

ABSTRACT
Objective:
Tyrosinemia type III (HT III) is the rarest form of tyrosinemia, and the full clinical spectrum of this disorder is still unknown. The neurological involvement varies, including intellectual impairment and attention deficit disorder with hyperactivity (ADHD). We report the case of two siblings diagnosed with HT III at different ages.
Case description:
The index case was diagnosed by newborn screening for endocrine and metabolic disorders, starting a low-protein diet immediately, with a consistent decrease in tyrosine levels. By the age of three, the child displayed a hyperactive behavior, starting treatment for ADHD two years later. At seven years of age, he shows a slight improvement in terms of behavior and attention span and has a cognitive performance slightly lower than his peers, despite maintaining acceptable tyrosine levels. His sister, who had a history of ADHD since age five, was diagnosed with HT III after family screening at the age of eight. Despite initiating a dietetic treatment, her behavior did not improve, and she has a mild intellectual impairment.
Comments:
This is the first case report describing siblings with HT III who underwent nutritional treatment with a low-protein diet in different phases of life, with a better neurological and behavioral evaluation in the patient who started treatment earlier.
Keywords: Tyrosine, Tyrosinemias, Attention deficit disorder with hyperactivity, Metabolism
RESUMO
Objetivo:
A tirosinemia tipo III (TT III) é a forma mais rara das tirosinemias e o espectro clínico desta entidade não está totalmente esclarecido. O envolvimento neurológico é variável, incluindo o atraso cognitivo ou transtorno do déficit de atenção com hiperatividade (TDAH). Descrevemos o caso de dois irmãos que foram diagnosticados com TT III em idades diferentes.
Descrição dos casos:
O caso índice foi diagnosticado no contexto do rastreio endócrino-metabólico neonatal, tendo iniciado imediatamente dieta hipoproteica, com redução consistente dos níveis de tirosina. Por volta dos três anos, foi detectado um comportamento hiperativo, tendo iniciado dois anos depois tratamento para o TDAH. Aos sete anos, apresenta leve melhora de comportamento e da atenção e avaliação cognitiva levemente inferior ou pouco abaixo quando comparado a crianças da mesma faixa etária, apesar de manter níveis aceitáveis de tirosina. A sua irmã, com história de TDAH desde os cinco anos, foi diagnosticada de TT III aos oito anos no contexto do rastreio de familiares. Apesar de iniciar tratamento dietético, nenhum efeito foi notado em termos de comportamento e a doente apresenta leve atraso cognitivo.
Comentários:
Este é o primeiro caso clínico descrito de irmãos com TT III que iniciaram terapêutica dietética com dieta hipoproteica em diferentes fases da vida, com melhor avaliação em termos neurológicos e comportamentais no doente que iniciou tratamento mais precocemente.
Palavras-chave: Tirosina, Tirosinemias, Transtorno do déficit de atenção com hiperatividade, Metabolismo
INTRODUCTION
Tyrosine is a non-essential amino acid, obtained directly from diet or the hydroxylation of phenylalanine. It is a precursor in the synthesis of catecholamines, thyroxine, and melanin. 1
Tyrosinemia type III (OMIM 276710) is a rare inborn error of tyrosine metabolism caused by mutations in the gene encoding the enzyme 4-hydroxyphenylpyruvate dioxygenase (HPPD), which catalyzes the conversion of 4-hydroxyphenylpyruvate to homogentisate, the second step in the tyrosine catabolic pathway. It is the rarest form of tyrosinemia, being transmitted in an autosomal recessive form. This metabolic disorder is characterized by elevated levels of serum tyrosine and increased excretion of phenolic metabolites [4-hydroxyphenylpyruvate (4-HPP), 4-hydroxyphenyllactate (4-HPL) and hydrophenylacetate] in the urine. Only a few case reports have been described in the literature, with a wide clinical phenotype spectrum: some patients presented neurodevelopmental symptoms while others were diagnosed by newborn screening, being asymptomatic. 1 , 2 , 3
We report the case of two siblings with tyrosinemia type III who were diagnosed and started treatment at different ages, presenting their clinical outcomes. A brief literature review is also presented.
CASE DESCRIPTION
Case 1
The male patient is the third child from a Portuguese consanguineous couple (third cousins), with an unremarkable family history. He was born at term by vaginal delivery following an uneventful pregnancy, with an Apgar score of 9 and 10 at 1 and 5 minutes, respectively. His birth weight was 3,510 g (89th percentile), length 50 cm (76th percentile), and head circumference 35.5 cm (93rd percentile).
The newborn screening performed at the fifth day of life revealed elevated tyrosine levels (526 µmol/L; cut-off values: <248 µmol/L). A control sample taken at the 29th day showed the persistence of elevated plasma tyrosine levels (680 µmol/L), with high urinary excretion of 4-HPL and 4-HPP and presence of N-acetyl-tyrosine and vanillactic acid.
The child started a protein-restricted diet supplemented with a tyrosine- and phenylalanine-free amino acid mixture at 1-month-old. Subsequent metabolic controls showed a consistent decrease in tyrosine levels: 280 µmol/L at 1.5 months, 262 µmol/L at 7 months, and 165 µmol/L at 12 months.
The diagnosis of tyrosinemia type III was confirmed by a genetic analysis performed by amplifying exons one to 14 of the HPD gene using polymerase chain reaction followed by DNA sequencing, which demonstrated a homozygous mutation p.A33T (c.97G>A).
His physical examination was normal, particularly with no ocular or skin involvement, and presenting typical growth (weight curve at the 85‒97th percentile until 30 months and then at the 50‒85th percentile; height curve at the 50th percentile until 30 months and then at the 50‒85th percentile; normal head circumference). His neurological examination was normal, and he had a brain magnetic resonance imaging at the age of 30 months showing no abnormalities. His blood test analysis (full blood count, liver and renal function, and electrolytes) was unremarkable.
His early psychomotor development was normal, with head control at less than two months, sitting alone without support at approximately eight months, and walking alone at 12 months. First words were spoken at 12 months, and he constructed simple sentences at the age of 18 months. However, after the age of three, language development progressed slowly, with sound articulation problems.
The parents also reported a hyperactive behavior, with impulsivity and inability to follow orders by the age of three. Throughout his pre-school years, he was repeatedly considered incapable of following the level of learning of his classmates.
A formal developmental assessment using the Griffiths Mental Development Scale at 32 months showed a global developmental quotient (GDQ) of 88.9, and the test was repeated at 54 months showing a GDQ of 87 (slightly lower score compared to the children in his age group, with more evident difficulties on the sub-scale of hearing and language).
At the age of five, the neurodevelopmental unit diagnosed him with Attention Deficit Disorder with Hyperactivity (ADHD) with a combined subtype (DSM-V criteria), and he initiated treatment with methylphenidate. He also started speech therapy sessions.
Currently, at the age of seven, he shows a slight improvement in terms of behavior and attention span and still undergoes speech therapy. He attends elementary school (2nd grade) with no learning difficulties, and at his last formal developmental assessment (Wechsler Intelligence Scale for Children®, third edition, Portuguese version), he had an intelligence quotient (IQ) of 78, a verbal IQ of 81, and a performance IQ of 82 (Figure 1).
Figure 1. WISC-III subtest profile of patient 1.
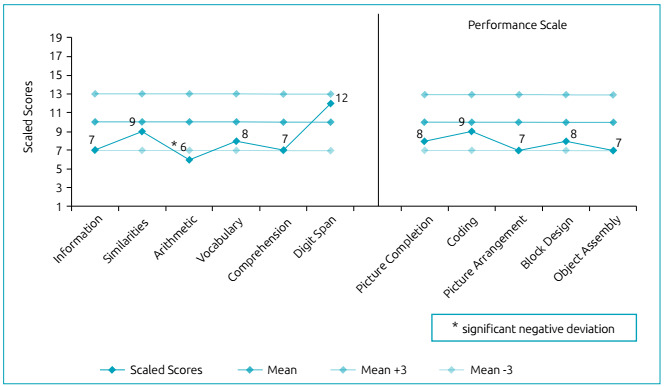
His tyrosine levels have been consistently below 300 µmol/L. On his last nutritional status evaluation performed at the age of seven, he presented a daily natural protein intake of 1 g/kg, supplemented with 1.1 g of amino acids/kg from phenylalanine- and tyrosine-free amino acid mixtures. Body composition analysis was considered adequate.
Case 2
A 15-year-old female, the eldest sibling of patient 1, was diagnosed with tyrosinemia type III at eight years of age, after a family screening following her brother’s diagnosis.
She was born at term by cesarean section (pelvic presentation) following an uneventful pregnancy, with an Apgar score of 8 and 9 at 1 and 5 minutes, respectively. Her birth weight was 4,030 g (87th percentile), length 53 cm (87th percentile), and head circumference 36 cm (83rd percentile). Her neonatal period was unremarkable, as was her early psychomotor development. She had a history of primary nocturnal enuresis and vesical instability, treated with desmopressin and oxybutynin hydrochloride since the age of six.
She was diagnosed with ADHD at the age of five and treated with methylphenidate and risperidone. She presented learning difficulties when she attended elementary school (2nd grade), and was included in a special education program at the age of ten.
At the time of diagnosis, her initial tyrosine level was 1,769 µmol/L. On the first nutritional status evaluation, her daily natural protein intake was 1.4 g/kg. This value dropped to 1.08 g/kg, and she started taking supplementation with 0.7 g of amino acids/kg from phenylalanine- and tyrosine-free amino acid mixtures, enabling a consistent decrease of tyrosine levels below 300 µmol/L.
Her physical examination and growth were normal (weight and height at the 50‒85th percentile), with an unremarkable neurological examination. The genetic study revealed the same mutation as her brother in homozygosity.
After initiating a low-protein diet, her behavior and school performance did not improve. The minimum level of daily natural protein intake reached was 0.65 g/kg at 14 years of age. She is currently in 7th grade, still in the special education program, and under treatment with methylphenidate. Her latest formal developmental assessment (Wechsler Intelligence Scale for Children®, third edition, Portuguese version) revealed a global IQ score of 68, with a verbal IQ of 68 and a performance IQ of 77 (Figure 2).
Figure 2. WISC-III subtest profile of patient 2.
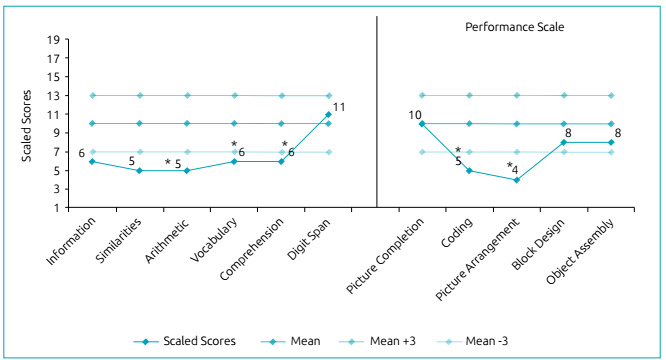
She slowly began diet liberalization and is now with a daily natural protein intake of 0.9 g/kg, combined with 0.8 g of amino acids/kg, maintaining tyrosine levels below 300 µmol/L nevertheless.
DISCUSSION
The symptoms of tyrosinemia type III are not well characterized, and there is no apparent correlation between tyrosine serum levels, the clinical phenotype, and the mutation type. As in the two cases presented, many patients have neurodevelopmental manifestations, including intellectual impairment, learning difficulties, dyslexia, attention deficit, hyperactivity, behavioral disturbance, ataxia, microcephaly, hypotonia, and seizures, but a classical phenotype has not been described. 1 , 2 , 3
Contrary to tyrosinemia types I and II, patients do not demonstrate any evidence of hepato-renal dysfunction or skin or eye lesions. In 2015, an 11-year-old girl with normal development and no neurological signs was diagnosed with tyrosinemia type III following the investigation of recurrent proteinuria (9-17 mg/L); in this case, laboratory tests revealed elevated serum tyrosine levels, which led to the diagnosis of the disease. However, it was not clear if the nephrological complications were associated with tyrosinemia type III. 4
In a 2001 review, which included 13 patients, the most common long-term complication was intellectual impairment (75% patients). Five patients were diagnosed by newborn screening, three of whom started a low-tyrosine and -phenylalanine diet after diagnosis; among them, two had no symptoms and presented normal development at 13 months and five years and five months, respectively, while the third had a delayed psychomotor development but demonstrated an average developmental quotient at the age of four (Griffiths Mental Development Scale). One patient only started treatment at 8 months of age, when developmental retardation was detected, and another who was not treated showed intellectual impairment. Eight patients were diagnosed after the neonatal period, seven of them because of neurologic signs and one due to developmental delay. Only one patient had normal development at 17 years of age. 2
The etiology of the neurological manifestations is not known but could be related to hypertyrosinemia as in tyrosinemia types I and II. Tyrosine and/or its derivatives seemed to be neurotoxic metabolites, and mental delay was associated with increased plasma concentrations of these substances. Several studies revealed that hypertyrosinemia inhibits the functioning of respiratory chain complexes, compromises the Krebs cycle, and decreases creatine kinase and pyruvate kinase activities, inducing an oxidative stress status and an impairment of energy metabolism in the cerebral cortex of rats. 5 , 6 , 7 , 8
Patients detected by newborn screening appear to have fewer neurological symptoms and a lower degree of cognitive impairment compared to those diagnosed later in life. 2 , 3 However, it is unclear if the clinical outcome is determined by the decrease in plasma tyrosine levels, and the neurological evolution can vary despite similar tyrosine levels. 2 Besides, there are asymptomatic patients diagnosed later in childhood or adolescence who never developed neurological or behavioral symptoms. 4
Another proposed hypothesis is that neurological impairment could be explained by an excessive nitric oxide release, which could contribute to neuronal damage. 9
Although it is unclear if lowering tyrosine levels can alter the natural history of the disease, treating it with a low-protein diet, at least in early childhood, to maintain tyrosine levels between 200 and 500 µmol/L has been considered reasonable. 10
Both our patients started nutritional treatment after diagnosis, maintaining tyrosinemia levels below 300 µmol/L afterward. However, while one was detected by newborn screening, the other was eight years old at the time of diagnosis and already symptomatic with learning difficulties. Clinically, they both presented ADHD requiring pharmacological treatment, but patient 1 had a better cognitive outcome despite being below average compared to the healthy population group. He attends the 2nd grade with a normal curriculum.
In patient 2, despite initiating treatment, her behavior did not improve, and she has a mild intellectual impairment. A decision to gradually increase natural protein intake was made, and she has maintained acceptable tyrosine levels, although she still takes phenylalanine- and tyrosine-free amino acid supplements to satisfy her nitrogen needs. These findings have been reported, and many patients seem to be able to maintain these levels after childhood, without diet control. 2
Currently, a significant number of metabolic disorders related to major psychiatric diseases remain underdiagnosed for years before more specific organic signs become evident. Metabolic diseases most associated with ADHD are succinic semialdehyde dehydrogenase deficiency, phenylketonuria, X-linked ichthyosis, and mucopolysaccharidosis type III (Sanfilippo syndrome). 11
Establishing a genotype-phenotype correlation in tyrosinemia is difficult since the literature has very few cases described on the subject, and only some of them identify distinct mutations. Tables 1 and 2 summarize the main clinical and analytical characteristics of patients known to date.
Table 1. Summary of patients with tyrosinemia type III detected by neonatal screening.
| Case-study | Gender | Age at diagnosis | Family history | Form of presentation | Neurological abnormalities | Brain image | Plasma tyrosinemia at diagnosis | Genetic study | Treatment | Follow-up |
|---|---|---|---|---|---|---|---|---|---|---|
| Patient 1 | Male | Neonatal screening | Consanguineous parents | Asymptomatic | Speech delay, ADHD | Normal | 526 µmol/L | Homozygous for A33T mutation in the HPD gene | Low-Phe/Tyr diet | “Borderline” development (7 y) |
| 1- Preece et al. 12 | Female | Neonatal screening | Crouzon syndrome | Asymptomatic | Developmental delay at 8 mo | Not described/performed | 355 µmol/L | Not described/performed | Low-Phe/Tyr diet | Learning difficulties (7 y) Crouzon syndrome |
| 2- Standing et al. 13 and Rüetschi et al. 14 | Female | Neonatal screening | First cousin of patient 3 (Table 1) Consanguineous parents | Asymptomatic | Mild jitteriness, brisk tendon reflexes | Abnormal appearance in subcortical and brainstem white matter (30 mo) | 1094 µmol/L | Homozygous for Y258X mutation in the HPD gene | Low-Phe/Tyr diet | Delayed psychomotor development (5 y) |
| 3- Standing et al. 13 and Rüetschi et al. 14 | Male | Neonatal screening | First cousin of patient 2 (Table 1) Consanguineous parents | Neonatal hepatitis | None | Not described/performed | Range: 500-1000 µmol/L in the first 19 mo | Homozygous for Y258X mutation in the HPD gene | Normal diet | Mild intellectual impairment (17 y) |
| 4- Ellaway et al. 15 | Not described | Neonatal screening | Not relevant | Asymptomatic | None | Not described/performed | 937 µmol/L | Not described/performed | Low-Phe/Tyr diet | Normal development (13 mo) |
| 5- Ellaway et al. 15 | Female | Neonatal screening | Not relevant | Asymptomatic | None | Not described/performed | 581 µmol/L | Not described/performed | Low-Phe/Tyr diet | Normal development (5 y 5 mo) |
| 6- Heylen et al. 10 | Male | Neonatal screening | Consanguineous parents | Asymptomatic | None | Not described/performed | 398 µmol/L | Homozygous splice donor mutation in intron 11, IVS11+1G>A in the HPD gene | Low-Phe/Tyr diet | Normal development (30 mo) |
ADHD: attention deficit disorder with hyperactivity; HPD: hydroxyphenylpyruvate dioxygenase; mo: months; Phe: phenylalanine; Tyr: tyrosine; y: years.
Table 2. Summary of patients with tyrosinemia type III detected after the neonatal period.
| Case-study | Gender | Age at diagnosis | Family history | Form of presentation | Neurological abnormalities | Brain image | Plasma tyrosinemia at diagnosis | Genetic study | Treatment | Follow-up |
|---|---|---|---|---|---|---|---|---|---|---|
| Patient 2 | Female | 8 y | Consanguineous parents | Learning difficulties | Psychomotor retardation, ADHD | Not described/performed | 1769 µmol/L | Homozygous for A33T mutation in the HPD gene | Low-Phe/Tyr diet | Mild intellectual impairment (15 y) |
| 1- Endo et al. 16 | Male | Infancy | Parents were siblings mother had hypertyrosinemia | Seizures and pneumonia at day 21 of life | Seizures, encephalopathy | Mild cerebral atrophy | 640 µmol/L | Not described/performed | Low-Phe/Tyr diet | Died due to accidental asphyxiation (day 105) |
| 2- Giardini et al. 17 and D’Eufemia et al. 18 , 19 | Female | 17 mo | Not relevant | Acute ataxia, confusion, motor incoordination, hypotonia, absent tendon reflexes | Recovered from all symptoms | Not described/performed | 624 µmol/L | Not described/performed | Low-protein diet until 3 y | Autoimmune thyroiditis (9 y) Normal development (17 y) |
| 3- Cerrone et al. 3 and Rüetschi et al. 14 | Male | 3.5 y | Not relevant | Developmental delay from 8 mo | Psychomotor retardation, hyperactivity, self-injurious behavior, severe speech delay | Normal | 532 µmol/L | Heterozygous for Y200X and I335M mutations in the HPD gene | Low-Phe/Tyr diet until 12 y | Severe intellectual impairment (14 y) |
| 4- Tomoeda et al. 20 | Male | 7 weeks | Not relevant | Neonatal restlessness, reduced head control | Microcephaly | Not described/performed | 594 µmol/L during treatment | Homozygous for A268V mutation in the HPD gene | Low Phe/Tyr diet until 17 y, then low-protein diet | Moderate intellectual impairment (19 y) |
| 5- Rüetschi et al. 14 | Male | 7.5 y | Brother of patient 4 (Table 2) Consanguineous parents | Urolithiasis | Essential tremor | Not described/performed | 830 µmol/L | Homozygous for Y160C mutation in the HPD gene | Normal diet | Mild intellectual impairment (7.5 y) Lost to follow-up |
| 6- Rüetschi et al. 14 | Male | 18 y | Brother of patient 5 (Table 2) Consanguineous parents | Psychomotor retardation | Essential tremor | Not described/performed | 262 µmol/L | Homozygous for Y160C mutation in the HPD gene | Normal diet | Lost to follow-up |
| 7- Ellaway et al. 15 | Male | 14 y | Consanguineous parents | Mild psychomotor retardation, generalized seizures | Suspected seizures since 2 y | Not described/performed | 913 µmol/L | Not described/performed | Low-protein diet initially, then ceased | Intellectual impairment (18 y) |
| 8- Ellaway et al. 15 | Not described | 18 mo | Not relevant | Developmental delay | None | Not described/performed | 1305 µmol/L | Not described/performed | Low-Phe/Tyr diet | Mild global developmental delay (6 y) |
| 9- Tahiroglu et al. 21 | Male | 2 y | Not described | Developmental delay from 15 mo | Psychomotor retardation, hyperactivity, autistic symptoms | Not described/performed | “Elevated levels” | Not described/performed | Low-Tyr diet | Severe intellectual impairment (9 y) Persistence of hyperactivity |
| 10- Szymanska et al. 4 | Female | 11 y | Not described | Recurrent proteinuria since 7 y | None | Not described/performed | 439 µmol/L | Homozygous for T160C mutation in the HPD gene | Normal diet | Normal development (11 y) |
ADHD: attention deficit disorder with hyperactivity; HPD: hydroxyphenylpyruvate dioxygenase; mo: months; Phe: phenylalanine; Tyr: tyrosine; y: years.
To our knowledge, this is the first case report of siblings with tyrosinemia type III who underwent nutritional treatment with a low-protein diet in different life stages, with the one who started it earlier and during an asymptomatic phase showing better results in terms of neurological and behavioral outcomes.
Both patients presented ADHD as a neurological manifestation. Therefore, we emphasize the importance of conducting a metabolic study in children with this disorder who do not respond adequately to pharmacological treatment.
Although the pathophysiology of neuronal injury in tyrosinemia type III is not completely explained by the accumulation of tyrosine in the central nervous system, a restrictive tyrosine and phenylalanine diet is recommended during childhood. Further studies and collection of information on these patients are necessary to understand the consequences of HPPD deficiency, the mechanisms of brain injury, and the long-term outcome in patients with this rare form of tyrosinemia.
ACKNOWLEDGMENTS
We thank the parents of the patients for their kind cooperation. Furthermore, we thank the contribution of the medical-laboratory assistants of our institutes.
Funding
This study did not receive funding.
REFERENCES
- 1.Russo PA, Mitchell GA, Tanguay RM. Tyrosinemia: a review. Pediatr Dev Pathol. 2001;4:212–221. doi: 10.1007/s100240010146. [DOI] [PubMed] [Google Scholar]
- 2.Ellaway CJ, Holme E, Standing S, Preece MA, Green A, Ploechl E, et al. Outcome of tyrosinemia type III. J Inherit Metab Dis. 2001;24:824–832. doi: 10.1023/a:1013936107064. [DOI] [PubMed] [Google Scholar]
- 3.Cerone R, Holme E, Schiaffino MC, Caruso U, Maritano L, Romano C. Tyrosinemia type III: diagnosis and ten-year follow-up. Acta Paediatr. 1997;86:1013–1015. doi: 10.1111/j.1651-2227.1997.tb15192.x. [DOI] [PubMed] [Google Scholar]
- 4.Szymanska E, Sredzinska M, Ciara E, Piekutowska-Abramczuk D, Ploski R, Rokicki D, et al. Tyrosinemia type III in an asymptomatic girl. Mol Genet Metab Rep. 2015;5:48–50. doi: 10.1016/j.ymgmr.2015.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Andrade RB, Gemelli T, Rojas DB, Funchal C, Dutra-Filho CS, Wannmacher CM. Tyrosine inhibits creatine kinase activity in cerebral cortex of young rats. Metab Brain Dis. 2011;26:221–227. doi: 10.1007/s11011-011-9255-9. [DOI] [PubMed] [Google Scholar]
- 6.Macedo LG, Carvalho-Silva M, Ferreira GK, Vieira JS, Olegario N, Goncalves RC, et al. Effect of acute administration of L-tyrosine on oxidative stress parameters in brain of young rats. Neurochem Res. 2013;38:2625–2630. doi: 10.1007/s11064-013-1180-3. [DOI] [PubMed] [Google Scholar]
- 7.De Prá SD, Ferreira GK, Carvalho-Silva M, Vieira JS, Scaini G, Leffa DD, et al. L-tyrosine induces DNA damage in brain and blood of rats. Neurochem Res. 2014;39:202–207. doi: 10.1007/s11064-013-1207-9. [DOI] [PubMed] [Google Scholar]
- 8.Teodorak BP, Scaini G, Carvalho-Silva M, Gomes LM, Teixeira LJ, Rebelo J, et al. Antioxidants reverse the changes in energy metabolism of rat brain after chronic administration of L.-tyrosine. Metab Brain Dis. 2017;32:557–564. doi: 10.1007/s11011-016-9936-5. [DOI] [PubMed] [Google Scholar]
- 9.D’Eufemia P, Finocchiaro R, Celli M, Raccio I, Properzi E, Zicari A. Increased nitric oxide release by neutrophils of a patient with tyrosinemia type III. Biomed Pharmacother. 2009;63:359–361. doi: 10.1016/j.biopha.2008.06.030. [DOI] [PubMed] [Google Scholar]
- 10.Heylen E, Scherer G, Vincent MF, Marie S, Fischer J, Nassogne MC. Tyrosinemia Type III detected via neonatal screening: management and outcome. Mol Genet Metab. 2012;107:605–607. doi: 10.1016/j.ymgme.2012.09.002. [DOI] [PubMed] [Google Scholar]
- 11.Simons A, Eyskens F, Glazemakers I, West D. Can psychiatric childhood disorders be due to inborn errors of metabolism? Eur Child Adolesc Psychiatry. 2017;26:143–154. doi: 10.1007/s00787-016-0908-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Preece MA, Rylance GW, MacDonald A, Green A, Gray RGF. A new case of tyrosinemia type III detected by neonatal screening. J Inherit Metab Dis. 1996;19(1):32–32. [Google Scholar]
- 13.Standing SJ, Dunger D, Rüetschi U, Holme E. Tyrosinemia type III detected by neonatal screening. J Inherit Metab Dis. 1998;21(2):25–25. [Google Scholar]
- 14.Rüetschi U, Cerone R, Pérez-Cerda C, Schiaffino MC, Standing S, Ugarte M, et al. Mutations in the 4-hydroxyphenylpyruvate dioxygenase gene (HPD) in patients with tyrosinemia type III. Hum Genet. 2000;106:654–662. doi: 10.1007/s004390000307. [DOI] [PubMed] [Google Scholar]
- 15.Ellaway CJ, Holme E, Standing S, Preece MA, Green A, Ploechl E, et al. Outcome of tyrosinemia type III. J Inherit Metab Dis. 2001;24:824–832. doi: 10.1023/A:1013936107064. [DOI] [PubMed] [Google Scholar]
- 16.Endo F, Kitano A, Uehara I, Nagata N, Matsuda I, Shinka T, et al. Four-Hydroxyphenylpyruvic acid oxidase de¢ciency with normal fumarylacetoacetase: a new variant of hereditary hypertyrosinaemia. Pediatr Res. 1983;17:92–96. doi: 10.1203/00006450-198302000-00002. [DOI] [PubMed] [Google Scholar]
- 17.Giardini O, Cantani A, Kennaway NG, D’Eufemia P. Chronic tyrosinemia associated with 4-hydroxyphenylpyruvate dioxygenase deficiency with acute intermittent ataxia and without visceral and bone involvement. Pediatr Res. 1983;17:25–29. doi: 10.1203/00006450-198301000-00005. [DOI] [PubMed] [Google Scholar]
- 18.D’Eufemia P, Giardini O, Cantani A, Martino F, Finocchiaro R. Autoimmune thyroiditis in a case of tyrosinemia III. J Inherit Metab Dis. 1992;15:861–862. doi: 10.1007/bf01800222. [DOI] [PubMed] [Google Scholar]
- 19.D’Eufemia P, Finocchiaro R, Celli M, Viozzi L, Giardini O. Immunological abnormalities in a patient with tyrosinemia type III. J Inherit Metab Dis. 1995;18:355–356. doi: 10.1007/bf00710429. [DOI] [PubMed] [Google Scholar]
- 20.Tomoeda K, Awata H, Matsuura T, Matsuda I, Ploechl E, Milovac T, et al. Mutations in the 4-Hydroxyphenylpyruvic acid dehydrogenase gene are responsible for tyrosinemia type III and hawkinisinuria. Mol Genet Metab. 2000;71:506–510. doi: 10.1006/mgme.2000.3085. [DOI] [PubMed] [Google Scholar]
- 21.Tahiroğlu AY, Mungan NÖ, Firat S, Avci A. Autism symptoms related to tyrosinemia type III: A case report. Turk J Endocrinol Metab. 2008;12:55–56. [Google Scholar]