

Abstract
Traumatic brain injury (TBI) is a major health problem in the United States, which affects about 1.7 million people each year. Glial cells, T-cells and mast cells perform specific protective functions in different regions of the brain for the recovery of cognitive and motor functions after central nervous system (CNS) injuries including TBI. Chronic neuroinflammatory responses resulting in neuronal death and the accompanying stress following brain injury predisposes or accelerates the onset and progression of Alzheimer’s disease (AD) in high-risk individuals. About 5.7 million Americans are currently living with AD. Immediately following brain injury, mast cells respond by releasing prestored and preactivated mediators and recruit immune cells to the CNS. Blood-brain barrier (BBB), tight junction and adherens junction proteins, neurovascular and gliovascular microstructural rearrangements and dysfunction associated with increased trafficking of inflammatory mediators and inflammatory cells from the periphery across the BBB leads to increase in the chronic neuroinflammatory reactions following brain injury. In this review, we advance the hypothesis that neuroinflammatory responses resulting from mast cell activation along with the accompanying risk factors such as age, gender, food habits, emotional status, stress, allergic tendency, chronic inflammatory diseases, and certain drugs can accelerate brain injury-associated neuroinflammation, neurodegeneration and AD pathogenesis.
Keywords: Alzheimer’s disease, blood brain barrier, brain injury, neuroinflammation, traumatic brain injury
Introduction
Innate immune cells including T-cells, macrophages, natural killer cells (NK cells) and mast cells seek and destroy foreign organisms and toxic substances by constant surveillance in all organs. During this process, they also attack some normal cells, cause death of these cells in the tissues and induce inflammation. Subsequent to these events, immune cells such as T-cells and mast cells play an important role in tissue repair, scar formation and generation of new cells to bring back the normal functions of the tissues or organs in the body (Filiano and others 2017). However, the magnitude of the immune response and tissue repair after an injury in the central nervous system (CNS) is naturally kept restricted (Negi and Das 2018). This is due to the limited regenerative capacity of neurons in the brain. CNS is also regularly under surveillance by the local and circulating immune cells in the cerebrospinal fluid (CSF) that entered the brain through choroid plexus and maintains brain plasticity in healthy condition and after an injury. Mild activation of glia provide neuroprotective effects. However, severe activation of neuroinflammatory pathways induce neuronal death (Nutma and others 2019). Endothelial cells and perivascular macrophages also contribute to the neuroinflammation.
Traumatic brain injury (TBI) can be classified into mild, moderate and severe types. Traumatic and non-traumatic brain injuries (non-TBI) are acquired and occur after birth due to tumors, stroke, seizure, infections, toxins, hypoxia and metabolic disorders. TBI may occur due to sports activities, vehicle accidents, falls, assaults, military activities, blasts, gunshots (Brain Injury Association of America, Vienna, VA). Combat military persons are exposed to explosive devices and blasts while civilians are targeted by terrorist blast attacks. It has been reported that 19.5 % of troops deployed in Operation Iraqi Freedom (OIF) and Operation Enduring Freedom (OEF) were subject to blast TBI (bTBI) (Bauer and others 2015). About 50% of patients with severe TBI usually die, while the remaining patients show severe short and long-term neurological complications. Football players subjected to repeated TBI show chronic traumatic encephalopathy (CTE) (Omalu and others 2005). CTE has been reported in former sports and ex-military personnel (Falcon and others 2019). CTE shows the presence of elevated hyperphosphorylated tau in the neurons, neurofibrillary tangles, astrocytes and cell processes around the blood vessels (Falcon and others 2019). It may take years to decades to show the signs and symptoms of CTE after the head injury (Bailes and others 2019). However, neuropathology of CTE is not yet clearly defined (Smith and others 2019). Neurotrophins such as brain-derived neurotrophic factor (BDNF), nerve growth factor (NGF), neurotrophin-3 (NT-3), and NT-4 provide neuroplasticity in protecting neurons from secondary injury in the injured brains. Brain injuries or TBI induces immune responses to protect the brain from any further injury. However, over and sustained immune response leads to neuroinflammation-mediated neurodegeneration leading to Alzheimer’s disease (AD) (Kempuraj and others 2016; McManus and Heneka 2017). Neuroinflammation refers to the neuroinflammatory response that originates in the brain or the spinal cord following an injury or infection associated with increased accumulation and activation of microglia and astrocytes. Activated glia and peripheral immune cells recruited into the brain after brain injury cross talk to induce neuroinflammatory responses that accelerates the pathogenesis of AD (Puntambekar and others 2018).
AD is a progressive neurodegenerative disease with clinical complication of dementia, eating problems, infections, and poor quality of life. Currently, about 5.7 million Americans are living with AD. About 44 million people were affected by AD and dementia in 2016 worldwide (Laurent and others 2018). The prevalence of AD will rise as life expectancy constantly increases worldwide. The prevalence of AD among people aged 65 to 84 years is about 10 to 19% and over 47% for people above 85 years. Older African Americans have nearly double the risk to have AD and dementia as compared to older whites (Alzheimer’s Association, Chicago, IL). The brain atrophy starts in temporal lobe and progresses to the frontal and parietal regions before extending to the occipital lobes. Presence of amyloid-beta (Aβ) plaques and hyper-phosphorylated tau containing neurofibrillary tangles (NFTs) in the brain are the pathological hallmarks of AD. AD patients show initial learning and memory disorders followed by language and global cognitive disorders. AD is the most common dementia that is linked with TBI. AD specific therapies are unavailable as the disease mechanisms are not clearly understood (Umar and Hoda 2018). The immune mechanisms in brain injury-associated AD pathogenesis are not yet known. In the present hypothesis paper, we advance the hypothesis that neuroinflammatory response and factors such as age, gender, emotional status, stress, allergic tendency, chronic inflammatory diseases, certain food and drugs can affect brain injury-induced neuroinflammation that mediates AD pathogenesis in the high-risk individuals.
Brain Region Specific Immune and Inflammatory Response
Normal innate and adaptive immune networks are essential for the development and functions of the CNS. The immune system is actively involved in physio pathological states of the brain and during the recovery after an injury (Filiano and others 2017). T-cells are essential in learning behavior and in stressful conditions. T-cells and mast cells constantly patrol the meningeal spaces and at the blood-brain-barrier (BBB) structures to prevent the entry of foreign organisms or antigens into the brain. Brain parenchymal cells are protected from the peripheral system by the tight junction and selective BBB permeability. The subarachnoid space, CSF and vasculature have leukocytes, macrophages, T-cells and dendritic cells but the entry of these cells into the brain parenchyma is prevented by the BBB. There are no lymphocytes in the naïve CNS parenchyma but T-cells that reside in the meninges execute its social, behavioral and cognition functions by releasing specific cytokines such as interferon-gamma (IFN-γ), neurotransmitters, neuromodulators and growth factors. These T-cells enter the meninges through blood vessels in leptomeninges. Naïve T-cells are recruited into the brain by chemoattractant signals from activated neurons, glia and endothelial cells. Brain injury causes influx of T-cells from the blood vessels by chemokines and upregulation of adhesion molecules in the endothelial cells and integrins on T-cells (Filiano and others 2017). Specific type of brain injury also influences differently on T-cell number, its recruitment, proliferation and responses.
Microglia are the innate immune cells in the brain. During normal conditions, microglia are in resting state (ramified) with small body and elongated thin processes that moves around. Microglial processes are the fastest moving structures in the brain. Resting microglia move their processes without moving the cell body. Activation changes the shape of microglia into amoeboid form to facilitate movement of microglia along with expression of several activation specific receptors, cytokines and chemokines. The injured site is surrounded by microglial processes within an hour in the brain. These processes can elongate and retract to scan its territory. Brain structures are completely scanned by microglial processes once every several hours. Microglia migrate and perform phagocytosis and present antigen in the normal brain. Microglia can remove the entire cells or cellular structures. Microglia recognize the cells that will undergo programmed cell death; once recognized, microglia migrate to the site before or during the programmed cell death for removal (Wolf and others 2017). Microglia also influence neuronal precursor cell proliferation, neuronal survival, synaptic plasticity and cognition. Microglia express receptor CX3CR1 for fractalkine (CX3CL1), a chemokine released by neurons; pattern recognition receptors (PRRs) that detect microbes and pathogen-associated molecular patterns (PAMS). Microglia show gender variations in rodents. Astrocytes are more ramified and has more activated phenotype of microglia in preoptic area in male (Wolf and others 2017). Microglia and neurons communicate bidirectional through CX3CL1-CX3CR1, CD200- CD200R, C3-CR3 and DAP12 signaling pathways for the formation and maintenance of neuronal circuits. It is clear that immune system plays an important role in the restoration of normal tissue homeostasis after the brain injury (Table 1 and Fig. 1).
Table 1.
Normal neuroprotective immunosurveillance by innate immune cells in the normal healthy brain
|
Normal immunosurveillance in the brain Microglia: acts as house keepers - resident innate immune cell - constantly perform immunusurveillance by migration, phagocytic and antigen presentation - release cytotoxic hydrogen peroxide and nitric oxide - cytokines and chemokines & maintain normal brain homeostasis - tissue repair mechanisms - influence neuronal survival - neurotransmitter release - maintenance of synapses - promote synaptogenesis - synaptic scaling - regulate BBB Astrocytes: A2 astrocytes acts as supporting cells and protect neurons, astrocytic process support BBB formation and its integrity - regulate endothelial transport of substances - guide axon and neurite outgrowth - secrete growth factors for neurons in embryonic stage and after an injury – synaptogenesis - synaptic plasticity, neurotransmitter synthesis - regulate cerebral blood flow - hormone synthesis - cell-cell communication. Naïve astrocytes express GFAP, S100 β, GLT-1, GLAST, reactive astrocytes express Nestin, MMP13, MMP2, Axin2, and scar forming astrocyte express Sox9, Acan, Pcan, Slit 2, Chst11, Cdh2 |
|
Neuroprotective cells, gliovascular unit (BBB + glia), neurovascular unit (BBB + neurons) in the brain Microglia normal (ramified M2) – A2 astrocytes - neutrophils – macrophage - mast cells - T-cells - endothelial cells - intact BBB and BCSFB |
|
Cells and neuroprotection associated mediators and receptors in the brain Microglia (M2): DAMPs (ATP, DNA, RNA), CCL2, IL-1α, IL-4, IL-10, IL-13, TGF-β, VEGF, BDNF, PDGF, progranulin, CX3CR1 R, PAMPs, TLRs, PRR, CXCL1-CX3CR1, CD200R A2 Astrocytes: neurotrophin, CCL2, TREM2 Neurons: CD200, fractalkine (CX3CL1) Endothelial cells: ICAM-1, VCAM-1 Neutrophils: Phagocytosis, myeloperoxidase, TGF-β, NGF, GM-CSF, VEGF, IL-4, IL-10,ROS, MMP9, neutrophil and microglianeutrophils Mast cells: Important for wound healing and scar formation after an injury. TNF-α, histamine, proteases, GM-CSF, IL-4, serotonin, heparin and tryptase are prestored in granules. Newly synthetized are TNF-α, IL-1α, IL-6, IL-33, CCL2, VEGF (angiogenesis), substance P, CRH, TGF-β, NGF, prostaglandins, growth factors, leukotrienes T-cells: IL-4 Macrophages (M2): CCL2, TNF-α, IL-1α, IL-6 |
|
Acute protective immune response in the brain Neutrophils recruitment, removal of dead cells and debris, wound healing and tissue remodeling, and angiogenesis |
Abbreviations: BBB, blood-brain barrier; BCSFB, BDNF, brain-derived neurotrophic factor; blood cerebrospinal fluid barrier; CCL2, chemokine (C-C motif) ligand 2; CRH, corticotropin releasing hormone; CXCR1, CXC3, chemokine receptor 1 and 3; DAMPs, damage-associated molecular patterns; IL-1apha, interleukin-1α; ICAM-1, intercellular adhesion molecule-1; PAMPs, pathogen-associated molecular pattern; PDGF, platelet-derived growth factor; PRR, pattern recognition receptors; TGF-beta, transforming growth factor-β; TLRs, toll-like receptors; TNF-α, tumor necrosis factor-alpha; GM-CSF, granulocyte-macrophage colony-stimulating factor; VCAM-1, vascular cell adhesion molecule 1; VEGF A, vascular endothelial growth factor A.
Figure 1.
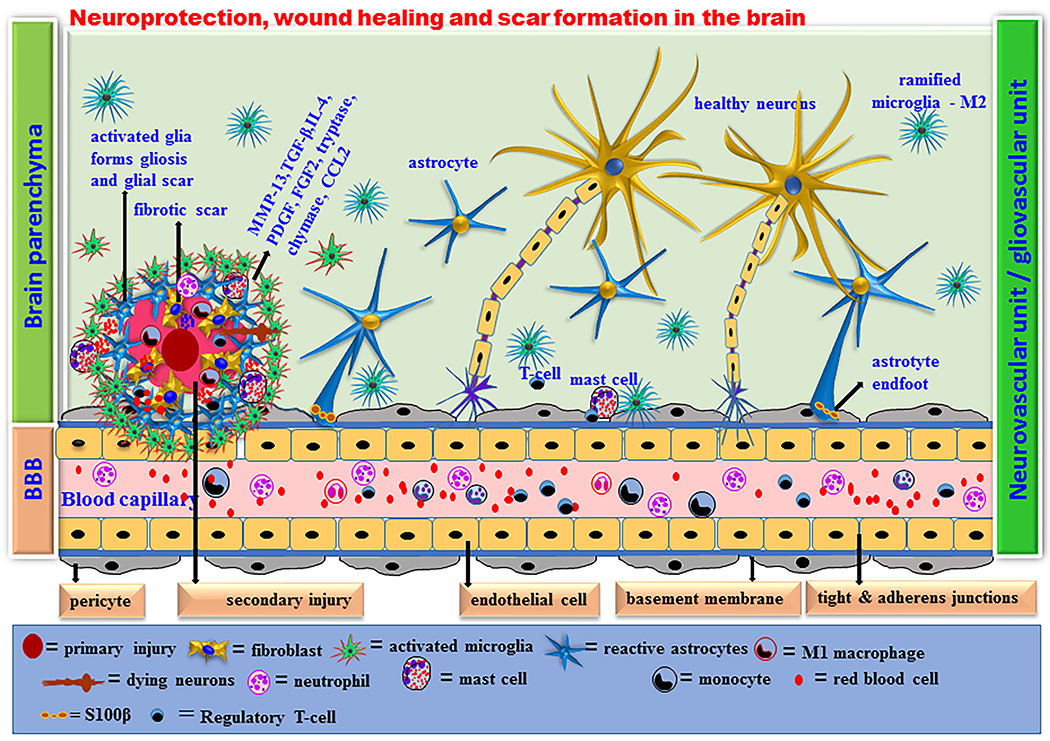
Acute neuroprotective immune response in the brain. Brain is not an immunologically privileged organ. Brain regions are continuously under surveillance by the innate immune cells to detect cell death, debris and infective organisms. Microglia are the resident innate immune cells that are neuroprotective (M2 phenotype) in the brain. Microglia are phagocytic and antigen presenting cells that release anti-inflammatory cytokines, chemokines and other mediators to maintain the brain homeostasis. Astrocytes (A2 phenotype) also play important role in neuroprotection by physically supporting the brain cells and BBB as well as by releasing neurotrophins and growth factors. Neutrophils infiltrate the site of infection or injury and further activate microglia and astrocytes in the brain. Mast cells are also necessary for brain development and neurobehavioral activities by releasing prestored and preactivated TNF-α and proteases within seconds followed by other neuroprotective cytokines and chemokines including IL-1α, IL-4, IL-10, IL-33, CCL2, TGF-β, histamine, tryptase and chymase. Mast cells are crucial in wound healing and tissue repair processes by promoting fibroblast growth, proliferation and its functions. Activated microglia and astrocytes release neuroprotective chemokines and cytokines within few hours whenever needed in the brain. These mediators induce recruitment of innate immune cells such as neutrophils and macrophages to the site of injury from the peripheral blood. The activated innate immune system protects the brain from further acute injury by removing dead cells and debris, initiating wound healing and axonal regrowth processes, neurodegeneration, fibrosis, protecting endothelial cells and BBB system in physiological conditions.
Brain Injury and Initial Neuroprotective Immune Response
The Centers for Disease Control and Prevention (CDC, Atlanta, GA) defines TBI as “a disruption in the normal function of the brain that can be caused by a bump, blow, or jolt to the head, or penetrating head injury” resulting in apoptotic and necrotic cell death of neurons and glia, disruption of BBB with subsequent infiltration of the injured brain by peripherally-derived immune cell populations such as neutrophils and macrophages” (Puntambekar and others 2018). This is accompanied by activation of astrocytes and microglia. TBI is an important cause of long-term adverse effects on general health, behavior, cognition, morbidity and mortality. TBI affects millions of people worldwide every year (World Health Organization, Geneva, Switzerland). In the United States army, the incidence of TBI has increased greatly in recent years. TBI is now recognized as a major health concern in the United States which affects over 1.7 million people each year (Johnson and others 2015). TBI significantly increases Medicare cost as the adverse effects continues for days, weeks, months, years and even decades (Najem and others 2018). Though TBI initiates immune responses in the brain that involves immune cells in the brain and the peripheral system, administration of anti-inflammatory drugs immediately after the injury was not clinically effective (Russo and McGavern 2016). Early suppression of immune and inflammatory responses can cause chronic pathological changes and lesion spread. Moreover, anti-inflammatory drugs are not effective against important inflammatory mediators or cells due to difficulties in drug delivery methods. Drug delivery to the CNS after TBI is complicated due to the BBB, blood-cerebrospinal-fluid barrier (BCSFB) and other problems (Weeks and others 2018). Intranasal drug delivery to the brain may improve the therapeutic effects (Drews and others 2019). Thus, currently there is no effective therapy for TBI patients though some drugs provide symptomatic relief.
Microglia, monocytes, macrophages, neutrophils, T-cells and mast cells are associated with the clearing of dead cells, support barrier systems, wound healing mechanisms and regeneration in the CNS as shown in Fig 1. As microglia express high levels of chemokine (C-C motif) ligand 2 (CCL2), they can quickly identify and respond to damage-associated products such as adenosine triphosphate (ATP), deoxyribonucleic acid (DNA), ribonucleic acid (RNA) and send their processes towards damaged site within minutes and also inhibit capillary leakages (JassamIzzy and others 2017). Infiltrated neutrophils in the brain recruit peripheral monocytes into the brain within a day or two that are necessary for the tissue repair process after the injury. Brain injury also induces T-cell and mast cell responses and recruit CD4+ T-cells that produce interleukin-4 (IL-4) which induces neuroprotection and axonal regrowth after an injury (Russo and McGavern 2016). Regulatory T cells (Tregs) are important in wound healing, remodeling and neurotoxic astrogliosis in the CNS after an injury (Ito and others 2019). The danger-associated molecules released from the site of brain injury determine T-cell response as either neuroprotective or neuro destructive. Treg cells suppress T-cell response through anti-inflammatory cytokines. Generally, T-cells provide neuroprotective effects after brain injuries that are controlled by Treg cells. The initial inflammatory responses after the brain injury are beneficial. However, chronic immune and inflammatory responses following the primary injury induce secondary brain injury by positive feedback and neuroinflammatory responses (JassamIzzy and others 2017). Diversity in the outcome of TBI is due to the differences in the extent of injury and site of injury (JassamIzzy and others 2017). Since neuroimmune and inflammatory responses could be neuroprotective or detrimental, it is important to know the ongoing anti-inflammatory or proinflammatory mechanisms before treating with anti-inflammatory drugs. Treating with anti-inflammatory drugs may worsen when it is in neuroprotective and anti-inflammatory status. Therefore, understanding the timings and the immune responses are crucial for the therapeutic benefit after the TBI (JassamIzzy and others 2017). This study suggests investigating innate immune cells after TBI that moves away from the current M1/M2 phenotypes of microglial activation. Naïve astrocytes express glial fibrillary acidic protein (GFAP), S100 β, glutamate transporter 1 (GLT-1), glutamate aspartate transporter (GLAST), and AldhL1 (aldehyde dehydrogenase 1 family, member L1). Reactive hypertrophic astrocytes proliferate, migrate and express Axin2, MMP2, MMP13, Nestin, cadherin associated protein beta1 (Ctnnb1) and Plaur. Scar forming astrocyte express Sox9, Acan, Pcan, cadherin 2 (Cdh2), secreted glycoprotein 2 (Slit 2), xylosyltransferase (Xylt1), chondroitin 4-0-sulfotransferase-1 (Chst11) and chondroitin sulfate N-acetylgalactosaminyltransferase-1 (Csgalnact1). There are two distinct types of reactive astrocytes such as A1 neurotoxic and A2 neuroprotective phenotypes in neuroinflammation and ischemic brain injury conditions (Clark and others 2019).
The immune system performs sequestration of tissue-damaging irritants, removal of cellular debris and enhancing wound-healing process following TBI. TBI provokes the release of DAMPs and alarmins such as IL-1α and IL-33. Recognition of DAMPs and alarmins induces local release of cytokines and chemokines that cause activation, proliferation and recruitment of immune cells into the site of injury. Neutrophil recruitment also requires increased expression of intercellular adhesion molecule-1 (ICAM-1) or CD54. Innate immune cells include neutrophils, macrophages, microglia, dendritic cells, eosinophils, basophils, mast cells, T lymphocytes. These cells use toll-like receptors (TLRs), nucleotide-binding oligomerization domain (NOD)-like receptors and scavenger receptors to sense pathogens and danger signals that can cause immune response (PrabhuDas and others 2017). The choroid plexus epithelium detect the danger signals or proinflammatory mediators such as tumor necrosis factor-alpha (TNF-α) from the damaged brain cells and induce neutrophil migration to the choroid plexus. These neutrophils circulate with CSF close to the injured areas that could migrate to the brain parenchyma through BCSFB. Neutrophils can migrate from blood vessels at ipsilateral leptomeninges and choroid plexus within 4 hours following TBI. Then these neutrophils infiltrate the injured site, which peaks within 24 to 48 hours.
Activated mast cells release specific proinflammatory and anti-inflammatory mediators such as IL-1β, IL-4, IL-6, IL-8, IL-33, IL-36, TNF-α, CCL2, CCL3, CCL5, granulocyte macrophage colony-stimulating factor (GM-CSF), vascular endothelial growth factor (VEGF), matrix metalloproteinases (MMPs), reactive oxygen species (ROS), substance P, nerve growth factor (NGF), transforming growth factor-beta (TGF-β), corticotrophin-releasing hormone (CRH), neurotensin, histamine, prostaglandin D2 (PGD2), leukotrienes (LTs), stem cell factor (SCF), tryptase and chymase as we and others have reported previously (Gallenga and others 2019; Kempuraj and others 2017b; Mukai and others 2018). These mediators released may vary depends upon the type of mast cells stimulated, nature of stimuli and tissue microenvironments. Mast cells can respond and protect the body from mold (fungi), bacteria, and viral infections and promote wound healing and fibrotic events (Kritas and others 2018; Mukai and others 2018; Ocak and others 2018). Several reports indicate that mast cells mediate neuroinflammation and neurodegeneration by releasing specific multifunctional inflammatory and neurotoxic mediators in the CNS as reported previously (Caraffa and others 2018; Kempuraj and others 2019). Mast cells act as both sensors and effectors and communicate with nervous, vascular and immune systems through direct cell-to-cell interactions through CD40, CD40L, neuroactive and vasoactive molecules such as CCL2, IL-8, TNF-α and VEGF (Kempuraj and others 2017a; Kempuraj and others 2017b). Mast cells respond to cell injury by detecting IL-33 released from injured cells. Further, mouse mast cell proteases activate glia and neurons to release IL-33 that in turn can activate mast cells (Kempuraj and others 2018b). Several reports indicate that mast cells are the first cells that recognize and respond to brain injury by releasing prestored and preactivated TNF-α and proteases and recruit other immune cells such as neutrophils within hours of injury (Dong and others 2014; Hendriksen and others 2017). TNF-α released from mast cells can upregulate the expression of endothelial leukocyte adhesion molecule 1 (ELAM-1), intercellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule 1 (VCAM-1) in vascular endothelial cells that influences adhesion and recruitment of neutrophils and other leukocytes into the site of injury (Meyer and others 2018; Mukai and others 2018). VEGFs released at the site of injury can increase mast cell accumulation through VEGF receptors (VEGFR) (Varricchi and others 2018). Transforming growth factor-beta (TGF-β) plays an important role in fibrosis, angiogenesis and tissue repair mechanisms and inhibits the functions of immune cells. Rapidly released TGF-β from mast cell granules can inhibit mast cells in an autocrine and paracrine manner. The first immune response after TBI is the infiltration and degranulation of mast cells with release of prestored and preactivated neuroprotective mediators in the brain (Moretti and others 2016).
Mast cells, microglia and endothelial cells release TNF-α within few seconds, 4 hours and 48 hours, respectively after the brain injury (Lindsberg and others 2010). Mast cells are early responders after hypoxia-ischemia and in other brain injuries (Lindsberg and others 2010). Rapid recruitment of TNF-α-positive mast cells within 1 hour of hypoxic–ischemia has been demonstrated (Jin and others 2009). Mast cells that are located near the hippocampus regulate emotionality and cognition functions. Mast cells enter the CNS during development through blood vessels and remain there perivascularly, and are present on the abluminal side of the blood vessels (Lindsberg and others 2010). Mast cells release preformed granular mediators within minutes and newly synthesized mediators in the next several hours and days and amplify vasoactive, neuroactive and immunoactive cellular and molecular responses in the CNS. Mast cells can activate microglia and neurons through mast cell specific tryptase, and protease-activated receptor-2 (PAR-2) that is expressed on microglia and neurons (Kempuraj and others 2018a; Skaper and others 2018). These cells remove cellular debris and damaged cells from the site of injury and promote angiogenesis and wound healing mechanisms including fibrosis (Mukai and others 2018). Mast cell tryptase is a strong proangiogenic protease that induces activation, proliferation and migration of endothelial cells and fibroblasts (Atiakshin and others 2018). Mast cells are localized close to capillary sprouting sites indicating its role in new blood vessel formation. Mast cells in association with as well as neuroinflammation are like double-edged sword. Inflammatory response is neuro protective on one side and neuro destructive on the other side including in acute TBI (Morganti-Kossmann and others 2002). Mast cells perform both protective and harmful functions in human health and disease throughout the lifespan. Mast cells are important in wound healing as they enhance acute inflammation, angiogenesis, fibroblast proliferation and scar formation.
Neurotrophic factors and anti-inflammatory progranulin attenuates neuronal damage and microglial activation after brain injury in mice, which suggests that recombinant progranulin, could be used to treat TBI (Menzel and others 2017). Brain injury causes the migration of monocytes from blood to the site of injury in the brain, mature into macrophages, and remain there until inflammation subsides. Peripheral CCL2-positive monocytes recruited into the brain can cause hippocampal damage in cortical brain injury. Neurotoxic and disruptive astrocytic phenotype (A1) increases after the brain injury (Clark and others 2019). Microglia mediate neuroprotective effects in early TBI. However, chronic microglial activation leads to classical activation and release of inflammatory mediators that mediate neurodegeneration. TBI can immediately activate microglia and the activation persists for years. Sites of microglia activation correlates with the extent of degeneration. Activated microglia change their shape from normal ramified to hypertrophic phenotype. The activation states are M1 or classically activated and M2 or alternatively activated. M1 type is proinflammatory and induces neuroinflammation and neurodegeneration by releasing various inflammatory mediators (Kanazawa and others 2017). M2 type is associated with the release of neurotrophic factors that promote repair and phagocytic role. However, in chronic diseases microglia are of mixed phenotype consisting of both M1 and M2, and thus difficult to clearly differentiate between distinct M1 or M2 phenotypes since they are in a transition state.
There are three phenotypic microglia in the CNS such as resting (ramified), activated non-phagocytic (antigen-presenting like) and reactive (phagocytic) microglia Transitory state of microglia are commonly seen in TBI. The extent and duration of adverse effects due to brain injury vary based upon the site and extent of injury in the brain. This is partially due to the difference in immune responses at different regions and depends upon the speed of immunocytes recruitment to the site of injury. Additionally, the distribution of immune cells vary from one region to other in the brain. Moreover, injury at some site is more dangerous and fatal than others. Accumulation of amyloid beta (Aβ) peptide and tau increases with age and alters macrophage response to TBI. Microglia are “primed” after the initial injury and become hyper-active in subsequent immune challenge and worsen the recovery mechanisms (Kokiko-Cochran and Godbout 2018).
MMP-9 regulate neutrophil migration to the site of injury. After 3 days of injury the number of neutrophils decrease with increased recruitment of peripheral immune cells such as T-cells, NK cells and dendritic cells (DCs), followed by the activation of local microglia and astrocytes. The recruited immune cells disappear in two weeks post injury. Neutrophils can initiate AD-like symptoms and play a role in the formation of amyloid plaques (APs) (LiuLi and Dai 2018). However, the activated astrocytes and microglia with elevated levels of inflammatory cytokines remain there for months to years after the TBI incident. This indicates that immune and inflammatory responses including presence of neutrophils persists for a long time in the tissue after the initial injury (McKee and Lukens 2016). Neutrophils can survive over a long period, and were detected one year after the induction in the CNS. Neutrophils can activate microglia to secrete several mediators including ROS, MMP9, TNF-α, IL-1β, IL-6 (Liu and others 2018). Whether the immune response contributes to neuroprotection or further destruction depends on the nature, duration, and magnitude of the immunological events that develop in response to TBI. Triggering receptor expressed on myeloid cells2 (TREM2) is expressed by several cells including macrophages, dendritic cells, granulocytes and microglia and is essential for the removal of Aβ contributes to and neuroprotection after brain injury (Kober and Brett 2017). Development of blood-based biomarker for AD was previously attempted and is being further explored. In conclusion, brain injury can initiate neuroprotective immune responses in the brain that can protect the brain from further damage and initiate wound healing processes (Fig. 1).
Brain Injury and Adverse Inflammatory Response in the Brain
Most of the adverse effects after brain injury or TBI are due to immune and inflammatory responses (McKee and Lukens 2016). Primary injury after TBI may continue for months and years that leads to secondary injury and ultimately induces neurological impairment and neurodegeneration (McKee and Lukens 2016). Therefore, targeting specific proinflammatory pathways after TBI may be an effective treatment option in these patients. TBI-mediated neuroinflammation is strongly associated with several neurological disorders including AD, Parkinson’s disease (PD), anxiety, depression, post-traumatic stress disorder (PTSD), CTE), apathy, and amyotrophic lateral sclerosis (ALS) (McKee and Lukens 2016). Though TBI is associated with several neurological disorders, there are no disease specific and disease-modifying treatment options currently available to treat these patients. Uncontrolled immune response leads to neuroinflammation-mediated neuronal death, tissue loss, BBB break down, edema, increased levels of inflammatory mediators, gliosis and immune cell infiltration in the CNS.
Immune cells involved in the TBI pathogenesis include neutrophils, macrophages, microglia, T-cells and mast cells. These cells specifically contribute to the immune and inflammatory consequences of TBI including activation of glia as shown in Fig. 2. Brain injury leads to the proliferation of microglia after 24 hours of injury and continue to proliferate for several weeks based upon the site and the extent of injury. TBI not only shows site-specific effect but also show neuronal type-specific effects in the CNS. Some neuronal types are more vulnerable than others in the same region of the injured brain (Brizuela and others 2017). Type of neurons and their responses vary according to mild, moderate and severe conditions of the injury. Therefore, mild injury may not affect all types of neurons in the injured area that might be affected by moderate injury. Currently, laser capture microdissection (LCM) method is useful to selectively collect specific neurons in specific brain regions affected by TBI for pathologic and gene analysis.
Figure 2.
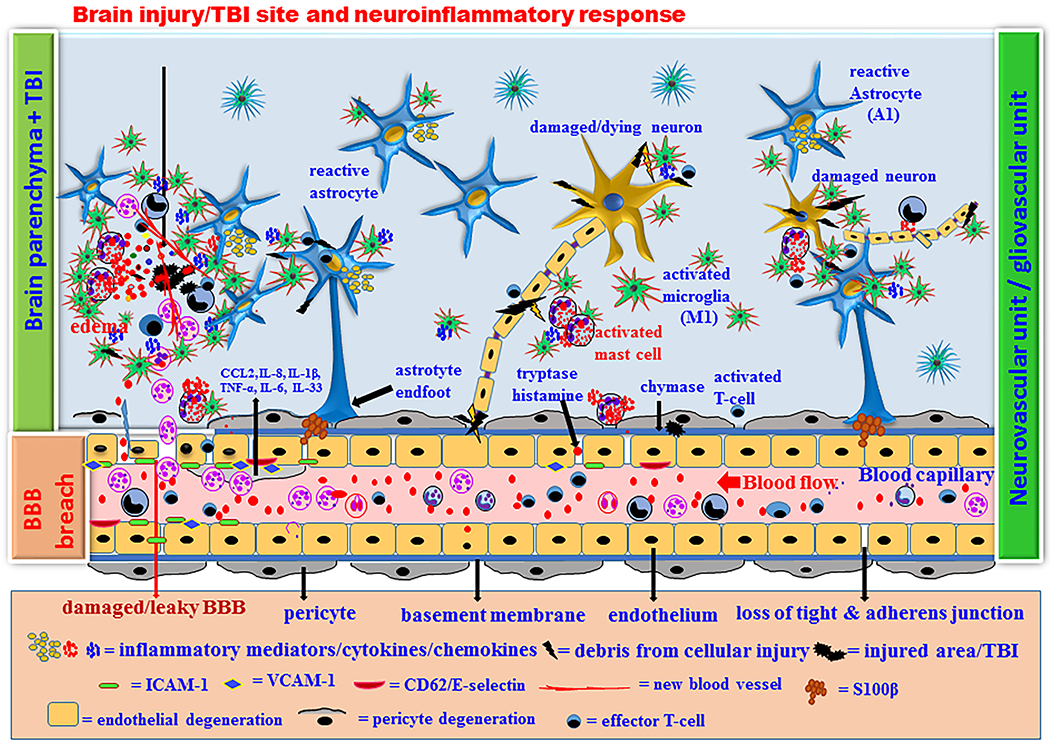
Brain injury/TBI-mediated chronic neuroinflammatory response and neurodegeneration. Acute neuroprotective immune response fails to control the neuroinflammatory response if the brain injury is more and if the patient has other risk factor(s). Chronic immune response activates innate immune cells and microglia in the brain as well as in the peripheral system. Innate immune cells such as neutrophils, mast cells and some progenitor cells migrate from peripheral blood to the site of injury and further activate neuroglia and endothelial cells in the brain. Increased activation of microglia, macrophage, T-cells, astrocytes and mast cells increase the release of neuroinflammatory mediators such as CCL2, IL-1β, IL-6, IL-8, TNF-α, GM-CSF, VEGF, MMP-9, ROS and proteases with increased activation of NF-kB, AP-1, MAPKs, TREM2 and ApoE4 in the brain cells and immune cells. Brain injury increases M1 and A1 neurotoxic and proinflmmatory microglia and astrocytes respectively in the brain. High levels of these inflammatory mediators in the brain and in the peripheral blood causes chronic BBB and BCSFB dysfunctions, degeneration of pericytes and endothelial cells, neuroinflammation and neuronal death in vicious fashion that further leads to secondary damage in the brain. Several mast cell mediators including tryptase and chymase play a significant role in the BBB breach in neuroinflammation. TNF-α, tryptase, CCL2, VEGF, IL-1β, IL-6, IL-8, ROS and MMP-3 from activated mast cells contribute to the chronic neuroinflammation. Chronic neuroinflammation leads to neuronal death and initiates onset, progression and severity of AD in certain high-risk category people.
The reactive astrocytes and activated microglia remain at the site of injury for years after the first incident of TBI (McKee and Lukens 2016). M1 macrophages/microglia with the marker CD16/32 increases in 3–7 days after injury. Micro particles (MPs)-derived from microglia-mediate neuroinflammation as well as participate in the peripheral inflammation (Kumar and others 2017). Increased IL-1α precedes IL-β production after injury and-induces neuroinflammation and neurodegeneration. IL-1 increases BBB permeability, glial activation, immune cell recruitment and neurodegeneration after TBI (Chodobski and others 2011).
Plasma IL-6 level is increased in severe TBI than in mild TBI patients (McKee and Lukens 2016). IL-6 plays an important role in neurovascular abnormalities, neuroinflammation and neurodegeneration. The level of TNF-α increase after TBI, and plays both protective as well as a detrimental role in acute and chronic phases, respectively. Immediately after the brain injury, TNF-α plays a crucial protective chemoattractant role for recruiting inflammatory cells to the site of injury. TNF-α level increases before IL-1β following TBI and these cytokines promote the expression of ICAM-1 and VCAM1 on the endothelial cells that promote BBB permeability and neuroinflammation (Sweeney and others 2019). Anti-chemokine intervention showed decreased immune cell recruitment into the brain from blood and reduced the inflammation after brain injury. Chemokines are immediately released from astrocytes, microglia, endothelium, mast cells and recruited neutrophils at the injured site in the brain. CCL2 is transported from abluminal to luminal side in the endothelium influences BBB permeability, edema formation and the recruitment of immune cells after brain injury. The extent of immune cell recruitment and the edema formation depends upon the extent of brain injury. The recruited neutrophils are toxic to neurons and increase BBB permeability by producing cytokines such as TNF-α, reactive oxygen species (ROS), proteolytic enzymes elastase and MMP9. Neutrophils secrete lysosomal enzymes, free radicals, decrease blood flow by direct microvascular occlusion and increase vascular permeability. Though leukocytes migrate across the brain, there are some specific sites where the migration is high after TBI in rodents (Carlos and others 1997). These includes periconstusional leptomeninges and subarachnoid CSF space adjacent to the injured area and cistern of velum interpostium. Glia maturation factor (GMF), a brain protein that was first discovered, sequenced and sub cloned in our laboratory has been shown to be implicated in the activation of brain cells, innate immune cells and neuroinflammation-mediated neurodegeneration in AD, PD and Multiple Sclerosis (MS) and its animal model experimental autoimmune encephalomyelitis (EAE) (Zaheer and others 2007). We have shown that GMF mediates neuroinflammation, neurodegeneration in PD, and is associated with APs and NFTs in AD (Kempuraj and others 2018a; Thangavel and others 2018). Since GMF plays a significant role in the neurodegeneration in the above mentioned disease conditions, we presume it could also play an important role in the pathogenesis of brain injury/TBI.
Brain injury chronically activates nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) pathway and contributes to the elevated inflammatory mediators in the CNS (Jones and Kounatidis 2017). The complement system also contribute to the inflammatory pathways in TBI. GM-CSF is an important cytokine that causes proliferation and mobilization of immune cells from bone marrow. GM-CSF could play a neuroprotective role after TBI, aiding in cognitive recovery, and increase neurogenesis in the hippocampus (Song and others 2016). Elevated level of IL-10 could mediate neuroprotection in TBI patients. Moreover, IL-10 suppresses NF-kB activation that is elevated in AD patients and TBI (Ju Hwang and others 2019). Exercise induces the release of IL-10 that may provide protective effects in patients such as with AD. Aspirin, a non-steroidal anti-inflammatory drug (NSAID) inhibit NF-κB activation, increases IL-10 and enhances the recruitment of immune cells (Medeiros and others 2013). Similarly, TGF-β also increases after TBI in patients. Additionally, apolipoprotein E4 (Apoε4) and TREM2 are implicated in inflammation, TBI-mediated neuroinflammation and impaired cognition (Andreasson and others 2016). Proinflammatory mediators and innate immune mechanisms increases C-reactive protein (CRP) in inflammatory conditions, psychiatric disorders, including in age-associated AD and TBI patients (Anada and others 2018; De Berardis and others 2017).
Mast cells located on the abluminal side induces BBB permeability, increased extravasation, brain edema, hemorrhage and local inflammation after cerebral ischemic injury (Ocak and others 2018; Ribatti 2015). Ischemic stroke, hemorrhage or TBI-induces increase in BBB permeability and its dysfunction (Yang and others 2019). This allows the entry of several blood-borne factors such as fibrinogen, thrombin and albumin that act on neurovascular unit and brain cells in the injured brain. Factors that enter into the brain through defective BBB can activate microglia and increase its phagocytic activity and chemokine release (Sweeney and others 2019). Fibrinogen can inhibit neurite outgrowth and neuronal repair mechanism after TBI (Jenkins and others 2018). Thrombin affects endothelium, induces edema formation, and oxidative stress after TBI. Albumin activates microglia and astrocytes to release inflammatory cytokines by increasing Ca+ influx and mitogen-activated protein kinases (MAPKs) activation after the brain injury.
Increased BBB permeability after the brain injury is due to several factors including TGF-β, glutamate, ROS, MMPs (MMP2, MMP3 and MMP9) and vascular endothelial growth factor A (VEGF A) (Takeshita and Ransohoff 2012). Immediately after the brain injury, the levels of cytokines such as TNF-α and IL-1β and chemokines increases followed by increase in cell adhesion molecules on endothelium. These factors increase the influx of immune cells from the blood to the brain through dysfunctional BBB. We have previously shown that mast cell activation induces BBB breakdown (Esposito and others 2001; Theoharides and others 2008). Neutrophils and monocytes also migrate through blood-CSF barrier (BCSFB).In addition to CCL2, CCL20 is also involved in the recruitment of macrophage to the ischemic brain injury site and release of TNF-α and IL-1β (Rayasam and others 2018). IL-33 can influence local macrophage to release IL-4 to induce recovery from stroke (Korhonen and others 2015). In conclusion, brain injury can initiate adverse and prolonged neuroinflammatory responses if the immune system cannot control the initial injury. This inflammatory response leads to neuroinflammation-mediated neurodegeneration in the brain (Table 2 and Fig. 2).
Table 2.
Brain injury-mediated chronic neuroinflammatory response and neurodegeneration after the brain injury/TBI
|
Brain injury
Causes: Direct blow to the skull, penetrating injury, acceleration, deceleration, rotational forces, Ischemic, hemorrhagic or traumatic injuries Injured brain cells: Neurons, astrocytes, microglia, ependymal cells, oligodendrocytes, endothelial cells Clinical complications: Hematoma, subarachnoid hemorrhage, contusion, diffuse axonal injury, cerebral ischemia, increased intracranial pressure, neuroinflammation, neurodegeneration, sickness behavior |
|
Neuroinflammatory responses in brain injury Disruption of tight junction proteins and integrity of basement membrane Increased BBB permeability, edema, neuroinflammation Neutrophil, macrophage, T-cell and mast cells recruitment into the injured site Incomplete removal of dead cells and debris Partial wound healing and tissue remodeling Insignificant axonal regrowth Aβ 1–42 deposition, amyloid plaque and NFTs formation in the brain |
|
Neuroinflammatory cells and their functional status in brain injury Microglia (M1 proinflammatory), resting ramified microglia to Reactive/activated microglia (amoeboid), migration, phagocytosis, antigen presentation Astrocytes (A1 proinflammatory), reactive astrocytes, reactive gliosis, retraction of process from synapsis, increased GFAP, S100β and vimentin, increase in metabolic enzymes Neutrophils – phagocytosis, MMPs release. Mast cells – degranulated and activated mast cell types T-cells – activated T-cells. Endothelial cells – activated cells Macrophage (M1-like) – activated cells BBB, BCSFB – increased permeability |
|
Brain injury and expression of neuroinflammatory mediators and receptors in the CNS/immune cells Microglia (M1) - activation, proliferation, phagocytosis, DAMPs (ATP, DNA, RNA), CCL2, IL-1β, TNF-α, IL-6, IL-8, IL-12, IL-23, IL-33, ROS, glutamate, MMPs, TLRs, PAR-2, P2, NO, TREM2 Astrocytes (A1) - CCL2, IL-1β, TNF-α, IL-6, GM-CSF, ROS, glutamate, MMPs, VEGFA, GFAP, S100β Neurons - MMPs, VEGFA, P2, arginine vasopressin, PAR-2 Endothelial cells – ICAM-1, VCAM-1, CCL2, NO, MMPs, VEGFR1, VEGFR2, TLRs, PTY2, arginine vasopressin Mast cells - degranulation - prestored TNF-α, histamine, proteases, GM-CSF, IL-4 Mast cells – activated - newly formed prostaglandins, leukotrienes, growth factors, TNF-α, IL-1α, IL-6, IL-8, IL-18, IL-23, IL-33, CCL2, VEGF, neurotrophins, substance P, CRH T-cells – IL-4, IL-21 Platelets – TGF-β Neutrophils - MMP9, TNF-α, ROS, elastase (ELANE) Astrocytes, neurons and inflammatory cells- thrombin, albumin, fibrinogen, Ca++ increase, ERK1/2, p38, JNK MAPKs; p13K/Akt, NF-kB, AP-1; TREM2, Apoε4, PAR1–3 and 4, ATP, UTP, purinoceptors |
Abbreviations: AP-1, activator protein 1; Apoε4, apolipoprotein E4; BBB, blood-brain barrier; BCSFB, BDNF, brain-derived neurotrophic factor; blood cerebrospinal fluid barrier; CCL2, chemokine (C-C motif) ligand 2; CRH, corticotropin releasing hormone; CXCR1, CXC3 chemokine receptor 1; DAMPs, damage-associated molecular patterns; ERK1/2, extracellular signal-regulated kinases; GFAP, glial fibrillary acidic protein; GM-CSF, granulocyte-macrophage colony-stimulating factor; IL-1apha, interleukin-1α; ICAM-1, intercellular adhesion molecule-1; JNK, c-jun N-terminal kinase; MAPKs, mitogen-activated protein kinase; MMP9, matrix metalloproteinase 9; NO, NF-κB, nuclear factor kappa-B; NO, nitric oxide; NFTs, neurofibrillary tangles; PAMs, pathogen-associated molecular pattern; PDGF, platelet-derived growth factor; PAR, protease activated receptor; ROS, reactive oxygen species; PRR, pattern recognition receptors; TGF-beta, transforming growth factor-β; TLRs, toll-like receptors; TNF-α, tumor necrosis factor-alpha; TREM2, Triggering receptor expressed on myeloid cells 2; VCAM-1, vascular cell adhesion molecule 1; VEGF A, vascular endothelial growth factor A; VEGFR, vascular endothelial growth factor receptor.
Animal Models of Brain Injury
Animal models are used to understand the pathogenesis of TBI. Animal models of TBI, concussion and CTE are widely used to understand the consequences of brain injury (McAteer and others 2017; Wojnarowicz and others 2017). In contusion models of TBI, focal TBI may be due to a blow to the head, weight drop, and fluid percussion (FP), rigid indentation and dynamic cortical deformation. Models of traumatic axonal injury (TAI) show diffuse axonal injury (DAI). TAI animal models include gyrencephalic models and rodent models. Animal model of blast-injured TBI is significantly important due to its association with military activities. Models of acute hematoma show intracranial hematoma due to tearing or rupture of veins. Animal models of TBI provide invaluable information on the pathological effects of brain injury particularly biomarker discovery and pre-clinical screening of drugs (Estrada-Rojo and others 2018). However, translation of research findings from animal models of TBI into human patients remain extremely difficult due to immunological, anatomical and functional differences between the CNS of human and small animals (Sorby-Adams and others 2018). Moreover, the diverse nature of TBI in humans makes it very difficult to develop appropriate clinically relevant single animal model and thus requires several types of TBI models including focal and diffuse type models. Thus, use of larger animals such as non-human primates, swine, and sheep may improve clinical translation. Currently, there is no single “gold standard” animal model for TBI that is close to human TBI pathology. In addition to animal models, in vitro cell culture models of TBI also provide valuable information to understand the interactions of brain cells and immune cells in the pathogenesis of TBI. The in vitro cell culture models of TBI include compression, transection, barotrauma, acceleration, hydrodynamic, chemical injury, and cell-stretch methods (Kumaria 2017). Cell culture systems include brain-on-a-chip, primary cell cultures, and immortalized cell lines, co-culture of various brain cells, acute preparations and organotypic cultures. An in vitro cell culture model of CTE may provide additional information on CTE pathogenesis (Jaber and Polster 2017; Jayakumar and others 2017). Further, replacement of in vivo studies with in vitro studies reduce the use of animals for the experiments. In conclusion, animal models are useful to investigate the pathogenesis of TBI but there is no single animal model that closely resembles to human TBI with reference to immune response, pathologies and behavioral abnormalities.
Brain Injury Accelerates AD pathogenesis in High-Risk Individuals
TBI is associated with increased risk for psychiatric disorders, PTSD and AD pathogenesis (Al-Dahhak and others 2018; Becker and others 2018). Even a single incident of TBI can be associated with risk for AD (Kokiko-Cochran and Godbout 2018). A recent study suggests that post traumatic chronic and progressive neuroinflammation is associated with neurodegeneration after the TBI (Xiong and others 2018). However, a recent report also indicate that biomarker analysis failed to confirm the association between TBI and AD dementia (Katsumoto and others 2018). BBB breakdown and loss of tight junction proteins are frequent problems in AD, which could be worsened by TBI. TBI induces brain swelling, neuronal injury, hypoxia, BBB and tight junction injury and dysfunction, oxidative stress, neuroinflammation, neurodegeneration and cognitive decline and predisposes to AD pathogenesis later in life (Sulhan and others 2018; Sweeney and others 2018). Moreover, TBI patients show increased frequency of AD pathogenesis (Breunig and others 2013). A recent study showed increased head injury or TBI in AD patients (Ilmaniemi and others 2019). TBI patients show the presence of AD hallmarks Aβ and NFTs in the brain immediately after the injury (Johnson and others 2012). Additionally TBI also increases amyloid precursor protein (APP) that is necessary for Aβ production in the brain. Moderate to severe TBI is a potential risk factor for dementia and AD, but whether mild TBI alters AD pathogenesis is not yet clearly known (Cheng and others 2018; Collins-Praino and Corrigan 2017). Additionally, some findings do not support a link between TBI and AD (Lo and others 2018). Meta-analysis of the hypothesized TBI-AD link have shown mixed results and in fact, one-report shows that TBI was not even a significant risk factor for AD pathogenesis (Xu and others 2015). This study suggests that mild TBI has minimal effects on Aβ metabolism, but age can influence the outcomes. Moreover, brain injury at old age has been shown to be associated with severe problems and poorer recovery than at young age (Cheng and others 2018). Mild TBI can cause temporary dysfunction of cells in the brain. About 80% of all TBI cases are classified as mild head injuries. TBI induces short-term and long-term cognitive impairment. In one self-reported TBI, a history of TBI with loss of consciousness was not associated with AD, dementia or neuropathological features of AD. In the second report using Veterans Administration medical records, they examined the history of TBI with cognitive impairments or changes in brain on magnetic resonance imaging (MRI) and Aβ deposition in the positron emission tomography (PET) scans. The results showed that TBI history did not correlate with impaired cognition and AD biomarkers. These studies suggest that TBI may not increase the risk of AD (Weiner and others 2017). Similar to AD, late life TBI also increases the risk of PD (Gardner and others 2015). Further evidence shows that repeated injuries are generally associated with high risk of developing chronic neurodegenerative diseases (Donat and others 2017). GFAP, tau and Aβ42 are increased in TBI and are proposed as diagnostic and prognostic biomarkers in TBI (Bogoslovsky and others 2017). Increased expression of GFAP can be lethal and can induce AD pathogenesis. Further investigations are necessary to conclusively determine the association of TBI to cognitive decline in the aged people.
Dysregulated immune responses have been shown to be responsible for the neuroinflammation and neurological disorders observed after TBI. Brain injury induces increased release and deposition of Aβ and phosphorylated tau that forms APs and NFTs to mediate AD pathogenesis specifically early in AD progression (Puntambekar and others 2018). Postmortem brains show increased Aβ and tau deposition that are hallmarks of TBI. Increased Aβ leads to the formation of plaques in the brain following TBI. Brain injury- mediated tau deposition varies with the severity, type, time-point, and number of brain injuries (Kokiko-Cochran and Godbout 2018). Tau deposition has been reported in a single-incident of CCI, weight-drop, FPI and bTBI conditions (Gabbita and others 2005). Carrying an allele of Apoε4 or a mutation in TREM2 show significantly increased rate of AD in humans and that manipulation of these could affect the severity of neurological dysfunction after TBI (Van Cauwenberghe and others 2016). TREM2 may control acute macrophage migration to brain but it also upregulates neurodegeneration in chronic TBI. TREM2 also regulates innate immune pathways in AD (Li and Zhang 2018). Brain injury leads to cognitive impairment, increased accumulation of APP, extracellular Aβ peptide and intracellular NFTs containing tau protein and inflammatory cytokine and chemokine release (Kokiko-Cochran and others 2016).About 30% of TBI patient’s brains show the presence of Aβ peptides (Roberts and others 1994). This Aβ deposition was found within 24 hours after TBI indicating the association of AD specific Aβ peptide in the TBI pathogenesis (Ikonomovic and others 2004).
Mast cells are suggested to play an important role in the pathogenesis of AD similar to microglia (Kempuraj and others 2017a; Shaik-Dasthagirisaheb and Conti 2016; Skaper and others 2017). Since mast cells are implicated in neuroinflammation, brain injury or TBI, these cells could accelerate brain injury/TBI-mediated pathogenesis of AD (Fig. 3) (Kempuraj and others 2017a; Shaik-Dasthagirisaheb and Conti 2016; Skaper and others 2018). Mast cells are able to detect the generation of amyloid plaques (APs) in AD. Immediately after the brain injury, mast cells infiltrate and degranulate releasing neuroprotective and chemo attractive mediators including TNF-α in the brain (Hendriksen and others 2017). However, mast cell number increases for days and weeks and contributes to the brain damage by releasing inflammatory mediators such as TNF-α and IL-9 in perinatal hypoxic-ischemia and that inhibition of mast cell activation decreased the brain damage in the immature rat brains (Ziemka-Nalecz and others 2017). Mild TBI is associated with persistent dura mast cell degranulation for few weeks and in chronic conditions this may compromise BBB integrity for immune cell recruitment to the brain (Skaper and others 2018). TBI induces axonal injury and affects brain networks that cause behavioral abnormalities. Axons in the corpus callosum, fornix, internal capsule, superior and inferior longitudinal fasciculi and cerebral fibers are particularly vulnerable to injury in TBI. TBI patients also show white matter abnormalities with defective memory and learning skills that increase with repetitive concussions Disturbances in the thalamic-frontal connections lead to attention deficits. The corpus callosum with more white matter fibers is a common site of damage in TBI leading to significant cognitive deficits. White matter injury has been considered as a marker of TBI severity (Hayes and others 2016). Damage to the hubs that link several neuronal networks may severely contribute to the long-term irreversible cognitive dysfunction in TBI patients and in TBI-mediated AD patients. From the available reports, we conclude that TBI may increase the risk for the development of AD in high-risk people.
Figure 3.
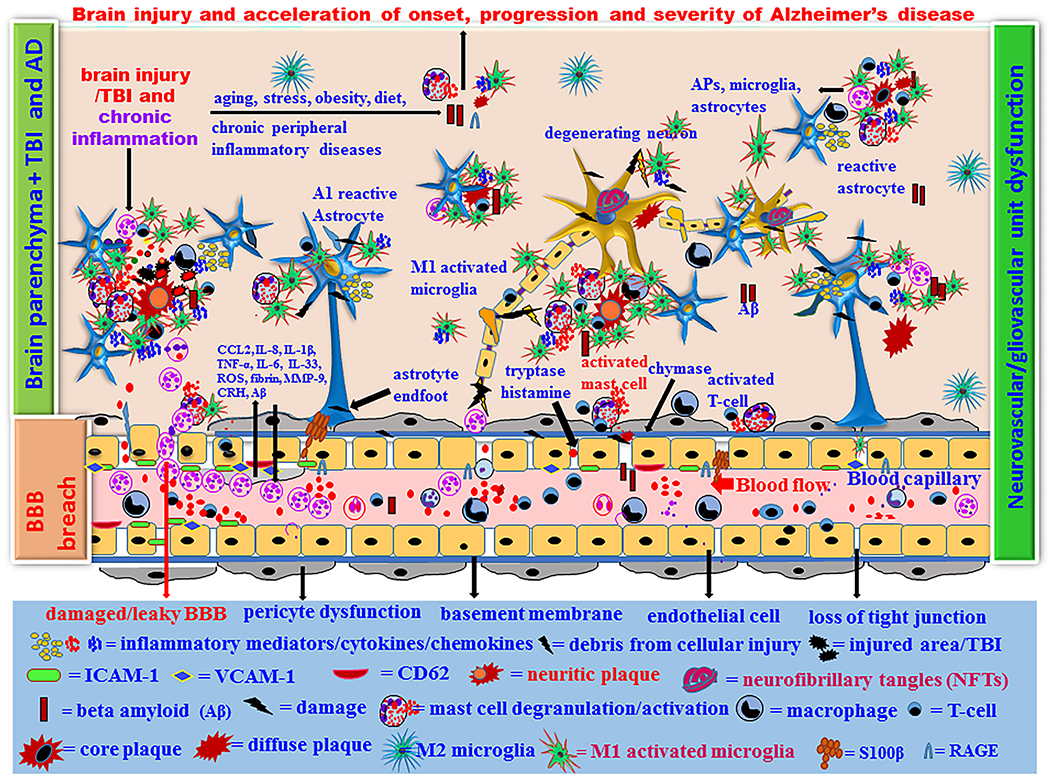
Proposed brain injury/TBI-associated onset, progression and severity of AD in high-risk people. Brain injury leads to microbleeds and acute primary damage of brain cells. If acute immune response continues chronically, that leads to secondary damage of brain cells due to over activation of immune cells associated with s significantly higher levels of inflammatory and neurotoxic mediator release in a vicious fashion in the brain and peripheral immune system. Increased number of neurotoxic and inflammatory M1 microglia and A1 astrocytes exacerbates neuroinflammation and neurodegeneration in the injured brain. Normal aging process and brain injury generate APP and Aβ1–42 in the brain and these chronically activate neuroglia and immune cells in the brain that release several neuroinflammatory and neurotoxic mediators. Aβ and tau protein generate the formation of extracellular APs, and intracellular NFTs in the neurons, respectively. Immune system fails to remove the excess Aβ peptide and NFTs and thus the levels of these increases continuously. This is associated with the degeneration of pericytes and endothelial cells, increased neuroinflammation and neurodegeneration in the brain. This ultimately cause cognitive dysfunction and the onset of AD. Brain injury/TBI in high-risk people with PD, anxiety, depression, PTSD, CTE, ALS, aging, obesity, emotion, chronic peripheral diseases, chronic allergic tendencies, genetic factors, environmental factors, and high fat diet can accelerate the pathogenesis of AD.
Influence of Personal Traits on TBI-mediated AD Pathogenesis - Age, Gender, Race, Emotion, Peripheral Diseases, Allergic Tendencies, Genetic Factors, Environmental Factors, and Nature of Regular Food can Influence TBI-mediated AD Pathogenesis
Aging increases the activation of glial cells and innate immune cells associated with neuroinflammation and neurodegeneration in the brain. Recent studies suggest that TBI at an early age has less influence on the neurodegenerative diseases. But, TBI in the aged could influence the onset, progression and the severity of specific neurodegenerative diseases such as AD or PD (Griesbach and others 2018). However, AD is known to cause dementia in both young as well as in older people. One recent study reported that aged TBI patients are at higher risk for developing long-term abnormalities than younger patients (Erlebach and others 2017). The immune system can strongly protect the brain after an injury at younger age. Decreased immune response in the old age leads to decreased neuroprotection and therefore TBI in old age can easily accelerate the pathogenesis of AD. Wound healing process at old age also takes more time and thus the inflammatory response continues for months and years in the older people and therefore are more prone to the onset of neurodegenerative diseases including AD.
TBI-mediated complications and mortality rates are less in females than in males (3 times higher) due to hormonal influence (MacQueen and others 2018). Ovarian hormones play an important role to protect women from stroke and neurodegenerative diseases (Villa and others 2016). Progesterone can shift M1 to M2 macrophage and microglia phenotypes. Studies have shown that females who show psychiatric and neurobehavioral symptoms, take long time-to recover from injury and to return to job after TBI (Woolhouse and others 2017). The number of US women soldiers involved in combat operations have increased since the Iraq and Afghanistan wars. Moreover, the number of women using the US Department of Veterans Affairs (VA) services has doubled since 2001 (Amoroso and Iverson 2017). The current US Department of Defense policy of allowing women in the combat roles will likely increase the risk for TBI-mediated and stress-induced AD in women (Amara and others 2014). Additionally, the recent wars have shown the need for women soldiers in combat zones, and thus increased the blast-related brain injuries in women soldiers (Amara and others 2014). Thus, it is crucial to design novel research strategies regarding the gender differences in TBI and gender-dependent treatment options for the future. Etiology of TBI in women veterans show multiple injuries due to blast (61%) and others causes such as fall and vehicular injuries (37%). Since women veterans show high risk for TBI, they should be screened for TBI during and after the military services (Amoroso and Iverson 2017).
Nutrition plays a key role in the cognitive functions, mental health, brain structure, neurotrophic factors, neuronal pathways, neuronal plasticity, and neurodegenerative diseases. Decreased cognitive function and increased risk for vascular dementia has been reported to be associated with lower milk consumption. Mediterranean-type diet has been associated with slower cognitive decline, reduced risk and mortality of AD patients (Otaegui-Arrazola and others 2014). Thus, certain race with specific food-habits show differences in the vulnerability of brain to the TBI-mediated complications. Low amount of regular wine consumption that provide anti-oxidant and anti-inflammatory properties were associated with reduced risk of AD pathogenesis in some individuals, particularly in young adults. However, as alcohol is neurotoxic and reduces cognitive functions, the same amount of alcohol consumption could be neurotoxic, particularly in the aged individuals (Chastain and Sarkar 2014). Thus, nutritional based approach may benefit AD patients. Natural compounds present in regular diet or tea could function as “nutraceuticals,” and inhibit microglial activation and thus demonstrate neuroprotective properties. Flavonoids are natural active compounds present in many fruits, vegetables and herbs. Several flavonoids show neuroprotective properties and are anti-amyloidogenic and thus useful as a therapeutic agent for many brain disorders. Flavonoids show anti-inflammatory, anti-ischemic, anti-cancer, anti-bacterial, and anti-viral and neuroprotective characteristics. Quercetin, a flavonoid has been shown to be neuroprotective in animal models of TBI, through reducing brain edema and neuronal apoptosis with improved motor function tests (Du and others 2018). Other flavonoids such as genistein and pycnogenol also show neuroprotective effect with functional recovery in animal models of TBI. Luteolin, a potent natural compound inhibited TBI-induced AD pathogenesis in Tg2576 mouse model of AD (Sawmiller and others 2014). Luteolin inhibited injury–mediated ROS level and increased neuronal survival and activation of the nuclear-factor-E2-related factor 2 (Nrf2)-ARE pathway in vitro. Plant based Resveratrol is reported to protect brain injury after TBI Flavonoid luteolin has been suggested to provide neuroprotective benefits in neurodegenerative disorders (Theoharides and Tsilioni 2018). Regular consumption of these nutritional supplements or diets containing these flavonoids can slow or prevent the onset of neurodegenerative diseases including the adverse effects observed post-TBI and TBI-mediated AD. Regarding gender, men are at twice the risk of developing AD-associated with previous TBI.
Accumulating evidence suggests that chronic stress is a string risk factor for cognitive decline and AD pathogenesis (Caruso and others 2018). Hippocampus is an important organ affected by AD. The hippocampus with high cortisol receptors is crucial for verbal memory and memory of context with respect to time and place of events. Chronic stress accelerates the aging process including neuronal loss in hippocampal pyramidal neurons in CA1 region. Stress affects hypothalamus as well as hippocampus and induces short-term memory, neuronal dysfunction, neuroinflammation, neurodegeneration and finally AD. Repeated stress can cause atrophy of dendrites or pyramidal neurons in the CA3 region of hippocampus. This is reversible but long-term stress can permanently damage hippocampal neurons. Chronic psychological stress is also considered as a risk factor for late-onset AD and cognitive deficits. Several stress conditions activate hypothalamic-pituitary-adrenal (HPA) axis and increase the release of glucocorticoids such as cortisol (corticosterone in rodents) that are elevated in AD patients (Caruso and others 2018; Escher and others 2019).
Stress can increase APP and tau phosphorylation in AD progression (Caruso and others 2018). Repetitive mild TBI-induced mechanical stress increases brain levels of Aβ1–42 peptide, tau and α-synuclein-mediated neurodegeneration in mice (Levy and others 2018). Restraint stress in AD mouse models (Tg2576) showed increased Aβ (1–42), tau hyper phosphorylation and cortical neuronal atrophy (Lee and others 2009). TBI-induces major depressive disorder (MDD) and PTSD and these are significantly associated with female, depression, unemployment, amnesia and memory of the traumatic event (Cnossen and others 2017). TBI is associated with PD, psychiatric disorders with increased risk of suicide and mortality. These symptoms can accelerate neuroinflammation and the onset and progression of AD after TBI in humans. Currently, it is very hard to compare the effect of stress in mice with humans regarding the pathogenesis of AD. The diagnosis of TBI and PTSD comorbidity increased recently in the United States Veterans. These veteran patients showed several neuropsychiatric symptoms such as insomnia, memory problem, anxiety, irritability, disrupted neuronal connections, neuronal circuitry dysfunctions, behavioral changes, memory impairment, neuroinflammation and oxidative damage. Mast cell activation is implicated in stress as well as in depression dependent inflammation and neurodegeneration (Georgin-Lavialle and others 2016; Kempuraj and others 2019; Kempuraj and others 2017a). Peripheral and brain mast cells are involved in the stress mediated effects including epithelial barrier dysfunction through corticotrophin releasing factor (CRF) and other mediators (Kempuraj and others 2017b; Vanuytsel and others 2014). Moreover, mast cells show sex specific differences in their activation after stress. Mast cells from female rodents show increased synthesis, storage and release of mast cell granule stored mediators independent of estrous cycle (Mackey and others 2016). Stress related diseases are more common in females and are associated with more mast cell activation in these patients. TBI induces stress in patients and mast cells play an important role in stress-mediated effects. Thus, mast cells could play a crucial role in TBI-mediated stress-induced neurodegeneration and AD pathogenesis. All these conditions increase neuroinflammation, neurodegeneration, neuronal death and worsen the quality of life of TBI-PTSD patients (Skaper and others 2018). Recently the prevalence of TBI in women has increased from 25% to 40% (Oyesanya and Ward 2016). Females show more stress reactions than males including women veterans. Moreover, TBI during pregnancy could increase the level of stress than in normal women.
Genetic factors also play important role in the pathogenesis of AD. Apoε4 in combination with TBI increases the risk of AD pathogenesis. Studies have shown that Apoε4-associated phospholipid dysregulation-mediates phosphorylation of tau after repetitive blast-induced TBI in mice. Apoε and its subset Apoε4 has been shown to be involved in Aβ toxicity, neuroinflammation and BBB disruption (Zhao and others 2018). Chronic allergic diseases such as asthma in which mast cells are increased and heavily activated are strongly linked to phosphorylation of tau, dementia, and severity of AD (Bolos and others 2017). Presence of chronic peripheral diseases and allergic diseases can increase TBI-mediated onset, progression and severity of AD. Certain drugs also affect TBI-induced AD pathogenesis. Antidepressant use is associated with consistently increased risk of falling and head injuries in community-dwelling persons with AD (Taipale and others 2017). Innate immune targeted therapy could improve AD symptoms.
Presence of peripheral chronic disease conditions also influence neuroinflammation and neurodegeneration in the brain such as in AD (Kempuraj and others 2017b; McManus and Heneka 2017). Aβ present in the APs induces TLR-dependent innate immune response that influences Aβ removal in AD (Scholtzova and others 2009). Studies have shown that peripheral inflammation induced by LPS administration induces Aβ formation, APs and impairment of cognitive functions in animal models of AD (Lee and others 2008). Similarly, CNS immune responses also affect peripheral immune responses. Recent studies have shown that diabetes mellitus, hypertension, hyperlipidemia, chronic kidney diseases, depression, stroke, periodontal diseases are clearly associated with cognitive impairment and immune response changes in the CNS (JassamIzzy and others 2017; Kempuraj and others 2017b). Chronic peripheral diseases cause BBB breach and increase the infiltration of immune cells and inflammatory mediators into the brain that in turn further activate glia and enhance neuroinflammation. Similarly, brain injury can cause leakage of debris and inflammatory mediators into periphery and induce systemic inflammatory response syndrome (SIRS) (JassamIzzy and others 2017). SIRS increases peripheral leukocyte numbers and its lifespan as well as the level of inflammatory mediators. Peripheral neutrophils arrive at the injured site in the brain within few hours. One study showed that both peripheral tissue generated and blood-derived Aβ can enter the brain and induce neurodegeneration and AD pathogenesis (Bu and others 2017). Increased duration of chronic periodontal diseases is associated with corresponding increased risk of AD pathogenesis (ChenWu and Chang 2017). The onset, progression and the severity of AD can be modified through preventive measures. Primary prevention may be achieved by reducing or eliminating specific risk factors and remain without AD. In secondary prevention, AD could be detected early before symptoms appear and treatment could delay or halt its progression. Tertiary prevention inhibits the severity of disease complications to improve the quality of life. From the above-mentioned reports, it is clear that there are several factors that could control the onset-progression and severity of TBI-mediated AD (Tables 3–4, Fig. 3).
Table 3.
Brain injury/TBI-associated onset, progression and severity of AD in high-risk people
 |
Abbreviations: Aβ, beta amyloid; AD, Alzheimer’s disease; BBB, blood-brain barrier; BCSFB, blood cerebrospinal fluid barrier; BDNF, brain-derived neurotrophic factor; NFTs, neurofibrillary tangles; TNF-α, tumor necrosis factor-alpha
Table 4.
Neuroimmune and neuroinflammatory responses after brain injury/TBI
| Timings | Brain injury/TBI-mediated onset, progression and severity of Alzheimer’s disease |
Factors accelerating AD pathogenesis Age, gender, race, emotion, peripheral disease, allergic tendencies, genetic factors, environmental factors, and nature of regular food intake can influence TBI-mediated AD pathogenesis |
|
Seconds & Minutes |
Injury to microglia, astrocytes, neurons and endothelial cells lead to direct BBB damage and increased permeability. Injured brain cells release DNA, RNA, and ATP. DAMPs causes mast cell degranulation and release of neuroprotective preactivated cytoplasmic granule stored TNF-α, IL-1α, tryptase, IL-4, IL-33, TGF-β and ROS. DAMPs activate microglia, astrocytes, and release pro and antiinflammatory mediators including IFN-γ, IL-1β, TNF-α, ROS, chemokines CCL2, CCL5, CXCL1, and IL-8. Physical damage to the BBB causes activation of microglia and astrocyte to release more chemokines CCL2, CCL5, CXCL1, CXCL2, and IL-8 to recruit immune cells such as neutrophils, macrophages, T-cells and mast cells. Mast cell released and other mediators increase vascular permeability and vasodilatation. | |
|
Hours |
Neutrophils release ROS, myeloperoxidase and perform phagocytosis. Increased phagocytosis and removal of cell debris by innate immune cells including neutrophils, microglia, macrophage and mast cells. Glial cell number increases (gliosis) to help removal of debris, promote BBB repair, and prevent spreading of injury related effects. Anti-inflammatory mediator release and neuroprotective responses. Mast cell chymase induces basement membrane breakdown. | |
|
Days |
Increased number of microglia, astrocytes, neutrophils, monocytes, T-cells and mast cells at the injury site. Increased expression of endothelial cell adhesion molecules that facilitate infiltration of immune cells from the periphery to the CNS. Tryptase, chymase and PGD2 induce collagen production and deposition. Fibroblast proliferation and growth. | |
|
Weeks |
Over immune and inflammatory responses with decreased removal of debris. Increased activation of astrocytes, microglia with increased activity of inflammatory signaling pathways. Increased release of neuroinflammatory mediators generated within and outside the brain. Wound contracture. Insufficient wound healing and repair process. Generation of Aβ and tau, phosphorylation of tau and its incomplete removal. VEGF induces angiogenesis and new blood vessel formation. Reduced clearance of Aβ from brain. Impaired mechanism of efflux of Aβ through BBB. | |
|
Months |
Vicious neuroinflammatory activation, chronic activation of neuroinflammatory pathways including microglia, astrocyte, T-cells and mast cell activations with peripheral influence. Leaky BBB and chronic disease influence on CNS diseases. Aβ1–42 activates glia and innate immune cells to release additional inflammatory mediators and induces neurodegeneration and neuronal death. | |
|
Years/decade(s) |
Increased amyloid plaques (APs) and neurofibrillary tangle (NFTs) formation in the brain. More activation of glia and immune cells at the vicinity of APs and NFTs. Increased GFAP expression. Chronic peripheral inflammatory diseases accelerate the neuroinflammatory mediators in the brain and increases the activation of glia and neurodegeneration that contribute to more neuroinflamation, neurodegeneration and cognitive decline. Chronic activation of glia, neurons and immune cells with neuroinflammation and neurodegeneration and continued cognitive decline. | |
| Brain injury/TBI accelerated Alzheimer’s disease pathogenesis | ||
Abbreviations: Aβ, beta amyloid; BBB, APs, amyloid plaques; ATP, adenosine triphosphate; blood-brain barrier; CNS, central nervous system; DAMPs, damage-associated molecular patterns; DNA, deoxyribonucleic acid; GFAP, glial fibrillary acidic protein; NFTs, neurofibrillary tangles; RNA, ribonucleic acid; TNF-α, tumor necrosis factor-alpha; IL-1α, interleukin-1alpha; TGF-β, tumor growth factor-beta; ROS, reactive oxygen species; IFN-γ, interferon gamma; CCL2, chemokine (C-C motif) ligand 2; CXCL1, C-X-C motif chemokine 1; PGD2, prostaglandins D2; VEGF, vascular endothelial growth factor
Immune and Inflammatory Pathways as Drug Targets for TBI-induced AD
TBI associated cognitive disorders, morbidity and mortality are still high in patients. It is estimated that about 5.3 million persons in the United States live with a TBI-related disability (the Centers for Disease Control and Prevention report) (Sulhan and others 2018). This is because the exact disease mechanisms are not clearly known. Moreover, studies have shown that neuroimmuine response is an important factor in the recovery of brain function after an injury (Kokiko-Cochran and Godbout 2018; Sun and others 2018). Additionally, the primed microglia in TBI are hyperactive for any subsequent immune challenge and this worsens the recovery in TBI. About 2% of the US population lives with various disabilities due to TBI (Hinson and others 2015). Certain drugs can improve cognitive dysfunctions, but significant improvement in cognitive dysfunction in chronic TBI patients is not yet clearly reported. Recent advances in the understanding of immune and inflammatory responses following brain injury or TBI led to investigate the neuroinflammatory response as a drug target to inhibit, prevent and to teat TBI patients. Neuroinflammatory response can induce secondary brain damage in TBI patients. However, it is not clearly known at what point the treatment should be effective to prevent the TBI mediated pathogenesis. Previous studies have shown clinical trials and therapeutic attempt to target the neuroimmune and neuroinflammatory responses after TBI. Results show that the neuroprotective and neuroinflammatory mechanisms overlap and make it difficult to treat due to its complexity. Several preclinical studies have been shown ineffective and unsuccessful since the exact immune and inflammatory response are still not clearly understood and due to the heterogeneity of patient population and TBI (JassamIzzy and others 2017; Sun and others 2018). Recent pre-clinical and clinical trials have shown significantly improved neuroprotective response in TBI patients. Therefore, it is very important to continue to understand the mechanism of immune and inflammatory response in TBI patients to develop significantly effective therapeutic options for TBI, and AD dementia.
Conclusions and Perspective
Brain injury is usually more common in the general population due to accidents, sports, and in the defense personnel in combat zone. Immune response mediated by innate immune cells such as neutrophils and mast cells prevent and limit inflammatory responses, remove dead cells and debris from the site of injury and promote the wound healing process. However, in severe and widespread injury, the protective innate immune response is insufficient to control the immune response and severe inflammatory response leads to the activation of glia and neuroinflammation. This chronic neuroinflammation leads to increased production of proinflammatory mediators in the CNS as well as in the peripheral systems, secondary tissue damage, and neurobehavioral changes with neuronal death. Mast cell activation in TBI and stress may increase the risk of AD pathogenesis in high-risk group. Increased neurodegeneration causes the onset and progression of AD in these individuals. Thus, TBI can accelerate the pathogenesis of AD. Even though few studies report that there may be no link between TBI and AD, most of the other reported studies do link TBI and AD. However, all the risk factors associated with this link are not yet clearly known and investigated. Moreover, the exact mechanism of this link is also not clearly known including the crucial contribution of the innate immune system. TBI-mediated adverse effects are very heterogeneous in humans. The severity and the duration of pathological changes depends upon the site, extent and the frequency of injury. Several factors including age, gender, diet, chronic peripheral diseases, allergic diseases, emotional characteristics, genetic factors, environmental factors, and certain drugs can affect or accelerate TBI-mediated AD pathogenesis. Additionally, future research should also focus on the emotional activities in an individual that can influence the neuroimmune responses, neurobehavioral characteristics and psychological stress. Effective therapeutic option should inhibit adverse neuroinflammatory pathways but not the immediate protective immune responses that specifically occur after TBI.
Acknowledgments
Funding
This work was funded by Veterans Affairs Merit Award I01BX002477, Veterans Affairs Research Career Scientist Award and National Institutes of Health Grants # AG048205 and NS073670 to Azgar Zaheer.
Footnotes
Declaration of Conflict of Interests
The authors declare no conflict of interest.
References
- Al-Dahhak R, Khoury R, Qazi E, Grossberg GT. 2018. Traumatic Brain Injury, Chronic Traumatic Encephalopathy, and Alzheimer Disease. Clin Geriatr Med 34(4):617–635. [DOI] [PubMed] [Google Scholar]
- Amara J, Iverson KM, Krengel M, Pogoda TK, Hendricks A. 2014. Anticipating the traumatic brain injury-related health care needs of women veterans after the Department of Defense change in combat assignment policy. Womens Health Issues 24(2):e171–6. [DOI] [PubMed] [Google Scholar]
- Amoroso T, Iverson KM. 2017. Acknowledging the Risk for Traumatic Brain Injury in Women Veterans. J Nerv Ment Dis 205(4):318–323. [DOI] [PubMed] [Google Scholar]
- Anada RP, Wong KT, Jayapalan JJ, Hashim OH, Ganesan D. 2018. Panel of serum protein biomarkers to grade the severity of traumatic brain injury. Electrophoresis 39(18):2308–2315. [DOI] [PubMed] [Google Scholar]
- Andreasson KI, Bachstetter AD, Colonna M, Ginhoux F, Holmes C, Lamb B and others. 2016. Targeting innate immunity for neurodegenerative disorders of the central nervous system. J Neurochem 138(5):653–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atiakshin D, Buchwalow I, Samoilova V, Tiemann M. 2018. Tryptase as a polyfunctional component of mast cells. Histochem Cell Biol. 149(5):461–477. [DOI] [PubMed] [Google Scholar]
- Bailes JE, Origenes AK, Alleva JT. 2019. Chronic traumatic encephalopathy. Dis Mon. pii: S0011-5029(19)30035–5. doi: 10.1016/j.disamonth.2019.02.008. [DOI] [PubMed] [Google Scholar]
- Bauer D, Tung ML, Tsao JW. 2015. Mechanisms of traumatic brain injury. Semin Neurol 35(1):e14–22. [DOI] [PubMed] [Google Scholar]
- Becker RE, Kapogiannis D, Greig NH. 2018. Does traumatic brain injury hold the key to the Alzheimer’s disease puzzle? Alzheimers Dement 14(4):431–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogoslovsky T, Wilson D, Chen Y, Hanlon D, Gill J, Jeromin A and others. 2017. Increases of Plasma Levels of Glial Fibrillary Acidic Protein, Tau, and Amyloid beta up to 90 Days after Traumatic Brain Injury. J Neurotrauma 34(1):66–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolos M, Perea JR, Avila J. 2017. Alzheimer’s disease as an inflammatory disease. Biomol Concepts 8(1):37–43. [DOI] [PubMed] [Google Scholar]
- Breunig JJ, Guillot-Sestier MV, Town T. 2013. Brain injury, neuroinflammation and Alzheimer’s disease. Front Aging Neurosci 5:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brizuela M, Blizzard CA, Chuckowree JA, Pitman KA, Young KM, Dickson T. 2017. Mild Traumatic Brain Injury Leads to Decreased Inhibition and a Differential Response of Calretinin Positive Interneurons in the Injured Cortex. J Neurotrauma 34(17):2504–2517. [DOI] [PubMed] [Google Scholar]
- Bu XL, Xiang Y, Jin WS, Wang J, Shen LL, Huang ZL and others. 2017. Blood-derived amyloid-beta protein induces Alzheimer’s disease pathologies. Mol Psychiatry. 23(9):1–9. [DOI] [PubMed] [Google Scholar]
- Caraffa A, Gallenga CE, Tettamanti L, Mastrangelo F and others. 2018. New concepts in neuroinflammation: mast cells pro-inflammatory and anti-inflammatory cytokine mediators. J Biol Regul Homeost Agents 32(3):449–454. [PubMed] [Google Scholar]
- Carlos TM, Clark RS, Franicola-Higgins D, Schiding JK, Kochanek PM. 1997. Expression of endothelial adhesion molecules and recruitment of neutrophils after traumatic brain injury in rats. J Leukoc Biol 61(3):279–85. [DOI] [PubMed] [Google Scholar]
- Caruso A, Nicoletti F, Mango D, Saidi A, Orlando R, Scaccianoce S. 2018. Stress as risk factor for Alzheimer’s disease. Pharmacol Res 132:130–134. [DOI] [PubMed] [Google Scholar]
- Chastain LG, Sarkar DK. 2014. Role of microglia in regulation of ethanol neurotoxic action. Int Rev Neurobiol 118:81–103. [DOI] [PubMed] [Google Scholar]
- Chen CK, Wu YT, Chang YC. 2017. Association between chronic periodontitis and the risk of Alzheimer’s disease: a retrospective, population-based, matched-cohort study. Alzheimers Res Ther 9(1):56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng WH, Stukas S, Martens KM, Namjoshi DR, Button EB, Wilkinson A and others. 2018. Age at injury and genotype modify acute inflammatory and neurofilament-light responses to mild CHIMERA traumatic brain injury in wild-type and APP/PS1 mice. Exp Neurol 301(Pt A):26–38. [DOI] [PubMed] [Google Scholar]
- Chodobski A, Zink BJ, Szmydynger-Chodobska J. 2011. Blood-brain barrier pathophysiology in traumatic brain injury. Transl Stroke Res 2(4):492–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark DPQ, Perreau VM, Shultz SR, Brady RD, Lei E, Dixit S and others. 2019. Inflammation in Traumatic Brain Injury: Roles for Toxic A1 Astrocytes and Microglial-Astrocytic Crosstalk. Neurochem Res. doi: 10.1007/s11064-019-02721-8. [DOI] [PubMed] [Google Scholar]
- Cnossen MC, Scholten AC, Lingsma HF, Synnot A, Haagsma J, Steyerberg PEW and others. 2017. Predictors of Major Depression and Posttraumatic Stress Disorder Following Traumatic Brain Injury: A Systematic Review and Meta-Analysis. J Neuropsychiatry Clin Neurosci 29(3):206–224. [DOI] [PubMed] [Google Scholar]
- Collins-Praino LE, Corrigan F. 2017. Does neuroinflammation drive the relationship between tau hyperphosphorylation and dementia development following traumatic brain injury? Brain Behav Immun 60:369–382. [DOI] [PubMed] [Google Scholar]
- De Berardis D, Serroni N, Campanella D, Marini S, Rapini G, Valchera A and others. 2017. Alexithymia, Suicide Ideation, C-Reactive Protein, and Serum Lipid Levels Among Outpatients with Generalized Anxiety Disorder. Arch Suicide Res 21(1):100–112. [DOI] [PubMed] [Google Scholar]
- Donat CK, Scott G, Gentleman SM, Sastre M. 2017. Microglial Activation in Traumatic Brain Injury. Front Aging Neurosci 9:208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong H, Zhang X, Qian Y. 2014. Mast cells and neuroinflammation. Med Sci Monit Basic Res 20:200–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drews HJ, Yenkoyan K, Lourhmati A, Buadze M, Kabisch D, Verleysdonk S and others. 2019. Intranasal Losartan Decreases Perivascular Beta Amyloid, Inflammation, and the Decline of Neurogenesis in Hypertensive Rats. Neurotherapeutics. doi: 10.1007/s13311-019-00723-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du G, Zhao Z, Chen Y, Li Z, Tian Y, Liu Z and others. 2018. Quercetin protects rat cortical neurons against traumatic brain injury. Mol Med Rep. 17(6):7859–7865. [DOI] [PubMed] [Google Scholar]
- Erlebach R, Pagnamenta A, Klinzing S, Stretti F, Cottini S, Schupbach R and others. 2017. Age-related outcome of patients after traumatic brain injury: a single-center observation. Minerva Anestesiol 83(11):1169–1177. [DOI] [PubMed] [Google Scholar]
- Escher CM, Sannemann L, Jessen F. 2019. Stress and Alzheimer’s disease. J Neural Transm (Vienna). doi: 10.1007/s00702-019-01988-z. [DOI] [PubMed] [Google Scholar]
- Esposito P, Gheorghe D, Kandere K, Pang X, Connolly R, Jacobson S and others. 2001. Acute stress increases permeability of the blood-brain-barrier through activation of brain mast cells. Brain Res 888(1):117–127. [DOI] [PubMed] [Google Scholar]
- Estrada-Rojo F, Martinez-Tapia RJ, Estrada-Bernal F, Martinez-Vargas M, Perez-Arredondo A, Flores-Avalos L and others. 2018. Models used in the study of traumatic brain injury. Rev Neurosci 29(2):139–149. [DOI] [PubMed] [Google Scholar]
- Falcon B, Zivanov J, Zhang W, Murzin AG, Garringer HJ, Vidal R and others. 2019. Novel tau filament fold in chronic traumatic encephalopathy encloses hydrophobic molecules. Nature. doi: 10.1038/s41586-019-1026-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filiano AJ, Gadani SP, Kipnis J. 2017. How and why do T cells and their derived cytokines affect the injured and healthy brain? Nat Rev Neurosci 18(6):375–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabbita SP, Scheff SW, Menard RM, Roberts K, Fugaccia I, Zemlan FP. 2005. Cleaved-tau: a biomarker of neuronal damage after traumatic brain injury. J Neurotrauma 22(1):83–94. [DOI] [PubMed] [Google Scholar]
- Gallenga CE, Pandolfi F, Caraffa A, Kritas SK, Ronconi G, Toniato E and others. 2019. Interleukin-1 family cytokines and mast cells: activation and inhibition. J Biol Regul Homeost Agents 33(1):1–6. [PubMed] [Google Scholar]
- Gardner RC, Burke JF, Nettiksimmons J, Goldman S, Tanner CM, Yaffe K. 2015. Traumatic brain injury in later life increases risk for Parkinson disease. Ann Neurol 77(6):987–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgin-Lavialle S, Moura DS, Salvador A, Chauvet-Gelinier JC, Launay JM, Damaj G and others. 2016. Mast cells’ involvement in inflammation pathways linked to depression: evidence in mastocytosis. Mol Psychiatry 21(11):1511–1516. [DOI] [PubMed] [Google Scholar]
- Griesbach GS, Masel BE, Helvie RE, Ashley MJ. 2018. The Impact of Traumatic Brain Injury on Later Life: Effects on Normal Aging and Neurodegenerative Diseases. J Neurotrauma 35(1):17–24. [DOI] [PubMed] [Google Scholar]
- Hayes JP, Bigler ED, Verfaellie M. 2016. Traumatic Brain Injury as a Disorder of Brain Connectivity. J Int Neuropsychol Soc 22(2):120–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendriksen E, van Bergeijk D, Oosting RS, Redegeld FA. 2017. Mast cells in neuroinflammation and brain disorders. Neurosci Biobehav Rev. 79:119–133. [DOI] [PubMed] [Google Scholar]
- Hinson HE, Rowell S, Schreiber M. 2015. Clinical evidence of inflammation driving secondary brain injury: a systematic review. J Trauma Acute Care Surg 78(1):184–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikonomovic MD, Uryu K, Abrahamson EE, Ciallella JR, Trojanowski JQ, Lee VM and others. 2004. Alzheimer’s pathology in human temporal cortex surgically excised after severe brain injury. Exp Neurol 190(1):192–203. [DOI] [PubMed] [Google Scholar]
- Ilmaniemi S, Taipale H, Tanskanen A, Tiihonen J, Hartikainen S, Tolppanen AM. 2019. Incidence of head injury and traumatic brain injury among people with Alzheimer’s disease. J Epidemiol Community Health. pii: jech-2018–211960. doi: 10.1136/jech-2018-211960. [DOI] [PubMed] [Google Scholar]
- Ito M, Komai K, Mise-Omata S, Iizuka-Koga M, Noguchi Y, Kondo T and others. 2019. Brain regulatory T cells suppress astrogliosis and potentiate neurological recovery. Nature 565(7738):246–250. [DOI] [PubMed] [Google Scholar]
- Jaber SM, Polster BM. 2017. An in vitro model yields ‘importin’ new insights into chronic traumatic encephalopathy: damaged astrocytes stop ‘thrombospondin’ to the injury: An Editorial Highlight for ‘Defective synthesis and release of astrocytic thrombospondin-1 mediates the neuronal TDP-43 proteinopathy, resulting in defects in neuronal integrity associated with chronic traumatic encephalopathy: in vitro studies’. J Neurochem 140(4):531–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jassam YN, Izzy S, Whalen M, McGavern DB, El Khoury J. 2017. Neuroimmunology of Traumatic Brain Injury: Time for a Paradigm Shift. Neuron 95(6):1246–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayakumar AR, Tong XY, Shamaladevi N, Barcelona S, Gaidosh G, Agarwal A and others. 2017. Defective synthesis and release of astrocytic thrombospondin-1 mediates the neuronal TDP-43 proteinopathy, resulting in defects in neuronal integrity associated with chronic traumatic encephalopathy: in vitro studies. J Neurochem 140(4):645–661. [DOI] [PubMed] [Google Scholar]
- Jenkins DR, Craner MJ, Esiri MM, DeLuca GC. 2018. Contribution of Fibrinogen to Inflammation and Neuronal Density in Human Traumatic Brain Injury. J Neurotrauma 35(19):2259–2271. [DOI] [PubMed] [Google Scholar]
- Jin Y, Silverman AJ, Vannucci SJ. 2009. Mast cells are early responders after hypoxia-ischemia in immature rat brain. Stroke 40(9):3107–12. [DOI] [PubMed] [Google Scholar]
- Johnson VE, Meaney DF, Cullen DK, Smith DH. 2015. Animal models of traumatic brain injury. Handb Clin Neurol 127:115–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson VE, Stewart W, Smith DH. 2012. Widespread tau and amyloid-beta pathology many years after a single traumatic brain injury in humans. Brain Pathol 22(2):142–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones SV, Kounatidis I. 2017. Nuclear Factor-Kappa B and Alzheimer Disease, Unifying Genetic and Environmental Risk Factors from Cell to Humans. Front Immunol 8:1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju Hwang C, Choi DY, Park MH, Hong JT. 2019. NF-kB as a mediator of brain inflammation in Alzheimer’s Disease. CNS Neurol Disord Drug Targets. 18(1):3–10. [DOI] [PubMed] [Google Scholar]
- Kanazawa M, Ninomiya I, Hatakeyama M, Takahashi T, Shimohata T. 2017. Microglia and Monocytes/Macrophages Polarization Reveal Novel Therapeutic Mechanism against Stroke. Int J Mol Sci 18(10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsumoto A, Takeuchi H, Takahashi K, Tanaka F. 2018. Microglia in Alzheimer’s Disease: Risk Factors and Inflammation. Front Neurol 9:978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kempuraj D, Mentor S, Thangavel R, Ahmed ME, Selvakumar GP, Raikwar SP and others. 2019. Mast Cells in Stress, Pain, Blood-Brain Barrier, Neuroinflammation and Alzheimer’s Disease. Front Cell Neurosci 13:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kempuraj D, Selvakumar GP, Thangavel R, Ahmed ME, Zaheer S, Kumar KK and others. 2018a. Glia Maturation Factor and Mast Cell-Dependent Expression of Inflammatory Mediators and Proteinase Activated Receptor-2 in Neuroinflammation. J Alzheimers Dis.66(3):1117–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kempuraj D, Selvakumar GP, Thangavel R, Ahmed ME, Zaheer S, Raikwar SP and others. 2017a. Mast Cell Activation in Brain Injury, Stress, and Post-traumatic Stress Disorder and Alzheimer’s Disease Pathogenesis. Front Neurosci 11:703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kempuraj D, Thangavel R, Natteru PA, Selvakumar GP, Saeed D, Zahoor H and others. 2016. Neuroinflammation Induces Neurodegeneration. J Neurol Neurosurg Spine 1(1). pii: 1003. [PMC free article] [PubMed] [Google Scholar]
- Kempuraj D, Thangavel R, Selvakumar GP, Ahmed ME, Zaheer S, Raikwar SP and others. 2018b. Mast Cell Proteases Activate Astrocytes and Glia-Neurons and Release Interleukin-33 by Activating p38 and ERK1/2 MAPKs and NF-kappaB. Mol Neurobiol. 56(3):1681–1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kempuraj D, Thangavel R, Selvakumar GP, Zaheer S, Ahmed ME, Raikwar SP and others. 2017b. Brain and Peripheral Atypical Inflammatory Mediators Potentiate Neuroinflammation and Neurodegeneration. Front Cell Neurosci 11:216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kober DL, Brett TJ. 2017. TREM2-Ligand Interactions in Health and Disease. J Mol Biol 429(11):1607–1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokiko-Cochran O, Ransohoff L, Veenstra M, Lee S, Saber M, Sikora M and others. 2016. Altered Neuroinflammation and Behavior after Traumatic Brain Injury in a Mouse Model of Alzheimer’s Disease. J Neurotrauma 33(7):625–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokiko-Cochran ON, Godbout JP. 2018. The Inflammatory Continuum of Traumatic Brain Injury and Alzheimer’s Disease. Front Immunol 9:672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korhonen P, Kanninen KM, Lehtonen S, Lemarchant S, Puttonen KA, Oksanen M and others. 2015. Immunomodulation by interleukin-33 is protective in stroke through modulation of inflammation. Brain Behav Immun 49:322–36. [DOI] [PubMed] [Google Scholar]
- Kritas SK, Gallenga CE, C DO, Ronconi G, Caraffa A, Toniato E and others. 2018. Impact of mold on mast cell-cytokine immune response. J Biol Regul Homeost Agents 32(4):763–768. [PubMed] [Google Scholar]
- Kumar A, Stoica BA, Loane DJ, Yang M, Abulwerdi G, Khan N and others. 2017. Microglial-derived microparticles mediate neuroinflammation after traumatic brain injury. J Neuroinflammation 14(1):47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumaria A 2017. In vitro models as a platform to investigate traumatic brain injury. Altern Lab Anim 45(4):201–211. [DOI] [PubMed] [Google Scholar]
- Laurent C, Buee L, Blum D. 2018. Tau and neuroinflammation: What impact for Alzheimer’s Disease and Tauopathies? Biomed J 41(1):21–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JW, Lee YK, Yuk DY, Choi DY, Ban SB, Oh KW and others. 2008. Neuro-inflammation induced by lipopolysaccharide causes cognitive impairment through enhancement of beta-amyloid generation. J Neuroinflammation 5:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KW, Kim JB, Seo JS, Kim TK, Im JY, Baek IS and others. 2009. Behavioral stress accelerates plaque pathogenesis in the brain of Tg2576 mice via generation of metabolic oxidative stress. J Neurochem 108(1):165–75. [DOI] [PubMed] [Google Scholar]
- Levy Nogueira M, Hamraz M, Abolhassani M, Bigan E, Lafitte O, Steyaert JM and others. 2018. Mechanical stress increases brain amyloid beta, tau, and alpha-synuclein concentrations in wild-type mice. Alzheimers Dement 14(4):444–453. [DOI] [PubMed] [Google Scholar]
- Li JT, Zhang Y. 2018. TREM2 regulates innate immunity in Alzheimer’s disease. J Neuroinflammation 15(1):107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsberg PJ, Strbian D, Karjalainen-Lindsberg ML. 2010. Mast cells as early responders in the regulation of acute blood-brain barrier changes after cerebral ischemia and hemorrhage. J Cereb Blood Flow Metab 30(4):689–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu YW, Li S, Dai SS. 2018. Neutrophils in traumatic brain injury (TBI): friend or foe? J Neuroinflammation 15(1):146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LoBue C, Cullum CM, Didehbani N, Yeatman K, Jones B, Kraut MA and others. 2018. Neurodegenerative Dementias After Traumatic Brain Injury. J Neuropsychiatry Clin Neurosci 30(1):7–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackey E, Ayyadurai S, Pohl CS, S DC, Li Y, Moeser AJ. 2016. Sexual dimorphism in the mast cell transcriptome and the pathophysiological responses to immunological and psychological stress. Biol Sex Differ 7:60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacQueen R, Fisher P, Williams D. 2018. A qualitative investigation of masculine identity after traumatic brain injury. Neuropsychol Rehabil:1–17. [DOI] [PubMed] [Google Scholar]
- McAteer KM, Turner RJ, Corrigan F. 2017. Animal models of chronic traumatic encephalopathy. Concussion 2(2):CNC32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKee CA, Lukens JR. 2016. Emerging Roles for the Immune System in Traumatic Brain Injury. Front Immunol 7:556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McManus RM, Heneka MT. 2017. Role of neuroinflammation in neurodegeneration: new insights. Alzheimers Res Ther 9(1):14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medeiros R, Kitazawa M, Passos GF, Baglietto-Vargas D, Cheng D, Cribbs DH and others. 2013. Aspirin-triggered lipoxin A4 stimulates alternative activation of microglia and reduces Alzheimer disease-like pathology in mice. Am J Pathol 182(5):1780–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menzel L, Kleber L, Friedrich C, Hummel R, Dangel L, Winter J and others. 2017. Progranulin protects against exaggerated axonal injury and astrogliosis following traumatic brain injury. Glia 65(2):278–292. [DOI] [PubMed] [Google Scholar]
- Meyer PF, Savard M, Poirier J, Labonte A, Rosa-Neto P, Weitz TM and others. 2018. Bi-directional Association of Cerebrospinal Fluid Immune Markers with Stage of Alzheimer’s Disease Pathogenesis. J Alzheimers Dis 63(2):577–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moretti R, Chhor V, Bettati D, Banino E, De Lucia S, Le Charpentier T and others. 2016. Contribution of mast cells to injury mechanisms in a mouse model of pediatric traumatic brain injury. J Neurosci Res 94(12):1546–1560. [DOI] [PubMed] [Google Scholar]
- Morganti-Kossmann MC, Rancan M, Stahel PF, Kossmann T. 2002. Inflammatory response in acute traumatic brain injury: a double-edged sword. Curr Opin Crit Care 8(2):101–5. [DOI] [PubMed] [Google Scholar]
- Mukai K, Tsai M, Saito H, Galli SJ. 2018. Mast cells as sources of cytokines, chemokines, and growth factors. Immunol Rev 282(1):121–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Najem D, Rennie K, Ribecco-Lutkiewicz M, Ly D, Haukenfrers J, Liu Q and others. 2018. Traumatic Brain Injury: Classification, Models and Markers. Biochem Cell Biol. 96(4):391–406. [DOI] [PubMed] [Google Scholar]
- Negi N, Das BK. 2018. CNS: Not an immunoprivilaged site anymore but a virtual secondary lymphoid organ. Int Rev Immunol 37(1):57–68. [DOI] [PubMed] [Google Scholar]
- Nutma E, Willison H, Gianvito M, Amor S. 2019. Neuroimmunology - The Past, Present and Future. Clin Exp Immunol. doi: 10.1111/cei.13279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ocak U, Ocak PE, Wang A, Zhang JH, Boling W, Wu P and others. 2018. Targeting mast cell as a neuroprotective strategy. Brain Inj:1–11. [DOI] [PubMed] [Google Scholar]
- Omalu BI, DeKosky ST, Minster RL, Kamboh MI, Hamilton RL, Wecht CH. 2005. Chronic traumatic encephalopathy in a National Football League player. Neurosurgery 57(1):128–34; discussion 128–34. [DOI] [PubMed] [Google Scholar]
- Otaegui-Arrazola A, Amiano P, Elbusto A, Urdaneta E, Martinez-Lage P. 2014. Diet, cognition, and Alzheimer’s disease: food for thought. Eur J Nutr 53(1):1–23. [DOI] [PubMed] [Google Scholar]
- Oyesanya TO, Ward EC. 2016. Mental Health in Women With Traumatic Brain Injury: A Systematic Review on Depression and Hope. Health Care Women Int 37(1):45–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PrabhuDas MR, Baldwin CL, Bollyky PL, Bowdish DME, Drickamer K, Febbraio M and others. 2017. A Consensus Definitive Classification of Scavenger Receptors and Their Roles in Health and Disease. J Immunol 198(10):3775–3789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puntambekar SS, Saber M, Lamb BT, Kokiko-Cochran ON. 2018. Cellular players that shape evolving pathology and neurodegeneration following traumatic brain injury. Brain Behav Immun. 71:9–17. [DOI] [PubMed] [Google Scholar]
- Rayasam A, Hsu M, Kijak JA, Kissel L, Hernandez G, Sandor M and others. 2018. Immune responses in stroke: how the immune system contributes to damage and healing after stroke and how this knowledge could be translated to better cures? Immunology. 154(3):363–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribatti D 2015. The crucial role of mast cells in blood-brain barrier alterations. Exp Cell Res 338(1):119–25. [DOI] [PubMed] [Google Scholar]
- Roberts GW, Gentleman SM, Lynch A, Murray L, Landon M, Graham DI. 1994. Beta amyloid protein deposition in the brain after severe head injury: implications for the pathogenesis of Alzheimer’s disease. J Neurol Neurosurg Psychiatry 57(4):419–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo MV, McGavern DB. 2016. Inflammatory neuroprotection following traumatic brain injury. Science 353(6301):783–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawmiller D, Li S, Shahaduzzaman M, Smith AJ, Obregon D, Giunta B and others. 2014. Luteolin reduces Alzheimer’s disease pathologies induced by traumatic brain injury. Int J Mol Sci 15(1):895–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholtzova H, Kascsak RJ, Bates KA, Boutajangout A, Kerr DJ, Meeker HC and others. 2009. Induction of toll-like receptor 9 signaling as a method for ameliorating Alzheimer’s disease-related pathology. J Neurosci 29(6):1846–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaik-Dasthagirisaheb YB, Conti P. 2016. The Role of Mast Cells in Alzheimer’s Disease. Adv Clin Exp Med 25(4):781–7. [DOI] [PubMed] [Google Scholar]
- Skaper SD, Facci L, Zusso M, Giusti P. 2017. Neuroinflammation, Mast Cells, and Glia: Dangerous Liaisons. Neuroscientist 23(5):478–498. [DOI] [PubMed] [Google Scholar]
- Skaper SD, Facci L, Zusso M, Giusti P. 2018. An Inflammation-Centric View of Neurological Disease: Beyond the Neuron. Front Cell Neurosci 12:72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DH, Johnson VE, Trojanowski JQ, Stewart W. 2019. Chronic traumatic encephalopathy - confusion and controversies. Nat Rev Neurol 15(3):179–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song S, Kong X, Acosta S, Sava V, Borlongan C, Sanchez-Ramos J. 2016. Granulocyte colony-stimulating factor promotes behavioral recovery in a mouse model of traumatic brain injury. J Neurosci Res 94(5):409–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorby-Adams AJ, Vink R, Turner RJ. 2018. Large animal models of stroke and traumatic brain injury as translational tools. Am J Physiol Regul Integr Comp Physiol. 315(2):R165–R190. [DOI] [PubMed] [Google Scholar]
- Sulhan S, Lyon KA, Shapiro LA, Huang JH. 2018. Neuroinflammation and blood-brain barrier disruption following traumatic brain injury: Pathophysiology and potential therapeutic targets. J Neurosci Res. doi: 10.1002/jnr.24331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun M, McDonald SJ, Brady RD, O’Brien TJ, Shultz SR. 2018. The influence of immunological stressors on traumatic brain injury. Brain Behav Immun 69:618–628. [DOI] [PubMed] [Google Scholar]
- Sweeney MD, Sagare AP, Zlokovic BV. 2018. Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat Rev Neurol 14(3):133–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweeney MD, Zhao Z, Montagne A, Nelson AR, Zlokovic BV. 2019. Blood-Brain Barrier: From Physiology to Disease and Back. Physiol Rev 99(1):21–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taipale H, Koponen M, Tanskanen A, Lavikainen P, Sund R, Tiihonen J and others. 2017. Risk of head and traumatic brain injuries associated with antidepressant use among community-dwelling persons with Alzheimer’s disease: a nationwide matched cohort study. Alzheimers Res Ther 9(1):59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeshita Y, Ransohoff RM. 2012. Inflammatory cell trafficking across the blood-brain barrier: chemokine regulation and in vitro models. Immunol Rev 248(1):228–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thangavel R, Bhagavan SM, Ramaswamy SB, Surpur S, Govindarajan R, Kempuraj D and others. 2018. Co-Expression of Glia Maturation Factor and Apolipoprotein E4 in Alzheimer’s Disease Brain. J Alzheimers Dis 61(2):553–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theoharides TC, Rozniecki JJ, Sahagian G, Jocobson S, Kempuraj D, Conti P and others. 2008. Impact of stress and mast cells on brain metastases. J Neuroimmunol 205(1–2):1–7. [DOI] [PubMed] [Google Scholar]
- Theoharides TC, Tsilioni I. 2018. Tetramethoxyluteolin for the treatment of neurodegenerative diseases. Curr Top Med Chem. 18(21):1872–1882. [DOI] [PubMed] [Google Scholar]
- Umar T, Hoda N. 2018. Alzheimer’s Disease: A Systemic Review Of Substantial Therapeutic Targets And The Leading Multi-Functional Molecules. Curr Top Med Chem. 17(31):3370–3389. [DOI] [PubMed] [Google Scholar]
- Van Cauwenberghe C, Van Broeckhoven C, Sleegers K. 2016. The genetic landscape of Alzheimer disease: clinical implications and perspectives. Genet Med 18(5):421–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanuytsel T, van Wanrooy S, Vanheel H, Vanormelingen C, Verschueren S, Houben E and others. 2014. Psychological stress and corticotropin-releasing hormone increase intestinal permeability in humans by a mast cell-dependent mechanism. Gut 63(8):1293–9. [DOI] [PubMed] [Google Scholar]
- Varricchi G, Loffredo S, Galdiero MR, Marone G, Cristinziano L, Granata F and others. 2018. Innate effector cells in angiogenesis and lymphangiogenesis. Curr Opin Immunol 53:152–160. [DOI] [PubMed] [Google Scholar]
- Villa A, Vegeto E, Poletti A, Maggi A. 2016. Estrogens, Neuroinflammation, and Neurodegeneration. Endocr Rev 37(4):372–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weeks JJ, Carlson LJ, Radabaugh HL, de la Tremblaye PB, Bondi CO, Kline AE. 2018. Intermittent treatment with haloperidol or quetiapine does not disrupt motor and cognitive recovery after experimental brain trauma. Behav Brain Res 340:159–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner MW, Crane PK, Montine TJ, Bennett DA, Veitch DP. 2017. Traumatic brain injury may not increase the risk of Alzheimer disease. Neurology 89(18):1923–1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojnarowicz MW, Fisher AM, Minaeva O, Goldstein LE. 2017. Considerations for Experimental Animal Models of Concussion, Traumatic Brain Injury, and Chronic Traumatic Encephalopathy-These Matters Matter. Front Neurol 8:240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf SA, Boddeke HW, Kettenmann H. 2017. Microglia in Physiology and Disease. Annu Rev Physiol 79:619–643. [DOI] [PubMed] [Google Scholar]
- Woolhouse R, McKinlay A, Grace RC. 2017. Women in Prison With Traumatic Brain Injury: Prevalence, Mechanism, and Impact on Mental Health. Int J Offender Ther Comp Criminol:306624X17726519. [DOI] [PubMed] [Google Scholar]
- Xiong Y, Mahmood A, Chopp M. 2018. Current understanding of neuroinflammation after traumatic brain injury and cell-based therapeutic opportunities. Chin J Traumatol. 21(3):137–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Tan L, Wang HF, Jiang T, Tan MS, Tan L and others. 2015. Meta-analysis of modifiable risk factors for Alzheimer’s disease. J Neurol Neurosurg Psychiatry 86(12):1299–306. [DOI] [PubMed] [Google Scholar]
- Yang C, Hawkins KE, Dore S, Candelario-Jalil E. 2019. Neuroinflammatory mechanisms of blood-brain barrier damage in ischemic stroke. Am J Physiol Cell Physiol 316(2):C135–C153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaheer A, Sahu SK, Wu Y, Zaheer A, Haas J, Lee K and others. 2007. Diminished cytokine and chemokine expression in the central nervous system of GMF-deficient mice with experimental autoimmune encephalomyelitis. Brain Res 1144:239–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao N, Liu CC, Qiao W, Bu G. 2018. Apolipoprotein E, Receptors, and Modulation of Alzheimer’s Disease. Biol Psychiatry 83(4):347–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziemka-Nalecz M, Jaworska J, Zalewska T. 2017. Insights Into the Neuroinflammatory Responses After Neonatal Hypoxia-Ischemia. J Neuropathol Exp Neurol 76(8):644–654. [DOI] [PubMed] [Google Scholar]