

Abstract
Self-assembly of short peptides gives rise to versatile nanoassemblies capable of promoting efficient catalysis. We have semi-rationally designed a series of seven-residue peptides that form hemin-binding catalytic amyloids to facilitate enantioselective cyclopropanation with efficiencies that rival those of engineered hemin proteins. These results demonstrate that: 1. Catalytic amyloids can bind complex metallocofactors to promote practically important multisubstrate transformations; 2. Even essentially flat surfaces of amyloid assemblies can impart a substantial degree of enantioselectivity without the need for extensive optimization; 3. The ease of peptide preparation allows for straightforward incorporation of unnatural amino acids and preparation of peptides made of D-amino acids with complete reversal of enantioselectivity.
Keywords: self-assembly, peptide, amyloid, catalysis, cyclopropanation
Graphical Abstract
Catalysis through self-assembly: Self-assembled nanostructures formed by semi-rationally designed seven-residue peptides and hemin catalyze efficient formation of cyclopropanes in an enantioselective manner.
Self-assembly is a powerful tool to create large supramolecular structures with a wide range of practically important applications ranging from biomineralization to catalysis.[1] Peptides, having functional groups that provide hydrogen bonding, electrostatic interactions, aromatic stacking and van der Waals forces, are particularly well suited to serve as building blocks for larger supramolecular structures.[2] Recently, discovery of highly efficient and functionally versatile catalytic amyloids showed great potential for development of new, easy to make, stable and robust catalysts, for a variety of chemical transformations.[3]
Catalytic amyloids show catalytic efficiency that rival those of natural enzymes by weight[4] and can facilitate hydrolytic and redox transformations as well as tandem reactions.[5] At the same time none of the successful examples of catalytic amyloids reported have yet to tap into the incredible potential of complex metal-containing cofactors. Porphyrin derivatives are widely used in nature for a diverse set of applications ranging from light harvesting to respiration and C-H bond activation, thus we decided to explore the potential of self-assembling peptides to accommodate hemin in a functional manner. Despite the outstanding potential of the porphyrin-based cofactors to promote catalysis examples of its use in self-assembled peptide systems remain sparse and are limited to peroxidase activity.[3i, 6]
Here we set out to explore whether catalytic amyloids can effectively bind hemin to efficiently promote catalysis in multisubstrate reactions. Multisubstrate reactions constitute a major challenge in de novo designed catalytic systems as productive association of different substrates with the active sites is necessary for efficient turnover. Moreover, we are interested in reactions that are bond-forming and resulting in formation of complex products. Finally, in this work we probed whether fundamentally chiral peptides that nonetheless form fairly flat assemblies[4c] are capable of enantioselective catalysis. In this study we focused on cyclopropanation of aromatic styrenes (Scheme 1); this practically important reaction results in formation of carbon-carbon bonds with new stereocenters.[7] Engineered heme-containing proteins have been shown to promote this transformation with high efficiency and enantioselectivity,[8] but the question whether peptide assemblies can support this transformation remained unanswered.
Scheme 1.
Schematic representation of hemin association with catalytic amyloids to promote cyclopropanation.
Previously we have developed a series of seven-residue peptides capable of self-assembly in the presence of transition metal ions to form catalytic amyloids that promote hydrolytic and redox transformations.[4a, 4b, 5] First, we have tested the ability of these peptides to coordinate hemin. The Soret band offers a sensitive and informative tool for quantitative characterization of association of various metalloporphyrins with exogenous ligands. Interestingly, the peptides that showed the highest propensity to promote hydrolysis in the presence of Zn2+ (Ac-IHIHIQI-CONH2 and Ac-IHIHIYI-CONH2) did not show any appreciable binding of hemin (Figure S1j-k, Supporting Information). Similarly, the valine core peptides that assemble rapidly both in the presence and absence of transition metals did not associate with hemin. At the same time, a distinct shift in Soret band was observed after 24 hours of incubation of hemin at pH 7 with Ac-LHLHLQL-NH2 (Figure S1l, Supporting Information), a peptide that relies of the leucine core for self-assembly with morphology different from that of Ac-IHIHIQI-CONH2, indicating some degree of histidine-iron coordination.
Encouraged by this result, we investigated the role of polar residues in position 6 of the sequence on hemin binding properties. Specifically, we replaced Gln to Phe to achieve two goals: a) increase hydrophobicity of the assembly thus making it more likely to interact with both hydrophobic cofactor and, ultimately, hydrophobic substrates; b) favor hemin binding to fibrils by introducing stacking interactions between the aromatic system of the cofactor and the side chain of the newly introduced amino acid. The resulting peptide Ac-LHLHLFL-NH2 forms β-sheet assemblies (Figure 1B) which bind hemin as shown by a significant (more than 20 nm) red shift of the Soret band maximum to 422 nm after 24 hours of incubation (Figure 1A) and the emergence of the corresponding bifurcated CD signal confirming the chiral environment around the porphyrin ring (Figure 1B, Inset).[9]
Figure 1.
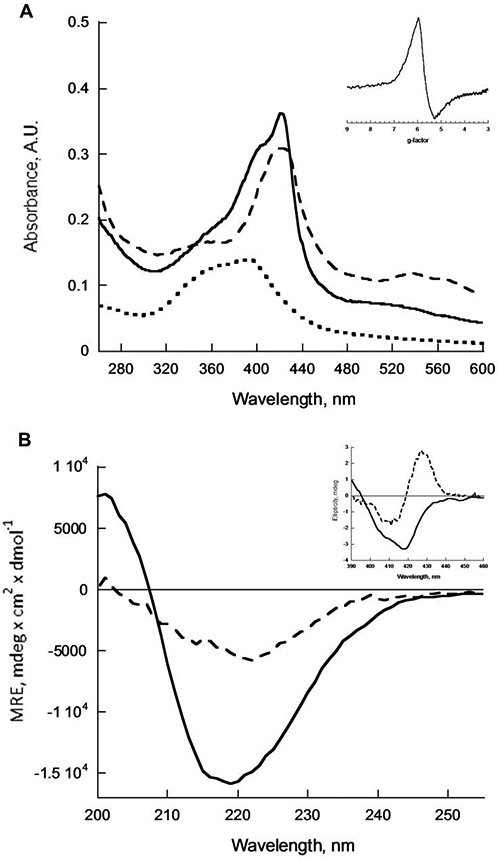
(A) UV-Vis spectra of 4 μM hemin (dotted trace), mixture of 30 μM Ac-LHLHLFL-NH2 with 4 μM hemin (dashed trace) and 30 μM Ac-LILHLFL-NH2 with 4 μM hemin (solid trace). Inset: EPR spectrum of 750 μM Ac-LILHLFL-NH2 with 100 μM hemin (B) Circular Dichroism spectra of 30 μM Ac-LHLHLFL-NH2 (dashed trace) and 30 μM Ac-LILHLFL-NH2 (solid trace). Inset: CD of Soret band of LHLHLFL-NH2 with 4 μM hemin (dashed trace) and 30 μM Ac-LILHLFL-NH2 with 4 μM hemin (solid trace). All measurements were taken in 100 mM phosphate buffer at pH 7 after 24 hours of incubation.
Excitingly, the resulting assemblies are capable of promoting diastereo- and enantiospecific cyclopropanation, albeit with modest yields and selectivities (Table 1). The diasteromeric ratio is fairly similar (within 10%) to the values shown by the hemin alone consistent with the overall reaction mechanism.
Table 1.
Catalytic activity and selectivity of peptide-hemin complexes in cyclopropanation of 4-(trifluoromethyl)styrene with EDA.[a]
| Peptide | Yield, %[b] | de, % [c] | eecis, %[d] | eetrans, %[d] |
|---|---|---|---|---|
| LHLHLFL | 39 | 76 | 4 | 6 |
| LHLALFL | 22 | 81 | 4 | 3 |
| LALHLFL | 28 | 69 | 6 | 7 |
| LFLHLFL | 42 | 77 | <1 | <1 |
| LLLHLFL | 48 | 72 | 1 | 6 |
| LTLHLFL | 30 | 72 | 0 | 1 |
| LILHLFL | 52 | 68 | −12 | 40 |
| LGLHLFL | 31 | 66 | 9 | 5 |
| LVLHLFL | 16 | 75 | 5 | 25 |
| LMLHLFL | 26 | 65 | 5 | 16 |
| LHLH(L-NMe)FL | 16 | 80 | 0 | 0 |
| LIL(3Me-H)LFL | 18 | 77 | 0 | 0 |
| AHAHAFA | 71 | 77 | 0 | <1 |
| Hemin | 15 | 70 | 0 | 0 |
Reaction conditions: 20 μM hemin with 150 μM peptide, 8 mM styrene, 24 mM EDA, 10 mM sodium dithionite, 100 mM phosphate buffer pH 7, 1 h. The catalysts were incubated for 24 hours prior to the experiments.
Yields were determined using GC.
Diastereomeric excess (de) values were calculated using the formula de = 100 x ([trans] − [cis])/([trans] + [cis])
Enantiomeric excess (ee) values were calculated using the formulae: eecis = 100 x ([1S,2R] − [1R,2S])/([1S,2R] + [1R,2S]); eetrans = 100 x ([1S,2S] − [1R,2R])/([1S,2S] + [1R,2R]).
In the next round of the design we focused on the residues responsible for metal coordination. Since only one histidine residue is required for hemin coordination, next we determined which one is best suited for functional incorporation of hemin into fibrils. We prepared two peptides where either of the histidine was replaced with alanine (Ac-LALHLFL-NH2 and Ac-LHLALFL-NH2) and spectroscopically monitored their association with hemin (Figure S1a-b, Supporting Information). Both peptides bound hemin, but the Soret band red shift is slightly more pronounced in the former case.
Both peptides promoted cyclopropanation of 4-(trifluoromethyl)styrene with ethyl diazoacetate (EDA). Importantly, Ac-LALHLFL-NH2-hemin showed enantioselectivity favoring the cis-1S,2R and 3% trans-1S,2S products (Table 1). Having identified that the binding of hemin by histidine in the fourth position is important for enantioselectivity, we subsequently probed the effect of different residues in position 2. In agreement with our original findings we observed that introduction of hydrophobic moieties further improved binding and reactivity of the self-assembling catalysts, whereas our attempts to introduce hydrogen bond donors did not produce any improvement in yields/selectivity of cyclopropanation reactions (Table 1). Considering that all peptides bound hemin and formed β-sheet assemblies (Figures S1, S5, S6, Supporting Information) this observation suggests that minor changes in sequence result in substantial changes in reactivity.
Ac-LILHLFL-NH2 showed the highest enantioselectivity and was characterized in more detail. Transmission electron microscopy (TEM) of Ac-LILHLFL-NH2 in complex with hemin (10:1 peptide:hemin ratio, Figure 2) shows well-resolved amyloid-like assemblies with morphologies that are distinctly different to those formed by Ac-LHLHLFL-NH2 (Figure S2a-b Supporting Information). The CD spectra of the fibrils are consistent with β-sheet secondary structure and the emergence of the CD signature in the Soret band region demonstrates hemin association with the catalysts (Figure 1A, B). Interestingly, the Soret band CD signal shows a lesser degree of exciton coupling as compared to LHLHLFL-hemin consistent with a slightly different cofactor microenvironment. UV-Vis titration studies, including a Job plot, of hemin association with peptide assemblies demonstrate ~4:1 peptide monomer to hemin stoichiometry (Figure S3, Supporting Information) with an apparent Kd of 2 ± 0.7 μM. The EPR studies of the Ac-LILHLFL-NH2 – hemin complex show a typical axial high spin Fe(III) heme signature consistent with a single histidine coordination of the iron (Figure 1A). This observation can also serve as indirect evidence that most of the cofactors are located on the surface of the fibrils and are easily accessible for the substrate.
Figure 2.

Representative TEM images of nanostructures formed by Ac-LILHLFL-NH2 with hemin (peptide:hemin is 10:1) in 100 mM pH 7 phosphate buffer and incubated for 24 hours.
Given the ease of incorporation of unnatural amino acids into short peptide sequences we probed the effects of substituting the hemin-binding histidine in Ac-LILHLFL-NH2 with 3-methyl-histidine (3Me-H). 3Me-H has been previously shown to improve catalytic properties in engineered myoglobins.[10] While introduction of the unnatural amino acid did not disrupt the assembly as confirmed by CD spectra (Figure S5n, Supporting Information) the hemin Soret band position was significantly different from the one in the hemin-Ac-LILHLFL-NH2 assemblies (Figure S6f, Supporting Information). Moreover, Ac-LIL(3Me-H)LFL-NH2-hemin complex showed no enantioselectivity in cyclopropanation (Table 1). This observation suggests potential importance of the hydrogen bonding in the histidine side chain for binding of the cofactor in the catalytically competent conformation.
In order to evaluate the effects of self-assembly on peptide reactivity and enantioselectivity we tested cyclopropanation promoted by hemin itself as well as hemin in the complex with peptides that do not form assemblies in the presence of the cofactor. Hemin on its own promotes cyclopropanation quite poorly giving a 70% de and no enantiomeric excess (Table 1). We have also prepared Ac-LHLH(L-NMe)FL-NH2 which had methylated backbone nitrogen, as well as a related peptide Ac-AHAHAFA-NH2 that lacks a hydrophobic core. These modifications render both peptides incapable of self-assembly as well as forming secondary structures, although they still bind hemin to some extent (Figure S1f, S6e, Supporting Information). Ac-LHLH(L-NMe)FL-NH2 shows the activity levels comparable to those of the uncoordinated hemin, whereas its more hydrophilic analog Ac-AHAHAFA-NH2 demonstrated improvement in activity, in fact it was the most active peptide of the series, yet showed no enantioselectivity. These results have two important implications: axial hemin coordination with a concomitant breakup of hemin aggregates leads to a large improvement in ability of the cofactor to promote cyclopropanation and self-assembly to create higher order supramolecular structures is absolutely essential for the enantioselectivity of the resulting catalysts.
Having identified Ac-LILHLFL-NH2 as the peptide that imparts the greatest enantioselectivity, we performed a substrate scope studies (Table 2). In all cases tested, which included both electron deficient and electron rich substrates, as well as substrates with various substitution patterns we observed cyclopropanation with good product yields (Table 2). Moreover, in all cases the catalyst was enantioselective, albeit with a different degree of preference for the trans-1S,2S products.
Table 2.
Substrate scope in cyclopropanation reaction catalyzed by peptide-hemin -assemblies.[a]
| Substrate | Catalyst | Yiel, %[b] |
eecis, %[c] |
eetrans, %[c] |
|---|---|---|---|---|
| styrene | Ac-LILHLFL-NH2 | 68 | −12 | 19 |
| (D)-Ac-LILHLFL-NH2 | 70 | 13 | −19 | |
| 4-chlorostyrene | Ac-LILHLFL-NH2 | 80 | −13 | 38 |
| (D)-Ac-LILHLFL-NH2 | 74 | 13 | −39 | |
| 4-(trifluoromethyl) styrene |
Ac-LILHLFL-NH2 | 52 | −12 | 40 |
| (D)-Ac-LILHLFL-NH2 | 50 | 11 | −40 | |
| 4-vinylphenylacetate | Ac-LILHLFL-NH2 | 71 | −8 | 26 |
| (D)-Ac-LILHLFL-NH2 | 88 | 7 | −27 | |
| 3-methylstyrene | Ac-LILHLFL-NH2 | 65 | −20 | 28 |
| (D)-Ac-LILHLFL-NH2 | 61 | 19 | −26 | |
| α-methylstyrene | Ac-LILHLFL-NH2 | 95 | −7 | 24 |
| (D)-Ac-LILHLFL-NH2 | 91 | 7 | −25 |
Reaction conditions: 20 μM hemin with 150 μM peptide, 8 mM styrene, 24 mM EDA, 10 mM dithionite, 100 mM pH 7 phosphate buffer, 1 h.
Yields were based on GC analysis of the worked up reaction mixture. Enantiomeric excess (ee) values were calculated using the formulae: eecis = 100 x ([1S,2R] − [1R,2S])/([1S,2R] + [1R,2S]); eetrans = 100 x ([1S,2S] − [1R,2R])/([1S,2S] + [1R,2R]).
We chose two representative peptides for in depth kinetic characterization: the most enantioselective Ac-LILHLFL-NH2 as well as the “middle of the pack” Ac-LALHLFL-NH2 capable of binding the hemin productively, but not showing high selectivity. Global fit of the initial rate dependence on substrate concentrations (Figure 3, Figure S4) was in agreement with the ping-pong two substrate kinetic model. The corresponding kinetic parameters are given in Table 3. Comparison of the self-assembled peptides with engineered heme proteins is instructive (although it should be noted that the experimental conditions vary somewhat between the studies and global fitting was not employed for the proteins). The turnover rates shown by the short peptides are comparable to the ones shown by heme enzymes, consistent with the fact that the properly ligated cofactor itself is primarily responsible for the reactivity. Interestingly, the Michaelis constants for EDA are significantly lower for the self-assembled system when compared to the proteins, a result that is perhaps unexpected for a system with fairly open access to heme and that potentially speaks to much potential in further improving enzymatic cavity in myoglobin and cytochromes P450 for the first step of the reaction. The Michaelis constants for styrenes are comparable between the peptides and enzymes.
Figure 3.
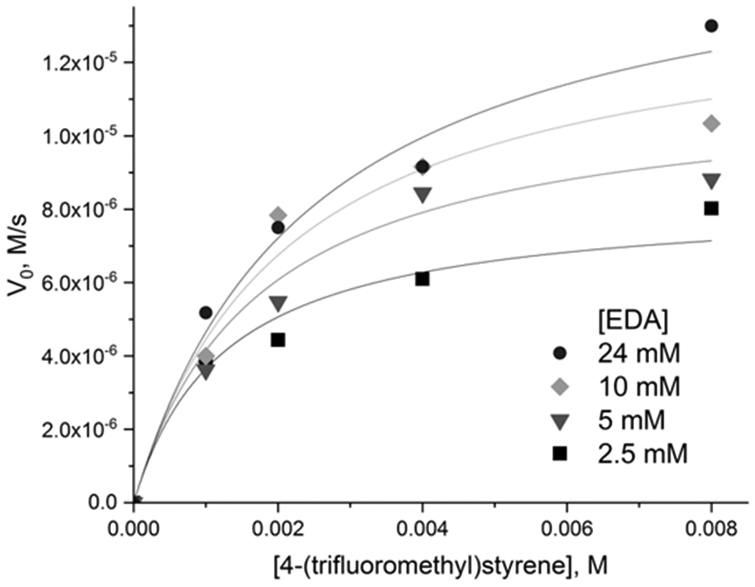
Initial rates of 4-(trifluoromethyl)styrene cyclopropanation with EDA catalyzed by Ac-LILHLFL-NH2-hemin complex (20 μM hemin mixed with 150 μM peptide) at pH 7 in 100 mM phosphate buffer.
Table 3.
Kinetic parameters for cyclopropanation promoted by hemin-bound self-assembled peptides and representative engineered heme proteins.
| Catalyst | kcat, s−1 | KM(EDA), mM |
KM(Styrene), mM |
Ref. |
|---|---|---|---|---|
| LILHLFL - hemin[a] | 0.905 ± 0.068 | 2.96 ± 0.55 | 2.76 ± 0.42 | this work |
| LALHLFL-hemin[a] | 0.600 ± 0.004 | 3.06 ± 0.45 | 2.01 ± 0.26 | this work |
| P450BM3-heme-CIS[b] | 1.7 ± 0.4 | 5.2 ± 3.5 | 1.4 ± 0.5 | [13] |
| Mb H64V/ V68A[c] |
- | 5 | 2 | [11a] |
Reaction conditions: 20 μM hemin with 150 μM peptide, 1 − 8 mM 4-(trifluoromethyl)styrene, 2.5 − 24 mM EDA, 10 mM dithionite, 100 mM pH 7 phosphate buffer.
Reaction was performed at pH 8 with 0.2-20 mM EDA and 30 mM styrene or 0.2-15 mM styrene and 20 mM EDA
Reaction was performed at pH 7 with styrene as substrate.
While the observed enantioselectivities are modest compared to those obtained with engineered hemin proteins, short peptides are very easy to prepare and modify. This feature allows for a straightforward preparation of the opposite enantiomers of the catalysts, a feat that is nearly impossible to do with proteins limiting their use in practical applications. We prepared (D)-Ac-LILHLFL-NH2, an enantiomer of the most selective catalysts and tested its ability to promote enantioselective cyclopropanation (Table 2). Excitingly, the enantioselectivities of (D)-Ac-LILHLFL-NH2 are exactly (within the error of measurement) opposite to those of Ac-LILHLFL-NH2 demonstrating the ease with which catalysts with opposite enantioselectivities can be prepared whilst and showing that the selectivity of the catalyst is fundamentally linked to the chirality of the monomeric peptides. As expected, the bulk morphological properties and hemin affinities of (D)-Ac-LILHLFL-CONH2 are identifcal to those of its enantiomer (Figure S5o, S6h Supporting Information).
In summary, while efficient and enantioselective cyclopropanation has been considered a prerogative of complex proteins, our studies showed for the first time that catalytic amyloids formed by very short peptides can bind complex metallocofactor in a highly sequence specific, functional manner to create efficient and enantioselective catalysts of cyclopropanation. This result was achieved by a conservative introduction of only two changes into the sequence of a catalytic amyloid-forming peptide incapable of binding hemin. The efficiencies of these catalysts are comparable to some engineered proteins[11] and demonstrate the ease with which hemin can be employed to promote cyclopropanation in general, at the same time the observed enantioselectivities are modest, consistent with the lack of a well-defined enclosed catalytic site found in globular proteins. Nonetheless, it is quite striking that self-assembly of short peptides into essentially flat surfaces results in any enantioselectivity at all and this observation suggests more subtle effects of the assembly on the transition state of the reaction. From a standpoint of Origin of Life, a rapid emergence of enantioselective catalysis in simple assemblies that can be formed abiotically lends credence to the “amyloids-first” hypothesis.[12] Finally, we have shown the potential for catalytic amyloids to promote multi-substrate reactions that require interactions of the catalysts with substrate with different physico-chemical properties. Together with ease of preparation and chemical modification as well as the simplicity of stereochemistry flipping, short self-assembling peptides demonstrate unique and versatile catalytic properties with great potential for customization.
Supplementary Material
Acknowledgements
This work was supported by the NIH (Grants No. GM119634, GM103521), the CRDF (Grant No. OISE-18-63891-0) and the Alexander von Humboldt Foundation. We thank Prof. Nancy Totah for providing access to a GC instrument. We thank Dr. Boris Dzikovski for help with acquisition of the EPR spectra. We thank Dr. Min Chul Kim and Ngoc T. Huynh for assistance in peptide purification and GC analysis.
Footnotes
Supporting information and the ORCID identification number(s) for the author(s) of this article can be found under http://dx.doi.org/10.1002/anie.201602480
References
- [1]a).Boncheva M, Whitesides GM, MRS Bull. 2011, 30, 736–742 [Google Scholar]; b) Hamley IW, Interface Focus 2017, 7, 20170062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2]a).Mendes AC, Baran ET, Reis RL, Azevedo HS, WIREs Nanomed. Nanobi 2013, 5, 582–612 [DOI] [PubMed] [Google Scholar]; b) Cherny I, Gazit E, Angew. Chem. Int. Ed 2008, 47, 4062–4069 [DOI] [PubMed] [Google Scholar]; c) Du X, Zhou J, Shi J, Xu B, Chem. Rev 2015, 115, 13165–13307 [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Habibi N, Kamaly N, Memic A, Shafiee H, Nano Today 2016, 11, 41–60 [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Lupas A, Trends Biochem. Sci 1996, 21, 375–382 [PubMed] [Google Scholar]; f) Zhao X, Pan F, Lu JR, Prog. Nat. Sci - Mater. Int 2008, 18, 653–660 [Google Scholar]; g) Dolan MA, Basa PN, Zozulia O, Lengyel Z, Lebl R, Kohn EM, Bhattacharya S, Korendovych IV, ACS Nano 2019, 13, 9292–9297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3]a).Zozulia O, Dolan MA, Korendovych IV, Chem. Soc. Rev 2018, 47, 3621–3639 [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Al-Garawi ZS, McIntosh BA, Neill-Hall D, Hatimy AA, Sweet SM, Bagley MC, Serpell LC, Nanoscale 2017, 9, 10773–10783 [DOI] [PubMed] [Google Scholar]; c) Friedmann MP, Torbeev V, Zelenay V, Sobol A, Greenwald J, Riek R, PLOS ONE 2015, 10, e0143948. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Heier JL, Mikolajczak DJ, Böttcher C, Koksch B, J. Pept. Sci 2017, 108, e23003. [DOI] [PubMed] [Google Scholar]; e) Luong TQ, Erwin N, Neumann M, Schmidt A, Loos C, Schmidt V, Fändrich M, Winter R, Angew. Chem. Int. Ed 2016, 55, 12412–12416 [DOI] [PubMed] [Google Scholar]; f) Monasterio O, Nova E, Diaz-Espinoza R, Biochem. Biophys. Res. Commun 2017, 482, 1194–1200 [DOI] [PubMed] [Google Scholar]; g) Omosun TO, Hsieh M-C, Childers WS, Das D, Mehta AK, Anthony NR, Pan T, Grover MA, Berland KM, Lynn DG, Nat. Chem 2017, 9, 805–809 [DOI] [PubMed] [Google Scholar]; h) Singh N, Kumar M, Miravet JF, Ulijn RV, Escuder B, Chem. Eur. J 2017, 23, 981–993 [DOI] [PubMed] [Google Scholar]; i) Solomon LA, Kronenberg JB, Fry HC, J. Am. Chem. Soc 2017, 139, 8497–8507 [DOI] [PubMed] [Google Scholar]; j) Zhang C, Shafi R, Lampel A, MacPherson D, Pappas CG, Narang V, Wang T, Maldarelli C, Ulijn RV, Angew. Chem. Int. Ed 2017, 56, 14511–14515 [DOI] [PubMed] [Google Scholar]; k) Makam P, Yamijala SSRKC, Tao K, Shimon LJW, Eisenberg DS, Sawaya MR, Wong BM, Gazit E, Nat. Catal 2019, 2, 977–985 [DOI] [PMC free article] [PubMed] [Google Scholar]; l) Makhlynets OV, Korendovych IV, Nat. Catal 2019, 2, 949–950. [Google Scholar]
- [4]a).Rufo CM, Moroz YS, Moroz OV, Stöhr J, Smith TA, Hu X, DeGrado WF, Korendovych IV, Nat. Chem 2014, 6, 303. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Makhlynets OV, Gosavi PM, Korendovych IV, Angew. Chem. Int. Ed 2016, 55, 9017–9020 [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Lee M, Wang T, Makhlynets OV, Wu Y, Polizzi NF, Wu H, Gosavi PM, Stöhr J, Korendovych IV, DeGrado WF, Hong M, Proc. Natl. Acad. Sci. U.S.A 2017, 114, 6191–6196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Lengyel Z, Rufo CM, Moroz YS, Makhlynets OV, Korendovych IV, ACS Catal. 2018, 8, 59–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6]a).Wang Q, Yang Z, Ma M, Chang CK, Xu B, Chem. Eur. J 2008, 14, 5073–5078 [DOI] [PubMed] [Google Scholar]; b) Atamna H, Boyle K, Proc. Natl. Acad. Sci. U.S.A 2006, 103, 3381–3386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7]a).Hayashi T, Tinzl M, Mori T, Krengel U, Proppe J, Soetbeer J, Klose D, Jeschke G, Reiher M, Hilvert D, Nat. Catal 2018, 1, 578–584 [Google Scholar]; b) Wong HNC, Hon MY, Tse CW, Yip YC, Tanko J, Hudlicky T, Chem. Rev 1989, 89, 165–198 [Google Scholar]; c) Burke AJ, Marques CS, Turner NJ, Hermann GJ, Active Pharmaceutical Ingredients in Synthesis: Catalytic Processes in Research and Development, John Wiley & Sons, 2018. [Google Scholar]
- [8].Brandenberg OF, Fasan R, Arnold FH, Curr. Opin. Biotechnol 2017, 47, 102–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9]a).Blauer G, Sreerama N, Woody RW, Biochemistry 1993, 32, 6674–6679 [DOI] [PubMed] [Google Scholar]; b) Nagai M, Mizusawa N, Kitagawa T, Nagatomo S, Biophys. Rev 2018, 10, 271–284 [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Nagai M, Nagai Y, Imai K, Neya S, Chirality 2014, 26, 438–442 [DOI] [PubMed] [Google Scholar]; d) Korendovych IV, Senes A, Kim YH, Lear JD, Fry HC, Therien MJ, Blasie JK, Walker FA, DeGrado WF, J. Am. Chem. Soc 2010, 132, 15516–15518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Carminati DM, Fasan R, ACS Catal. 2019, 9, 9683–9697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11]a).Bordeaux M, Tyagi V, Fasan R, Angew. Chem 2015, 127, 1764–1768 [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Villarino L, Splan KE, Reddem E, Alonso-Cotchico L, Gutiérrez de Souza C, Lledós A, Maréchal J-D, Thunnissen A-MWH, Roelfes G, Angew. Chem. Int. Ed 2018, 57, 7785–7789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12]a).Carny O, Gazit E, FASEB J. 2005, 19, 1051–1055 [DOI] [PubMed] [Google Scholar]; b) Greenwald J, Riek R, J. Mol. Biol 2012, 421, 417–426 [DOI] [PubMed] [Google Scholar]; c) Maury CPJ, Orig. Life Evol. Biosph 2009, 39, 141–150. [DOI] [PubMed] [Google Scholar]
- [13]a).Coelho PS, Wang ZJ, Ener ME, Baril SA, Kannan A, Arnold FH, Brustad EM, Nat. Chem. Biol 2013, 9, 485–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.