, Brenner M, Carpen O, Siren A. Differential cellular expression of tumor necrosis factor-alpha and type I tumor necrosis factor receptor after transient global forebrain ischemia. J Neurol Sci. 2001;186:87–99. doi: 10.1016/S0022-510X(01)00508-1. [DOI] [PubMed] [Google Scholar]
- 53.Scheinfeld N. A comprehensive review and evaluation of the side effects of the tumor necrosis factor alpha blockers etanercept, infliximab and adalimumab. J Dermatolog Treat. 2004;15:280–294. doi: 10.1080/09546630410017275. [DOI] [PubMed] [Google Scholar]
- 54.Smith CJ, Hulme S, Vail A, Heal C, Parry-Jones AR, Scarth S, Hopkins K, Hoadley M, Allan SM, Rothwell NJ, et al. SCIL-STROKE (subcutaneous Interleukin-1 receptor antagonist in ischemic Stroke): a randomized controlled phase 2 trial. Stroke. 2018;49:1210–1216. doi: 10.1161/STROKEAHA.118.020750. [DOI] [PubMed] [Google Scholar]
- 55.Sotgiu S, Zanda B, Marchetti B, Fois ML, Arru G, Pes GM, Salaris FS, Arru A, Pirisi A, Rosati G. Inflammatory biomarkers in blood of patients with acute brain ischemia. Eur J Neurol. 2006;13:505–513. doi: 10.1111/j.1468-1331.2006.01280.x. [DOI] [PubMed] [Google Scholar]
- 56.Sumbria RK, Boado RJ, Pardridge WM. Brain protection from stroke with intravenous TNFalpha decoy receptor-Trojan horse fusion protein. J Cereb Blood Flow Metab. 2012;32:1933–1938. doi: 10.1038/jcbfm.2012.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Svensson EH, Soderholm M, Abul-Kasim K, Engstrom G. Tumor necrosis factor receptor 1 and 2 are associated with risk of Intracerebral hemorrhage. Stroke. 2017;48:2710–2715. doi: 10.1161/STROKEAHA.117.017849. [DOI] [PubMed] [Google Scholar]
- 58.Taoufik E, Tseveleki V, Chu SY, Tselios T, Karin M, Lassmann H, Szymkowski DE, Probert L. Transmembrane tumour necrosis factor is neuroprotective and regulates experimental autoimmune encephalomyelitis via neuronal nuclear factor-kappaB. Brain. 2011;134:2722–2735. doi: 10.1093/brain/awr203. [DOI] [PubMed] [Google Scholar]
- 59.Tarkowski E, Rosengren L, Blomstrand C, Wikkelso C, Jensen C, Ekholm S, Tarkowski A. Early intrathecal production of interleukin-6 predicts the size of brain lesion in stroke. Stroke. 1995;26:1393–1398. doi: 10.1161/01.STR.26.8.1393. [DOI] [PubMed] [Google Scholar]
- 60.Tobinick E, Kim NM, Reyzin G, Rodriguez-Romanacce H, DePuy V. Selective TNF inhibition for chronic stroke and traumatic brain injury: an observational study involving 629 consecutive patients treated with perispinal etanercept. CNS drugs. 2012;26:1051–1070. doi: 10.1007/s40263-012-0013-2. [DOI] [PubMed] [Google Scholar]
- 61.Tomimoto H, Akiguchi I, Wakita H, Kinoshita A, Ikemoto A, Nakamura S, Kimura J. Glial expression of cytokines in the brains of cerebrovascular disease patients. Acta Neuropathol (Berl) 1996;92:281–287. doi: 10.1007/s004010050519. [DOI] [PubMed] [Google Scholar]
- 62.Tuttolomondo A, Di Sciacca R, Di Raimondo D, Serio A, D'Aguanno G, La Placa S, Pecoraro R, Arnao V, Marino L, Monaco S, et al. Plasma levels of inflammatory and thrombotic/fibrinolytic markers in acute ischemic strokes: relationship with TOAST subtype, outcome and infarct site. J Neuroimmunol. 2009;215:84–89. doi: 10.1016/j.jneuroim.2009.06.019. [DOI] [PubMed] [Google Scholar]
- 63.Um JY, An NH, Kim HM. TNF-alpha and TNF-beta gene polymorphisms in cerebral infarction. J Mol Neurosci. 2003;21:167–171. doi: 10.1385/JMN:21:2:167. [DOI] [PubMed] [Google Scholar]
- 64.Um JY, Moon KS, Lee KM, Yun JM, Cho KH, Moon BS, Kim HM. Association of interleukin-1 alpha gene polymorphism with cerebral infarction. Brain Res Mol Brain Res. 2003;115:50–54. doi: 10.1016/S0169-328X(03)00179-7. [DOI] [PubMed] [Google Scholar]
- 65.van Sloten TT, Sedaghat S, Laurent S, London GM, Pannier B, Ikram MA, Kavousi M, Mattace-Raso F, Franco OH, Boutouyrie P, et al. Carotid stiffness is associated with incident stroke: a systematic review and individual participant data meta-analysis. J Am Coll Cardiol. 2015;66:2116–2125. doi: 10.1016/j.jacc.2015.08.888. [DOI] [PubMed] [Google Scholar]
- 66.Vila N, Castillo J, Davalos A, Chamorro A. Proinflammatory cytokines and early neurological worsening in ischemic stroke. Stroke. 2000;31:2325–2329. doi: 10.1161/01.STR.31.10.2325. [DOI] [PubMed] [Google Scholar]
- 67.Vila N, Filella X, Deulofeu R, Ascaso C, Abellana R, Chamorro A. Cytokine-induced inflammation and long-term stroke functional outcome. J Neurol Sci. 1999;162:185–188. doi: 10.1016/s0022-510x(98)00319-0. [DOI] [PubMed] [Google Scholar]
- 68.Wang X, Li X, Currie RW, Willette RN, Barone FC, Feuerstein GZ. Application of real-time polymerase chain reaction to quantitate induced expression of interleukin-1beta mRNA in ischemic brain tolerance. J Neurosci Res. 2000;59:238–246. doi: 10.1002/(SICI)1097-4547(20000115)59:2<238::AID-JNR10>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 69.Wytrykowska A, Prosba-Mackiewicz M, Nyka WM. IL-1beta, TNF-alpha, and IL-6 levels in gingival fluid and serum of patients with ischemic stroke. J Oral Sci. 2016;58:509–513. doi: 10.2334/josnusd.16-0278. [DOI] [PubMed] [Google Scholar]
- 70.Yli-Karjanmaa M, Clausen BH, Degn M, Novrup HG, Ellman DG, Toft-Jensen P, Szymkowski DE, Stensballe A, Meyer M, Brambilla R, et al. Topical administration of a soluble TNF inhibitor reduces infarct volume after focal cerebral ischemia in mice. Front Neurosci. 2019;13:781. doi: 10.3389/fnins.2019.00781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zaremba J, Losy J. Early TNF-alpha levels correlate with ischaemic stroke severity. Acta Neurol Scand. 2001;104:288–295. doi: 10.1034/j.1600-0404.2001.00053.x. [DOI] [PubMed] [Google Scholar]
- 72.Zeng L, Wang Y, Liu J, Wang L, Weng S, Chen K, Domino EF, Yang GY. Pro-inflammatory cytokine network in peripheral inflammation response to cerebral ischemia. Neurosci Lett. 2013;548:4–9. doi: 10.1016/j.neulet.2013.04.037. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Supplementary Figs. 1–9 and Supplementary Table 1.
Data Availability Statement
All data are hosted at Open Patient data Explorative Network (OPEN; https://open.rsyd.dk/), and requests to access datasets should be directed to klambertsen@health.sdu.dk.