

Abstract
In diabetic patients, treatment with insulin and pramlintide (an amylin analogue) is more effective than treatment with insulin only. But because mixtures of insulin and pramlintide are unstable and have to be injected separately, amylin analogues are only used by 1.5% of diabetics needing rapid-acting insulin. Here, we show that the supramolecular modification of insulin and pramlintide with cucurbit[7]uril-conjugated polyethylene glycol improves the pharmacokinetics of the dual-hormone therapy and enhances post-prandial glucagon suppression in diabetic pigs. The co-formulation is stable for over 100 hours at 37 ºC under continuous agitation, whereas commercial formulations of insulin analogues aggregate after 10 hours under similar conditions. In diabetic rats, the administration of the stabilized co-formulation increased the area-of-overlap ratio of the pharmacokinetic curves of pramlintide and insulin to 0.7 ± 0.1 from 0.4 ± 0.2 (mean ± s.d.) for the separate administration of the hormones. The co-administration of supramolecularly stabilized insulin and pramlintide better mimics the endogenous kinetics of co-secreted insulin and amylin, and holds promise as a dual-hormone replacement therapy.
There are over 422 million people living with diabetes worldwide, and 5-10% of these people have type 1 diabetes.[1] These patients often suffer from severe side effects such as renal failure, heart disease, vision loss, and limb amputation, which can all be prevented with tight glycemic control. Type 1 diabetes occurs after an autoimmune response resulting in the destruction of pancreatic β cells responsible for production and secretion of metabolically active hormones including insulin and amylin. Patients with type 1 diabetes, therefore, cannot produce the insulin required for glucose uptake by cells. Amylin complements the action of insulin to regulate blood glucose levels by acting centrally to slow gastric emptying, suppress postprandial glucagon secretion, and decrease food intake by increasing satiety (Figure 1a).[2–10] Similar to insulin, amylin production is insufficient at diagnosis and deteriorates with on-going autoimmune destruction in individuals with type 1 diabetes. Further, patients with type 1 diabetes experience additional loss of metabolic signaling such as suppression of post-prandial glucagon secretion, resulting in glucagon-driven glycogenolysis that compounds meal-time hyperglycemic excursions (Figure 1a).
Fig. 1 |. CB[7]-PEG binds to insulin and pramlintide and alters diffusion rates in formulation.
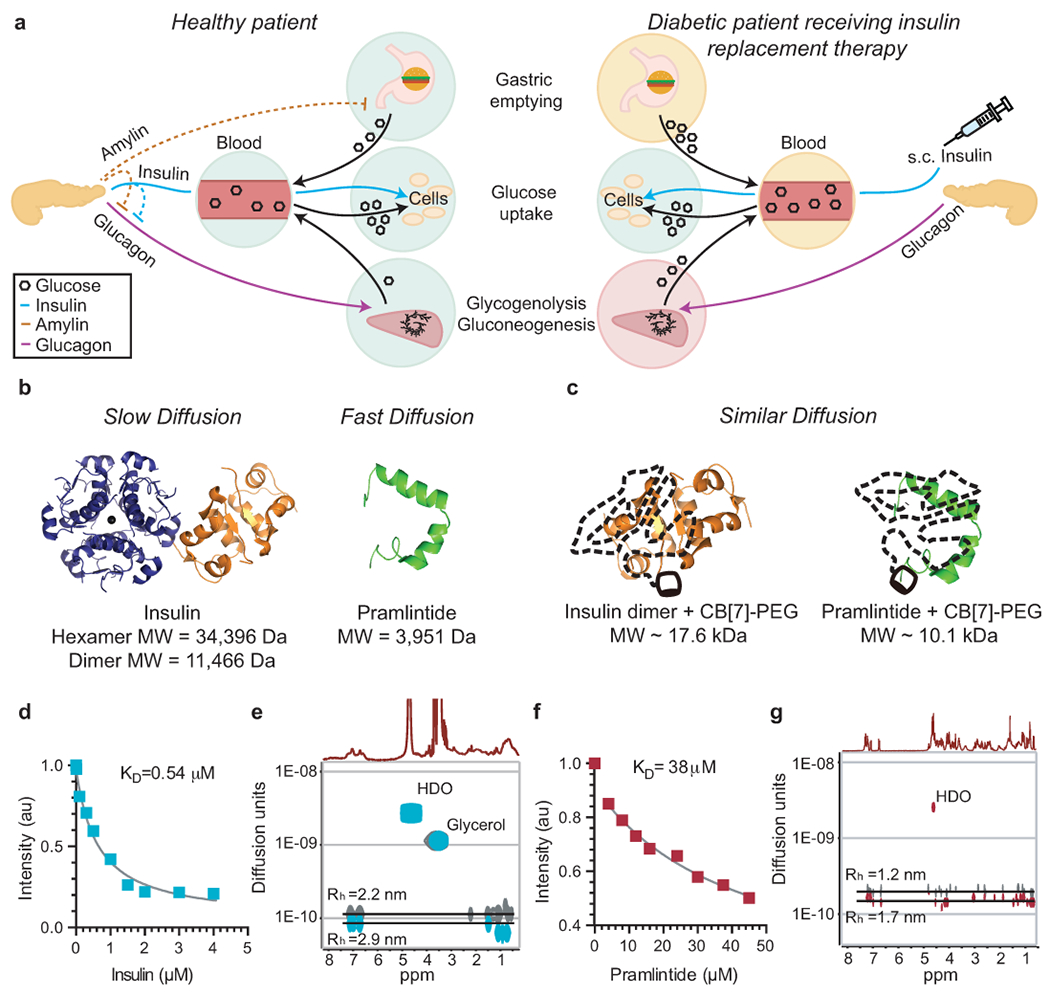
Scheme of post-mealtime metabolic signaling pathways in a, non-diabetic people and type 1 diabetic people receiving insulin replacement therapy. In non-diabetic people, endogenous insulin promotes cellular glucose uptake and acts with amylin to locally suppress post-prandial glucagon, thus decreasing glycogenolysis & gluconeogenesis. In contrast, treatment of diabetic patients with s.c. insulin alone cannot restore glucagon suppression. Amylin replacement is critical to fully restore metabolic signaling and constitute a true hormone replacement therapy. b, c, Scheme demonstrating how molecular weight affects diffusion rates, which directly impacts absorption kinetics following s.c. administration. b, Standard insulin formulations comprise a mixture of insulin aggregation states (i.e., hexamers and dimers) that exhibit extended duration of insulin action when injected into the s.c. space. In contrast, the pramlintide monomer is rapidly absorbed into the blood. c, After complexation with CB[7]-PEG such that only insulin dimers exist in formulation, insulin and pramlintide have more similar molecular weights and diffusion rates to one another. Acridine orange competitive binding assay of d, aspart (n=1 independent experiment) and f, pramlintide (n=1 independent experiment), indicating binding of CB[7] to both proteins. Diffusion-ordered NMR Spectroscopy (DOSY) provides insight into the formation of protein/CB[7]-PEG complexes and their rates of diffusion in formulation. In these studies, e, Aspart/CB[7]-PEG complex (cyan) exhibits a 30% reduction in diffusion rate when compared to standard dimeric aspart (grey). Moreover, g, Pramlintide/CB[7]-PEG complex (red) exhibits approximately a two-fold reduction in diffusion rate when compared to pramlintide alone (grey). Complexation of the two proteins with CB[7]-PEG results in a ratio in diffusion rates of pramlintide/CB[7]-PEG to aspart/CB[7]-PEG of only 1.6, compared to 2.3 for pramlintide and aspart in typical formulations, indicating the proteins have more similar diffusivities in co-formulation.
Insulin replacement therapy has been the focus of diabetes treatment for over 80 years, yet amylin has largely been overlooked. Current treatments use subcutaneous injections or infusion from pumps to deliver insulin. A true hormone replacement therapy for patients with type 1 diabetes would simultaneously deliver amylin and insulin. Amylin replacement therapy is critical to regain suppression of post-prandial glucagon, which cannot be achieved with subcutaneous insulin delivery alone (Figure 1, Figure S1). Amylin replacement therapy has proven to be challenging because amylin is highly unstable in formulation and rapidly aggregates into amyloid fibrils,[11] prompting the development of the amylin analogue, pramlintide, which acts through similar mechanisms to amylin in vivo. Pramlintide differs from amylin by alterations to three amino acids that suppress amyloid fibrillation and enable its stable formulation at pH~4.[2–9] Unfortunately, insulin and its analogues are typically formulated at pH~7.4, meaning that insulin and pramlintide must be administered in two separate injections. Patients treated with a combination of insulin and pramlintide at mealtimes have been shown to have improved glycemic control, observed as a 0.3% decrease in HbA1c levels, when compared with patients treated with insulin alone.[4, 5, 7, 8, 12–14] Despite the increased efficacy of dual-hormone treatment, by 2012 only 29,000 patients of over 2,100,000 patients who would potentially benefit from such a treatment had adopted it due to the burdensome requirement for administration in two separate injections.[15]
In addition to formulation challenges, the pharmacokinetics of insulin and pramlintide in current formulations are highly dissimilar and the resulting lack of pharmacokinetic overlap does not mimic their natural mode of action. In non-diabetic people, insulin and amylin are co-secreted at a fixed ratio from the β-cells in the pancreas and act with similar kinetics.[16] In contrast, current “rapid-acting” insulin analogue formulations Humalog (insulin lispro) and Novolog (insulin aspart) exhibit delayed onset of action of ~20-30 min, peak action at ~60-90 min and total duration of action of ~3-4 hours,[4, 6, 17, 18] while Symlin (pramlintide) begins to act almost immediately, exhibits peak action at ~20 min and total duration of action of ~90 min. This large dissimilarity in pharmacokinetics arises from the distinct aggregation states of the proteins in formulation and the resulting impact on absorption behaviour. These insulin formulations contain a mixture of hexamers, dimers and monomers, which, upon subcutaneous injection, dissociate and are absorbed at different rates resulting in the delayed onset and long duration of action of these formulations (Figure 1b).[19–21] In contrast, the pramlintide monomer is absorbed rapidly from the subcutaneous space (Figure 1b). The lack of overlap between insulin and pramlintide pharmacokinetics in current treatment strategies hinders the synergistic effects of pramlintide and insulin action. Recent clinical studies are moving towards evaluating the benefits of delivering a fixed ratio of insulin and pramlintide using two separate pumps to better simulate endogenous insulin-pramlintide secretion.[22–24] While the use of two separate pumps can deliver a fixed ratio of pramlintide with insulin,[24] this method is overly burdensome outside of a research setting and does not address the poor pharmacokinetic overlap of these two hormones following subcutaneous administration.
A new class of excipients are needed for protein formulation to address concerns surrounding aggregation and denaturation over time.[25, 26] Covalent PEGylation has been successful as a strategy to stabilize insulin and amylin in formulation;[27–29] however, covalent modification of proteins often interferes with their activity, typically extends their pharmacokinetics in vivo, and can lead to increased immunogenicity.[30] Recent research has shown that non-covalent modification of proteins can enhance their stability in formulation.[31, 32] In particular, cucurbit[n]urils (CB[n]) are a family of macrocyclic hosts that exhibit strong binding affinities for aromatic amino acids,[33–36] and have a reassuring safety profile.[37–39] Conjugation of a polyethylene glycol (PEG) chain to CB[7] creates a designer excipient (CB[7]-PEG) for non-covalent PEGylation of protein therapeutics. Insulin has an N-terminal phenylalanine and pramlintide has an amidated C-terminal tyrosine, making them ideal targets for supramolecular modification using the CB[7]-PEG system.[31] In this work, we exploit CB[7]-PEG for simultaneous supramolecular PEGylation of insulin and pramlintide to stabilize the two hormones in a co-formulation whereby the therapeutic ratio is defined in the formulation. We demonstrate that this dual-hormone therapy can be administered in a single injection, thus reducing burden, and that increased overlap of the pharmacokinetics of the two pharmaceuticals restores post-prandial glucagon suppression in a swine model of insulin-deficient diabetes for tighter glycemic control, and enhanced diabetes management.
Results
Characterization of CB[7]-PEG binding.
CB[7]-PEG with varying PEG molecular weights has been shown to bind to recombinant human insulin with micromolar affinities, increasing its stability in formulation and enabling simple tuning of the duration of insulin action in a mouse model of insulin-deficient diabetes through modulation of the PEG molecular weight.[31] In this study, we chose to work with CB[7]-PEG5k on account of its demonstrated capacity to stabilize recombinant human insulin in formulation without significantly extending insulin duration of action in vivo. We aimed to not only stabilize insulin and pramlintide together in formulation, but to also use co-formulation as an opportunity to simultaneously alter the pharmacokinetics of the two hormones in vivo to more closely match one another. Through a combination of insulin hexamer disruption by removal of formulation zinc and simultaneous complexation of insulin and pramlintide with CB[7]-PEG, the effective hydrodynamic size of both components become similar to one another (Figure 1b,c). We hypothesized this similarity in hydrodynamic size, which directly impacts absorption following subcutaneous administration, would promote greater overlap between the pharmacokinetic profiles of the two therapeutics.
We evaluated an insulin analogue, aspart, because it is the active ingredient in the most commonly used commercial rapid-acting insulin formulation, Novolog. We determined the binding affinity of CB[7] to aspart and pramlintide using a competitive binding assay with acridine orange (Figure 1d,f). The binding of CB[7] to aspart was determined to be 0.54 μM, which is similar to values previously reported for binding to recombinant insulin,[31] while the binding to pramlintide was determined to be 38 μM. The higher binding affinity of CB[7] to insulin compared to pramlintide is due to the well-documented higher binding affinity of CB[7] to N-terminal aromatic amino acids on account of the hydrophobic guest being flanked by a protonated amine group.[31] Circular dichroism confirmed that binding of both aspart and pramlintide with CB[7]-PEG did not affect protein structure (See Supplemental Information, Figure S2).
We then used diffusion-ordered NMR spectroscopy (DOSY) to provide insight into the hydrodynamic size and diffusion characteristics of the protein/CB[7]-PEG complexes (Figure 1e,g; Figure S3, S4). In these studies, aspart was formulated with CB[7]-PEG and ethylenediaminetetraacetic acid (EDTA) to remove formulation zinc. EDTA forms strong complexes with zinc (KD ~ 10×10−18 M)[40, 41] and addition of one molar equivalent of EDTA relative to zinc found in insulin formulations rapidly sequesters the zinc, preventing it from interacting with the insulin and disrupting insulin hexamer formation in solution. In DOSY experiments, CB[7]-PEG and aspart were found to diffuse together, verifying the binding interaction observed previously using competitive binding assays. The aspart dimer exhibited a diffusion rate of D~1.2×10−10 m2 s−1, while the complex of aspart/CB[7]-PEG exhibited a 30% lower diffusion rate of D~8.7×10−11 m2 s−1. The Stokes-Einstein relationship specifies that the diffusion rate, D, is inversely proportional to the size of the species in solution, whereby a 50% increase in the molecular weight is expected to decrease the diffusion rate by roughly 1/3, as observed in this study. We use this relationship to approximate the hydrodynamic radius (Rh) to be 2.2 nm for dimeric aspart and 2.9 nm for the aspart/CB[7]-PEG complex. For comparison, the insulin hexamer has a hydrodynamic radius of approximately 2.8 nm.[42]
Similarly, the diffusion rate for pramlintide decreases from D~2×10−10 m2 s−1 for the protein alone to D~1.4×10−10 m2 s−1 for the pramlintide/CB[7]-PEG complex, corresponding to a change in Rh from 1.2 nm to 1.7 nm. The degree of diffusion rate increase after the addition of CB[7]-PEG to pramlintide is less than that observed for aspart, likely on account of the weaker and more dynamic binding. We observed that the ratio of the diffusion rates for the pramlintide/CB[7]-PEG and aspart/CB[7]-PEG complexes is ~1.6, while the ratio of diffusion rates for pramlintide alone and insulin in a standard formulation is ~2.3. These observations suggested that the zinc-free co-formulation of the two protein/CB[7]-PEG complexes makes the two hormones more similar in hydrodynamic size than is possible with standard formulation approaches.
Formulation stability in vitro.
To determine if CB[7]-PEG stabilizes pramlintide in combination with insulin at physiological pH, insulin and pramlintide aggregation was assessed under stressed conditions (37ºC with continuous agitation) over time. As insulin and pramlintide destabilize, they form amyloid fibrils that are insoluble, inactive, and often immunogenic.[43–45] These aggregates are large and scatter light, and thus the degree of aggregation can be evaluated by measuring the change in transmittance over time.[31]
Commercial Novolog and Humalog both aggregate in these stressed aging conditions after 10 ± 1 hrs and 6 ± 0.2 hrs respectively, but are both stabilized for over 100 hours when formulated with CB[7]-PEG (See Supplemental Information, Figure S5). Pramlintide formulated in sodium acetate buffer (pH=4; similar to the commercial formulation Symlin) was stable for over 100 hours under stressed conditions (Figure 2a); however, when formulated in PBS (pH=7.4), pramlintide aggregated after only 15 ± 4 hrs, indicating a dramatic reduction in stability at physiologic pH. In contrast, when formulated with CB[7]-PEG in PBS (pH=7.4), pramlintide remained stable for over a 100 hours under stressed conditions.
Fig. 2 |. Formulation with CB[7]-PEG stabilizes a co-formulation of Novolog or Humalog and pramlintide at physiological pH.
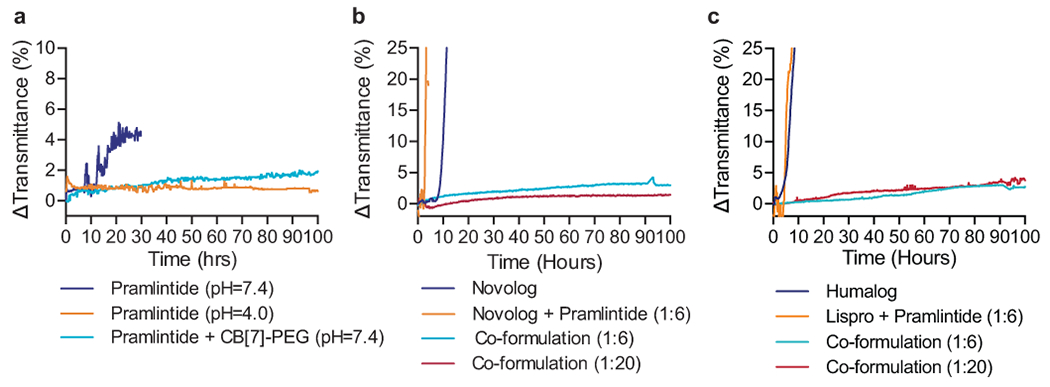
a, In vitro stability of pramlintide formulations at various pH values with and without CB[7]-PEG. b, In vitro stability of pramlintide-aspart (1:6 and 1:20 molar ratio) co-formulations with CB[7]-PEG at physiological pH. c, In vitro stability of pramlintide-lispro (1:6 and 1:20 molar ratio) co-formulations with CB[7]-PEG at physiological pH. Co-formulations were compared to controls of commercial Novolog or Humalog, and mixtures of the incompatible aspart+pramlintide or lispro+pramlintide in the absence of CB[7]-PEG. These assays assess the aggregation of proteins in formulation over time during stressed aging (i.e., continuous agitation at 37ºC) by monitoring changes in transmittance at 540nm. These experiments demonstrate that formulation with CB[7]-PEG prevents protein aggregation over the 100h period assayed, even when commercial formulations aggregate within 10h. Data shown are average transmittance traces for n = 3 samples per group.
In addition to stabilizing pramlintide and insulin analogues separately, CB[7]-PEG also facilitated the development of a stable insulin-pramlintide co-formulation (Figure 2b–c). Pramlintide co-formulated with either aspart or lispro in PBS (pH=7.4) in the absence of CB[7]-PEG aggregated after only 2.9 ± 0.2 hrs (aspart+pramlintide) or 4.9 ± 0.3 hrs (lispro+pramlintide) under stressed conditions, while co-formulation with CB[7]-PEG in the same buffer conditions was completely stable for the duration of the 100-hour kinetic study. These results demonstrate that simultaneous supramolecular PEGylation of pramlintide with either aspart or lispro and CB[7]-PEG enables the development of a viable dual-hormone co-formulation.
Pharmacodynamics and pharmacokinetics in diabetic rats.
Having established the stability of the insulin–pramlintide co-formulation, we evaluated our co-formulation in vivo by measuring blood glucose as well as insulin and pramlintide pharmacokinetics in a well-studied rat model of insulin-deficient diabetes.[46] We hypothesized that increasing the overlap between insulin and pramlintide pharmacokinetics will enable the development of a more physiologically-relevant dual-hormone treatment. Here Novolog refers to administration of the available commercial formulation, whereas insulin aspart formulations contain isolated zinc-free insulin aspart. In these studies, aspart-pramlintide co-formulations (PBS at pH=7) comprising zinc-free aspart (1.5U/kg), CB[7]-PEG (5 equivalents relative to insulin), and pramlintide (either 1:15, 1:8 or 1:2 equivalents relative to insulin) were compared to commercial Novolog alone (1.5U/kg) and to the clinically relevant combination of Novolog (1.5U/kg) and pramlintide (sodium acetate buffer at pH=4) administered in separate injections (Figure 3a–c). The rationale of pramlintide concentrations is discussed in the supplemental information.
Fig. 3 |. Aspart and pramlintide pharmacokinetics following different administration routes in diabetic rats.
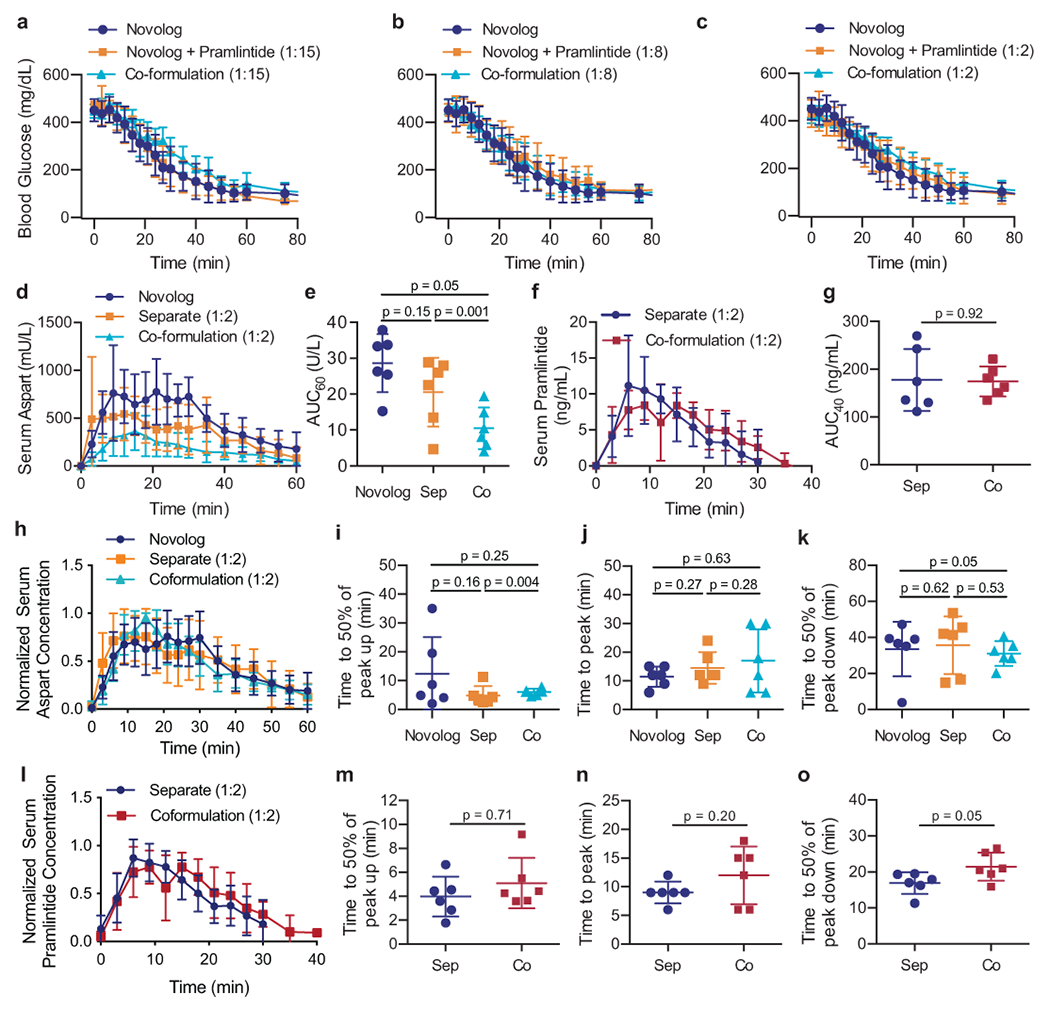
Fasted diabetic male rats (n=6) received subcutaneous administration of therapies comprising either (i) commercial Novolog, (ii) commercial Novolog and pramlintide (pH=4) delivered in separate injections, or (iii) aspart-pramlintide co-formulation with CB[7]-PEG. All treatment groups received 1.5U/kg of insulin. Blood glucose levels were evaluated at several ratios of pramlintide to aspart: a, 1:15, b, 1:8, c, 1:2. All pharmacokinetic studies were evaluated with pramlintide at 1:2 aspart:pramlintide ratio. Pharmacokinetics of d, insulin aspart in mU/L or f, pramlintide in ng/mL. The area under the pharmacokinetic curves (AUC) of e, aspart (**p=0.0012) and g, pramlintide for the first 60 minutes or 40 minutes, respectively, after subcutaneous injection. Pharmacokinetics for each rat were individually normalized to peak serum and normalized values were averaged for h, aspart or l, pramlintide concentration for each treatment group. Time to reach 50% of peak i, aspart or m, pramlintide serum concentration (onset). Time to reach peak j, aspart or n, pramlintide serum concentration. Time for k, aspart or o, pramlintide depletion to 50% of peak serum concentration (*p=0.047). Error bars indicate mean ± s.d. with n=6 animals for all groups. Statistical significance was determined by a two-tailed student’s t-test.
The rate of blood glucose depletion following administration in fasted diabetic rats was similar between all treatment groups and a blood glucose drop from 448 ± 17 mg/dL (t=0) to 116 ± 17 mg/dL (t=60 min) was observed across all treatment groups. The molar ratio of pramlintide to Novolog had no effect on the rate or degree of blood glucose depletion.
Serum concentrations of insulin and pramlintide were measured over time by ELISA following subcutaneous administration of each of the treatment groups outlined above to assess the degree of overlap between the pharmacokinetic profiles of the two hormones. Aspart area under the curve (AUC) following the administration in co-formulation with pramlintide (10 ± 6 mU/mL) was significantly lower than when administered alone in commercial Novolog (29 ± 8 mU/mL) (t=4.47; df=10; 95% CI [−27203, −9099]; p=0.0012) (Figure 3e). These results suggest that pramlintide affects aspart serum concentrations and that this effect is amplified when the dual-hormone therapy is administered in a co-formulation treatment rather than in two separate injections (See Supplemental Information). The “onset” rate of fast-acting insulins is often determined using two metrics: (i) time-to-50% normalized peak height up, and (ii) time-to-peak insulin serum concentration. Normalized serum concentration data was used to compare the time-to-peak aspart concentrations between treatment groups (Figure 3h). No difference was seen in aspart time to peak, or time-to-50% normalized peak height up following administration of commercial Novolog alone, Novolog alongside a separate injection to pramlintide, or administration in a single co-formulation injection (Figure 3i–j). There was also no significant difference in aspart duration-of-action, determined by measuring the terminal time-to-50% normalized peak height, between treatment groups (Figure 3k).
When evaluating pramlintide pharmacokinetics, no significant differences were seen in pramlintide AUC following different treatments (Figure 3f–g). No significant difference was seen in pramlintide time-to-onset or peak action when pramlintide and insulin are administered in separate injections compared to the insulin-pramlintide co-formulation (Figure 3m–n). In contrast, pramlintide duration-of-action was extended from 17 ± 3 min for separate administrations to 21 ± 4 min for the co-formulation (t=2.26; df=10; 95% CI [0.07, 9.065]; p=0.047) (Figure 3o). As hypothesized, extended pramlintide duration of action resulted in increased overlap between aspart and pramlintide pharmacokinetics (t=4.01; df=10; 95% CI [0.15, 0.52]; p=0.0025). The overlap of the pharmacokinetic profiles can be represented by the ratio of AUC of serum pramlintide to serum aspart, which was increased from 0.4 ± 0.2 when these proteins are delivered separately to 0.7 ± 0.1 when delivered in co-formulation (Figure 4).
Fig. 4 |. Administration of aspart and pramlintide as a co-formulation in diabetic rats enhances pharmacokinetic overlap.
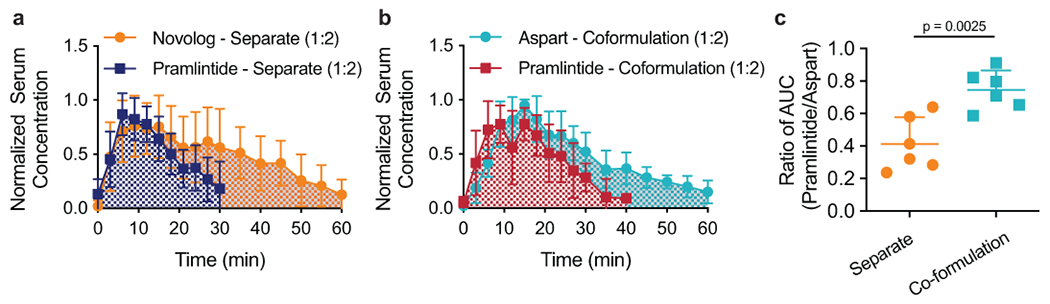
Mean normalized serum concentration (normalized for each individual rat) of Novolog and Pramlintide when administered as a, two separate injections or b, pramlintide-aspart co-formulation with CB[7]-PEG at physiologic pH. c, Ratio of the area under the curve (AUC) of the pharmacokinetic profiles of pramlintide and aspart for administration as separate injections and as a co-formulation (**p=0.0025). Error bars indicate mean ± s.d. with n=6 animals for all groups. Statistical significance was determined by a two-tailed student’s t-test.
Pharmacodynamics and pharmacokinetics in diabetic pigs.
To assess translationally relevant pharmacokinetics and post-prandial treatment benefits (See Supplemental Information) of insulin-pramlintide co-formulations, we conducted studies in a swine model of insulin-deficient diabetes. Initially, fasted diabetic swine were treated with either (i) commercial Humalog (4U; 0.13 U/kg), (ii) separate injections of commercial Humalog (4U; 0.13 U/kg) and pramlintide (pH=4; pramlintide:insulin ratio of 1:6), or (iii) lispro-pramlintide co-formulation (4U insulin; pramlintide:insulin ratio of 1:6; molar ratio of 3:1 CB[7]-PEG:lispro+pramlintide) (Figure S7). While pigs who received a meal, but no insulin showed the expected increase in blood glucose levels, no difference in blood glucose was observed between each of the formulations tested, and no post-prandial glucose excursions were observed in treated groups (Figure S6). Serum or plasma concentrations of lispro and pramlintide were then measured over time by ELISA following a meal given simultaneously with subcutaneous administration of each of the treatment groups outlined above (Figure 5). The AUC of the lispro pharmacokinetic curve when delivered as a part of the co-formulation was lower than when delivered in a separate injection from insulin. Consistent with the data in rats, no differences were observed in lispro time-to-onset, peak action, or duration-of-action between all treatment groups (Figure 5e–h). Pramlintide pharmacokinetics demonstrated increased time-to-onset (t=2.53; df=24; 95% CI [0.67, 6.55]; p=0.018) and prolonged duration-of-action (t=2.53; df=24; 95% CI [2.06, 20.47]; p=0.019) when administered as a part of the co-formulation (onset: 9 ± 3 minutes; duration: 58 ± 7 minutes) compared to administration in a separate injection (onset: 6 ± 4 minutes; duration: 47 ± 14 minutes) (Figure 5i–l). These observations corroborate the observations on pramlintide pharmacokinetics made in rats. The modulation of the pramlintide pharmacokinetics was confirmed by an increase in overlap between insulin and pramlintide pharmacokinetic curves when administered as a co-formulation compared to administration in separate injections (Figure 6a–c). The ratio of the overlap time over the total time at half-peak height was determined to be 0.67 ± 0.29 for the co-formulation and 0.42 ± 0.30 for separate injections (t=2.15; df=24; 95% CI [0.010, 0.487]; p=0.042).
Fig. 5 |. Lispro and pramlintide pharmacokinetics following different administration routes in diabetic pigs.
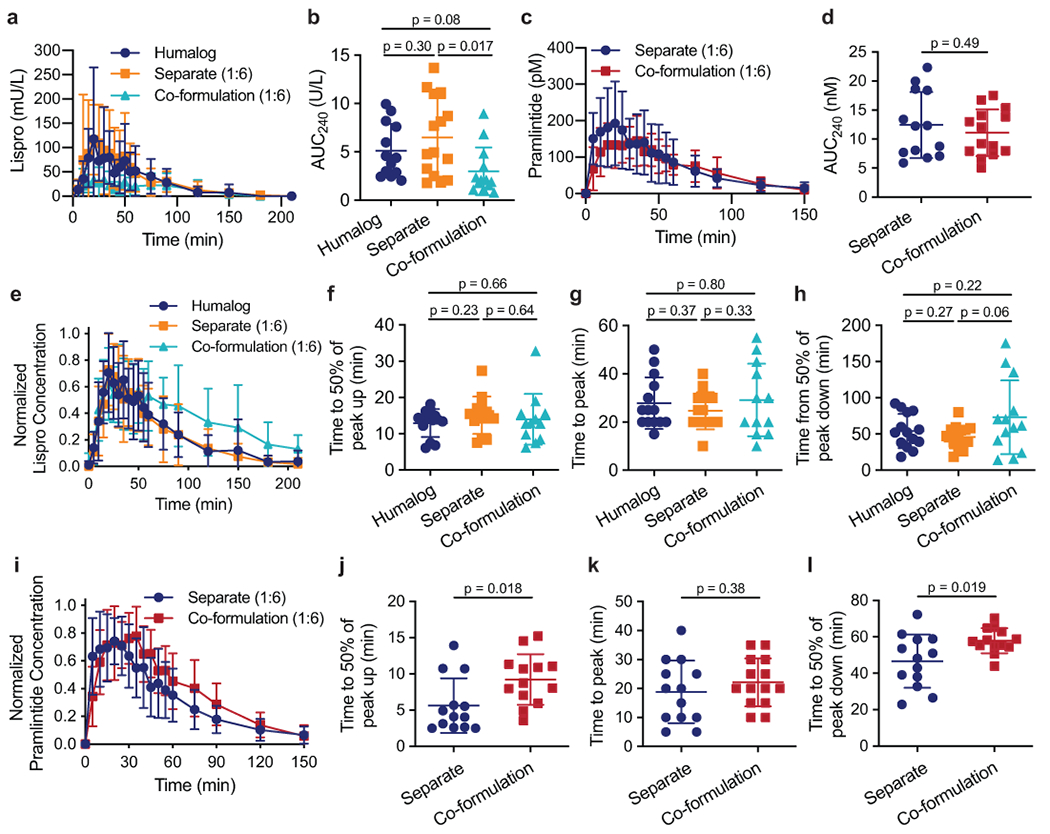
Diabetic female pigs received subcutaneous administration of therapies comprising either (i) commercial Humalog, (ii) commercial Humalog and pramlintide (pH=4) delivered in separate injections, or (iii) lispro-pramlintide co-formulation with CB[7]-PEG. Treatments were administered simultaneously with a 200g meal. All treatment groups received 4U insulin and pramlintide groups received a molar ratio of 1:6 pramlintide to lispro. Pharmacokinetics of a, insulin lispro in mU/L lispro (Humalog n=14; Separate n=15; Co-formulation n=13) or c, pramlintide in pM (Separate n=14; Co-formulation n=14). The area under the pharmacokinetic curves (AUC) of b, lispro (Humalog n=12; Separate n=14; Co-formulation n=13) and d, pramlintide (Separate n=13; Co-formulation n=14) for the first 240 minutes after subcutaneous injection. Pharmacokinetics for each pig were individually normalized to peak concentrations and normalized values were averaged for e, lispro (Humalog n=14; Separate n=15; Co-formulation n=13) or i, pramlintide (Separate n=14; Co-formulation n=14) concentration for each treatment group. Time to reach 50% of peak f, lispro (Humalog n=13;Separate n=14; Co-formulation n=12) or j, pramlintide concentration (onset) (Separate n=13; Co-formulation n=13). Time to reach peak g, lispro (Humalog n=14; Separate n=14; Co-formulation n=12) or k, pramlintide concentration (Separate n=13; Co-formulation n=14). Time for h, lispro (Humalog n=14; Separate n=15; Co-formulation n=13) or l, pramlintide depletion to 50% of peak concentration (Separate n=13; Co-formulation n=13). The specified sample size n refers to a cohort of 11 pigs who received each treatment group an equal number of times. Error bars, mean ± s.d. The Grubbs’ outlier test (alpha=0.05) was used to remove outliers. Statistical significance was determined by a two-tailed student’s t-test.
Fig. 6 |. Overlap between pharmacokinetic curves of lispro and pramlintide and glucagon suppression following treatment with different formulations in diabetic pigs.
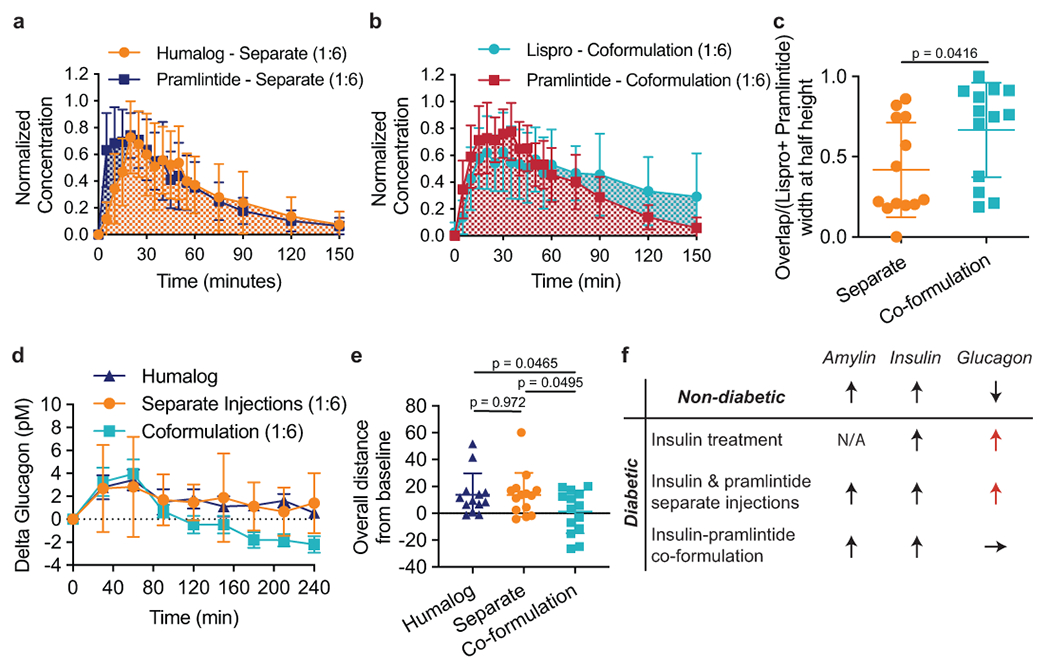
Pharmacokinetics of lispro and pramlintide after injection with Humalog and pramlintide as separate injections and as a lispro-pramlintide co-formulation. Mean normalized concentration (normalized individually for each pig) of lispro and pramlintide when administered as a, two separate injections (Humalog n=15; Pramlintide n=13) or b, as a co-formulation (Lispro n=13; Pramlintide n=14) with CB[7]-PEG. The overlap between curves was evaluated as the time during which both lispro and pramlintide concentrations were greater than 0.5 (width at half peak height), shown as a ratio of c, overlap time over the total width of both peaks (Overlap/(Lispro + Pramlintide - Overlap)) (Separate n=13; Co-formulation n=13). Pharmacokinetics of glucagon after a meal and treatment with Humalog alone, Humalog and pramlintide as separate injections or as a lispro-pramlintide co-formulation. Glucagon is plotted as d, change in glucagon concentrations from baseline over 4-hours following treatment administration e, overall distance from baseline by treatment group (sum of individual points) (Humalog n=13; Separate n=14; Co-formulation n=14). The co-formulation reduced glucagon levels compared to Humalog (*p=0.0465) and separate administrations of Humalog and pramlintide (*p=0.0495). f, A summary schematic of how treatment affects post-prandial glucagon. The specified sample size n refers to a cohort of 11 pigs who received each treatment group an equal number of times. Error bars indicate mean ± s.d. The ROUT test (Q=1%) was used to remove outliers. Statistical significance was determined by a two-tailed student’s t-test.
We hypothesized that increased overlap of insulin and pramlintide pharmacokinetics in our insulin-pramlintide co-formulation would be advantageous for treatment outcomes. Indeed, co-formulation resulted in suppressed post-prandial glucagon levels (1 ± 16 pM) compared to both Humalog alone (14 ± 16 pM) (t=2.09; df=25; 95% CI [−25.39, −0.21]; p=0.0465) as well as insulin and pramlintide delivered in two separate injections (14 ± 17 pM) (t=2.06; df=26; 95% CI [−25.13, −0.03]; p=0.0495) (Figure 6d–e). Separate injections did not result in statistically significant differences in post-prandial glucagon suppression compared to Humalog alone (t=0.04; df=25; 95% CI [−12.97, 12.53]; p=0.97). These results suggest that co-formulation improves restoration of metabolic signaling compared to separate delivery of insulin and pramlintide.
Biocompatibility of CB[7]-PEG.
As CB[7]-PEG is a new chemical entity, we sought to assess its biocompatibility by using blood chemistry and histopathology to look for negative effects on the liver or kidney. Healthy Sprague Dawley rats (n=4) received daily injections of CB[7]-PEG (at a dose equivalent to what would be administered in an insulin injection) for six weeks. Blood chemistry was monitored biweekly and single-blinded assessment of the histopathology of the liver and kidney was conducted at the endpoint of the study (See Supplemental Information, Figure S8). No differences were observed between treated animals and untreated controls during these studies. Blood chemistry in diabetic pigs who received intermittent injections of the insulin-pramlintide co-formulation (1:6) containing CB[7]-PEG corroborated the findings in rats (Figure S9).
Discussion
Natural insulin secretion results in insulin levels that are several times higher in the liver than in the peripheral tissues on account of first pass insulin absorption from the portal vein. While subcutaneous insulin replacement therapy successfully stimulates glucose uptake in the peripheral tissues, it does not suppress hepatic glucose secretion to the same degree as endogenous insulin on account of differential pharmacokinetics, pharmacodynamics, and biodistribution. In turn, the reduction in hepatic signaling results in unrestricted glycogen mobilization in the post-prandial period. A physiological replacement therapy for amylin in diabetic patients may play an important role in improving the efficacy of insulin treatments since amylin and its analogues act synergistically to inhibit glycogen mobilization from hepatic tissues by suppressing post-prandial glucagon.[47] Yet, co-formulation of biopharmaceuticals is difficult on account of their poor stability and potential for differential solubility, and traditional formulation approaches to prepare an insulin-pramlintide co-formulation have been unsuccessful.
In this study, a co-formulation of insulin and pramlintide was created using an approach that utilizes simultaneous supramolecular PEGylation of the two hormones with CB[7]-PEG to stabilize pramlintide in combination with insulin analogues such as aspart or lispro in the absence of formulation zinc. We demonstrated the utility of this simple, excipient-based approach to simultaneously endow otherwise incompatible proteins, insulin and pramlintide, with PEG chains to inhibit protein aggregation.[31] This approach exploits the specific and strong binding of the macrocyclic host molecule CB[7] to aromatic amino acids, including the N-terminal phenylalanine on insulin and the amidated C-terminal tyrosine on pramlintide,[33−36] through simple mixing as these interactions are non-covalent. CB[7]-PEG exhibits binding affinities for these proteins in the micromolar range such that over 98% of the complexes are bound at typical formulation concentrations, yet less than 1% are bound upon dilution following administration in the body. This feature affords the automatic release of authentic, unmodified therapeutic proteins upon administration and overcomes the limitations of traditional approaches to covalent grafting of polymers onto proteins, which include reduced activity.[28, 48] This approach thereby offers a broadly useful and modular excipient strategy for formulation of unmodified protein drugs to enhance their formulation shelf life and alter pharmacokinetics.
We hypothesized that simultaneous supramolecular PEGylation of insulin and pramlintide would not only enable their co-formulation at physiologic pH by enhancing the stability of the two proteins, but would also facilitate the modification of insulin-pramlintide pharmacokinetics to more closely mimic endogenous hormone secretion and restore meal-time glucagon suppression. When injected separately according to the current clinical model, fast-acting insulin analogues and pramlintide have reduced overlap between their pharmacokinetic curves resulting from the slower absorption of insulin as traditionally formulated (i.e., consisting of a combination of monomers, dimers, and hexamers) from the subcutaneous space than the pramlintide, which only exists in a monomeric form. Using a translationally relevant porcine model of insulin-deficient diabetes, we demonstrated that meal-time administration of an insulin-pramlintide co-formulation leads to increased overlap of insulin and pramlintide pharmacokinetics and restoration of mealtime glucagon suppression when compared with the clinical standard of separate administration of the hormones. While separate delivery of pramlintide has been clinically shown to suppress meal-time glucagon at high doses, we show that insulin-amylin co-formulation exhibits potent glucagon suppression at lower doses than can be achieved with separate administrations. Co-formulation, therefore, captures the synergistic effects of amylin and insulin and shows promise as a true biomimetic dual-hormone replacement therapy with greater physiological relevance than insulin alone. Moreover, the ability of this biomimetic dual-hormone treatment therapy to be administered in a single injection will reduce patient burden and potentially enable more broad adoption by patients who would benefit from such a therapy.
Outlook
In this study we have demonstrated that simultaneous non-covalent PEGylation of insulin and pramlintide imbue enhanced stability and enable their co-formulation at physiologic pH. This dual-hormone co-formulation exhibits pharmacokinetics that more closely mimic endogenous co-secretion of these two hormones from the healthy pancreas and restores post-prandial glucagon suppression compared to insulin and pramlintide delivered separately. Further, with the development of automated insulin delivery systems, the co-administration of insulin and amylin analogues together in a single formulation could play a major role in allowing for a fully automated closed-loop system without the need for meal announcement or meal boluses.[22−24] Future studies will require comprehensive assessment of the biocompatibility and immunogenicity of CB[7]-PEG and the complete insulin-pramlintide co-formulation to facilitate the clinical translation of this co-formulation.
Methods
This section should not contain figures or tables, and should be structured in sub-sections, with appropriate specific headings such as Study design, Cell culture, Materials, Device fabrication, and Statistical analyses. The last subheading should be Reporting summary. Any supplementary methods, discussion, figures and tables should be included as SI.
Materials.
CB[7]-PEG was prepared according to published protocols,[31] with method modification to enable copper “click” chemistry following reported protocols.[49] Novolog (Novo Nordisk), Humalog (Eli Lilly) and pramlintide (BioTang) were purchased and used as received. For pig studies lispro was isolated using PD MidiTrap G-10 gravity columns (GE Healthcare) and then concentrated using Amino Ultra 3K centrifugal units (Millipore). All other reagents were purchased from Sigma-Aldrich unless otherwise specified.
NMR-DOSY.
1H 2D DOSY spectra were recorded at a protein concentration (aspart or pramlintide) of 6 mg/mL in 200 mM phosphate buffer, pH 7, in D2O. 1D H1-NMR of the complex showed a broadening of both insulin and CB[7]-PEG signals (Figure S3). This was exacerbated with an increasing ratio of CB[7]-PEG to insulin (Figure S4). As such, an optimum ratio of CB[7]-PEG to insulin for DOSY was established to 1.25 mol. A Varian Inova 600 MHz NMR instrument was used to acquire the data. Magnetic field strengths ranging from 2 to 57 G cm−1. The DOSY time and gradient pulse were set at 132 ms (∆) and 3 ms (δ) respectively. All NMR data were processed using MestReNova 11.0.4 software.
Acridine orange binding affinity.
For these studies, unmodified CB[7] was purchased from Strem Chemicals and Acridine Orange (AO) was purchased from Sigma-Aldrich. Binding of CB[7] to Novolog and pramlintide was assessed using the AO dye displacement assay, as previously described.[31] Briefly, 6 μM of CB[7] and 8 μM AO (for Novolog assay) or 2 μM AO (for pramlintide assay) were combined with 100μL of either Novolog or Pramlintide samples. Novolog samples were diluted to concentrations of 0, 0.01, 0.1, 0.3, 0.5, 1, 1.5, 2, 3, 4 μM in H2O. Pramlintide samples were diluted to concentrations of 0, 4, 8, 12, 18, 24, 30, 37.5, 40 μM in H2O. Samples were incubated overnight in light-free conditions, and fluorescent spectra were collected on an BioTek SynergyH1 microplate reader, exciting at 485 nm and collecting the resulting fluorescent spectra from 495 to 650 nm. The decay in the peak of AO fluorescent signal was fit to a one-site competitive binding model (GraphPad Prism, version 6.0), using the CB[7]•AO equilibrium constant reported previously (Keq = 2×105 M−1),[50] to determine binding constants of unmodified CB[7] to insulin and pramlintide.
Circular dichroism.
Circular dichroism was used to validate that binding between CB[7]-PEG insulin aspart and pramlintide resulted in changes to the secondary structure of the protein. Novolog was diluted to 0.2 mg/mL in PBS (pH=7.4) and was evaluated (i) alone, (ii) with EDTA at a 1:1 molar ratio to zinc, (iii) with CB[7]-PEG at a 5:1 molar excess to insulin (1.1 mg/mL), and (iv) with both EDTA and CB[7]-PEG. The concentration of CB[7]-PEG in formulation results in 93% of insulin bound. Pramlintide was evaluated (i) alone in PBS at 0.5 mg/mL and (ii) with an excess of CB[7]-PEG at a concentration of 1.1 mg/mL. After mixing, samples were left to equilibrate for 15 minutes at room temperature. Near-UV circular dichroism spectroscopy was performed at 20ºC with a J-815 CD Spectropolarimeter (Jasco Corporation) over a wavelength range of 185-250 nm using a 0.1 cm path-length cell.
In vitro stability.
Methods for aggregation assays for recombinant human insulin were adapted from previous studies.[31] Briefly, formulation samples were plated at 150 μL per well (n = 3/group) in a clear 96-well plate and sealed with optically clear and thermally stable seal (VWR). The plate was immediately placed into a plate reader and incubated with continuous shaking 37ºC. Absorbance readings were taken every 10 minutes at 540 nm for 100 h (BioTek SynergyH1 microplate reader). The aggregation of insulin leads to light scattering, which results in reduction of sample transmittance. The time for aggregation was defined as a >10% increase in transmittance from the transmittance at time zero. Controls included: (i) Novolog, (ii) Humalog, (iii) zinc-free Novolog (1:1 EDTA) , (iv) zinc-free Humalog (1:1 EDTA), (v) pramlintide (sodium acetate buffer at pH=4), (vi) pramlintide (PBS at pH=7), (vii) aspart + pramlintide (PBS at pH=7.4), (viii) lispro + pramlintide (PBS at pH=7.4). Zinc(II) was removed from the insulin through competitive binding by addition of ethylenediaminetetraacetic acid (EDTA), which exhibits a dissociation binding constant approaching attomolar concentrations (KD~10−18 M).[40, 41] EDTA was added to formulations (1 eq with respect to zinc) to sequester zinc from the formulation. The stability of formulations mixed with CB[7]-PEG evaluated were: (i) zinc-free aspart (100U/mL) + CB[7]-PEG (5 eq), (ii) zinc-free lispro (100U/mL) + CB[7]-PEG (5 eq), (iii) pramlintide (PBS at pH=7) + CB[7]-PEG (5 eq), (iv) zinc-free aspart + pramlintide (1:20 molar ratio pramlintide:insulin) + CB[7]-PEG (5 eq), (v) zinc-free lispro + pramlintide (1:20 molar ratio pramlintide:insulin) + CB[7]-PEG (5 eq), (vi) zinc-free aspart + pramlintide (1:6 molar ratio pramlintide:insulin) + CB[7]-PEG (5 eq), (vii) zinc-free lispro + pramlintide (1:6 molar ratio pramlintide:insulin) + CB[7]-PEG (5 eq), (viii) zinc-free lispro + CB[7]-PEG (3 eq), (iix) zinc-free lispro + pramlintide (1:6 molar ratio pramlintide:insulin) + CB[7]-PEG (3 eq).
Streptozotocin induced model of diabetes in rats.
Male Sprague Dawley rats (Charles River) were used for experiments. Animal studies were performed in accordance with the guidelines for the care and use of laboratory animals; all protocols were approved by the Stanford Institutional Animal Care and Use Committee. The protocol used for STZ induction adapted from the protocol by Kenneth K. Wu and Youming Huan.[51] Briefly, male Sprague Dawley rats 160-230g (8-10 weeks) were weighed and fasted 6-8 hours prior to treatment with STZ. STZ was diluted to 10mg/mL in the sodium citrate buffer immediately before injection. STZ solution was injected intraperitoneally at 65mg/kg into each rat. Rats were provided with water containing 10% sucrose for 24 hours after injection with STZ. Rat blood glucose levels were tested for hyperglycemia daily after the STZ treatment via tail vein blood collection using a handheld Bayer Contour Next glucose monitor (Bayer). Diabetes was defined as having 3 consecutive blood glucose measurements >400 mg/dL in non-fasted rats.
Streptozotocin induced diabetes in swine.
Female Yorkshire pigs (Pork Power) were used for experiments. Animal studies were performed in accordance with the guidelines for the care and use of laboratory animals and all protocols were approved by the Stanford Institutional Animal Care and Use Committee. Type-1-like diabetes was induced in pigs (25-30 kg) using streptozotocin (STZ) (MedChemExpress). STZ was infused intravenously at a dose of 125 mg/kg and animals were monitored for 24 hours. Food and administration of 5% dextrose solution was given as needed to prevent hypoglycemia. Diabetes was defined as fasting blood glucose greater than 300 mg/dL.
In vivo pharmacokinetics and pharmacodynamics in diabetic rats.
Diabetic rats were fasted for 6-8 hours. Rats were injected subcutaneously with the following formulations: (i) Novolog (1.5 U/kg), (ii) separate injections of Novolog (1.5 U/kg) and pramlintide, (iii) insulin-pramlintide co-formulation (zinc-free aspart at 1.5 U/kg; pramlintide at 2.3 μg/kg) with CB[7]-PEG (5 eq). For blood glucose measurements formulations (ii) and (iii) were evaluated at three different pramlintide ratios (a) 1:15 pramlintide to aspart; 2.3 μg/kg, (b) 1:8 pramlintide to aspart; 4.4 μg/kg, and (c) 1:2 pramlintide to aspart; 17.5μg/kg. For pharmacokinetic studies only 1:2 pramlintide to aspart ratios were tested due to resolution needed for ELISA. Before injection, baseline blood glucose was measured. Rats with a baseline blood glucose between 400 mg/dL-500mg/dL were selected for the study. After injection, blood was sampled every 3 minutes for the first 30 minutes, then every 5 minutes for the next 30 minutes, then at 75, 90, 120, 150, and 180 minutes. Blood glucose was measured using a handheld blood glucose monitor and additional blood was collected in serum tubes (Starstedt) for analysis with ELISA. Serum pramlintide concentrations were quantified using a human amylin ELISA kit (Phoenix Pharmaceuticals) with pure pramlintide as standards. Serum Novolog concentrations were quantified using a Human Insulin & Insulin Analogs ELISA kit (Alpha Diagnostics International) with Novolog standards.
In vivo pharmacokinetics and pharmacodynamics in diabetic swine.
Diabetic pigs were fasted for 4-6 hours. Simultaneously with their morning meal (200 g Teklad Miniswine Diet 8753; 66 g carbohydrates), pigs were injected subcutaneously with a 4U dose (0.13 U/kg) of the following formulations: (i) no treatment (pigs received food only) (ii) Humalog (100U/mL, Eli Lilly), (iii) separate administrations of Humalog and pramlintide (pH=4) (1:6 pramlintide to lispro, 0.5 μg/kg), (iv) lispro-pramlintide co-formulation (zinc-free lispro at 0.13 U/kg; pramlintide at 0.5 μg/kg) with CB[7]-PEG (3 eq to insulin + pramlintide). Co-formulations with 3 eq. CB[7]-PEG were as stable as current commercial insulin formulations (Figure S9). Insulin lispro was chosen for these studies due to greater availability of insulin lispro at the time of experiments and was formulated as previously described.[52] Briefly, EDTA was removed from formulations using a desalting column and then concentrated to formulate with excipients (phosphate buffer with glycerol (2.6%) and phenoxyethanol (0.85%)) at 100 U/mL. Before injection, baseline blood was sampled from an intravenous catheter line and measured using a handheld glucose monitor (Bayer Contour Next). After injection, blood was sampled from the intravenous catheter line every 5 minutes for the first 60 minutes, then every 30 minutes up to 4 hours. Blood glucose was measured using a handheld blood glucose monitor and additional blood was collected in serum tubes (Starstedt) or K2EDTA plasma tubes (Greiner-BioOne) for analysis with ELISA. Serum and plasma lispro concentrations were quantified using an iso-insulin ELISA kit or lispro-NL ELISA kit (Mercodia), serum and plasma pramlintide was quantified using a human amylin ELISA kit (Millipore Sigma), and serum and plasma glucagon was quantified with a Glucagon ELISA kit (Mercodia). If the ELISA of a sample was run multiple times the averages of the values was taken for analysis.
Biocompatibility.
Healthy 10-week old rats (n=4) were administered subcutaneously with CB[7]-PEG (0.2 mg/kg) in PBS (pH=7.4) once daily for six weeks. The experimental dose was equivalent to the CB[7]-PEG concentration in the insulin formulations used for studies in diabetic rats. Blood was collected for blood chemistry tests on day 14, 28, and 42. Chemistry analysis was performed on a Siemens Dimension Xpand analyzer. A medical technologist performed all testing, including dilutions and repeat tests as indicated, and reviewed all of the data. At the end of the six week experiment, the rats were euthanized and tissues (kidney and liver) were collected for histology. Harvested tissue was fixed and then transverse sections of the left lateral lobe and right medial lobe of the liver and longitudinal sections of the kidney were taken for blinded histological analysis by a professional pathologist (n=2). Hematoxylin & Eosin and Masson’s Trichrome staining were performed by Histo-tec Laboratory. Similar to the studies described for rats, diabetics pigs were dosed with the insulin-pramlintide co-formulation containing CB[7]-PEG at 10-13 meals over the course of six weeks. Blood chemistry was analyzed as described above on blood samples taken 3-4 days following the induction of diabetes and again at the endpoint of the study.
Statistics.
All results are expressed as a mean ± standard deviation. Comparisons between two groups were conducted using a two-tailed Student’s t-test with GraphPad Prism. Statistical significance was considered as p < 0.05. The ROUT method (Q=5%) or Grubb’s method was used to remove outliers when specified.
Reporting summary.
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
All data supporting the results in this study are available within the Article and its Supplementary Information. The broad range of raw datasets acquired and analysed (or any subsets of it), which for reuse would require contextual metadata, are available from the corresponding author on reasonable request.
Supplementary Material
Acknowledgements
This work was funded in part by NIDDK R01 (NIH grant #R01DK119254) and a Pilot and Feasibility funding from the Stanford Diabetes Research Center (NIH grant #P30DK116074) and the Stanford Child Health Research Institute, as well as a Research Starter Grant from the PhRMA Foundation. C.L.M. was supported by the NSERC Postgraduate Scholarship and the Stanford BioX Bowes Graduate Student Fellowship. A.A.A.S. was funded by grant NNF18OC0030896 from the Novo Nordisk Foundation and the Stanford Bio-X Program, and also funded by the Danish Council of Independent Research (Grant No. DFF5054-00215). The authors thank the Stanford Animal Diagnostic Lab and the Veterinary Service Centre staff for their technical assistance.
Footnotes
Competing interests
E.A.A., B.A.B., D.M.M., C.L.M., and G.A.R. are inventors on a patent filing describing the work reported in this manuscript.
Supplementary information is available for this paper at https://doi.org/10.1038/s41551-020-0555-4.
Reprints and permissions information is available at www.nature.com/reprints. Correspondence and requests for materials should be addressed to
References
- 1.WHO. Diabetes: Key Facts. World Health Organization; (2017). [Google Scholar]
- 2.Borm AK et al. The effect of pramlintide (amylin analogue) treatment on bone metabolism and bone density in patients with type 1 diabetes mellitus. Horm. Metab. Res 31, 472–475 (1999). [DOI] [PubMed] [Google Scholar]
- 3.Gottlieb A et al. Pramlintide as an adjunct to insulin therapy improved glycemic and weight control in people with type 1 diabetes during treatment for 52 weeks. Diabetes. 49, A109–A109 (2000). [Google Scholar]
- 4.Ryan GJ, Jobe LJ & Martin R Pramlintide in the treatment of type 1 and type 2 diabetes mellitus. Clin. Ther 27, 1500–1512 (2005). [DOI] [PubMed] [Google Scholar]
- 5.Edelman S et al. A double-blind, placebo-controlled trial assessing pramlintide treatment in the setting of intensive insulin therapy in type 1 diabetes. Diabetes Care. 29, 2189–2195 (2006). [DOI] [PubMed] [Google Scholar]
- 6.Jones MC Therapies for diabetes: pramlintide and exenatide. Am Fam Physician. 75, 1831–1835 (2007). [PubMed] [Google Scholar]
- 7.Rodriguez LM et al. The role of prandial pramlintide in the treatment of adolescents with type 1 diabetes. Pediatr. Res 62, 746–749 (2007). [DOI] [PubMed] [Google Scholar]
- 8.Weinzimer SA et al. Effect of pramlintide on prandial glycemic excursions during closed-loop control in adolescents and young adults with type 1 diabetes. Diabetes Care. 35, 1994–1999 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grunberger G Novel therapies for the management of type 2 diabetes mellitus: part 1. pramlintide and bromocriptine-QR. J. Diabetes 5, 110–117 (2013). [DOI] [PubMed] [Google Scholar]
- 10.Hay DL et al. Amylin: pharmacology, physiology, and clinical potential. Pharmacol. Rev 67, 564 (2015). [DOI] [PubMed] [Google Scholar]
- 11.Wang H et al. Rationally designed, nontoxic, nonamyloidogenic analogues of human islet amyloid polypeptide with improved solubility. Biochemistry-US. 53, 5876–5884 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ratner R et al. Adjunctive therapy with pramlintide lowers HbA1c without concomitant weight gain and increased risk of severe hypoglycemia in patients with type 1 diabetes approaching glycemic targets. Exp Clin Endocrinol Diabetes. 113, 199–204 (2005). [DOI] [PubMed] [Google Scholar]
- 13.Whitehouse F et al. A randomized study and open-label extension evaluating the long-term efficacy of pramlintide as an adjunct to insulin therapy in type 1 diabetes. Diabetes Care. 25, 724–730 (2002). [DOI] [PubMed] [Google Scholar]
- 14.Ratner RE et al. Amylin replacement with pramlintide as an adjunct to insulin therapy improves long-term glycaemic and weight control in type 1 diabetes mellitus: a 1-year, randomized controlled trial. Diabetic Med. 21,1204–1212 (2004). [DOI] [PubMed] [Google Scholar]
- 15.Hampp C et al. Use of antidiabetic drugs in the U.S., 2003–2012. Diabetes Care. 37, 1367–1374 (2014). [DOI] [PubMed] [Google Scholar]
- 16.Martin C The physiology of amylin and insulin: maintaining the balance between glucose secretion and glucose uptake. Diabetes Educator. 32,101S–104S (2006). [DOI] [PubMed] [Google Scholar]
- 17.Heptulla RA et al. The role of subcutaneous pramlintide infusion in the treatment of adolescents with type 1 diabetes. Diabetes. 54, A110–A111 (2005). [Google Scholar]
- 18.Want LL & Ratner R Exenatide and pramlintide: new therapies for diabetes. Int. J. Clin. Pract 60,1522–1523 (2006). [DOI] [PubMed] [Google Scholar]
- 19.Mathieu C et al. Insulin analogues in type 1 diabetes mellitus: getting better all the time. Nat. Rev. Endocrinol 13, 385–399 (2017). [DOI] [PubMed] [Google Scholar]
- 20.Holleman F & Hoekstra JBL Insulin Lispro. New Engl. J. Med 337, 176–183 (1997). [DOI] [PubMed] [Google Scholar]
- 21.Gast K et al. Rapid-acting and human insulins: hexamer dissociation kinetics upon dilution of the pharmaceutical formulation. Pharm. Res 34, 2270–2286 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Riddle MC et al. Fixed ratio dosing of pramlintide with regular insulin before a standard meal in patients with type 1 diabetes. Diabetes Obes. Metab 17, 904–907 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Haidar A et al. Insulin-plus-pramlintide artificial pancreas in type 1 diabetes—randomized controlled trial. Diabetes. 67, (2018). [Google Scholar]
- 24.Riddle MC et al. Control of postprandial hyperglycemia in type 1 diabetes by 24-hour fixed-dose coadministration of pramlintide and regular human insulin: a randomized, two-way crossover study. Preprint at 10.2337/dc18-1091 (2018). [DOI] [PubMed] [Google Scholar]
- 25.Manning MC et al. Stability of protein pharmaceuticals: an update. Pharm. Res 27, 544–575 (2010). [DOI] [PubMed] [Google Scholar]
- 26.Mitragotri S, Burke PA & Langer R Overcoming the challenges in administering biopharmaceuticals: formulation and delivery strategies. Nat. Rev. Drug Discov 13, 655–672 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang C, Lu D & Liu Z How PEGylation enhances the stability and potency of insulin: a molecular dynamics simulation. Biochemistry-US. 50, 2585–2593 (2011). [DOI] [PubMed] [Google Scholar]
- 28.Guerreiro LH et al. Preparation and characterization of PEGylated amylin. AAPS PharmSciTech. 14, 1083–1097 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sisnande T et al. Monoconjugation of human amylin with methylpolyethyleneglycol. PLoS One. 10, e0138803 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Veronese FM & Mero A The impact of PEGylation on biological therapies. BioDrugs. 22, 315–29 (2008). [DOI] [PubMed] [Google Scholar]
- 31.Webber MJ et al. Supramolecular PEGylation of biopharmaceuticals. P. Natl. Acad. Sci. USA 113, 14189–14194 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hirotsu T et al. Self-assembly PEGylation retaining activity (SPRA) technology via a host-guest interaction surpassing conventional PEGylation methods of proteins. Mol. Pharm 14, 368–376 (2017). [DOI] [PubMed] [Google Scholar]
- 33.Bush M, Bouley N & Urbach AR Charge-mediated recognition of N-terminal tryptophan in aqueous solution by a synthetic host. J. Am. Chem. Soc 127, 14511–14517 (2005). [DOI] [PubMed] [Google Scholar]
- 34.Heitmann LM et al. Sequence-specific recognition and cooperative dimerization of N-terminal aromatic peptides in aqueous solution by a synthetic host. J. Am. Chem. Soc 128,12574–12581 (2006). [DOI] [PubMed] [Google Scholar]
- 35.Rajgariah P & Urbach AR Scope of amino acid recognition by cucurbit[8]uril. J. Incl. Phenom. Macro 62, 251–254 (2008). [Google Scholar]
- 36.Reczek JJ et al. Multivalent recognition of peptides by modular self-assembled receptors. J. Am. Chem. Soc 131, 2408–2415 (2009). [DOI] [PubMed] [Google Scholar]
- 37.Yin H & Wang R Applications of cucurbit[n]urils (n=7 or 8) in pharmaceutical sciences and complexation of biomolecules. Isr. J. Chem 58, 188–198 (2018). [Google Scholar]
- 38.Walker S et al. The Potential of Cucurbit[n]urils in Drug Delivery. Isr. J. Chem 51, 616–624 (2011). [Google Scholar]
- 39.Kuok KI et al. Cucurbit[7]uril: an emerging candidate for pharmaceutical excipients. Ann. NY. Acad. Sci 1398, 108–119 (2017). [DOI] [PubMed] [Google Scholar]
- 40.Berthon G Handbook Of Metal-ligand Interactions in Biological Fluids: Bioinorganic chemistry. (New York: Marcel Dekker, 1995). [Google Scholar]
- 41.Waters RS et al. EDTA chelation effects on urinary losses of cadmium, calcium, chromium, cobalt, copper, lead, magnesium, and zinc. Biol. Trace. Elem. Res 83, 207–221 (2001). [DOI] [PubMed] [Google Scholar]
- 42.Hvidt S Insulin association in neutral solutions studied by light scattering. Biophys. Chem 39, 205–213 (1991). [DOI] [PubMed] [Google Scholar]
- 43.Fineberg SE et al. Immunological responses to exogenous insulin. Endocr. Rev 28, 625–652 (2007). [DOI] [PubMed] [Google Scholar]
- 44.Woods RJ et al. Intrinsic fibrillation of fast-acting insulin analogs. J. Diabetes Sci. Technol 6, 265–276 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.da Silva DC et al. Amyloidogenesis of the amylin analogue pramlintide. Biophys. Chem 219,1–8 (2016). [DOI] [PubMed] [Google Scholar]
- 46.Like AA & Rossini AA Streptozotocin-induced pancreatic insulitis: new model of diabetes mellitus. Science. 193, 415–417 (1976). [DOI] [PubMed] [Google Scholar]
- 47.Gedulin BR, Rink TJ & Young AA Dose-response for glucagonostatic effect of amylin in rats. Metabolis. 46, 67–70 (1997). [DOI] [PubMed] [Google Scholar]
- 48.Knadler MP et al. Addition of 20-kDa PEG to insulin lispro alters absorption and decreases clearance in animals. Pharm. Res 33, 2920–2929 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zou L, Braegelman AS and Webber MJ Dynamic Supramolecular Hydrogels Spanning an Unprecedented Range of Host–Guest Affinity. ACS Appl. Mater. Inter 11, 5695–5700 (2019). [DOI] [PubMed] [Google Scholar]
- 50.Chinai JM et al. Molecular recognition of insulin by a synthetic receptor. J. Am. Chem. Soc 133, 8810–8813 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wu KK, & Huan Y Streptozotocin-induced diabetic models in mice and rats. Curr. Protoc. Pharmacol 5, 1–14 (2008). [DOI] [PubMed] [Google Scholar]
- 52.Maikawa CL et al. Stable monomeric insulin formulations enabled by supramolecular PEGylation of insulin analogues. Adv. Therap 3, 1900094 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data supporting the results in this study are available within the Article and its Supplementary Information. The broad range of raw datasets acquired and analysed (or any subsets of it), which for reuse would require contextual metadata, are available from the corresponding author on reasonable request.