

Abstract
Rationale:
The gene encoding transcription factor TCF21 has been linked to coronary artery disease (CAD) risk by human genome wide association studies (GWAS) in multiple racial ethnic groups. In murine models, Tcf21 is required for phenotypic modulation of smooth muscle cells (SMC) in atherosclerotic tissues and promotes a fibroblast phenotype in these cells. In humans, TCF21 expression inhibits risk for CAD. The molecular mechanism by which TCF21 regulates SMC phenotype is not known.
Objective:
To better understand how TCF21 affects SMC phenotype, we sought to investigate the possible mechanisms by which it regulates the lineage determining myocardin (MYOCD)-serum response factor (SRF) pathway.
Methods and Results:
Modulation of TCF21 expression in HCASMC revealed that TCF21 suppresses a broad range of SMC markers, as well as key SMC transcription factors MYOCD and SRF, at the RNA and protein level. We conducted chromatin immunoprecipitation (ChIP)-sequencing to map SRF binding sites in HCASMC, showing that binding is colocalized in the genome with TCF21, including at a novel enhancer in the SRF gene, and at the MYOCD gene promoter. In vitro genome editing indicated that the SRF enhancer CArG box regulates transcription of the SRF gene, and mutation of this conserved motif in the orthologous mouse SRF enhancer revealed decreased SRF expression in aorta and heart tissues. Direct TCF21 binding and transcriptional inhibition at co-localized sites were established by reporter gene transfection assays. Chromatin immunoprecipitation and protein co-immunoprecipitation studies provided evidence that TCF21 blocks MYOCD and SRF association by direct TCF21-MYOCD interaction.
Conclusions:
These data indicate that TCF21 antagonizes the MYOCD-SRF pathway through multiple mechanisms, further establishing a role for this CAD associated gene in fundamental SMC processes and indicating the importance of smooth muscle response to vascular stress and phenotypic modulation of this cell type in CAD risk.
Keywords: Atherosclerosis, smooth muscle, genetics, genomics, differentiation, myocardin, differentiation, gene expression, regulation, coronary circulation
Subject Terms: Atherosclerosis, Coronary Artery Disease, Gene Expression and Regulation, Smooth Muscle Cell Proliferation and Differentiation, Vascular Disease
Graphical Abstract
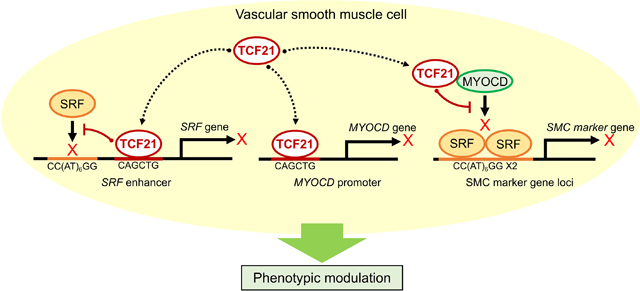
TCF21 is a highly validated coronary artery disease (CAD) gene that is protective toward coronary artery disease risk. We have shown previously that this gene promotes de-differentiation, proliferation, and migration of SMC into the developing lesion, where they contribute to the protective fibrous cap. A critical question is how does TCF21 mediate these effects on the SMC? Our hypothesis was that TCF21 functions, at least in part, by blocking the MYOCD-SRF lineage determining pathway. By performing chromatin-immunoprecipitation-sequencing we were able to show that TCF21 binds in many of the same loci as MYOCD-SRF. Further, TCF21 binding was associated with decreased binding of MYOCD-SRF, suggesting an epigenetic effect. Interestingly, TCF21 was found to bind at a novel enhancer in the SRF gene and in the promoter region of MYOCD, and binding of TCF21 to these sequences inhibited transcription. Finally, TCF21 was found to disrupt the interaction of MYOCD and SRF in solution, thus blocking the ability of this complex to bind DNA. These studies identify a regulatory pathway in SMC that modulates the risk for CAD, thus providing a possible mechanism of directly inhibiting atherosclerosis through targeting SMC phenotype.
INTRODUCTION
There is a growing appreciation for the role of vascular smooth muscle cells (SMCs) in coronary artery disease (CAD) risk1–6. In response to atherogenic stimuli in culture, SMCs de-differentiate, proliferate, display increased migratory behavior, and adopt a “synthetic” phenotype in which they secrete extracellular matrix components and inflammatory cytokines in a process known as “phenotypic modulation”7–11. In mouse models of atherosclerosis, contractile SMCs de-differentiate and migrate into the developing lesion, where they comprise as many as 30% of all cells in the lesion12. In this context, phenotypically modulated SMCs have been shown to contribute to multiple areas of the lesion including the fibrous cap that is thought to protect against plaque rupture and myocardial infarction11. However, the effect of these SMC cell state changes on human atherosclerosis risk has not been elucidated.
We have recently shown that TCF21, a basic helix-loop-helix transcription factor and causal CAD gene, is required for de-differentiation, proliferation and migration of medial SMC into mouse model atherosclerotic lesions, and is also expressed by human coronary artery SMC undergoing phenotypic modulation5, 6, 13–16. Human genetic data indicates that TCF21 expression is protective toward human CAD risk, and by inference suggests that these functions in SMC inhibit primary disease processes6. Although TCF21 has a marked effect on SMC phenotype, the mechanisms by which it exerts this control are not known. Myocardin (MYOCD) is a potent transcriptional co-activator found primarily in SMCs. MYOCD is unable to bind DNA on its own, but instead binds to the widely-expressed DNA-binding transcription factor serum response factor (SRF) and this complex regulates the fundamental SMC gene expression program17. In particular, MYOCD has been shown to function as a master regulator of SMC differentiation. Its expression is correlated with the quiescent SMC phenotype and ectopic expression of MYOCD activates expression of SMC differentiation markers such as myosin heavy chain 11 (MYH11), calponin1 (CNN1) and transgelin (TAGLN)18–21.
Given the marked effect of TCF21 on SMC phenotypic modulation, we hypothesized that TCF21 exerts its effect by inhibiting the MYOCD-SRF pathway. In this study, we demonstrate that TCF21 inhibits expression of multiple SMC differentiation markers, that TCF21 and SRF binding are co-localized in HCASMC genome wide, and that TCF21 inhibits SRF and MYOCD expression. Further, we show that TCF21 blocks MYOCD-induced transactivation of SMC target genes, and provide evidence that TCF21 inhibits association of the MYOCD-SRF transcriptional complex, possibly through a direct interaction with MYOCD. Through this work, we have identified several molecular mechanisms by which a highly replicated CAD-associated gene modulates SMC phenotype to influence risk of disease.
METHODS
Data availability.
SRF ChIP-seq data has been deposited to the Gene Expression Omnibus (GEO) database, accession number GSE124011.
Detailed experimental materials and methods are described in the Online Supplement.
RESULTS
TCF21 regulates expression of SMC differentiation markers in vitro.
As a first step to defining the extent to which TCF21 is able to block the SMC contractile phenotype, we have investigated in detail its effect on expression levels for a number of SMC lineage markers. We conducted both TCF21 siRNA knockdown and TCF21 overexpression in human coronary artery smooth muscle cells (HCASMC) and assessed these effects at both the RNA and protein levels (Figs. 1A–1C). We found that TCF21 overexpression led to decreased SM22α (TAGLN), SM α-actin (ACTA2), Calponin (CNN1), and SM MHC (MYH11) mRNA (Fig. 1D) and protein levels (Fig. 1E). Conversely, we confirmed that TCF21 knockdown resulted in increased expression of these markers (Figs. 1F, 1G). For these data and all subsequent analyses where representative images and other types of data are presented, examples were chosen that represent values closest to the mean.
Figure 1. TCF21 suppresses SMC lineage marker expression.
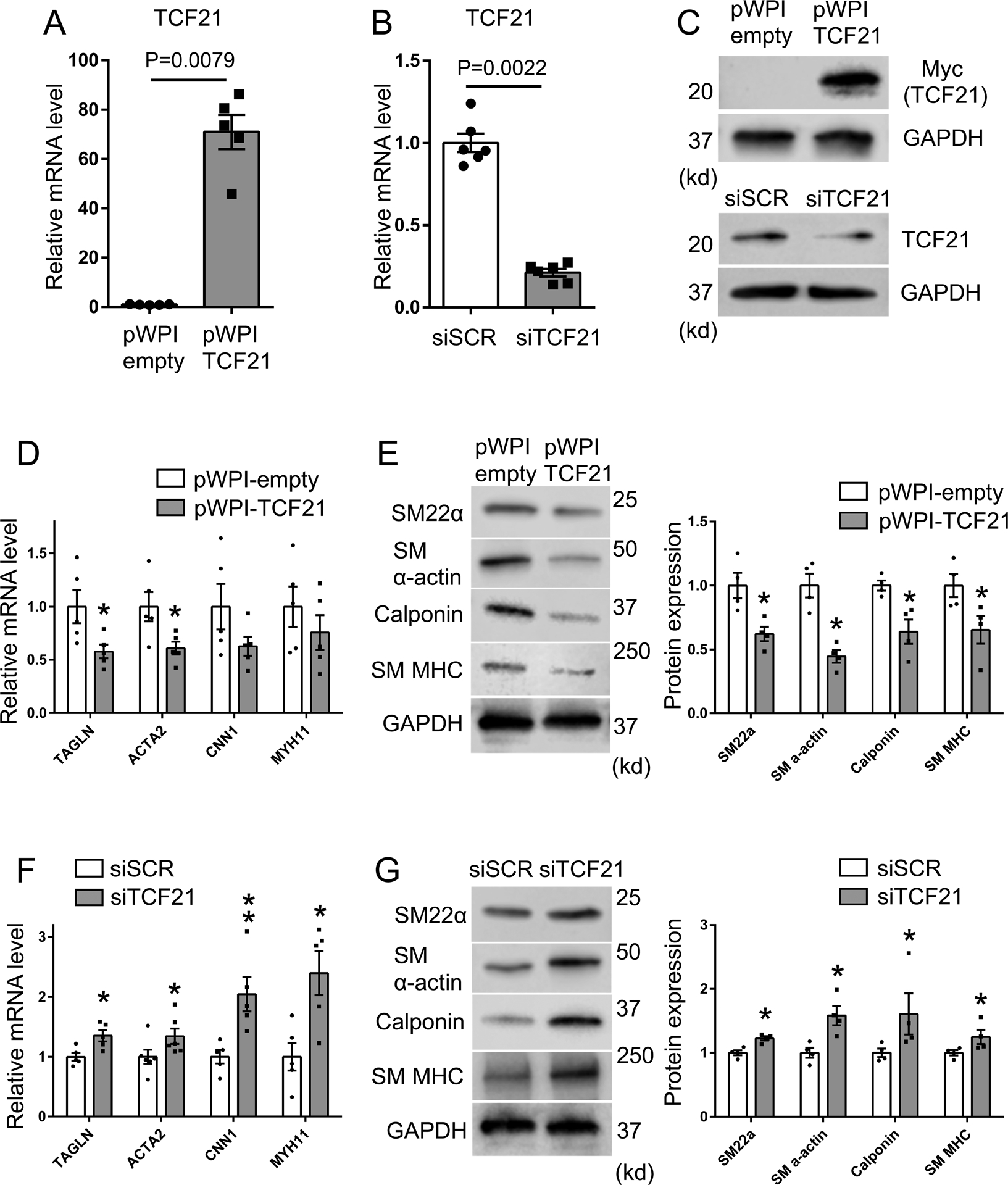
A-C) For overexpression or knockdown of TCF21, HCASMC were transfected with TCF21 overexpressing lentivirus (pWPI-TCF21) or siTCF21, respectively. mRNA and protein expression of TCF21 was evaluated by qPCR or western blot. D) HCASMC were treated with TCF21 overexpressing lentivirus (pWPI-TCF21) or empty control lentivirus (pWPI-empty) for 24 hours, and mRNA was evaluated by qPCR for expression of SMC markers TAGLN, ACTA2, CNN1, MYH11 (n=5), (p* values, TAGLN; 0.0317, ACTA2; 0.0317). E) HCASMC were treated with TCF21 overexpressing lentivirus (pWPI-TCF21) or empty lentivirus (pWPI-empty) and protein expression of SMC markers evaluated by western blot. Band intensity of SMC markers was quantified by image J software and normalized to GAPDH (n=4) (all p* values; 0.0286). F) HCASMC underwent TCF21 knockdown with transfection of siTCF21 or control siSCR regulatory RNAs and SMC marker expression evaluated by qPCR (n=5–6) (p* values, TAGLN; 0.0317, ACTA2; 0.026, MYH11; 0.0317; p** value, CNN1; 0.0079). G) HCASMC underwent TCF21 knockdown with transfection of siTCF21 or control siSCR regulatory RNAs and protein expression of SMC markers evaluated by western blot. Band intensity was quantified by image J software and normalized to GAPDH (n=4) (all p* values; 0.0286). Data analyzed by Mann-Whitney U test.
SRF co-localizes with TCF21 at regions of SMC open chromatin.
To investigate the mechanism by which TCF21 inhibits SMC marker expression, we investigated the interaction of TCF21 with known regulators of SMC differentiation, MYOCD and SRF. We hypothesized that TCF21 preferentially colocalizes in SRF target loci where it independently inhibits expression of SMC contractile state genes, or regulates binding of MYOCD-SRF. To address this hypothesis, we performed chromatin immunoprecipitation-sequencing (ChIP-seq) in HCASMC to define SRF target regions of the genome, where it primarily interacts with MYOCD to promote SMC-specific gene expression18, 19, 22, 23. While SRF ChIP-seq has been performed with other cell types24, it has not been performed in HCASMC. HCASMC have a unique origin from mesodermal cells in the pro-epicardial organ, exhibit unique physiological features25, and are the specific cell type that has been implicated in the causal mechanism of the TCF21 effect on disease risk2, 4, 6, 26, 27. We thus reasoned that SRF ChIP-seq in these cells was required for comparison to TCF21 binding. With SRF ChIP-seq, we obtained 2779 significant peaks with p-value cutoff 1e-5. 22.5% of the peaks were distributed in promoter regions while 36.9% were located in introns and 34.9% in intergenic regions. Most peaks, 72%, were located in open chromatin regions of HCASMC as identified by previous genome-wide Assay for Transposase-Accessible Chromatin using sequencing (ATACseq)2.
Intersection of SRF and TCF21 ChIP-seq data revealed significant co-localization in HCASMC regions of open chromatin. This is illustrated with a genome-wide heatmap centered on the SRF ChIP-seq peaks (Fig. 2A, left panel), which are aligned with previously identified TCF21 binding regions (Fig. 2A, middle panel)28, 29, and previously identified ATACseq regions of open chromatin (Fig. 2A, right panel)2. Intersection of peaks from the TCF21 and SRF datasets showed that 25.7% of the SRF binding sites localized within 200 bp of the nearest TCF21 peak (p=1.75e-25) (Fig. 2B), and 22.6% of the SRF sites co-localized with at least one base pair of peak overlap (p=1.65e-319, for > 1 bp overlap) (Fig. 2C). Scanning of SRF only peaks for known transcription factor binding sites found the SRF cognate binding motif termed a CArG box in 43% of SRF peaks, an approximately 20-fold enrichment over background (Fig. 2D). Performing the same analysis for intersected peaks showed a 15-fold enrichment for SRF sites and a 3-fold enrichment for TCF21 sites (Fig. 2E). To compare and investigate the pathways enriched for genes in the target loci, we used the Genomic Regions Enrichment of Annotations Tool (GREAT)30 to assign genes to the common target SRF-TCF21 binding regions and investigated the pathways that were found to be associated with this gene list. The majority of the SRF loci genes were identified in regions that also bound TCF21 (93%, Fig. 2F).
Figure 2. SRF and TCF21 co-localize genome-wide in HCASMC and regulate HCASMC pathways.
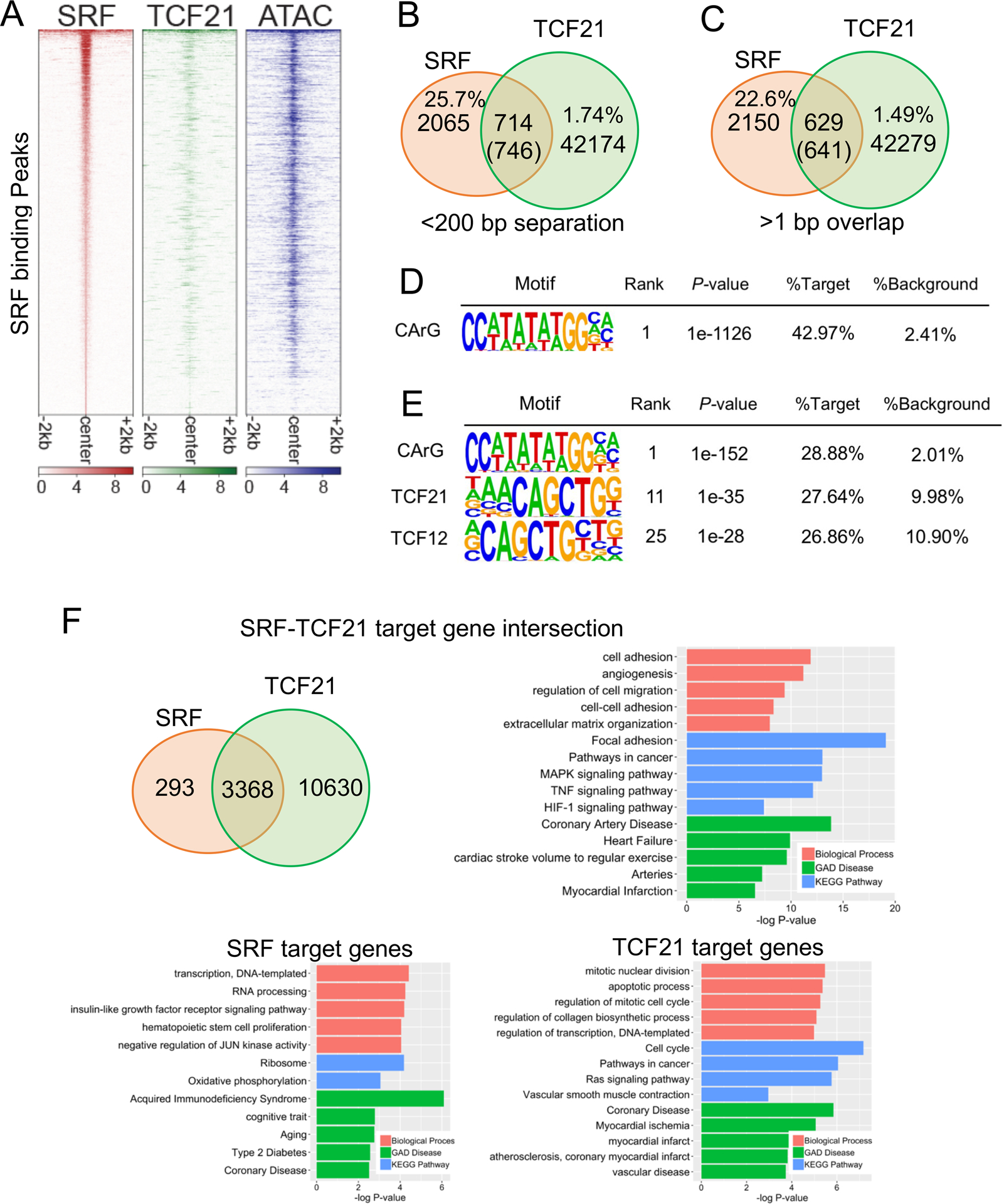
A) Heatmap distribution of SRF binding compared to previously generated TCF21 ChIP-seq and open chromatin regions (ATAC-seq), centered on SRF peaks in HCASMC within a 4-kb window. B) Venn diagram showing the number of overlaps of TCF21 and SRF peaks in the single nucleosome (<200bp separation). C) Venn diagram showing the number of direct overlaps (>1bp) of TCF21 and SRF peaks. D) Homer known motif analysis of SRF peaks identified CArG as the top motif. E) Homer known motif analysis of SRF-TCF21 joint loci showing the co-enrichment of TCF21 E-boxes and SRF CArG binding motifs. F) Venn diagram showing the number of overlapped target genes of TCF21 and SRF. Barplots show the DAVID Gene Ontology analysis of SRF target genes (bottom left), TCF21 target genes (bottom right), or intersected target genes (top right). Genes were assigned by GREAT.
Gene ontology analysis with the DAVID algorithm for genes located at SRF and TCF21 co-regulated loci identified biological process terms “cell adhesion,” “regulation of cell migration,” and “cell-cell adhesion” (Fig. 2F). The identified KEGG pathways were also relevant for vascular smooth muscle cell functions, and included “focal adhesion,” and “MAPK signaling pathway”. Most importantly, the disease terms included “coronary artery disease,” “arteries,” and “myocardial infarction.” While some of these terms were also identified in the analyses with SRF peak genes and TCF21 peak genes alone, the terms identified for the co-localized regions were more appropriate for the cell type function and disease relevance. To further characterize the pathways co-regulated by SRF and TCF21, we investigated the overlap of genes that are differentially regulated by TCF21, as identified by RNAseq of siRNA knockdown in HCASMC31, with genes that are direct targets of SRF as identified by ChIP-seq studies reported here. There were 486 genes identified. Gene Ontology analysis provided highly significant terms related to blood vessel development, including “blood vessel development,” “blood vessel morphogenesis,” “angiogenesis.” Genes identified through these analyses provide information regarding how genes regulated by both factors are important for both CAD and developmental processes, and the fundamental link between these has been highlighted with CAD GWAS studies14.
SRF and TCF21 binding appear counter-regulatory at colocalizing sites.
To further investigate a possible relationship between binding of SRF and TCF21 in colocalizing target regions, we performed an analysis aimed to compare simultaneous binding of these two TFs. We analyzed the relative binding of each factor by normalizing the number of reads for peaks in SRF and TCF21 ChIP-seq datasets compared to background counts in the region4. We focused on those binding sites which showed a greater than two-fold difference in normalized read counts, as a measure of relative binding (Fig. 3). For the directly overlapping ChIP-seq binding sites for SRF and TCF21, more than half of the sites showed greater than a 2-fold discrepancy in binding. Out of 469 biased binding sites, 170 (36.2%) showed higher relative binding of TCF21 and 68 (14.5%) showed higher relative binding for SRF (Fig. 3). This pattern of SRF compared to TCF21 binding at shared loci is much different than expected for factors that bind with equal affinity, as we have shown previously for this type of analysis4. These findings suggest that SRF and TCF21 interact at these loci in a fashion that is counter-inhibitory or showing opposing responses to external signaling pathways.
Figure 3. SRF and TCF21 show opposing binding behavior in many colocalized regions of the genome.
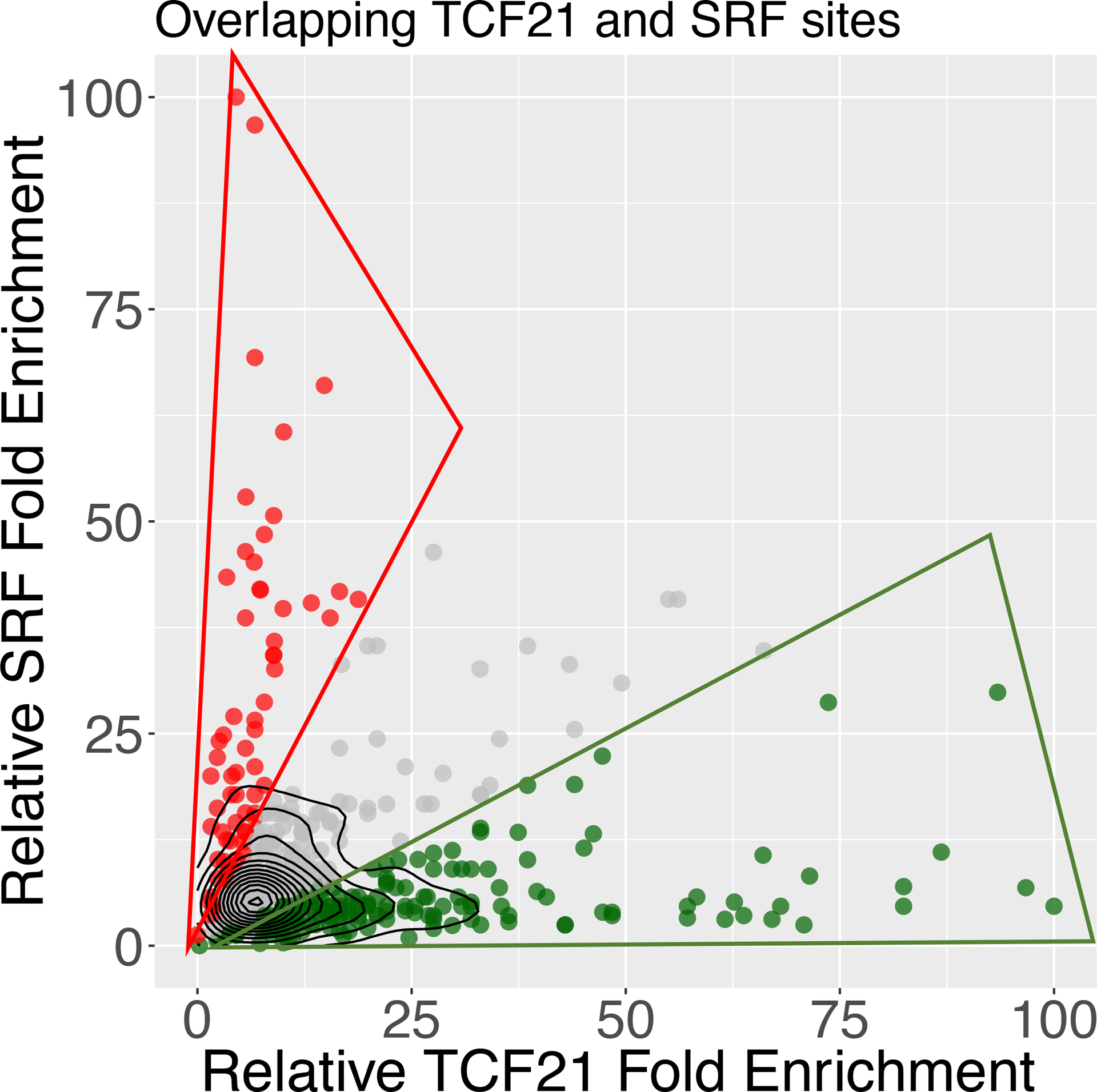
SRF and TCF21 joint binding regions were evaluated for degree of occupancy by each factor. DNA binding was assessed by comparing peak reads normalized to background reads at each locus and this variable graphed. Most of the colocalized binding regions were biased for either SRF or TCF21 binding, with a greater than 2-fold number of normalized reads for one or the other factor.
Characterization of an auto-regulatory enhancer in the SRF gene that is inhibited by TCF21.
Analysis of overlapping regions of binding by SRF and TCF21 identified an enhancer region in the second intron of the SRF gene (Chr6: 43141913–43142212, hg19) (Fig. 4A, Online Fig. I). This enhancer was identified by H3K27ac chromatin modification and ATACseq chromatin accessibility datasets developed previously in HCASMC2, 4. Analysis of this region identified a single SRF canonical CC(AT)6GG (CArG) binding sequence located 71 base-pairs from the characterized TCF21 E-box binding site as defined by previous ChIP-seq studies (Online Fig. I)6, 28. Interestingly, there is only one CC(AT)6GG found in this region, while typically CArG boxes function in pairs to promote heterodimer binding18, 19, 22, 23. However, the high degree of sequence conservation in mammalian species ranging from human to opossum (Fig.4B) is consistent with this CArG sequence being functional.
Figure 4. Identification of an intragenic autoregulatory SRF enhancer region inhibited by TCF21.
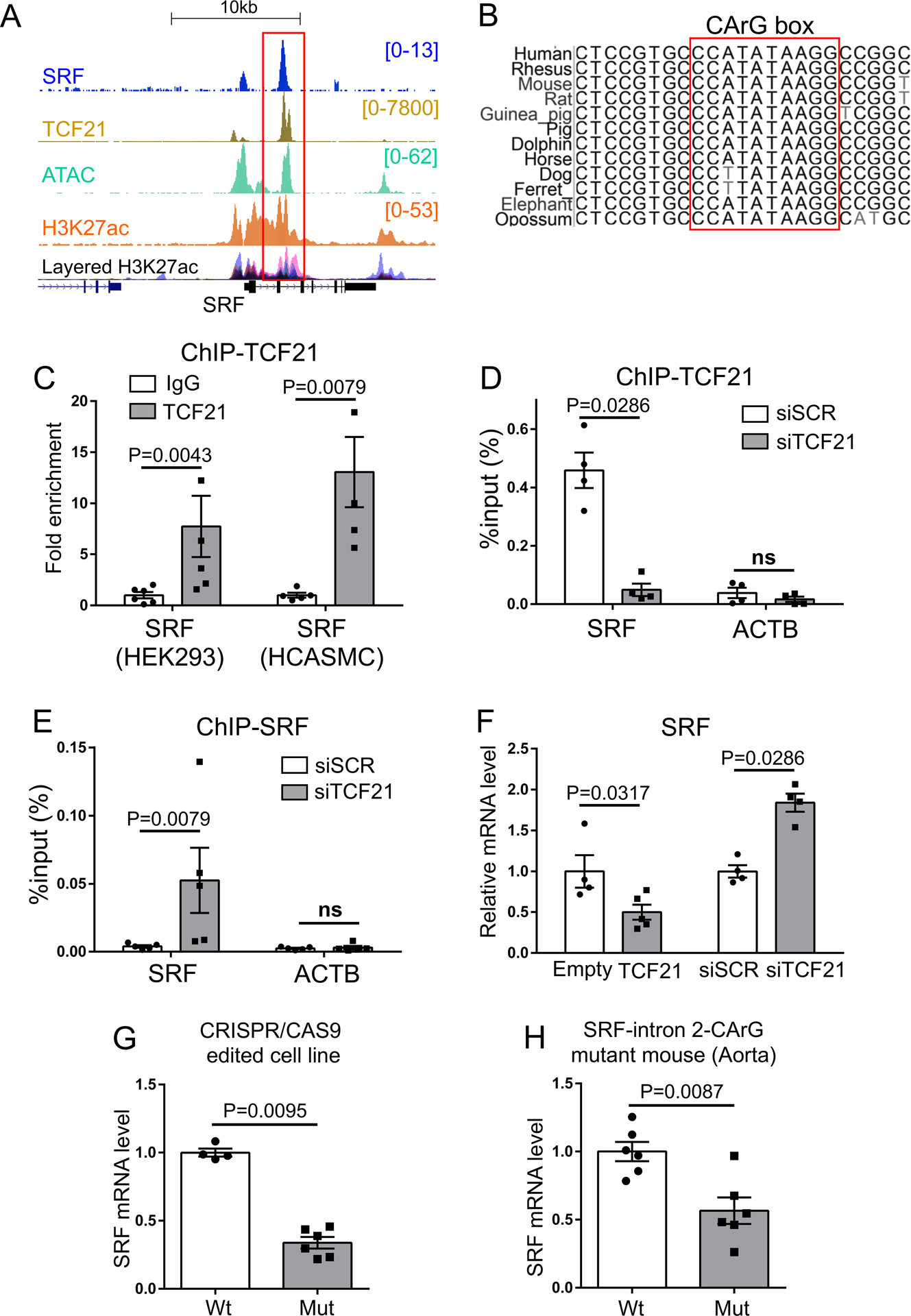
A) Pattern of ChIP-seq mapping of SRF binding as well previously determined TCF21, H3K27ac histone modification and ATAC-seq identification of open chromatin in HCASMC at the human SRF locus. ENCODE H3K27ac data are shown as well. B) Phylogenetic conservation of DNA sequence in the second intron of the SRF enhancer at the CArG box. C) Chromatin immunoprecipitation PCR (ChIP-qPCR) for TCF21 binding at the SRF enhancer in HEK293 overexpressing TCF21 or HCASMC (n=5–6). D) ChIP-qPCR for TCF21 binding (n=4) or E) SRF binding (n=5) at the SRF enhancer in HCASMC treated with siTCF21 or scrambled siRNA (siSCR). ACTB, β-actin transcription start site, was used as a negative control. F) Relative SRF expression level evaluated by qPCR with TCF21 over-expression (TCF21) compared to control (Empty) (n=4–5), and SRF expression level with TCF21 knockdown (siTCF21) or control (siSCR) transfection in HCASMC (n=4). G) SRF expression levels evaluated in wild type (WT) or CRISPR/Cas9 edited mutant CArG box HEK293 cells (Mut) at the SRF enhancer (Wt vs Mut =4 vs 6). H) qPCR analysis of SRF in wild-type (Wt) vs homozygous mutant CArG box (Mut) at the SRF enhancer in mouse aorta (n=6). C-H) Data analyzed by Mann-Whitney U test.
Using ChIP-qPCR with both HEK293 cells transfected with TCF21 and HCASMC that express native TCF21, we verified that TCF21 binds this SRF enhancer region (Fig. 4C). Because SRF and TCF21 co-localize at this SRF enhancer, we determined the effect of TCF21 knockdown on SRF binding by ChIP-qPCR. Interestingly, TCF21 knockdown resulted in a significant increase in SRF binding (Fig. 4D, 4E), suggesting that TCF21 inhibits the ability of SRF to bind to this intronic enhancer. These protein binding data correlated with decreased SRF mRNA expression in HCASMC expressing increased TCF21, and increased SRF mRNA expression in cells with decreased TCF21 (Fig. 4F). To further validate the functionality of the CArG box in this enhancer region, we performed genome editing in HEK293 cells with the CRISPR/Cas9 approach. A single guide RNA was co-transfected along with a CRISPR/Cas9 plasmid using standard methods, and clones analyzed for genome editing and mRNA expression. The six clones investigated in detail showed various deletions in and around the CArG motif of the SRF enhancer along with significantly decreased expression levels of SRF (Fig. 4G, Online Fig. II). Finally, to investigate the in vivo role of this enhancer region we took advantage of the high degree of phylogenetic conservation, and used CRISPR/Cas9 genome editing to generate a transgenic mouse line with a mutated Srf enhancer CArG box, and the mutant allele bred to homozygosity (Online Fig. III). The germline gtgcCCATATAAGG sequence was mutated to gcGGGGATATAAGG, with a resulting decrease in Srf expression in aorta and heart, but no significant change in expression in other tissues including bladder and skeletal muscle (Fig. 4H, Online Fig. IV). Taken together these data suggest that the SRF enhancer is a functional mechanism of autoregulation for this gene, and is likely modulated by binding of TCF21.
TCF21 binds the MYOCD promoter and inhibits its expression.
We next investigated TCF21 binding and transcriptional regulation at the MYOCD gene locus. We identified a region of TCF21 ChIP-seq binding within the MYOCD promoter, and found that this peak localized to a region of open chromatin as determined by colocalization with our previously generated ATAC-seq, H3K27Ac histone modification, and TCF21 ChIP-seq data2, 4, 6, 28 (Fig. 5A, Online Fig. V). ChIP-qPCR analysis confirmed that recombinant TCF21 bound this MYOCD promoter region in transfected HEK293 cells and similar results were obtained with native TCF21 in HCASMC (Fig. 5B). siTCF21 knockdown attenuated TCF21 binding to the MYOCD promoter region by ChIP-qPCR (Fig. 5C), reinforcing the specificity of our initial ChIP-seq findings at this site. To determine the effect of TCF21 on MYOCD expression, we perturbed TCF21 expression in HCASMC and measured MYOCD expression at the mRNA level. Overexpression of TCF21 in HCASMC with lentiviral transduction resulted in a decrease in MYOCD mRNA (Fig. 5D). On the other hand, siTCF21 knockdown in HCASMC resulted in an increase in MYOCD expression (Fig. 5E). Interestingly, there was no evidence for SRF binding at the MYOCD locus (Fig. 5A). Together, these experiments provide evidence that TCF21 binds to the MYOCD gene promoter region and inhibits MYOCD expression.
Figure 5. TCF21 binds the MYOCD promoter and suppresses its expression.
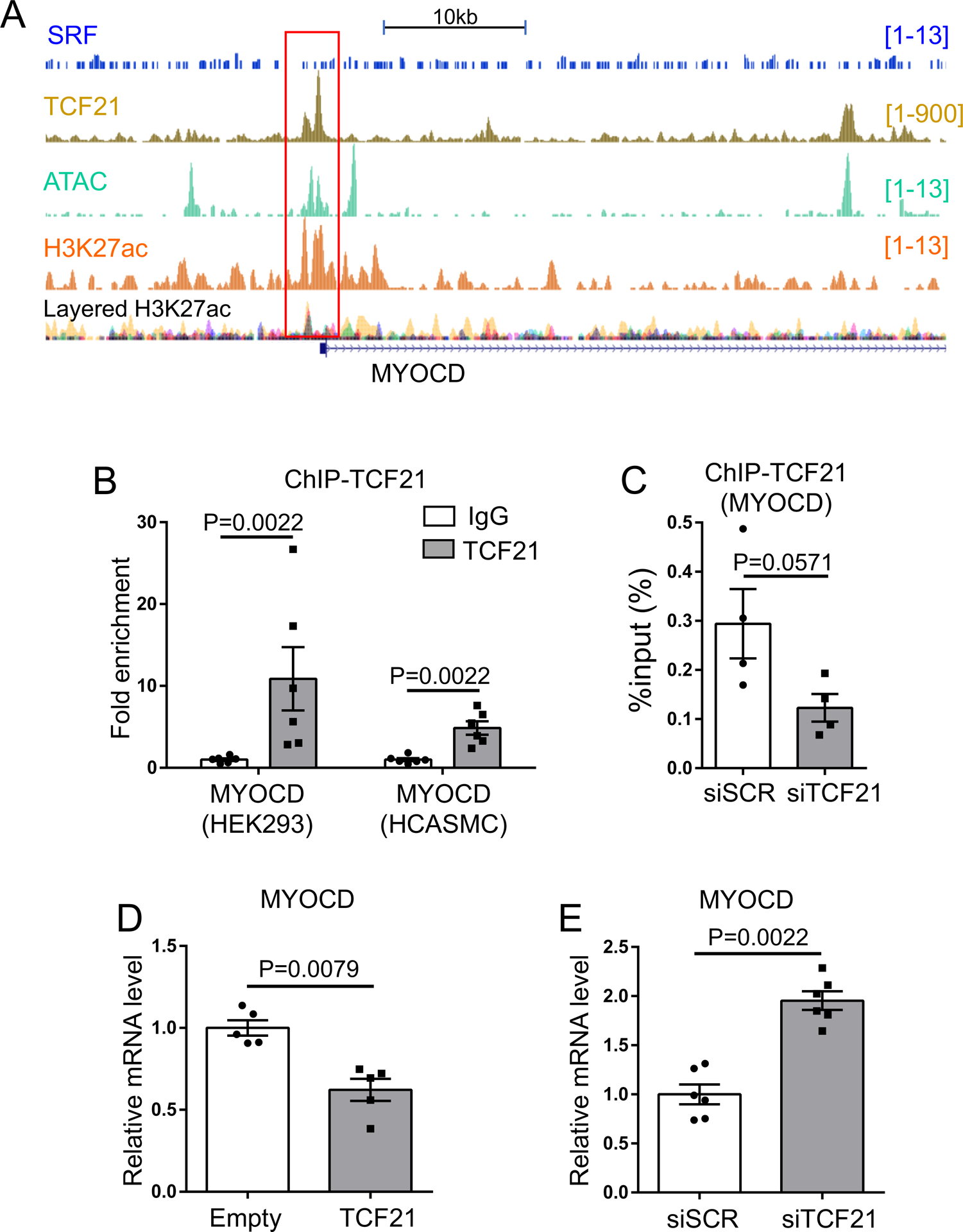
A) Pattern of TCF21 binding at the MYOCD locus, as identified by previously determined TCF21 ChIP-seq, H3K27ac histone modification and ATAC-seq identification of open chromatin in HCASMC. B) Chromatin immunoprecipitation for TCF21 binding to the MYOCD promoter in HEK293 overexpressing TCF21 and HCASMC (n=5–6).C) Chromatin immunoprecipitation for TCF21 binding to the MYOCD promoter in HCASMC treated with siTCF21 or siSCR (n=4). D, E) MYOCD gene expression analyzed by qPCR. For overexpression or knockdown of TCF21, HCASMC were treated with TCF21 overexpressing lentivirus (pWPI-TCF21) or siTCF21, respectively. B-E) Data analyzed by Mann-Whitney U test.
TCF21 inhibits MYOCD transactivation of SMC genes.
In addition to its effect on MYOCD expression, we sought to determine whether TCF21 affects the ability of MYOCD to transactivate its target genes. Thus, we conducted reporter gene transfection studies. We transfected different amounts of TCF21 expression construct DNA, along with a MYOCD expression construct and an SRF enhancer reporter plasmid into A7R5 cells. Transfection of MYOCD produced a robust activation of reporter expression as measured by dual luciferase assay (Fig. 6A, Online Table II), and this effect was abrogated when the CArG box was deleted from the reporter (Online Fig. VI, Online Table III). Expression of TCF21 resulted in a dose-dependent decrease in SRF enhancer reporter expression. The inhibitory effect was also observed in cultured HCASMC treated with the highest concentration of TCF21 DNA (800ng) (Fig. 6B, Online Table II). This effect was not due to changes in MYOCD levels, as shown by evaluating the transfected cells by western blot with an antibody validated to detect recombinant protein in these cells (Online Fig. VII). The inhibitory effect of TCF21 was also seen at endogenous levels, as MYOCD transactivation of SRF-luc was perturbed by knocking down native TCF21 in HCASMC (Online Fig. VIII). Similar experiments were conducted with reporter gene constructs containing the SMC-restricted promoters of the TAGLN and CNN1 genes (Methods)32–34. The reporters for these genes showed similar ability to respond to MYOCD mediated stimulation of transcription with a similar inhibition produced by TCF21 concomitant expression in both A7R5 and HCASMC (Fig. 6C–F, Online Table II).
Figure 6. TCF21 blocks transcriptional regulation by MYOCD at the SRF enhancer, the TAGLN promoter and the CNN1 promoter.
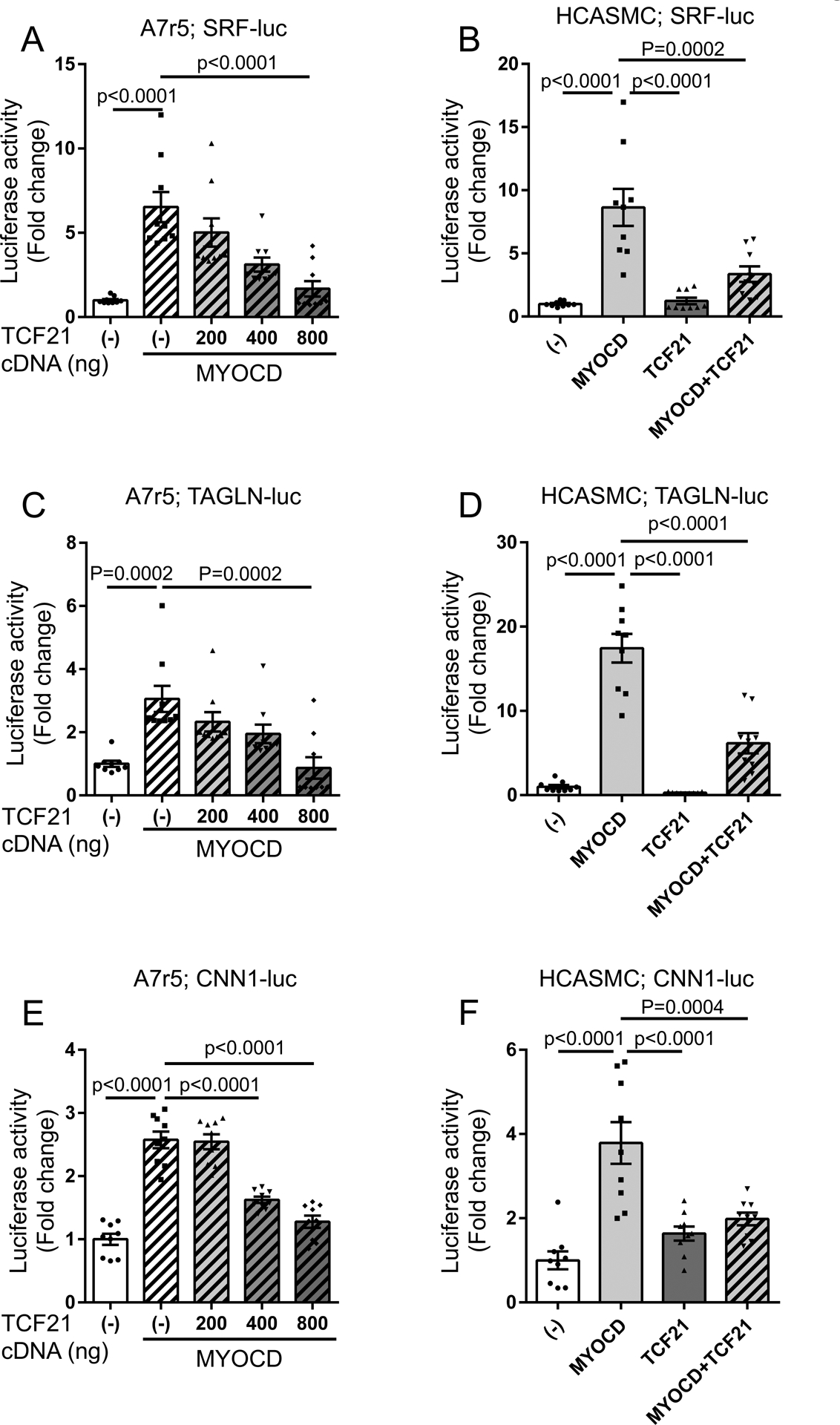
A, B) Either one or both MYOCD (pcDNA3.1-MYOCD) and TCF21 (pCMV6-AC-TCF21) expression plasmids were co-transfected with a reporter construct containing the SRF enhancer (SRF-luc) into A7r5 (A) and HCASMC (B). Dual luciferase assays were performed, using Renilla luciferase reporter plasmid as an internal control of transfection efficiency. C) Dual luciferase assays were conducted using a reporter construct containing the TAGLN promoter (TAGLN-luc) transfected into A7r5cells, and D) HCASMC with the same methodology. E) Dual luciferase assays were conducted using a reporter construct containing the CNN1 promoter (CNN1-luc) transfected into A7r5cells, and F) HCASMC with the same methodology. All experiments, (n>8). Each group was compared to “TCF21(−) with MYOCD” (A,C,E) or “MYOCD” (B,D,F) using one-way ANOVA followed by Dunnett’s post-test (B,D-F), or Kruskal-Wallis test, followed by Dunn’s post-test (A,C).
The fact that TCF21 was able to block the transcriptional activity of the TAGLN promoter was surprising, given that the TAGLN promoter sequence in the reporter plasmid did not contain a classical TCF21 binding site28. To investigate further the possibility that TCF21 was affecting reporter gene expression in the absence of its canonical binding sequence, we deleted the TCF21 binding motif (CAGCTG) from the SRF enhancer-luciferase construct. Co-transfection of MYOCD and TCF21 with this modified luciferase reporter vector showed a significant reduction in enhancer activity, with the 20-fold effect of MYOCD decreasing to ~3-fold in HCASMC (Online Fig. IX, Online Table IV). These results are not conclusive, but suggest that the inhibitory effect of TCF21 is independent of DNA binding and may exert its affect by interacting directly with the MYOCD-SRF complex to block assembly in solution or its binding to DNA.
TCF21 directly interferes with formation and function of the MYOCD-SRF complex.
To further investigate mechanisms by which TCF21 interacts with MYOCD and SRF to regulate gene expression, we investigated whether expression of TCF21 interfered with their known protein-protein interaction. In the first experiments, HEK293 cells were transfected with plasmid constructs encoding MYOCD, SRF, and increasing amounts of Myc-tagged TCF21 plasmid DNA (0, 1, 2 μg). In the presence of increasing concentrations of TCF21, we demonstrated a reduced concentration of SRF in the protein fraction immunoprecipitated by the anti-MYOCD antibody (Fig. 7A). As shown in these studies, expression of TCF21 did not affect expression levels of MYOCD or SRF, so we speculated that the decrease in co-immunoprecipitated SRF was due to a disruption of the MYOCD-SRF complex. We thus pursued further experiments to investigate the possibility that TCF21 mediates this effect by directly interacting with MYOCD or SRF. For these studies, we expressed recombinant MYOCD and Myc-tagged TCF21 in HEK293 cells and found that immunoprecipitated MYOCD was accompanied by Myc-tagged TCF21 as detected by western blot (Fig. 7B). Further, the immunoprecipitation of Myc-tagged TCF21 was accompanied by MYOCD immunoreactivity by western blot (Fig. 7C). To ensure this interaction is found at endogenous levels of MYOCD, the experiments were repeated in A7r5 cells, which showed Myc-tagged TCF21 co-immunoprecipitated with native Myocd (Fig. 7D). Similar experiments failed to provide evidence for MycTCF21 interacting with Srf (Fig. 7E). Further, we have performed ChIP-PCR assays using MYOCD antibody and showed there was approximately a 3-fold increase in MYOCD binding at the SRF enhancer in HCASMC with siTCF21 knockdown (Fig. 7F). Taken together, these data are suggestive that TCF21 interacts with MYOCD to block interaction of MYOCD and SRF in solution and binding to the CArG motif in SMC (Fig. 7G).
Figure 7. TCF21 binds MYOCD and disrupts the functional MYOCD-SRF complex.
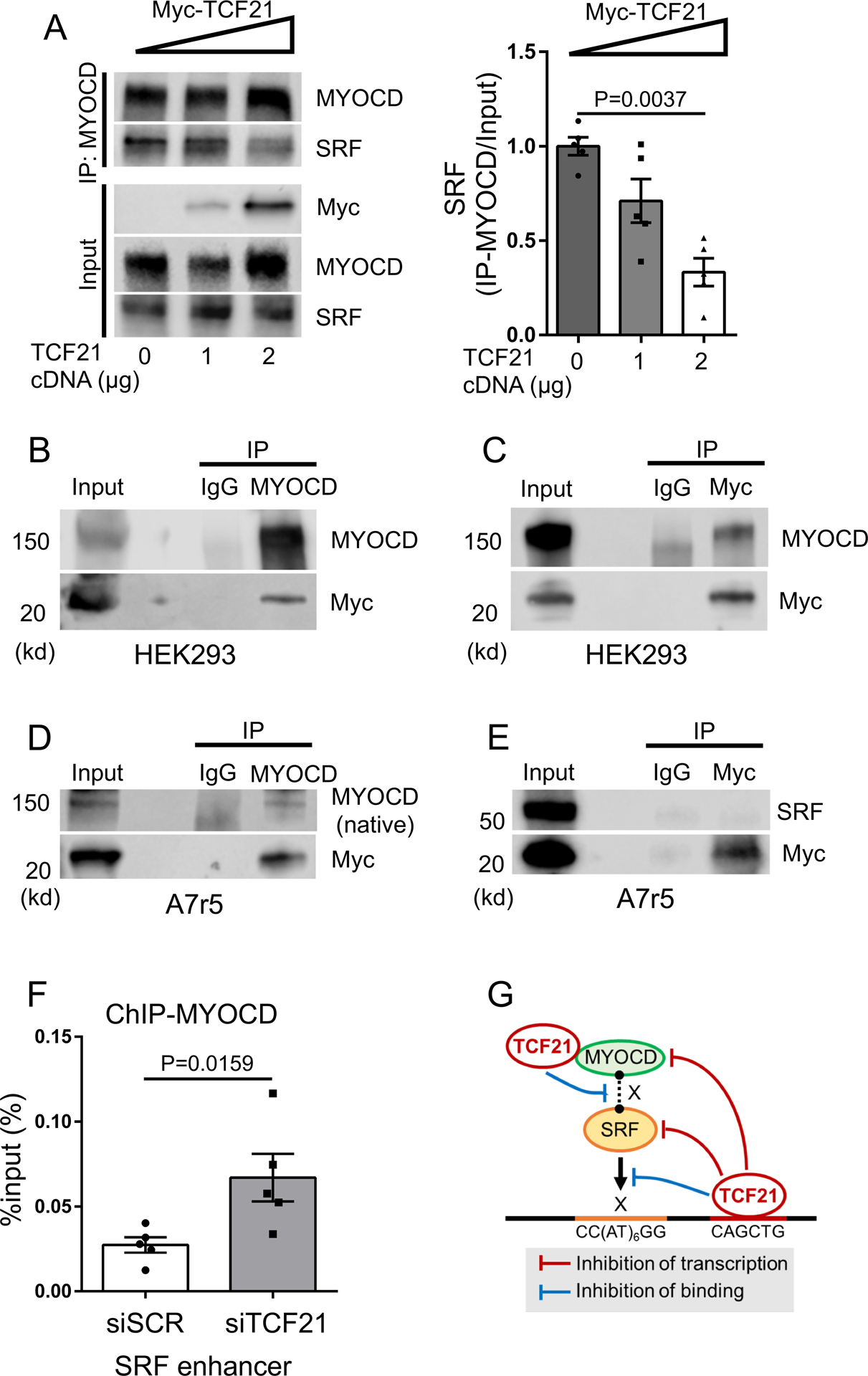
A) HEK293 cells were transduced with plasmid constructs encoding MYOCD, SRF, and increasing amounts of Myc-tagged TCF21 plasmid DNA (0, 1, 2 μg). Immunoprecipitation was then performed with anti-MYOCD antibody, followed by western blot with antibodies as indicated. Band intensity of SRF was quantified using image J software and normalized to the input band of SRF (n=5). Data analyzed by Kruskal-Wallis test, followed by Dunn’s post-test. B, C) HEK293 cells were transfected with MYOCD and Myc-tagged TCF21 expression plasmids and immunoprecipitation was performed with anti-MYOCD antibody or anti-Myc antibody, followed by western blot with antibodies as indicated. D, E) To test the physical interaction of TCF21 with native rat Myocd (D) or Srf (E), A7r5 cells were transfected with Myc-tagged TCF21 expression plasmid and immunoprecipitation was performed with anti-MYOCD or anti-Myc antibody, followed by western blot with antibodies as indicated. F) ChIP-qPCR with anti-MYOCD antibody at the SRF enhancer in HCASMC treated with siTCF21 or scrambled siRNA (siSCR) (n=5). Data analyzed by Mann-Whitney U test. G) TCF21 binding is enriched in SRF targeted loci genome-wide, and binds and inhibits transcription in both the SRF and MYOCD loci. TCF21 also blocks SRF binding to DNA at an autoregulatory enhancer region in the SRF gene. Further, direct interaction of TCF21 with MYOCD disrupts the MYOCD-SRF complex leading to decreased MYOCD-SRF association.
Unfortunately, we were not able to detect reproducible native TCF21 binding to native MYOCD, due in part to the anti-correlated expression patterns of the two proteins in HCASMC (Online Fig. X) and the inability of available MYOCD antibodies to reliably detect endogenous MYOCD protein at the expected molecular weight in HCASMC35. Nevertheless, our data do show convincing evidence for native Myocd interacting with recombinant TCF21 in the A7R5 cell line, and knockdown of native TCF21 was shown to increase the binding of native MYOCD to the SRF enhancer in the HCASMC nucleus by ChIP-PCR. The data showing co-immunoprecipitation of recombinant MYOCD and TCF21 are suggestive that protein-protein interaction of these two factors impairs their association in solution, and thus the ability of the complex to bind DNA.
DISCUSSION
To date, GWAS have identified over 160 genomic loci associated with CAD13–15. Through replication of these findings in an independent cohort and evidence for a validated mechanism in vascular cells, these findings provide an unparalleled opportunity to identify causal mechanisms for this disease process in the context of vascular wall pathology. However, because the majority of these associations reflect allelic differences in gene expression levels, due to complex enhancer gene-promoter interactions that are affected by functional differences between the causal and protective variants, progress in understanding mechanisms of genetic CAD risk remains quite limited2, 36. The genomic locus at 6q23.2 is one of the most highly replicated CAD loci, in multiple racial ethnic groups13–15, 37, and extensive genomic and genetic studies have identified TCF21 as the causal gene in this region of the human genome5, 16, 27. Using lineage tracing and single cell RNA sequencing, we have recently characterized the transcriptomic phenotype of modulated SMC in vivo in mice with SMC-specific knockout of Tcf21, and showed that loss of Tcf21 expression leads to fewer modulated SMC within the lesion and the protective fibrous cap6. TCF21 expression was also strongly associated with SMC phenotypic modulation in diseased human coronaries. In human CAD-relevant tissues,TCF21 expression was associated with decreased CAD risk, establishing a protective role for both TCF21 and SMC phenotypic modulation in this disease. Studies described here were aimed at investigating the mechanism by which this transcription factor regulates the de-differentiated SMC phenotype, focusing on the SMC lineage determining MYOCD-SRF pathway.
Our data suggest that a critical component of TCF21 effects on HCASMC phenotype, and likely the disease process in the vessel wall, are related to inhibition of SMC lineage gene expression, mediated in part by inhibition of the MYOCD-SRF pathway. We had previously shown that TCF21 binds the ACTA2 locus and inhibits expression of this gene, thus indicating that it directly blocks expression of some SMC genes31. However, there was no evidence for TCF21 binding in many of the SMC genes that were differentially regulated by TCF21 expression28. Thus, we initiated studies to investigate whether TCF21 affects SMC gene expression indirectly by blocking MYOCD and SRF signaling, which is responsible in large part for determining the SMC differentiated phenotype18, 19, 21–23.
These studies suggest multiple mechanisms by which TCF21 blocks MYOCD-SRF function in SMC, underscoring the importance of this interaction in the regulation of SMC phenotype (Fig. 7G). First, we mapped SRF binding genome-wide in HCASMC and intersected the binding sites with those identified by similar studies for TCF2128, with the expectation of finding SMC target regions where TCF21 might bind and independently regulate expression of genes that are activated by MYOCD-SRF, or TCF21 might regulate MYOCD-SRF binding and function. This overlap was highly statistically significant. Interestingly, SRF was found to colocalize with TCF21 in the second intron of the SRF gene, in a region of HCASMC open chromatin that is also marked by an enhancer mark, H3K27ac histone modification. This finding suggests a positive feedback autoregulatory capability of SRF that is inhibited by TCF21. Importantly, knockdown of TCF21 led to increased binding of SRF at the intronic enhancer and SRF expression. CRISPR/Cas9 genome editing studies in cultured cells, and in a mouse model in the highly conserved murine Srf enhancer, showed that mutation of the CArG motif led to decreased Srf expression. Also, TCF21 was shown to bind at the MYOCD gene promoter and inhibit its gene expression. Previous study of the regulatory mechanisms for Myocd expression identified an enhancer ~30 kilobases upstream of the regulatory region identified in these studies, which also failed to bind SRF but rather was shown to be regulated by Mef2, Tead and Foxo factors38. Interestingly, this enhancer conferred an autoregulatory function on the Myocd gene, similar to the SRF enhancer described in these studies.
The inhibitory effects of TCF21 on transcription at the SRF and MYOCD loci are consistent with TCF21 altering the local epigenetic landscape by recruiting inhibitory histone deacetylases39, 40 as we have shown for the effect of TCF21 on SMAD3 binding in loci where they colocalize4, 29. Alternatively, at the SRF enhancer TCF21 and MYOCD-SRF bind 71 bp apart and are potentially associated with the same nucleosome. Thus the opposing effect of TCF21 on SRF binding might reflect the physical competition for the space necessary to establish DNA interaction. Also, the fact that TCF21 blocks MYOCD transactivation of the TAGLN promoter and the mutated SRF enhancer, which do not contain TCF21 binding sequences, is most consistent with direct protein-protein interactions that block MYOCD-SRF binding and/or interaction with the basal transcription apparatus. Our data suggest that direct TCF21 interaction with MYOCD, i.e. protein-protein interaction, that disrupts the MYOCD-SRF complex may be responsible for some of the SMC gene expression inhibitory effects of TCF21. In this regard, TCF21 is similar to the Hippo signaling molecule TEAD1, which also directly interacts with MYOCD and blocks MYOCD-SRF complex formation41. We have shown direct protein-protein interaction for recombinant MYOCD and TCF21 in heterologous HEK 293 cells by over-expression, and interaction of native MYOCD with recombinant TCF21 in A7R5 SMC, which do not normally express TCF21. We were also able to show by ChIP-PCR that TCF21 suppresses binding of native MYOCD in HCASMC at the SRF enhancer.
It has been difficult to understand whether phenotypic modulation of HCASMC in the coronary artery promotes or diminishes the risk for CAD. On one hand, de-differentiated cells migrate into the plaque where they secrete stabilizing matrix proteins and contribute to the stabilizing fibrous cap11, 42. On the other hand, these cells are also believed to contribute to the macrophage like cells in the plaque, promoting the inflammatory milieu that destabilizes the vessel wall11, 43. Through molecular interaction of the MYOCD-SRF pathway with the CAD associated gene TCF21, studies reported here provide evidence for involvement of basic HCASMC processes such as lineage restricted gene expression and phenotypic modulation in the risk for CAD. Human genetic and genomic data indicate that expression of TCF21 promotes protection from CAD6, 27, and thus by inference suggest that phenotypic modulation is protective, while factors that promote differentiation, such as the MYOCD-SRF pathway promotes CAD risk5. This directionality is strengthened by the fact that CAD associated factors with pro-differentiation effects on HCASMC, such as SMAD3 and TGFβ1, appear to promote risk2, 4. However, this paradigm is not consistent with ApoE knockout model data indicating that Myocd negatively regulates SMC inflammatory activation and atherogenesis. The basis of this disparity in directionality of effect is not understood and will require additional study in both animal disease models and humans44.
In summary, TCF21 is associated with CAD risk, with protective alleles of this gene being linked to increased expression in HCASMC2, 5, 27. In keeping with its embryonic role where expression in the pro-epicardial organ and epicardium inhibits differentiation of coronary artery SMC precursor cells45, 46, re-activation of expression of this factor in adult SMC is associated with increased proliferation, migration, and suppressed expression of classical SMC lineage markers, the process of phenotypic modulation6, 31. One molecular mechanism by which it mediates this effect is through inhibition of the lineage determining MYOCD-SRF pathway. TCF21 binds in some of the same loci as SRF, including in an intronic enhancer in the SRF gene where it inhibits positive autoregulation. It binds the MYOCD promoter and inhibits its expression, and also blocks transcriptional regulation by MYOCD. Together, these studies provide evidence for epigenetic and transcriptional mechanisms by which TCF21 regulates MYOCD-SRF expression and function. Finally, TCF21 may possibly interact directly with MYOCD to disrupt its functional association with SRF and transcriptional regulation of SMC genes.
Supplementary Material
NOVELTY AND SIGNIFICANCE.
What Is Known?
Smooth muscle cells constitute an important component of the blood vessel wall and have been implicated in vascular disease because of their loss of mature markers and expansion in the lesion.
The transcription factor TCF21 has been shown to promote these disease-associated features in SMC, and inhibit disease through this process, but the mechanism of this effect in these cells has not been identified.
To potentially target this pathway for therapeutic purposes, the molecular mechanism must be examined.
What New Information Does This Article Contribute?
This work shows that TCF21 binds in the same regions of the genome as the myocardin (MYOCD)-serum response factor (SRF) complex that promotes a mature phenotype in SMC.
This work has also shown that TCF21 binds in an autoregulatory enhancer in the SRF gene as well as the promoter of the MYOCD gene to block expression of these factors.
Further, these studies show that TCF21 directly blocks the ability of MYOCD and SRF proteins to associate in solution and bind to DNA, thus further inhibiting the ability of these factors to direct a mature phenotype in the SMC.
SOURCES OF FUNDING
This work was supported by National Institutes of Health grants R01HL109512 (TQ), R01HL134817 (TQ), R33HL120757 (TQ), R01DK107437 (TQ), R01HL139478 (TQ), R01HL138987 (JMM), R01HL147476 (JMM), and a grant from the Chan Zuckerberg Foundation - Human Cell Atlas Initiative (TQ).
Nonstandard Abbreviations and Acronyms:
- TCF21
transcription factor 21
- SRF
serum response factor
- MYOCD
myocardin
- SMC
smooth muscle cell
- HCASMC
human coronary artery smooth muscle cells
- CAD
coronary artery disease
- ATAC-seq
(Assay for Transposase-Accessible Chromatin using sequencing)
- ChIP-seq
chromatin immunoprecipitation sequencing
Footnotes
DISCLOSURES
None.
REFERENCES
- 1.Nanda V, Wang T, Pjanic M, Liu B, Nguyen T, Matic LP, Hedin U, Koplev S, Ma L, Franzen O, Ruusalepp A, Schadt EE, Bjorkegren JLM, Montgomery SB, Snyder MP, Quertermous T, Leeper NJ, Miller CL. Functional regulatory mechanism of smooth muscle cell-restricted lmod1 coronary artery disease locus. PLoS Genet. 2018;14:e1007755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Miller CL, Pjanic M, Wang T, Nguyen T, Cohain A, Perisic L, Hedin U, Betsholtz C, Ruusalepp A, Franzen O, Assimes TL, Montgomery SB, Schadt EE, Bjorkegren JLM, Quertermous T. Integrative functional genomics identifies regulatory mechanisms at coronary artery disease loci. Nature communications. 2016:12092–12108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kojima Y, Downing K, Kundu R, Miller C, Dewey F, Lancero H, Raaz U, Perisic L, Hedin U, Schadt E, Maegdefessel L, Quertermous T, Leeper NJ. Cyclin-dependent kinase inhibitor 2b regulates efferocytosis and atherosclerosis. J Clin Invest. 2014;124:1083–1097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Iyer D, Zhao Q, Wirka R, Naravane A, Nguyen T, Liu B, Nagao M, Cheng P, Miller CL, Kim JB, Pjanic M, Quertermous T. Coronary artery disease genes smad3 and tcf21 promote opposing interactive genetic programs that regulate smooth muscle cell differentiation and disease risk. PLoS Genet. 2018;14:e1007681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu B, Pjanic M, Wang T, Nguyen T, Gloudemans M, Rao A, Castano VG, Nurnberg S, Rader DJ, Elwyn S, Ingelsson E, Montgomery SB, Miller CL, Quertermous T. Genetic regulatory mechanisms of smooth muscle cells map to coronary artery disease risk loci. Am J Hum Genet. 2018;103:377–388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wirka R, W D., Paik DT, Pjanic M, Nguyen AT, Miller CJ, Kundu R, Nagao M, Coller J, Koyano TK, Fong R, Woo YJ, Liu B, Montgomery SB, Wu JC, Zhu K, Chang R, Alamprese M, Tallquist MD, Kim JB, Quertermous T. Single cell analysis of smooth muscle cell phenotypic modulation in vivo reveals a critical role for coronary disease gene tcf21 in mice and humans. Nat Med. 2108;in press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sandison ME, Dempster J, McCarron JG. The transition of smooth muscle cells from a contractile to a migratory, phagocytic phenotype: Direct demonstration of phenotypic modulation. J Physiol. 2016;594:6189–6209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pidkovka NA, Cherepanova OA, Yoshida T, Alexander MR, Deaton RA, Thomas JA, Leitinger N, Owens GK. Oxidized phospholipids induce phenotypic switching of vascular smooth muscle cells in vivo and in vitro. Circ Res. 2007;101:792–801 [DOI] [PubMed] [Google Scholar]
- 9.Sjolund M, Madsen K, von der Mark K, Thyberg J. Phenotype modulation in primary cultures of smooth-muscle cells from rat aorta. Synthesis of collagen and elastin. Differentiation. 1986;32:173–180 [DOI] [PubMed] [Google Scholar]
- 10.Thyberg J, Nilsson J, Palmberg L, Sjolund M. Adult human arterial smooth muscle cells in primary culture. Modulation from contractile to synthetic phenotype. Cell and tissue research. 1985;239:69–74 [DOI] [PubMed] [Google Scholar]
- 11.Bennett MR, Sinha S, Owens GK. Vascular smooth muscle cells in atherosclerosis. Circ Res. 2016;118:692–702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shankman LS, Gomez D, Cherepanova OA, Salmon M, Alencar GF, Haskins RM, Swiatlowska P, Newman AA, Greene ES, Straub AC, Isakson B, Randolph GJ, Owens GK. Klf4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nat Med. 2015;21:628–637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Klarin D, Zhu QM, Emdin CA, Chaffin M, Horner S, McMillan BJ, Leed A, Weale ME, Spencer CCA, Aguet F, Segre AV, Ardlie KG, Khera AV, Kaushik VK, Natarajan P, Consortium CAD, Kathiresan S. Genetic analysis in uk biobank links insulin resistance and transendothelial migration pathways to coronary artery disease. Nat Genet. 2017;49:1392–1397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nelson CP, Goel A, Butterworth AS, Kanoni S, Webb TR, Marouli E, Zeng L, Ntalla I, Lai FY, Hopewell JC, Giannakopoulou O, Jiang T, Hamby SE, Di Angelantonio E, Assimes TL, Bottinger EP, Chambers JC, Clarke R, Palmer CNA, Cubbon RM, Ellinor P, Ermel R, Evangelou E, Franks PW, Grace C, Gu D, Hingorani AD, Howson JMM, Ingelsson E, Kastrati A, Kessler T, Kyriakou T, Lehtimaki T, Lu X, Lu Y, Marz W, McPherson R, Metspalu A, Pujades-Rodriguez M, Ruusalepp A, Schadt EE, Schmidt AF, Sweeting MJ, Zalloua PA, AlGhalayini K, Keavney BD, Kooner JS, Loos RJF, Patel RS, Rutter MK, Tomaszewski M, Tzoulaki I, Zeggini E, Erdmann J, Dedoussis G, Bjorkegren JLM, Consortium E-C, CardioGramplusC4D, group UKBCCCw, Schunkert H, Farrall M, Danesh J, Samani NJ, Watkins H, Deloukas P. Association analyses based on false discovery rate implicate new loci for coronary artery disease. Nat Genet. 2017;49:1385–1391 [DOI] [PubMed] [Google Scholar]
- 15.van der Harst P, Verweij N. The identification of 64 novel genetic loci provides an expanded view on the genetic architecture of coronary artery disease. Circ Res. 2017;122:433–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Miller CL, Anderson DR, Kundu RK, Raiesdana A, Nurnberg ST, Diaz R, Cheng K, Leeper NJ, Chen CH, Chang IS, Schadt EE, Hsiung CA, Assimes TL, Quertermous T. Disease-related growth factor and embryonic signaling pathways modulate an enhancer of tcf21 expression at the 6q23.2 coronary heart disease locus. PLoS Genet. 2013;9:e1003652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang D, Chang PS, Wang Z, Sutherland L, Richardson JA, Small E, Krieg PA, Olson EN. Activation of cardiac gene expression by myocardin, a transcriptional cofactor for serum response factor. Cell. 2001;105:851–862 [DOI] [PubMed] [Google Scholar]
- 18.Yoshida T, Sinha S, Dandre F, Wamhoff BR, Hoofnagle MH, Kremer BE, Wang DZ, Olson EN, Owens GK. Myocardin is a key regulator of carg-dependent transcription of multiple smooth muscle marker genes. Circ Res. 2003;92:856–864 [DOI] [PubMed] [Google Scholar]
- 19.Pipes GC, Creemers EE, Olson EN. The myocardin family of transcriptional coactivators: Versatile regulators of cell growth, migration, and myogenesis. Genes Dev. 2006;20:1545–1556 [DOI] [PubMed] [Google Scholar]
- 20.Wang DZ, Olson EN. Control of smooth muscle development by the myocardin family of transcriptional coactivators. Current opinion in genetics & development. 2004;14:558–566 [DOI] [PubMed] [Google Scholar]
- 21.Chen J, Kitchen CM, Streb JW, Miano JM. Myocardin: A component of a molecular switch for smooth muscle differentiation. J Mol Cell Cardiol. 2002;34:1345–1356 [DOI] [PubMed] [Google Scholar]
- 22.Miano JM. Myocardin in biology and disease. Journal of biomedical research. 2015;29:3–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Du KL, Ip HS, Li J, Chen M, Dandre F, Yu W, Lu MM, Owens GK, Parmacek MS. Myocardin is a critical serum response factor cofactor in the transcriptional program regulating smooth muscle cell differentiation. Mol Cell Biol. 2003;23:2425–2437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang M, Fang H, Zhou J, Herring BP. A novel role of brg1 in the regulation of srf/mrtfa-dependent smooth muscle-specific gene expression. J Biol Chem. 2007;282:25708–25716 [DOI] [PubMed] [Google Scholar]
- 25.Patel S, Shi Y, Niculescu R, Chung EH, Martin JL, Zalewski A. Characteristics of coronary smooth muscle cells and adventitial fibroblasts. Circulation. 2000;101:524–532 [DOI] [PubMed] [Google Scholar]
- 26.Kim BJ, Pjanic M, Nguyen T, Miller CL, Liu B, Wang T, Sazonova O, Carcamo-Orive I, Perisic L, Maegdefessel L, Hedin U, Quertermous T. Tcf21 and the aryl-hydrocarbon receptor cooperate to activate a pro-atherosclerotic gene expression program. PLoS Genet. 2017;13:1006750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Miller CL, Haas U, Diaz R, Leeper NJ, Kundu RK, Patlolla B, Assimes TL, Kaiser FJ, Perisic L, Hedin U, Maegdefessel L, Schunkert H, Erdmann J, Quertermous T, Sczakiel G. Coronary heart disease-associated variation in tcf21 disrupts a mir-224 binding site and mirna-mediated regulation. PLoS Genet. 2014;10:e1004263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sazonova O, Zhao Y, Nurnberg S, Miller C, Pjanic M, Castano VG, Kim JB, Salfati EL, Kundaje AB, Bejerano G, Assimes TL, Yang X, Quertermous T. Characterization of tcf21 downstream target regions identifies a transcriptional network linking multiple independent coronary artery disease loci. PLoS Genet. 2015;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhao Q, Wirka R, Nguyen T, Nagao M, Cheng P, Miller CL, Kim JB, Pjanic M, Quertermous T. Tcf21 and ap-1 interact through epigenetic modifications to regulate coronary artery disease gene expression. Genome Med. 2019;11:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, Bejerano G. Great improves functional interpretation of cis-regulatory regions. Nat Biotechnol. 2010;28:495–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nurnberg ST, Cheng K, Raiesdana A, Kundu R, Miller CL, Kim JB, Arora K, Carcamo-Oribe I, Xiong Y, Tellakula N, Nanda V, Murthy N, Boisvert WA, Hedin U, Perisic L, Aldi S, Maegdefessel L, Pjanic M, Owens GK, Tallquist MD, Quertermous T. Coronary artery disease associated transcription factor tcf21 regulates smooth muscle precursor cells that contribute to the fibrous cap. PLoS Genet. 2015;11:e1005155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Long X, Slivano OJ, Cowan SL, Georger MA, Lee TH, Miano JM. Smooth muscle calponin: An unconventional carg-dependent gene that antagonizes neointimal formation. Arterioscler Thromb Vasc Biol. 2011;31:2172–2180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Han Y, Slivano OJ, Christie CK, Cheng AW, Miano JM. Crispr-cas9 genome editing of a single regulatory element nearly abolishes target gene expression in mice--brief report. Arterioscler Thromb Vasc Biol. 2015;35:312–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Miano JM, Carlson MJ, Spencer JA, Misra RP. Serum response factor-dependent regulation of the smooth muscle calponin gene. J Biol Chem. 2000;275:9814–9822 [DOI] [PubMed] [Google Scholar]
- 35.Lyu Q, Dhagia V, Han Y, Guo B, Wines-Samuelson ME, Christie CK, Yin Q, Slivano OJ, Herring P, Long X, Gupte SA, Miano JM. Crispr-cas9-mediated epitope tagging provides accurate and versatile assessment of myocardin-brief report. Arterioscler Thromb Vasc Biol. 2018;38:2184–2190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Maurano MT, Humbert R, Rynes E, Thurman RE, Haugen E, Wang H, Reynolds AP, Sandstrom R, Qu H, Brody J, Shafer A, Neri F, Lee K, Kutyavin T, Stehling-Sun S, Johnson AK, Canfield TK, Giste E, Diegel M, Bates D, Hansen RS, Neph S, Sabo PJ, Heimfeld S, Raubitschek A, Ziegler S, Cotsapas C, Sotoodehnia N, Glass I, Sunyaev SR, Kaul R, Stamatoyannopoulos JA. Systematic localization of common disease-associated variation in regulatory DNA. Science. 2012;337:1190–1195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lu X, Wang L, Chen S, He L, Yang X, Shi Y, Cheng J, Zhang L, Gu CC, Huang J, Wu T, Ma Y, Li J, Cao J, Chen J, Ge D, Fan Z, Li Y, Zhao L, Li H, Zhou X, Chen L, Liu D, Chen J, Duan X, Hao Y, Wang L, Lu F, Liu Z, Yao C, Shen C, Pu X, Yu L, Fang X, Xu L, Mu J, Wu X, Zheng R, Wu N, Zhao Q, Li Y, Liu X, Wang M, Yu D, Hu D, Ji X, Guo D, Sun D, Wang Q, Yang Y, Liu F, Mao Q, Liang X, Ji J, Chen P, Mo X, Li D, Chai G, Tang Y, Li X, Du Z, Liu X, Dou C, Yang Z, Meng Q, Wang D, Wang R, Yang J, Schunkert H, Samani NJ, Kathiresan S, Reilly MP, Erdmann J, Peng X, Wu X, Liu D, Yang Y, Chen R, Qiang B, Gu D. Genome-wide association study in han chinese identifies four new susceptibility loci for coronary artery disease. Nat Genet. 2012;44:890–894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Creemers EE, Sutherland LB, McAnally J, Richardson JA, Olson EN. Myocardin is a direct transcriptional target of mef2, tead and foxo proteins during cardiovascular development. Development. 2006;133:4245–4256 [DOI] [PubMed] [Google Scholar]
- 39.Hong CY, Gong EY, Kim K, Suh JH, Ko HM, Lee HJ, Choi HS, Lee K. Modulation of the expression and transactivation of androgen receptor by the basic helix-loop-helix transcription factor pod-1 through recruitment of histone deacetylase 1. Mol Endocrinol. 2005;19:2245–2257 [DOI] [PubMed] [Google Scholar]
- 40.Tandon P, Miteva YV, Kuchenbrod LM, Cristea IM, Conlon FL. Tcf21 regulates the specification and maturation of proepicardial cells. Development. 2013;140:2409–2421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu F, Wang X, Hu G, Wang Y, Zhou J. The transcription factor tead1 represses smooth muscle-specific gene expression by abolishing myocardin function. J Biol Chem. 2014;289:3308–3316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cherepanova OA, Gomez D, Shankman LS, Swiatlowska P, Williams J, Sarmento OF, Alencar GF, Hess DL, Bevard MH, Greene ES, Murgai M, Turner SD, Geng YJ, Bekiranov S, Connelly JJ, Tomilin A, Owens GK. Activation of the pluripotency factor oct4 in smooth muscle cells is atheroprotective. Nat Med. 2016;22:657–665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Misra A, Feng Z, Chandran RR, Kabir I, Rotllan N, Aryal B, Sheikh AQ, Ding L, Qin L, Fernandez-Hernando C, Tellides G, Greif DM. Integrin beta3 regulates clonality and fate of smooth muscle-derived atherosclerotic plaque cells. Nature communications. 2018;9:2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ackers-Johnson M, Talasila A, Sage AP, Long X, Bot I, Morrell NW, Bennett MR, Miano JM, Sinha S. Myocardin regulates vascular smooth muscle cell inflammatory activation and disease. Arterioscler Thromb Vasc Biol. 2015;35:817–828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Braitsch CM, Combs MD, Quaggin SE, Yutzey KE. Pod1/tcf21 is regulated by retinoic acid signaling and inhibits differentiation of epicardium-derived cells into smooth muscle in the developing heart. Dev Biol. 2012;368:345–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Acharya A, Baek ST, Huang G, Eskiocak B, Goetsch S, Sung CY, Banfi S, Sauer MF, Olsen GS, Duffield JS, Olson EN, Tallquist MD. The bhlh transcription factor tcf21 is required for lineage-specific emt of cardiac fibroblast progenitors. Development. 2012;139:2139–2149 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
SRF ChIP-seq data has been deposited to the Gene Expression Omnibus (GEO) database, accession number GSE124011.
Detailed experimental materials and methods are described in the Online Supplement.