

A cryo-EM structure and functional study show that Arp2/3 complex activation requires NPF-mediated delivery of actin to both Arps.
Abstract
Actin-related protein (Arp) 2/3 complex nucleates branched actin networks that drive cell motility. It consists of seven proteins, including two actin-related subunits (Arp2 and Arp3). Two nucleation-promoting factors (NPFs) bind Arp2/3 complex during activation, but the order, specific interactions, and contribution of each NPF to activation are unresolved. Here, we report the cryo–electron microscopy structure of recombinantly expressed human Arp2/3 complex with two WASP family NPFs bound and address the mechanism of activation. A cross-linking assay that captures the transition of the Arps into the activated filament-like conformation shows that actin binding to NPFs favors this transition. Actin-NPF binding to Arp2 precedes binding to Arp3 and is sufficient to promote the filament-like conformation but not activation. Structure-guided mutagenesis of the NPF-binding sites reveals their distinct roles in activation and shows that, contrary to budding yeast Arp2/3 complex, NPF-mediated delivery of actin at the barbed end of both Arps is required for activation of human Arp2/3 complex.
INTRODUCTION
Actin filament nucleators catalyze the rate-limiting step in actin polymerization, i.e., nucleation (1, 2). Actin-related protein (Arp) 2/3 complex is the only nucleator that can generate branched actin networks, an architecture that plays key roles in motile processes such as vesicular trafficking, endocytosis/exocytosis, and cell motility (3). Arp2/3 complex–dependent assembly also drives the movement of metastatic cancer cells and numerous pathogens. The complex consists of seven proteins, including two Arps (Arp2 and Arp3) that act as a pseudo-actin dimer during nucleation and five scaffolding subunits (ArpC1 to ArpC5) that hold the Arps in place (4) and mediate interactions with the mother actin filament in the branch (5). Isolated mammalian Arp2/3 complex is inactive, with the Arps splayed apart (4). In the presence of actin monomers and filaments, the complex is activated by a family of nucleation-promoting factors (NPFs) that perform three main functions: (i) they promote a conformational change that repositions the Arps into a filament-like (short-pitch) conformation (5–9), (ii) they recruit actin subunits to the barbed end of the Arps to form the initial polymerization seed (10, 11), and (iii) they promote binding of the complex to the side of a preexisting (mother) filament to form a branch (daughter) filament that emerges at a ~70° angle (5, 12, 13). The NPFs are generally unrelated, except for their C-terminal WCA region, comprising one to three WASP-homology 2 (WH2) domains (W) that bind actin, and Central (C) and Acidic (A) segments that bind Arp2/3 complex (14–18). The WCA region of NPFs is necessary and sufficient to activate Arp2/3 complex in vitro, whereas their N-terminal domains typically diverge and are involved in regulation/localization, allowing for the spatiotemporal control of Arp2/3 complex activity in cells (3, 19).
Studies using engineered dimeric WCA peptides (11, 20), analytical ultracentrifugation (11), chemical cross-linking (11, 21–23), isothermal titration calorimetry (24, 25), and fluorescence energy transfer (9, 25) have shown that Arp2/3 complex binds two NPFs during activation. Although monomeric WCA can activate the complex, the engineered WCA dimers are more potent activators (11, 20, 26). Several WCA-binding models have been proposed (11, 23–25), and although these models differ in important ways, they generally suggest that one NPF binds subunits Arp2 and ArpC1 and the other binds Arp3 to deliver actin subunits at the barbed end of Arp2 and Arp3, respectively (referred to here as the Arp2-ArpC1 and Arp3 pathways). However, the order of binding of each NPF, their specific interactions with Arp2/3 complex, the conformational changes they each produce in the complex, and their individual contributions to activation are poorly understood. Here, we address these questions using recombinantly expressed human Arp2/3 complex and a combination of biochemical and structural approaches. We show that actin binding to NPFs shifts the equilibrium of Arp2/3 complex toward a filament-like conformation and that actin-NPF binding to Arp2 precedes binding to Arp3 and is sufficient to promote the short-pitch conformation but not activation. A cryo–electron microscopy (cryo-EM) structure reveals the binding sites of NPFs on Arp2/3 complex and informs mutagenesis studies that demonstrate that NPF-mediated delivery of actin to both Arps is required for activation.
RESULTS
Expression of human Arp2/3 complex and implementation of short-pitch reporter assay
Most mutagenesis studies have previously focused on Saccharomyces cerevisiae (budding yeast) Arp2/3 complex (referred to here as yeast Arp2/3 complex, unless otherwise specified), which differs substantially from the mammalian complex in sequence and activity (27) (as also shown here). We developed a method for the expression of recombinant human Arp2/3 complex using the biGBac system (Fig. 1A), in which all the subunits of Arp2/3 complex are assembled into a single baculovirus-insect cell expression vector (28). A key advantage of this system is to increase expression yields by reducing cell stress from infection with multiple viruses and as a result of the stoichiometric expression of complex subunits. Three of the seven subunits of mammalian Arp2/3 complex have isoform variants, ArpC1 (ArpC1A and ArpC1B), ArpC5 (ArpC5 and ArpC5L), and Arp3 (Arp3A and Arp3B). The complex expressed here contains the most abundant isoforms found in tissue-purified samples (ArpC1B, ArpC5, and Arp3A) (4).
Fig. 1. Factors controlling the short-pitch transition in recombinant human Arp2/3 complex.
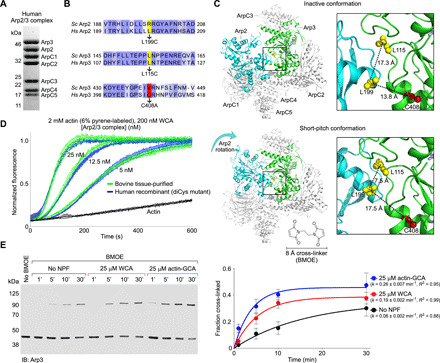
(A) SDS-PAGE of recombinantly expressed human Arp2/3 complex. (B) Sequence alignment of human and budding yeast Arp2 and Arp3, showing the three amino acids mutated to generate the diCys mutant used in the cross-linking assay by analogy with the yeast complex (29). (C) Illustration of the cross-linking assay; Arp2 L199 and Arp3 L115 (both mutated to cysteine residues) are within BMOE cross-linking distance (~8 Å) only in the short-pitch conformation, in which Arp2 is moved alongside Arp3, analogous to two adjacent subunits of the actin filament. The C408A mutation in Arp3 was introduced to prevent spurious cross-linking. (D) Time course of actin polymerization by diCys Arp2/3 complex mutant and tissue-purified bovine Arp2/3 complex over a range of concentrations (n = 3, per concentration). (E) Western blot analysis (immunoblotting: Arp3) and densitometric quantification (n = 3) of the cross-linked fraction of diCys Arp2/3 complex in the absence of NPFs and with either WCA or actin-GCA and different BMOE treatment times (as indicated). Data were fit to a first-order exponential function to obtain the kinetic parameters.
The ability to obtain large quantities of pure human Arp2/3 complex opened the way to mutagenesis studies and the implementation of a cross-linking assay that had been previously used to assess the short-pitch transition in yeast Arp2/3 complex (29). By analogy with the yeast complex, two amino acids were selected, one in Arp2 (L199) and one in Arp3 (L115), which are ~17 Å apart (inter-Cα distance) in the inactive complex but are predicted to be ~7 Å apart in the short-pitch conformation (Fig. 1, B and C). These two amino acids were mutated to cysteine for cross-linking with the 8.0-Å disulfide cross-linker bismaleimidoethane (BMOE). Different from the yeast complex, human Arp3 has an endogenous cysteine residue (C408) that in the inactive conformation is ~13.8 Å apart from Arp2 L199 (Fig. 1C). To prevent spurious cross-linking, C408 was mutated to alanine. The resulting Arp2/3 complex triple mutant (Arp2 L199C, Arp3 L115C, and Arp3 C408A) is referred to here as the diCys mutant and had comparable nucleation activity to that of tissue-purified bovine Arp2/3 complex over a range of concentrations (Fig. 1D).
The short-pitch transition can now be covalently captured by cross-linking, and the fraction of cross-linked Arp2/3 complex can be quantified by Western blot analysis using anti-Arp3 and anti-Arp2 antibodies (Fig. 1E and fig. S1A). In this way, cross-linking time courses were obtained in the absence of WCA and with saturating amounts of N-WASP WCA (25 μM). In each case, an Arp2-Arp3 cross-linking band appeared in the gel at ~100 kDa and increased with the time after BMOE treatment. This band reacted with both anti-Arp2 and anti-Arp3 antibodies and was not observed with untreated (no BMOE added) human Arp2/3 complex diCys mutant or with wild-type (WT) human Arp2/3 complex treated with BMOE for 60 min (Fig. 1E and fig. S1A). Quantification of several experiments showed that the cross-linking reaction reaches a plateau after ~30 min, probably after BMOE reacts independently with both cysteine residues, which can no longer form a cross-link. A cross-linking time of 30 min is thus used in other experiments of this study. It is important to note, however, that cross-linking (Fig. 1E) is a much slower reaction than nucleation (Fig. 1D); i.e., the short-pitch transition happens at a much faster rate than the cross-linking reaction, which only captures a small fraction of such transitions. Judging from the half times, WCA substantially increases the speed of the cross-linking reaction (~3.2-fold) and the total amount of cross-linked complex at 30 min. In the presence of WCA, the total cross-linked fraction is similar for human and yeast Arp2/3 complex, but the cross-linking rate is substantially faster for the budding yeast complex (8), consistent with its lower activation barrier (27).
The concentration of actin monomers in cells is >100 μM, such that NPFs are likely preloaded with actin, which binds to the WH2 domain with a Kd < 1.0 μM (14, 17, 30). Therefore, we explored the role of actin binding to NPFs for the short-pitch transition. However, because nucleation is transient and NPFs are released immediately after nucleation (13), we used in these experiments GCA instead of WCA. GCA is a construct in which the WH2 domain of WCA is replaced with gelsolin segment-1 based on structural similarities in the way the two domains interact with actin monomers (fig. S1, B to D). Gelsolin segment-1 is also larger in size and has higher affinity for actin (31) than the WH2 domain (14, 17, 30). In this way, GCA binds actin stably and sterically blocks the addition of actin subunits to the polymerization nucleus and is thus used here to “trap” the actin-NPF-Arp2/3 transitional complex for biochemical studies (25). The WCA to GCA substitution does not influence the conformation of Arp2/3 complex itself, since the CA region, but not the WH2 domain, contacts Arp2/3 complex (fig. S1D) (14, 16). Compared to WCA alone, the presence of saturating amounts (25 μM) of prepurified actin-GCA leads to an additional increase in the speed of the reaction and the total amount of cross-linked Arp2/3 complex at 30 min (Fig. 1E). Together, the results suggest that (i) in the absence of NPFs, human Arp2/3 complex transiently visits the short-pitch conformation, but this does not lead to activation like in the yeast complex (8, 27); (ii) NPFs shift the equilibrium toward the short-pitch conformation, similar to the yeast complex (8); and (iii) actin binding to NPFs further shifts the equilibrium of Arp2/3 complex toward a short-pitch conformation, which is also similar to the yeast complex (29).
Actin-NPF binding to a single high-affinity site induces the short-pitch transition
The binding affinities of the two NPF-binding sites of Arp2/3 complex differ >10-fold, whether alone or with actin bound (24, 25), suggesting that the two sites are occupied in a stepwise manner. We used the cross-linking assay to ask whether the transition into the short-pitch conformation was incrementally achieved with the binding of one versus two actin-GCA to Arp2/3 complex. Because of their different affinities, only one actin-GCA remains bound to Arp2/3 complex by gel filtration (25) or glycerol gradient separation (Fig. 2A and fig. S2, A and B), resulting in a half-saturated 1:1:1 actin:GCA:Arp2/3 complex. The cross-linking kinetics of this prepurified 1:1:1 complex is indistinguishable from that of Arp2/3 complex in the presence of saturating amounts of actin-GCA (2:2:1 complex; 25 μM actin-GCA; 1 μM Arp2/3 complex) (Fig. 2B). This result suggests that binding of actin-NPF to the first, high-affinity site is sufficient to shift the equilibrium toward a short-pitch conformation, with no additional gain from binding to the second site.
Fig. 2. Actin-GCA binding to high-affinity site induces the short-pitch transition but is insufficient for activation.
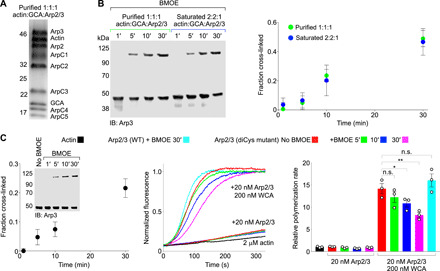
(A) SDS-PAGE of diCys Arp2/3 complex with actin-GCA bound to a single, high-affinity site after glycerol gradient fractionation (1:1:1 complex, see also fig. S2, A and B). (B) Western blot analysis (IB: Arp3) and densitometric quantification (n = 3) of the cross-linked fraction of diCys Arp2/3 complex as a function of BMOE treatment time in the 1:1:1 complex or in the presence of a high molar excess (25 μM) of actin-GCA (2:2:1 complex). (C) Time course of actin polymerization by diCys Arp2/3 complex (middle) as a function of cross-linking time (left) and in the presence or the absence of N-WASP WCA (as indicated). (Right) Relative polymerization rates were calculated by determining the first derivative of the curve and normalizing the maximum derivative value to that of actin alone (n = 3). The statistical significance of the measurements was determined using an unpaired two-sided Student’s t test (n.s., P ≥ 0.05; *, P < 0.05; **, P ≤ 0.01).
The short-pitch transition is insufficient for activation of mammalian Arp2/3 complex
We then asked whether the short-pitch transition was sufficient to activate human Arp2/3 complex, bypassing the need for NPFs, as observed with yeast Arp2/3 complex (8). The Arp2/3 complex diCys mutant was cross-linked in the absence of NPFs for 1, 5, 10, and 30 min, to trap its spontaneous transition into the short-pitch conformation (Fig. 2C). The total cross-linked fraction at 30 min was ~22%, which is similar to the cross-linked fraction used in the yeast Arp2/3 complex study (~28% at 5 min) (8). The polymerization activity was then assessed in the absence or the presence of N-WASP WCA using the pyrene-actin polymerization assay (Fig. 2C). In the absence of WCA, no increase in actin polymerization was observed as a function of BMOE treatment time, whereas in the presence of 200 nM WCA, the activity decreased with cross-linking time. This decrease in activity is possibly due to both cysteine residues being independently reacted with BMOE, which may sterically hinder the short-pitch transition. It is also possible that the conformation of the cross-linked complex is not exactly the same as that of the native short-pitch state. Other factors, such as nonspecific cross-linking, can be ruled out since the activity of WT human Arp2/3 complex was unaffected by BMOE treatment (Fig. 2C). Yet, the most notable observation is that unlike yeast Arp2/3 complex, which is hyperactivated by stabilization of a short-pitch conformation through this cross-link (8), human Arp2/3 complex is not.
The actin-NPF–binding site on Arp2-ArpC1 is the high-affinity site
While actin-NPF binding to a single, high-affinity site is sufficient to induce a short-pitch conformation (Fig. 2B), interpretation of the existing evidence has led to disagreement as to whether this site is on Arp2-ArpC1 (10, 25, 32) or Arp3 (8, 24, 33). To identify the high-affinity site occupied in the 1:1:1 actin:GCA:Arp2/3 complex, we used EM of negatively stained bovine Arp2/3 complex specimens, cross-linked and purified using a glycerol gradient (fig. S2, A and B). Because of the low resolution inherent to this approach, the analysis here focuses on comparisons of the overall shapes and dimensions of representative two-dimensional (2D) class averages with models of actin-GCA at the barbed end of either Arp2 or Arp3 and their 2D projections (figs. S2, C and D, and S3, A and B). Actin-GCA can be added to Arp2 either in the inactive conformation or in the short-pitch conformation, obtained by moving Arp2 to form a filament-like heterodimer with Arp3. Both models result in similar overall dimensions (~15 nm). In contrast, actin-GCA can only be added to Arp3 in the short-pitch conformation (since Arp2 occludes the barbed end of Arp3 in the inactive conformation), and the dimensions of this model are substantially smaller (~12 nm) than those of the Arp2 models (fig. S3A). The 2D class averages of the prepurified and cross-linked 1:1:1 complex have shapes and dimensions that more closely resemble those of the models of actin-GCA on Arp2 (fig. S3B). To remove any remaining ambiguity, we also analyzed a sample containing maltose-binding protein (MBP) added N-terminally to GCA (fig. S2, E and F). MBP extends the theoretical dimensions of the Arp2 and Arp3 models to ~20 and ~15 nm, respectively (fig. S3C). Consistently, the 2D class averages obtained with this sample were more elongated, featuring a ~4-nm extra density likely belonging to MBP, and their shapes and dimensions more closely resemble those of the models of actin-MBP-GCA on Arp2 (fig. S3D). These results allow us to conclude that the high-affinity NPF-binding site is the one along Arp2-ArpC1, a conclusion further supported by mutagenesis analysis (see below).
Cryo-EM structure reveals the two CA-binding sites on Arp2/3 complex
To understand the specific contribution of each NPF-binding site to activation, we obtained a cryo-EM structure of WCA-bound human Arp2/3 complex. A total of 627,163 particles were used for reference-free 2D classification, and 123,198 particles were selected for 3D reconstruction with a global resolution of 3.8 Å (Fig. 3A, fig. S4, and table S1). The cryo-EM map displays the inactive conformation observed in crystal structures (4) and was fit with the crystal structure of glia maturation factor (GMF)-bound Arp2/3 complex [Protein Data Bank (PDB) ID: 4JD2] in which Arp2 was fully resolved (Fig. 3B and movie S1). While WCA enhances the likelihood of a short-pitch transition in solution (Fig. 1E), a short-pitch structural class could not be resolved using particle subtraction, targeted classification, or multibody refinement. The complex subunits were individually fit to the map using rigid-body refinement and, while some side chains had to be repositioned to avoid clashes, no attempt was made to refine their positions at this resolution. The map is well defined for most of the structure, revealing some loops that were unresolved in the crystal structures. For Arp2, however, the density is somewhat more diffuse (Fig. 3A), consistent with it being the most flexible subunit of the complex (not fully visualized in most crystal structures of the inactive complex) and the one undergoing the major conformational change during activation. Unattributed density was observed on Arp2-ArpC1 and Arp3 and was assigned to the CA portion of two WCA peptides bound; the WH2 domain does not bind Arp2/3 complex (14, 16) and was not resolved.
Fig. 3. Cryo-EM structure of human Arp2/3 complex with bound N-WASP CA.
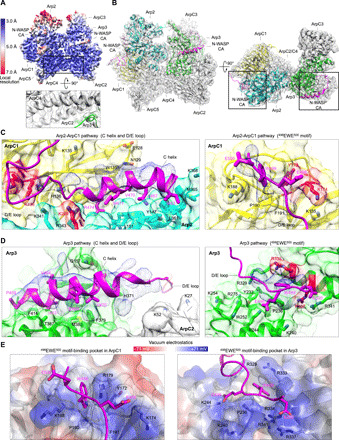
(A) Cryo-EM map of human Arp2/3 complex with bound N-WASP CA colored by resolution (as indicated in the side bar). The inset illustrates the presence of side-chain densities in the core region of the map. Arp2 is more mobile and therefore less well defined. (B) Two perpendicular orientations of the cryo-EM map fit with the Arp2/3 complex structure (subunits are labeled), showing two CA chains (magenta), one bound to Arp2 (cyan) and ArpC1 (yellow), and the other bound to Arp3 (green). See also movie S1. (C and D) Close-up views of the difference map (Arp2-ArpC1 pathway, 3.8 σ; Arp3 pathway, 4.0 σ) showing specific interactions of the C helix, D/E loop, and 498EWE500 motif along each CA pathway. Amino acids mutated in this study are highlighted red (see also fig. S6). (E) Electrostatic surface representation of the 498EWE500 motif-binding pockets in ArpC1 and Arp3. In both cases, the conserved tryptophan of this motif is sandwiched in between a proline and an arginine residue, and positively charged side chains surround the pocket, favoring the interaction with negatively charged amino acids of the A region.
The observed density for CA at both binding sites is fragmented and is composed of an elongated portion on each of the Arps that was assigned to the helix of the C region (C helix) and a separate patch assigned to the C-terminal 498EWE500 motif of the A region (Fig. 3, B to D). While we cannot reliably fit the side chains of the CA region into this density, we can propose a model of the C helix based on the NMR structure of the autoinhibited core of WASP, consisting of the C region bound intramolecularly to the GTPase–binding domain (GBD) (34). In this structure, most of the C region consists of a four-turn amphipathic α helix, whose hydrophobic side faces the GBD. Upon WASP activation by GTPase binding, the hydrophobic side of the C helix binds Arp2/3 complex and, accordingly, alanine substitutions of hydrophobic residues in this helix impair the ability of WCA to activate Arp2/3 complex (16). The C helix can be fit into the densities on each of the Arps, with its hydrophobic side (I463, V464, L467, and M471) facing the Arps (Fig. 3, C and D). Several models had placed the C helix near the barbed end of the Arps (10, 11, 23–25). While this idea is mostly correct, the structure reveals notable differences in the way the C helix binds to Arp2 and Arp3 (Fig. 4A). In Arp2, the C helix lies in front of the “hinge helix” (Q141-Q149), which functions as the axis for interdomain movements in the actin fold (35). The C helix also interacts with hydrophobic residues in an adjacent helix of Arp2 (M353-I364) and with L151 and L152 in the loop that follows the hinge helix (Fig. 4A). In Arp3, the hinge helix is one turn longer than in Arp2 (and actin), which would create a clash with the C helix of NPFs if it were to bind the way it binds to Arp2. Instead, the C helix in Arp3 binds in a hydrophobic cleft formed between the hinge helix and the adjacent helix A376-T387 (Fig. 4A). The interaction of the C helix with the Arps is also quite different from those of the WH2 domain and the G-actin binding domain (GAB) of vasodilator-stimulated phosphoprotein (VASP) with actin (Fig. 4B) (17, 36). Last, the C helix is longer and binds differently compared to the “protrusion helix” (Fig. 4B), a loop-helix-loop motif of ArpC1 that in the structure of the inactive complex is detached from the core of the β-propeller and makes a crystal contact with Arp3 (4). This crystal contact has been proposed to mimic both the interaction of the C helix with the Arps (24, 25) and contacts with the mother filament (4, 5).
Fig. 4. Interaction of the C helix with the Arps and comparison with related helices from other domains.
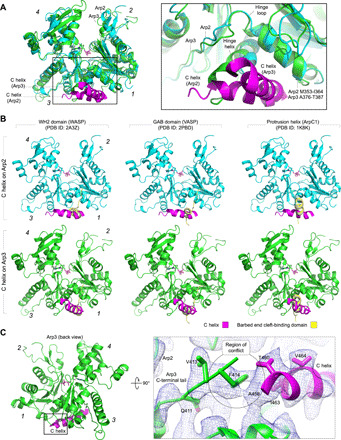
(A) Superimposition of Arp2 (cyan) and Arp3 (green), showing how the orientation of the C helix (magenta) of NPFs is different for the two Arps (numbers in italic designate subdomains 1 to 4). The inset shows that the hinge helix is one helical turn longer in Arp3 than in Arp2, which would create a clash with the C helix if it were to bind to Arp3 the way it binds to Arp2. (B) The interaction of the C helix with Arp2 (top row) and Arp3 (bottom row) differs from those of the WH2 domain (left column, yellow) and GAB domain (middle column, yellow) with actin (blue) and from the crystal contact made by the protrusion helix of ArpC1 with Arp3 in the structure of inactive Arp2/3 complex (right column, yellow). (C) The N-terminal end of the C helix on Arp3 clashes with the C-terminal tail of Arp3, which would have to move for the C helix to bind. The inset (right) shows a close-up view of this clash, with the cryo-EM map contoured at 6.6 σ.
In Arp3, there appears to exist a steric conflict between the N-terminal end of the C helix and the C-terminal tail of Arp3, according to the position of this tail in the crystal structure of inactive Arp2/3 complex (4). The cryo-EM map suggests that the binding of the C helix to Arp3 displaces the C-terminal tail of Arp3 (Fig. 4C), which is consistent with this tail functioning as an autoinhibitory element in Arp2/3 complex (8). The steric conflict between the C helix and the C-terminal tail of Arp3 may also explain why the affinity of the NPF-binding site on Arp3 is >10-fold lower than that on Arp2-ArpC1 (25).
The region between the C helix and the C-terminal 498EWE500 motif of NPFs is variable in length and sequence but is characterized by its overall negative charge resulting from the presence of several aspartic acid and glutamic acid side chains (fig. S5A). We refer to this region here as the D/E loop. This loop is not well-resolved in the structure and appears to be rather flexible, which is likely important for its role as a hinge that accommodates the large movement of Arp2 during the short-pitch transition. The cryo-EM map suggests a path for this loop in the inactive conformation that puts it in contact with numerous positive side chains on both Arp2-ArpC1 and Arp3 (Fig. 3, C and D, and movie S1). The involvement of some of these side chains in NPF interactions was tested here by mutagenesis (see below).
On the Arp3 side, the 498EWE500 motif binds in a pocket in subdomain 4 of Arp3 (Fig. 3, D and E), consistent with a previous structure revealing these three amino acids in crystals of bovine Arp2/3 complex soaked with a CA peptide (24). A strikingly similar pocket exists in ArpC1, formed by a long loop (A171-L194) inserted between the last two β strands of the fourth blade of the β-propeller (Fig. 3, C to E). A patch of density associated with this pocket was assigned to the 498EWE500 motif of the WCA peptide bound along the Arp2-ArpC1 pathway. In crystal structures of bovine Arp2/3 complex, this pocket is partially masked by a crystal packing contact, possibly explaining why the interaction of the 498EWE500 motif with ArpC1 was not visualized in soaked crystals. In both ArpC1 and Arp3, the conserved tryptophan of the A region appears to be sandwiched in between an arginine residue (ArpC1 R179 and Arp3 R333) and a proline residue (ArpC1 P190 and Arp3 P236), and its indole nitrogen appears to be hydrogen-bonded by Y172 in ArpC1 and D330 in Arp3. A net positive charge surrounds both pockets, favoring the interaction with the acidic side chains that border the tryptophan in the sequence of NPFs (Fig. 3E).
Two of the three variable isoforms of Arp2/3 complex interact with NPFs (Arp3 and ArpC1), which could affect the nucleation activity of the complex, as suggested by the finding that the ArpC1A-containing complex has lower nucleation activity than the ArpC1B-containing complex (37). We analyzed both CA-binding pathways for isoform variations that could cause these differences in activity (fig. S5, B and C). However, only a few amino acid variations between isoforms fall near the binding paths of the NPFs in the inactive conformation, and for the most part, these are conservative mutations that cannot readily explain the different activities of Arp2/3 complex isoform variants.
Mutagenesis analysis reveals a different role for each NPF-binding site
On the basis of the cryo-EM structure, five Arp2/3 complex mutants (M1 to M5) were generated to disrupt the two CA-binding sites and assess their individual roles in the short-pitch transition and activation (Fig. 5A and fig. S6). These mutants were generated in the background of the diCys complex used in the cross-linking assay (referred to in this experiment as WT). Three of the mutants target the CA-binding pathway along Arp2-ArpC1, including M1 (Arp2 K299E) and M2 (ArpC1 K138E and K139E), intended to disrupt interactions of the D/E loop, and M3 (ArpC1 Y172A and K174E), targeting the pocket that binds the 498EWE500 motif in ArpC1. Another ArpC1 mutant (M4) targets the protrusion helix and the loops tethering this helix to the β-propeller (residues 293 to 310) and consists of an 18–amino acid substitution of this segment by a poly-S/G loop of equal length (fig. S6). Last, M5 (Arp3 R333D and R337D) was designed to disrupt the pocket that binds the 498EWE500 motif in Arp3. The solubility of all the mutants was similar to that of WT Arp2/3 complex, and they also had similar gel filtration, SDS–polyacrylamide gel electrophoresis (PAGE), and native PAGE profiles (fig. S7, A to C), suggesting that the mutations do not compromise folding or assembly of Arp2/3 complex.
Fig. 5. Distinct roles of the two NPF-binding sites of Arp2/3 complex tested by mutagenesis.
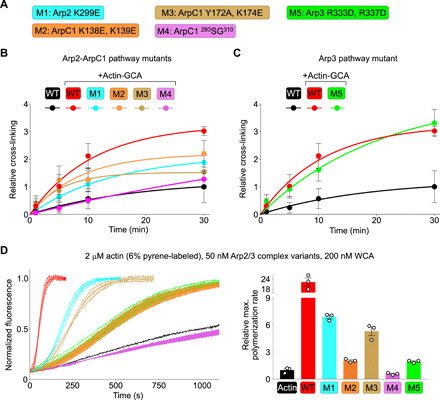
(A) Description of structure-inspired Arp2/3 complex mutants M1 to M5 targeting the two CA binding pathways (color-coded, see also fig. S6). These mutants were generated in the background of the diCys mutant (called WT in this figure for simplicity) used in cross-linking assays and whose activity is indistinguishable from that of tissue-purified bovine Arp2/3 complex (Fig. 1D). (B and C) Cross-linking of Arp2/3 complex (1 μM) mutants targeting the Arp2-ArpC1 and Arp3 pathways (color-coded as indicated) as a function of BMOE treatment time and in the presence of actin-GCA (2.5 μM), expressed relative to the spontaneous cross-linking of diCys Arp2/3 complex in the absence of actin-GCA (black trace). The data were fit to a first-order exponential function. (D) (Left) Time course of actin polymerization of Arp2/3 complex mutants (as indicated) under the conditions listed above the figure. (Right) Relative polymerization rates calculated by determining the first derivative of the curve and normalizing the maximum derivative value to that of actin alone (n = 3).
Using the cross-linking assay (Fig. 1C), all three mutants targeting the Arp2-ArpC1 pathway impaired the ability of Arp2/3 complex to undergo a short-pitch transition, with relative cross-linking at plateau (30 min) reduced by ~30% or more compared to the WT complex in the presence of actin-GCA (Fig. 5B and fig. S7D). Note that in this case, the extent of cross-linking is reported relative to that of the WT complex in the absence of actin-GCA, to reduce inconsistencies resulting from different Arp2/3 complex mutant preparations. The effect of these mutations on the short-pitch transition is consistent with a previous chemical cross-linking-mass spectrometry analysis of the WASP-binding sites on the yeast complex (23).
Mutant M4, targeting the protrusion helix of ArpC1, had the most severe effect in blocking the short-pitch transition (Fig. 5B). The protrusion helix has been implicated in interactions with both the mother filament (4, 5) and NPFs (38). However, the structure shows that this helix is too far apart from the two NPF-binding sites to support an interaction. Like in crystal structures, the protrusion helix and tethering loops are flexible and poorly resolved in the cryo-EM map, but by lowering the density threshold (6 σ) in the cryo-EM map before local contrast transfer function (CTF) correction (fig. S4), this region can be visualized and appears within contact distance to subdomain 4 of Arp2 in the model of the short-pitch conformation (fig. S8A). Thus, the cryo-EM map suggests that the reduced relative cross-linking of mutant M4 is due to destabilization of the short-pitch conformation and not to direct interaction with NPFs. In cosedimentation assays, mutant M4 also displayed reduced binding to F-actin compared to both WT Arp2/3 complex and mutant M5 (fig. S8B), supporting its role in mother filament binding (4, 5).
That mutant M3, intended to disrupt the 498EWE500-binding pocket in ArpC1, severely affects the short-pitch transition is consistent with the crucial role that the conserved tryptophan of the A region of NPFs plays in Arp2/3 complex binding and activation (14, 16, 32). In stark contrast, mutant M5, which was designed to disrupt the 498EWE500-binding pocket in Arp3, had no detectable effect in the short-pitch transition in the cross-linking assay (Fig. 5C). As explained above, mutant M5 appears properly folded (fig. S7, A to C), as further supported by a similar F-actin–binding affinity as WT Arp2/3 complex in cosedimentation assays (fig. S8B). Therefore, we conclude that the NPF-binding pathway along Arp3 is dispensable for the short-pitch transition in the mammalian complex, which is consistent with our observation that actin-GCA binding to a single, high-affinity site along Arp2-ArpC1 is sufficient to induce this transition (Fig. 2B).
All the mutants, including M5 on Arp3, severely impaired the nucleation activity in pyrene-actin polymerization assays and at two different Arp2/3 complex concentrations (Fig. 5D and fig. S9A). Thus, NPF binding along the Arp2-ArpC1 pathway is necessary for both the short-pitch transition and activation. In contrast, NPF binding to Arp3 does not promote the short-pitch transition but is crucial for activation. This result suggests that, contrary to the yeast complex (8), NPF-mediated delivery of actin to Arp3 is a necessary step in mammalian Arp2/3 complex activation.
NPF dimerization restores the activity of the NPF-binding–deficient Arp3 mutant
To further explore the role of actin-NPF binding to Arp3 in activation, we assessed the ability of dimeric WCA to activate mutant M5. In cells, NPFs accumulate at cellular membranes and/or dimerize, and consistently, WCA dimerization substantially increases the rate of Arp2/3 complex nucleation (11, 20, 26). We reasoned that within a dimeric WCA, one WCA arm could bind along the high-affinity site on Arp2-ArpC1 and deliver actin to Arp2, whereas the other WCA arm could deliver actin to Arp3, even if NPF binding to the Arp3 site was disrupted by mutagenesis (Fig. 6A). While we have used here construct WCA, comprising a single WH2 domain, N-WASP has two WH2 domains. Thus, for direct comparison with published work (11, 20), we used in this experiment construct WWCA, comprising the two WH2 domains of N-WASP, and its dimeric variant obtained through the N-terminal addition of glutathione S-transferase (GST-WWCA) (Fig. 6A and fig. S9B). Consistent with previous observations (21), monomeric WWCA and WCA similarly stimulated nucleation by WT Arp2/3 complex (Fig. 6B). However, like WCA, monomeric WWCA did not promote nucleation by mutant M5, suggesting that actin delivery to Arp3 cannot occur from tandem WH2 domains bound along the high-affinity Arp2-ArpC1 pathway. In contrast, dimeric GST-WWCA substantially increased the nucleation activity of WT Arp2/3 complex (Fig. 6B), as previously reported (11, 20). What is more, GST-WWCA partially restored the nucleation activity of mutant M5 (Fig. 6B). That the activity is not restored to the same level as WT Arp2/3 complex demonstrates that NPF dimerization is not sufficient to optimally position an actin subunit at the barbed end of Arp3 and emphasizes the importance of proper NPF binding to the Arp3 site. These results further support the idea that NPF-mediated delivery of actin to both Arps is necessary for activation of actin nucleation by mammalian Arp2/3 complex.
Fig. 6. NPF-mediated actin delivery to both Arps is required for mammalian Arp2/3 complex activation.
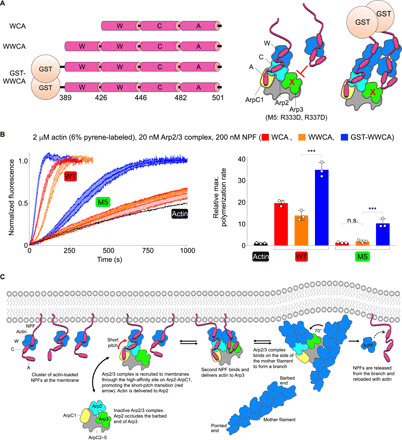
(A) (Left) Design of the dimeric N-WASP WWCA construct (see also fig. S9B). (Right) Schematic illustration of the hypothetical delivery of an actin subunit at the barbed end of Arp3 in mutant M5 (with a disrupted 498EWE500 motif-binding pocket in Arp3) through GST-mediated NPF dimerization, which cannot be accomplished with monomeric NPFs. (B) (Left) Time course of actin polymerization by Arp2/3 complex (WT and M5) in the presence of N-WASP constructs WCA, WWCA, and GST-WWCA. (Right) Relative polymerization rates calculated by determining the first derivative of the curve and normalizing the maximum derivative value to that of actin alone (n = 3). The statistical significance of the measurements was determined using an unpaired two-sided Student’s t test (n.s., P ≥ 0.05; ***P < 0.001). (F) Model of Arp2/3 complex activation and branch formation steps (left to right) by clusters of NPFs at membranes.
DISCUSSION
For many years, Arp2/3 complex was thought to be activated by one NPF. Then, it became increasingly clear that the complex binds two NPFs and that NPF dimerization, which more closely reflects the situation in cells, leads to more efficient activation (11, 20). However, what was unknown, and this study reveals, is that the two NPF-binding sites do not incrementally enhance each other’s activating capacity, but rather both NPFs are required and interdependent since each plays a different role and one cannot activate Arp2/3 complex without the other (Fig. 6C). Furthermore, actin binding to NPFs and its delivery at the barbed end of each of the Arps is a necessary component of the activation reaction in the mammalian complex.
The direct visualization of the two NPF-binding sites in the cryo-EM structure offers conclusive evidence of their existence (Fig. 3) and, together with the ability to express and mutagenize human Arp2/3 complex, has allowed us to separate the distinct roles of each NPF pathway in activation (Fig. 6C). Thus, the biochemical and structural data suggest that the first, high-affinity NPF-binding site engages Arp2-ArpC1 (fig. S3), shifting the equilibrium toward a short-pitch conformation in which Arp2 moves alongside Arp3, and delivering the first actin subunit at the barbed end of Arp2 (Fig. 2B and fig. S3). This first step is required for activation, and accordingly, mutations that disrupt this pathway (Fig. 5) and small molecules that impede the movement of Arp2 (29) inhibit nucleation. The movement of Arp2 also frees the barbed end of Arp3 that in the inactive structure is partially occluded by Arp2 (4). Then, the second NPF, which interacts exclusively with Arp3 (Fig. 3), can bind and deliver the second actin subunit at the barbed end of Arp3. NPF binding to Arp3 seems to require that the C-terminal tail of Arp3 be slightly displaced (Fig. 4C), which could explain the lower affinity of this NPF-binding site (25). Direct, NPF-mediated delivery of an actin subunit at the barbed end of Arp3 is a crucial step for activation of mammalian Arp2/3 complex (Figs. 5 and 6) but not of the budding yeast complex (8). While this may seem like an unexpected result, considering the conflicting evidence with the budding yeast complex, previous work already suggested this possibility. A WCA heterodimer lacking one of the two WH2 domains was previously shown to have very low activating capacity with mammalian (bovine) Arp2/3 complex (11), leading these authors to suggest that actin delivery by both NPFs is necessary for optimal activation. In light of our results, these authors were likely observing compromised actin delivery to Arp3, since the intact WCA in their heterodimer was probably bound to the high-affinity site on Arp2-ArpC1, with >10-fold higher affinity than the Arp3 site with or without actin (25).
Perhaps unexpectedly, the cryo-EM structure of NPF-bound human Arp2/3 complex revealed the inactive conformation of the complex, despite the ability of NPFs to shift the equilibrium toward a short-pitch conformation (Fig. 1E). This observation is nevertheless consistent with the short-pitch conformation being a transient state that relaxes quickly toward the energetically more favorable inactive conformation (12). Moreover, a key factor in promoting the short-pitch conformation, actin (Fig. 1E), was not part of this structure. Activation is a complex multistep reaction with several factors contributing together toward the formation of a branch, including nucleotide, NPFs, actin, and the mother filament (1).
While binding to the mother filament is not directly addressed here, we note that the movement of Arp2 into a short-pitch conformation appears to put it in contact with the protrusion helix in ArpC1 (fig. S8A), which is also implicated in mother filament binding (fig. S8B) (4, 5). Arp2 also contacts the mother filament directly when in the short-pitch position (5). These connections may increase the affinity of the activated complex for the mother filament and stabilize the short-pitch conformation, whereas the binding of actin at the barbed end of the first two actin subunits and nucleotide-dependent conformational changes in the Arps may be some of the factors contributing to the release of the NPFs after nucleation (13).
MATERIALS AND METHODS
Proteins
Skeletal α-actin was purified from rabbit skeletal muscle and Arp2/3 complex was purified from bovine brain as we have previously described (10). The cDNA-encoding mouse N-WASP (UniProt ID: Q91YD9) was obtained from the American Type Culture Collection (ATCC). Construct N-WASP WCA was amplified by polymerase chain reaction and cloned between the Nde I and Eco RI sites of vector pTYB12 [New England BioLabs (NEB)], comprising a chitin-binding domain for affinity purification and an intein domain for self-cleavage after purification. The cDNA-encoding human gelsolin (UniProt ID: P06396) was purchased from ATCC, and gelsolin segment 1 (GS1, residues 52 to 179) was cloned into vector pTYB12 as above. The hybrid construct GCA was obtained by introducing silent mutations in the GS1 and CA genes that introduced Nps I restriction sites for hybridization in the reverse and forward primers, respectively. The ligation product was then cloned into vector pTYB12. The MBP-GCA fusion construct was made by cloning GCA between the Bam HI and Hind III site of vector pMAL-C5X (NEB). This cloning strategy resulted in a 15–amino acid linker (ISHMSMGGRDIVDGS) being inserted between MBP and GCA.
The GCA and MBP-GCA constructs were expressed in BL21(DE3) cells (Invitrogen) grown in Terrific Broth medium at 37°C until the optical density at 600 nm (OD600) reached a value of 1.5 to 2. Expression was induced with 0.5 mM isopropylthio-β-d-thiogalactoside and carried out for 16 hours at 19°C. Cells were harvested by centrifugation, resuspended in 20 mM Hepes (pH 7.5), 500 mM NaCl, and 1 mM phenylmethanesulfonyl fluoride (PMSF), and lysed using a microfluidizer (Microfluidics). GCA and MBP-GCA were first purified through chitin affinity (NEB) and amylose affinity columns (NEB), followed by purification on a Superose 6 size exclusion column (GE Healthcare). Complexes of GCA and MBP-GCA with actin were made in the presence of a 1.1 molar excess of actin in 20 mM Hepes (pH 7.5) and 150 mM NaCl at room temperature (RT) for 1 hour. Polymerized actin was removed by centrifugation (278,000g for 30 min at 4°C) in a Beckman TLA-100 ultracentrifuge. The complexes were then purified on a Superose 6 size exclusion column.
All the subunits of Arp2/3 complex were coexpressed in Sf9 cells using a single baculovirus-expression vector (biGBac), which increases expression yields by reducing cell stress from infection with multiple viruses and from the substoichiometric expression of complex subunits. The set of biGBac cloning vectors were a gift from B. Schulman (Max Planck Institute of Biochemistry, Martinsried, Germany). The Arp2/3 complex subunits were cloned into the pBIG2abc vector following the Schulman’s laboratory published protocol (28). The genes of Arp2/3 complex subunits were codon optimized for expression in Sf9 cells (Biomatik). A C-terminal FLAG tag was added to subunit ArpC3 on the basis of its solvent accessibility in the inactive crystal structure (4). All the genes were individually cloned between the Bam HI and Hind III sites of vector pLib. Gibson assembly was used to insert subgroups of Arp2/3 complex subunits into three different vectors (pBIG1a to pBIG1c) using CasI, CasII, and CasIII primers: Arp2 and Arp3 (vector pBIG1a), ArpC1 to ArpC3 (vector pBIG1b), and ArpC4 and ArpC5 (vector pBIG1c). These three gene cassettes were then released with Pme I digestion and Gibson-assembled into a single vector (pBIG2abc). The final product was sequence-verified using Arp2/3 complex gene-specific primers. Arp2/3 complex mutants were generated before insertion into vectors pBIG1a to pBIG1c, using the Q5 site-directed mutagenesis kit (NEB). Bacmids were generated via a translocation reaction in DH10Bac competent cells (Thermo Fisher Scientific). Sf9 cells (Thermo Fisher Scientific) were infected at log phase (3 × 106 to 7 × 106 cells ml−1) with high titer virus and maintained in ESF 921 insect cell culture medium (Expression Systems). Cells were lysed in 20 mM Hepes (pH 7.5), 200 mM NaCl, 1% Triton X-100, 2 mM PMSF, and cOmplete EDTA-free protease inhibitor cocktail (MilliporeSigma). The lysate was loaded into an anti-FLAG antibody resin (GenScript), washed with 20 mM Hepes (pH 7.5), 200 mM NaCl, and eluted with 0.3 mg ml−1 3×FLAG peptide (Biomatik). Arp2/3 complex was concentrated using a 100-kDa Amicon Ultra centrifugal filter (MilliporeSigma) and loaded onto a Superose 6 size exclusion column in Arp-buffer [20 mM Hepes (pH 7.5), 100 mM, NaCl, and 1 mM dithiothreitol (DTT)]. Arp2/3 complex fractions were collected, pooled, and concentrated to ~10 to 20 μM based on the UV absorbance at 280 nm or the Bradford protein assay (Bio-Rad). Arp2/3 complex was flash-frozen in liquid nitrogen and stored at −80°C.
Glycerol gradient assembly of Arp2/3 complex with bound NPFs
Glycerol gradient fixation was performed similarly to Kastner et al. (39). Arp2/3 complex (5 μM) was mixed with rabbit skeletal α-actin (15 μM) and GCA (20 μM). The same molar ratio was used to assemble Arp2/3 complex with MBP-GCA and actin. Proteins were incubated on ice for 30 min before ultracentrifugation. Samples in Arp buffer supplemented with 0.4 mM MgCl2 and 0.2 mM adenosine 5′-triphosphate (ATP) were sedimented at 40,000 rpm for ~16 hours in a 5 to 30% glycerol gradient at 4°C using a Beckman SW 60 Ti rotor. Glycerol gradients were prepared using a Gradient Master device (BioComp Instruments). When cross-linking was used for EM analysis, 0.125% (v/v) glutaraldehyde was added to the 30% glycerol solution before gradient preparation. Samples were then fractionated with a Piston Gradient Fractionator (BioComp Instruments). Cross-linking reactions were quenched by the addition of glycine-HCl buffer (pH 7.5) to a final concentration of 40 mM. Before EM analysis, Arp2/3 complex was dialyzed into Arp buffer supplemented with 0.4 mM MgCl2 and 0.2 mM ATP to remove glycerol.
EM and image analysis of negatively stained Arp2/3 complex
Bovine Arp2/3 complex (alone or with bound actin-GCA or actin-MBP-GCA) samples were adsorbed onto negatively glow-discharged carbon film grids (CF200-Cu mesh, Electron Microscopy Sciences) at 15 μg ml−1. Samples were incubated for 60 s, washed with 2% uranyl formate, blotted, and dried. Micrographs were collected at ×42,000 magnification (image sampling of 2.6 Å pixel−1) on an FEI Tecnai T12 microscope operated at 120 kV and equipped with an Orius SC1000 CCD camera, with a defocus range of 0.6 to 1.5 μm and operated in low-dose mode (average dose of ~20 e− Å−2).
Images were CTF-corrected using CTFFIND4 (40). Particles were boxed semiautomatically using the e2boxer subroutine of the EMAN2 software suite (41). Boxes were extracted and normalized with Relion v2.1 (42), using a box size of 150 pixels. 2D class averages of groups of ~100 particles were generated using 2D classification in Relion. The Arp2/3 complex alone, Arp2/3 complex with actin-GCA, and Arp2/3 complex with actin-MBP-GCA datasets initially consisted of 5764, 5727, and 5980 particles, respectively.
Models of actin-NPFs bound along the Arp2-ArpC1 and Arp3 pathways were generated (fig. S3) and converted to density using EMAN2’s subroutine e2pdb2mrc. The resulting maps were back-projected using e2project3d at Euler angle increments of 10°, with a box size of 150 pixels and a Gaussian low-pass filter of 2 nm. The EMageFit protocol from the Integrative Modeling Platform suite (43) was used to determine the projections that more closely represented the 2D class averages. The class averages were assigned to the best-fitting projection on the basis of correlation coefficients calculated with the sximgstat subroutine of the program SPARX (44).
Cryo-EM sample preparation and data collection
To prepare cryo-EM grids, 1 μM Arp2/3 complex was incubated with 25 μM WCA at RT for 30 min in KMEI buffer [10 mM imidazole (pH 7.0), 1 mM MgCl2, 1 mM EGTA, 50 mM KCl, 0.2 mM ATP, and 0.5 mM TCEP]. The sample was buffer-exchanged to Arp buffer supplemented with 0.4 mM MgCl2 and 0.2 mM ATP and concentrated to 5 mg ml−1 using a 100-kDa Amicon Ultra centrifugal filter. Before plunge freezing, NP-40 was added to a final concentration of 0.0025% (v/v) to broaden the particle orientation distribution. The sample (2 μl) was then applied to glow-discharged (1 min, easiGlow, Pelco) R1.2/1.3 200-mesh or R2/2 300-mesh Quantifoil holey carbon grids (Electron Microscopy Sciences). The grids were subsequently blotted for 2 s using Whatman Grade 41 filter paper (Sigma-Aldrich) and flash-frozen in liquid ethane with a Leica EM CPC manual plunger (Leica Microsystems). EM grids were prepared in batches and the freezing conditions were optimized by screening on an FEI TF20 microscope operating at 200 kV and equipped with an FEI Falcon III direct electron detection camera.
Cryo-EM specimens were imaged at the National Cryo-Electron Microscopy Facility (Frederick National Laboratory) using an FEI Titan Krios transmission electron microscope operating at 300 kV and equipped with a K2 Summit direct electron detector (Gatan), with an energy quantum filter (Gatan) and at a nominal magnification of ×165,000 in super-resolution mode (pixel size of 0.418 Å). A total of 5004 images were taken during two different data collections: 1648 images with R1.2/1.3 200-mesh grid and 3356 images with R2/2 300-mesh grid. The exposure time was 7 s, divided into 40 frames, at a dose rate of 4.02 e− s−1 pixel−1, resulting in a nominal dose of 40 e− Å−2.
Image processing and 3D reconstruction
The cryo-EM data were processed and maps were calculated with cryoSPARC v2.12.4 (fig. S4) (45). Datasets were motion-corrected separately using cryoSPARC and patch motion–corrected and combined for CTF correction with CTFFIND4 (40). Blob-based picking with cryoSPARC was used to produce a small subset of particles for the generation of 2D references. A total of 627,163 particles were picked by template-based picking, and two rounds of reference-free 2D classification were performed to remove particles that lacked clear features, resulting in a subset of ~70,000 particles. Ab initio reconstruction with cryoSPARC, followed by homogenous refinement, and nonuniform refinement of 67,418 particles resulted in a 4.1-Å-resolution map with clear density for both NPFs.
The 4.1-Å-resolution map was used as a reference for a single round of heterogeneous classification on the original 627,163 particles, from which two classes totaling 310,587 particles were selected (map 4 and map 5, fig. S4). These particles were then subjected to four rounds of heterogeneous refinement, after which a single class of 123,198 particles with clear density for the CA region of both NPFs was selected. This class was subjected to homogeneous refinement, local motion correction, and nonuniform refinement in cryoSPARC. The resulting 3.9-Å-resolution map was then further improved by local CTF refinement and nonuniform refinement, resulting in a 3.8-Å-resolution map. The 3.8-Å map had substantially improved density for the two CA regions and well-defined side chains in the core region of Arp2/3 complex. The local resolution of the map (Fig. 3A) was determined using Fourier shell correlation (FSC) (cutoff of 0.5) with blocres (46).
Model building and refinement
The resulting 3.8-Å map was fit with the crystal structure of GMF-bound bovine Arp2/3 complex (PDB ID: 4JD2) in Coot (47). GMF was removed and the Arp2/3 complex subunits were fit individually as rigid bodies. The sequence was substituted for that of human Arp2/3 complex in Coot (47). Some side chains were remodeled to remove clashes created during rigid-body fitting and guided by their densities in the map. Some side chains in the newly built regions of the structure (not present in the crystal structure) were modeled as alanine if density was not available for modeling.
The densities corresponding to the two CA regions were visualized by subtracting the fit model from the map at 6.0 Å resolution using the EMAN2 subroutine e2pdb2mrc (41) and UCSF Chimera (Fig. 3, C and D, and movie S1). The C helix of both NPFs was modeled on the basis of the NMR structure of the C region bound to the GTPase-binding domain of human WASP (PDB ID: 1EJ5) and was fit such that its hydrophobic side faced the hydrophobic clefts of the Arps and guided by the densities of side chains in the 3.8-Å-resolution cryo-EM map for azimuthal fitting. The 498EWE500 motif of the A region bound in similar pockets of Arp3 and ArpC1 and was modeled according to a crystal structure of Arp2/3 complex that shows this motif bound to Arp3 (PDB ID: 3RSE). The D/E loops between these two regions were less well defined in the density and are not included in the deposited model. Structural models were visualized and figures were prepared using Coot (47), Chimera (48), and PyMOL (Schrödinger, LLC).
Short-pitch cross-linking assay
Purified Arp2/3 complex (±NPFs and actin) were incubated for 10 min at RT in KMEI buffer at the concentrations listed in the figures. BMOE (Thermo Fisher Scientific) was added to a final concentration of 16 μM. BMOE was prepared fresh in dimethyl sulfoxide before every reaction. Reactions were performed at 21°C and quenched at the specified time points indicated in the figures with the addition of fresh beta-mercaptoethanol to a final concentration of 50 mM. For Western blot analysis, 2× SDS-loading buffer (LI-COR Biosciences) was added to quench the reactions. Samples were loaded onto 4 to 15% SDS-PAGE gradient gels (Bio-Rad), transferred onto nitrocellulose membranes (Criterion Blotter, Bio-Rad), blocked with Odyssey Blocking Buffer (LI-COR Biosciences), and probed with anti-Arp2 (1:1000 dilution, Cell Signaling Technologies) and anti-Arp3 (1:200 dilution Santa Cruz Biotechnology) antibodies. Secondary IRDye 680RD goat anti-rabbit and IRDye 800CW goat anti-mouse antibodies (LI-COR Biosciences) were used at dilutions of 1:15,000 to 1:40,000. Membranes were imaged on an Odyssey CLx Near-Infrared Fluorescence Imaging System (LI-COR Biosciences). Densitometric analysis of the bands with Image Studio software (LI-COR Biosciences) was used to determine the fraction of cross-linked product. Mean and SD values were calculated from three or more independent reactions. The data were fit to a first-order exponential curve to determine the kinetics of the reaction.
Actin polymerization assays
Pyrene-actin polymerization assays were carried out using a Cary Eclipse fluorescence spectrophotometer (Varian) as previously described (10). Before data acquisition, 2 μM Mg-ATP-actin (6% pyrene-labeled) was mixed with the indicated concentrations of Arp2/3 complex and NPFs in F-buffer [10 mM Tris (pH 7.5), 1 mM MgCl2, 50 mM KCl, 1 mM EGTA, 0.1 mM NaN3, and 0.2 mM ATP]. Relative polymerization rates were calculated by determining the first derivative of each dataset with the program Prism v7.0 (GraphPad Software) and normalizing the maximum derivative value to that of actin alone. Mean and SD values were calculated using at least three measurements. Graphs and Student’s t test statistical analysis were performed using Prism v7.0.
Actin filament cosedimentation assays
Cosedimentation assays were performed as previously described (49). Briefly, actin (20 μM) in G-buffer [2 mM Tris (pH 7.4), 0.2 mM CaCl2, 0.2 mM ATP, 0.5 mM DTT, and 1 mM NaN3] was polymerized with the addition of 100 mM KCl, 2 mM MgCl2, and 1 mM EGTA for 60 min at RT. Before incubation with F-actin, Arp2/3 complex samples were centrifuged for 10 min at 278,000g using a Beckman TLA-100 ultracentrifuge to remove potential aggregates. Arp2/3 complex (1 μM) was mixed with various concentrations of F-actin (0 to 12 μM) in a 50-μl volume of Arp buffer. The mixtures were then incubated overnight at 4°C, followed by 100,000g centrifugation for 30 min. Pellets were washed in 200 μl of Arp buffer. Equal volumes of supernatant and resuspended pellet fractions were analyzed by SDS-PAGE. Gels were imaged on a G:Box gel scanner (Syngene International) and densitometric analysis was performed using ImageJ (50). The percentage of Arp2/3 complex bound to F-actin in the pellet was determined from the relative intensities of the ArpC2 band in the supernatant and pelleted fractions. Mean and SD values were determined from three independent experiments. The observed dissociation constant (Kobs) was calculated by global fitting of the data with Prism v7.0, using a one-site binding model. Graphs and Student’s t test statistical analysis were performed using Prism v7.0.
Statistical methods
The statistical significance of the measurements was determined using an unpaired two-way Student’s t test in Prism v7.0. Further details are reported in figure legends.
Supplementary Material
Acknowledgments
We thank M. Ostap for insightful discussions. Funding: This work was supported by NIH grants R01 GM073791 and R01 MH087950 to R.D., NIH grant GM123233 to K.M., NIH training grant T32-GM008275 to A.Z. and T.V.E., and NIH grant F31-HL146077 to A.Z. and by the National Cancer Institute’s National Cryo-EM Facility under contract HSSN261200800001E. Computational resources were supported by NIH Project Grant S10OD023592. Author contributions: R.D. conceived and directed the project. A.Z., T.V.E., M.B., G.R., K.M., and R.D. designed and performed experiments and analyzed the data. A.Z. and R.D. wrote the paper and prepared the figures for publication. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors. The cryo–electron density map was deposited in the Electron Microscopy Data Bank (EMDB-20770). The atomic coordinates were deposited in the PDB (accession code: 6UHC).
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/23/eaaz7651/DC1
REFERENCES AND NOTES
- 1.Pollard T. D., Regulation of actin filament assembly by Arp2/3 complex and formins. Annu. Rev. Biophys. Biomol. Struct. 36, 451–477 (2007). [DOI] [PubMed] [Google Scholar]
- 2.Dominguez R., The WH2 domain and actin nucleation: Necessary but insufficient. Trends Biochem. Sci. 41, 478–490 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Molinie N., Gautreau A., The Arp2/3 regulatory system and its deregulation in cancer. Physiol. Rev. 98, 215–238 (2018). [DOI] [PubMed] [Google Scholar]
- 4.Robinson R. C., Turbedsky K., Kaiser D. A., Marchand J.-B., Higgs H. N., Choe S., Pollard T. D., Crystal structure of Arp2/3 complex. Science 294, 1679–1684 (2001). [DOI] [PubMed] [Google Scholar]
- 5.Rouiller I., Xu X. P., Amann K. J., Egile C., Nickell S., Nicastro D., Li R., Pollard T. D., Volkmann N., Hanein D., The structural basis of actin filament branching by the Arp2/3 complex. J. Cell Biol. 180, 887–895 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goley E. D., Rodenbusch S. E., Martin A. C., Welch M. D., Critical conformational changes in the Arp2/3 complex are induced by nucleotide and nucleation promoting factor. Mol. Cell 16, 269–279 (2004). [DOI] [PubMed] [Google Scholar]
- 7.Rodal A. A., Sokolova O., Robins D. B., Daugherty K. M., Hippenmeyer S., Riezman H., Grigorieff N., Goode B. L., Conformational changes in the Arp2/3 complex leading to actin nucleation. Nat. Struct. Mol. Biol. 12, 26–31 (2005). [DOI] [PubMed] [Google Scholar]
- 8.Rodnick-Smith M., Luan Q., Liu S.-L., Nolen B. J., Role and structural mechanism of WASP-triggered conformational changes in branched actin filament nucleation by Arp2/3 complex. Proc. Natl. Acad. Sci. U.S.A. 113, E3834–E3843 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Espinoza-Sanchez S., Metskas L. A., Chou S. Z., Rhoades E., Pollard T. D., Conformational changes in Arp2/3 complex induced by ATP, WASp-VCA, and actin filaments. Proc. Natl. Acad. Sci. U.S.A. 115, E8642–E8651 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boczkowska M., Rebowski G., Petoukhov M. V., Hayes D. B., Svergun D. I., Dominguez R., X-ray scattering study of activated Arp2/3 complex with bound actin-WCA. Structure 16, 695–704 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Padrick S. B., Doolittle L. K., Brautigam C. A., King D. S., Rosen M. K., Arp2/3 complex is bound and activated by two WASP proteins. Proc. Natl. Acad. Sci. U.S.A. 108, E472–E479 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Smith B. A., Daugherty-Clarke K., Goode B. L., Gelles J., Pathway of actin filament branch formation by Arp2/3 complex revealed by single-molecule imaging. Proc. Natl. Acad. Sci. U.S.A. 110, 1285–1290 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Smith B. A., Padrick S. B., Doolittle L. K., Daugherty-Clarke K., Correa I. R. Jr., Xu M.-Q., Goode B. L., Rosen M. K., Gelles J., Three-color single molecule imaging shows WASP detachment from Arp2/3 complex triggers actin filament branch formation. eLife 2, e01008 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marchand J.-B., Kaiser D. A., Pollard T. D., Higgs H. N., Interaction of WASP/Scar proteins with actin and vertebrate Arp2/3 complex. Nat. Cell Biol. 3, 76–82 (2001). [DOI] [PubMed] [Google Scholar]
- 15.Miki H., Takenawa T., Direct binding of the verprolin-homology domain in N-WASP to actin is essential for cytoskeletal reorganization. Biochem. Biophys. Res. Commun. 243, 73–78 (1998). [DOI] [PubMed] [Google Scholar]
- 16.Panchal S. C., Kaiser D. A., Torres E., Pollard T. D., Rosen M. K., A conserved amphipathic helix in WASP/Scar proteins is essential for activation of Arp2/3 complex. Nat. Struct. Biol. 10, 591–598 (2003). [DOI] [PubMed] [Google Scholar]
- 17.Chereau D., Kerff F., Graceffa P., Grabarek Z., Langsetmo K., Dominguez R., Actin-bound structures of Wiskott–Aldrich syndrome protein (WASP)-homology domain 2 and the implications for filament assembly. Proc. Natl. Acad. Sci. U.S.A. 102, 16644–16649 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Higgs H. N., Blanchoin L., Pollard T. D., Influence of the C terminus of Wiskott-Aldrich syndrome protein (WASp) and the Arp2/3 complex on actin polymerization. Biochemistry 38, 15212–15222 (1999). [DOI] [PubMed] [Google Scholar]
- 19.Kast D. J., Zajac A. L., Holzbaur E. L. F., Ostap E. M., Dominguez R., WHAMM directs the Arp2/3 complex to the ER for autophagosome biogenesis through an actin comet tail mechanism. Curr. Biol. 25, 1791–1797 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Padrick S. B., Cheng H.-C., Ismail A. M., Panchal S. C., Doolittle L. K., Kim S., Skehan B. M., Umetani J., Brautigam C. A., Leong J. M., Rosen M. K., Hierarchical regulation of WASP/WAVE proteins. Mol. Cell 32, 426–438 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zalevsky J., Lempert L., Kranitz H., Mullins R. D., Different WASP family proteins stimulate different Arp2/3 complex-dependent actin-nucleating activities. Curr. Biol. 11, 1903–1913 (2001). [DOI] [PubMed] [Google Scholar]
- 22.Weaver A. M., Heuser J. E., Karginov A. V., Lee W.-l., Parsons J. T., Cooper J. A., Interaction of cortactin and N-WASp with Arp2/3 complex. Curr. Biol. 12, 1270–1278 (2002). [DOI] [PubMed] [Google Scholar]
- 23.Luan Q., Zelter A., MacCoss M. J., Davis T. N., Nolen B. J., Identification of Wiskott-Aldrich syndrome protein (WASP) binding sites on the branched actin filament nucleator Arp2/3 complex. Proc. Natl. Acad. Sci. U.S.A. 115, E1409–E1418 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ti S.-C., Jurgenson C. T., Nolen B. J., Pollard T. D., Structural and biochemical characterization of two binding sites for nucleation-promoting factor WASp-VCA on Arp2/3 complex. Proc. Natl. Acad. Sci. U.S.A. 108, E463–E471 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Boczkowska M., Rebowski G., Kast D. J., Dominguez R., Structural analysis of the transitional state of Arp2/3 complex activation by two actin-bound WCAs. Nat. Commun. 5, 3308 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Higgs H. N., Pollard T. D., Activation by Cdc42 and PIP(2) of Wiskott-Aldrich syndrome protein (WASp) stimulates actin nucleation by Arp2/3 complex. J. Cell Biol. 150, 1311–1320 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wen K.-K., Rubenstein P. A., Acceleration of yeast actin polymerization by yeast Arp2/3 complex does not require an Arp2/3-activating protein. J. Biol. Chem. 280, 24168–24174 (2005). [DOI] [PubMed] [Google Scholar]
- 28.Weissmann F., Petzold G., VanderLinden R., Huis In 't Veld P. J., Brown N. G., Lampert F., Westermann S., Stark H., Schulman B. A., Peters J.-M., biGBac enables rapid gene assembly for the expression of large multisubunit protein complexes. Proc. Natl. Acad. Sci. U.S.A. 113, E2564–E2569 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hetrick B., Han M. S., Helgeson L. A., Nolen B. J., Small molecules CK-666 and CK-869 inhibit actin-related protein 2/3 complex by blocking an activating conformational change. Chem. Biol. 20, 701–712 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee S. H., Kerff F., Chereau D., Ferron F., Klug A., Dominguez R., Structural basis for the actin-binding function of missing-in-metastasis. Structure 15, 145–155 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Way M., Pope B., Gooch J., Hawkins M., Weeds A. G., Identification of a region in segment 1 of gelsolin critical for actin binding. EMBO J. 9, 4103–4109 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pan F., Egile C., Lipkin T., Li R., ARPC1/Arc40 mediates the interaction of the actin-related protein 2 and 3 complex with Wiskott-Aldrich syndrome protein family activators. J. Biol. Chem. 279, 54629–54636 (2004). [DOI] [PubMed] [Google Scholar]
- 33.Nolen B. J., Pollard T. D., Structure and biochemical properties of fission yeast Arp2/3 complex lacking the Arp2 subunit. J. Biol. Chem. 283, 26490–26498 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim A. S., Kakalis L. T., Abdul-Manan N., Liu G. A., Rosen M. K., Autoinhibition and activation mechanisms of the Wiskott-Aldrich syndrome protein. Nature 404, 151–158 (2000). [DOI] [PubMed] [Google Scholar]
- 35.Dominguez R., Holmes K. C., Actin structure and function. Annu. Rev. Biophys. 40, 169–186 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ferron F., Rebowski G., Lee S. H., Dominguez R., Structural basis for the recruitment of profilin-actin complexes during filament elongation by Ena/VASP. EMBO J. 26, 4597–4606 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Abella J. V. G., Galloni C., Pernier J., Barry D. J., Kjaer S., Carlier M. F., Way M., Isoform diversity in the Arp2/3 complex determines actin filament dynamics. Nat. Cell Biol. 18, 76–86 (2016). [DOI] [PubMed] [Google Scholar]
- 38.Balcer H. I., Daugherty-Clarke K., Goode B. L., The p40/ARPC1 subunit of Arp2/3 complex performs multiple essential roles in WASp-regulated actin nucleation. J. Biol. Chem. 285, 8481–8491 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kastner B., Fischer N., Golas M. M., Sander B., Dube P., Boehringer D., Hartmuth K., Deckert J., Hauer F., Wolf E., Uchtenhagen H., Urlaub H., Herzog F., Peters J. M., Poerschke D., Luhrmann R., Stark H., GraFix: Sample preparation for single-particle electron cryomicroscopy. Nat. Methods 5, 53–55 (2008). [DOI] [PubMed] [Google Scholar]
- 40.Rohou A., Grigorieff N., CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J. Struct. Biol. 192, 216–221 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tang G., Peng L., Baldwin P. R., Mann D. S., Jiang W., Rees I., Ludtke S. J., EMAN2: An extensible image processing suite for electron microscopy. J. Struct. Biol. 157, 38–46 (2007). [DOI] [PubMed] [Google Scholar]
- 42.Kimanius D., Forsberg B. O., Scheres S. H. W., Lindahl E., Accelerated cryo-EM structure determination with parallelisation using GPUs in RELION-2. eLife 5, e18722 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Russel D., Lasker K., Webb B., Velázquez-Muriel J., Tjioe E., Schneidman-Duhovny D., Peterson B., Sali A., Putting the pieces together: Integrative modeling platform software for structure determination of macromolecular assemblies. PLoS Biol. 10, e1001244 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hohn M., Tang G., Goodyear G., Baldwin P. R., Huang Z., Penczek P. A., Yang C., Glaeser R. M., Adams P. D., Ludtke S. J., SPARX, a new environment for cryo-EM image processing. J. Struct. Biol. 157, 47–55 (2007). [DOI] [PubMed] [Google Scholar]
- 45.Punjani A., Rubinstein J. L., Fleet D. J., Brubaker M. A., CryoSPARC: Algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017). [DOI] [PubMed] [Google Scholar]
- 46.Cardone G., Heymann J. B., Steven A. C., One number does not fit all: Mapping local variations in resolution in cryo-EM reconstructions. J. Struct. Biol. 184, 226–236 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Emsley P., Lohkamp B., Scott W. G., Cowtan K., Features and development of Coot. Acta Crystallogr. D 66, 486–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., Ferrin T. E., UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004). [DOI] [PubMed] [Google Scholar]
- 49.Madasu Y., Yang C., Boczkowska M., Bethoney K. A., Zwolak A., Rebowski G., Svitkina T., Dominguez R., PICK1 is implicated in organelle motility in an Arp2/3 complex-independent manner. Mol. Biol. Cell 26, 1308–1322 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B., Tinevez J.-Y., White D. J., Hartenstein V., Eliceiri K., Tomancak P., Cardona A., Fiji: An open-source platform for biological-image analysis. Nat. Methods 9, 676–682 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/23/eaaz7651/DC1