

Abstract
Adventitial fibroblasts (AFs) are major contributors to vascular remodeling and maladaptive cascades associated with arterial disease, where AFs both contribute to and respond to alterations in their surrounding matrix. The impact of matrix stiffness on AFs is not well understood, and we investigated relationships between matrix modulus and human aortic AF (AoAF) function using PEG-based hydrogels designed with MMP-sensitive and integrin-binding peptides. Initial equilibrium shear storage moduli for the substrates examined were 0.33, 1.42, and 2.90 kPa; after 42 days of culture, all hydrogels exhibited similar storage moduli (0.3–0.7 kPa) regardless of initial modulus, with encapsulated AoAFs spreading and proliferating. In 10wt% and 7.5wt% hydrogels, modulus decreased monotonically throughout culture; however, in 5wt% hydrogels, modulus increased after an initial 7 days of culture, accompanied by increased myofibroblast transdifferentiation and increased expression of collagen I and III through day 28. Thereafter, significant reductions in both collagens occurred, with increased MMP-9 and decreased TIMP-1/−2 production. Releasing cytoskeletal tension or inhibiting cellular protein secretion in 5wt% hydrogels blocked the stiffening of the polymer matrix. Our results indicate that encapsulated AoAFs initiate cell-mediated matrix remodeling and demonstrate the utility of dynamic 3D systems to elucidate the complex interactions between cell behavior and substrate properties.
Keywords: hydrogel, fibroblast, cytokine, mechanical history, collagen
1. Introduction
Despite significant advances in treatment, arterial diseases remain major causes of morbidity and mortality worldwide. Recently, adventitial fibroblasts (AFs) have been implicated as major contributors in the maladaptive cascades occurring in arterial disease, in vessel remodeling, and in restenosis after vascular interventions.[1] In fact, AFs are the first vascular wall cell to exhibit signs of activation following physiologic stresses.[2] Activated AFs can transdifferentiate to myofibroblasts and secrete a multitude of signaling factors, including cytokines, growth factors, and proteases, that alter the behaviors of other resident adventitial cells and ultimately disturb vascular pathobiology.[2–3] AFs alter the function and structure of vessel walls in a variety of ways, especially by balancing the production of extracellular matrix (ECM) proteins and ECM-degrading proteases, such as matrix metalloproteinases (MMPs), to alter tissue stiffness.[1a, 1b, 4] In turn, matrix stiffness can dramatically alter AF gene expression and cytokine production;[5] thus, AFs both alter substrate stiffness and respond to it in the context of adventitial function and vascular disease progression.
The complex interplay between cell-mediated alterations of the cellular microenvironment and the potentially reflexive effects of microenvironmental properties on normal and pathologic cell function are only partially understood. The development of in vitro cell culture platforms has been critical for understanding the response of vascular cells to tissue stiffness.[5a] Pivotal reports from the field of cell mechanotransduction have demonstrated that matrix stiffness can largely influence cytoskeletal organization, proliferation, differentiation, and protein secretion.[6] Our group has previously utilized poly(ethylene glycol) (PEG)-based hydrogels to evaluate the effects of substrate stiffness on vascular AFs in a 3D cell culture model.[5b] This work, in conjunction with other reports,[5a, 7] supports the notion that substrate stiffness is a powerful regulator of vascular cell activities, including proliferation, gene expression profiles, and differentiation status.
While most studies have utilized static in vitro cultures to elucidate the biophysical cues that regulate cell fate, investigators have more recently designed a variety of synthetic cell culture platforms that can either soften or stiffen over time.[8] The development of constructs that undergo spontaneous, cell-mediated, or user-defined transitions in matrix mechanics have proven beneficial for understanding the biological implications of dynamic stiffness alterations towards cell populations.[9] Moreover, dynamic systems may provide pivotal information regarding cellular mechanical memory about past physical culture conditions that influence future behaviors.[10] In the context of vascular adventitial cell populations, cell culture systems that enable dynamic, cell-mediated alterations in stiffness, which are known to accompany vascular development, regeneration, and disease progression, will enable researchers to gain an improved understanding of the dynamic interactions between AFs and their substrate, and how these interactions might be manipulated to drive beneficial cell phenotypes.
In this study, we utilized PEG hydrogel platforms to investigate the impact of matrix modulus on human aortic AF (AoAF) activation in a 3D culture model, across a range of substrate moduli (0.3–3.2 kPa) that are physiologically relevant to the arterial adventitium.[11] Further, we investigated AoAF remodeling of their surrounding microenvironment and the impact of cell-mediated, local modifications on cell fate. Our findings substantiate the associations between substrate physical properties and cell behavior, while highlighting the influence of initial hydrogel conditions, as well as subsequent dynamic changes, in guiding AoAF cell phenotype. Our results also elaborate the linkages between cell fate, substrate physical characteristics, and mechanisms of arterial disease that may provide opportunities for advanced therapies.
2. Results and Discussion
2.1. Design and Characterization of Hydrogel Models
To study the role of matrix stiffness on AoAF fate, 3D cell-laden hydrogels were formed under physiological conditions via a Michael-type addition reaction using thiol end-functionalized PEG macromers (PEG-SH4) and bis-maleimide end-functionalized, MMP-sensitive crosslinker peptides (PQ-MI2) to obtain highly elastic hydrogels that accommodated cell-mediated remodeling of the matrix via protease degradation (Figure 1A). As collagen is the predominant structural and cell-adhesive protein found in arterial adventitia and a key mediator of AoAF activities,[12] 1 mM mono-maleimide pendent RGD peptide (RGD-MI), an adhesive motif found within collagen, was incorporated into the polymer matrix to facilitate AoAF interactions with the hydrogel network. The viscoelastic properties of the hydrogels, tuned by varying the polymer concentration over a narrow range, were measured via oscillatory shear rheology, using dynamic time sweep assays in the linear viscoelastic regime. The equilibrium shear storage moduli for acellular 5wt%, 7.5wt%, and 10wt% hydrogels were 0.38 ± 0.10 kPa, 1.38 ± 0.18 kPa, and 2.80 ± 0.23 kPa, respectively, after 24 hrs of incubation at 37°C in stromal cell growth medium (SCGM) (Figure 1B). As expected for PEG-based materials,[13] concentration-dependent swelling varied with formulation with capacities of 96.4% ± 0.1%, 93.9% ± 0.2%, and 92.0% ± 0.1% observed for 5wt%, 7.5wt%, and 10wt% hydrogels, respectively (Table S1). Complete hydrogel degradation occurred following treatment with 0.1% collagenase type II and 0.05% trypsin solution, confirming that maleimide-modification of the peptides did not inhibit proteolytic susceptibility of the hydrogels (Figure S1). The rate of hydrogel degradation showed an inverse correlation with polymer crosslink density, which is consistent with previous work correlating increased stiffness, decreased swelling, and degradation rate characteristics.[14]
Figure 1.
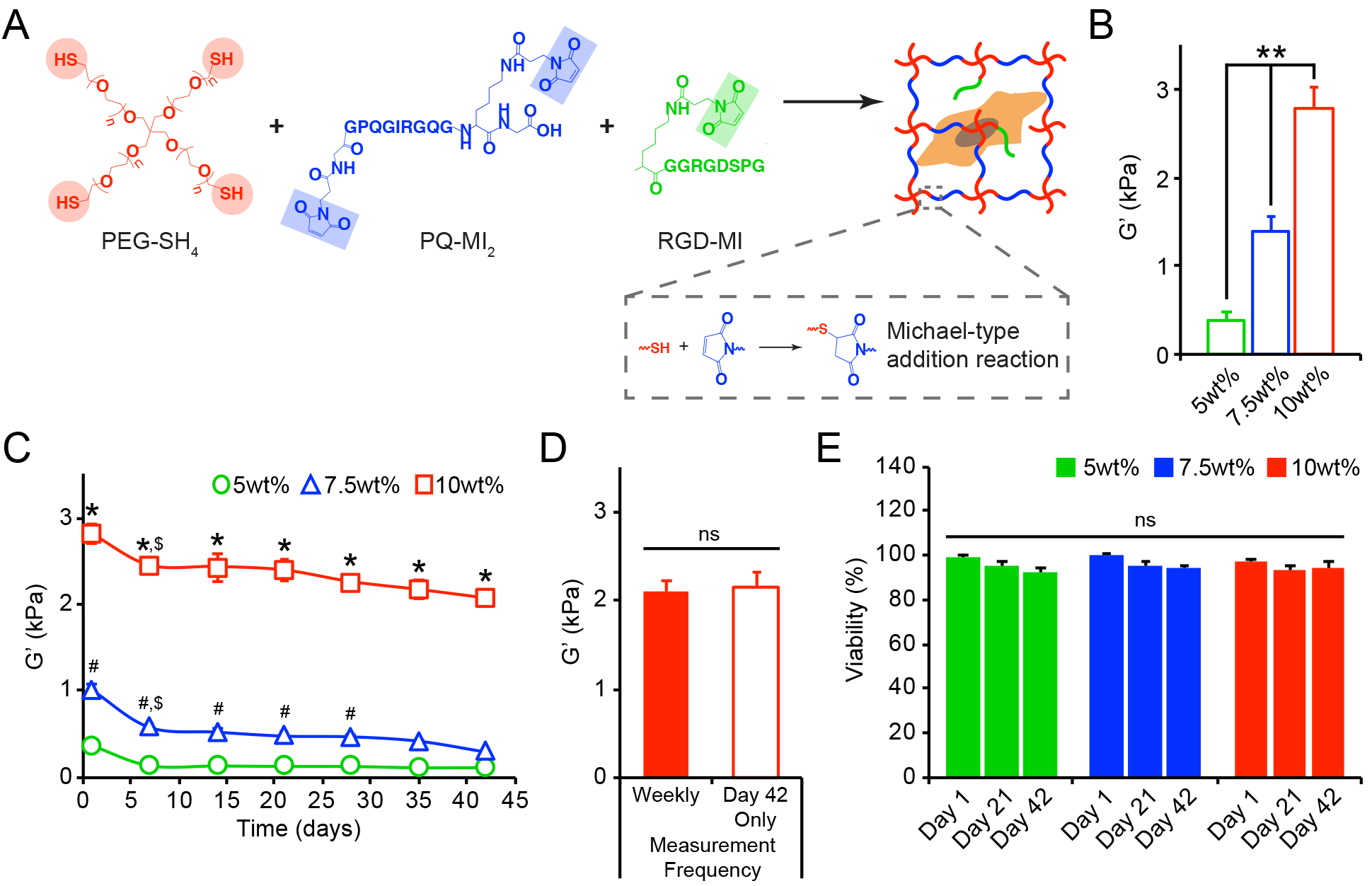
(A) Reaction scheme for enzymatically degradable hydrogel formation. (B) The initial equilibrium storage moduli (G’, kPa), evaluated by oscillatory shear rheology after 24 hrs, significantly increased with increasing wt%. (C) The bulk shear storage moduli (G’, kPa) of acellular 5wt%, 7.5wt%, and 10wt% (w/v%) hydrogels were evaluated once per week by oscillatory shear rheology over 42 days. (D) No differences in the final storage moduli were observed between hydrogels that endured repeated measurements (performed once per week) and those that were measured only once after 42 days of incubation. (E) Cell viability, assessed using live/dead staining, was not compromised by repeated rheological measurements (performed once per week) of cell-laden 5wt%, 7.5wt%, and 10wt% hydrogels. In B-E, data are represented as the mean ± SEM, with n = 3 biological replicates per condition. For B: a one-way ANOVA, followed by a Tukey HSD post hoc test, was used to detect statistical significance, **p<0.0001. For C: a two-way ANOVA, followed by a Tukey HSD post hoc test, was used to detect statistical significance, *p<0.0001 for 10wt% relative to 5wt% and 7.5wt% at a given time point, #p<0.05 for 7.5wt% relative to 5wt% at a given time point, $p<0.05 for a given time point relative to day 1, within an individual hydrogel formulation. For D-E: no statistical significance (ns) was indicated following (D) a student’s t-test or (E) a two-way ANOVA.
The use of PEG-based hydrogels formed via a Michael-type addition allowed for stable crosslinks to be created under physiological conditions that are amenable for cell encapsulation.[14–15] The encapsulation of AoAFs (deidentified 53 yo male donor) did not significantly alter the initial equilibrium shear storage moduli of the hydrogels, and cell-laden hydrogels exhibited stiffnesses of 0.33 ± 0.03, 1.42 ± 0.08, and 2.90 ± 0.29 kPa at 5wt%, 7.5wt%, and 10wt%, respectively (Figure S2). Following encapsulation, cells were >95% viable (Figure S3A) and homogenously distributed within hydrogel volumes. After 24 hrs, widefield fluorescence imaging demonstrated that cells had a rounded morphology (circularity: 0.81–0.86) and were of similar size (maximal cross-sectional area: 242.4–296.2 μm2), independent of initial shear storage modulus of the encapsulating hydrogel (Figure S3B–C).
To begin understanding the complex relationships between cell-mediated alterations of the hydrogel microenvironment and the corresponding effects of the microenvironment on cell function, the mechanical properties of hydrogels over time in culture were measured using oscillatory shear rheology, an approach that has been well validated in previous studies.[16] Acellular hydrogels were subjected to repeated rheological measurements and remained intact and stable over 42 days (Figure 1C). Decreases in shear storage moduli were observed for all hydrogels over time, indicative of a decrease in hydrogel crosslink density likely due to endogenous enzymes present in the serum or retro-Michael addition reactions [17]. The repeated measurements, conducted once per week over 42 days, neither compromised the mechanical integrity of the substrates (Figure 1D) nor impacted the viability of encapsulated cells (Figure 1E, Figure S4), establishing this technique for correlating alterations in the bulk mechanical properties of hydrogels with cell behavior.
2.2. Early AoAF Responses to Altered Matrix Stiffness in 3D Hydrogels
We next sought to determine whether differences in initial matrix stiffness provoked AoAF activation just after culture initiation by assessing cell proliferation and secretion of key cytokines, including the angiogenic growth factor vascular endothelial growth factor (VEGF-A), and the inflammatory cytokines interleukin-6 (IL-6) and monocyte chemoattractant protein-1 (MCP-1). Elevated proliferation was observed in soft 5wt% hydrogels in the initial 7 days following encapsulation and proliferation decreased as hydrogel stiffness increased (Figure 2A), similar to previous reports with fibroblasts in 3D.[5b, 18] We further observed that elevated AoAF proliferation in 5wt% hydrogels corresponded with enhanced production of the angiogenic growth factor, VEGF-A (detected via enzyme-linked immunosorbent assay (ELISA)), compared to 7.5wt% and 10wt% hydrogels (Figure S5A), which is consistent with in vivo reports where increased adventitial cell proliferation corresponded to increased VEGF-A production.[19]
Figure 2.
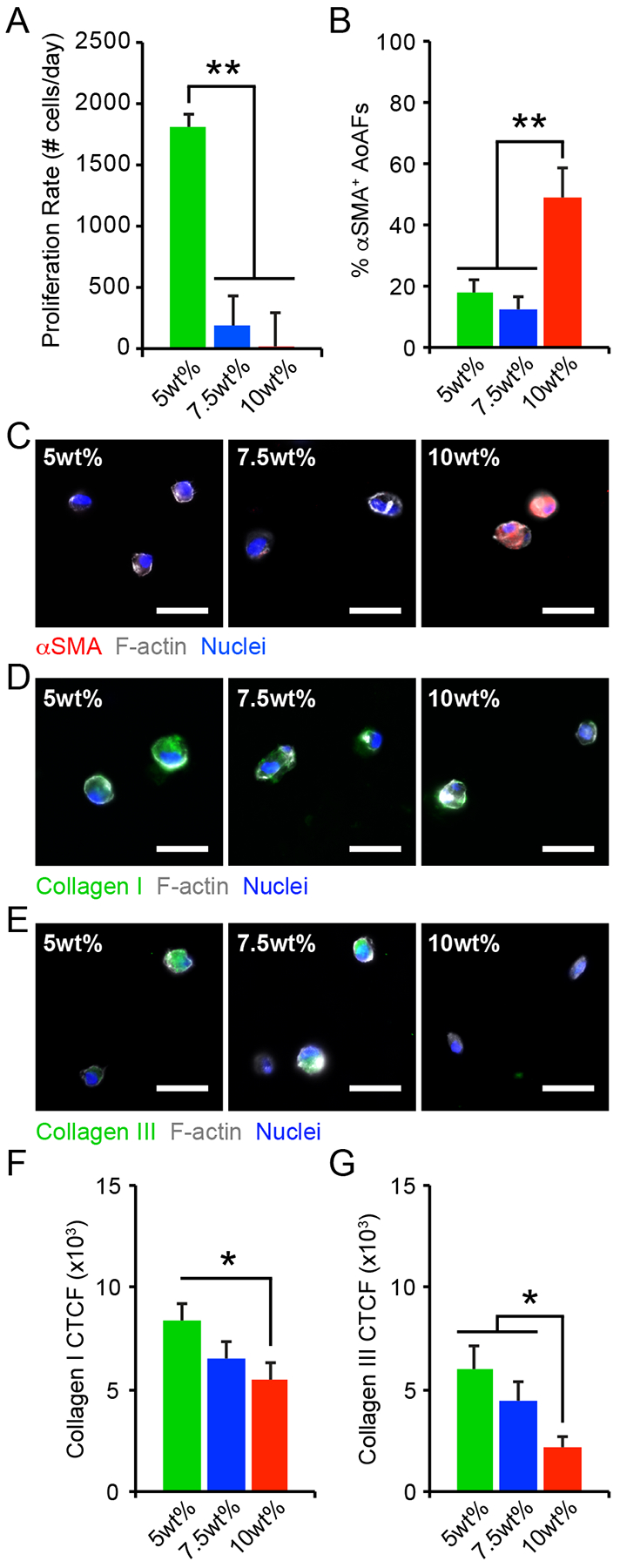
(A) The proliferation rate of AoAFs encapsulated in 5wt%, 7.5wt%, and 10wt% hydrogels significantly decreased with increasing weight percent over 7 days. (B-C) The percentage of αSMA+ AoAFs was significantly increased in 10wt% hydrogels compared to softer 5wt% and 7.5wt% substrates after 1 day, as confirmed by immunocytochemistry. Blue: Hoechst 33258 (nuclei), Grey: F-actin, Red: αSMA. Scale bar = 25 μm. (D-E) AoAFs produced (D) collagen I or (E) collagen III, detected via immunocytochemistry, following encapsulation in 5wt%, 7.5wt%, and 10wt% hydrogels for 1 day. Blue: Hoechst 33258 (nuclei), Grey: F-actin, Green: collagen I or collagen III. Scale bar = 25 μm. (F-G) Expression of (F) collagen I and (G) collagen III expression, measured by corrected total cell fluorescence, decreased with increasing weight percent. Data represented as the mean ± SEM, where in A: n = 3 biological replicates per condition and in C & F-G, n = 20 cells from 3 images from each of 3 hydrogels per condition. For A, C, & F-G: a one-way ANOVA, followed by a Tukey HSD post hoc test, was used to detect statistical significance, *p<0.05, **p<0.01.
Under physiologic and pathophysiologic conditions, alterations to the adventitia are often accompanied by the appearance of proliferative cells expressing α-smooth muscle actin (αSMA), a marker of fibroblast to myofibroblast transition, and increased matrix deposition.[20] Though AoAFs in the 5wt% hydrogels exhibited enhanced proliferation, immunostaining revealed that few cells (<20%) expressed αSMA in 5wt% and 7.5wt% hydrogels 24 hrs post-encapsulation (Figure 2B–C). On the other hand, ~50% of AoAFs in the stiffer 10wt% hydrogels expressed diffuse αSMA staining after 24 hrs. While our results differ from previous observations in 3D hydrogels, where fibroblast activation decreased with increased stiffness,[21] they are consistent with in vivo observations of pathologic tissue stiffening driving myofibroblast transdifferentiation.[22] Further, αSMA expression in 10wt% hydrogels was accompanied by the secretion of the inflammatory proteins, IL-6 and MCP-1 (measured via ELISA; Figure S5B–C). Interestingly, significantly increased IL-6 and MCP-1 production by AoAFs encapsulated in 5wt% hydrogels, compared to 7.5wt% hydrogels, suggests that inflammatory cytokine production by activated AoAFs may be regulated through pathways distinct from those driving myofibroblast transdifferentiation. The concept that substrate stiffness has complex effects on AoAF phenotype is also supported by previous work from our group.[5b]
Within native tissues and tissue mimetic substrates, cells actively respond to their surroundings by synthesizing and secreting ECM proteins.[23] Accordingly, we assessed the distribution of collagen I and collagen III, which are the main collagen constituents in the adventitia.[24] Following encapsulation, collagen I and collagen III immunofluorescent staining was primarily localized to the intracellular or pericellular regions in all cultures (Figure 2D–E). Quantification of collagen levels by corrected total cell fluorescence (CTCF) revealed that collagen I expression was increased among AoAFs in 5wt% hydrogels after 1 day, but then decreased as hydrogel stiffness increased (Figure 2F). Collagen III expression was also increased in 5wt% hydrogels, compared to cells in 7.5% and 10wt% hydrogels (Figure 2G). Overall, these results are in agreement with previous findings where substrate stiffness regulates ECM protein production,[23a, 25] and support the notion that initial hydrogel conditions strongly impact the composition of the secreted ECM.[26]
2.3. Fibroblasts Remodel Hydrogel Matrix to a Preferential Modulus Range in Association with Protease and Protease Inhibitor Secretion
To investigate the degree to which AoAFs remodel encapsulating PEG-based hydrogels of different initial shear storage moduli, we utilized oscillatory shear rheology to probe alterations in macroscale shear rheological properties over time.[16] Temporal alterations in cell-laden hydrogel shear storage moduli were observed across 5wt%, 7.5wt% and 10wt% hydrogels (Figure 3A). These changes depended on the presence of encapsulated cells and did not occur in acellular hydrogels (Figure S6), indicating that encapsulated AoAFs initiate cell-mediated matrix remodeling of these PEG-based materials. Cell-mediated reductions in stiffness were observed for all hydrogels over the first 7 days of culture, but thereafter, changes were dependent on the initial shear storage modulus. Specifically, the modulus of 10wt% hydrogels decreased throughout the duration of the study; whereas, shear storage moduli stabilized after 7 days in 7.5wt% hydrogels, and 5wt% hydrogels were observed to stiffen after 7 days.
Figure 3.
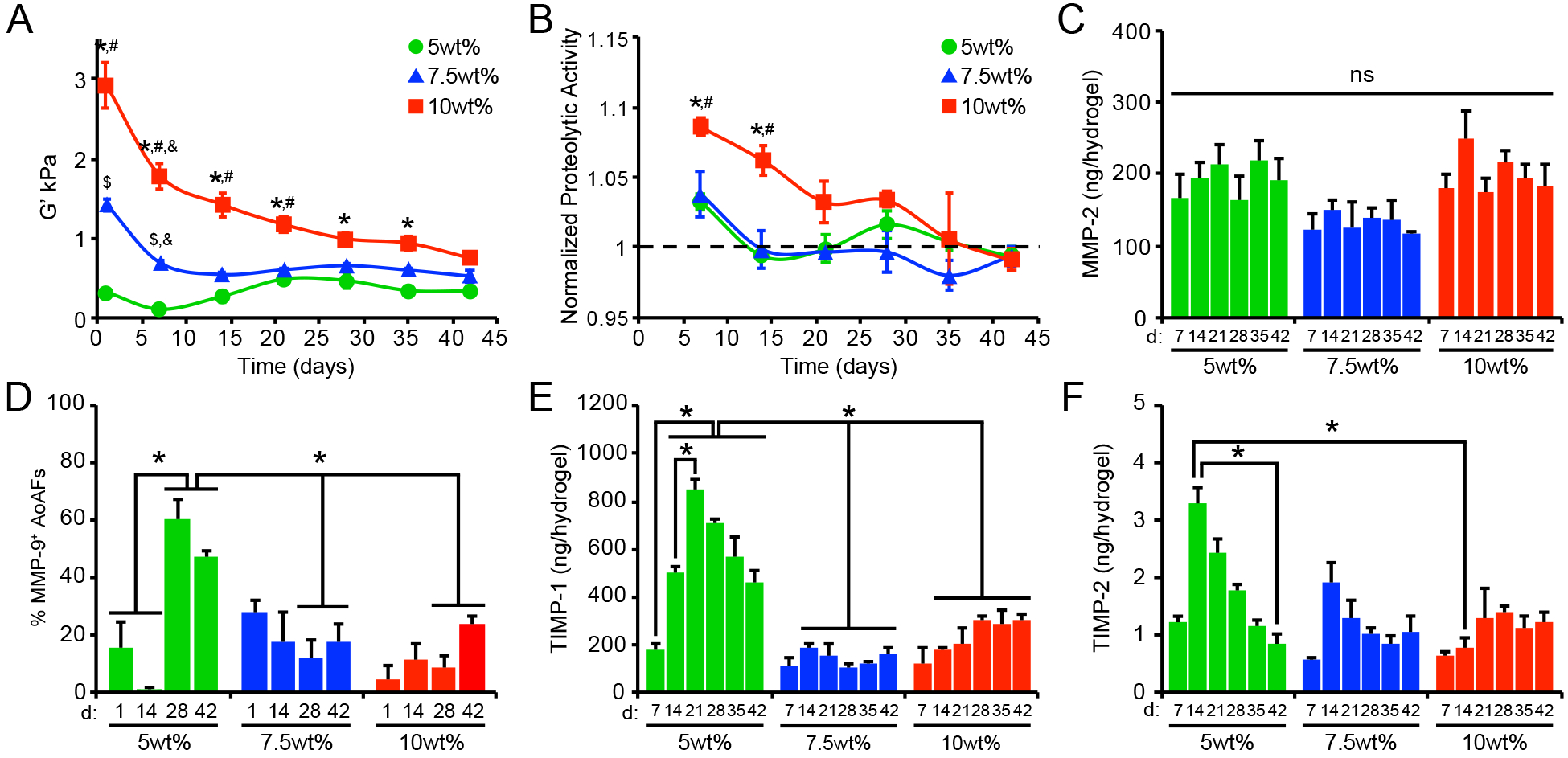
(A) Alterations in swollen storage modulus, measured using oscillatory shear rheology, were observed in AoAF-laden 5wt%, 7.5wt%, and 10wt%, hydrogels over time. (B) Increased proteolytic activity was observed in hydrogels undergoing degradation (i.e. decreased bulk shear storage modulus. Data are mean proteolytic activity relative to acellular hydrogels (dashed line). (C) MMP-2 production by encapsulated AoAFs was similar among hydrogel formulations and over time. (D) Quantification of the percentage of MMP-9-positive cells in AoAF-laden 5wt%, 7.5wt%, and 10wt% hydrogels over the 42 day culture. (E-F) Quantification of the tissue inhibitors of metalloproteinases (E) TIMP-1 and (F) TIMP-2 in the medium collected from AoAF-laden 5wt%, 7.5wt%, and 10wt% hydrogels during the 42 day culture period. Data are mean ± SEM, where in A: n = 6 per condition, in B, D-F: n = 3 biological replicates per condition, and in C: n > 30 cells from 3 images from each of 3 hydrogels per condition. In A-F: a two-way ANOVA, followed by a Tukey HSD post hoc test, was used to detect statistical significance. In A-B: *p<0.01 for 10wt% relative to 5wt%, #p<0.05 for 10wt% relative to 7.5wt%, $p<0.05 for 7.5wt% relative to 5wt%, at a given time point; &p<0.01 for a given time point relative to day 1, within an individual hydrogel formulation. In C: no statistical significance (ns) was indicated following a two-way ANOVA. In D-F: *p<0.05 for indicated comparisons.
Interestingly, after 42 days, all AoAF-laden hydrogels exhibited similar shear storage moduli (0.3–0.7 kPa) regardless of the initial conditions, suggesting that the cells remodeled the hydrogels toward a preferential modulus range. To evaluate whether the trend to a specific modulus was a characteristic of the particular cell donor, we obtained AoAFs from an unrelated second donor (an unidentified, 19 yo female; Lonza). Similar remodeling profiles were observed when AoAFs from the second donor were evaluated (Figure S7). After 42 days, the final shear storage moduli of these hydrogels clustered together, as was observed for the 53 yo male donor, although the range of moduli was lower (0.07–0.3 kPa) for the remodeled matrix from the cells of the younger donors. As cells and tissue stiffen during aging in vivo,[27] the observed disparity in the range of the final shear storage moduli may not be unexpected. However, further work utilizing age and gender-matched donors is required to identify the implications of these variables on matrix remodeling.
To better understand the processes by which the AoAFs modified the PEG-based hydrogel matrices, we investigated cellular mechanisms of ECM modification. During vascular development, tissue homeostasis, and disease, cells express and secrete a variety of proteases and protease inhibitors involved in matrix remodeling.[4a] To investigate the role of increased proteolytic activity in decreasing the stiffness of the current hydrogel formulations, we measured protease activity in the medium collected from hydrogels using a dye quenched gelatin (DQ-gelatin), which exhibits drastic changes in fluorescence intensity after proteolytic degradation.[28] Increased proteolytic activity was detected in the medium collected from all AoAF-laden hydrogels on day 7 (Figure 3B). Thereafter, little-to-no proteolytic activity was detected in the 5wt% and 7.5wt% hydrogel cultures; however, increased proteolytic activity was observed in the 10wt% hydrogel cultures through 28 days. Importantly, proteolytic activity in 7.5% and 10wt% hydrogels was positively correlated with hydrogel stiffness over time (Pearson’s correlation; r=0.60, p=0.002 for 10wt%; r=0.64, p=0.002; Table S2), while a negative correlation was observed for 5wt% hydrogels (r=0.53, p=0.024; Table S2).
We next evaluated whether differences in proteolytic activity were attributable to altered production of the proteases MMP-2 and MMP-9, which are ubiquitous gelatinases secreted by AoAFs and associated with arterial ECM remodeling during arteriogenesis and disease progression.[29] MMP-2 was detected, via ELISA, in the conditioned medium at similar levels across all hydrogel formulations and over time (Figure 3C). Significant levels of MMP-9 were not detected in the medium of any sample (data not shown). This finding is not surprising given that decreased production of MMP-9, compared to MMP-2, has been reported for fibroblast-like cells.[30] As the availability of secreted cell products in the local microenvironment may differ from that detected in the culture supernatant,[31] we utilized immunofluorescence microscopy to further visualize MMP production at the cell exterior. MMP-2 was readily observed in the pericellular regions and no differences in the percentage of MMP-2+ cells were detected across hydrogel formulations after 42 days of culture (Figure S8). Although MMP-9 was not detected in the collected medium, we did visualize MMP-9 in pericellular regions (Figure 3D, Figure S9). The percentage of cells expressing pericellular MMP-9 increased over time in 5wt% hydrogels. In comparison, AoAFs encapsulated in 7.5wt% and 10wt% hydrogels produced significantly less pericellular MMP-9, consistent with previous reports demonstrating decreased MMP-9 expression with increased substrate stiffness.[32] Importantly, variations in secreted protease activity did not correlate with MMP-2 or MMP-9 expression in any hydrogel formulation (Table S2).
We also investigated the expression of two tissue inhibitors of metalloproteinases (TIMP-1 and TIMP-2), which are inhibitors of MMP-2 and MMP-9 and main protease inhibitors associated with maintaining arterial wall structure during maladaptive remodeling.[33] Both TIMP-1 and TIMP-2 were produced at higher levels in the 5wt% hydrogel cultures over time, compared to the other hydrogels (Figure 3E–F), suggesting that pathways that inhibit proteolytic matrix degradation were activated in the softer hydrogels. However, neither TIMP-1 nor TIMP-2 production by encapsulated AoAFs correlated well with proteolytic activity in any hydrogel formation (Table S2), and it is likely that encapsulated cells utilized additional means to regulate proteolytic activity. Together these findings suggest that while the production of MMPs may not be significantly impacted by altered microenvironmental stiffness,[26, 32] the activation of these enzymes, or the production of counteracting inhibitors, may be controlled by cells to regulate cell-mediated tissue remodeling.[32]
2.4. Alterations in Fibroblast Phenotype are Associated with Cell-Mediated Matrix Remodeling
Previous studies have indicated that vascular fibroblasts alter their phenotype in response to changes in microenvironmental stiffness.[5a, 5b, 21] To determine whether temporal changes in matrix modulus observed within our hydrogels subsequently altered cell behavior, we evaluated multiple characteristics of the encapsulated AoAFs over time, including cell shape and size (aspect ratio and area of optical cross sections from widefield fluorescent images), cell proliferation, the degree of conversion to a myofibroblast, the expression of inflammatory cytokines, and ECM protein production. Cell shape changed over time, with decreased cell circularity observed in all hydrogels (circularity: 0.48–0.57 after 42 d), indicating the cells adopted an elongated shape (Figure S10). However, significant increases in cell size were only detected in 5wt% and 7.5wt% hydrogels, where average cell areas increased by 2.5- and 3.5-fold, respectively, after 42 days of culture (Figure 4A). No significant increases in cell area were observed in 10wt% hydrogels; however, as cell spreading is dependent on the local degradability of the network in 3D,[34] decreased spreading in the 10wt% hydrogels was likely associated with the smaller mesh size (calculated at 10.3 nm after 42 days (Table S3)).
Figure 4.
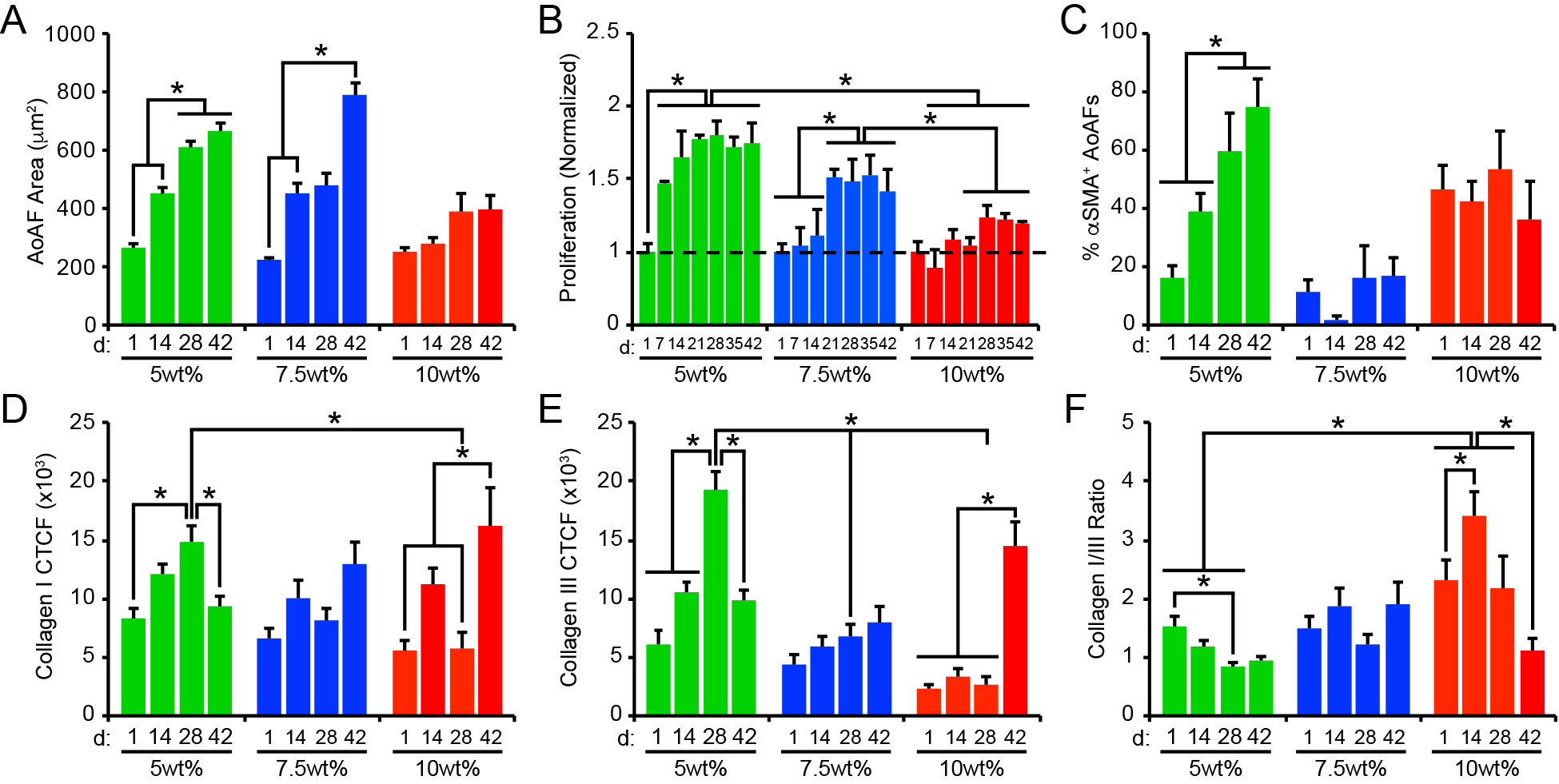
(A) Cell area increased over time in 5wt% and 7.5wt% hydrogels, but not in 10wt% substrates. (B) Similarly, AoAF proliferation increased in 5wt% and 7.5wt% hydrogels over time. Data are mean DNA content relative to DNA content in hydrogels after 1 day of culture (dashed line) ± SEM, n = 3 biological replicates per condition. (C) The percentage of αSMA` AoAFs was significantly increased in 5wt% hydrogels over time, but alterations were not observed in 7.5wt% and 10wt% hydrogels. Expression of (D) collagen I and (E) collagen III, measured via corrected total cell fluorescence, by AoAFs encapsulated in 5wt%, 7.5wt%, and 10wt% hydrogels was altered over time. (F) Ratio of collagen I to collagen III, determined from total collagen signal obtained from the corrected total cell fluorescence, in AoAF-laden 5wt%, 7.5wt%, and 10wt% hydrogels over time. In A-F, data are represented as mean ± SEM, n = 3 biological replicates per condition. A two-way ANOVA, followed by a Tukey HSD post hoc test, was used to detect statistical significance, *p<0.05, **p<0.01.
Physical-constraint limitations were also observed in measures of cell proliferation. In 5wt% hydrogels exhibiting a shear storage modulus of 0.4 kPa and a calculated mesh size of 10.6 nm (Figure 3A, Table S3), proliferation was immediately observed (Figure 4B). Interestingly, proliferation in the 5wt% hydrogels ceased by day 21, corresponding to a stiffening of the hydrogel matrix to >0.5 kPa (with a calculated mesh size of < 10.5 nm). AoAF proliferation in 5wt% hydrogels was associated with enhanced production of VEGF-A (Figure S11). Conversely, significant levels of proliferation were not observed in the 7.5wt% hydrogels until day 21, when the bulk storage modulus was reduced to <0.55 kPa (corresponding to a calculated mesh size of 10.5 nm). AoAFs encapsulated in 10wt% hydrogels exhibited limited proliferation over 42 days. The lack of proliferation observed in 10wt% hydrogels occurred in the context of a final bulk storage modulus and calculated mesh size of 0.7 kPa and 10.3 nm, respectively, after 42 days. Unlike the 5wt% hydrogels, enhanced production of VEGF-A was not observed for either the 7.5wt% or 10wt% hydrogels (Figure S11) indicating that microenvironmental conditions may differ across hydrogel systems, associated with altered proliferation and cell morphology.
Consistent with previous reports,[18, 21] our results collectively illustrate that fibroblast spreading and proliferation depended on the biophysical properties of the encapsulating substrate. Our results further demonstrate that alterations in the shear storage modulus of the hydrogel, due to degradation of the polymer network, induced changes in cell shape and proliferation, where encapsulated AoAFs began to spread and proliferate only after the substrate reached a shear storage modulus <0.55 kPa and the calculated mesh size increased above ~10.5 nm. This observation is consistent with previous reports demonstrating enhanced cell proliferation in PEG hydrogels exhibiting low moduli and mesh sizes greater than 10 nm.[35]
To evaluate differences in myofibroblast transdifferentiation within AoAF populations over time, we stained hydrogel cultures with an antibody to αSMA to identify myofibroblasts. A low percentage of AoAFs (<20%) expressed αSMA initially following encapsulation in 5wt% hydrogels; however, the percentage of αSMA+ AoAFs significantly increased over time (Figure 4C, Figure S12). Importantly, we observed that the percentage of αSMA+ AoAFs present in 5wt% hydrogels tracked with hydrogel modulus (Pearson’s correlation, r=0.6677, p=0.0192; Table S4), similar to the effects observed during pathologic tissue stiffening in vivo.[22] Conversely, the percentage of αSMA+ AoAFs was not altered over time in 7.5wt% and 10wt% hydrogels, where ~20% of AoAFs in 7.5wt% hydrogels and ~45% of AoAFs in 10wt% hydrogels expressed diffuse αSMA staining after 42 days in culture, similar to day 1. Unlike the 5wt% hydrogels, the percentage of αSMA+ AoAFs did not correlate with bulk shear moduli in 7.5wt% and 10wt% hydrogels (Table S4). Interestingly, the increased presence of αSMA+ cells in 5wt% hydrogels did not correspond to enhanced production of the inflammatory cytokines IL-6 and MCP-1, which are associated with myofibroblast-mediated inflammation and tissue stiffening in vivo (Table S4).[36] Indeed, production of IL-6 by AoAFs in 5wt% hydrogels did not vary significantly over time, while MCP-1 secretion generally decreased between 14d and 42d as the percentage of myofibroblasts increased in these materials (Figure S13). Similarly, AoAFs encapsulated in 7.5wt% or 10wt% hydrogels did not alter their production of IL-6 over time, while MCP secretion generally decreased over time in 7.5wt% hydrogels. Taken together, these data suggest that transdifferentiation of AoAFs to myofibroblasts in 5wt% hydrogels resulted in a biosynthetic, but not an inflammatory phenotype.
We next investigated whether the production of collagen I or collagen III, key components of the arterial adventitium that regulate matrix stiffness in vivo,[37] were altered in conjunction with the observed differences in cell-mediated alterations of hydrogel stiffness. Collagen I and collagen III were observed in all constructs over time, with primary localization to intracellular and pericellular compartments (Figure S14). In soft 5wt% hydrogels, levels of both ECM proteins increased through day 28 (Figure 4D–E); this pattern paralleled the enhancement of cell proliferation and myofibroblast transdifferentiation observed both in our study and in other published reports.[25, 38] Thereafter, significant reductions in collagen I and III levels were noted, likely attributable to increased MMP-9 and decreased TIMP-1/−2 production (Figure 3D–F), which have been associated with ECM destabilization.[39] Regardless, collagen I and III protein levels in soft 5wt% hydrogels were correlated with storage modulus (for collagen I: r=−0.3052, p=0.3347; for collagen III: r=0.0.7012, p=0.0111; Table S4). In 7.5wt% hydrogels, collagen I and III generally increased over time and inversely correlated with substrate stiffness (for collagen I: r=−0.4395, p=0.1528; for collagen III: r=−0.6014, p=0.0386; Table S4), although the change in collagen levels over time was not statistically significant. In 10wt% hydrogels, collagen I and III levels exhibited bimodal distributions, with significant increases in both collagens observed on day 42. The pattern of collagen expression in 10wt% hydrogels corresponded to decreased gelatinolytic activity (Figure 3B); however, this finding is inconsistent with the observed stable expression of MMP-2/−9 and TIMP-1/−2 in 10wt% cultures, suggesting that the activities of additional enzymes may be responsible for collagen I remodeling. Interestingly, collagen expression by AoAFs in 10wt% hydrogels was not significantly correlated with reduced stiffness over time (for collagen I: r=−0.3984, p=0.1996; for collagen III: r=−0.5182, p=0.0844; Table S4).
As fibroblast function is affected by both the presence of specific collagen types and by their relative abundance,[40] we examined the ratio of collagen I to collagen III in our systems. In 5wt% hydrogels, the collagen I/III ratio decreased over time (Figure 4F), corresponding to stiffening of the hydrogel matrix (Figures 3A). In 7.5wt% hydrogels, where substrate modulus was minimally altered by AoAFs over time, no changes in the collagen I/III ratio were observed. Importantly, in both 5wt% and 7.5wt% hydrogels, the collagen I/III ratio remained below the reported value for the native adventitia (6.1:1),[41] indicating increased production of unorganized, immature collagen III even after 42 days of culture, compared to the native adventitia. In 10wt% hydrogels, at early time points, the collagen I/III ratio approached that of the native adventitia. However, the collagen I/III ratio decreased on day 42, due to an increase in collagen III expression. Importantly, our results indicate that differential collagen production by encapsulated AoAFs is strongly impacted by alterations in hydrogel composition.
2.5. Fibroblasts Control Stiffening of Hydrogel Modulus Using Cellular Traction Pathways
As we observed the synthesis and pericellular accumulation of both collagen I and collagen III by encapsulated AoAFs, we sought to determine whether secreted ECM contributed to substrate stiffening by AoAFs in 5wt% hydrogel cultures after day 7. Increased substrate stiffness due to secretion of collagens by cells encapsulated in hydrogels has been previously reported.[42] Thus, we used hydroxyproline-based assays to estimate collagen levels in our system and detected consistently low levels (<1 μg/hydrogel) of total collagen across 5wt%, 7.5wt%, and 10wt% hydrogels over time (Figure S15). The low levels of collagen produced by encapsulated AoAFs suggest that increased collagen production was not the sole cause of matrix stiffening in 5wt% hydrogel cultures.
Previous studies implicate the exertion of tractive forces by fibroblasts and fibroblast networks attached to collagen fibrils as a mechanism to contract compliant collagen matrices in vitro.[43] In our 5wt% hydrogels, increased shear storage moduli were associated with extensive cell spreading (Figure S16) and decreased bulk hydrogel dimensions (Table S3) were observed by 14 days, but not in the other hydrogel formulations. Accordingly, we set out to evaluate the presence of multi-cellular fibroblast networks and the presence of cell-dependent traction forces.
Cell-matrix interactions and cell traction are mediated through cell surface and secreted proteins. To evaluate the potential effects of cell traction on the polymer matrix, we exposed cells to 20 μM Exo-1, an inhibitor that impedes secretion of the extracellular proteins mediating cell attachment (Figure 5A).[44] No detectable effect on cell viability or the stability of multicellular, interconnected cellular networks was observed following the 7 day Exo-1 treatment (Figure S17A); however, treatment with Exo-1 abolished integrin β1 and collagen I staining on the surface of the encapsulated AoAFs (Figure 5B). As decreased integrin activity has been shown to reduce the fibroblast mechanoactivation and traction force generation,[45] we were unsurprised to observe that Exo-1 treatment blocked the decreases in swollen volume and increases in shear storage modulus observed in untreated cultures prepared in parallel (Figure 5C–D). These findings indicated that cell surface proteins were required to facilitate hydrogel contraction and stiffening of the polymer matrix.
Figure 5.
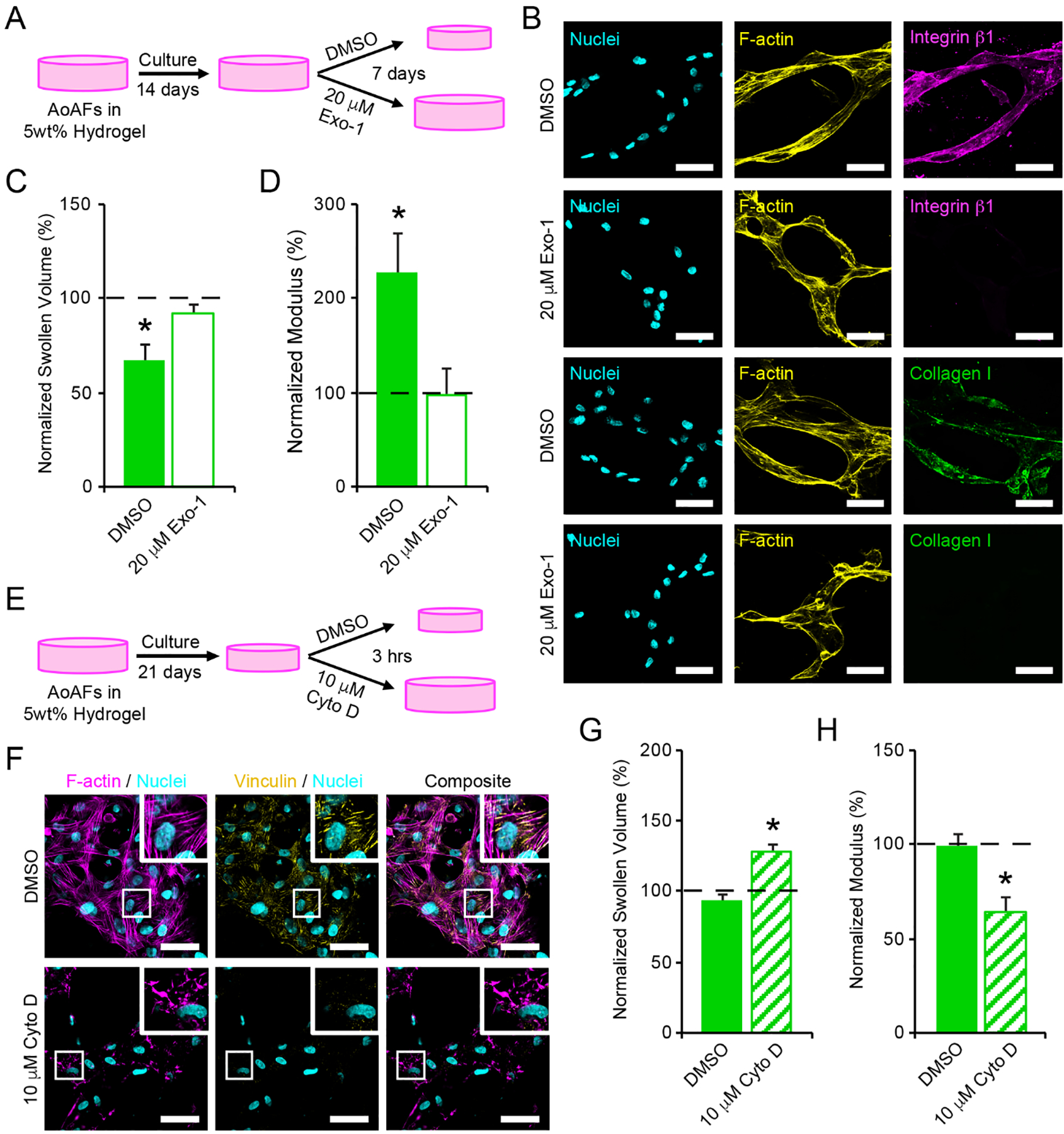
(A) To investigate whether cell interactions with the hydrogel network were responsible for contraction of 5wt% hydrogels, encapsulated AoAFs were cultured for 14 days prior to an additional 7 days of culture in medium supplemented with or without 20 μM Exo-1. (B) Treatment with 20 μM Exo-1 for 7 days decreased integrin β1 and collagen I expression by AoAFs in 5wt% hydrogels. Cyan: Hoechst 33258 (nuclei), yellow: F-actin, magenta: integrin β1, green: collagen I. Scale bar = 50 μm. (C-D) Hydrogels treated with 20 μM Exo-1 for 7 days did not exhibit (C) decreased swollen volume or (D) increased hydrogel modulus. (E) To assess the importance of cytoskeletal tension in maintaining hydrogel contraction, encapsulated AoAFs were cultured in 5wt% hydrogels for 21 days and then exposed to medium with and without 10 μM Cyto D for 3 hrs. (F) Treatment with 10 μM Cyto D for 3 hrs decreased F-actin polymerization and focal adhesion formation, as indicated by vinculin, by AoAFs in 5wt% hydrogels. Cyan: Hoechst 33258 (nuclei), magenta: F-actin, yellow: vinculin. Scale bar = 50 μm. (G-H) Following exposure to 10 μM Cyto D, (G) swollen volume of 5wt% hydrogels increased and (H) hydrogel modulus decreased. Data are mean ± SEM, n = 3 biological replicates per condition. For C-D & G-H: A repeated measures student’s t-test, followed by a Tukey’s HSD post hoc test, was used to detect statistical significance from day 14 measurements (dotted line) and from DMSO treated controls. In C-D: *p<0.05 for DMSO control relative to pre-treatment measurements on day 14 and to Exo-1-treated scaffolds on day 21. In G-H: *p<0.05 for Cyto D-treated scaffolds relative to pre-treatment measurements (dotted line) and to DMSO treated controls.
We further investigated the role of cytoskeletal tension in maintaining increased substrate modulus in 5wt% hydrogels following contraction of the polymer network. After 21 days of culture, confocal microscopy indicated that AoAFs in soft hydrogels exhibited actin stress fibers and vinculin-positive focal adhesions, which were elongated in the direction of their associated stress fibers (Figure 5F). Parallel cultures of AoAF-laden 5wt% hydrogels were treated with 10 μM cytochalasin D (Cyto D), an inhibitor that disrupts actin filaments and relieves cytoskeletal tension (Figure 5E).[46] Treatment with Cyto D for 3 hrs had no detectable effect on cell viability (Figure S17B). However, Cyto D decreased filamentous actin and abolished focal adhesions in AoAFs (Figure 5F). The disappearance of focal adhesion complexes in conjunction with the dismantling of actin filaments by Cyto D is not surprising as disruption of actin filaments has been shown to secondarily interrupt the integrity of focal adhesion complexes.[47] Further, alterations in the actomysin cytoskeleton resulted in an increase in swollen volume of the hydrogel and a corresponding reduction in matrix stiffness, compared to controls (Figure 5G–H). The abrogation of hydrogel contraction observed following Cyto D treatment indicates that actin-mediated AoAF contraction is a key contributor to hydrogel stiffening, a phenomenon which has been previously observed for lung fibroblasts in 3D collagen hydrogels.[48] Together, our observations suggest that AoAFs within soft hydrogels can modulate their local environment to stiffen their encapsulating matrix through integrin-mediated adhesion, actin-mediated cytoskeletal tension, and subsequent contraction of the polymer matrix and emphasize the importance of cellular tension on microenvironmental remodeling within these soft materials.
3. Conclusion
Our results indicate that the initial stiffness of 3D hydrogel cell culture matrices dictates the phenotype of encapsulated AoAFs and simple adjustment of the starting hydrogel modulus alters the phenotype among encapsulated cells in hydrogel cultures over a narrow range of bulk stiffnesses. While others have reported that decreased matrix mechanics can lead to reversal of myofibroblast phenotype in hepatic cells,[49] our data suggest that AoAFs, similar to lung fibroblasts,[50] exhibit memory of initial mechanical conditions, enabling retention of their phenotype even after a reduction in matrix stiffness. In soft 5wt% hydrogels, AoAFs transdifferentiated and stiffened the polymer network using pathways dependent on cell surface proteins and actin-based cytoskeletal tension; however, in 7.5wt% hydrogels, the soft substrate modulus was maintained, and the AoAFs remained undifferentiated. AoAFs in the stiffer 10wt% hydrogels expressed elevated αSMA, but did not alter their expression of αSMA as the substrate softened, indicating that cells retained mechanical history, which differed from that observed in the 7.5wt% condition. Clearly, temporal changes in the mechanics of the soft 5wt% hydrogels invoked alteration of the phenotypes of the AoAFs, where an override of cell memory occurs upon reaching a specific stiffness or following alterations in matrix mechanics. The generalizability of the temporal regulation of matrix stiffness observed in our studies was verified using cells obtained from multiple donors in this study; however, further work examining proliferation and protein secretion across different donors is required to establish widespread implications of cell-mediated matrix remodeling. Overall, these results demonstrate that the use of degradable 3D hydrogels allows for dynamic remodeling of the polymer matrix and highlight the complex relationship between cell phenotype and matrix mechanical properties. This in vitro dynamic system is particularly valuable in the study of vascular development and disease as it may be manipulated to drive beneficial cell phenotypes.
4. Experimental Section
Hydrogel Formation:
Hydrogels were formed by crosslinking of four-arm thiol-functionalized poly(ethylene glycol) (PEG-SH4; f=4, Mn 10,000 g/mol, JenKem Technology, Allen, TX), with the PQ-MI2 and RGD-MI peptides. Hydrogels were prepared at concentrations of 5wt%, 7.5wt%, and 10wt% (w/v%) by altering the concentrations of PEG-SH4 and PQ-MI2, while the concentration of RGD-MI was held constant at 1 mM, independent of hydrogel formulation. Hydrogels were crosslinked at a thiol to maleimide residue ratio of 1:1 for all formulations. As such, hydrogels of a concentration of 5wt% comprised 3.9 mM PEG-SH4 and 7.2 mM PQ-MI2; 7.5wt% hydrogels comprised 5.8 mM PEG-SH4 and 11.1 mM PQ-MI2; and 10wt% hydrogels comprised 7.8 mM PEG-SH4 and 15.0 mM PQ-MI2. To form hydrogels, PEG-SH4, PQ-MI2, and RGD-MI were independently dissolved in buffer (10 mM sodium phosphate monobasic monohydrate, 5 mM citric acid trisodium salt (anhydrous), 140 mM sodium chloride, pH 4.8) and sterilized by passage through a 0.2 μm PVDF filter. Maleimide- and thiol-containing solutions, along with 10 mM HEPES in HBSS (1/5 of the final hydrogel volume; pH 7.4), were mixed together via simple pipetting.
Adventitial Fibroblast Culture and Encapsulation in Hydrogels:
Human aortic adventitial fibroblasts (AoAF; deidentified 53 yo male donor; Lonza, Allendale, NJ) were cultured in stromal cell growth medium (SCGM), which comprised fibroblast basal medium supplemented with 5% FBS, basic fibroblast growth factor, and insulin (all from Lonza). Cells were maintained at 37°C with 5% CO2 and used between passage numbers 5 and 7 for all assays. AoAF phenotype was characterized by examining cell morphology, proliferative capacity, and verifying that fewer than 10% of the cells were positive for α-smooth muscle actin, a marker of myofibroblasts, at each passage.
To encapsulate cells in hydrogels, AoAFs were suspended in 10 mM HEPES in HBSS and added to the hydrogel precursor solutions to achieve 1×106 cells/mL hydrogel, unless otherwise noted. Once mixed, 30 μL volumes of hydrogel were added to a cylindrical mold (diameter = 4.6 mm, thickness = 1.8 mm) and allowed to crosslink for 20 mins at room temperature. AoAF-laden hydrogels were ejected from the molds, subsequently immersed in SCGM (1.5 mL), and incubated at 37°C with 5% CO2. Medium was replaced once per week over the duration of these experiments. Conditioned medium and hydrogels were collected on days 1, 7, 14, 21, 28, 35, and 42, and stored at −80°C for further analysis. For inhibitor studies, hydrogels were immersed in SCGM supplemented with Cytochalasin D (10 μM; Sigma-Aldrich, St. Louis, MO) for 3 hrs or Exo-1 (20 μM; Sigma) for 7 days.
Rheological Characterization of Hydrogels:
The mechanical properties of the hydrogels were characterized via bulk oscillatory rheology (AR-G2, TA instruments). Briefly, the hydrogel precursor solutions, with and without AoAFs, were added to a cylindrical mold (30 μL; diameter = 4.6 mm, thickness = 1.8 mm) and allowed to gel for 15 mins at room temperature. Hydrogels were subsequently immersed in SCGM (1.5 mL) and incubated at 37°C with 5% CO2. The equilibrium shear storage moduli of the swollen hydrogels were measured on days 1, 7, 14, 21, 28, 35, and 42, using a 20-mm diameter stainless steel, parallel plate geometry. Time sweep measurements were obtained within the linear viscoelastic regime using 2% constant strain and 2 rad/s angular frequency. A normal force of 0.2 N was applied to prevent slip during measurement.
Proliferation:
At indicated time points, the hydrogel cultures were washed with PBS, flash frozen in liquid nitrogen, and stored at −20°C until use. A Quant-iT™ Picogreen® dsDNA assay (Life Technologies) was used to measure total DNA content of the harvested samples. Briefly, after thawing, the samples were digested overnight at 65°C with papain (180 μg/mL; Sigma) diluted in PBS containing ethylenediaminetetraacetic acid (4 mmol/L; Sigma) and L-cysteine hydrochloride (10 mmol/L; Sigma). Samples were cooled to room temperature and centrifuged at 0.1 × g for 1 min to remove debris, before collecting the supernatant for analysis via Picogreen dsDNA assay, prepared as instructed by the manufacturer. Samples and DNA standards were measured in this assay in triplicate. The number of AoAFs within each hydrogel at each time point was determined from the total amount of DNA in each sample. For the 3D culture samples, the fluorescence signals obtained from blank hydrogels (i.e. without cells, cultured and processed similarly to those with cells) were used as background values.
Protein Quantification:
MMP-2 and MMP-9 levels in the collected medium were quantified via Quantikine ELISA kits (R&D Systems, Minneapolis, MN). TIMP-1 and TIMP-2 levels in collected medium were measured with a MSD human Cytokine Multiplex kit, while IL-6, MCP-1, and VEGF-A were measured with a MSD human U-PLEX kit (Mesoscale Discovery, Rockville, MD). All assays were used in accordance with the manufacturers’ instructions. To account for variance in the number of AoAF cells in different hydrogel cultures, the amount of IL-6, MCP-1, and VEGF-A detected in each sample of culture medium was normalized to the amount of DNA in the corresponding hydrogel.
Immunostaining:
Hydrogels were cryosectioned by the Histochemistry and Tissue Processing Core Laboratory of Nemours-A.I. duPont Hospital for Children using a protocol adapted from Ruan et al.[51] Briefly, PEG hydrogels were removed from culture medium and fixed with paraformaldehyde in PBS (4%; pH 7.4) overnight, rinsed, and then infiltrated with Tissue-Tek® Optimal Cutting Temperature embedding medium (OCT; Sakura-Finetek, Torrance, CA) overnight at 4°C. Samples were rinsed with fresh OCT and placed in a chilled well of the Precision Cryoembedding System (Pathology Innovations, Wyckoff, NJ) within a −21°C cryostat (Leica CM 3050S). The well was filled with additional OCT and topped with a cryostat chuck. Samples were stored at −80°C and 10 μm cryosections were obtained from the frozen blocks using a cryostat (Leica CM 3050S) at −21°C.
Sections were permeabilized with Triton X-100 (0.1%; Sigma) for 15 min and blocked with bovine serum albumin (BSA) in PBS (3%; Sigma) for 30 min. Samples were stained for 1 hr at room temperature with primary antibodies without shaking, washed, and incubated with secondary antibodies for 1 hr at room temperature without shaking (Table S5). Samples were counterstained with Phalloidin-546 (1:100; Life Technologies) and the nuclear stain Hoechst 33258 (1:1000; Life Technologies) for 1 hr at room temperature. Cells were visualized using an Olympus BX-60 fluorescence microscope with a 20x Oil UPlanApo 0.8 N.A. objective and an Evolution QEi monochrome 12-bit digital camera (Media Cybernetics) controlled by Image Pro Plus software (version 7.0; Media Cybernetics). ImageJ was utilized to measure CTCF and was defined as CTCF = integrated density of selected cell – (area of selected cell x mean fluorescence of background readings). Morphological changes of AoAFs were analyzed via ImageJ using images taken at selected time points. Area represents the total pixel area measured and converted to μm for analysis. Circularity was calculated using the following equation: 4π*area/perimeter2, where a value of 1 corresponds to a perfect circle, and a value approaching 0 indicates an increasingly elongated shape. Shape descriptors were calculated using >30 cells per treatment group.
For the assessment of cells in inhibitor studies, PEG hydrogels were removed from culture medium and fixed with paraformaldehyde in PBS (4%; pH 7.4) for 1 hr, permeabilized with the addition of Triton X-100 (0.2%) for 1 hr, and blocked with goat serum (10%; Invitrogen) in PBS for 2 hrs. Samples were then stained for 48 hrs at room temperature with primary antibodies with shaking, washed extensively with PBS containing Tween-20 (0.05%), and incubated with secondary antibodies, Phalloidin-546 (1:100), and the nuclear stain Hoechst 33258 (1:1000) for 12 hrs at room temperature with shaking (Table S5). Cells were visualized using a Zeiss 880 confocal microscope with a 40x C-Apochromat 1.2 N.A. objective.
Proteolytic Activity:
Proteolytic activity in the collected medium was assessed via Enzchek Gelatinase/Collagenase Assays (ThermoFisher Scientific), following the manufacturer’s instructions. Collected medium (100 μL) was added directly to DQ-gelatin substrate (10 μg/mL) in reaction buffer and incubated at 37°C for 18 hrs. The fluorescence intensity of the substrate was measured using a PerkinElmer plate reader at ex/em wavelengths of 490 nm/535 nm. Baseline levels were determined using medium collected from acellular hydrogels.
Statistical Analysis:
Data are expressed as the mean ± standard error of the mean, unless otherwise noted. Specific numbers of replicates (n ≥ 3) are noted for each experiment within the figure captions. Details regarding the specific test utilized to evaluate each experiment are indicated in figure captions. In brief, assumptions of residual normality (Shapiro-Wilks test) and homoscedasticity (Bartlett’s test) were first verified for each group. Following confirmation that assumptions for parametric analysis were met, statistical significance was analyzed by performing either a Student’s t‐test, one-way ANOVA, or two-way ANOVA, as appropriate, where p < 0.05 was considered significant. In cases where a one-way or two-way ANOVA was used, if the F-test revealed significant statistical differences at the 0.05 level, pairwise comparisons were made using Tukey HSD post-hoc. Statistical interpretations were made using JMP Pro 14 (SAS Institute Inc.).
Supplementary Material
Acknowledgements
The authors would like to thank Nile Bunce and Christopher McGann for technical help with peptide synthesis. This research was made possible by funds from the Nemours Foundation and by support from the NHLBI (R01 HL108110, F32 HL127983). Instrumentation and other resources were provided by support from the NIGMS (P30 GM110758, U54 GM104941, and S10 OD016361). The views and conclusions in this manuscript do not necessarily represent the views of the funding agencies.
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
References
- [1].(a) Stenmark KR, Davie N, Frid M, Gerasimovskaya E, Das M, Physiology, 2006, 21, 134; [DOI] [PubMed] [Google Scholar]; (b) Cecelja M, Chowienczyk P, J. R. Soc. Med. Cardiovasc. Dis, 2012, 1, cvd.2012.012016; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Kuwabara JT, Tallquist MD, Arterioscler. Thromb. Vasc. Biol, 2017, 37, 1598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Sartore S, Chiavegato A, Faggin E, Franch R, Puato M, Ausoni S, Pauletto P, Circ. Res, 2001, 89, 1111. [DOI] [PubMed] [Google Scholar]
- [3].Davie NJ, Gerasimovskaya EV, Hofmeister SE, Richman AP, Jones PL, Reeves JT, Stenmark KR, Am. J. Pathol, 2006, 168, 1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].(a) Galis Z, Khatri JJ, Circ. Res, 2002, 90, 251; [PubMed] [Google Scholar]; (b) van Popele NM, Grobbee DE, Bots ML, Asmar R, Topouchian J, Reneman RS, Hoeks AP, van der Kuip DA, Hofman A, Witteman JC, Stroke, 2001, 32, 454. [DOI] [PubMed] [Google Scholar]
- [5].(a) Robinson KG, Nie T, Baldwin AD, Yang EC, Kiick KL, Akins RE Jr., J. Biomed. Mater. Res., Part A, 2012, 100A, 1356; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Scott RA, Kharkar PM, Kiick KL, Akins RE, Biomaterials, 2017, 137, 1; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Li M, Riddle SR, Frid MG, El Kasmi KC, McKinsey TA, Sokol RJ, Strassheim D, Meyrick B, Yeager ME, Flockton AR, McKeon BA, Lemon DD, Horn TR, Anwar A, Barajas C, Stenmark KR, J. Immunol, 2011, 187, 2711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].(a) Engler AJ, Sen S, Sweeney HL, Discher DE, Cell, 2006, 126, 677; [DOI] [PubMed] [Google Scholar]; (b) Paszek MJ, Zahir N, Johnson KR, Lakins JN, Rozenberg GI, Gefen A, Reinhart-King CA, Margulies SS, Dembo M, Boettiger D, Hammer DA, Weaver VM, Cancer Cell, 2005, 8, 241. [DOI] [PubMed] [Google Scholar]
- [7].Mahadevaiah S, Robinson KG, Kharkar PM, Kiick KL, Akins RE, Biomaterials, 2015, 62, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].(a) Lutolf MP, Hubbell JA, Biomacromolecules, 2003, 4, 713; [DOI] [PubMed] [Google Scholar]; (b) Kloxin AM, Kasko AM, Salinas CN, Anseth KS, Science, 2009, 324, 59; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Khetan S, Guvendiren M, Legant WR, Cohen DM, Chen CS, Burdick JA, Nat. Mater, 2013, 12, 458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].DeForest CA, Anseth KS, Nat. Chem, 2011, 3, 925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Yang C, Tibbitt MW, Basta L, Anseth KS, Nat. Mater, 2014, 13, 645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].(a) Akhtar R, Graham HK, Derby B, Sherratt MJ, Trafford AW, Chadwick RS, Gavara N, J. Mech. Behav. Biomed. Mater, 2016, 64, 10; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Grant CA, Twigg PC, ACS Nano, 2013, 7, 456; [DOI] [PubMed] [Google Scholar]; (c) Rafuse M, Xu X, Stenmark K, Neu CP, Yin X, Tan W, J. Biomech, 2019, 88, 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Farquharson C, Robins SP, Histochem. J, 1989, 21, 172. [DOI] [PubMed] [Google Scholar]
- [13].Scott R, Marquardt L, Willits RK, J. Biomed. Mater. Res., Part A, 2010, 93A, 817. [DOI] [PubMed] [Google Scholar]
- [14].Kharkar PM, Kiick KL, Kloxin AM, Polym. Chem, 2015, 6, 5565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Liang Y, Coffin MV, Manceva SD, Chichester JA, Jones RM, Kiick KL, J. Biomed. Mater. Res., Part A, 2016, 104, 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Kharkar PM, Scott RA, Olney LP, LeValley PJ, Maverakis E, Kiick KL, Kloxin AM, Adv. Healthcare Mater, 2017, 6, 1700713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].(a) Akkapeddi P, Azizi S-A, Freedy AM, Cal PMSD, Gois PMP, Bernardes GJL, Chem. Sci, 2016, 7, 2954; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Vining KH, Scherba JC, Bever AM, Alexander MR, Celiz AD, Mooney DJ, Adv. Mater, 2018, 30, 10704486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Bott K, Upton Z, Schrobback K, Ehrbar M, Hubbell JA, Lutolf MP, Rizzi SC, Biomaterials, 2010, 31, 8454. [DOI] [PubMed] [Google Scholar]
- [19].(a) Li XD, Hong MN, Chen J, Lu YY, Ye MQ, Ma Y, Zhu DL, Gao PJ, Cardiovasc. Res, 2019, 10.1093/cvr/cvz159; [DOI] [PubMed] [Google Scholar]; (b) Lu Y, Azad N, Wang L, Iyer AKV, Castranova V, Jiang BH, Rojanasakul Y, Am. J. Respir. Cell Mol. Biol, 2010, 42, 432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Yang B, Janardhanan R, Vohra P, Greene EL, Bhattacharya S, Withers S, Roy B, Nieves Torres EC, Mandrekar J, Leof EB, Mukhopadhyay D, Misra S, Kidney Int, 2014, 85, 289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Mabry KM, Schroeder ME, Payne SZ, Anseth KS, ACS Appl. Mater. Interfaces, 2016, 8, 21914. [DOI] [PubMed] [Google Scholar]
- [22].Southern BD, Grove LM, Rahaman SO, Abraham S, Scheraga RG, Niese KA, Sun H, Herzog EL, Liu F, Tschumperlin DJ, Egelhoff TT, Rosenfeld SS, Olman MA, J. Biol. Chem, 2016, 291, 6083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].(a) Ferreira SA, Faull PA, Seymour AJ, Yu TTL, Loaiza S, Auner HW, Snijders AP, Gentleman E, Biomaterials, 2018, 176, 13; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ferreira SA, Motwani MS, Faull PA, Seymour AJ, Yu TTL, Enayati M, Taheem DK, Salzlechner C, Haghighi T, Kania EM, Oommen OP, Ahmed T, Loaiza S, Parzych K, Dazzi F, Varghese OP, Festy F, Grigoriadis AE, Auner HW, Snijders AP, Bozec L, Gentleman E, Nat. Commun, 2018, 9, 4049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].(a) Silver FH, Horvath I, Foran DJ, Crit. Rev. Biomed. Eng, 2001, 29, 279; [DOI] [PubMed] [Google Scholar]; (b) Xu J, Shi GP, Biochim. Biophys. Acta, Mol. Basis Dis, 2014, 1842, 2106. [Google Scholar]
- [25].Woods K, Thigpen C, Wang JP, Park H, Hielscher A, BMC Cell Biol, 2017, 18, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Nicodemus GD, Skaalure SC, Bryant SJ, Acta Biomater, 2011, 7, 492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].(a) Schulze C, Wetzel F, Kueper T, Malsen A, Muhr G, Jaspers S, Blatt T, Wittern KP, Wenck H, Kas JA, Biophys. J, 2010, 99, 2434; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Sicard D, Haak AJ, Choi KM, Craig AR, Fredenburgh LE, Tschumperlin DJ, Am. J. Physiol. Lung Cell. Mol. Physiol, 2018, 314, L946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Ugarte-Berzal E, Vandooren J, Bailón E, Opdenakker G, García-Pardo A, J. Biol. Chem, 2016, 291, 11751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Nagase H, Visse R, Murphy G, Cardiovasc. Res, 2006, 69, 562. [DOI] [PubMed] [Google Scholar]
- [30].Pereira RF, Barrias CC, Bártolo PJ, Granja PL, Acta Biomater, 2018, 66, 282. [DOI] [PubMed] [Google Scholar]
- [31].Valdez J, Cook CD, Ahrens CC, Wang AJ, Brown A, Kumar M, Stockdale L, Rothenberg D, Renggli K, Gordon E, Lauffenburger D, White F, Griffith L, Biomaterials, 2017, 130, 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Lachowski D, Cortes E, Rice A, Pinato D, Rombouts K, del Rio Hernandez A, Sci. Rep, 2019, 9, 7299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Raffetto JD, Khalil RA, Biochem. Pharmacol, 2008, 75, 346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Schultz KM, Kyburz KA, Anseth KS, Proc. Natl. Acad. Sci. U. S. A, 2015, 112, E3757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Russell LN, Lampe KJ, ACS Biomater. Sci. Eng, 2017, 3, 3459. [DOI] [PubMed] [Google Scholar]
- [36].(a) Cieslik KA, Trial J, Entman ML, FASEB J, 2015, 29, 3160; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Meléndez Giselle C, McLarty Jennifer L, Levick Scott P, Du Y, Janicki Joseph S, Brower Gregory L, Hypertension, 2010, 56, 225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Diez J, Adv. Cardiol, 2007, 44, 76. [DOI] [PubMed] [Google Scholar]
- [38].McLane JS, Ligon LA, Biophys. J, 2015, 109, 249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Herum KM, Choppe J, Kumar A, Engler AJ, McCulloch AD, Mol. Biol. Cell, 2017, 28, 1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Tracy LE, Minasian RA, Caterson EJ, Adv. Wound Care, 2016, 5, 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Murata K, Motayama T, Kotake C, Atherosclerosis, 1986, 60, 251. [DOI] [PubMed] [Google Scholar]
- [42].(a) Bryant SJ, Anseth KS, J. Biomed. Mater. Res, 2002, 59, 63; [DOI] [PubMed] [Google Scholar]; (b) Bryant SJ, Bender RJ, Durand KL, Anseth KS, Biotechnol. Bioeng, 2004, 86, 747. [DOI] [PubMed] [Google Scholar]
- [43].(a) Lotz C, Schmid FF, Oechsle E, Monaghan MG, Walles H, Groeber-Becker F, ACS Appl. Mater. Interfaces, 2017, 9, 20417; [DOI] [PubMed] [Google Scholar]; (b) Han YL, Ronceray P, Xu G, Malandrino A, Kamm RD, Lenz M, Broedersz CP, Guo M, Proc. Natl. Acad. Sci. U. S. A, 2018, 115, 4075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Feng Y, Yu S, Lasell TKR, Jadhav AP, Macia E, Chardin P, Melancon P, Roth M, Mitchison T, Kirchhausen T, Proc. Natl. Acad. Sci. U. S. A, 2003, 100, 6469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].(a) Fiore VF, Wong SS, Tran C, Tan C, Xu W, Sulchek T, White ES, Hagood JS, Barker TH, JCI Insight, 2018, 3, e97597; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Puig M, Lugo R, Gabasa M, Giménez A, Velásquez A, Galgoczy R, Ramírez J, Gómez-Caro A, Busnadiego Ó, Rodríguez-Pascual F, Gascón P, Reguart N, Alcaraz J, Mol. Cancer Res, 2015, 13, 161. [DOI] [PubMed] [Google Scholar]
- [46].Brown SS, Spudich JA, J. Cell. Biol, 1979, 83, 657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Soong HK, B. D, Lee B, J. Ocul. Pharmacol. Ther, 1990, 6, 113. [DOI] [PubMed] [Google Scholar]
- [48].Bogatkevich GS, Tourkina E, Silver RM, Ludwicka-Bradley A, J. Biol. Chem, 2001, 276, 45184. [DOI] [PubMed] [Google Scholar]
- [49].Caliari SR, Perepelyuk M, Soulas EM, Lee GY, Wells RG, Burdick JA, Integr. Biol, 2016, 8, 720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Balestrini JL, Chaudhry S, Sarrazy V, Koehler A, Hinz B, Integr. Biol, 2012, 4, 410. [DOI] [PubMed] [Google Scholar]
- [51].Ruan JL, Tulloch NL, Muskheli V, Genova EE, Mariner PD, Anseth KS, Murry CE, Tissue Eng., Part C, 2013, 19, 794. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.