

 This work is licensed under a
This work is licensed under a Abstract
Summary
We describe the case of a 56 year-old woman with the almost simultaneous appearance of diffuse idiopathic pulmonary neuroendocrine cell hyperplasia (DIPNECH) and a carotid body paraganglioma. Of interest, 6 years earlier, the patient underwent total thyroidectomy due to papillary thyroid carcinoma and, in the meantime, she was submitted to mastectomy to treat an invasive ductal carcinoma of the breast. In order to explain these lesions, an extensive genetic study was performed. Results showed positivity for the presence of the tumor suppressor gene PALB2, whose presence had already been detected in a niece with breast cancer. The patient underwent different procedures to treat the lesions and currently she is symptom-free over 2 years of follow-up.
Learning points:
The presence of two rare neoplasms in a single person should raise the suspicion of a common etiology.
To the best of our knowledge, this is the first case that shows the coexistence of DIPNECH and paraganglioma.
The contribution of the PALB2 gene in the etiology of these rare neoplasms is a possibility.
Patient Demographics: Adult, Female, White, Portugal
Clinical Overview: Adrenal, Adrenal, Thyroxine (T4), Neuroendocrine tumour, Paraganglioma, Papillary thyroid cancer, Breast cancer*, DIPNECH*
Diagnosis and Treatment: Dyspnoea, Wheezing, CT scan, Biopsy, Pulmonary biopsy*, PET scan, Synaptophysin, Haematoxylin and eosin staining, Histopathology, Chromogranin A, Immunohistochemistry, S-100 protein*, Cytokeratins*, Molecular genetic analysis, Resection of tumour, Levothyroxine
Related Disciplines: Oncology
Publication Details: New disease or syndrome: presentations/diagnosis/management, May, 2020
Background
Paragangliomas are uncommon neuroendocrine tumors that originate from the extraadrenal autonomic paraganglia, with the ability to release catecholamines in the circulation. Sympathetic paragangliomas typically secrete catecholamines and their location is in the sympathetic paravertebral ganglia of thorax, abdomen, and pelvis. On the contrary, most parasympathetic paragangliomas are nonfunctional and located along the glossopharyngeal and vagal nerves in the neck and at the base of the skull.
Paragangliomas and pheochromocytomas are predictable to arise in about 2–8 of 1 million persons per year and, in about 0.1% of hypertensive patients, this is the cause that justifies hypertension (1).
Diffuse idiopathic pulmonary neuroendocrine cell hyperplasia (DIPNECH) is a relatively recent and rare disease that is frequently misdiagnosed. It is characterized by being a pre-invasive generalized proliferation of pulmonary neuroendocrine cells and is regarded as a precursor lesion for pulmonary neuroendocrine tumors (NETs). It includes tumorlets, which extend beyond the basement membrane and form lesions <5 mm in greatest dimension and nodules >5 mm. The typical patient is a non-smoking, middle-aged women and is linked with a mostly obstructive ventilatory pattern on pulmonary function tests. Most of the patients present with nonproductive cough and exertional dyspnea that are normally misdiagnosed for bronchial asthma or chronic bronchitis. Treatments included systemic and inhaled corticosteroids, bronchodilators, and lung resection (2).
This case is unique because, to the best of our knowledge, it is the first case that reflects the coexistence of these two rare pathologies. In addition, it raises the suspicion of a genetic predisposition to the etiology of the tumors, not only due to the presence of these two rare entities but also by the presence of two more malignant neoplasms. A variant of the PALB2 tumor suppressor gene was identified in this patient.
Case presentation
We report the case of a 59-year-old female, with history of obesity, hypertension, dyslipidemia, non-allergic asthma, and bronchiectasis. Nine years ago, she was submitted to total thyroidectomy due to a papillary thyroid cancer. Currently, she is controlled with levothyroxine 125 μg/day, and she has no evidence of persistent or metastatic disease (thyroglobulin levels less than 0.5 μg/L after TSH stimulation). The patient also underwent mastectomy 2 years ago to treat a breast tumor. Histology showed an invasive ductal carcinoma, Grade 3; Estrogen Receptor negative, Progesterone Receptor negative, HER2 negative; Stage I - pT1bN0M0. Her medical medical history was relevant for breast cancer in a niece. Patient denied current or previous smoking or alcoholic habits.
The patient complained about frequent episodes of dyspnea and wheezing without flushing, cough, or diarrhea. She had no palpitations, hyperhidrosis, or headaches. In a follow-up CT scan to evaluate her pulmonary disease, a dimensional increase of nodularities already reported in previous CT scans (from 4/5 mm to 7/9 mm) was detected, as also the presence of new ones (Fig. 1). Due to her previous cancer history, the possibility of secondary lesions was raised and it was decided to perform biopsy to better characterize the lesions.
Figure 1.

The thorax CT scan which revealed several nodularities (maximum size 9 mm).
Therefore, the patient underwent biopsy of a pulmonary nodule, whose report described a neuroendocrine tumor (NET), although definitive diagnosis, namely differential diagnosis between typical and atypical carcinoid, could only be made in the excision piece. Subsequently, she performed Ga-68 DOTANOC PET/CT that demonstrated at the level of the left superior cervical region, in the axial plane of the angle of the mandible, near the left carotid, a mass that could translate a paraganglioma (Fig. 2). In order to evaluate whether the paraganglioma was a catecholamine-secretor or not, the urinary metanephrines were measured and the results were within the normal range. Laboratory findings were also negative for hyperparathyroidism.
Figure 2.

A Ga-68 DOTANOC PET/CT which suggest a carotid body paraganglioma.
The patient was then submitted to excision of the cervical lesion (Fig. 3). Immunohistochemistry studies were performed and staining was positive for synaptophysin, chromogranin, and S-100 protein and negative for Cytokeratin 8/18. Ki67 Proliferative Index was between 1 and 3%. Results revealed a well differentiated paraganglioma with a GAPP score of 2.
Figure 3.
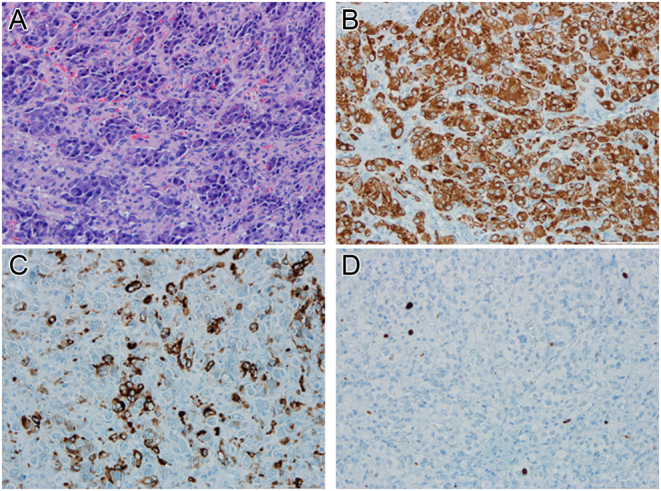
Paraganglioma. (A) A tumor with a zellballen pattern; the tumor cells are uniform with a polygonal shape and abundant cytoplasm. Mitosis or necrosis are absent (hematoxylin-eosin stained slide, 200×). (B) A diffuse expression of synaptophysin by neoplastic cells (200×). (C) Focal expression of chromogranin by neoplastic cells (200×). (D) A proliferative index of approximately 2% (ki67 stained slide, 400×).
Months later, the patient underwent pulmonary surgery. The histological piece showed DIPNECH with tumorlets and two typical NETs-pT2a (m) NxR0 (Fig. 4).
Figure 4.
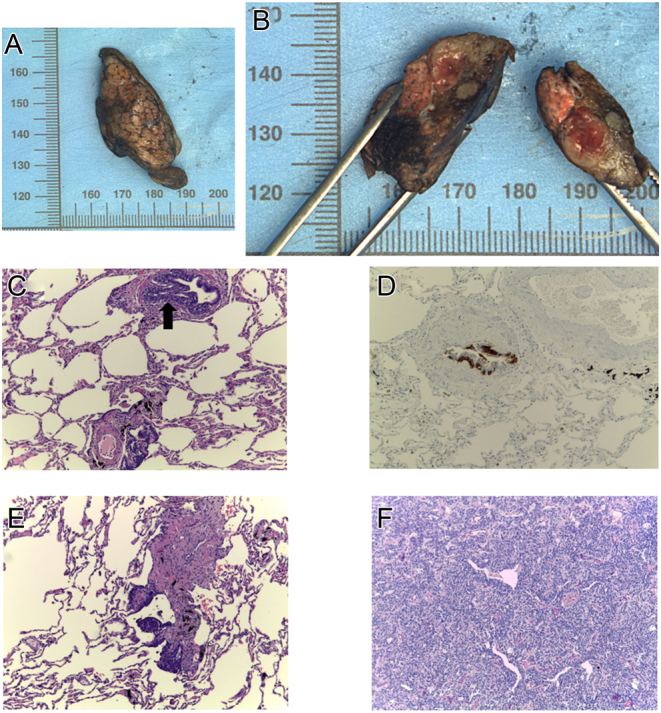
DIPNECH. (A and B) A lung, medial segment of the middle lobe: nodular lesion, compact and yellow, with 1.2 × 1.0 × 0.8 cm. (C) An intramucosal aggregate of pulmonary neuroendocrine cells elevating the epithelium in a terminal bronchiole (hematoxylin-eosin stained slide, 100×). (D) Neuroendocrine cells hyperplasia (chromogranin stained slide, 100×). (E) The proliferation of pulmonary neuroendocrine cells around a terminal bronchiole, spreading into the surrounding parenchyma and forming a tumorlet (hematoxylin-eosin stained slide, 100×). (F) A typical carcinoid: a tumor with organoid pattern and fine vascular stroma; the tumor cells are uniform, with a polygonal shape, finely granular nuclear chromatin, and moderate eosinophilic cytoplasm. Absence of mitosis or necrosis. (hematoxylin-eosin stained slide, 100×).
The genetic study for the mutations of succinate dehydrogenase (SDH)-B, D, and C subunits, MYC-associated factor X (MAX), Transmembrane protein 127 (TMEM 127), and Von Hippel-Lindau (VHL) were requested. Results were negative. PALB2 gene variant c.1408A>G, p (Thr470Ala) with unknown meaning was found when a genetic cause for her breast cancer was studied. This variation was investigated because it was identified in a niece with breast cancer at young age.
Currently, the patient is symptom and disease free over 2 years of follow-up.
Investigation
Chest CT scan described the dimensional increase of nodularities already reported in previous images and also the presence of new ones (Fig. 1).
Biopsy of a pulmonary nodule revealed a neuroendocrine tumor (NET), although definitive diagnosis, namely differential diagnosis between typical and atypical carcinoid, could only be made in the excision piece.
Ga-68 DOTANOC PET/CT that demonstrated at the level of the left superior cervical region, in the axial plane of the angle of the mandible, near the left carotid, a mass that could translate a paraganglioma (Fig. 2).
The biochemistry results are given in Table 1.
Table 1.
The biochemistry results table.
| Parameter | Value | Reference range |
|---|---|---|
| Urinary normetanephrines, μg/24 h | 412 | <800 |
| Urinary metanephrines, μg/24 h | 176 | <400 |
| Urinary 5-HIAA, mg/24 h | 3.5 | 0.7–8.2 |
| Calcium, mmol/L | 2.5 | 2.1–2.6 |
| Albumin, g/L | 44 | 35–55 |
| Ionized calcium, mmol/L | 1.32 | 1.13–1.32 |
| Phosphate, mg/dL | 3.5 | 2.7–4.5 |
| Magnesium, mmol/L | 0.8 | 0.78–1.02 |
| Parathyroid hormone, pg/mL | 39.6 | 10–65 |
| TSH, μUI/mL | 0.53 | 0.35–4.94 |
| Free T4, ng/dL | 0.86 | 0.7–1.48 |
| Thyroglobulin, ng/mL | <0.2 | 0–55 |
| Thyroglobulin antibody, UI/mL | 0.7 | <4.1 |
| Peroxidase antibody, UI/mL | 2.7 | <5.6 |
5-HIAA, 5-Hydroxyindole acetic acid.
Immunohistochemistry studies in the excision piece of the cervical paraganglioma were positive for synaptophysin, chromogranin, and S-100 protein and negative for Cytokeratin 8/18. Ki67 Proliferative Index was between 1–3% (Fig. 3).
The histological piece of the pulmonary nodeule showed DIPNECH with tumorlets and two typical NETs-pT2a (m) NxR0 (Fig. 4).
Genetic study was negative for SDH-B, D, and C subunits, MAX, TMEM 127, and VHL. It was found to be a PALB2 gene variant.
Treatment
Long-term biochemical monitoring of levothyroxine doses is performed by the endocrine clinic. Long-term follow-up in the pulmonology clinic regarding lung disease is also done with periodic chest CT-scans.
Outcome and follow-up
Currently, the patient is symptom-free over 2 years of follow-up and there is no evidence of breast and thyroid cancer recurrence.
Discussion and conclusions
The clinical presentation of paragangliomas is heterogeneous, depending on several factors like tumor localization or catecholamine secretion. Most of parasympathetic ganglia-derived paragangliomas are located in the neck and skull base. The majority of these are non-functioning and the symptoms usually manifest due to the mass effect. Conversely, sympathetic paragangliomas arise mainly in the abdomen, most of them are functional and present with catecholamine hypersecretion. Hypertension is the most common feature. Hypertensive crises are commonly manifested by episodic headache, sweating, and tachycardia/palpitations, referred to as the ‘classic triad.’ Other symptoms associated with functioning paragangliomas include palpitations, tremor, pallor, dyspnea, generalized weakness, or panic-attack-type symptoms (1).
Two different clinical presentations have been described for DIPNECH. In a subgroup of patients, the disease is accidentally diagnosed in the course of the study of another entity, usually a neoplasm. In another, patients have a clinical history characterized by cough and dyspnea that have worsened slowly over years (3). A minority may also present with hemoptysis or wheezing (2). The reason why some are symptomatic while others are not is essentially unknown (3).
In our case report, the cervical mass turned out to be an incidentaloma when we were studying the pulmonary nodules. Our patient had no symptoms suggestive of inappropriate secretion of catecholamines, which suits the location of the tumor, or complained of symptoms indicative of local mass effect. However, wheezing and dyspnea were present, which could be in relation with the pulmonary NET. Wheezing, as mentioned, does not correspond to the main clinical manifestation of this entity but may be a less common form of presentation.
Even with the most recent genetic testing, the majority of paragangliomas appear to be sporadic. In about one-third to one-half of cases, a paraganglioma is a component of an inherited syndrome (4).
Most of hereditary paragangliomas arising in the skull base and neck have been linked to mutations in the genes encoding different subunits of the succinate dehydrogenase (SDH) enzyme complex. Furthermore, propensity to pheochromocytomas and paragangliomas is an established component of several genetic syndromes, which include: Multiple Endocrine Neoplasia types 2A and 2B (MEN2), neurofibromatosis type 1 (NF1), and VHL. Most cases of hereditary paraganglioma are accounted for mutations in SDHD, SDHB, and SDHC, VHL, and NF1 (5).
Pulmonary NETs usually occur sporadically. Infrequently, they arise in association with Multiple Endocrine Neoplasia type 1 (MEN1) (6).
The appearance and almost simultaneous presence of four tumors in this patient (Table 2) promptly raised the hypothesis of a genetic etiology, especially when two of these neoplasms are rare. Only the patient’s age was not suggestive of a genetic etiology.
Table 2.
List of the patient’s tumors and age of diagnosis.
| Tumors | Age of diagnosis |
|---|---|
| Papillary thyroid cancer | 50 |
| Breast invasive ductal carcinoma | 55 |
| Cervical Paraganglioma | 57 |
| DIPNECH | 57 |
In the literature, there is no reference to the coexistence of paraganglioma and DIPNECH as part of a particular genetic syndrome. Due to the fact that DIPNECH is a relatively recent condition and there is still not much information about a genetic etiology, the initial genetic study focused on the major genetic causes of paraganglioma. The requested study (see Case presentation) was, however, negative.
MEN hypothesis was also raised. It is known that MEN1 can be associated with lung carcinoid tumors (~2%) and pheochromocytoma (~1%) (6). In addition, there is a case of coexistence of paraganglioma with MEN1 (7). However, this hypothesis seems very unlikely. Not only because the neoplasms of this patient appear in a minority of MEN1 cases but also because the pathologies that define this syndrome were absent, namely the presence of primary hyperparathyroidism, which affects almost all patients in the age group of our patient (6). The hypothesis of MEN2 was also considered but seemed very unlikely because the patient did not present any of the pathologies that define the syndrome.
PALB2 is a tumor suppressor gene. This action is accomplished by mediating DNA damage repair via homologous recombination repair of dsDNA breaks via the BRCA-associated pathway. Its role in the genetic etiology of breast cancer is now well established and it was in this context that a variant was identified in our patient. The literature also claims clear evidence of the involvement of this gene in the etiology of ovarian and pancreatic cancers (8) and as part of germline mutations among individuals with thyroid cancer (9); however, little is known about its role in other neoplasms. There is a report of the possible involvement of the PALB2 gene in the etiology of paragangliomas (10) as well as a report of a clinical case where a PALB2 mutation in metastatic pancreatic adenocarcinoma and neuroendocrine tumor was described (11). Based on this evidence, we cannot draw conclusions; however, it can be hypothesized that the PALB2 gene may be involved in the etiology of these rare neoplasms in our patient.
With this report, we intend to make known our case, in order to encourage further studies not only regarding the coexistence of these tumors but also the possible involvement of the PALB2 tumor suppressor gene in its etiology. Given that DIPNECH is recent and an increasingly recognized entity, it will not be surprising that more and more published cases of this condition will be identified.
Declaration of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of this case report.
Funding
This work did not receive any specific grant from any funding agency in the public, commercial, or not-for-profit sector.
Patient consent
Written informed consent has been obtained from the patient.
Authors contribution statement
J Pedro wrote this case report and was involved in the patient care. F Cunha, V Neto, V Hespanhol, D F Martins, S Guimarães, and A Varela were involved in the patient care and revised the draft. D Carvalho revised the draft.
References
- 1.Lenders JWM, Duh QY, Eisenhofer G, Gimenez-Roqueplo AP, Grebe SKG, Murad MH, Naruse M, Pacak K, Young WF. & Endocrine Society. Pheochromocytoma and paraganglioma: an Endocrine Society Clinical Practice Guideline. Journal of Clinical Endocrinology and Metabolism 2014. 99 1915–1942. ( 10.1210/jc.2014-1498) [DOI] [PubMed] [Google Scholar]
- 2.Ofikwu G, Mani VR, Rajabalan A, Adu A, Ahmed L, Vega D. A rare case of diffuse idiopathic pulmonary neuroendocrine cell hyperplasia. Case Reports in Surgery 2015. 2015 318175 ( 10.1155/2015/318175) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Trisolini R, Valentini I, Tinelli C, Ferrari M, Guiducci GM, Parri SNF, Dalpiaz G, Cancellieri A. DIPNECH: association between histopathology and clinical presentation. Lung 2016. 194 243–247. ( 10.1007/s00408-016-9854-7) [DOI] [PubMed] [Google Scholar]
- 4.Fishbein L, Merrill S, Fraker DL, Cohen DL, Nathanson KL. Inherited mutations in pheochromocytoma and paraganglioma: why all patients should be offered genetic testing. Annals of Surgical Oncology 2013. 20 1444–1450. ( 10.1245/s10434-013-2942-5) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Burnichon N, Brière JJ, Libé R, Vescovo L, Rivière J, Tissier F, Jouanno E, Jeunemaitre X, Bénit P, Tzagoloff A, et al. SDHA is a tumor suppressor gene causing paraganglioma. Human Molecular Genetics 2010. 19 3011–3020. ( 10.1093/hmg/ddq206) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kamilaris CDC, Stratakis CA. Multiple endocrine neoplasia type 1 (MEN1): an update and the significance of early genetic and clinical diagnosis. Frontiers in Endocrinology 2019. 10 339 ( 10.3389/fendo.2019.00339) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jamilloux Y, Favier J, Pertuit M, Delage-Corre M, Lopez S, Teissier MP, Mathonnet M, Galinat S, Barlier A, Archambeaud F. A MEN1 syndrome with a paraganglioma. European Journal of Human Genetics 2014. 22 283–285. ( 10.1038/ejhg.2013.128) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nepomuceno TC, De Gregoriis G, de Oliveira FMB, Suarez-Kurtz G, Monteiro AN, Carvalho MA. The role of PALB2 in the DNA damage response and cancer predisposition. International Journal of Molecular Sciences 2017. 18 1–20. ( 10.3390/ijms18091886) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kamihara J, LaDuca H, Dalton E, Speare V, Garber JE, Black MH. Germline mutations in cancer predisposition genes among patients with thyroid cancer. Journal of Clinical Oncology 2017. 35 (15_Supplement) 1581–1581. ( 10.1200/jco.2017.35.15_suppl.1581) [DOI] [Google Scholar]
- 10.Burnichon N, Buffet A, Gimenez-Roqueplo AP. Pheochromocytoma and paraganglioma: molecular testing and personalized medicine. Current Opinion in Oncology 2016. 28 5–10. ( 10.1097/CCO.0000000000000249) [DOI] [PubMed] [Google Scholar]
- 11.Chan D, Clarke S, Gill AJ, Chantrill L, Samra J, Li BT, Barnes T, Nahar K, Pavlakis N. Pathogenic PALB2 mutation in metastatic pancreatic adenocarcinoma and neuroendocrine tumour: a case report. Molecular and Clinical Oncology 2015. 3 817–819. ( 10.3892/mco.2015.533) [DOI] [PMC free article] [PubMed] [Google Scholar]