

Three salts containing the 4-(4-fluorophenyl)piperazin-1-ium cation have been prepared and structurally characterized.
Keywords: piperazines, piperazinium salts, crystal structure, molecular conformation, hydrogen bonding, supramolecular assembly
Abstract
Three salts containing the 4-(4-fluorophenyl)piperazin-1-ium cation have been prepared and structurally characterized. In 4-(4-fluorophenyl)piperazin-1-ium 2-hydroxy-3,5-dinitrobenzoate, C10H14FN2 +·C7H3N2O7 −, (I), the anion contains an intramolecular O—H⋯O hydrogen bond, and it has a structure similar to that of the picrate ion. The cations and anions are linked into [001] chains of rings by a combination of two three-centre N—H⋯(O)2 hydrogen bonds. The anion in 4-(4-fluorophenyl)piperazin-1-ium hydrogen oxalate, C10H14FN2 +·C2HO4 −, (II), is planar, and the cations and anions are linked into (100) sheets by multiple hydrogen bonds including two-centre N—H⋯O, three-centre N—H⋯(O)2, O—H⋯O, C—H⋯O and C—H⋯π(arene) types. In 4-(4-fluorophenyl)piperazin-1-ium hydrogen (2R,3R)-tartrate monohydrate, C10H14FN2 +·C4H5O6 −·H2O, (III), the anion exhibits an approximate non-crystallographic twofold rotation symmetry with antiperiplanar carboxyl groups. A combination of eight hydrogen bonds, encompassing two- and three-centre N—H⋯O systems, O—H⋯O and C—H⋯π(arene) types, link the independent components into a three-dimensional framework. Comparisons are made with some related structures.
Chemical context
N-(4-fluorophenyl)piperazine (4-FPP) is a major metabolite (Keane et al., 1982 ▸; Sanjuan et al., 1983 ▸) of the sedative and hypnotic drug niaprazine (N-{4-[4-(4-fluorophenyl)piperazin-1-yl]butan-2-yl}pyridine-3-carboxamide), used in the treatment of autistic disorders (Rossi et al., 1999 ▸). 4-FPP itself has mildly psychedelic and euphorigenic properties and, in this respect, it exhibits effects similar to those of the related compound N-(4-methoxyphenyl)piperazine (MeOPP), also used as a recreational drug (Nagai et al., 2007 ▸).
We have recently reported the structure of MeOPP and those of a number of salts derived from it (Kiran Kumar et al., 2019 ▸, 2020 ▸). With the similarities of action between MeOPP and 4-FPP in mind, we have now prepared and structurally characterized a selection of salts derived from 4-FPP, namely 4-(4-fluorophenyl)piperazin-1-ium 2-hydroxy-3,5-dinitrobenzoate (I), 4-(4-fluorophenyl)piperazin-1-ium hydrogenoxalate (II) and 4-(4-fluorophenyl)piperazin-1-ium (2R,3R)-hydrogentartrate, which crystallizes from ethyl acetate as a monohydrate (III).
Structural commentary
Compounds (I)–(III) are all 1:1 salts (Figs. 1 ▸–3 ▸ ▸) in which a single proton has been transferred from the diprotic acid component to the 4-(4-fluorophenyl)piperazine component: of these, (I) and (II) both crystallize in solvent-free form, but (III) crystallizes as a monohydrate. Since a single enantiomer of tartaric acid, the (2R,3R) form, was used in the synthesis of (III), which occurred under very mild conditions unlikely to induce any stereochemical changes, only a single enantiomer is present in the product, which therefore crystallizes in a Sohncke space group containing neither inversion nor reflection (mirror or glide) operations, here P212121.
Figure 1.

The independent components of compound (I) showing the atom-labelling scheme and the hydrogen bonds (drawn as dashed lines) within the asymmetric unit. Displacement ellipsoids are drawn at the 30% probability level.
Figure 2.

The independent components of compound (II) showing the atom-labelling scheme and the hydrogen bonds (drawn as dashed lines) within the asymmetric unit. Displacement ellipsoids are drawn at the 30% probability level.
Figure 3.

The independent components of compound (III) showing the atom-labelling scheme and the hydrogen bonds (drawn as dashed lines) within the asymmetric unit. Displacement ellipsoids are drawn at the 30% probability level.
In each of (I)–(III), the piperazine ring adopts an almost perfect chair conformation, with the 4-fluorophenyl substituent occupying an equatorial site. The value of the ring-puckering angle θ (Cremer & Pople, 1975 ▸), calculated for the atom sequence (N1,C2,C3,N4,C5,C6), ranges from to 2.0 (4)° in (III) to 4.85 (12)° in (II), very close to the ideal value of zero for a perfect chair form (Boeyens, 1978 ▸).
In the anions in each of compounds (I)–(III), the location of the remaining acidic H atom was initially deduced from difference-Fourier maps, and then confirmed by refinement of the atomic coordinates, reinforced by inspection of the final difference-Fourier map and of the relevant C—O bond lengths, where the single and double bonds have distances entirely typical of their types (Allen et al., 1987 ▸).
In the anion of compound (I) (Fig. 1 ▸), it is the phenolic proton that has been transferred rather than the carboxyl proton; this was confirmed as described above. The other bond lengths in this anion show some interesting features. Firstly, the distance C32—O33, 1.2719 (18) Å, is much closer to the values typically found in cyclohexanones (mean value, 1.211 Å) than to those found in phenols (mean value 1.362 Å); secondly, the bond lengths C31—C32 and C32—C33, 1.441 (2) and 1.4318 (19) Å, respectively, are much longer than the other C—C distances in this ring, which lie in the range from 1.368 (2) to 1.388 (2) Å. The bond lengths in the anion, taken together, thus indicate extensive delocalization of the negative charge away from atom O33 and onto the aromatic ring atoms C31,C33,C34,C35,C36 (cf. Scheme), as has been observed in picrate (2,4,6-trinitrophenolate) anions (Sagar et al., 2017 ▸; Shaibah et al., 2017a ▸,b ▸). However, this anion is not completely planar: the substituents at atoms C31, C33 and C35 make dihedral angles with the plane of the ring of 7.62 (16), 9.31 (12), and 10.9 (2)°, respectively.
By contrast, the anion in compound (II) (Fig. 2 ▸) is planar: the r.m.s. deviation from the mean plane through the non-H atoms is only 0.014 Å, with a maximum individual deviation from this plane of 0.0186 (6) Å for atom O34. In the anion of (III), the carboxyl and carboxylate groups are antiperiplanar, as shown by the value of −178.81 (10)° for the torsional angle C31—C32—C33—C34, while the disposition of the two hydroxyl groups is indicated by the value of −66.5 (3)° for the torsional angle O33—C32—C33—O34. Together with the torsional angles O31—C32—C33—C34 and O36—C34—C33—C32, 64.7 (4)° and 59.5 (3)°, respectively, the torsional angles overall indicate that the non-H atoms in this anion exhibit approximate, although non-crystallographic, two-fold rotation symmetry.
Supramolecular features
Within the selected asymmetric unit for compound (I) (Fig. 1 ▸), the anion contains an intramolecular O—H⋯O hydrogen bond (Table 1 ▸), generating an S(6) motif (Etter, 1990 ▸; Etter et al., 1990 ▸; Bernstein et al., 1995 ▸), and the cation and anion are linked by a three-centre N—H⋯(O)2 system to form an  (6) motif. Ion pairs of this type, which are related by the c glide plane at y = 0.25, are linked by a second, rather asymmetric, three-centre system via an (4) motif to form a chain of rings running parallel to [001] (Fig. 4 ▸). There is also a short C—H⋯O contact (Table 1 ▸), which lies within the chain of rings: however, the small C—H⋯O angle indicates that the interaction energy is likely to be very small (Wood et al., 2009 ▸), so that this is probably best regarded as an adventitious contact of little structural significance.
(6) motif. Ion pairs of this type, which are related by the c glide plane at y = 0.25, are linked by a second, rather asymmetric, three-centre system via an (4) motif to form a chain of rings running parallel to [001] (Fig. 4 ▸). There is also a short C—H⋯O contact (Table 1 ▸), which lies within the chain of rings: however, the small C—H⋯O angle indicates that the interaction energy is likely to be very small (Wood et al., 2009 ▸), so that this is probably best regarded as an adventitious contact of little structural significance.
Table 1. Hydrogen-bond geometry (Å, °) for (I) .
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| N1—H11⋯O33 | 0.90 (2) | 2.014 (19) | 2.7968 (18) | 144.8 (15) |
| N1—H11⋯O34 | 0.90 (2) | 2.352 (19) | 3.049 (2) | 134.4 (14) |
| N1—H12⋯O31i | 0.912 (19) | 2.075 (19) | 2.959 (2) | 163.0 (17) |
| N1—H12⋯O32i | 0.912 (19) | 2.487 (18) | 3.1576 (19) | 130.7 (15) |
| O32—H32⋯O33 | 0.97 (3) | 1.55 (3) | 2.4676 (17) | 157 (3) |
| C2—H2B⋯O35ii | 0.97 | 2.51 | 3.313 (2) | 140 |
Symmetry codes: (i)  ; (ii)
; (ii)  .
.
Figure 4.

Part of the crystal structure of compound (I) showing the formation of a hydrogen-bonded chain of rings running parallel to [001]. Hydrogen bonds are drawn as dashed lines and, for the sake of clarity, the H atoms bonded to C atoms have been omitted.
The component ions in compound (II) (Fig. 2 ▸) are linked by a single N—H⋯O hydrogen bond (Table 2 ▸). The ion pairs, which are related by a 21 screw axis along (0.5, y, 0.25), are linked by a combination of an asymmetric three-centre N—H⋯(O)2 hydrogen bond and a two-centre O—H⋯O hydrogen bond (Table 2 ▸) to form a complex chain of rings running parallel to the [010] direction (Fig. 5 ▸). This chain is reinforced by two C—H⋯O hydrogen bonds, involving methylene atoms C2 and C6 as the donors. However, the combination of the C—H⋯O hydrogen bond having methylene atom C5 as the donor and the C—H⋯π(arene) hydrogen bond having atom C2 as the donor links ion pairs, which are related by the c glide plane at y = 0.75, to form a second chain of rings, this time running parallel to the [001] direction (Fig. 6 ▸). The combination of chains along [010] and [001] generates a complex sheet lying parallel to (100). There is a fairly short O⋯C contact between inversion-related anions, with a distance O31⋯C32i [symmetry code: (i) 1 − x, 1 − y, 2 − z] of 3.0108 (14) Å, but it is unclear whether this has any structural significance.
Table 2. Hydrogen-bond geometry (Å, °) for (II) .
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| N1—H11⋯O32 | 0.918 (16) | 1.896 (16) | 2.7769 (14) | 160.2 (15) |
| N1—H12⋯O31i | 0.920 (16) | 1.902 (17) | 2.7507 (14) | 152.6 (15) |
| N1—H12⋯O34i | 0.920 (16) | 2.354 (16) | 2.9588 (14) | 123.1 (13) |
| O34—H34⋯O32ii | 0.908 (17) | 1.712 (17) | 2.6102 (12) | 170.0 (17) |
| C2—H2A⋯O33iii | 0.97 | 2.54 | 3.4454 (15) | 155 |
| C5—H5A⋯O32iv | 0.97 | 2.45 | 3.3849 (15) | 163 |
| C6—H6B⋯O31v | 0.97 | 2.50 | 3.4259 (15) | 159 |
| C2—H2B⋯Cg1vi | 0.97 | 2.65 | 3.6124 (14) | 170 |
Symmetry codes: (i)  ; (ii)
; (ii)  ; (iii)
; (iii)  ; (iv)
; (iv)  ; (v)
; (v)  ; (vi)
; (vi)  .
.
Figure 5.

Part of the crystal structure of compound (II) showing the formation of a hydrogen-bonded chain of rings running parallel to the [010] direction. Hydrogen bonds are drawn as dashed lines and, for the sake of clarity, the H atoms bonded to C atoms have been omitted.
Figure 6.

Part of the crystal structure of compound (II) showing the formation of a chain of rings running parallel to the [001] direction and built from C—H⋯O and C—H⋯π(arene) hydrogen bonds. Hydrogen bonds are drawn as dashed lines and, for the sake of clarity, the H atoms bonded to the C atoms not involved in the motif shown have been omitted.
The supramolecular assembly in the monohydrate (III) is more complex than that in either (I) or (II), and it is three-dimensional as opposed to the one- and two-dimensional assembly in (I) and (II), respectively. However, the three-dimensional assembly in (III) can readily be analysed in terms of some simpler sub-structures (Ferguson et al., 1998a
▸,b
▸; Gregson et al., 2000 ▸). Within the asymmetric unit (Fig. 3 ▸), the components are linked by two N—H⋯O hydrogen bonds and one O—H⋯O hydrogen bond (Table 3 ▸), forming a compact aggregate containing an  (11) motif (Fig. 3 ▸). The inter-aggregate hydrogen bonds having atoms O36 and O41 as the donors link aggregates related by translation to form a sheet lying parallel to (001) in the domain 0.5 < z < 1.0 (Fig. 7 ▸). A second sheet of this type, related to the first by the 21 screw axes parallel to [100], lies in the domain 0 < z < 0.5 and adjacent sheets of this type are linked into a bilayer by a combination of N—H⋯O and O—H⋯O hydrogen bonds (Table 3 ▸). Finally, the bilayers are linked into a continuous three-dimensional structure by a single C—H⋯π(arene) hydrogen bond: in combination with the N—H⋯O hydrogen bond linking the ion pairs within the asymmetric unit, this C—H⋯π interaction generates a chain running parallel to the [001] direction (Fig. 8 ▸), thereby linking adjacent bilayers.
(11) motif (Fig. 3 ▸). The inter-aggregate hydrogen bonds having atoms O36 and O41 as the donors link aggregates related by translation to form a sheet lying parallel to (001) in the domain 0.5 < z < 1.0 (Fig. 7 ▸). A second sheet of this type, related to the first by the 21 screw axes parallel to [100], lies in the domain 0 < z < 0.5 and adjacent sheets of this type are linked into a bilayer by a combination of N—H⋯O and O—H⋯O hydrogen bonds (Table 3 ▸). Finally, the bilayers are linked into a continuous three-dimensional structure by a single C—H⋯π(arene) hydrogen bond: in combination with the N—H⋯O hydrogen bond linking the ion pairs within the asymmetric unit, this C—H⋯π interaction generates a chain running parallel to the [001] direction (Fig. 8 ▸), thereby linking adjacent bilayers.
Table 3. Hydrogen-bond geometry (Å, °) for (III) .
Cg1 represents the centroid of the ring (C21–C26).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| N1—H11⋯O36 | 0.87 (4) | 2.31 (4) | 2.929 (4) | 128 (3) |
| N1—H11⋯O35i | 0.87 (4) | 2.17 (4) | 2.855 (4) | 136 (3) |
| N1—H12⋯O41 | 0.92 (4) | 1.83 (4) | 2.740 (5) | 169 (3) |
| O33—H33⋯O34ii | 0.80 (4) | 2.10 (4) | 2.805 (3) | 146 (3) |
| O34—H34⋯O31ii | 0.81 (4) | 2.07 (4) | 2.806 (3) | 151 (4) |
| O36—H36⋯O32iii | 0.95 (4) | 1.53 (4) | 2.470 (3) | 175 (3) |
| O41—H41⋯O31 | 0.96 (5) | 1.82 (5) | 2.771 (4) | 178 (5) |
| O41—H42⋯O33iv | 0.78 (5) | 2.00 (5) | 2.754 (4) | 163 (5) |
| C25—H25⋯Cg1v | 0.93 | 2.86 | 3.649 (5) | 144 |
Symmetry codes: (i) 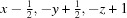 ; (ii)
; (ii)  ; (iii) ; (iv)
; (iii) ; (iv)  ; (v)
; (v)  .
.
Figure 7.

Part of the crystal structure of compound (III) showing the formation of a hydrogen-bonded sheet lying parallel to (001). Hydrogen bonds are drawn as dashed lines and, for the sake of clarity, the H atoms bonded to C atoms have been omitted.
Figure 8.

Part of the crystal structure of compound (III) showing the formation of a hydrogen-bonded chain of cations and anions running parallel to the [001] direction. Hydrogen bonds are drawn as dashed lines and, for the sake of clarity, the water molecules and the H atoms not involved in the motif shown have been omitted.
Related structures
It is of interest briefly to compare the structures reported here with those of some closely related compounds. An obvious comparison is between compound (I), reported here and the analogous salt (IV) derived from MeOPP (Kiran Kumar et al., 2019 ▸). Although (I) and (IV) both crystallize in space-group type P21/c, their unit-cell dimensions are very different, as is the manner of their supramolecular assembly. Thus, in the structure of (IV), a combination of N—H⋯O and C—H⋯O hydrogen bonds links the component ions into a chain of centrosymmetric rings in which rings of  (10) and
(10) and  (16) types alternate, with chains of this type linked by C—H⋯π(arene) hydrogen bonds to form a three-dimensional network, as compared with the one-dimensional assembly in (I). Thus a change in one small passive substituent between compounds (I) and (IV) is associated with a considerable change in the crystal structure. The constitution of compound (II) has some resemblance to the hydrogensuccinate (V) and hydrogenfumarate (VI) salts of MeOPP, in both of which anions exhibits some disorder (Kiran Kumar et al., 2019 ▸). In each of (V) and (VI) the component ions are linked by a combination of O—H⋯O and N—H⋯O hydrogen bonds to form sheets, which are in turn linked into a three-dimensional assembly by C—H⋯π(arene) hydrogen bonds, as compared to the two dimensional assembly in (II). We also note that structures have been reported for 4-[bis(4-fluorophenyl)methyl)piperazine (VII) (Dayananda et al., 2012a
▸), and for its 1-acetyl derivative (VIII) (Dayananda et al., 2012b
▸), both of which are intermediates on the synthetic pathway to the calcium-channel blocker flunarizine, 1-[bis(4-fluorophenyl)methyl]-4-cinnamyl-piperazine (IX) (Prasanna & Row, 2001 ▸).
(16) types alternate, with chains of this type linked by C—H⋯π(arene) hydrogen bonds to form a three-dimensional network, as compared with the one-dimensional assembly in (I). Thus a change in one small passive substituent between compounds (I) and (IV) is associated with a considerable change in the crystal structure. The constitution of compound (II) has some resemblance to the hydrogensuccinate (V) and hydrogenfumarate (VI) salts of MeOPP, in both of which anions exhibits some disorder (Kiran Kumar et al., 2019 ▸). In each of (V) and (VI) the component ions are linked by a combination of O—H⋯O and N—H⋯O hydrogen bonds to form sheets, which are in turn linked into a three-dimensional assembly by C—H⋯π(arene) hydrogen bonds, as compared to the two dimensional assembly in (II). We also note that structures have been reported for 4-[bis(4-fluorophenyl)methyl)piperazine (VII) (Dayananda et al., 2012a
▸), and for its 1-acetyl derivative (VIII) (Dayananda et al., 2012b
▸), both of which are intermediates on the synthetic pathway to the calcium-channel blocker flunarizine, 1-[bis(4-fluorophenyl)methyl]-4-cinnamyl-piperazine (IX) (Prasanna & Row, 2001 ▸).
Synthesis and crystallization
All starting materials were obtained commercially, and all were used as received. For the preparation of compounds (I)–(III), N-(4-fluorophenyl)piperazine (100 mg, 0.55 mmol) was dissolved in methanol (10 ml) and a solution of the appropriate acid (0.55 mmol) in methanol (10 ml) [2-hydroxy-3,5-dinitrobenzoic acid, 125.5 mg for (I), oxalic acid, 49.5 mg for (II), and (2R,3R)-tartaric acid, 82.5 mg for (III)] was then added; the mixtures were briefly stirred at 323 K before being set aside at ambient temperature to crystallize. After two days, the resulting solid products were collected by filtration and dried in air. Crystals suitable for single-crystal X-ray diffraction were grown by slow evaporation, at ambient temperature and in the presence of air, of solutions in ethyl acetate for (I) and (III), or in methanol for (II): m.p. (I) 460–463 K, (II) 421–425 K, (III) 437–441 K.
Refinement
Crystal data, data collection and refinement details are summarized in Table 4 ▸. All H atoms were located in difference-Fourier maps. The H atoms bonded to C atoms were then treated as riding atoms in geometrically idealized positions with C—H distances 0.93 Å (aromatic), 0.97 Å (CH2), or 0.98 Å (aliphatic C—H) and with U iso(H) = 1.2U eq(C). The H atoms bonded to N or O atoms were refined with U iso(H) = 1.2U eq(N) or 1.5U eq(O), giving the N—H and O—H distances shown in Tables 1 ▸–3 ▸ ▸. In the absence of significant resonant scattering in compound (III), the Flack x parameter (Flack, 1983 ▸) was indeterminate (Flack & Bernardinelli, 2000 ▸): thus the value of x, calculated (Parsons et al., 2013 ▸) using 683 quotients of type [(I +) − (I −)]/[(I +) + (I −)], was −1.5 (7). Since a single enantiomer, the (2R,3R) form, of tartaric acid was used in the preparation of compound (III), the absolute configuration in the crystal of (III) was set on this basis.
Table 4. Experimental details.
| (I) | (II) | (III) | |
|---|---|---|---|
| Crystal data | |||
| Chemical formula | C10H14FN2 +·C7H3N2O7 − | C10H14FN2 +·C2HO4 − | C10H14FN2 +·C4H5O6 −·H2O |
| M r | 408.35 | 270.26 | 348.33 |
| Crystal system, space group | Monoclinic, P21/c | Monoclinic, P21/c | Orthorhombic, P212121 |
| Temperature (K) | 293 | 293 | 293 |
| a, b, c (Å) | 10.6829 (6), 13.1701 (6), 13.5563 (7) | 17.0606 (6), 5.7820 (2), 12.5815 (5) | 7.0961 (4), 7.4967 (4), 30.757 (2) |
| α, β, γ (°) | 90, 108.970 (5), 90 | 90, 102.761 (4), 90 | 90, 90, 90 |
| V (Å3) | 1803.71 (17) | 1210.44 (8) | 1636.19 (17) |
| Z | 4 | 4 | 4 |
| Radiation type | Mo Kα | Mo Kα | Mo Kα |
| μ (mm−1) | 0.13 | 0.12 | 0.12 |
| Crystal size (mm) | 0.50 × 0.44 × 0.34 | 0.34 × 0.34 × 0.28 | 0.40 × 0.22 × 0.10 |
| Data collection | |||
| Diffractometer | Oxford Diffraction Xcalibur with Sapphire CCD | Oxford Diffraction Xcalibur with Sapphire CCD | Oxford Diffraction Xcalibur with Sapphire CCD |
| Absorption correction | Multi-scan (CrysAlis RED; Oxford Diffraction, 2009 ▸) | Multi-scan (CrysAlis RED; Oxford Diffraction, 2009 ▸) | Multi-scan (CrysAlis RED; Oxford Diffraction, 2009 ▸) |
| T min, T max | 0.874, 0.958 | 0.877, 0.966 | 0.904, 0.988 |
| No. of measured, independent and observed [I > 2σ(I)] reflections | 7194, 3905, 2845 | 4450, 2596, 2237 | 4553, 3036, 2347 |
| R int | 0.011 | 0.009 | 0.019 |
| (sin θ/λ)max (Å−1) | 0.656 | 0.656 | 0.656 |
| Refinement | |||
| R[F 2 > 2σ(F 2)], wR(F 2), S | 0.041, 0.108, 1.03 | 0.033, 0.089, 1.03 | 0.045, 0.085, 1.14 |
| No. of reflections | 3905 | 2596 | 3036 |
| No. of parameters | 271 | 182 | 238 |
| H-atom treatment | H atoms treated by a mixture of independent and constrained refinement | H atoms treated by a mixture of independent and constrained refinement | H atoms treated by a mixture of independent and constrained refinement |
| Δρmax, Δρmin (e Å−3) | 0.29, −0.20 | 0.32, −0.14 | 0.18, −0.21 |
| Absolute structure | – | – | Flack x determined using 683 quotients [(I +)-(I -)]/[(I +)+(I -)] (Parsons et al., 2013 ▸) |
Supplementary Material
Crystal structure: contains datablock(s) global, I, II, III. DOI: 10.1107/S2056989020006398/wm5557sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S2056989020006398/wm5557Isup2.hkl
Structure factors: contains datablock(s) II. DOI: 10.1107/S2056989020006398/wm5557IIsup3.hkl
Structure factors: contains datablock(s) III. DOI: 10.1107/S2056989020006398/wm5557IIIsup4.hkl
Supporting information file. DOI: 10.1107/S2056989020006398/wm5557Isup5.cml
Additional supporting information: crystallographic information; 3D view; checkCIF report
Acknowledgments
CHC thanks University of Mysore for research facilities.
supplementary crystallographic information
4-(4-Fluorophenyl)piperazin-1-ium 2-hydroxy-3,5-dinitrobenzoate (I). Crystal data
| C10H14FN2+·C7H3N2O7− | F(000) = 848 |
| Mr = 408.35 | Dx = 1.504 Mg m−3 |
| Monoclinic, P21/c | Mo Kα radiation, λ = 0.71073 Å |
| a = 10.6829 (6) Å | Cell parameters from 3905 reflections |
| b = 13.1701 (6) Å | θ = 2.6–27.8° |
| c = 13.5563 (7) Å | µ = 0.13 mm−1 |
| β = 108.970 (5)° | T = 293 K |
| V = 1803.71 (17) Å3 | Block, yellow |
| Z = 4 | 0.50 × 0.44 × 0.34 mm |
4-(4-Fluorophenyl)piperazin-1-ium 2-hydroxy-3,5-dinitrobenzoate (I). Data collection
| Oxford Diffraction Xcalibur with Sapphire CCD diffractometer | 3905 independent reflections |
| Radiation source: Enhance (Mo) X-ray Source | 2845 reflections with I > 2σ(I) |
| Graphite monochromator | Rint = 0.011 |
| ω scans | θmax = 27.8°, θmin = 2.6° |
| Absorption correction: multi-scan (CrysAlis RED; Oxford Diffraction, 2009) | h = −13→8 |
| Tmin = 0.874, Tmax = 0.958 | k = −17→12 |
| 7194 measured reflections | l = −11→17 |
4-(4-Fluorophenyl)piperazin-1-ium 2-hydroxy-3,5-dinitrobenzoate (I). Refinement
| Refinement on F2 | Primary atom site location: difference Fourier map |
| Least-squares matrix: full | Hydrogen site location: mixed |
| R[F2 > 2σ(F2)] = 0.041 | H atoms treated by a mixture of independent and constrained refinement |
| wR(F2) = 0.108 | w = 1/[σ2(Fo2) + (0.0493P)2 + 0.4487P] where P = (Fo2 + 2Fc2)/3 |
| S = 1.02 | (Δ/σ)max < 0.001 |
| 3905 reflections | Δρmax = 0.29 e Å−3 |
| 271 parameters | Δρmin = −0.20 e Å−3 |
| 0 restraints |
4-(4-Fluorophenyl)piperazin-1-ium 2-hydroxy-3,5-dinitrobenzoate (I). Special details
| Geometry. All esds (except the esd in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell esds are taken into account individually in the estimation of esds in distances, angles and torsion angles; correlations between esds in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell esds is used for estimating esds involving l.s. planes. |
4-(4-Fluorophenyl)piperazin-1-ium 2-hydroxy-3,5-dinitrobenzoate (I). Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| N1 | 0.23326 (15) | 0.37624 (11) | 0.56770 (10) | 0.0410 (3) | |
| H11 | 0.2339 (18) | 0.3081 (15) | 0.5648 (13) | 0.049* | |
| H12 | 0.1870 (18) | 0.3961 (14) | 0.6103 (14) | 0.049* | |
| C2 | 0.16795 (18) | 0.41660 (13) | 0.46103 (12) | 0.0458 (4) | |
| H2A | 0.0754 | 0.3969 | 0.4369 | 0.055* | |
| H2B | 0.2098 | 0.3883 | 0.4135 | 0.055* | |
| C3 | 0.17888 (16) | 0.53003 (12) | 0.46196 (13) | 0.0446 (4) | |
| H3A | 0.1373 | 0.5561 | 0.3920 | 0.054* | |
| H3B | 0.1328 | 0.5583 | 0.5066 | 0.054* | |
| N4 | 0.31711 (13) | 0.56110 (10) | 0.49976 (10) | 0.0386 (3) | |
| C5 | 0.38008 (18) | 0.52566 (14) | 0.60598 (13) | 0.0507 (4) | |
| H5A | 0.3366 | 0.5558 | 0.6515 | 0.061* | |
| H5B | 0.4721 | 0.5466 | 0.6302 | 0.061* | |
| C6 | 0.37196 (18) | 0.41196 (14) | 0.61064 (15) | 0.0527 (4) | |
| H6A | 0.4238 | 0.3820 | 0.5712 | 0.063* | |
| H6B | 0.4093 | 0.3898 | 0.6825 | 0.063* | |
| C21 | 0.34479 (15) | 0.66117 (12) | 0.47504 (12) | 0.0369 (3) | |
| C22 | 0.29655 (18) | 0.69322 (13) | 0.37175 (13) | 0.0481 (4) | |
| H22 | 0.2418 | 0.6502 | 0.3218 | 0.058* | |
| C23 | 0.3279 (2) | 0.78703 (14) | 0.34172 (15) | 0.0545 (5) | |
| H23 | 0.2948 | 0.8074 | 0.2724 | 0.065* | |
| C24 | 0.40826 (18) | 0.84938 (13) | 0.41541 (16) | 0.0505 (4) | |
| F24 | 0.44076 (13) | 0.94182 (8) | 0.38489 (10) | 0.0748 (4) | |
| C25 | 0.45600 (18) | 0.82219 (13) | 0.51707 (16) | 0.0533 (5) | |
| H25 | 0.5101 | 0.8664 | 0.5660 | 0.064* | |
| C26 | 0.42392 (17) | 0.72812 (13) | 0.54787 (13) | 0.0464 (4) | |
| H26 | 0.4557 | 0.7097 | 0.6179 | 0.056* | |
| C37 | 0.15341 (16) | 0.07140 (13) | 0.28960 (12) | 0.0422 (4) | |
| O31 | 0.11601 (13) | 0.02065 (10) | 0.21079 (8) | 0.0549 (3) | |
| O32 | 0.19930 (15) | 0.16315 (11) | 0.28736 (10) | 0.0644 (4) | |
| H32 | 0.224 (3) | 0.1886 (19) | 0.358 (2) | 0.097* | |
| C31 | 0.15560 (14) | 0.03293 (11) | 0.39367 (11) | 0.0336 (3) | |
| C32 | 0.21010 (14) | 0.09577 (11) | 0.48464 (11) | 0.0328 (3) | |
| O33 | 0.24279 (12) | 0.18739 (8) | 0.47606 (8) | 0.0461 (3) | |
| C33 | 0.22155 (15) | 0.04718 (11) | 0.58166 (11) | 0.0346 (3) | |
| C34 | 0.17610 (15) | −0.04917 (12) | 0.58678 (12) | 0.0384 (4) | |
| H34 | 0.1844 | −0.0782 | 0.6511 | 0.046* | |
| C35 | 0.11827 (15) | −0.10288 (12) | 0.49668 (12) | 0.0379 (3) | |
| C36 | 0.10914 (14) | −0.06281 (12) | 0.39986 (12) | 0.0365 (3) | |
| H36 | 0.0717 | −0.1008 | 0.3396 | 0.044* | |
| N33 | 0.28212 (14) | 0.09929 (11) | 0.68062 (10) | 0.0456 (3) | |
| O34 | 0.33859 (15) | 0.17925 (10) | 0.68299 (10) | 0.0654 (4) | |
| O35 | 0.2769 (2) | 0.05901 (14) | 0.75965 (10) | 0.0944 (6) | |
| N35 | 0.06646 (15) | −0.20338 (11) | 0.50384 (13) | 0.0497 (4) | |
| O36 | 0.09063 (16) | −0.24215 (11) | 0.58996 (12) | 0.0768 (5) | |
| O37 | −0.00086 (15) | −0.24444 (10) | 0.42356 (12) | 0.0668 (4) |
4-(4-Fluorophenyl)piperazin-1-ium 2-hydroxy-3,5-dinitrobenzoate (I). Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| N1 | 0.0566 (9) | 0.0317 (7) | 0.0400 (7) | −0.0031 (6) | 0.0228 (6) | −0.0008 (6) |
| C2 | 0.0570 (10) | 0.0409 (9) | 0.0380 (8) | −0.0130 (8) | 0.0132 (7) | −0.0001 (7) |
| C3 | 0.0416 (9) | 0.0372 (9) | 0.0506 (9) | −0.0068 (7) | 0.0088 (7) | 0.0042 (7) |
| N4 | 0.0405 (7) | 0.0348 (7) | 0.0378 (7) | −0.0064 (6) | 0.0091 (5) | 0.0011 (5) |
| C5 | 0.0513 (10) | 0.0503 (10) | 0.0426 (9) | −0.0115 (8) | 0.0044 (8) | 0.0038 (8) |
| C6 | 0.0504 (10) | 0.0503 (11) | 0.0544 (10) | 0.0019 (8) | 0.0131 (8) | 0.0131 (9) |
| C21 | 0.0373 (8) | 0.0338 (8) | 0.0422 (8) | −0.0041 (6) | 0.0166 (6) | −0.0040 (6) |
| C22 | 0.0613 (11) | 0.0398 (9) | 0.0434 (9) | −0.0120 (8) | 0.0171 (8) | −0.0032 (7) |
| C23 | 0.0682 (12) | 0.0464 (10) | 0.0528 (10) | −0.0056 (9) | 0.0251 (9) | 0.0068 (8) |
| C24 | 0.0522 (10) | 0.0331 (9) | 0.0740 (13) | −0.0064 (8) | 0.0313 (9) | 0.0020 (8) |
| F24 | 0.0864 (9) | 0.0408 (6) | 0.1059 (10) | −0.0168 (6) | 0.0432 (7) | 0.0086 (6) |
| C25 | 0.0485 (10) | 0.0404 (10) | 0.0705 (12) | −0.0149 (8) | 0.0187 (9) | −0.0135 (9) |
| C26 | 0.0464 (9) | 0.0439 (10) | 0.0466 (9) | −0.0088 (8) | 0.0121 (7) | −0.0059 (7) |
| C37 | 0.0451 (9) | 0.0506 (10) | 0.0317 (8) | 0.0017 (8) | 0.0138 (7) | 0.0028 (7) |
| O31 | 0.0654 (8) | 0.0674 (8) | 0.0314 (6) | −0.0048 (7) | 0.0151 (5) | −0.0039 (6) |
| O32 | 0.1018 (11) | 0.0559 (8) | 0.0392 (7) | −0.0167 (8) | 0.0279 (7) | 0.0062 (6) |
| C31 | 0.0340 (7) | 0.0384 (8) | 0.0304 (7) | 0.0037 (6) | 0.0132 (6) | 0.0013 (6) |
| C32 | 0.0346 (7) | 0.0330 (8) | 0.0337 (7) | 0.0025 (6) | 0.0152 (6) | 0.0016 (6) |
| O33 | 0.0662 (8) | 0.0337 (6) | 0.0416 (6) | −0.0063 (5) | 0.0219 (5) | 0.0001 (5) |
| C33 | 0.0364 (8) | 0.0376 (8) | 0.0312 (7) | 0.0025 (6) | 0.0126 (6) | −0.0002 (6) |
| C34 | 0.0393 (8) | 0.0419 (9) | 0.0370 (8) | 0.0051 (7) | 0.0165 (7) | 0.0095 (7) |
| C35 | 0.0362 (8) | 0.0323 (8) | 0.0476 (9) | 0.0007 (6) | 0.0169 (7) | 0.0039 (7) |
| C36 | 0.0341 (8) | 0.0374 (8) | 0.0387 (8) | 0.0005 (6) | 0.0130 (6) | −0.0037 (6) |
| N33 | 0.0534 (8) | 0.0503 (9) | 0.0327 (7) | 0.0013 (7) | 0.0133 (6) | 0.0002 (6) |
| O34 | 0.0900 (11) | 0.0529 (8) | 0.0464 (7) | −0.0153 (7) | 0.0128 (7) | −0.0104 (6) |
| O35 | 0.1494 (17) | 0.0992 (13) | 0.0325 (7) | −0.0423 (11) | 0.0265 (8) | 0.0008 (7) |
| N35 | 0.0475 (8) | 0.0385 (8) | 0.0662 (10) | −0.0035 (6) | 0.0228 (7) | 0.0050 (7) |
| O36 | 0.0962 (11) | 0.0551 (9) | 0.0759 (10) | −0.0183 (8) | 0.0235 (8) | 0.0238 (8) |
| O37 | 0.0731 (9) | 0.0487 (8) | 0.0777 (10) | −0.0215 (7) | 0.0232 (8) | −0.0112 (7) |
4-(4-Fluorophenyl)piperazin-1-ium 2-hydroxy-3,5-dinitrobenzoate (I). Geometric parameters (Å, º)
| N1—C6 | 1.481 (2) | C24—F24 | 1.3664 (19) |
| N1—C2 | 1.485 (2) | C25—C26 | 1.386 (2) |
| N1—H11 | 0.898 (19) | C25—H25 | 0.9300 |
| N1—H12 | 0.912 (19) | C26—H26 | 0.9300 |
| C2—C3 | 1.498 (2) | C37—O31 | 1.2125 (19) |
| C2—H2A | 0.9700 | C37—O32 | 1.308 (2) |
| C2—H2B | 0.9700 | C37—C31 | 1.492 (2) |
| C3—N4 | 1.456 (2) | O32—H32 | 0.97 (3) |
| C3—H3A | 0.9700 | C31—C36 | 1.368 (2) |
| C3—H3B | 0.9700 | C31—C32 | 1.441 (2) |
| N4—C21 | 1.4147 (19) | C32—O33 | 1.2719 (18) |
| N4—C5 | 1.454 (2) | C32—C33 | 1.4318 (19) |
| C5—C6 | 1.502 (2) | C33—C34 | 1.368 (2) |
| C5—H5A | 0.9700 | C33—N33 | 1.4578 (19) |
| C5—H5B | 0.9700 | C34—C35 | 1.372 (2) |
| C6—H6A | 0.9700 | C34—H34 | 0.9300 |
| C6—H6B | 0.9700 | C35—C36 | 1.388 (2) |
| C21—C26 | 1.387 (2) | C35—N35 | 1.450 (2) |
| C21—C22 | 1.391 (2) | C36—H36 | 0.9300 |
| C22—C23 | 1.376 (2) | N33—O34 | 1.2089 (19) |
| C22—H22 | 0.9300 | N33—O35 | 1.2129 (18) |
| C23—C24 | 1.361 (3) | N35—O37 | 1.2187 (19) |
| C23—H23 | 0.9300 | N35—O36 | 1.2224 (19) |
| C24—C25 | 1.353 (3) | ||
| C6—N1—C2 | 111.24 (13) | C24—C23—H23 | 120.6 |
| C6—N1—H11 | 108.3 (12) | C22—C23—H23 | 120.6 |
| C2—N1—H11 | 108.8 (11) | C25—C24—C23 | 121.80 (16) |
| C6—N1—H12 | 109.8 (11) | C25—C24—F24 | 119.70 (17) |
| C2—N1—H12 | 109.5 (11) | C23—C24—F24 | 118.50 (17) |
| H11—N1—H12 | 109.1 (16) | C24—C25—C26 | 119.69 (16) |
| N1—C2—C3 | 109.76 (13) | C24—C25—H25 | 120.2 |
| N1—C2—H2A | 109.7 | C26—C25—H25 | 120.2 |
| C3—C2—H2A | 109.7 | C25—C26—C21 | 120.47 (16) |
| N1—C2—H2B | 109.7 | C25—C26—H26 | 119.8 |
| C3—C2—H2B | 109.7 | C21—C26—H26 | 119.8 |
| H2A—C2—H2B | 108.2 | O31—C37—O32 | 120.48 (15) |
| N4—C3—C2 | 110.53 (14) | O31—C37—C31 | 123.05 (16) |
| N4—C3—H3A | 109.5 | O32—C37—C31 | 116.42 (14) |
| C2—C3—H3A | 109.5 | C37—O32—H32 | 106.3 (15) |
| N4—C3—H3B | 109.5 | C36—C31—C32 | 122.05 (13) |
| C2—C3—H3B | 109.5 | C36—C31—C37 | 118.46 (13) |
| H3A—C3—H3B | 108.1 | C32—C31—C37 | 119.46 (13) |
| C21—N4—C5 | 117.95 (13) | O33—C32—C33 | 124.33 (13) |
| C21—N4—C3 | 116.41 (13) | O33—C32—C31 | 120.87 (13) |
| C5—N4—C3 | 110.38 (13) | C33—C32—C31 | 114.80 (13) |
| N4—C5—C6 | 110.31 (14) | C34—C33—C32 | 122.36 (13) |
| N4—C5—H5A | 109.6 | C34—C33—N33 | 116.65 (13) |
| C6—C5—H5A | 109.6 | C32—C33—N33 | 120.98 (13) |
| N4—C5—H5B | 109.6 | C33—C34—C35 | 119.82 (14) |
| C6—C5—H5B | 109.6 | C33—C34—H34 | 120.1 |
| H5A—C5—H5B | 108.1 | C35—C34—H34 | 120.1 |
| N1—C6—C5 | 111.34 (15) | C34—C35—C36 | 121.23 (14) |
| N1—C6—H6A | 109.4 | C34—C35—N35 | 118.82 (14) |
| C5—C6—H6A | 109.4 | C36—C35—N35 | 119.95 (14) |
| N1—C6—H6B | 109.4 | C31—C36—C35 | 119.54 (14) |
| C5—C6—H6B | 109.4 | C31—C36—H36 | 120.2 |
| H6A—C6—H6B | 108.0 | C35—C36—H36 | 120.2 |
| C26—C21—C22 | 117.66 (15) | O34—N33—O35 | 121.56 (15) |
| C26—C21—N4 | 123.34 (14) | O34—N33—C33 | 120.20 (13) |
| C22—C21—N4 | 118.92 (14) | O35—N33—C33 | 118.22 (15) |
| C23—C22—C21 | 121.61 (16) | O37—N35—O36 | 123.20 (15) |
| C23—C22—H22 | 119.2 | O37—N35—C35 | 118.20 (15) |
| C21—C22—H22 | 119.2 | O36—N35—C35 | 118.59 (15) |
| C24—C23—C22 | 118.74 (17) | ||
| C6—N1—C2—C3 | −54.64 (19) | O32—C37—C31—C32 | −1.7 (2) |
| N1—C2—C3—N4 | 58.34 (18) | C36—C31—C32—O33 | −174.27 (14) |
| C2—C3—N4—C21 | 160.93 (13) | C37—C31—C32—O33 | 7.8 (2) |
| C2—C3—N4—C5 | −61.10 (18) | C36—C31—C32—C33 | 4.9 (2) |
| C21—N4—C5—C6 | −163.70 (14) | C37—C31—C32—C33 | −173.01 (13) |
| C3—N4—C5—C6 | 59.05 (19) | O33—C32—C33—C34 | 175.03 (14) |
| C2—N1—C6—C5 | 53.7 (2) | C31—C32—C33—C34 | −4.1 (2) |
| N4—C5—C6—N1 | −55.5 (2) | O33—C32—C33—N33 | −4.5 (2) |
| C5—N4—C21—C26 | −2.9 (2) | C31—C32—C33—N33 | 176.36 (13) |
| C3—N4—C21—C26 | 131.79 (17) | C32—C33—C34—C35 | 0.6 (2) |
| C5—N4—C21—C22 | 173.56 (16) | N33—C33—C34—C35 | −179.81 (14) |
| C3—N4—C21—C22 | −51.7 (2) | C33—C34—C35—C36 | 2.4 (2) |
| C26—C21—C22—C23 | 1.3 (3) | C33—C34—C35—N35 | −177.75 (14) |
| N4—C21—C22—C23 | −175.35 (16) | C32—C31—C36—C35 | −2.2 (2) |
| C21—C22—C23—C24 | 0.1 (3) | C37—C31—C36—C35 | 175.71 (14) |
| C22—C23—C24—C25 | −1.1 (3) | C34—C35—C36—C31 | −1.6 (2) |
| C22—C23—C24—F24 | 178.99 (16) | N35—C35—C36—C31 | 178.55 (14) |
| C23—C24—C25—C26 | 0.6 (3) | C34—C33—N33—O34 | 171.04 (15) |
| F24—C24—C25—C26 | −179.47 (16) | C32—C33—N33—O34 | −9.4 (2) |
| C24—C25—C26—C21 | 0.9 (3) | C34—C33—N33—O35 | −7.2 (2) |
| C22—C21—C26—C25 | −1.8 (2) | C32—C33—N33—O35 | 172.40 (16) |
| N4—C21—C26—C25 | 174.71 (15) | C34—C35—N35—O37 | 169.81 (15) |
| O31—C37—C31—C36 | −2.3 (2) | C36—C35—N35—O37 | −10.4 (2) |
| O32—C37—C31—C36 | −179.71 (15) | C34—C35—N35—O36 | −9.1 (2) |
| O31—C37—C31—C32 | 175.74 (15) | C36—C35—N35—O36 | 170.72 (15) |
4-(4-Fluorophenyl)piperazin-1-ium 2-hydroxy-3,5-dinitrobenzoate (I). Hydrogen-bond geometry (Å, º)
| D—H···A | D—H | H···A | D···A | D—H···A |
| N1—H11···O33 | 0.90 (2) | 2.014 (19) | 2.7968 (18) | 144.8 (15) |
| N1—H11···O34 | 0.90 (2) | 2.352 (19) | 3.049 (2) | 134.4 (14) |
| N1—H12···O31i | 0.912 (19) | 2.075 (19) | 2.959 (2) | 163.0 (17) |
| N1—H12···O32i | 0.912 (19) | 2.487 (18) | 3.1576 (19) | 130.7 (15) |
| O32—H32···O33 | 0.97 (3) | 1.55 (3) | 2.4676 (17) | 157 (3) |
| C2—H2B···O35ii | 0.97 | 2.51 | 3.313 (2) | 140 |
Symmetry codes: (i) x, −y+1/2, z+1/2; (ii) x, −y+1/2, z−1/2.
4-(4-Fluorophenyl)piperazin-1-ium hydrogen oxalate (II). Crystal data
| C10H14FN2+·C2HO4− | F(000) = 568 |
| Mr = 270.26 | Dx = 1.483 Mg m−3 |
| Monoclinic, P21/c | Mo Kα radiation, λ = 0.71073 Å |
| a = 17.0606 (6) Å | Cell parameters from 2596 reflections |
| b = 5.7820 (2) Å | θ = 3.3–27.8° |
| c = 12.5815 (5) Å | µ = 0.12 mm−1 |
| β = 102.761 (4)° | T = 293 K |
| V = 1210.44 (8) Å3 | Block, colourless |
| Z = 4 | 0.34 × 0.34 × 0.28 mm |
4-(4-Fluorophenyl)piperazin-1-ium hydrogen oxalate (II). Data collection
| Oxford Diffraction Xcalibur with Sapphire CCD diffractometer | 2596 independent reflections |
| Radiation source: Enhance (Mo) X-ray Source | 2237 reflections with I > 2σ(I) |
| Graphite monochromator | Rint = 0.009 |
| ω scans | θmax = 27.8°, θmin = 3.3° |
| Absorption correction: multi-scan (CrysAlis RED; Oxford Diffraction, 2009) | h = −16→22 |
| Tmin = 0.877, Tmax = 0.966 | k = −7→5 |
| 4450 measured reflections | l = −16→9 |
4-(4-Fluorophenyl)piperazin-1-ium hydrogen oxalate (II). Refinement
| Refinement on F2 | Hydrogen site location: mixed |
| Least-squares matrix: full | H atoms treated by a mixture of independent and constrained refinement |
| R[F2 > 2σ(F2)] = 0.033 | w = 1/[σ2(Fo2) + (0.0443P)2 + 0.346P] where P = (Fo2 + 2Fc2)/3 |
| wR(F2) = 0.089 | (Δ/σ)max < 0.001 |
| S = 1.03 | Δρmax = 0.32 e Å−3 |
| 2596 reflections | Δρmin = −0.14 e Å−3 |
| 182 parameters | Extinction correction: SHELXL, Fc*=kFc[1+0.001xFc2λ3/sin(2θ)]-1/4 |
| 0 restraints | Extinction coefficient: 0.0084 (12) |
| Primary atom site location: difference Fourier map |
4-(4-Fluorophenyl)piperazin-1-ium hydrogen oxalate (II). Special details
| Geometry. All esds (except the esd in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell esds are taken into account individually in the estimation of esds in distances, angles and torsion angles; correlations between esds in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell esds is used for estimating esds involving l.s. planes. |
4-(4-Fluorophenyl)piperazin-1-ium hydrogen oxalate (II). Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| N1 | 0.34076 (6) | 0.76074 (19) | 0.62909 (9) | 0.0310 (2) | |
| H11 | 0.3629 (9) | 0.731 (3) | 0.7011 (13) | 0.037* | |
| H12 | 0.3813 (9) | 0.831 (3) | 0.6035 (12) | 0.037* | |
| C2 | 0.27227 (7) | 0.9240 (2) | 0.62039 (10) | 0.0316 (3) | |
| H2A | 0.2911 | 1.0681 | 0.6566 | 0.038* | |
| H2B | 0.2324 | 0.8584 | 0.6559 | 0.038* | |
| C3 | 0.23469 (8) | 0.9699 (2) | 0.50098 (10) | 0.0324 (3) | |
| H3A | 0.1896 | 1.0749 | 0.4950 | 0.039* | |
| H3B | 0.2739 | 1.0411 | 0.4660 | 0.039* | |
| N4 | 0.20732 (6) | 0.75101 (17) | 0.44718 (8) | 0.0268 (2) | |
| C5 | 0.27693 (7) | 0.5982 (2) | 0.45117 (10) | 0.0319 (3) | |
| H5A | 0.3154 | 0.6735 | 0.4163 | 0.038* | |
| H5B | 0.2595 | 0.4561 | 0.4120 | 0.038* | |
| C6 | 0.31618 (8) | 0.5431 (2) | 0.56812 (11) | 0.0347 (3) | |
| H6A | 0.2788 | 0.4590 | 0.6017 | 0.042* | |
| H6B | 0.3629 | 0.4461 | 0.5705 | 0.042* | |
| C21 | 0.15689 (7) | 0.7661 (2) | 0.34045 (9) | 0.0276 (3) | |
| C22 | 0.10736 (8) | 0.5776 (2) | 0.30382 (11) | 0.0364 (3) | |
| H22 | 0.1070 | 0.4520 | 0.3499 | 0.044* | |
| C23 | 0.05858 (8) | 0.5743 (3) | 0.19977 (12) | 0.0424 (3) | |
| H23 | 0.0262 | 0.4472 | 0.1753 | 0.051* | |
| C24 | 0.05932 (8) | 0.7625 (3) | 0.13391 (11) | 0.0418 (3) | |
| F24 | 0.01133 (7) | 0.75923 (18) | 0.03205 (7) | 0.0688 (3) | |
| C25 | 0.10587 (9) | 0.9541 (3) | 0.16709 (11) | 0.0418 (3) | |
| H25 | 0.1044 | 1.0806 | 0.1210 | 0.050* | |
| C26 | 0.15542 (8) | 0.9549 (2) | 0.27143 (10) | 0.0344 (3) | |
| H26 | 0.1877 | 1.0826 | 0.2950 | 0.041* | |
| C31 | 0.47968 (7) | 0.58033 (19) | 0.86963 (9) | 0.0239 (2) | |
| C32 | 0.43856 (7) | 0.3385 (2) | 0.84980 (9) | 0.0258 (2) | |
| O31 | 0.55249 (5) | 0.58853 (15) | 0.91087 (7) | 0.0319 (2) | |
| O32 | 0.43316 (5) | 0.74960 (14) | 0.84049 (7) | 0.0315 (2) | |
| O33 | 0.36759 (5) | 0.31730 (17) | 0.81152 (8) | 0.0417 (2) | |
| O34 | 0.48994 (5) | 0.16802 (15) | 0.87815 (7) | 0.0320 (2) | |
| H34 | 0.4648 (10) | 0.029 (3) | 0.8663 (13) | 0.048* |
4-(4-Fluorophenyl)piperazin-1-ium hydrogen oxalate (II). Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| N1 | 0.0245 (5) | 0.0331 (6) | 0.0310 (5) | −0.0043 (4) | −0.0037 (4) | 0.0048 (4) |
| C2 | 0.0321 (6) | 0.0295 (6) | 0.0305 (6) | −0.0019 (5) | 0.0008 (5) | −0.0036 (5) |
| C3 | 0.0355 (6) | 0.0246 (6) | 0.0324 (6) | 0.0032 (5) | −0.0025 (5) | −0.0014 (5) |
| N4 | 0.0250 (5) | 0.0250 (5) | 0.0273 (5) | 0.0024 (4) | −0.0007 (4) | −0.0001 (4) |
| C5 | 0.0302 (6) | 0.0289 (6) | 0.0345 (6) | 0.0046 (5) | 0.0025 (5) | −0.0019 (5) |
| C6 | 0.0301 (6) | 0.0278 (6) | 0.0411 (7) | 0.0033 (5) | −0.0032 (5) | 0.0027 (5) |
| C21 | 0.0245 (5) | 0.0300 (6) | 0.0265 (6) | 0.0042 (5) | 0.0019 (4) | −0.0010 (5) |
| C22 | 0.0349 (7) | 0.0339 (7) | 0.0363 (7) | −0.0028 (5) | −0.0008 (5) | 0.0016 (5) |
| C23 | 0.0370 (7) | 0.0411 (8) | 0.0424 (7) | −0.0024 (6) | −0.0058 (6) | −0.0082 (6) |
| C24 | 0.0402 (7) | 0.0471 (8) | 0.0303 (6) | 0.0124 (6) | −0.0093 (5) | −0.0052 (6) |
| F24 | 0.0811 (7) | 0.0650 (6) | 0.0406 (5) | 0.0116 (5) | −0.0291 (5) | −0.0051 (5) |
| C25 | 0.0503 (8) | 0.0382 (7) | 0.0326 (7) | 0.0108 (6) | −0.0001 (6) | 0.0061 (6) |
| C26 | 0.0363 (6) | 0.0300 (6) | 0.0338 (6) | 0.0018 (5) | 0.0011 (5) | 0.0003 (5) |
| C31 | 0.0273 (5) | 0.0224 (5) | 0.0215 (5) | 0.0006 (4) | 0.0042 (4) | −0.0009 (4) |
| C32 | 0.0269 (6) | 0.0248 (6) | 0.0241 (5) | −0.0004 (4) | 0.0026 (4) | −0.0004 (4) |
| O31 | 0.0246 (4) | 0.0274 (4) | 0.0405 (5) | −0.0014 (3) | 0.0006 (3) | −0.0036 (4) |
| O32 | 0.0324 (5) | 0.0224 (4) | 0.0360 (5) | 0.0037 (3) | −0.0002 (4) | 0.0008 (3) |
| O33 | 0.0272 (5) | 0.0349 (5) | 0.0560 (6) | −0.0030 (4) | −0.0057 (4) | −0.0029 (4) |
| O34 | 0.0292 (4) | 0.0200 (4) | 0.0442 (5) | −0.0003 (3) | 0.0026 (4) | 0.0000 (4) |
4-(4-Fluorophenyl)piperazin-1-ium hydrogen oxalate (II). Geometric parameters (Å, º)
| N1—C6 | 1.4854 (16) | C21—C26 | 1.3917 (18) |
| N1—C2 | 1.4872 (16) | C21—C22 | 1.3935 (17) |
| N1—H11 | 0.918 (16) | C22—C23 | 1.3875 (18) |
| N1—H12 | 0.920 (16) | C22—H22 | 0.9300 |
| C2—C3 | 1.5208 (16) | C23—C24 | 1.370 (2) |
| C2—H2A | 0.9700 | C23—H23 | 0.9300 |
| C2—H2B | 0.9700 | C24—F24 | 1.3600 (15) |
| C3—N4 | 1.4622 (15) | C24—C25 | 1.373 (2) |
| C3—H3A | 0.9700 | C25—C26 | 1.3956 (17) |
| C3—H3B | 0.9700 | C25—H25 | 0.9300 |
| N4—C21 | 1.4284 (14) | C26—H26 | 0.9300 |
| N4—C5 | 1.4723 (15) | C31—O31 | 1.2368 (13) |
| C5—C6 | 1.5098 (17) | C31—O32 | 1.2625 (13) |
| C5—H5A | 0.9700 | C31—C32 | 1.5597 (16) |
| C5—H5B | 0.9700 | C32—O33 | 1.2064 (14) |
| C6—H6A | 0.9700 | C32—O34 | 1.3148 (14) |
| C6—H6B | 0.9700 | O34—H34 | 0.908 (18) |
| C6—N1—C2 | 111.85 (9) | C5—C6—H6A | 109.7 |
| C6—N1—H11 | 110.9 (9) | N1—C6—H6B | 109.7 |
| C2—N1—H11 | 109.8 (9) | C5—C6—H6B | 109.7 |
| C6—N1—H12 | 110.0 (9) | H6A—C6—H6B | 108.2 |
| C2—N1—H12 | 109.5 (9) | C26—C21—C22 | 118.75 (11) |
| H11—N1—H12 | 104.5 (13) | C26—C21—N4 | 123.95 (11) |
| N1—C2—C3 | 109.65 (10) | C22—C21—N4 | 117.30 (11) |
| N1—C2—H2A | 109.7 | C23—C22—C21 | 121.10 (13) |
| C3—C2—H2A | 109.7 | C23—C22—H22 | 119.4 |
| N1—C2—H2B | 109.7 | C21—C22—H22 | 119.4 |
| C3—C2—H2B | 109.7 | C24—C23—C22 | 118.41 (13) |
| H2A—C2—H2B | 108.2 | C24—C23—H23 | 120.8 |
| N4—C3—C2 | 109.16 (10) | C22—C23—H23 | 120.8 |
| N4—C3—H3A | 109.8 | F24—C24—C23 | 118.36 (13) |
| C2—C3—H3A | 109.8 | F24—C24—C25 | 119.02 (13) |
| N4—C3—H3B | 109.8 | C23—C24—C25 | 122.63 (12) |
| C2—C3—H3B | 109.8 | C24—C25—C26 | 118.56 (13) |
| H3A—C3—H3B | 108.3 | C24—C25—H25 | 120.7 |
| C21—N4—C3 | 116.55 (9) | C26—C25—H25 | 120.7 |
| C21—N4—C5 | 112.49 (9) | C21—C26—C25 | 120.53 (12) |
| C3—N4—C5 | 109.32 (9) | C21—C26—H26 | 119.7 |
| N4—C5—C6 | 109.92 (10) | C25—C26—H26 | 119.7 |
| N4—C5—H5A | 109.7 | O31—C31—O32 | 126.93 (11) |
| C6—C5—H5A | 109.7 | O31—C31—C32 | 118.43 (10) |
| N4—C5—H5B | 109.7 | O32—C31—C32 | 114.64 (9) |
| C6—C5—H5B | 109.7 | O33—C32—O34 | 125.62 (11) |
| H5A—C5—H5B | 108.2 | O33—C32—C31 | 122.08 (10) |
| N1—C6—C5 | 109.77 (10) | O34—C32—C31 | 112.29 (9) |
| N1—C6—H6A | 109.7 | C32—O34—H34 | 110.9 (10) |
| C6—N1—C2—C3 | −55.37 (14) | N4—C21—C22—C23 | −177.69 (12) |
| N1—C2—C3—N4 | 58.86 (13) | C21—C22—C23—C24 | −0.9 (2) |
| C2—C3—N4—C21 | 168.31 (10) | C22—C23—C24—F24 | −179.84 (13) |
| C2—C3—N4—C5 | −62.77 (13) | C22—C23—C24—C25 | −0.5 (2) |
| C21—N4—C5—C6 | −166.31 (10) | F24—C24—C25—C26 | −179.45 (13) |
| C3—N4—C5—C6 | 62.56 (13) | C23—C24—C25—C26 | 1.2 (2) |
| C2—N1—C6—C5 | 54.78 (14) | C22—C21—C26—C25 | −0.68 (19) |
| N4—C5—C6—N1 | −57.70 (14) | N4—C21—C26—C25 | 178.36 (12) |
| C3—N4—C21—C26 | 23.67 (17) | C24—C25—C26—C21 | −0.6 (2) |
| C5—N4—C21—C26 | −103.72 (14) | O31—C31—C32—O33 | 178.81 (11) |
| C3—N4—C21—C22 | −157.27 (11) | O32—C31—C32—O33 | −1.35 (16) |
| C5—N4—C21—C22 | 75.34 (14) | O31—C31—C32—O34 | −1.92 (14) |
| C26—C21—C22—C23 | 1.42 (19) | O32—C31—C32—O34 | 177.92 (10) |
4-(4-Fluorophenyl)piperazin-1-ium hydrogen oxalate (II). Hydrogen-bond geometry (Å, º)
| D—H···A | D—H | H···A | D···A | D—H···A |
| N1—H11···O32 | 0.918 (16) | 1.896 (16) | 2.7769 (14) | 160.2 (15) |
| N1—H12···O31i | 0.920 (16) | 1.902 (17) | 2.7507 (14) | 152.6 (15) |
| N1—H12···O34i | 0.920 (16) | 2.354 (16) | 2.9588 (14) | 123.1 (13) |
| O34—H34···O32ii | 0.908 (17) | 1.712 (17) | 2.6102 (12) | 170.0 (17) |
| C2—H2A···O33iii | 0.97 | 2.54 | 3.4454 (15) | 155 |
| C5—H5A···O32iv | 0.97 | 2.45 | 3.3849 (15) | 163 |
| C6—H6B···O31v | 0.97 | 2.50 | 3.4259 (15) | 159 |
| C2—H2B···Cg1vi | 0.97 | 2.65 | 3.6124 (14) | 170 |
| C23—H23···Cg1vii | 0.93 | 2.94 | 3.5865 (16) | 128 |
Symmetry codes: (i) −x+1, y+1/2, −z+3/2; (ii) x, y−1, z; (iii) x, y+1, z; (iv) x, −y+3/2, z−1/2; (v) −x+1, y−1/2, −z+3/2; (vi) x, −y+3/2, z+1/2; (vii) −x, y−1/2, −z+1/2.
4-(4-Fluorophenyl)piperazin-1-ium hydrogen (2R,3R)-tartrate monohydrate (III) . Crystal data
| C10H14FN2+·C4H5O6−·H2O | Dx = 1.414 Mg m−3 |
| Mr = 348.33 | Mo Kα radiation, λ = 0.71073 Å |
| Orthorhombic, P212121 | Cell parameters from 3036 reflections |
| a = 7.0961 (4) Å | θ = 2.7–27.8° |
| b = 7.4967 (4) Å | µ = 0.12 mm−1 |
| c = 30.757 (2) Å | T = 293 K |
| V = 1636.19 (17) Å3 | Needle, yellow |
| Z = 4 | 0.40 × 0.22 × 0.10 mm |
| F(000) = 736 |
4-(4-Fluorophenyl)piperazin-1-ium hydrogen (2R,3R)-tartrate monohydrate (III) . Data collection
| Oxford Diffraction Xcalibur with Sapphire CCD diffractometer | 3036 independent reflections |
| Radiation source: Enhance (Mo) X-ray Source | 2347 reflections with I > 2σ(I) |
| Graphite monochromator | Rint = 0.019 |
| ω scans | θmax = 27.8°, θmin = 2.7° |
| Absorption correction: multi-scan (CrysAlis RED; Oxford Diffraction, 2009) | h = −8→7 |
| Tmin = 0.904, Tmax = 0.988 | k = −7→9 |
| 4553 measured reflections | l = −39→20 |
4-(4-Fluorophenyl)piperazin-1-ium hydrogen (2R,3R)-tartrate monohydrate (III) . Refinement
| Refinement on F2 | Primary atom site location: difference Fourier map |
| Least-squares matrix: full | Hydrogen site location: mixed |
| R[F2 > 2σ(F2)] = 0.045 | H atoms treated by a mixture of independent and constrained refinement |
| wR(F2) = 0.085 | w = 1/[σ2(Fo2) + (0.0206P)2 + 0.5183P] where P = (Fo2 + 2Fc2)/3 |
| S = 1.14 | (Δ/σ)max < 0.001 |
| 3036 reflections | Δρmax = 0.18 e Å−3 |
| 238 parameters | Δρmin = −0.21 e Å−3 |
| 0 restraints | Absolute structure: Flack x determined using 683 quotients [(I+)-(I-)]/[(I+)+(I-)] (Parsons et al., 2013) |
4-(4-Fluorophenyl)piperazin-1-ium hydrogen (2R,3R)-tartrate monohydrate (III) . Special details
| Geometry. All esds (except the esd in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell esds are taken into account individually in the estimation of esds in distances, angles and torsion angles; correlations between esds in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell esds is used for estimating esds involving l.s. planes. |
4-(4-Fluorophenyl)piperazin-1-ium hydrogen (2R,3R)-tartrate monohydrate (III) . Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| N1 | 0.6296 (4) | 0.2270 (5) | 0.60097 (11) | 0.0454 (8) | |
| H11 | 0.688 (5) | 0.200 (5) | 0.5771 (12) | 0.054* | |
| H12 | 0.623 (5) | 0.349 (5) | 0.5995 (12) | 0.054* | |
| C2 | 0.7310 (4) | 0.1802 (5) | 0.64079 (12) | 0.0464 (9) | |
| H2A | 0.8493 | 0.2450 | 0.6418 | 0.056* | |
| H2B | 0.7597 | 0.0537 | 0.6405 | 0.056* | |
| C3 | 0.6174 (5) | 0.2235 (5) | 0.68063 (11) | 0.0424 (9) | |
| H3A | 0.6852 | 0.1847 | 0.7063 | 0.051* | |
| H3B | 0.5998 | 0.3516 | 0.6826 | 0.051* | |
| N4 | 0.4346 (3) | 0.1361 (4) | 0.67912 (8) | 0.0359 (7) | |
| C5 | 0.3323 (5) | 0.1901 (5) | 0.64017 (11) | 0.0464 (9) | |
| H5A | 0.3106 | 0.3178 | 0.6410 | 0.056* | |
| H5B | 0.2107 | 0.1311 | 0.6396 | 0.056* | |
| C6 | 0.4402 (5) | 0.1435 (6) | 0.59966 (11) | 0.0521 (10) | |
| H6A | 0.4530 | 0.0150 | 0.5974 | 0.063* | |
| H6B | 0.3720 | 0.1855 | 0.5743 | 0.063* | |
| C21 | 0.3285 (5) | 0.1373 (4) | 0.71803 (10) | 0.0350 (7) | |
| C22 | 0.3867 (5) | 0.2237 (5) | 0.75550 (11) | 0.0439 (9) | |
| H22 | 0.4981 | 0.2890 | 0.7551 | 0.053* | |
| C23 | 0.2824 (6) | 0.2149 (5) | 0.79359 (12) | 0.0554 (11) | |
| H23 | 0.3243 | 0.2707 | 0.8188 | 0.066* | |
| C24 | 0.1175 (6) | 0.1230 (5) | 0.79317 (12) | 0.0541 (10) | |
| F24 | 0.0137 (4) | 0.1146 (4) | 0.83031 (7) | 0.0923 (9) | |
| C25 | 0.0551 (5) | 0.0358 (6) | 0.75739 (12) | 0.0547 (11) | |
| H25 | −0.0575 | −0.0276 | 0.7582 | 0.066* | |
| C26 | 0.1605 (5) | 0.0422 (5) | 0.71956 (12) | 0.0456 (9) | |
| H26 | 0.1185 | −0.0178 | 0.6949 | 0.055* | |
| C31 | 0.9787 (4) | 0.8324 (4) | 0.56468 (10) | 0.0317 (7) | |
| C32 | 1.0513 (4) | 0.6417 (4) | 0.56228 (10) | 0.0269 (7) | |
| H32A | 1.0143 | 0.5796 | 0.5890 | 0.032* | |
| C33 | 0.9627 (4) | 0.5450 (4) | 0.52373 (9) | 0.0270 (7) | |
| H33A | 0.8260 | 0.5415 | 0.5281 | 0.032* | |
| C34 | 1.0350 (4) | 0.3543 (4) | 0.52206 (10) | 0.0285 (7) | |
| O31 | 0.8084 (3) | 0.8554 (3) | 0.57208 (8) | 0.0416 (6) | |
| O32 | 1.0978 (3) | 0.9520 (3) | 0.55833 (9) | 0.0510 (7) | |
| O33 | 1.2503 (3) | 0.6346 (3) | 0.55866 (8) | 0.0381 (6) | |
| H33 | 1.279 (5) | 0.722 (5) | 0.5451 (12) | 0.057* | |
| O34 | 0.9995 (3) | 0.6363 (3) | 0.48486 (7) | 0.0397 (6) | |
| H34 | 1.101 (6) | 0.610 (5) | 0.4746 (12) | 0.060* | |
| O35 | 1.1228 (3) | 0.2964 (3) | 0.49167 (8) | 0.0453 (6) | |
| O36 | 0.9946 (3) | 0.2667 (3) | 0.55754 (7) | 0.0341 (5) | |
| H36 | 1.040 (5) | 0.148 (5) | 0.5570 (11) | 0.051* | |
| O41 | 0.5786 (4) | 0.5889 (4) | 0.60508 (10) | 0.0566 (8) | |
| H41 | 0.659 (7) | 0.681 (6) | 0.5942 (14) | 0.085* | |
| H42 | 0.477 (7) | 0.607 (6) | 0.5968 (15) | 0.085* |
4-(4-Fluorophenyl)piperazin-1-ium hydrogen (2R,3R)-tartrate monohydrate (III) . Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| N1 | 0.0359 (17) | 0.061 (2) | 0.0389 (18) | −0.0067 (17) | 0.0065 (15) | −0.0021 (18) |
| C2 | 0.0323 (18) | 0.058 (2) | 0.049 (2) | −0.0006 (17) | 0.0024 (18) | 0.004 (2) |
| C3 | 0.0353 (19) | 0.054 (2) | 0.038 (2) | −0.0045 (17) | −0.0052 (17) | −0.0010 (19) |
| N4 | 0.0317 (14) | 0.0420 (16) | 0.0339 (15) | −0.0036 (14) | 0.0002 (13) | −0.0030 (14) |
| C5 | 0.0322 (17) | 0.070 (3) | 0.037 (2) | −0.0069 (18) | −0.0011 (17) | −0.004 (2) |
| C6 | 0.0374 (19) | 0.080 (3) | 0.039 (2) | −0.017 (2) | 0.0033 (17) | −0.010 (2) |
| C21 | 0.0416 (17) | 0.0331 (17) | 0.0304 (18) | 0.0017 (17) | 0.0000 (16) | −0.0018 (16) |
| C22 | 0.050 (2) | 0.041 (2) | 0.040 (2) | −0.0004 (18) | −0.0025 (19) | −0.0011 (18) |
| C23 | 0.078 (3) | 0.056 (2) | 0.032 (2) | 0.007 (2) | −0.004 (2) | −0.005 (2) |
| C24 | 0.069 (3) | 0.061 (3) | 0.032 (2) | 0.010 (2) | 0.013 (2) | 0.010 (2) |
| F24 | 0.101 (2) | 0.131 (3) | 0.0449 (14) | 0.0048 (19) | 0.0275 (15) | 0.0101 (16) |
| C25 | 0.048 (2) | 0.066 (3) | 0.050 (2) | −0.005 (2) | 0.010 (2) | 0.010 (2) |
| C26 | 0.045 (2) | 0.053 (2) | 0.040 (2) | −0.0076 (19) | 0.0030 (18) | −0.0018 (19) |
| C31 | 0.0327 (17) | 0.0241 (15) | 0.0382 (19) | 0.0000 (14) | −0.0036 (15) | 0.0015 (14) |
| C32 | 0.0241 (14) | 0.0230 (14) | 0.0337 (18) | −0.0003 (13) | −0.0011 (13) | 0.0030 (15) |
| C33 | 0.0237 (14) | 0.0263 (14) | 0.0311 (17) | 0.0013 (14) | 0.0003 (14) | 0.0038 (15) |
| C34 | 0.0276 (14) | 0.0281 (15) | 0.0298 (17) | −0.0001 (15) | −0.0028 (14) | −0.0002 (16) |
| O31 | 0.0319 (12) | 0.0327 (12) | 0.0604 (16) | 0.0040 (11) | 0.0029 (11) | 0.0000 (13) |
| O32 | 0.0352 (12) | 0.0217 (10) | 0.096 (2) | −0.0011 (11) | 0.0024 (14) | 0.0034 (14) |
| O33 | 0.0244 (11) | 0.0261 (11) | 0.0639 (17) | −0.0006 (10) | −0.0059 (11) | 0.0078 (13) |
| O34 | 0.0400 (13) | 0.0421 (13) | 0.0371 (14) | 0.0082 (12) | 0.0024 (11) | 0.0137 (12) |
| O35 | 0.0533 (15) | 0.0436 (14) | 0.0391 (14) | 0.0126 (13) | 0.0113 (13) | −0.0048 (12) |
| O36 | 0.0399 (13) | 0.0205 (10) | 0.0419 (13) | 0.0022 (10) | 0.0052 (11) | 0.0037 (11) |
| O41 | 0.0408 (16) | 0.0624 (18) | 0.0666 (19) | −0.0045 (14) | −0.0111 (14) | 0.0216 (15) |
4-(4-Fluorophenyl)piperazin-1-ium hydrogen (2R,3R)-tartrate monohydrate (III) . Geometric parameters (Å, º)
| N1—C2 | 1.463 (4) | C23—H23 | 0.9300 |
| N1—C6 | 1.483 (4) | C24—C25 | 1.355 (5) |
| N1—H11 | 0.87 (4) | C24—F24 | 1.361 (4) |
| N1—H12 | 0.92 (4) | C25—C26 | 1.384 (5) |
| C2—C3 | 1.503 (5) | C25—H25 | 0.9300 |
| C2—H2A | 0.9700 | C26—H26 | 0.9300 |
| C2—H2B | 0.9700 | C31—O31 | 1.241 (4) |
| C3—N4 | 1.454 (4) | C31—O32 | 1.248 (4) |
| C3—H3A | 0.9700 | C31—C32 | 1.522 (4) |
| C3—H3B | 0.9700 | C32—O33 | 1.418 (3) |
| N4—C21 | 1.414 (4) | C32—C33 | 1.525 (4) |
| N4—C5 | 1.458 (4) | C32—H32A | 0.9800 |
| C5—C6 | 1.503 (5) | C33—O34 | 1.402 (3) |
| C5—H5A | 0.9700 | C33—C34 | 1.519 (4) |
| C5—H5B | 0.9700 | C33—H33A | 0.9800 |
| C6—H6A | 0.9700 | C34—O35 | 1.204 (4) |
| C6—H6B | 0.9700 | C34—O36 | 1.306 (4) |
| C21—C22 | 1.385 (4) | O33—H33 | 0.80 (4) |
| C21—C26 | 1.390 (5) | O34—H34 | 0.81 (4) |
| C22—C23 | 1.387 (5) | O36—H36 | 0.95 (4) |
| C22—H22 | 0.9300 | O41—H41 | 0.95 (5) |
| C23—C24 | 1.358 (6) | O41—H42 | 0.78 (5) |
| C2—N1—C6 | 111.5 (3) | C21—C22—H22 | 119.3 |
| C2—N1—H11 | 115 (2) | C23—C22—H22 | 119.3 |
| C6—N1—H11 | 108 (3) | C24—C23—C22 | 118.4 (4) |
| C2—N1—H12 | 108 (2) | C24—C23—H23 | 120.8 |
| C6—N1—H12 | 112 (3) | C22—C23—H23 | 120.8 |
| H11—N1—H12 | 102 (3) | C25—C24—C23 | 122.3 (4) |
| N1—C2—C3 | 111.5 (3) | C25—C24—F24 | 118.9 (4) |
| N1—C2—H2A | 109.3 | C23—C24—F24 | 118.8 (4) |
| C3—C2—H2A | 109.3 | C24—C25—C26 | 119.3 (4) |
| N1—C2—H2B | 109.3 | C24—C25—H25 | 120.3 |
| C3—C2—H2B | 109.3 | C26—C25—H25 | 120.3 |
| H2A—C2—H2B | 108.0 | C25—C26—C21 | 120.6 (3) |
| N4—C3—C2 | 110.8 (3) | C25—C26—H26 | 119.7 |
| N4—C3—H3A | 109.5 | C21—C26—H26 | 119.7 |
| C2—C3—H3A | 109.5 | O31—C31—O32 | 126.0 (3) |
| N4—C3—H3B | 109.5 | O31—C31—C32 | 118.0 (3) |
| C2—C3—H3B | 109.5 | O32—C31—C32 | 116.0 (3) |
| H3A—C3—H3B | 108.1 | O33—C32—C31 | 112.1 (2) |
| C21—N4—C3 | 116.4 (3) | O33—C32—C33 | 109.4 (2) |
| C21—N4—C5 | 115.4 (3) | C31—C32—C33 | 110.2 (2) |
| C3—N4—C5 | 110.2 (3) | O33—C32—H32A | 108.4 |
| N4—C5—C6 | 111.3 (3) | C31—C32—H32A | 108.4 |
| N4—C5—H5A | 109.4 | C33—C32—H32A | 108.4 |
| C6—C5—H5A | 109.4 | O34—C33—C34 | 111.6 (2) |
| N4—C5—H5B | 109.4 | O34—C33—C32 | 110.7 (2) |
| C6—C5—H5B | 109.4 | C34—C33—C32 | 109.5 (2) |
| H5A—C5—H5B | 108.0 | O34—C33—H33A | 108.3 |
| N1—C6—C5 | 109.9 (3) | C34—C33—H33A | 108.3 |
| N1—C6—H6A | 109.7 | C32—C33—H33A | 108.3 |
| C5—C6—H6A | 109.7 | O35—C34—O36 | 125.5 (3) |
| N1—C6—H6B | 109.7 | O35—C34—C33 | 122.7 (3) |
| C5—C6—H6B | 109.7 | O36—C34—C33 | 111.8 (3) |
| H6A—C6—H6B | 108.2 | C32—O33—H33 | 105 (3) |
| C22—C21—C26 | 117.9 (3) | C33—O34—H34 | 112 (3) |
| C22—C21—N4 | 123.3 (3) | C34—O36—H36 | 113 (2) |
| C26—C21—N4 | 118.8 (3) | H41—O41—H42 | 108 (4) |
| C21—C22—C23 | 121.4 (4) | ||
| C6—N1—C2—C3 | −54.1 (4) | C23—C24—C25—C26 | −1.0 (6) |
| N1—C2—C3—N4 | 55.9 (4) | F24—C24—C25—C26 | −179.4 (3) |
| C2—C3—N4—C21 | 168.2 (3) | C24—C25—C26—C21 | −0.2 (6) |
| C2—C3—N4—C5 | −57.9 (4) | C22—C21—C26—C25 | 0.4 (5) |
| C21—N4—C5—C6 | −166.5 (3) | N4—C21—C26—C25 | 178.2 (3) |
| C3—N4—C5—C6 | 59.1 (4) | O31—C31—C32—O33 | −173.2 (3) |
| C2—N1—C6—C5 | 54.1 (4) | O32—C31—C32—O33 | 7.7 (4) |
| N4—C5—C6—N1 | −56.8 (4) | O31—C31—C32—C33 | 64.7 (4) |
| C3—N4—C21—C22 | 3.3 (5) | O32—C31—C32—C33 | −114.4 (3) |
| C5—N4—C21—C22 | −128.2 (3) | O33—C32—C33—O34 | −66.5 (3) |
| C3—N4—C21—C26 | −174.3 (3) | C31—C32—C33—O34 | 57.2 (3) |
| C5—N4—C21—C26 | 54.1 (4) | O33—C32—C33—C34 | 56.9 (3) |
| C26—C21—C22—C23 | 0.6 (5) | C31—C32—C33—C34 | −179.4 (2) |
| N4—C21—C22—C23 | −177.1 (3) | O34—C33—C34—O35 | 4.2 (4) |
| C21—C22—C23—C24 | −1.7 (6) | C32—C33—C34—O35 | −118.7 (3) |
| C22—C23—C24—C25 | 2.0 (6) | O34—C33—C34—O36 | −177.5 (2) |
| C22—C23—C24—F24 | −179.6 (3) | C32—C33—C34—O36 | 59.5 (3) |
4-(4-Fluorophenyl)piperazin-1-ium hydrogen (2R,3R)-tartrate monohydrate (III) . Hydrogen-bond geometry (Å, º)
Cg1 represents the centroid of the ring (C21–C26).
| D—H···A | D—H | H···A | D···A | D—H···A |
| N1—H11···O36 | 0.87 (4) | 2.31 (4) | 2.929 (4) | 128 (3) |
| N1—H11···O35i | 0.87 (4) | 2.17 (4) | 2.855 (4) | 136 (3) |
| N1—H12···O41 | 0.92 (4) | 1.83 (4) | 2.740 (5) | 169 (3) |
| O33—H33···O32 | 0.80 (4) | 2.19 (4) | 2.614 (3) | 113 (3) |
| O33—H33···O34ii | 0.80 (4) | 2.10 (4) | 2.805 (3) | 146 (3) |
| O34—H34···O35 | 0.81 (4) | 2.41 (4) | 2.702 (3) | 102 (3) |
| O34—H34···O31ii | 0.81 (4) | 2.07 (4) | 2.806 (3) | 151 (4) |
| O36—H36···O32iii | 0.95 (4) | 1.53 (4) | 2.470 (3) | 175 (3) |
| O41—H41···O31 | 0.96 (5) | 1.82 (5) | 2.771 (4) | 178 (5) |
| O41—H42···O33iv | 0.78 (5) | 2.00 (5) | 2.754 (4) | 163 (5) |
| C25—H25···Cg1v | 0.93 | 2.86 | 3.649 (5) | 144 |
Symmetry codes: (i) x−1/2, −y+1/2, −z+1; (ii) x+1/2, −y+3/2, −z+1; (iii) x, y−1, z; (iv) x−1, y, z; (v) −x, y−1/2, −z+3/2.
Funding Statement
This work was funded by University Grants Commission grant .
References
- Allen, F. H., Kennard, O., Watson, D. G., Brammer, L., Orpen, A. G. & Taylor, R. (1987). J. Chem. Soc. Perkin Trans. 2, pp. S1–S19.
- Bernstein, J., Davis, R. E., Shimoni, L. & Chang, N.-L. (1995). Angew. Chem. Int. Ed. Engl. 34, 1555–1573.
- Boeyens, J. C. A. (1978). J. Cryst. Mol. Struct. 8, 317–320.
- Cremer, D. & Pople, J. A. (1975). J. Am. Chem. Soc. 97, 1354–1358.
- Dayananda, A. S., Dutkiewicz, G., Yathirajan, H. S., Ramesha, A. R. & Kubicki, M. (2012a). Acta Cryst. E68, o2817. [DOI] [PMC free article] [PubMed]
- Dayananda, A. S., Yathirajan, H. S., Keeley, A. C. & Jasinski, J. P. (2012b). Acta Cryst. E68, o2237. [DOI] [PMC free article] [PubMed]
- Etter, M. C. (1990). Acc. Chem. Res. 23, 120–126.
- Etter, M. C., MacDonald, J. C. & Bernstein, J. (1990). Acta Cryst. B46, 256–262. [DOI] [PubMed]
- Ferguson, G., Glidewell, C., Gregson, R. M. & Meehan, P. R. (1998a). Acta Cryst. B54, 129–138.
- Ferguson, G., Glidewell, C., Gregson, R. M. & Meehan, P. R. (1998b). Acta Cryst. B54, 139–150.
- Flack, H. D. (1983). Acta Cryst. A39, 876–881.
- Flack, H. D. & Bernardinelli, G. (2000). J. Appl. Cryst. 33, 1143–1148.
- Gregson, R. M., Glidewell, C., Ferguson, G. & Lough, A. J. (2000). Acta Cryst. B56, 39–57. [DOI] [PubMed]
- Keane, P. E., Benedetti, M. S. & Dow, J. (1982). Neuropharmacology, 21, 163–169. [DOI] [PubMed]
- Kiran Kumar, H., Yathirajan, H. S., Foro, S. & Glidewell, C. (2019). Acta Cryst. E75, 1494–1506. [DOI] [PMC free article] [PubMed]
- Kiran Kumar, H., Yathirajan, H. S., Harish Chinthal, C., Foro, S. & Glidewell, C. (2020). Acta Cryst. E76, 488–495. [DOI] [PMC free article] [PubMed]
- Nagai, F., Nonaka, R. & Kamimura, K. S. H. (2007). Eur. J. Pharmacol. 559, 132–137. [DOI] [PubMed]
- Oxford Diffraction (2009). CrysAlis CCD and CrysAlis RED. Oxford Diffraction Ltd, Abingdon, Oxfordshire, England.
- Parsons, S., Flack, H. D. & Wagner, T. (2013). Acta Cryst. B69, 249–259. [DOI] [PMC free article] [PubMed]
- Prasanna, M. D. & Row, T. N. G. (2001). J. Mol. Struct. 562, 55–61.
- Rossi, P. G., Posar, A., Parmeggiani, A., Pipitone, E. & D’Agata, M. (1999). J. Child Neurol. 14, 547–550. [DOI] [PubMed]
- Sagar, B. K., Girisha, M., Yathirajan, H. S., Rathore, R. S. & Glidewell, C. (2017). Acta Cryst. E73, 1320–1325. [DOI] [PMC free article] [PubMed]
- Sanjuan, M., Rovei, V., Dow, J. & Benedetti, R. S. (1983). Int. J. Mass Spectrom. Ion Phys. 48, 93–96.
- Shaibah, M. A. E., Sagar, B. K., Yathirajan, H. S., Kumar, S. M. & Glidewell, C. (2017a). Acta Cryst. E73, 1513–1516. [DOI] [PMC free article] [PubMed]
- Shaibah, M. A. E., Yathirajan, H. S., Kumar, S. M., Byrappa, K. & Glidewell, C. (2017b). Acta Cryst. E73, 1488–1492. [DOI] [PMC free article] [PubMed]
- Sheldrick, G. M. (2015a). Acta Cryst. A71, 3–8.
- Sheldrick, G. M. (2015b). Acta Cryst. C71, 3–8.
- Spek, A. L. (2020). Acta Cryst. E76, 1–11. [DOI] [PMC free article] [PubMed]
- Wood, P. A., Allen, F. H. & Pidcock, E. (2009). CrystEngComm, 11, 1563–1571.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablock(s) global, I, II, III. DOI: 10.1107/S2056989020006398/wm5557sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S2056989020006398/wm5557Isup2.hkl
Structure factors: contains datablock(s) II. DOI: 10.1107/S2056989020006398/wm5557IIsup3.hkl
Structure factors: contains datablock(s) III. DOI: 10.1107/S2056989020006398/wm5557IIIsup4.hkl
Supporting information file. DOI: 10.1107/S2056989020006398/wm5557Isup5.cml
Additional supporting information: crystallographic information; 3D view; checkCIF report