

RING1B co-operates with ERα to regulate estrogen-mediated gene and enhancer activities and chromatin reorganization.
Abstract
RING1B, a core Polycomb repressive complex 1 subunit, is a histone H2A ubiquitin ligase essential for development. RING1B is overexpressed in patients with luminal breast cancer (BC) and recruited to actively transcribed genes and enhancers co-occupied by the estrogen receptor α (ERα). Whether ERα-induced transcriptional programs are mediated by RING1B is not understood. We show that prolonged estrogen administration induces transcriptional output and chromatin landscape fluctuations. RING1B loss impairs full estrogen-mediated gene expression and chromatin accessibility for key BC transcription factors. These effects were mediated, in part, by RING1B enzymatic activity and nucleosome binding functions. RING1B is recruited in a cyclic manner to ERα, FOXA1, and GRHL2 cobound sites and regulates estrogen-induced enhancers and ERα recruitment. Last, ChIP exo revealed multiple binding events of these factors at single-nucleotide resolution, including RING1B occupancy approximately 10 base pairs around ERα bound sites. We propose RING1B as a key regulator of the dynamic, liganded-ERα transcriptional regulatory circuit in luminal BC.
INTRODUCTION
The steroid hormones 17β-estradiol (E2) and progesterone (P4) are the major female sex hormones (1). E2 plays essential roles during development of the mammary glands and the reproductive system and is required for brain, skin, and bone homeostasis (2). More than 70% of all human breast cancers express the estrogen receptor α (ERα), and most of these are E2 dependent for growth (3). Upon E2 stimulation, liganded ERα translocates into the nucleus and is recruited to chromatin through multiple mechanisms, including binding to a cognate DNA sequence known as estrogen response elements (EREs). ERα directly regulates genes involved in cell proliferation and cell cycle by interacting with a plethora of chromatin remodelers, epigenetic regulators, and transcription factors (TFs) (4). Despite the extensive literature on how E2 and ERα cooperate to induce expression of pro-oncogenic regulators of cell growth and survival, a greater understanding of the mechanisms underpinning this process is required to develop new therapeutic strategies for treatment of luminal [ER-positive (ER+)] breast cancer.
Although EREs are found in both promoters and enhancers, ERα predominantly binds to EREs at enhancers (5, 6). This observation suggests that liganded ERα regulates gene expression via modulation of enhancer-promoter interactions. Cancer cells have permissive chromatin accessibility and an enhancer landscape that instruct oncogenes and cell cycle genes to induce aberrant cell proliferation (7, 8). Enhancer-bound ERα is essential for the expression of E2-induced genes (9, 10). Most of our understanding of how hormones regulate gene transcription and chromatin architecture has been derived from studying the effects of acute hormone administration, typically within minutes of exposure to E2 in steroid-deprived cells (11, 12). The impact of prolonged hormone exposure on gene expression and chromatin landscape in breast cancer cells is less studied. In addition, it is still not fully understood how ERα target genes and enhancer activity are linked to the maintenance thereof and how the epigenetic landscape is modified following E2 addition, both immediately and in the long term. Addressing these central questions might provide a better understanding of how estrogen influences breast cancer initiation and progression over time.
Polycomb repressive complex 1 and 2 (PRC1 and PRC2) are essential regulators of development and are strongly implicated in cancer (13). Although PRC1/2 are mostly associated with gene repression, increasing evidence indicates that they can also be recruited to actively transcribed genes in multiple biological processes (14). We recently demonstrated that PRC1 complexes are recruited to active enhancers and promoters in several cancer types, including ER+ and triple-negative breast cancer (TNBC). Enhancers containing PRC1 are also co-occupied by ERα in ER+ breast cancer cells (15). Independent studies showed that the PRC1 subunits PCGF2 (Polycomb Group Ring Ringer 2) and CBX8 (Chromobox 8) also play important roles in breast cancer: PCGF2 positively regulates expression of ESR1 (encoding ERα), and CBX8 regulation of gene expression in breast cancer is both dependent and independent of its association with other PRC1 subunits (16, 17). How PRC1 proteins are recruited to chromatin and the molecular mechanisms that regulate their novel roles as activators of gene transcription in breast cancer remain to be determined.
Here, we show that comprehensive analyses of accessible chromatin and transcriptome landscapes identify unique chromatin stages that are dynamically assembled in response to E2. We found that RING1B is an essential epigenetic factor required for both initiation and maintenance of such chromatin stages both dependently and independently of its enzymatic activity and binding to nucleosomes. Mechanistically, RING1B is recruited to FOXA1-GRHL2-ERα–bound active enhancers and genes in response to E2 stimulation, is required for full engagement of ERα, and is required for E2-induced chromatin opening genome-wide. We further show that RING1B depletion induces an epigenetic reprogramming that results in changes in the enhancer landscape. We also demonstrate that RING1B depletion blocks cell proliferation and diminishes cell fitness. Last, we identified RING1B binding events at single-nucleotide resolution co-occupied by ERα and TFs functionally involved in estrogen signaling including FOXA1 and GRHL2. We propose RING1B as a critical factor regulating the E2-ERα–mediated epigenetic changes that are required for breast cancer cell proliferation.
RESULTS
Transcription profiles and accessible chromatin landscape in response to prolonged estrogen stimulation
Hormones induce alterations in the chromatin structure that are accompanied by massive changes in the transcriptional landscape (18). Nevertheless, most studies to date determined the immediate effects in gene transcription and chromatin accessibility after acute administration, typically minutes, of E2 and P4, among others (11, 18). Our previous studies indicated that RING1B regulated genes and enhancers bound by ERα in luminal breast cancer cells when cultured in the presence of serum that contains low levels of E2 [referred hereafter as full media (FM)] (fig. S1A) (15). To precisely determine transcription and chromatin accessibility mediated by liganded ERα, we deprived cells of E2 by culturing them in media containing charcoal-stripped serum [hereafter defined as hormone-deprived (HD) media] for 72 hours. HD induces growth arrest of ER+ cells, and proliferation can be stimulated by administration of E2 (19). For our studies, we stimulated the cells with 10 nM E2 for 4, 8, 12, and 24 hours and then examined gene expression profile changes by RNA sequencing (RNA-seq) and mapped the chromatin landscape by assay of transposase accessible chromatin sequencing (ATAC-seq). These experiments were performed using control and RING1B-depleted cells from two independent transductions and E2 inductions (fig. S1A).
We first delineated the effects of prolonged administration of E2 in regulating gene expression changes and chromatin accessibility in control cells. RNA-seq revealed that distinct gene expression patterns emerged along the E2 time course [fold change (FC) > 2, q value < 0.01]. While ~100 genes were down-regulated (group 1), ~1200 genes were dynamically up-regulated during the course of E2 (groups 2 to 5). Unexpectedly, a small number of genes was continuously up-regulated from HD to 24 hours after E2 (group 2), with most genes being transcriptionally induced at the 12- and 24-hour time points (groups 3 and 5) (fig. S1B). Notably, a large set of genes was up-regulated specifically at 12 hours and then down-regulated at 24 hours (group 3), suggesting that massive chromatin architecture changes may occur between 8 and 24 hours after E2 administration. Genes up-regulated in each of the clusters were well-known E2-responsive genes including CXCL12 and FMN1 (early response) as well as E2F1 and CCNA2 (late response) (fig. S1C) (20). Gene set enrichment analysis (GSEA) confirmed successful E2 stimulation, since the induced genes were enriched for the early and late E2 response pathways and were also cell cycle and E2F targets (fig. S1D). These results indicated that E2 induced gene expression changes in a time-dependent manner that requires exquisite orchestration and coordination of dynamic changes in gene transcription to induce proliferation of luminal breast cancer cells.
Chromatin is extensively remodeled upon hormone administration (18, 21, 22). We next sought to determine chromatin accessibility changes after estrogen induction and how these changes correlated with gene expression. Distribution of genome-wide ATAC-seq peaks indicated that chromatin was most dynamic at promoters and intergenic regions in response to E2 (fig. S1E). In agreement with the massive changes in gene transcription observed between 8 and 24 hours upon E2 administration (fig. S1B, clusters 3 to 5), TF binding sites of key breast cancer TFs such as FOXA1/2, JUNB, SP1, and GRHL2 (23), as well as the chromatin organization factor CTCF (24), became increasingly accessible after 4 and 8 hours (fig. S1F), suggesting that chromatin accessibility changes primarily occur during the first 8 hours of E2 induction. By clustering genome-wide ATAC-seq peaks (fig. S1G), we confirmed that the most changes in accessibility occur at 8 hours. To determine whether gene expression correlated with chromatin accessibility, we interrogated ATAC-seq peaks ±2.5 kb from genome-wide transcription start sites (TSS). The greatest changes in chromatin accessibility around TSS were observed after 8 hours of E2 induction (fig. S1H), suggesting that changes in chromatin landscape occur before differences in gene transcription observed after 12 and 24 hours of E2 administration. However, we did not observe complete correlation between transcription of E2-induced genes and chromatin accessibility changes. For instance, CXCL12 and FMN1 genes, which belong to group 2 in the RNA-seq classification (fig. S1B), exhibited diverse ATAC-seq profiles (fig. S1I). The TSS of CCND1, a gene that was strongly induced at 12 hours but decreased at 24 hours, demonstrated significant accessibility at 8 hours, while GREB1, which was induced at both 12 and 24 hours, was most accessible at 24 hours. Overall, these results indicate that (i) E2 dynamically modulates genetic programs, (ii) massive chromatin accessibility changes occur during the first 8 hours of E2 exposure, and (iii) global correlation of chromatin opening with gene up-regulation was modest, possibly because of secondary effects of the estrogen response.
RING1B is required for de novo estrogen-induced gene expression and chromatin accessibility
We recently noted that RING1B may be functionally involved in E2-mediated gene regulation (15). Whether RING1B is required for E2-induced gene expression changes and chromatin accessibility is not known. Analysis of all and unique differentially expressed genes induced by E2 (FC > 2, q value < 0.05) in control cells compared to RING1B-depleted cells (fig. S2A) revealed that E2-mediated gene regulation strongly depends on RING1B (Fig. 1, A and B). RING1B depletion predominantly down-regulated early and late E2-responsive genes, epithelial-to-mesenchymal transition, G2M checkpoints, as well as E2F and MYC targets (Fig. 1C). These results were further confirmed by reverse transcription quantitative polymerase chain reaction (RT-qPCR), by both stable short hairpin RNA (shRNA) and acute (small interfering RNA) RING1B depletion, and also in MCF7 cells, another ER+ breast cancer cell line (fig. S2, B to D). Interferon-α and interferon-γ response were the only pathways up-regulated after RING1B depletion (Fig. 1C). However, interferon genes were not occupied by RING1B or ERα, suggesting that RING1B does not directly regulate the interferon pathway.
Fig. 1. RING1B is required for estrogen-induced gene expression and chromatin accessibility.

(A) RNA-seq heat maps of all deregulated genes in control and RING1B-depleted T47D cells. Fold change > 2, q value < 0.05. N = 2. (B) Genome browser screenshots of RNA-seq tracks at TFF1 and GREB1 loci in control and RING1B KD cells. (C) GSEAs of RING1B-depleted cells compared to control cells. NES, normalized enrichment score. (D) Western blot analysis after replacement of RING1B with shRNA-resistant and HA-tagged RING1B mutants. VINCULIN was used as a loading control. RT-qPCR analysis of endogenous RING1B normalized to the housekeeping gene RPO in shCTR and shRING1B cells expressing HA-RING1BR98A or HA-RING1BI53A. N = 2. (E) Volcano plots (adjusted P value) of deregulated genes in T47D-shCTR (RING1BWT) and cells expressing RING1B mutants after 24 hours of E2. (F) Venn diagram of up-regulated genes after 24 hours of E2 in the three cell lines from (E). (G) Western blot of ERα, RING1B, and HA, from shCTR and shRING1B cells before and after HA-RING1BWT expression. VINCULIN was used as a loading control. Volcano plots (adjusted P value) of deregulated genes in the RING1B rescue cells after 24 hours of E2. (H) GSEA of RING1B rescue cells 24 hours after 24 hours of E2. (I) Binary ATAC-seq heat map in control and RING1B-depleted cells during E2 administration. (J) Genome browser screenshots of ATAC-seq peaks at the TFF1 locus in control and RING1B KD cells. (K) ATAC-seq signals in control and RING1B KD cells in HD condition and the E2 time course. (L) Genome browser screenshots of ATAC-seq peaks at the GREB1 locus in control and RING1B KD cells.
RING1B is an E3 ligase that can also bind to the histone H2A/H2B dimer. These functions are dictated by specific amino acids on the RING1B protein. Specifically, isoleucine at position 53 (I53) interacts with the E2-ligase, UBCH5C, to ubiquitinate its substrate (25), and mutation to alanine (I53A) disrupts RING1B E3 ligase activity. Similarly, arginine 98 (R98) inserts into an acidic pocket of H2A residues, and mutation of R98 to alanine (R98A) results in a 50-fold decrease in RING1B interaction with the nucleosome concomitant with reduced H2A ubiquitination (26). Thus, we wondered whether these functions are required for the E2-mediated transcriptional response. We generated T47D cells expressing hemagglutinin (HA)–tagged versions of RING1B with an alanine at position 53 in place of isoleucine (RING1BI53A) or an alanine at position 98 in place of arginine (RING1BR98A). Cells were transduced with lentiviruses expressing HA-RING1BI53A or HA-RING1BR98A (resistant to shRNA against RING1B) containing an internal ribosomal entry site mCherry. Fluorescence-activated cell sorting (FACS)–sorted mCherry+ cells were then transduced with shRNA-RING1B lentivirus to deplete endogenous RING1B (Fig. 1D). In agreement with our previous results, neither RING1B depletion nor the expression of both RING1B mutants affected global H2AK119ub1 levels (fig. S2E). Cellular fractionation assays showed that HA-RING1BR98A was displaced from the insoluble chromatin. Similarly to HA-RING1BWT, HA-RING1BI53A remained at both soluble and insoluble chromatin fractions (fig. S2F). These results confirm that the R98 residue of RING1B is required for strong association of RING1B to chromatin. T47D cells expressing endogenous and wild-type RING1B (RING1BWT), HA-RING1BI53A, or HA-RING1BR98A were cultured in HD media for 72 hours (Fig. 1, A and B, and fig. S1, A and B), and E2 was administered for 24 hours. Cells expressing the RING1B mutants only partially responded to E2 compared to WT, as demonstrated by global gene expression changes (Fig. 1E). Specifically, RING1BR98A mutation significantly down-regulated the E2-mediated transcriptional response compared to RING1BI53A cells (Fig. 1E), suggesting that RING1B nucleosomal binding is more functionally important than its enzymatic activity in mediating the estrogen response. Only ~20% of the genes up-regulated in RING1BWT (171 of 835) were also up-regulated in the mutant cells (Fig. 1F), indicating that some E2-induced genes do not require RING1B enzymatic activity (360 of 427) nor binding to nucleosomes (192 of 223). About 50% of genes up-regulated in RING1BWT (454 of 835) were not induced in the RING1B mutants, confirming that RING1B enzymatic activity and interaction with histone H2A/H2B dimers were required for their transcriptional activation. RNA-seq experiments performed in RING1B rescue cells (shRING1B + HA-RING1BWT) before and after 24 hours of E2 administration showed a similar gene expression profile compared to shCTR cells in the presence of 24 hours of E2 (Fig. 1G and fig. S1D). GSEA revealed a full functional rescue when HA-RING1BWT was ectopically expressed in shRING1B cells (compare Fig. 1H and fig. S1D).
E2 induced dynamic changes in chromatin accessibility and gene transcription (fig. S1). Since RING1B depletion hampered expression of E2-responsive genes, we expected to detect reduced accessibility at these regulatory sites. Thousands of de novo sites that demonstrated increased accessibility upon E2 administration were dependent on RING1B (Fig. 1, I to L). These effects of RING1B loss were not due to defects in cell cycle or proliferation (fig. S2, G and H), further suggesting that RING1B is required for the initiation and maintenance of gene transcription induced by E2 in luminal breast cancer cells.
RING1B loss impairs E2-induced de novo enhancers
Chromatin is heavily remodeled during E2 administration (fig. S1); therefore, we sought to determine the enhancer landscape generated upon E2 induction. We first interrogated whether E2 administration following RING1B depletion affected global levels of histone modifications associated with Polycomb-mediated repression (H2AK119ub1 and H3K27me3), gene activation (H3K4me3), and active enhancers (H3K4me1 and H3K27ac). In agreement with our previous report, we did not detect changes in global H2AK119ub1 after RING1B depletion (15), irrespective of E2 induction (Fig. 2A and fig. S2). Although H3K27me3 levels remained constant, accumulation of global H3K27ac following E2 stimulation was hindered in RING1B-depleted cells (Fig. 2A). However, RING1B depletion did not affect either the expression or the protein level of EP300, the main histone acetyltransferase that deposits H3K27ac (fig. S3, A and B) (27). The general genome-wide distribution of H3K27ac was mostly unchanged upon 24 hours of E2 in control and RING1B-depleted cells (Fig. 2B), suggesting a potential role of RING1B in regulating a specific set of enhancers that are mediated by E2.
Fig. 2. Enhancer dynamics mediated by RING1B.
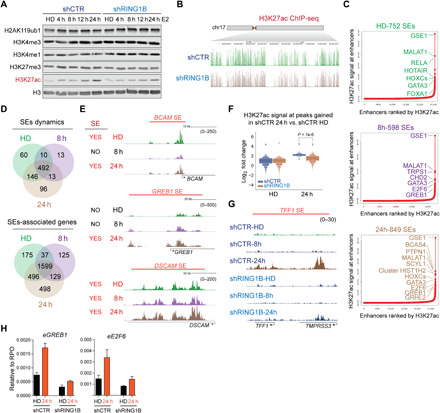
(A) Western blots of histone modifications in control and RING1B-depleted cells in HD condition and upon E2 administration. Histones were extracted using sulfuric acid. (B) H3K27ac ChIP-seq signal across the right arm of the chromosome 17 in control and RING1B-depleted cells. (C) SEs identified in each E2 time point. (D) Venn diagram of SEs and genes associated with SEs in each E2 time point. (E) Genome browser screenshots of H3K27ac ChIP-seq in SEs identified in the HD and 24 hours of E2 condition (BCAM SE), only after 24 hours of E2 (GREB1 SE), and at all the time points analyzed (DSCAM SE). (F) H3K27ac signal in control and RING1B KD cells at sites that acquired H3K27ac after 24 hours of E2 in control cells. Significance was determined by Mann-Whitney U test. (G) Genome browser screenshots of H3K27ac in control and RING1B KD cells before and after 8 and 24 hours of E2. (H) RT-qPCR analyses of enhancer RNA expression at the GREB1 and E2F6 SEs in control and RING1B-depleted cells in the HD condition and after 24 hours of E2. mRNA expression was normalized to the housekeeping gene RPO. N = 3.
Because super-enhancers (SEs) regulate oncogenic pathways in cancer (28), we focused our attention to SE dynamics (gain and loss) in response to E2. Potential target genes of the 752 SEs identified in hormone-deprived cells included key breast cancer TFs such as FOXA1 and GATA3 (Fig. 2C). In estrogen-treated cells, 598 and 859 SEs were identified with potential target genes including GREB1 and E2F6 (Fig. 2C). There was a relatively high overlap in the three experimental conditions between SEs and SE target genes (Fig. 2, D and E), suggesting that a unique subset of SEs are dynamically regulated upon E2 administration. Genome-wide H3K27ac at de novo enhancers gained in response to E2 was significantly reduced in RING1B-depleted cells compared to control (Fig. 2, F and G) concomitant with a reduction of enhancer RNA levels (Fig. 2H). RING1B depletion did not reduce H3K27ac at sites that were already decorated with high levels of H3K27ac before E2 stimulation (fig. S3, C and D), indicating that RING1B functions primarily at sites of de novo enhancers following E2 induction.
RING1B is recruited to enhancers and promoters in response to E2 induction
How the Polycomb group of proteins are recruited to chromatin is under constant examination (14, 29). Our previous studies identified co-occupancy of RING1B and ERα at enhancers and promoters containing EREs and FOXA1 motifs in luminal breast cancer cells (15). We thus hypothesized that E2 administration may regulate deposition of RING1B at chromatin. Since ERα is recruited to chromatin within minutes of E2 addition (9), we mapped genome-wide ERα, FOXA1, and RING1B binding following 45 min of E2 stimulation as well as at 8 and 24 hours to investigate potential binding dynamics over prolonged E2 exposure. To minimize potential secondary effects on gene transcription due to stable RING1B depletion, we generated new T47D cells with doxycycline-inducible RING1B knockdown (fig. S4, A and B). RING1B depletion was initiated 48 hours before hormone deprivation for 72 hours and E2 stimulation.
Notably, RING1B binding at chromatin was dependent on estrogen (Fig. 3A). As expected, E2 treatment led to massive ERα localization to chromatin (6, 30–32), while recruitment of FOXA1 was mostly independent of E2 (33) (Fig. 3A). Upon E2 induction, RING1B was recruited to a large majority of ERα/FOXA1 cotargets (Fig. 3, B and C). We also observed RING1B occupancy at genomic sites not cobound by ERα/FOXA1 that lose RING1B binding after 45 min of E2 induction (Fig. 3, B to D), indicating a redistribution of RING1B during the early estrogen response. Specifically, we found 455 peaks corresponding to 245 genes occupied by RING1B in the absence of E2, while 4212 peaks corresponding to 2092 genes were found to be RING1B targets after 45 min of E2 induction. In agreement with previous reports, the number of ERα binding sites increased ~5-fold after 45 min of E2, while the number of FOXA1 binding sites modestly increased (Fig. 3E).
Fig. 3. RING1B recruitment to chromatin and role in ERα And FOXA1 recruitment upon 45 min of E2 administration.
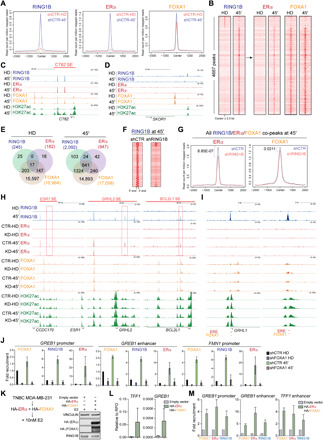
(A) RING1B, ERα, and FOXA1 ChIP-seq signals in control cells before (HD) and after 45′ of E2. Number of RING1B peaks in HD = 455, after 45′ of E2 = 4212. ERα peaks in HD = 328, after 45′ of E2 = 2015. FOXA1 peaks in HD = 102,304, after 45′ of E2 = 140,846. (B) ChIP-seq heat maps of RING1B, ERα and FOXA1 ChIP-seq signal before and after 45′ of E2. Heat maps are clustered by RING1B occupancy. (C and D) Genome browser screenshots of RING1B, ERα, FOXA1, and H3K27ac in control cells at CT62 (C) and SKOR1 (D) before and after 45′ of E2. (E) Venn diagrams of target genes before (HD) and after 45′ of E2. (F) ChIP-seq heat maps of RING1B in control and RING1B-depleted cells. (G) RING1B, ERα, and FOXA1 ChIP-seq signals in control cells before (HD) and after 45′ of E2. Significance was determined by Mann-Whitney U test. (H and I) Genome browser screenshots of ChIP-seqs at ESR1, BCL2L1, and BCL2L1 SEs (H) and GRHL1 (I). (J) FOXA1, RING1B, and ERα ChIP-qPCR of RING1B, ERα, and FOXA1 cobound sites in control and FOXA1-depleted cells. N = 2. (K) Western blots of ERα, FOXA1, and RING1B in MDA-MB-231 cells expressing HA-ERα and HA-FOXA1 and after E2 administration. VINCULIN was used as a loading control. (L) RT-qPCR analyses of TFF1 and GREB1 in MDA-MB-231 as in (K). (M) ChIP-qPCR of ERα, RING1B, and FOXA1 cotargets in T47D. Results are presented as fold recruitment over cells not transfected with HA-ERα and HA-FOXA1. N = 2.
We then asked whether RING1B depletion regulated recruitment of ERα and FOXA1. To this end, we first confirmed that knockdown of RING1B diminished chromatin-bound RING1B genome-wide (Fig. 3F). Analysis of RING1B, ERα, and FOXA1 co-occupied sites after 45 min of E2 administration revealed that loss of RING1B did not alter FOXA1 binding nor H3K27ac deposition but significantly reduced ERα recruitment (Fig. 3G and fig. S4, C and D). We then divided the RING1B chromatin immunoprecipitation sequencing (ChIP-seq) signal in quartiles to determine whether ERα and FOXA1 recruitment depended on RING1B binding levels. ERα occupancy levels strongly correlated with that of RING1B and were significantly reduced following RING1B depletion in the first two quartiles, whereas FOXA1 occupancy remained relatively unchanged in all four quartiles (fig. S4E). Significant reduction of ERα binding was observed at the promoters and enhancers of genes with key oncogenic functions in breast cancer including ESR1, GRHL2, and BCL2L1 (Fig. 3H). We also found that genes co-occupied by RING1B and FOXA1, but not ERα, contained EREs (P value of 1 × 10−100) (Fig. 3I), suggesting that RING1B can be recruited to ERα binding motifs in the absence of ERα. Last, we performed ChIP-seq of the RING1B mutants to determine whether their chromatin occupancy was altered. In agreement with the lack of response to E2 of both cell lines expressing HA-RING1B mutants in a stable shRING1B background (Fig. 1, G and H), neither RING1BI53A nor RING1BR98A was stably associated with chromatin (fig. S4F). These results suggest that interaction of RING1B with the nucleosomes and its enzymatic activity to nonhistone substrates are required for its stabilization to chromatin both in the absence and presence of E2.
The pioneer factor FOXA1 is a key determinant of ERα recruitment and function. Since E2 administration also induces massive RING1B recruitment to chromatin, we next asked whether FOXA1 served as a pioneer factor for RING1B. FOXA1 depletion by shRNA (fig. S4G) strongly impaired recruitment of both ERα and RING1B (Fig. 3J), indicating that FOXA1 binding is also required for E2-mediated RING1B recruitment to chromatin. Moreover, ectopic expression of HA-tagged ERα and FOXA1 in the TNBC cell line MDA-MB-231, in which FOXA1 is repressed by RING1B (15), was sufficient to induce expression of GREB1 and TFF1 concomitant with recruitment of RING1B, ERα, and FOXA1 to their promoters and enhancers (Fig. 3, K to M). These results confirm that expression of ERα and FOXA1 in TNBC cells can induce expression of estrogen-responsive genes following E2 induction with concomitant RING1B recruitment at the regulatory sites of these genes. These observations highlight the cooperative interplay between RING1B and ERα/FOXA1 in regulating E2-induced genes.
E2-mediated RING1B and ERα recruitment to chromatin is dynamic
ERα binding to chromatin during the early estrogen response is cyclic (30); thus, we next sought to determine whether RING1B is recruited to chromatin in a similar manner and whether dynamic chromatin cycling occurs during prolonged E2 stimulation. While FOXA1 binding profiles remained relatively similar along the time course of E2 induction (fig. S5A), RING1B and ERα demonstrated dynamic chromatin occupancy following hours of E2 administration (Fig. 4, A and B). We identified six clusters of distinct RING1B binding profiles containing EREs and GRHL2 binding motifs (Fig. 4A) concomitant with significant up-regulation of the associated RING1B target genes after 8 hours of E2 administration (fig. S5B). Notably, cluster 6, which exhibited stable RING1B occupancy at all time points following E2 exposure, also contained genes that were stably up-regulated in response to E2 (Fig. 4A and fig. S5B). We then interrogated the RING1B occupancy at genes that were transcriptionally active in HD and became repressed during E2 administration (Fig. 1A). Only 12% of these genes (23 of 184) were occupied by RING1B. This result indicated that RING1B was not playing a major role as a transcriptional repressor in T47D cells and suggested that the canonical repressive function of PRC1 is mediated by RING1A.
Fig. 4. Dynamics of RING1B and ERα during E2 administration.
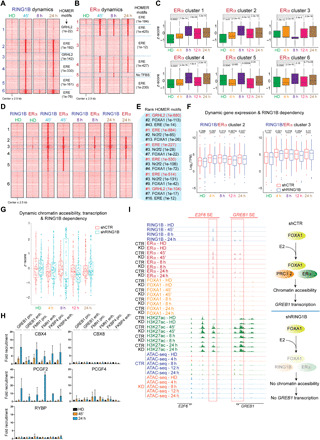
(A and B) ChIP-seq heat maps of RING1B (A) and ERα (B) signals before (HD) and after 45′, 8 hours, and 24 hours of E2. Six ChIP-seq clusters were identified from 7053 peaks (A) and 5100 peaks (B). The top-enriched motif in each cluster is shown. (C) E2-induced expression changes of genes associated with peaks within each ERα ChIP-seq cluster. Box plots are represented by z score. (D) Heat map clustering analysis of RING1B and ERα ChIP-seq before (HD) and after 45′, 8 hours, and 24 hours of E2. Number of peaks: cluster 1 = 2196, cluster 2 = 1397, cluster 3 = 540, cluster 4 = 1721, cluster 5 = 1960, cluster 6 = 4339. (E) TF motif analysis of the six clusters. (F) Box plots of RNA-seq signal in control and RING1B KD cells at the different E2 time points. TPM, transcripts per million. (G) ATAC-seq peak intensity and dynamics upon E2 at RING1B and ERα cotargets in control and RING1B-depleted cells. (H) PRC1 subunits ChIP-qPCR of RING1B/ERα cotarget genes before (HD) and after 45′ and 24 hours of E2. N = 3. (I) Genome browser screenshots of RING1B, ERα, FOXA1, and H3K27ac ChIP-seq and ATAC-seq signals at E2F6 and GREB1 SEs in control and RING1B-depleted cells during E2 administration.
ERα followed a similar recruitment pattern as RING1B (ERα clusters 1, 2, 4, and 5) but demonstrated more dynamic occupancy profiles at 45 min (cluster 6), 8 hours (cluster 5), and 24 hours (cluster 3). As expected, EREs were strongly enriched at ERα target genes (Fig. 4B). Genes stably occupied by ERα along the E2 time course, similar to cluster 6 of RING1B occupancy, were significantly up-regulated (Fig. 4C, clusters 1 and 2). Genes with ERα bound only at 24 hours (cluster 3) exhibited small changes, albeit significant, in gene expression, whereas genes occupied by ERα only at 45 min and 24 hours (clusters 4 and 6) demonstrated significant up-regulation at these time points. The small set of genes containing ERα only at 8 hours (cluster 5) appeared to be repressed when compared to the HD condition, suggesting that ERα also facilitates gene repression (Fig. 4C) (4, 34).
We then wondered whether RING1B and ERα bound the same genomic targets during the E2 response, so we grouped RING1B and ERα binding patterns into six clusters (Fig. 4D). Four of these clusters (clusters 2, 3, 4, and 5) contained genomic sites targeted by both RING1B and ERα at some point during the E2 time course and contained EREs as the number one TF binding motif (Fig. 4E). Contrastingly, clusters 1 and 6, which have little to no ERα binding, primarily contained GRHL2 and FOXA1 binding motifs with substantially less enrichment for EREs (Fig. 4E).
We next determined the impact of RING1B loss on the expression of genes occupied by RING1B and ERα containing the highest ERE and FOXA1 binding motifs. We first identified RING1B and ERα cotargets up-regulated in shCTR cells after addition of E2 (clusters 2 to 5) and determined their expression following RING1B depletion. We found that RING1B directly regulated genes in clusters 1, 2, and 3 (Fig. 4F and fig. S5C), but RING1B depletion was not sufficient to significantly affect expression of genes in clusters 4 and 5 (fig. S5C). These results indicate that in a set of genes co-occupied by RING1B, ERα, and FOXA1 (not shown), RING1B is required for their full transcriptional activation upon E2 administration. Among the down-regulated genes in clusters 2 and 3 were key genes involved in breast cancer progression (e.g., GREB1, FMN1, TFF1, and FKBP4) (Fig. 1, A to D) (35–37).
We then assessed whether RING1B and ERα induces the expression of their direct targets in response to E2 by increasing chromatin accessibility at these sites. To this end, we restricted our analysis of ATAC-seq peaks to those located at the promoters and TSS of genes up-regulated at any time point during E2 stimulation that are RING1B and ERα cotargets. We found that overall chromatin accessibility at E2-stimulated genes in control cells is dynamic with cyclical opening and closing during the E2 response from 0 to 24 hours (Fig. 4G), similar to the cycling of ERα on and off the chromatin (30). In contrast, RING1B depletion abrogates this cyclical trend of chromatin accessibility, with a significant reduction in accessibility compared to shCTR at 8 and 12 hours after E2 addition. A cluster of genes gained significant accessibility at 8 hours in RING1B-depleted cells (Fig. 4G). Nonetheless, these results suggest that correlation between chromatin accessibility and gene expression during the estrogen response is gene specific and time dependent.
Once we established the recruitment pattern of RING1B and ERα and the impact of RING1B loss on gene transcription and chromatin accessibly, we next asked whether RING1B was recruited to chromatin in a PRC1 context. We recently showed that in T47D cells cultured in FM containing constant E2, RING1B only associated with cPRC1 subunits. Whether RING1B was recruited to chromatin as part of a PRC1 complex upon acute E2 administration was not known. We thus performed ChIP-qPCR experiments using antibodies against cPRC1 (CBX4 and CBX8) and ncPRC1 (RYBP) subunits and PCGF2 and PCGF4 orthologs that can be part of both cPRC1 and ncPRC1 complexes. PCGF2 and CBX4 were recruited to both promoters and enhancers co-occupied by RING1B, ERα, and FOXA1 after 24 hours of E2 administration, but not after 45 min of E2. RYBP was not recruited to any of the regulatory sites analyzed, indicating that RING1B only associated with a cPRC1 complex containing CBX4 and PCGF2 24 hours after E2 administration (Fig. 4H). We then sought to determine the role of RING1B in the recruitment of ERα and FOXA1 during prolonged E2 administration. We found that after 24 hours of E2 induction, RING1B recruitment was reduced by ~70% in RING1B-depleted cells, and ERα recruitment was significantly reduced at the GREB1 SE, concomitant with drastic reduction of ATAC-seq signal and gene expression (Figs. 1B, and 4I and fig. S5D). These results suggest that RING1B may have a strong impact on late ERα binding events or stable recruitment during extended exposure to estrogen.
Molecular and biological RING1B dependencies after prolonged E2 administration
We then interrogated the genome-wide RING1B dependency on ERα and FOXA1 recruitment to chromatin at 8 and 24 hours after E2 induction. Since GRHL2 binding sites were strongly enriched in two of the RING1B binding clusters (Fig. 4A), we also asked whether GRHL2 recruitment was dependent on RING1B (Fig. 5A). GRHL2 was recently demonstrated to be bound to FOXA1-occupied enhancers in ER+ cancer cells (38). Similar to FOXA1 binding profiles, GRHL2 was observed to be already bound to chromatin in the absence of E2, and RING1B depletion did not affect its occupancy at randomly selected RING1B-FOXA1-GRHL2 cotarget genes (Fig. 5B).
Fig. 5. RING1B regulates ERα recruitment, cell fitness, and cell proliferation.
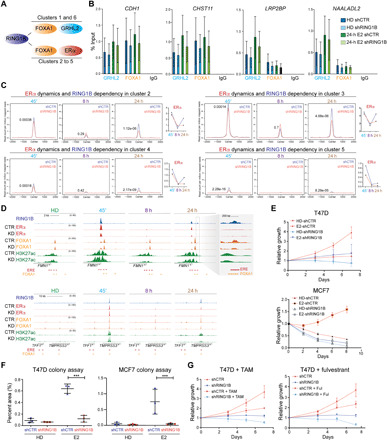
(A) RING1B is recruited to clusters containing either FOXA1 and GRHL2 or FOXA1 and ER. (B) FOXA1 and GRHL2 ChIP-qPCR of FOXA1/GRHL2 cobound genes in control and RING1B-depleted cells. Immunoglobulin G (IgG) was used as a negative control. N = 2. (C) Dynamics of ERα ChIP-seq signals during E2 and effect in ERα recruitment upon RING1B depletion at clusters 2 to 5 identified in Fig. 4D. Significance was determined by Mann-Whitney U test. (D) Genome browser screenshots of RING1B, ERα, FOXA1, and H3K27ac ChIP-seq at FMN1 and TFF1 loci in HD, 45′, 8 hours, and 24 hours of E2 in control and RING1B-depleted cells. The gray box is a zoomed-in view of a genomic region upstream of the TSS of FMN1 containing an ERE and a FOXA1 binding site. (E) Growth curve of T47D and MCF7 control and RING1B KD cells cultured in HD media or HD supplemented with E2. N = 3. ***P < 0.001, two-tailed t-test. (F) Colony formation assay of T47D and MCF7 control and RING1B-depleted cells cultured in HD media or HD supplemented with E2 for 14 days (T47D) and 21 days (MCF7). N = 3. (G) Growth curve of T47D control and RING1B KD cells treated with tamoxifen (TAM; 100 ng/ml) and fulvestrant (30 ng/ml) for 7 days. N = 3.
We next determined whether RING1B modulated FOXA1 and ERα recruitment during prolonged exposure to E2 at RING1B-FOXA1-ERα cotargets. While FOXA1 recruitment to chromatin was modestly affected by the loss of RING1B (fig. S6A), ERα recruitment was diminished by ~50% at 24 hours in clusters 2 to 4 (Fig. 5C). Moreover, clusters 2 and 3 exhibited the strongest ERα recruitment (Fig. 5C) and contained genes that were significantly deregulated upon RING1B depletion (Fig. 4F). ERα recruitment was severely affected at key genes such as FMN1 and TFF1 upon RING1B depletion (Fig. 5D). The requirement of RING1B in ERα recruitment was time dependent: At FMN1, RING1B was required for full ERα recruitment after 45 min and 24 hours of E2 treatment, whereas at the TFF1 enhancer, ERα recruitment at 45 min was not affected by the loss of RING1B but was strongly affected at 24 hours. RING1B was also recruited to EREs not occupied by ERα and with low FOXA1 occupancy upstream of the FMN1 promoter 24 hours after E2 induction (Fig. 5D, right). Reduced ERα binding (Figs. 3 and 5), enhancer regulation (Fig. 2), chromatin accessibility, and lack of full response to E2 (Fig. 1) in RING1B-depleted cells resulted in a reduced proliferation over time (Fig. 5E) and decreased cellular fitness (Fig. 5F). Notably, TCGA data from 1082 patients with breast invasive carcinoma showed a positive correlation of RNF2 (encoding RING1B) expression with ESR1 (encoding ERα), ERα cofactors such as GATA3, FOXA1, EP300, and KMT2C, and RING1B/ERα cotarget genes (e.g., TFF1 and FMN1) (fig. S7, A to C). Moreover, RING1B depletion enhanced the negative effect in cell proliferation mediated by tamoxifen and fulvestrant, selective ER modulator and downregulator, respectively, further supporting a cooperative role of RING1B in regulating the ER pathway. In FOXA1-depleted cells where RING1B exhibits reduced chromatin binding (Fig. 3G), we also observed a strong impairment in cell fitness, corroborating the importance of RING1B and ERα in maintaining the cellular identity of luminal breast cancer cells (fig. S6B).
Single-nucleotide resolution profiles of RING1B, ERα, FOXA1, and GRHL2
Standard ChIP experiments performed with cross-linking agents do not discriminate between direct and indirect binding of proteins to DNA. While TFs are typically recruited to chromatin through the recognition of cognate DNA motifs, they can also occupy sites devoid of these motifs via interaction with other factors. Moreover, most epigenetic machineries do not directly bind DNA and are recruited to specific genomic locations via interaction with TFs, RNA molecules, or histone modifications. Although we did not observe interaction of RING1B with ERα or FOXA1, our ChIP-seq experiments revealed a high degree of chromatin colocalization of RING1B with ERα, FOXA1, and GRHL2 in the presence of E2 (Figs. 3 to 5). To determine how these factors associate and network functionally at chromatin during E2 stimulation and to detect all their possible protein-protein-DNA and protein-DNA interaction events, we performed ChIP-exo experiments (39) and applied the ChExMix pipeline (40). ChIP-exo greatly improves the resolution of binding sites from hundreds of base pairs to a single-nucleotide resolution by including a 5′-3′ exonuclease that degrades the DNA protruding from the occupied binding site (41). We performed RING1B, ERα, FOXA1, and GRHL2 ChIP-exo (to the best of our knowledge, RING1B and GRHL2 ChIP-exo are not yet reported) in two biological replicates (reads merged for downstream analysis). In agreement with our ChIP-seq experiments (Fig. 3), ChIP-exo tags for ERα and RING1B were strongly enriched at chromatin upon E2 induction, while FOXA1 enrichment was similar between HD and E2 conditions (Fig. 6, A and B). Also, GRHL2 binding to chromatin was not dependent on E2 stimulation (Fig. 5B and Fig. 6, A to B). Genome browser screenshots of RING1B, FOXA1, and ERα ChIP-seq and ChIP-exo assays showed a comparable enrichment following E2 administration (Fig. 6C).
Fig. 6. Single-nucleotide binding resolution of ERα, RING1B, FOXA1, and GRHL2 identified by ChIP-exo.
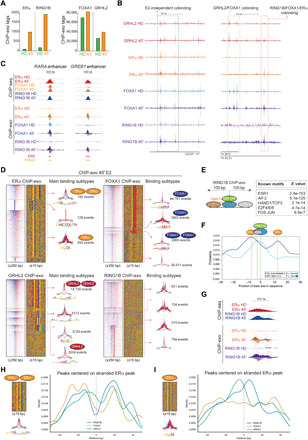
(A) Number of ERα, RING1B, FOXA1, and GRHL2 ChIP-exo tags identified in HD and after 45′ of E2. (B) Genome browser screenshots of GRHL2, RING1B, ERα, and FOXA1 ChIP-exo at CDC27 and MYC loci in HD and after 45′ of E2. (C) Genome browser screenshots of RING1B, ERα, and FOXA1 ChIP-seq and ChIP-exo at RARA and GREB1 enhancers in HD and after 45′ of E2. (D) Heat maps and sequence color plots of binding subtypes identified in ERα, FOXA1, GRHL2, and RING1B ChIP-exo after 45′ of E2. On the right of each sequence color plot, see the distribution of the ChIP-exo tag patterns at each of the main subclass binding events identified in ERα, RING1B, FOXA1, and GRHL2 ChIP-exo experiments and the number of events. (E) MEME analysis of known TF motifs identified within 100 bp from the summit of the RING1B tags. (F) Distribution of ESR1 (ERα) and JUN cognate sequences respective to the submit of the RING1B tags. (G) Genome browser screenshots of RING1B and ERα ChIP-seq and ChIP-exo in HD and after 45′ of E2. Boxes represent distance between the submit of ChIP-exo tags. (H) Distribution of FOXA1, GRHL2, and RING1B ChIP-exo tags relative to stranded ERα tags containing a full ERE motif [ERα subtype 1 in (D)]. (I) Distribution of FOXA1, GRHL2, and RING1B ChIP-exo tags relative to stranded ERα tags containing half ERE motif [ERα subtype 3 in (D)].
We then identified binding event subtypes for each of the four factors upon E2 administration. ERα ChIP-exo tags (~80%) contained EREs, of which 182 and 592 harbored full EREs and half EREs, respectively, suggesting that ERα was recruited to chromatin as a homodimer in ~30% of the binding events (Fig. 6D, upper left). We found 128 events in which ERE was not detected, suggesting that ERα may potentially bind a novel motif. In addition, most of the FOXA1 and GRHL2 were bound at their cognate sequences (both single and double motifs), albeit with subtle differences between the subtypes (Fig. 6D, upper right and bottom left). However, we were unable to determine the existence of a RING1B cognate DNA binding motif, although we identified four RING1B binding types based on the shape of the tags, suggesting that RING1B is not recruited to a specific DNA sequence but rather is recruited by RNA or by multiple TFs, or both (Fig. 6D, bottom right). Next, we determined the binding motifs located within 100 base pairs (bp) upstream and downstream of the RING1B ChIP-exo tags to identify potential TF corecruitment with RING1B. In agreement with our model, we found significant enrichment of ERE (ESR1) motifs, indicative of a potential ERα binding as well as motifs of known ERα cofactors such FOS/JUN, E2F, and AP-2 families (Fig. 6E). Notably, the JUN binding motif was only 3 bp from the RING1B binding sites, and ERE (ESR1) motifs were found approximately 10 bp around RING1B (Fig. 6F). We confirmed the binding prediction of ERα ~10 bp next to RING1B (Fig. 6G), which was not possible to achieve with standard-resolution ChIP-seq. Last, we determined the tag distribution of RING1B, FOXA1, and GRHL2 relative to the main ERα binding subtypes, as a homodimer or a monomer. Notably, the distribution of RING1B, FOXA1, and GRHL2 binding is influenced by the ERα binding pattern. All three factors can be recruited up to 30 bp upstream and downstream of the palindromic ERα homodimer motif (Fig. 6H). However, since the ERα monomer motif is directional, we could determine the relative binding localization of these factors in a strand-specific manner, which reveals that most of the RING1B recruitment is downstream and GRHL2 upstream of ERα monomers (Fig. 6I). FOXA1 binding does not appear to be influenced by ERα as its distribution with respect to ERα is relatively even compared to that of RING1B and GRHL2. This finding suggests that at half ERE sites, FOXA1 plays a crucial role in recruiting ERα to the chromatin, which is in line with prior findings (42). However, when ERα binds as a homodimer at full ERE sites, the binding distribution of all three factors—RING1B, GRHL2, and FOXA1—seems to be heavily influenced by ER binding (Fig. 6H). This suggests that at full ERE sites, ERα can influence FOXA1, GRHL2, and RING1B binding. Together, these results demonstrate a high-resolution view of the intimate binding profiles of RING1B with breast cancer TFs including ERα, FOXA1, and GRHL2, further supporting our overall finding of functional cooperativity between these factors in the estrogen response of luminal breast cancer.
DISCUSSION
Despite knowing for over 80 years that estrogen drives breast cancer proliferation, the exact molecular mechanisms of liganded ERα and its effects on gene regulation and chromatin organization are still not well understood. A deeper understanding is needed to uncover novel therapeutic strategies for treating ERα-dependent breast cancers and other estrogen-regulated human diseases. Much effort has been dedicated toward characterizing the intricate network of functional interactions between ER, oncogenic TFs, and epigenetic machineries, with particular emphasis on how these factors are assembled upon acute E2 exposure (4, 43, 44). Nevertheless, there is a limited understanding of the hierarchical events that occur at the genomic and epigenomic level following hours of exposure to estrogens. Given the plasticity of the breast cancer genome during hormone-stimulated proliferation (22), it is crucial to uncover the changes in chromatin organization and epigenetic events that occur during prolonged periods of estrogen exposure. Our results reveal that the Polycomb protein RING1B is at the core of the epigenetic factory that positively regulates the transcriptional response to estrogen in ER+ breast cancer cells (see model, fig. S8).
The mechanisms that regulate the tethering of Polycomb proteins to chromatin are under constant investigation (13). Although we know much about PRC1 and PRC2 recruitment mechanisms in embryonic stem cells, very little is known in adult stem cells and cancer cells (14, 29). There is a significant gap in knowledge of how PRC1 complexes regulate genes during initiation and progression in cancer (45) and how different PRC1 variants are dynamically assembled and recruited to chromatin. In ESCs, PRC1 complexes are mainly involved in maintaining repression of developmental genes (46), but recent studies indicate that PRC1 acquires dual functions during early cell specification and in adult stem cells. While PRC1 complexes still repress lineage-specific genes, they also facilitate gene transcription. Examples are found during neuronal and mesodermal differentiation, in epidermal and intestinal stem cells, as well as in breast cancer and melanoma (17, 47–51). At least two outstanding questions remain: (i) Why do differentiating cells and somatic stem cells require PRC1 complexes to regulate both gene expression and repression? (ii) What is driving this functional switch? It would be fascinating to determine whether PRC1 complexes acquire gene activating functions in premalignant cells as a by-product of tumor evolution or whether PRC1 drives cancer development by gaining novel activating functions. It is not yet known whether RING1B exhibits activating functions by regulating active genes and enhancers in differentiating adult mammary stem cells (MaSC) during mammary gland development and whether these functions differ during in MaSC self-renewal.
In ER+ breast cancer cells, ERα is rapidly recruited to chromatin and ubiquitinated (52) after ligand binding. Ubiquitinated ERα cycles on and off ERE sites to activate target gene transcription (30, 53). A large number of ERα cofactors are recruited to chromatin in a tightly coordinated and dynamic manner within minutes after ligand administration (10, 54). Our results expand upon this knowledge and indicate that bursts of ERα-driven transcriptional activity continue to occur many hours following estrogen stimulation. A large proportion of these transcriptional changes occur independently of chromatin accessibility. Instead, we propose that chromatin accessibility may be also required for recruitment of factors involved in gene repression (55, 56). Nevertheless, future analysis of both gene transcription and chromatin accessibility at the single-cell level will be instrumental to delineate in greater detail how hormone-induced transcriptional changes are coupled to changes in chromatin structure.
Previous study from our lab has shown that cPRC1 colocalizes with ERα at active genes and enhancers to regulate their expression (15). However, the molecular mechanism by which RING1B regulates ERα function is not known. Here, we propose that minutes after E2 administration, RING1B is recruited to chromatin by RNA molecules and/or ERα cofactors in a cPRC1-independent context and that upon prolonged and constant E2 administration, a cPRC1 complex, containing CBX4 and PCGF2, is engaged to chromatin to maintain the transcriptional activity of enhancers and promoters required for proliferation of luminal breast cancer cells. Our efforts aimed to determine whether the RING1B enzymatic activity or interaction with the nucleosome is required for the regulation of estrogen-induced genes revealed a much more complex scenario than previously anticipated. Whether RING1B is an E3 ligase of nonhistone substrates in luminal breast cancer cells is not known. We hypothesize that in a subset of RING1B-ERα cotarget genes and enhancers, RING1B binds to nucleosomes and ubiquitinates either a TF (e.g., ERα, FOXA1, and GRHL2) or an epigenetic factor to stabilize their function (4, 30, 57). Ligand-bound ERα is recruited to chromatin along with a number of E3 ligases (e.g., E6AP and BRCA1) that serve as coactivators not only to promote ERα-driven gene expression but also to mediate ERα ubiquitination and subsequent proteolysis through a mechanism known as activation-coupled ERα degradation (4, 58, 59). RING1B might prove to be another E3 ligase of ERα, although we did not detect RING1B-ERα direct interaction in our previous study under stringent pull-down conditions (15), which may not capture transient interaction or indirect interaction through other cofactors. Mechanistically, our results suggest that the enzymatic activity of RING1B is required for its stable binding to chromatin, supporting a role of RING1B in monoubiquitinating cofactors recruited to RING1B/ERα cotarget genes and enhancers. Further analyses are required to determine the exact role of RING1B’s activity in regulating specific sets of RING1B-ERα cotargets during estrogen stimulation. Nonetheless, we propose that RING1B is required for maintaining the positive feedback loop of ERα cycles in response to estrogen in luminal breast cancer.
We observed RING1B recruitment to EREs not occupied by ERα. EREs can be occupied by ERα and ERβ. ERβ has antiproliferative effects (60), is not expressed in T47D cells, and is not regulated by RING1B. These observations suggest that either RING1B is recruited to these sites after ERα displacement or, in contrast, RING1B is a sensor that dictates future ERα recruitment. We hypothesize the latter for two main reasons: (i) RING1B is recruited before the first on and off cycle of ERα recruitment and displacement, which occurs at around 120 min after E2 addition (30), and (ii) after 24 hours of E2 administration, ERα and RING1B are corecruited to sites that were previously only occupied by RING1B upon 45 min of estrogen exposure.
Last, our results establish the existence of relevant cooperative and functional interactions between RING1B, ERα, and other key TFs central in regulating the estrogen-mediated transcriptional program. We propose that a defined binding arrangement of these factors dictates their interrelationships, resulting in a dynamic gene-regulatory network deployed during the early and late stimulation with estrogen to ensure rapid and constant transcriptional programs in luminal breast cancer.
MATERIALS AND METHODS
Cell lines
MDA-MB-231, T47D, and MCF7 [American Type Culture Collection (ATCC) catalog #HTB-26, #HTB-133, and #HTB-22) were maintained at 37°C with 5% CO2 and split every 2 to 3 days according to ATCC recommendations. Culture media was supplemented with 1× penicillin/streptomycin and 1× glutaMAX, and complete culture media for each cell line were as follows: MDA-MB-231, Dulbecco’s modified Eagle’s medium with 10% fetal bovine serum (FBS); T47D, RPMI 1640 with 10% FBS and insulin (10 μg/ml); MCF7, Eagle’s Minimum Essential Medium (EMEM) with 10% FBS and insulin (10 μg/ml). When estrogen (10 nM E2, Sigma-Aldrich E2758-250MG) was added, cells were maintained in phenol-red free media and 5% charcoal-depleted FBS for 72 hours before treating with ethanol (vehicle) or E2. Cells were routinely tested to be free of mycoplasma infection.
Cell cycle analysis
For the 5-bromo-2′-deoxyuridine (BrdU) incorporation analysis, cells (2 × 105/ml) were incubated for 30 min in culture medium containing 10 μM BrdU. Then, cells were harvested, washed twice with 1× phosphate-buffered saline (PBS), and fixed in cold 70% ethanol overnight at 4°C. After removal of ethanol, DNA was denatured with 2 N HCl supplemented with 0.5% Triton X-100 for 30 min at room temperature, then neutralized with two washes of 0.1 M sodium tetraborate (pH 9), and resuspended in 70% ethanol. Then, cells were recovered by centrifugation, washed once with 1× PBS, and resuspended in 100 μl of blocking buffer [0.5% Tween 20 and 1% bovine serum albumin (BSA) in 1× PBS] containing 10 μl of mouse anti-BrdU antibody (Becton Dickinson, #347580), and incubated at room temperature for 30 min. After a wash with 1× PBS, cells were incubated 15 min at room temperature with goat anti-mouse Alexa 647 antibody (Thermo Fisher Scientific, #A21236) diluted in blocking buffer. Last, cells were washed with 1× PBS once and resuspended in 1× PBS containing propidium iodide (5 μg/ml) (Sigma) and analyzed using BD FACSCanto II (BD Biosciences) in the Flow Cytometry Shared Resource, Sylvester Comprehensive Cancer Center.
Cell proliferation was evaluated using the Cell Proliferation Dye eFluor 670 (Thermo Fisher Scientific) following the manufacturer’s specifications. Briefly, 5 million cells were incubated during 5 min at 37°C with 10 μM eFluor 670 in 1× PBS and 0.1% BSA. The reaction was stopped by adding complete media and incubated for 5 min at 37°C. After washing the cells once with complete media, the cells were cultured under normal conditions. After 24 hours, cell populations were evaluated using BD FACSCanto II (BD Biosciences).
Generation of stable and inducible shRNA cells
To produce shRNA lentiviruses, 2 × 106 human embryonic kidney 293T cells (ATCC #CRL-3216) were plated into a 10-cm2 plate and transfected 16 hours later with 8 μg of pLKO-shRNAs (Addgene, #10879 for CTR; Sigma-Aldrich, #TRCN0000033697 for RING1B; and Sigma-Aldrich, #TRCN0000014881 for FOXA1), 2 μg of pCMV-VSV-G, and 6 μg of pCMV-dR8.91 plasmids using calcium phosphate. Seventy-two hours after transfection, the viral supernatant was collected, passed through a 0.45 μM polyethersulfone filter, and used to transduce MDA-MB-231 and T47D cells. Specifically, 3 × 105 cells were plated into a six-well plate followed by the addition of viral media with polybrene (8 μg/ml; Millipore-Sigma, #TR-1003-G). Cells were centrifuged for 1 hour at 1000g at 32°C and then incubated overnight with fresh viral media. Viruses were removed and complete culture medium was added for cell recovery. Cells were selected 24 hours after recovery with puromycin (2 μg/ml; Biogems, #5855822) and were maintained in selection. All experiments were performed within 2 weeks after transduction. The shRING1B oligos were cloned into the pLKO-tet-on plasmid, and LT3GEPIR-shRenilla luciferase was used as the doxycycline-inducible control. Lentiviruses were produced from the two plasmids as described above. Cells were treated with doxycycline (100 ng/ml) for 2 days before culturing them in HD media for 72 hours.
Growth curves
Doxycycline-inducible T47D shCTR and shRING1B cells were treated with doxycycline (100 ng/ml) for 3 days. After induction, 4000 shCTR and shRING1B cells were plated into individual wells in a 96-well plate. The medium was replaced 1 day after plating, and the cells were treated with 100 nM tamoxifen (4-hydroxytamoxifen; Sigma-Aldrich, H7904-5MG) or 30 nM fulvestrant (ICI 182780; Tocris, catalog #1047). The treatment medium was changed every 2 days, and the number of viable cells in culture was measured on days 0, 3, 5, and 7 using the CellTiter-Glo Luminescent Cell Viability Assay (Promega, G7572).
Colony formation assays
Stable T47D and MCF7 shCTR and shRING1B cells were first cultured in hormone-deprived media for 3 days. After hormone deprivation, 3000 T47D shCTR and shRING1B cells and 2000 MCF7 shCTR and shRING1B cells were each plated into three individual wells on a six- well plate. Cells were cultured in 2 ml of media containing either vehicle (ethanol) or vehicle plus 10 nM estrogen. Medium was refreshed every 3 days. After 2 weeks of culture (T47D) and 3 weeks of culture (MCF7), medium was removed, and colonies were stained with crystal violet (0.25 g of Crystal Violet; Sigma-Aldrich, C3886-25G; 13.5 ml of 37% Formaldehyde; Sigma-Aldrich, 252549-100; 5 ml of methanol; VWR, BDH20864.400; 1× PBS to 500 ml; Sigma-Aldrich, P3813-10PAK) for 20 min. After staining, the wells were gently washed by dipping the plates into a tub of running tap water to avoid disturbing the colonies on the plate surface. The plates were air-dried after thorough washing, and the colonies were imaged using an Epson V750 Pro photoscanner at 1200 dpi resolution. Colonies were quantified and analyzed using the ImageJ Plugin ColonyArea.
Western blotting
Cells were lysed in high-salt buffer [300 nM NaCl, 50 mM tris-HCl (pH 8), 10% glycerol, and 0.2% NP-40] supplemented with protease inhibitors (Sigma-Aldrich, #04693132001) and sonicated 5 min at 4°C with a Bioruptor in 30 seconds ON-OFF cycles. After centrifugation at 16,000g for 15 min, soluble material was quantified by Bradford assay (Bio-Rad, #5000006). Western blotting was performed using standard protocols and imaged on an Odyssey CLx imaging system (Li-COR), and various exposures within the linear range were captured using Image Studio software. Images were rotated, resized, and cropped using Adobe Photoshop CC 2019 and imported into Adobe Illustrator CC 2019 to be assembled into figures.
Transient expression of ERα and FOXA1
MDA-MB-231 cells were grown to 50 to 60% confluency before dissociation with trypsin. A total of 5.6 × 106 cells were pelleted and washed with 1× PBS. Seven micrograms of HA-FOXA1-pCDNA3 and 8 μg of HA-ERα-pCDNA3 or 5 μg of GFP-pCDNA3 were added to the cell pellet. Pellet was resuspended in Resuspension Buffer (Neon Transfection System 100 μL Kit; Thermo Fisher Scientific, MPK10025) to a total volume of 100 μl. Cells were electroporated with a Neon Transfection System (Thermo Fisher Scientific, MPK5000) at 1400 V, 10-ms pulse width, and a pulse number of 4. A total of 1.12 × 107 transfected cells were plated onto one P150 plate. Medium was replaced 24 hours later with growth media containing 10 nM E2, and the cells were collected for Western blotting, RT-qPCR, and ChIP assays 48 hours later.
Histone extraction
A total of 6 × 105 freshly harvested cells were washed with cold 1× PBS and pelleted. Each pellet of cells was resuspended in 500 μl of cold lysis buffer [10 mM tris-HCl (pH 8), 50 mM sodium bisulfite, 1% Triton X-100, 10 mM MgCl2, 8.6% sucrose, and 10 mM sodium butyrate, adjusted to pH 6.5] before centrifugation at 20,000g for 15 min at 4°C. The supernatant was discarded and the pellet was kept on ice. Pellet was again resuspended in 500 μl of cold lysis buffer before centrifugation at 20,000g for 15 min at 4°C. The supernatant was also discarded. This step was repeated two more times. After a total of four rounds of lysis buffer treatment, the pellet was incubated for 1 hour at 4°C in 100 μl of 0.4 M H2SO4 and then centrifuged at 20,000g for 5 min. The supernatant was placed in a new microtube, and 900 μl of acetone was added to the supernatant and stored at −20°C overnight. The next day, samples were centrifuged at 20,000g for 10 min, and the supernatant was discarded. The pellet containing the histone was air-dried for 2 to 5 min and then resuspended in 30 to 50 μl of water. Concentration of histones was measured by a Bradford assay.
Preparation of ATAC reactions and libraries and analysis
ATAC-seq experiments were performed as we previously described (15). FASTQ data were processed using the ATAC-seq/ENCODE pipeline from the Kundaje lab (https://github.com/kundajelab/atac_dnase_pipelines) with default parameters and aligned to the hg19 genome. Homer annotatePeaks and findMotifsGenome were used for peak annotation and motif analysis, respectively. The TCseq Bioconductor package was used to visualize temporal patterns of ATAC-seq peaks. ATAC-seq heat maps and boxplots were created with R v.3.5.1 ComplexHeatmap and ggplot2 packages, respectively. Binary ATAC-seq heat maps were generated using Python v2.7.3 (https://gist.github.com/daler/07eb1a95f1e4639f22bd). Bedtools v2.26.0 was used to determine peak overlaps and NGS Plot v2.61 was used to generate density plots.
ChIP and ChIP-seq library preparation
Cells were grown to 70 to 80% confluency on 150-cm2 plates, and typically six plates were used. Before and after treatment with 10 nM E2 for 45 min, 8 hours, and 24 hours, cells were washed once with 1× PBS. Cells were then cross-linked for 10 min in 10 ml of 1× PBS with 1 ml of 11% formaldehyde buffer [50 mM Hepes-KOH (pH 7.5), 100 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 11% formaldehyde; Thermo Fisher Scientific, #28908] before quenching with 0.5 ml of 2.5 M glycine for 5 min. Cells were then washed two times with 1× PBS. Cross-linked cells were harvested and washed once more with cold 1× PBS, and the pellet was flash-frozen in liquid nitrogen and stored at −80°C. Magnetic beads were preblocked and antibody-bound before the addition of chromatin. For each ChIP, 50 μl of Dynabeads Protein G (Invitrogen, #10004D) was washed three times with 1 ml of 0.5% BSA in 1× PBS (Sigma-Aldrich, A9418), using a magnetic stand to collect the magnetic beads in between washes. Beads were suspended in 250 μl of the BSA solution, and 5 μg (for nonhistone proteins) or 2 μg (for histone modifications) of antibody was added (RING1B: Active Motif, #39664; ERα: Diagenode, #15100066; FOXA1: Abcam, #ab23738; H3K27Ac: Abcam, #ab4729; GRHL2: Sigma, #HPA004820). For ChIP-seq experiments, 1 μg of spike-in antibody was also added (Active Motif, #61686). Beads were incubated on a rotating platform overnight at 4°C. The next day, the beads were washed three more times in the BSA solution before the chromatin was added. To prepare the chromatin, each pellet was resuspended in 2.5 ml of LB1 [50 mM Hepes-KOH (pH 7.5), 140 mM NaCl, 1 mM EDTA, 10% glycerol, 0.5% IGEPAL CA-630 (Sigma-Aldrich, I8896), and 10% Triton X-100] and rocked at 4°C for 10 min. After spinning down at 1350g for 5 min at 4°C, the pellets were resuspended in 2.5 ml of LB2 [10 mM tris-HCl (pH 8), 200 mM NaCl, 1 mM EDTA, and 0.5 mM EGTA] and rocked at room temperature for 10 min. Nuclei were pelleted at 1350g for 5 min at 4°C before resuspension in 2 ml of LB3 [10 mM tris-HCl (pH 8), 100 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 0.1% Na-deoxycholate, and 0.5% N-lauroylsarcosine] and sonication using the Bioruptor Pico (Diagenode, #B01060010) at 4°C in Bioruptor tubes with beads (Diagenode, #C01020031)—10 cycles of 30 s on, 30s off, repeat with brief vortex in between. Two hundred microliters of 10% Triton X-100 was added to the sonicated lysate, which was spun at 20,000g for 15 min at 4°C to pellet the debris. The chromatin (20 μl) was decross-linked in 80 μl of 1× PBS at 65°C for 3 hours, purified using the QIAquick PCR Purification Kit (Qiagen, #28106), and quantified using Qubit. Chromatin (30 μg) was added to the preblocked beads and gently mixed overnight on rotators at 4°C. Spike-in chromatin (50 ng; Active Motif, #53083) was also added. The next day, the supernatant was discarded and the beads were washed 5× with radioimmunoprecipitation assay buffer [50 mM Hepes-KOH (pH 7.5), 500 mM LiCl, 1 mM EDTA, 1% IGEPAL, and 0.7% Na-deoxycholate], collecting the beads on the magnetic rack in between washes. Beads were washed once more with 1 ml of TE with 50 mM NaCl and spun down at 960g for 3 min at 4°C to remove residual TE buffer. Beads were eluted in 210 μl of elution buffer [50 mM tris-HCl (pH 8), 10 mM EDTA, and 1% SDS] at 65°C for 15 min with brief vortexing every 2 min. Beads were spun down, and 200 μl of supernatant was transferred to a new tube and decross-linked overnight at 65°C with shaking at 1000 rpm. One percent of the chromatin input is also decross-linked in the same volume of elution buffer. The next day, 200 μl of TE is added to the decross-linked samples, which were treated for 2 hours at 37°C with ribonuclease A (0.2 μg/ml) followed by 2 hours of proteinase K (0.2 μg/ml; New England Biolabs, #P8107) at 55°C. The immunoprecipitated DNA was purified using the QIAquick PCR Purification Kit and quantified via Qubit. Immunoprecipitated DNA was used to either perform ChIP-qPCR or generate libraries using the NEBNext Ultra DNA Library Prep Kit for Illumina (New England Biolabs, #E7370) following the manufacturer’s instructions. Libraries were visualized on a Tapestation 2200 using D1000 DNA ScreenTape (Agilent Technologies, #5067–5582) and quantified on a Qubit 3 fluorometer with Qubit double-stranded DNA high-sensitivity reagents (Thermo Fisher Scientific, #Q32851) following the manufacturer’s instructions, then pooled and sequenced (single-end, 75 bp) on a NextSeq 500. Processed data were viewed using the University of California, Santa Cruz (UCSC) genome browser. ChIP-qPCR was performed on a Bio-Rad CFX96 Real-Time System with iTaq Universal SYBR Green Supermix (Bio-Rad, #1725124) and analyzed with CFX Manager software (Bio-Rad).
ChIP-seq analysis
All ChIP-seq data generated in this study were analyzed according to the following methodology: FASTQ data were processed with Trimmomatic v0.32 to remove low-quality reads and then aligned to the human genome hg19 using Burrows-Wheeler Aligner (BWA) v0.7.13 with the following parameters: aln -q 5 -l 32 -k 2. Duplicate reads were removed using Picard v1.126. Peaks were called using MACS2.1 with default parameters –shiftsize 160 –nomodel –p 0.01 for all data. Whole-cell extract (input) from the corresponding cells was used as controls. Peaks with signal (fold enrichment over input generated from MACS2) > 4 and a q value < 0.05 were used for downstream analysis. BigWig file output from MACS v 2.1.0.20150731 was visualized in the UCSC genome browser. Homer annotatePeaks v4.8.3 was used for peak annotation. Bedtools v2.26.0 intersect was used to determine peak overlaps. NGS Plot v2.61 was used to generate heat maps and density plots.
ChIP-exo
ChIP-exo was performed using the ChIP-exo 5.0 protocol (39) with minor adaptations. Protein A Mag Sepharose (GE Healthcare) beads were preblocked and bound with antibody before the ChIP. Ten microliters of beads was washed three times with 500 μl of 0.5% BSA in 1× PBS at 4°C. Five micrograms of antibody was added to the washed beads, and the beads were resuspended in 250 μl of the 0.5% BSA solution and allowed to incubate overnight on a rotating platform at 4°C. The next day, the beads were washed three times in 500 μl of 0.5% BSA solution. Sixty micrograms of chromatin was added to the beads, and the IP was performed in a total volume of 500 μl of IP dilution buffer [20 mM tris-HCl (pH 8.0), 2 mM EDTA, 150 mM NaCl, and 1% Triton X-100] with protease inhibitors (Roche) at 4°C overnight. A total of 120 μg of chromatin (two samples with 60 μg of chromatin) was used for each ChIP-exo experiment. In addition, NEBNext Multiplex Oligos for Illumina Index Primers and the Universal PCR Primer for Illumina were used instead of the ExA2_iNN and the ExA1-58 oligos. All other steps were identical to the original ChIP-exo 5.0 protocol.
ChIP-exo analysis
FASTQ data were processed with cutadapt v1.15 (--nextseq-trim=20 -m 10) to remove low-quality reads and then aligned to the human genome hg19 using BWA v0.7.13 (aln -q 5 -l 32 -k 2). Peaks and motif subtypes were determined using ChExMix v0.41 and MEME v5.0.5 after filtering blacklisted regions and enabling a probability-based duplicate filter (--readfilter). Motif matching and motif distributions were determined using MEME-chip v5.0.2 with the JASPAR 2016 motif database. Peak distributions were determined using gaussian kernel function estimates of ChExMix motif aligned peaks.
Identification of SEs
SE and typical enhancers were defined using the ROSE pipeline with default parameters using H3K27ac ChIP-seq peaks as input.
RNA-seq analysis
FASTQ data were processed with cutadapt v1.15 (--nextseq-trim=20 -m 18) to remove low-quality reads. Expected gene counts were obtained using RSEM v1.3.0 and STAR v2.5.3a alignment to the human hg19 transcriptome (GENCODE V19 annotation). RUVseq v1.12.0 was used to adjust gene counts by removing unwanted variance using exogenous ERCC spike-in RNA. Differential expression was determined using DESeq2 v1.18.1 and R (version 3.4.1) with a q value < 0.05 and an FC > 2 (Wald test). Heat maps were generated using variance stabilized gene counts from DESeq2. For GSEAs, the Wald statistic of each time point compared to hormone-deprived conditions was used as input for the Preranked module of GSEA v3.0 on Hallmark gene sets (-scoring_scheme weighted –nrom meandiv).
Statistical analysis
Significance was determined by Student’s t test, nonparametric Wilcoxon test, Mann-Whitney test, or Kolmogorov-Smirnov test. Error bars in figures represent SD of at least two independent experiments. The ggbloxplot function of the ggpubr package (https://cran.r-project.org/web/packages/ggpubr/index.html) and the geom_boxplot function of the ggplot2 package (https://cran.r-project.org/web/packages/ggplot2/index.html) were used to generate boxplots for data visualization.
Supplementary Material
Acknowledgments
We are indebted to members of the Morey laboratory and Dr. Gloria Mas for discussions and the Oncogenomics Core Facility at the Sylvester Comprehensive Cancer Center for performing high-throughput sequencing. We also thank the Flow Cytometry Core Facility for assistance with cell sorting. We are grateful to F. Beckedorff for assisting with the ATAC-seq analysis and M. J. Rossi for providing technical help with the ChIP-exo experiments. Funding: This work was supported by Sylvester Comprehensive Cancer Center funds, AACR-Bayer Innovation and Discovery grant (18-80-44-MORE), Flight Attendant Medical Research Institute (FAMRI) Breast Cancer Developmental Grant, American Cancer Society (ACS; IRG-17-183-16), Stanley J. Glaser Foundation Research Award (UM-SJG-2020-3), Leukemia and Lymphoma Society Specialized Center of Research grant (LLS-SCOR), and the Lampert Breast Cancer Research Fund to L.M. Research reported in this publication was supported by the National Cancer Institute of the National Institutes of Health under Award Number P30CA240139. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Author contributions: L.M. and Y.Z. designed the study and analyzed the experiments with input from J.M.S. and R.E.V. Y.Z. conducted all the experiments except histone extractions, growth curves, and expression profiles in MCF7 cells (L.G.-M.), ATAC-seq analysis and colony formation assays (H.L.C.), and cell cycle profiles and BrdU assays (N.W. in the laboratory of R.E.V.). Bioinformatics analyses were performed by Y.Z., H.L.C., and D.L.K. (Bioinformatics core, Sylvester Comprehensive Cancer Center). D.L.K. performed ChIP-exo analysis. L.M. supervised the experiments and provided intellectual support toward interpretation of the results. L.M. and H.L.C. wrote the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors. The genome-wide data of this study are deposited in the NCBI Gene Expression Omnibus (GEO) database: GSE137579.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/23/eaaz7249/DC1
REFERENCES AND NOTES
- 1.JavanMoghadam S., Weihua Z., Hunt K. K., Keyomarsi K., Estrogen receptor alpha is cell cycle-regulated and regulates the cell cycle in a ligand-dependent fashion. Cell Cycle 15, 1579–1590 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Couse J. F., Korach K. S., Estrogen receptor null mice: What have we learned and where will they lead us? Endocr. Rev. 20, 358–417 (1999). [DOI] [PubMed] [Google Scholar]
- 3.Riggins R. B., Schrecengost R. S., Guerrero M. S., Bouton A. H., Pathways to tamoxifen resistance. Cancer Lett. 256, 1–24 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhou W., Slingerland J. M., Links between oestrogen receptor activation and proteolysis: Relevance to hormone-regulated cancer therapy. Nat. Rev. Cancer 14, 26–38 (2014). [DOI] [PubMed] [Google Scholar]
- 5.Li W., Notani D., Ma Q., Tanasa B., Nunez E., Chen A. Y., Merkurjev D., Zhang J., Ohgi K., Song X., Oh S., Kim H. S., Glass C. K., Rosenfeld M. G., Functional roles of enhancer RNAs for oestrogen-dependent transcriptional activation. Nature 498, 516–520 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carroll J. S., Meyer C. A., Song J., Li W., Geistlinger T. R., Eeckhoute J., Brodsky A. S., Keeton E. K., Fertuck K. C., Hall G. F., Wang Q., Bekiranov S., Sementchenko V., Fox E. A., Silver P. A., Gingeras T. R., Liu X. S., Brown M., Genome-wide analysis of estrogen receptor binding sites. Nat. Genet. 38, 1289–1297 (2006). [DOI] [PubMed] [Google Scholar]
- 7.Shlyueva D., Stampfel G., Stark A., Transcriptional enhancers: From properties to genome-wide predictions. Nat. Rev. Genet 15, 272–286 (2014). [DOI] [PubMed] [Google Scholar]
- 8.Whyte W. A., Orlando D. A., Hnisz D., Abraham B. J., Lin C. Y., Kagey M. H., Rahl P. B., Lee T. I., Young R. A., Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 153, 307–319 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gao W.-W., Xiao R. Q., Zhang W. J., Hu Y. R., Peng B. L., Li W. J., He Y. H., Shen H. F., Ding J. C., Huang Q. X., Ye T. Y., Li Y., Liu Z. Y., Ding R., Rosenfeld M. G., Liu W., JMJD6 licenses ERα-dependent enhancer and coding gene activation by modulating the recruitment of the CARM1/MED12 co-activator complex. Mol. Cell 70, 340–357.e8 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Murakami S., Nagari A., Kraus W. L., Dynamic assembly and activation of estrogen receptor α enhancers through coregulator switching. Genes. Dev. 31, 1535–1548 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Swinstead E. E., Miranda T. B., Paakinaho V., Baek S., Goldstein I., Hawkins M., Karpova T. S., Ball D., Mazza D., Lavis L. D., Grimm J. B., Morisaki T., Grøntved L., Presman D. M., Hager G. L., Steroid receptors reprogram FoxA1 occupancy through dynamic chromatin transitions. Cell 165, 593–605 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tan S. K., Lin Z. H., Chang C. W., Varang V., Chng K. R., Pan Y. F., Yong E. L., Sung W. K., Cheung E., AP-2γ regulates oestrogen receptor-mediated long-range chromatin interaction and gene transcription. EMBO J. 30, 2569–2581 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schuettengruber B., Bourbon H.-M., Di Croce L., Cavalli G., Genome regulation by polycomb and trithorax: 70 years and counting. Cell 171, 34–57 (2017). [DOI] [PubMed] [Google Scholar]
- 14.Chan H. L., Morey L., Emerging roles for polycomb-group proteins in stem cells and cancer. Trends Biochem. Sci. 44, 688–700 (2019). [DOI] [PubMed] [Google Scholar]
- 15.Chan H. L., Beckedorff F., Zhang Y., Garcia-Huidobro J., Jiang H., Colaprico A., Bilbao D., Figueroa M. E., Cava J. L., Shiekhattar R., Morey L., Polycomb complexes associate with enhancers and promote oncogenic transcriptional programs in cancer through multiple mechanisms. Nat. Commun. 9, 3377 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee J.-Y., Won H. Y., Park J. H., Kim H. Y., Choi H. J., Shin D. H., Kang J. H., Woo J. K., Oh S. H., Son T., Choi J. W., Kim S., Kim H. Y., Yi K., Jang K. S., Oh Y. H., Kong G., MEL-18 loss mediates estrogen receptor–α downregulation and hormone independence. J. Clin. Invest. 125, 1801–1814 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chung C.-Y., Sun Z., Mullokandov G., Bosch A., Qadeer Z. A., Cihan E., Rapp Z., Parsons R., Aguirre-Ghiso J. A., Farias E. F., Brown B. D., Gaspar-Maia A., Bernstein E., Cbx8 Acts non-canonically with Wdr5 to promote mammary tumorigenesis. Cell Rep. 16, 427–486 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Le Dily F., Sun Z., Mullokandov G., Bosch A., Qadeer Z. A., Cihan E., Rapp Z., Parsons R., Aguirre-Ghiso J. A., Farias E. F., Brown B. D., Gaspar-Maia A., Bernstein E., Distinct structural transitions of chromatin topological domains correlate with coordinated hormone-induced gene regulation. Genes Dev. 28, 2151–2162 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Murphy C. S., Meisner L. F., Wu S. Q., Jordan V. C., Short- and long-term estrogen deprivation of T47D human breast cancer cells in culture. Eur. J. Cancer Clin. Oncol. 25, 1777–1788 (1989). [DOI] [PubMed] [Google Scholar]
- 20.Müller H., Helin K., The E2F transcription factors: Key regulators of cell proliferation. Biochim. Biophys. Acta 1470, M1–M12 (2000). [DOI] [PubMed] [Google Scholar]
- 21.Schmidt D., Schwalie P. C., Ross-Innes C. S., Hurtado A., Brown G. D., Carroll J. S., Flicek P., Odom D. T., A CTCF-independent role for cohesin in tissue-specific transcription. Genome Res. 20, 578–588 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fullwood M. J., Liu M. H., Pan Y. F., Liu J., Xu H., Mohamed Y. B., Orlov Y. L., Velkov S., Ho A., Mei P. H., Chew E. G. Y., Huang P. Y. H., Welboren W. J., Han Y., Ooi H. S., Ariyaratne P. N., Vega V. B., Luo Y., Tan P. Y., Choy P. Y., Wansa K. D. S. A., Zhao B., Lim K. S., Leow S. C., Yow J. S., Joseph R., Li H., Desai K. V., Thomsen J. S., Lee Y. K., Karuturi R. K. M., Herve T., Bourque G., Stunnenberg H. G., Ruan X., Cacheux-Rataboul V., Sung W.-K., Liu E. T., Wei C.-L., Cheung E., Ruan Y., An oestrogen-receptor-α-bound human chromatin interactome. Nature 462, 58–64 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tarulli G. A., Laven-Law G., Shakya R., Tilley W. D., Hickey T. E., Hormone-sensing mammary epithelial progenitors: Emerging identity and hormonal regulation. J. Mammary Gland Biol. Neoplasia 20, 75–91 (2015). [DOI] [PubMed] [Google Scholar]
- 24.Dixon J. R., Gorkin D. U., Ren B., Chromatin domains: The unit of chromosome organization. Mol. Cell 62, 668–680 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Buchwald G., van der Stoop P., Weichenrieder O., Perrakis A., van Lohuizen M., Sixma T. K., Structure and E3-ligase activity of the Ring-Ring complex of polycomb proteins Bmi1 and Ring1b. EMBO J. 25, 2465–2474 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McGinty R. K., Henrici R. C., Tan S., Crystal structure of the PRC1 ubiquitylation module bound to the nucleosome. Nature 514, 591–596 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pasini D., Malatesta M., Jung H. R., Walfridsson J., Willer A., Olsson L., Skotte J., Wutz A., Porse B., Jensen O. N., Helin K., Characterization of an antagonistic switch between histone H3 lysine 27 methylation and acetylation in the transcriptional regulation of Polycomb group target genes. Nucleic Acids Res. 38, 4958–4969 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hnisz D., Schuijers J., Lin C. Y., Weintraub A. S., Abraham B. J., Lee T. I., Bradner J. E., Young R. A., Convergence of developmental and oncogenic signaling pathways at transcriptional super-enhancers. Mol. Cell 58, 362–370 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Laugesen A., Højfeldt J. W., Helin K., Molecular mechanisms directing PRC2 recruitment and H3K27 methylation. Mol. Cell 74, 8–18 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shang Y., Hu X., DiRenzo J., Lazar M. A., Brown M., Cofactor dynamics and sufficiency in estrogen receptor-regulated transcription. Cell 103, 843–852 (2000). [DOI] [PubMed] [Google Scholar]
- 31.Burakov D., Crofts L. A., Chang C.-P. B., Freedman L. P., Reciprocal recruitment of DRIP/mediator and p160 coactivator complexes in vivo by estrogen receptor. J. Biol. Chem. 277, 14359–14362 (2002). [DOI] [PubMed] [Google Scholar]
- 32.Stanišić V., Lonard D. M., O’Malley B. W., Estrogen receptor-α: Molecular mechanisms and interactions with the ubiquitin proteasome system. Horm. Mol. Biol. Clin. Investig. 1, 1–9 (2010). [DOI] [PubMed] [Google Scholar]
- 33.Glont S.-E., Chernukhin I., Carroll J. S., Comprehensive Genomic Analysis Reveals that the Pioneering Function of FOXA1 Is Independent of Hormonal Signaling. Cell Rep. 26, 2558–2565.e3 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Delage-Mourroux R., Martini P. G. V., Choi I., Kraichely D. M., Hoeksema J., Katzenellenbogen B. S., Analysis of estrogen receptor interaction with a repressor of estrogen receptor activity (REA) and the regulation of estrogen receptor transcriptional activity by REA. J. Biol. Chem. 275, 35848–35856 (2000). [DOI] [PubMed] [Google Scholar]
- 35.Prest S. J., May F. E. B., Westley B. R., The estrogen-regulated protein, TFF1, stimulates migration of human breast cancer cells. FASEB J. 16, 592–594 (2002). [DOI] [PubMed] [Google Scholar]
- 36.Yang W. S., Moon H. G., Kim H. S., Choi E. J., Yu M. H., Noh D. Y., Lee C., Proteomic approach reveals FKBP4 and S100A9 as potential prediction markers of therapeutic response to neoadjuvant chemotherapy in patients with breast cancer. J. Proteome Res. 11, 1078–1088 (2012). [DOI] [PubMed] [Google Scholar]
- 37.Toy W., Shen Y., Won H., Green B., Sakr R. A., Will M., Li Z., Gala K., Fanning S., King T. A., Hudis C., Chen D., Taran T., Hortobagyi G., Greene G., Berger M., Baselga J., Chandarlapaty S., ESR1 ligand-binding domain mutations in hormone- resistant breast cancer. Nat. Genet. 45, 1439–1445 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jozwik K. M., Chernukhin I., Serandour A. A., Nagarajan S., Carroll J. S., FOXA1 directs H3K4 monomethylation at enhancers via recruitment of the methyltransferase MLL3. Cell Rep. 17, 2715–2723 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rossi M. J., Lai W. K. M., Pugh B. F., Simplified ChIP-exo assays. Nat. Commun. 9, 2842 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yamada N., Lai W. K. M., Farrell N., Pugh B. F., Mahony S., Characterizing protein-DNA binding event subtypes in ChIP-exo data. Bioinformatics 35, 903–913 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rhee H. S., Pugh B. F., Comprehensive genome-wide protein-DNA interactions detected at single-nucleotide resolution. Cell 147, 1408–1419 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hurtado A., Holmes K. A., Ross-Innes C. S., Schmidt D., Carroll J. S., FOXA1 is a key determinant of estrogen receptor function and endocrine response. Nat. Genet. 43, 27–33 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Feinberg A. P., Koldobskiy M. A., Göndör A., Epigenetic modulators, modifiers and mediators in cancer aetiology and progression. Nat. Rev. Genet. 17, 284–299 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Siersbæk R., Kumar S., Carroll J. S., Signaling pathways and steroid receptors modulating estrogen receptor α function in breast cancer. Genes Dev. 32, 1141–1154 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Koppens M., van Lohuizen M., Context-dependent actions of Polycomb repressors in cancer. Oncogene 35, 1341–1352 (2016). [DOI] [PubMed] [Google Scholar]
- 46.Morey L., Santanach A., Di Croce L., Pluripotency and epigenetic factors in mouse embryonic stem cell fate regulation. Mol. Cell. Biol. 35, 2716–2728 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Morey L., Helin K., Polycomb group protein-mediated repression of transcription. Trends Biochem. Sci. 35, 323–332 (2010). [DOI] [PubMed] [Google Scholar]
- 48.Morey L., Santanach A., Blanco E., Aloia L., Nora E. P., Bruneau B. G., di Croce L., Polycomb regulates mesoderm cell fate-specification in embryonic stem cells through activation and repression mechanisms. Cell Stem Cell 17, 300–315 (2015). [DOI] [PubMed] [Google Scholar]
- 49.Rai K., Akdemir K. C., Kwong L. N., Fiziev P., Wu C. J., Keung E. Z., Sharma S., Samant N. S., Williams M., Axelrad J. B., Shah A., Yang D., Grimm E. A., Barton M. C., Milton D. R., Heffernan T. P., Horner J. W., Ekmekcioglu S., Lazar A. J., Ernst J., Chin L., Dual roles of RNF2 in melanoma progression. Cancer Discov. 5, 1314–1327 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cohen I., Zhao D., Bar C., Valdes V. J., Dauber-Decker K. L., Nguyen M. B., Nakayama M., Rendl M., Bickmore W. A., Koseki H., Zheng D., Ezhkova E., PRC1 fine-tunes gene repression and activation to safeguard skin development and stem cell specification. Cell Stem Cell 22, 726–739.e7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pivetti S., Fernandez-Perez D., D’Ambrosio A., Barbieri C. M., Manganaro D., Rossi A., Barnabei L., Zanotti M., Scelfo A., Chiacchiera F., Pasini D., Loss of PRC1 activity in different stem cell compartments activates a common transcriptional program with cell type-dependent outcomes. Sci. Adv. 5, eaav1594 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nawaz Z., Lonard D. M., Dennis A. P., Smith C. L., O’Malley B. W., Proteasome-dependent degradation of the human estrogen receptor. Proc. Natl. Acad. Sci. U.S.A. 96, 1858–1862 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lonard D. M., Nawaz Z., Smith C. L., O’Malley B. W., The 26S proteasome is required for estrogen receptor-alpha and coactivator turnover and for efficient estrogen receptor-alpha transactivation. Mol. Cell 5, 939–948 (2000). [DOI] [PubMed] [Google Scholar]
- 54.Dasgupta S., Lonard D. M., O'Malley B. W., Nuclear receptor coactivators: Master regulators of human health and disease. Annu. Rev. Med. 65, 279–292 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pajoro A., Madrigal P., Muiño J. M., Matus J., Jin J., Mecchia M. A., Debernardi J. M., Palatnik J. F., Balazadeh S., Arif M., Ó’Maoiléidigh D. S., Wellmer F., Krajewski P., Riechmann J. L., Angenent G. C., Kaufmann K., Dynamics of chromatin accessibility and gene regulation by MADS-domain transcription factors in flower development. Genome Biol. 15, R41 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Natarajan A., Yardimci G. G., Sheffield N. C., Crawford G. E., Ohler U., Predicting cell-type-specific gene expression from regions of open chromatin. Genome Res. 22, 1711–1722 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Métivier R., Penot G., Hübner M. R., Reid G., Brand H., Koš M., Gannon F., Estrogen receptor-alpha directs ordered, cyclical, and combinatorial recruitment of cofactors on a natural target promoter. Cell 115, 751–763 (2003). [DOI] [PubMed] [Google Scholar]
- 58.Perissi V., Rosenfeld M. G., Controlling nuclear receptors: The circular logic of cofactor cycles. Nat. Rev. Mol. Cell Biol. 6, 542–554 (2005). [DOI] [PubMed] [Google Scholar]
- 59.Fan S., Wang J., Yuan R., Ma Y., Meng Q., Erdos M. R., Pestell R. G., Yuan F., Auborn K. J., Goldberg I. D., Rosen E. M., BRCA1 inhibition of estrogen receptor signaling in transfected cells. Science 284, 1354–1356 (1999). [DOI] [PubMed] [Google Scholar]
- 60.Paruthiyil S., Parmar H., Kerekatte V., Cunha G. R., Firestone G. L., Leitman D. C., Estrogen receptor β inhibits human breast cancer cell proliferation and tumor formation by causing a G2 cell cycle arrest. Cancer Res. 64, 423–428 (2004). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/23/eaaz7249/DC1