

Abstract
Rationale
Currently, there is no effective intervention available that can reduce brain damage following reperfusion. Clinical studies suggest a positive correlation between the increased influx of neutrophils and severity of brain injury following reperfusion. Integrin α9β1 is highly expressed on activated neutrophils and contributes to stable adhesion, but its role in stroke outcome has not been demonstrated to date.
Objective
We sought to determine the mechanistic role of myeloid-specific α9β1 in the progression of ischemic stroke in murine models with preexisting comorbidities.
Methods and Results
We generated novel myeloid-specific α9-deficient (α9−/−) wild-type (α9fl/flLysMCre+/−), hyperlipidemic (α9fl/flLysMCre+/−Apoe−/−) and aged (bone marrow chimeric) mice to evaluate stroke outcome. Susceptibility to ischemia/reperfusion injury was evaluated at 1, 7- and 28-days following reperfusion in two models of experimental stroke: filament and embolic. We found that peripheral neutrophils displayed elevated α9 expression following stroke. Irrespective of sex, genetic deletion of α9 in myeloid cells improved short and long-term stroke outcomes in the wild-type, hyperlipidemic, and aged mice. Improved stroke outcome and enhanced survival in myeloid-specific α9−/− mice was due to marked decrease in cerebral thrombo-inflammatory response as evidenced by reduced fibrin, platelet thrombi, neutrophil, NETosis and decreased phospho-NF-κB, TNFα, and IL-1β levels. α9−/− mice were less susceptible to FeCl3 injury-induced carotid artery thrombosis that was concomitant with improved regional CBF following stroke as revealed by laser speckle imaging. Mechanistically, fibronectin containing extra domain A, a ligand for integrin α9, partially contributed to α9-mediated stroke exacerbation. Infusion of a specific anti-integrin α9 inhibitor into hyperlipidemic mice following reperfusion significantly reduced infarct volume and improved short and long-term functional outcomes up to 28 days.
Conclusion
We provide genetic and pharmacologic evidence for the first time that targeting myeloid-specific integrin α9β1 improves short and long-term functional outcome in stroke models with preexisting comorbidities by limiting cerebral thrombosis and inflammation.
Keywords: Integrin α9β1, NETosis, stroke, arterial thrombosis, vascular inflammation, integrin, Inflammation, Ischemic Stroke, Thrombosis
Graphical Abstract
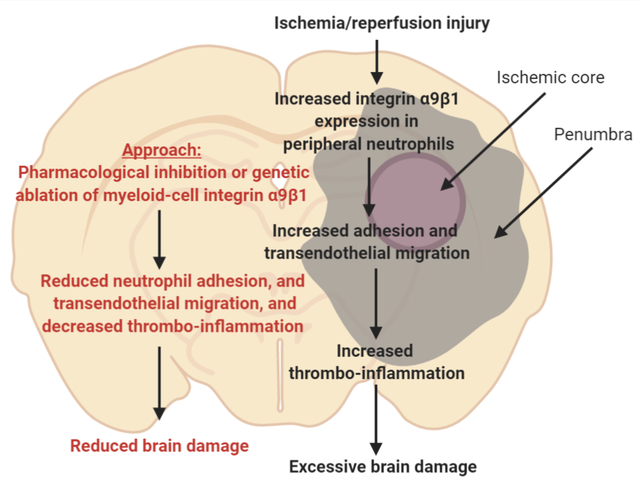
INTRODUCTION
Current treatments for ischemic stroke include thrombolysis using tissue plasminogen activator (tPA) and mechanical thrombectomy. While reperfusion of ischemic brain regions has shown promising clinical outcomes, evidence from clinical studies and animal models suggest that cerebral reperfusion promotes oxidative stress, secondary thrombosis, and vascular inflammation, which aggravate neuronal death in the ischemic penumbra.1–3 Antiplatelet agents, including aspirin, P2Y12 antagonists, and glycoprotein IIb/IIIa inhibitors can reduce the risk of secondary thrombosis, but they are associated with increased risk of hemorrhagic transformation.4 At present, there is no effective intervention available that can efficiently reduce brain damage during reperfusion. A better understanding of molecular mechanisms that facilitate ischemia/reperfusion injury may, therefore, result in the development of novel and effective therapeutic regimens for the treatment of acute ischemic stroke with minimal side-effects.
Since thrombosis and inflammation (thrombo-inflammation) mediate ischemia/reperfusion injury,5–7 an ideal therapeutic target would be the one that limits thrombo-inflammatory responses without causing significant bleeding. Studies have reported a positive correlation between the increased influx of neutrophils and severity of brain injury following reperfusion.3 Neutrophils potentiate neuronal injury by triggering capillary sludging, secreting inflammatory mediators, generating free radicals, and enhancing thrombosis via the formation of neutrophil-platelet aggregates and neutrophil extracellular traps (NETs).8–10 Integrin α9β1, which is highly expressed on activated neutrophils, stabilizes neutrophil adhesion to the activated endothelium in synergy with β2 integrin.11 β1 is only known subunit of α9. Besides neutrophils, integrin α9β1 is also expressed on several other cells, including monocytes, smooth muscle cells, hepatocytes, endothelial cells, and epithelial cells.12 Until a few years ago, signaling cascades involving α9β1 were largely overlooked; however, with recent discoveries linking α9β1 to essential processes in physiology and disease, this integrin is now receiving increased attention.13 The potential mechanistic role of α9β1 in stroke evolution has not been investigated.
Current Stroke Therapy Academic Industry Roundtable (STAIR) recommendations and the NIH-NINDS panel consensus for preclinical assessment of novel neuroprotective agents for stroke recommend evaluation of underlying mechanisms for stroke progression, with assessment of response to treatment in at least two different stroke models in both sexes with preexisting comorbidities that adequately mimic the human physiology.14, 15 Herein, we evaluated the role of myeloid-specific integrin α9 on stroke outcome following the current STAIR guidelines. Novel myeloid-specific α9−/− mice were generated on wild-type (WT) (α9fl/flLysMCre+/−), hyperlipidemic (α9fl/flLysMCre+/−Apoe−/−) and aged (bone marrow chimeric) background to determine the mechanistic role of integrin α9 in ischemic stroke outcome. We chose preexisting comorbidities including hyperlipidemia because these conditions are known to exacerbate stroke outcome16 and thereby enhance sensitivity to stroke and worsen sensorimotor deficit. We provide evidence for the first time that myeloid-specific integrin α9 exacerbates short and long-term stroke outcome in murine stroke model with preexisting comorbidity by promoting thrombo-inflammation.
METHODS
Primary data that support the findings of this study are available from the first author or corresponding author upon request.
A detailed description of materials and methods is available in the Online Data Supplement.
Mice
The University of Iowa Animal Care and Use Committee approved all the procedures and studies were performed according to the current Animal Research: Reporting of In Vivo Experiment guidelines (https://www.nc3rs.org.uk/arrive-guidelines). Details of generating myeloid specific α9-deficient mice on WT and hyperlipidemic background are provided in the Method section in the Online Data Supplements.
Cerebral ischemia and reperfusion injury
Focal cerebral ischemia was induced by transiently occluding the right middle cerebral artery for 30 or 60 minutes, as described.17, 18 See Method section in the Online Data Supplements for details.
Embolic middle cerebral artery occlusion
Embolic stroke model was performed as previously described.19 See Method section in the Online Data Supplements for details.
Statistical analysis
The statistical significance was assessed using either unpaired t-test or one-way or two-way ANOVA followed by Holm-Sidak’s or Fisher’s LSD multiple comparisons test (for normally distributed data) and Mann Whitney test or Kruskal-Wallis tests followed by Dunn’s multiple comparisons test (for not normally distributed data). No corrections for multiple testing were made across different statistical tests. P<0.05 was considered to be statistically significant. See Online Data Supplement for details.
RESULTS
Integrin α9-deficient neutrophils exhibit reduced adhesion and trans-endothelial migration
Evidence from clinical studies suggest rapid changes in gene expression profile in peripheral neutrophils after ischemic stroke.20, 21 To determine if integrin α9 expression on peripheral neutrophils changes after acute ischemic stroke, neutrophils were isolated from the wild-type (WT) mice after 1 hour of ischemia followed by 3, 6, and 24 hours of reperfusion (Figure 1A). Littermate mice with sham surgery were used as controls. Integrin α9 expression was observed to be significantly up-regulated following 3 and 6 hours of reperfusion, but not at 24 hours (Figure 1B). These results were confirmed in parallel by flow cytometry (Figure 1C). Given the early postnatal mortality in global α9−/− mice due to respiratory failure,22 we generated myeloid-specific integrin α9−/− (α9fl/flLysMCre+/−) mice (Online Figure I). Western blot and immunofluorescence analysis confirmed the lack of integrin α9 on neutrophils isolated from the bone marrow (BM) of α9fl/flLysMCre+/− mice (Figure 1D and E). Previously, it was shown that global deletion of α9 in mice results in a defect in granulopoiesis.23 In contrast, we found that complete blood counts (CBCs) were comparable between α9fl/flLysMCre+/− and α9fl/fl mice (Online Table I), suggesting that myeloid-specific deletion of α9 does not result in impaired granulopoiesis and these mutant mice could be used as a genetic model to determine the role of integrin α9 in ischemic stroke.
Figure 1. Integrin α9-deficient neutrophils exhibit reduced adhesion and trans-endothelial migration.

A. Schematic of experimental design. B. Quantification of α9 expression in peripheral neutrophils following ischemia/reperfusion injury by real-time PCR. n=3, 5, 5, 5. C. Left: Representative image of flow-cytometric analysis for each group. Right: Quantification of α9 expression in peripheral neutrophils following ischemia/reperfusion injury by flow-cytometry, n=5. D. Western blot analysis of integrin α9 from the bone-marrow-derived neutrophils of the α9fl/fl and α9fl/flLysMCre+/− mice. E. Representative images showing immunostaining for α9 expression (red) and counterstain by DAPI. Scale bar: 30μm. F. Peripheral neutrophils were isolated after 1 hour of ischemia and 3 hours reperfusion and subjected to adhesion assay on TNF-α stimulated brain microvascular endothelial cells. Left: Representative images of calcein-blue labeled neutrophils. Scale bar: 50μm. Right: Quantification of the adhered neutrophils. n=4, 5, 4, 5. G. Peripheral neutrophils were isolated after 1 hour of ischemia and 3 hours reperfusion and subjected to trans-migration assay on TNF-α-stimulated brain microvascular endothelial cells, n=5/group. Data represent mean ± SEM. Statistical analysis: 1-way ANOVA followed by Holm-Sidak multiple comparisons test (1B, 1C and 1F), unpaired t-test (1G).
Having observed elevated integrin α9 expression on peripheral neutrophil after acute ischemic stroke, we determined whether integrin α9-deficiency inhibits post-stroke peripheral neutrophil adhesion and trans-endothelial migration in vitro. Peripheral neutrophils were isolated after 1 hour of ischemia and 3 hours of reperfusion and assayed for adhesion to TNF-α activated mouse brain endothelial cells. We found that neutrophils from the α9fl/flLysMCre+/− mice exhibited reduced adhesion and trans-endothelial migration, compared to α9fl/fl littermate mice (Figure 1F and G). Together, these results suggest that post-stroke neutrophil integrin α9 is upregulated and contributes to adhesion and trans-endothelial migration.
Myeloid-specific α9−/− mice exhibit improved stroke outcome
Next, we determined stroke outcomes in α9fl/fl and α9fl/flLysMCre+/− mice on the WT background subjected to 1 hour of ischemia and 23 hours of reperfusion in the filament model. Both male and female α9fl/flLysMCre+/− mice exhibited smaller infarcts and better neurological outcome on day 1 when compared with α9fl/fl mice (Online Figure II), concomitant with reduced neutrophil infiltration in the infarcted brain (Online Figure III). Infarcts and neurological scores were comparable between α9fl/fl and α9+/+LysMCre+/− mice, thus ruling out non-specific effects of LysM-Cre recombinase expression (Online Figure IV) on stroke outcome. To determine whether integrin α9 deficiency on myeloid cells also protects during the ischemic phase, we quantified infarcts just before reperfusion. Infarcts were comparable between α9fl/fl and α9fl/flLysMCre+/− mice subjected to 1 hour of ischemia (Online Figure V). Together these results suggest that lack of integrin α9 on myeloid cells improves stroke outcome only in the setting of ischemia/reperfusion injury.
Based on this compelling phenotype in the WT mice, and according to updated STAIR recommendations, we next determined stroke outcome in mice with a preexisting comorbidity, hyperlipidemia, which is known to exacerbate ischemic damage by promoting endothelial injury, inflammation, oxidative stress and neuronal death.16 We generated α9fl/flLysMCre+/− mice on the hyperlipidemic apolipoprotein E-deficient (Apoe−/−) background. All the mice were fed a normal chow diet after weaning until 8–10 weeks, an age at which no significant vascular lesions are found (not shown), to minimize the potential confounding effects of advanced atherosclerotic lesions, which can impair collateral flow and indirectly influence the stroke outcome. Body weight, plasma cholesterol, triglycerides, and complete blood counts were comparable between these groups (Online Table II and III). Neutrophil count and surface expression levels of integrin α9 were comparable between WT and Apoe−/− mice, suggesting that hyperlipidemia does not modulate integrin α9 expression on neutrophils (Online Figure VI). To ensure rigor and confirm that the observed effect is generalizable in a broader context, susceptibility to ischemia/reperfusion injury was evaluated in the same mouse following 1, 3, and 7 days of reperfusion in two models of stroke: the filament model and the embolic model (Figure 2A). α9fl/flLysMCre+/−Apoe−/− mice exhibited smaller infarcts (~32%) and better neurological outcomes at day 1 when compared with α9fl/flApoe−/− mice (Figure 2B and C), in both male and female mice. Consistent with these results, at day 7, α9fl/flLysMCre+/−Apoe−/− mice showed a better survival rate (~70%) compared to α9fl/flApoe−/− mice (~35%, Figure 2D). Next, using the same mouse, we evaluated the modified Neurological Severity Score (mNSS) that rates neurological function based on spontaneous activity, symmetry in limb movement, forepaw outstretching, climbing, body proprioception, and responses to vibrissae touch (on the scale of 3 to 18; higher score indicates a better outcome). α9fl/flLysMCre+/−Apoe−/− mice demonstrated significantly improved neurological outcomes on days 1, 3, day 7 when compared with α9fl/flApoe−/− mice (Figure 2E). Consistent with these results, α9fl/flLysMCre+/−Apoe−/− mice exhibited smaller infarcts, a better survival rate and improved neurological outcomes in the embolic model reperfused with tPA (Figure 2F, G, H and I). Laser Doppler flow measurements (Online Table IV) and physiological parameters (Online Table V) were similar among groups before, during, and after ischemia. Moreover, no gross differences in cerebrovascular anatomy were observed between groups (Online Figure VII).
Figure 2. Deletion of integrin α9 in myeloid cells improves stroke outcome in preexisting comorbid condition of hyperlipidemia.

A. Schematic of experimental design. B. Left: Representative magnetic resonance imaging from one mouse of each genotype on day 1 in filament model. White is the infarct area. Right: Corrected mean infarct volumes of each genotype, n=8, 11, 9, 7. C. Neurological outcome (Bederson score) from each genotype as assessed on day 1 (higher score indicates worse outcome, n=17, 18). D. Survival rate between day 0 to day 7 after 60 min transient ischemia in filament model. E. Modified Neurological Severity Score (mNSS) at days 1, 3, and 7 in filament model based on spontaneous activity, symmetry in the movement of 4 limbs, forepaw outstretching, climbing, body proprioception and responses to vibrissae touch (higher score indicates a better outcome). n=17, 18, 12, 16, 8, 12. F, G H & I. Infarction (%, n=10, 10), Bederson score (n=10, 10), survival rate (%) and mNSS (n=10, 10, 4, 9, 3, 4) in embolic model. Only male mice were used in embolic model. Data are mean ± SEM (2B &2F) and median ± range (2C, 2G, 2E and 2I). Statistical analysis: two-way ANOVA followed by Fisher’s LSD multiple comparisons test (2B), unpaired t-test (2F), Mann-Whitney test (2C & 2G), Comparison of survival curves was evaluated by log-rank (Mantel-Cox) test (2D & 2H), Kruskal-Wallis test followed by Fisher’s LSD multiple comparisons test (2E & 2I).
Lack of Apoe in mice promotes blood-brain barrier (BBB) breakdown and neuronal death by activating the proinflammatory cyclophilin A (CypA)-nuclear factor kB-matrix metalloproteinase-9 pathway in pericytes.24 Since, blocking CypA with cyclosporin A reverses excessive BBB breakdown and neuronal death in Apoe−/− mice,24 we pre-treated α9fl/flLysMCre+/−Apoe−/− and α9fl/flApoe−/− mice with cyclosporin A and determined the protective effect of integrin α9 deficiency on stroke outcome, independent of BBB breakdown. At baseline, BBB leakage was comparable between α9fl/flApoe−/− and α9fl/flLysMCre+/−Apoe−/− mice (Online Figure VIII). Cyclosporin A treated α9fl/flLysMCre+/−Apoe−/− mice exhibited reduced infarct size and improved neurological outcome compared to cyclosporin A treated α9fl/flApoe−/− mice (Online Figure IX). Furthermore, α9 deficiency on myeloid cells significantly reduced infarct volume and improved neurological outcomes in another model of hyperlipidemia, low-density lipoprotein receptor deficient mice (Online Figure X). Together these results suggest that lack of α9 on myeloid cells improves stroke outcome in the setting of hyperlipidemia.
Myeloid-specific α9−/− mice exhibited improved local cerebral blood flow and reduced post-ischemia/reperfusion thrombo-inflammation
Both thrombosis and inflammation are known to contribute to the pathophysiology of ischemic stroke.1, 5, 17 To determine whether improved stroke outcome in the α9fl/flLysMCre+/−Apoe−/− mice was associated with reduced post-ischemia/reperfusion cerebral thrombosis and improved local cerebral blood flow, laser speckle imaging was performed at different time points. We found that regional CBF was improved at 60 and 90-minutes following reperfusion in α9fl/flLysMCre+/−Apoe−/− mice when compared with α9fl/flApoe−/− mice (Figure 3A). Consistent with these results, we observed significantly reduced intracerebral fibrin(ogen), platelet (CD41-positive) deposition and thrombotic index in the α9fl/flLysMCre+/−Apoe−/− mice when compared with α9fl/flApoe−/− mice (Figure 3B and C). These results led us to hypothesize that, following reperfusion, integrin α9 may promote thrombosis and thereby exacerbate stroke outcome. To test this hypothesis, age-matched male mice (same age that were used for stroke evolution) were subjected to experimental thrombosis (FeCl3 injury-induced carotid artery thrombosis). Using intravital microscopy, we found that α9fl/flLysMCre+/−Apoe−/− mice developed smaller thrombi (~40% occlusion) compared to α9fl/flApoe−/− mice (~80% occlusion), 12 minutes after FeCl3 injury-induced thrombosis (Figure 3D). The mean time to complete occlusion was significantly prolonged in the α9fl/flLysMCre+/−Apoe−/− mice (Figure 3D) suggesting that integrin α9 contributes to arterial thrombosis.
Figure 3. Deletion of integrin α9 in myeloid cells improves local cerebral blood flow and inhibits post-ischemia/reperfusion thrombosis.

A. Left: Representative images were taken using laser speckle imaging of regional cerebral blood flow (CBF) in the cortical region. Right: Quantification at different time points (5–90 minutes). n=6, 5. B. Brain homogenates from the infarcted and peri-infarcted area following 1-hour ischemia/23 hours reperfusion was processed for Western blotting: Representative Western blots and densitometric analysis of fibrin(ogen) and platelets (CD4-positive). β-Actin was used as a loading control. n=4/group. C. Left: Representative immunostaining images for platelet (CD41-positive, green) and fibrin(ogen) (red). Scale bar: 100 μm. Right: Thrombotic index, n=4/group D. Left: Representative microphotographs depicting percentage occlusion ∼12 minutes after FeCl3-injured carotid arteries as visualized by upright intravital microscopy. Platelets were labeled with calcein green. White lines delineate the arteries. Right: Mean time to complete occlusion of FeCl3 injured carotid artery, n=8, 8. Data are mean ± SEM. Statistical analysis: Two-way ANOVA followed by Holm-Sidak multiple comparisons test (3A), unpaired t-test (3B, 3C 3D, and 4D).
To determine if improved stroke outcome in the α9fl/flLysMCre+/−Apoe−/− mice was associated with reduced post-ischemia/reperfusion cerebral inflammation, neutrophil and monocyte influx was measured within the infarct and peri-infarct regions of the perfused brain after 1 hour of ischemia and 23 hours of reperfusion in the filament model. Using flow cytometry, we found a significant reduction in neutrophil influx in the α9fl/flLysMCre+/−Apoe−/− mice, while monocyte influx was unaltered in brain homogenates prepared from the infarct and peri-infarct regions when compared with α9fl/flApoe−/− mice (Figure 4A and B). In parallel, we confirmed these results using immunohistochemistry and found significantly reduced neutrophil influx (Ly6B.2-positive cells) in the α9fl/flLysMCre+/− Apoe−/− mice when compared with α9fl/flApoe−/− mice (Figure 4C). Total leukocyte counts were similar among genotypes (Online Table III). The neutrophil influx was absent in the non-ischemic region of the contralateral hemisphere or in sham-operated mice (not shown). In alignment with these results, we found a significant reduction in the inflammatory markers such as phospho-NFκB p65, TNF-α and IL-1β in brain homogenates prepared from the infarcted and surrounding areas of α9fl/flLysMCre+/−Apoe−/− mice when compared with α9fl/flApoe−/− mice (Figure 4D, E and F). Furthermore, we found a reduction in neuronal injury (Fluro-Jade C-positive) and ipsilateral edema in the α9fl/flLysMCre+/−Apoe−/− mice when compared with α9fl/flApoe−/− mice (Figure 4G and H). To determine whether integrin α9 modulates neutrophils extracellular traps formation (NETosis) and thereby exacerbates cerebral thromo-inflammation, we performed NET assay in vitro using stimuli Phorbol 12-myrisate 13-acetate (PMA). The percentage of cells releasing NETs was significantly reduced in α9fl/flLysMCre+/− Apoe−/− neutrophils stimulated with PMA. (Online Figure XI). Together, these results suggest that myeloid-specific integrin α9 exacerbates stroke outcome by promoting post-ischemia/reperfusion thrombo-inflammation.
Figure 4. Deletion of integrin α9 in myeloid cells inhibits post-ischemia/reperfusion inflammation.

A. Following one-hour ischemia and 23 hours after the reperfusion, perfused ipsilateral hemispheres were homogenized and processed for flow cytometry. Left panels show representative dot plots displaying neutrophils in isolated ipsilateral hemispheres from each genotype. The right panel shows quantification, n=6, 5. B. Left panels show representative dot plots displaying monocytes in isolated ipsilateral hemispheres from each genotype and right panel shows quantification, n=6, 5. C. Left: Representative immunostained images for neutrophils (brown Ly6B.2-positive cells indicated by red arrows). Boxed region (lower magnification). Inset in the boxed region is magnified and shown in microphotograph. Scale bar: 100 μm. Right: Quantification. n=5/group D. Brain homogenates from the infarcted and peri-infarcted area following 1-hour ischemia/23 hours reperfusion was processed for Western blotting. Representative Western blots and densitometric analysis of nuclear factor κ B p65 (NFκB p65). β-Actin was used as a loading control. n=4/group. E & F. Quantification of tumor necrosis factorα (TNFα) and interleukin 1β (IL-1β) levels by ELISA in brain homogenates. n=5 /group. G. Representative images for Fluro-jade C (green), and counter-stained with DAPI (stains nuclei, blue). Boxed region (lower magnification). Inset in the boxed region is magnified and shown in the microphotographs. Scale bar: 200 μm. Right: Quantification. Data are mean ± SEM, n=5,4. H. Ipsilateral edema quantified as Extent of edema = (volume of ipsilateral hemisphere – volume of contralateral hemisphere)/volume of contralateral hemisphere X 100, n=8,10. Data are mean ± SEM. Statistical analysis: unpaired t-test (4A, 4B, 4C, 4D, 4E, 4F, 4H).
Myeloid-specific integrin α9 exacerbates stroke outcome partially through its ligand extracellular matrix protein Fn-EDA
The extracellular matrix protein fibronectin containing extra domain A (Fn-EDA) is an endogenous ligand for integrin α9β1 that is known to mediate cell adhesion and migration.25 We have previously reported that Fn-EDA contributes to stroke excerbation.17, 26 To evaluate whether myeloid-specific integrin α9 exacerbates stroke outcome through Fn-EDA, we transplanted irradiated Apoe−/− and Fn-EDA+/+Apoe−/− mice with BM from either α9fl/flLysMCre+/−Apoe−/− or LysMCre+/−Apoe−/− mice (Figure 5A). The efficiency of BM transplant procedure was checked by genotyping 4-weeks after the procedure. Complete blood counts were comparable, suggesting that BMT did not affect the number of BM-derived blood cells (Online Table VI). Susceptibility to ischemia/reperfusion injury was evaluated in the same mouse following 1, 3, and 7 days of reperfusion using the filament model. We found significantly reduced infarcts in α9fl/flLysMCre+/−Apoe−/−-BM→Fn-EDA+/+Apoe−/− mice when compared to LysMCre+/−Apoe−/−-BM→Fn-EDA+/+Apoe−/− mice; reduction in infarct volume was concomitant with improved neurological outcome and survival rate at day 1, 3 and 7 (Figure 5B, C, D and E). On the other hand, Fn-EDA+/+Apoe−/− mice transplanted with BM of LysMCre+/−Apoe−/− mice (LysMCre+/−Apoe−/−-BM→Fn-EDA+/+Apoe−/−) exhibited increased infarcts and worse neurological outcomes compared to Apoe−/− mice transplanted with BM of LysMCre+/−Apoe−/− mice (LysMCre+/−Apoe−/−-BM→ Apoe−/−, Figure 5B and C). We speculate that stroke exacerbation in Fn-EDA+/+Apoe−/− mice (which constitutively express Fn-EDA) is due to elevated (3-fold increase) plasma Fn-EDA levels when compared to Apoe−/− mice.27 Together these results suggest that Fn-EDA partially contributes to α9-mediated stroke exacerbation.
Figure 5. Fn-EDA partially contributes to myeloid cell α9-mediated stroke exacerbation.

A. Schematic of experimental design. B. Left: Representative magnetic resonance imaging from 1 mouse of each group on day 1. White is the infarct area. Right: Corrected mean infarct volumes of each genotype. n=10, 10, 10, 11. C. Neurological outcome (Bederson score) from each genotype as assessed on day 1 (higher score indicates a worse outcome, n=10,10,10,11). D. The survival rate after 60 min transient ischemia. E. Modified Neurological Severity Score (mNSS) at days 1, 3, and 7 based on spontaneous activity, symmetry in the movement of 4 limbs, forepaw outstretching, climbing, body proprioception and responses to vibrissae touch (higher score indicates a better outcome). n=10, 10, 8, 9, 5, 6 and 10,11, 5, 10, 3, 6. Data are mean ± SEM (5B) and median ±range (5C, 5E). Statistical analysis: two-way ANOVA followed by Fisher’s LSD multiple comparisons test (5B), Mann-Whitney test (5C) Comparison of survival curves was evaluated by log-rank (Mantel-Cox) test (5D), Kruskal-Wallis test followed by Fisher’s LSD multiple comparisons test (5E).
Deficiency of integrin α9 on myeloid cells improves stroke outcome and enhances the sensorimotor recovery in the comorbid condition of aging
Aging is considered as one of the most important risk factors for acute ischemic stroke. To determine the detrimental effect of aging on stroke outcome, we compared 70-week-old aged mice reconstituted with WT bone marrow (WT-BM→ aged) versus 12-week-old young mice reconstituted with WT bone marrow (WT-BM→ young). We observed exacerbated stroke outcome in 70-week-old aged mice reconstituted with WT bone marrow (WT-BM→ aged) at day 1, 3 and 7 (Online Figure XII). Next, to test the hypothesis that myeloid-specific integrin α9 exacerbates stroke outcome in aged mice, we transplanted irradiated 70-week-old aged WT mice with BM from either α9fl/flLysMCre+/− or α9fl/fl mice (Figure 6A). The success of the BM transplant procedure was confirmed by genotyping and measuring complete blood counts (Online Table VII). Susceptibility to 60 minutes ischemia was evaluated on the same aged mice (76 weeks) following 1, 3, and 7 days of reperfusion in the filament model. We found significantly reduced infarct in the myeloid-specific integrin α9−/− aged WT mice that were concomitant with improved neurological outcomes and survival rate at day 1, 3 and 7 when compared with control aged WT mice (Figure 6B, C, D and E). In parallel, we evaluated post-stroke sensorimotor recovery in the same chimeric aged mice using the cylinder and accelerated rotarod tests. The cylinder test is a sensorimotor test to assess asymmetry in forelimb use during vertical exploratory behavior inside a glass cylinder, whereas the accelerated rotarod test evaluates motor coordination and balance. We found significantly enhanced sensorimotor recovery on day-7 in myeloid-specific integrin α9−/− aged WT mice compared to control (Figure 6F and G). We were not able to evaluate 28-day stroke outcomes in aged mice because of higher mortality observed in controls.
Figure 6: Deletion of integrin α9 on myeloid cells improves stroke outcome and enhances the sensorimotor recovery in aged mice.

A. Schematic of experimental design. B. Left: Representative magnetic resonance imaging from 1 mouse of each group on day 1. White is the infarct area. Right: Corrected mean infarct volumes of each genotype (n=13, 11). C. Neurological outcome (Bederson score) from each genotype as assessed on day 1 (higher score indicates a worse outcome, n=13, 11). D. The survival rate after 60 min transient ischemia. E. Modified neurological severity score (mNSS) at days 1, 3, and 7 based on spontaneous activity, symmetry in the movement of 4 limbs, forepaw outstretching, climbing, body proprioception and responses to vibrissae touch (higher score indicates a better outcome, n=13, 11, 6,10, 4, 7). F. Post-stroke neurological, behavioral recovery as analyzed by cylinder test on day 7, n=4, 7. G. Post-stroke motor function as analyzed by accelerated rotarod on 7, n=4, 7. Data are mean ± SEM (6B) and median ± range (6C, 6E, 6F and 6G). Statistical analysis: unpaired t test (6B), Mann-Whitney test (6C, 6F and 6G) Comparison of survival curves was evaluated by log-rank (Mantel-Cox) test (6D), Kruskal-Wallis test followed by Fisher’s LSD multiple comparisons test (6E).
Infusion of anti-integrin α9 antibody improves stroke outcome and enhances long-term sensorimotor recovery in a preexisting comorbid condition of hyperlipidemia
We next evaluated the therapeutic potential of targeting integrin α9β1 using the specific anti-α9 antibody 55A2C. This antibody is a well-characterized integrin α9-blocking antibody that has been shown to have an inhibitory effect on the progression of arthritis and multiple sclerosis in mouse models.13 Male Apoe−/− mice were randomly assigned to receive either 55A2C or control Ig and susceptibility to ischemia/reperfusion injury was evaluated following 30 minutes of ischemia and up to 4 weeks of reperfusion in the filament model. We decreased the time of ischemia from 60 minutes to 30 minutes in the intervention study because we have observed excessive mortality in Apoe−/− mice at day 7 following 60 minutes of transient ischemia. To mimic clinical conditions of acute stroke therapy, we infused the 55A2C antibody intravenously into male Apoe−/− mice 15 minutes after reperfusion. Infarct volumes were evaluated on day-1, and neurological/sensorimotor tests were performed from 1-week up to 4-weeks (Figure 7A). Both groups of mice appeared normal during and after treatment. Bodyweight and temperature were comparable among treated and control mice (not shown). A significant reduction in infarct volume (~30%) was observed in anti-integrin α9 antibody-treated Apoe−/− mice, compared to control Ig-treated Apoe−/− mice (Figure 7B). Consistent with this observation, anti-integrin α9 integrin antibody-treated Apoe−/− mice exhibited improved neurological outcome (mNSS) from 1-week up to 4-weeks when compared with control Ig-treated mice (Figure 7C). Importantly, sensorimotor recovery (as analyzed by cylinder test, accelerated rota-rod test, and adhesive tape removal test) was improved in the mice treated with the anti-integrin α9 antibody compared to control mice (Figure 7D, E, F and G).
Figure 7: Infusion of anti-integrin α9 antibody improves stroke outcome and enhances long-term sensorimotor activity in the comorbid condition of hyperlipidemia.

A. Schematic of experimental design. B. Left: Representative magnetic resonance imaging from 1 mouse of each group on day 1. White is the infarct area. Right: Corrected mean infarct volumes of each genotype (n=11,12). C. Modified Neurological Severity Score (mNSS) at weeks-1, 2, 3 and 4 based on spontaneous activity, symmetry in the movement of 4 limbs, forepaw outstretching, climbing, body proprioception and responses to vibrissae touch (higher score indicates a better outcome, n=7/group). Post-stroke sensorimotor behavioral recovery at week-1, 2, 3 and 4 as analyzed by D. cylinder test, n=7/group E. accelerated rotarod, n=7/group F. contact time in adhesive tape removal test, n=7/group and G. removal time in adhesive tape removal test, n=7/group. Data are mean ± SEM (7B) and median ± range (7C, 7D, 7E, 7F and 7G). Statistical analysis: unpaired t test (7B), two-way ANOVA followed by Fisher’s LSD multiple comparisons test (7C, 7D, 7E, 7F and 7G).
DISCUSSION
While a large number of neuroprotective agents have shown efficacy in preclinical studies, they have been unsuccessful in clinical studies. The lack of success of these neuroprotective agents from “bench to bedside” is likely due in part to the practice of performing preclinical evaluation of mechanisms underlying stroke progression and intervention in healthy male WT animals, whereas human stroke usually occurs during the pathophysiological progression of risk factors such as hyperlipidemia, hypertension, obesity, and influenced by age or biological sex. Therefore, STAIR criterion for preclinical stroke evolution recommends the determination of the associated mechanisms and response to the therapeutic intervention in animal models with the pre-existing comorbidities that adequately mimic the risk factors in human stroke.14, 15 Herein, we report a novel role of integrin α9 in the pathophysiology of ischemic stroke. Importantly, we have incorporated several STAIR-RIGOR criteria’s including; a) confirming the findings in two-stroke models: filament and embolic with pre-existing comorbidities such as hyperlipidemia and aging; b) utilizing male and female mice; c) predefining inclusion-exclusion criteria; d) conducting experiments in cohorts and blind fashion, and e) assessment of infarct volume, short and long-term functional outcomes on the same mouse. We believe that these findings may have a clinical significance for the following reasons: First, we have found that integrin α9 is upregulated on peripheral neutrophils following ischemic stroke and contributes to stable adhesion and transmigration. Second, we provide genetic evidence that irrespective of sex, targeting myeloid-specific integrin α9 improves short and long-term stroke outcomes in models with pre-existing comorbidities by limiting post-ischemia/reperfusion thrombo-inflammation. Third, as a potential intervention, infusion of specific anti-integrin α9 antibody following reperfusion improves short and long-term stroke outcome that was concomitant with enhanced long-term sensorimotor recovery.
Abrupt reperfusion following the mechanical thrombectomy or rtPA may promote microvascular dysfunction, oxidative stress and secondary thrombo-inflammation, which further aggravates neuronal death in the ischemic penumbra.1, 7 Early infiltration of neutrophils is a major hallmark of post-ischemic thrombo-inflammation.8 Neutrophils can potentiate thrombo-inflammation by several mechanisms including capillary sludging, releasing free radicals, secreting inflammatory mediators and neutrophil extracellular traps.8–10 Herein, we report that integrin α9 is up-regulated on peripheral neutrophils for up to six hours of cerebral ischemia/reperfusion injury. Importantly, we demonstrated that genetic deletion of integrin α9 on myeloid cells contribute to stable adhesion and trans-endothelial migration to stimulated brain microvascular endothelial cells. Our results are in agreement with previous in vitro studies that utilized pharmacological approach to demonstrate that integrin α9β1 is upregulated upon neutrophil activation and contributes to cell adhesion (human umbilical vein endothelial cells) and migration.28, 29
We demonstrated that myeloid-specific integrin α9−/− mice were less susceptible to ischemia/reperfusion injury in the filament and embolic stroke models, suggesting that myeloid-specific integrin α9 contributes to stroke exacerbation. Based on compelling evidence of improved local cerebral blood flow following reperfusion coupled with decreased intracerebral fibrin(ogen) and platelet deposition in myeloid-specific integrin α9−/− mice, we hypothesized that integrin α9 might exacerbate stroke outcome by potentiating post-ischemia/reperfusion thrombosis. Indeed, myeloid-specific integrin α9−/− mice on the hyperlipidemic background were less susceptible to experimental arterial thrombosis. Mechanistically, integrin α9 may enhance arterial thrombosis by promoting neutrophil recruitment, neutrophil-mediated platelet aggregation, and neutrophil extracellular traps. Indeed, we found that integrin α9 modulates neutrophil extracellular traps formation. In humans, acute ischemic stroke, the severity of the injury and neurological outcome correlates with a neutrophil influx in the infarcted and peri-infarcted region.30 Neutrophils promote local inflammation by producing reactive oxygen species, releasing proinflammatory cytokines and chemokines, and by activating the canonical NF-κB signaling pathway. We found that myeloid-specific integrin α9−/− mice exhibited reduced neutrophil-influx in the infarcted and peri-infarcted region that was associated with decreased phospho-NFκB expression and inflammatory cytokines TNF-α and IL-1β levels. To define the specific role of neutrophil integrin α9, we generated neutrophil-specific integrin α9−/− (α9fl/flMrp8cre+/−) mice. We found that neutrophil-specific integrin α9−/− mice also exhibited improved stroke outcome at day 1 (not shown). Together, these results suggest that myeloid-specific integrin α9, most likely neutrophil integrin α9, exacerbates stroke outcome by promoting post-ischemia/reperfusion inflammation in addition to thrombosis.
We also investigated the molecular mechanism by which myeloid cell integrin α9 promotes stroke exacerbation. α9β1 is known to bind with multiple ligands, including vascular cell adhesion protein 1 (VCAM-1), vascular endothelial growth factor (VEGF), extracellular matrix proteins including tenascin C, osteopontin, and Fn-EDA that are enriched at the sites of inflammation.31 Although α9β1 was shown to mediate neutrophil adhesion and transmigration via VCAM, evidence suggests that anti-VCAM-1 antibodies do not protect against I/R brain injury.32 Tenascin C is proinflammatory,33 and Fn-EDA is both pro-thrombotic and proinflammatory.27, 34, 35 The role of tenascin C in stroke evolution is not known. We hypothesized that the extracellular matrix protein Fn-EDA, might contribute to α9-mediated stroke exacerbation because of overlapping phenotype in genetic models, which have shown that Fn-EDA is pro-thrombotic and pro-inflammatory and exacerbates stroke and atherosclerosis. 17, 19, 27 Using a BM transplantation approach, we found that Fn-EDA partially contributes to α9-mediated stroke exacerbation, suggesting other ligands including tenascin C or other signaling pathways might also contribute to α9-mediated stroke exacerbation.
Currently, there is no effective intervention to reduce brain damage following reperfusion with tPA or mechanical thrombectomy. Several recent clinical trials utilizing mechanical thrombectomy have provided clear evidence that partial reperfusion is associated with adverse stroke outcomes.36, 37 Moreover, many patients treated with rtPA do not show any clinical improvement, despite early recanalization.38 Among many other reasons, post-ischemia/reperfusion increased cerebral thrombo-inflammation might be one of the reasons for the worse outcome. Therefore, there is an important need for the development of interventions that reduce thrombo-inflammation, following reperfusion. Consequently, we evaluated whether targeting integrin α9 with a specific inhibitor (anti-integrin α9 Ig 55A2C) will improve stroke outcome following reperfusion. We provide evidence that targeting integrin α9 with specific anti-integrin α9 Ig, 55A2C, after reperfusion significantly improved long-term functional outcome (up to 28 days) in stroke model with comorbidity by limiting infarct size and enhancing long-term sensorimotor recovery. The anti-α9 Ig 55A2C is known to inhibit the binding of α9/NIH cells to the synthetic peptides AEIDGIEL, the sequence similar to the EDGIHEL sequence present the Fn-EDA segment.25 We speculate that beneficial effects of the anti-α9 Ig 55A2C on stroke outcome might be partly due to the inhibition of integrin α9 binding to Fn-EDA.
While neuroprotection by targeting neutrophils has shown efficacy in preclinical settings, it has been plagued with unexpected failures. One such trial investigated the efficacy of targeting intercellular adhesion molecule-1 with R6.5 (enlimomab). Results of the trial demonstrated that treated patients were more likely to have a neurological disability with a trend towards excess mortality.39 This could be because enlimomab is a murine IgG2a mAb to human intercellular adhesion molecule-1, and it induces profound inflammation and enhances oxidative burst activity.40 Another two trials, using a humanized IgG1 antibody directed at human CD1840 and a recombinant neutrophil inhibitory factor41 did not report any efficacy in stroke outcome. The lack of success could be partially because these agents were not pre-tested in stroke model with co-morbidities or were not clinically evaluated after the successful reperfusion. Since clinical studies have shown a positive correlation between neutrophil influx and severity of brain injury following reperfusion, 3, 42 we suggest that neutrophil-targeted interventions to reduce reperfusion injury requires reevaluation by following current STAIR guidelines to minimize methodological shortcomings.
Strength and limitation
A particular strength of our study is the observation that post-stroke peripheral neutrophils exhibited increased integrin α9 expression levels and therapeutic targeting with a single dose of anti-integrin α9 antibody following reperfusion improved short and long-term functional outcome in stroke models with preexisting comorbidity. Despite this strength, our study has limitations. Integrin α9 is expressed in other cell types, including lymphatic, endothelial, and epithelial cells. The possibility of potential unexpected and adverse physiological side effects of targeting integrin α9 with a specific inhibitor cannot be ruled out. However, we speculate that such a scenario is unlikely because of the acute nature of the treatment. Extending these studies to other species, for example, hypertensive rats might further validate the translational feasibility of these novel findings in a broader context.
Conclusions
Our studies unequivocally support the mechanistic role of myeloid-specific integrin α9 in modulating short and long-term stroke outcome by promoting thrombo-inflammation. Targeting myeloid-specific integrin α9 may have implications to limit brain damage in stroke patients following reperfusion.
Supplementary Material
NOVELTY AND SIGNIFICANCE.
What Is Known?
Clinical studies have reported a positive correlation between the increased influx of neutrophils and severity of brain injury following reperfusion.
Integrin α9β1 is highly expressed on activated neutrophils, stabilizes neutrophil adhesion to the activated endothelium in synergy with β2 integrin. The role of α9β1 in the pathophysiology of stroke has not been demonstrated to date.
What New Information Does This Article Contribute?
Utilizing novel myeloid-specific α9-deficient mice, we provide evidence for the first time that integrin α9β1 contributes to ischemia/reperfusion injury in stroke models with pre-existing comorbidities by promoting thrombo-inflammation. Mechanistically, the extracellular matrix protein Fn-EDA partially contributes to α9-mediated stroke exacerbation.
Targeting integrin α9 with a specific antibody 55A2C following ischemia/reperfusion significantly improved short and long-term functional outcomes in stroke model with pre-existing comorbidity.
Although a large number of neuroprotective agents have shown efficacy in preclinical studies, they have been unsuccessful in clinical studies. The lack of success of these neuroprotective agents from “bench to bedside” is likely due in part to the practice of performing preclinical evaluation of mechanisms underlying stroke progression and intervention in healthy male WT animals, whereas human stroke usually occurs during the pathophysiological progression of risk factors such as hyperlipidemia, hypertension, obesity, and influenced by age or biological sex. In the current study, we demonstrate for the first time a novel role for integrin α9β1 in the pathophysiology of ischemic stroke. Importantly, using several STAIR criteria for preclinical stroke evolution, we demonstrate that targeting integrin α9 genetically or pharmacologically improved short and long-term functional outcomes in stroke models with pre-existing comorbidity. Based on these findings, we propose myeloid-specific integrin α9 as a therapeutic target to limit brain damage in ischemic stroke patients following reperfusion.
Acknowledgments
SOURCES OF FUNDING
The A.K.C lab is supported by grants from the National Institutes of Health grant (R35HL139926, R01NS109910 & U01NS113388) and by Established Investigator Award 18EIA33900009 from American Heart Association. N.D. is supported by the Scholar award from the American Society of Hematology.
Nonstandard Abbreviations and Acronyms
- NETs
neutrophil extracellular traps
- TNFα
tumor Necrosis Factor alpha
- IL-1β
interleukin 1 beta
- tPA
tissue plasminogen activator
- STAIR
stroke therapy academic industry roundtable
- WT
wild-type
- Fn-EDA
fibronectin containing extra domain A
- Ig
Antibody
- CBF
cerebral blood flow
- BMT
bone-marrow transplant
Footnotes
DISCLOSURES
None.
REFERENCES
- 1.Aronowski J, Strong R, Grotta JC. Reperfusion injury: Demonstration of brain damage produced by reperfusion after transient focal ischemia in rats. J Cereb Blood Flow Metab. 1997;17:1048–1056 [DOI] [PubMed] [Google Scholar]
- 2.Warach S, Latour LL. Evidence of reperfusion injury, exacerbated by thrombolytic therapy, in human focal brain ischemia using a novel imaging marker of early blood-brain barrier disruption. Stroke. 2004;35:2659–2661 [DOI] [PubMed] [Google Scholar]
- 3.Akopov SE, Simonian NA, Grigorian GS. Dynamics of polymorphonuclear leukocyte accumulation in acute cerebral infarction and their correlation with brain tissue damage. Stroke. 1996;27:1739–1743 [DOI] [PubMed] [Google Scholar]
- 4.Zinkstok SM, Beenen LF, Majoie CB, Marquering HA, de Haan RJ, Roos YB. Early deterioration after thrombolysis plus aspirin in acute stroke: A post hoc analysis of the antiplatelet therapy in combination with recombinant t-pa thrombolysis in ischemic stroke trial. Stroke. 2014;45:3080–3082 [DOI] [PubMed] [Google Scholar]
- 5.Desilles JP, Syvannarath V, Di Meglio L, Ducroux C, Boisseau W, Louedec L, Jandrot-Perrus M, Michel JB, Mazighi M, Ho-Tin-Noe B. Downstream microvascular thrombosis in cortical venules is an early response to proximal cerebral arterial occlusion. J Am Heart Assoc. 2018;7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.del Zoppo GJ. Inflammation and the neurovascular unit in the setting of focal cerebral ischemia. Neuroscience. 2009;158:972–982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stoll G, Nieswandt B. Thrombo-inflammation in acute ischaemic stroke - implications for treatment. Nat Rev Neurol. 2019;15:473–481 [DOI] [PubMed] [Google Scholar]
- 8.Aronowski J, Roy-O’Reilly MA. Neutrophils, the felons of the brain. Stroke. 2019;50:e42–e43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.del Zoppo GJ, Schmid-Schonbein GW, Mori E, Copeland BR, Chang CM. Polymorphonuclear leukocytes occlude capillaries following middle cerebral artery occlusion and reperfusion in baboons. Stroke; a journal of cerebral circulation. 1991;22:1276–1283 [DOI] [PubMed] [Google Scholar]
- 10.Laridan E, Denorme F, Desender L, Francois O, Andersson T, Deckmyn H, Vanhoorelbeke K, De Meyer SF. Neutrophil extracellular traps in ischemic stroke thrombi. Ann Neurol. 2017;82:223–232 [DOI] [PubMed] [Google Scholar]
- 11.Mambole A, Bigot S, Baruch D, Lesavre P, Halbwachs-Mecarelli L. Human neutrophil integrin alpha9beta1: Up-regulation by cell activation and synergy with beta2 integrins during adhesion to endothelium under flow. J Leukoc Biol. 2010;88:321–327 [DOI] [PubMed] [Google Scholar]
- 12.Singh P, Reimer CL, Peters JH, Stepp MA, Hynes RO, Van De Water L. The spatial and temporal expression patterns of integrin alpha9beta1 and one of its ligands, the eiiia segment of fibronectin, in cutaneous wound healing. J Invest Dermatol. 2004;123:1176–1181 [DOI] [PubMed] [Google Scholar]
- 13.Kon S, Uede T. The role of alpha9beta1 integrin and its ligands in the development of autoimmune diseases. J Cell Commun Signal. 2018;12:333–342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bosetti F, Koenig JI, Ayata C, Back SA, Becker K, Broderick JP, Carmichael ST, Cho S, Cipolla MJ, Corbett D, Corriveau RA, Cramer SC, Ferguson AR, Finklestein SP, Ford BD, Furie KL, Hemmen TM, Iadecola C, Jakeman LB, Janis S, Jauch EC, Johnston KC, Kochanek PM, Kohn H, Lo EH, Lyden PD, Mallard C, McCullough LD, McGavern LM, Meschia JF, Moy CS, Perez-Pinzon MA, Ramadan I, Savitz SI, Schwamm LH, Steinberg GK, Stenzel-Poore MP, Tymianski M, Warach S, Wechsler LR, Zhang JH, Koroshetz W. Translational stroke research: Vision and opportunities. Stroke; a journal of cerebral circulation. 2017;48:2632–2637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fisher M, Feuerstein G, Howells DW, Hurn PD, Kent TA, Savitz SI, Lo EH, Group S. Update of the stroke therapy academic industry roundtable preclinical recommendations. Stroke. 2009;40:2244–2250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ayata C, Shin HK, Dilekoz E, Atochin DN, Kashiwagi S, Eikermann-Haerter K, Huang PL. Hyperlipidemia disrupts cerebrovascular reflexes and worsens ischemic perfusion defect. J Cereb Blood Flow Metab. 2013;33:954–962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dhanesha N, Ahmad A, Prakash P, Doddapattar P, Lentz SR, Chauhan AK. Genetic ablation of extra domain a of fibronectin in hypercholesterolemic mice improves stroke outcome by reducing thrombo-inflammation. Circulation. 2015;132:2237–2247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dhanesha N, Prakash P, Doddapattar P, Khanna I, Pollpeter MJ, Nayak MK, Staber JM, Chauhan AK. Endothelial cell-derived von willebrand factor is the major determinant that mediates von willebrand factor-dependent acute ischemic stroke by promoting postischemic thrombo-inflammation. Arterioscler Thromb Vasc Biol. 2016;36:1829–1837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dhanesha N, Chorawala MR, Jain M, Bhalla A, Thedens D, Nayak M, Doddapattar P, Chauhan AK. Fn-eda (fibronectin containing extra domain a) in the plasma, but not endothelial cells, exacerbates stroke outcome by promoting thrombo-inflammation. Stroke. 2019;50:1201–1209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tang Y, Xu H, Du X, Lit L, Walker W, Lu A, Ran R, Gregg JP, Reilly M, Pancioli A, Khoury JC, Sauerbeck LR, Carrozzella JA, Spilker J, Clark J, Wagner KR, Jauch EC, Chang DJ, Verro P, Broderick JP, Sharp FR. Gene expression in blood changes rapidly in neutrophils and monocytes after ischemic stroke in humans: A microarray study. J Cereb Blood Flow Metab. 2006;26:1089–1102 [DOI] [PubMed] [Google Scholar]
- 21.Moore DF, Li H, Jeffries N, Wright V, Cooper RA Jr., Elkahloun A, Gelderman MP, Zudaire E, Blevins G, Yu H, Goldin E, Baird AE. Using peripheral blood mononuclear cells to determine a gene expression profile of acute ischemic stroke: A pilot investigation. Circulation. 2005;111:212–221 [DOI] [PubMed] [Google Scholar]
- 22.Huang XZ, Wu JF, Ferrando R, Lee JH, Wang YL, Farese RV Jr., Sheppard D. Fatal bilateral chylothorax in mice lacking the integrin alpha9beta1. Mol Cell Biol. 2000;20:5208–5215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen C, Huang X, Atakilit A, Zhu QS, Corey SJ, Sheppard D. The integrin alpha9beta1 contributes to granulopoiesis by enhancing granulocyte colony-stimulating factor receptor signaling. Immunity. 2006;25:895–906 [DOI] [PubMed] [Google Scholar]
- 24.Bell RD, Winkler EA, Singh I, Sagare AP, Deane R, Wu Z, Holtzman DM, Betsholtz C, Armulik A, Sallstrom J, Berk BC, Zlokovic BV. Apolipoprotein e controls cerebrovascular integrity via cyclophilin a. Nature. 2012;485:512–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liao YF, Gotwals PJ, Koteliansky VE, Sheppard D, Van De Water L. The eiiia segment of fibronectin is a ligand for integrins alpha 9beta 1 and alpha 4beta 1 providing a novel mechanism for regulating cell adhesion by alternative splicing. J Biol Chem. 2002;277:14467–14474 [DOI] [PubMed] [Google Scholar]
- 26.Khan MM, Gandhi C, Chauhan N, Stevens JW, Motto DG, Lentz SR, Chauhan AK. Alternatively-spliced extra domain a of fibronectin promotes acute inflammation and brain injury after cerebral ischemia in mice. Stroke. 2012;43:1376–1382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Doddapattar P, Gandhi C, Prakash P, Dhanesha N, Grumbach IM, Dailey ME, Lentz SR, Chauhan AK. Fibronectin splicing variants containing extra domain a promote atherosclerosis in mice through toll-like receptor 4. Arterioscler Thromb Vasc Biol. 2015;35:2391–2400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Taooka Y, Chen J, Yednock T, Sheppard D. The integrin alpha9beta1 mediates adhesion to activated endothelial cells and transendothelial neutrophil migration through interaction with vascular cell adhesion molecule-1. J Cell Biol. 1999;145:413–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shang T, Yednock T, Issekutz AC. Alpha9beta1 integrin is expressed on human neutrophils and contributes to neutrophil migration through human lung and synovial fibroblast barriers. J Leukoc Biol. 1999;66:809–816 [DOI] [PubMed] [Google Scholar]
- 30.Mo X, Li T, Ji G, Lu W, Hu Z. Peripheral polymorphonuclear leukocyte activation as a systemic inflammatory response in ischemic stroke. Neurol Sci. 2013;34:1509–1516 [DOI] [PubMed] [Google Scholar]
- 31.Hoye AM, Couchman JR, Wewer UM, Fukami K, Yoneda A. The newcomer in the integrin family: Integrin alpha9 in biology and cancer. Adv Biol Regul. 2012;52:326–339 [DOI] [PubMed] [Google Scholar]
- 32.Justicia C, Martin A, Rojas S, Gironella M, Cervera A, Panes J, Chamorro A, Planas AM. Anti-vcam-1 antibodies did not protect against ischemic damage either in rats or in mice. J Cereb Blood Flow Metab. 2006;26:421–432 [DOI] [PubMed] [Google Scholar]
- 33.Midwood K, Sacre S, Piccinini AM, Inglis J, Trebaul A, Chan E, Drexler S, Sofat N, Kashiwagi M, Orend G, Brennan F, Foxwell B. Tenascin-c is an endogenous activator of toll-like receptor 4 that is essential for maintaining inflammation in arthritic joint disease. Nat Med. 2009;15:774–780 [DOI] [PubMed] [Google Scholar]
- 34.Chauhan AK, Kisucka J, Cozzi MR, Walsh MT, Moretti FA, Battiston M, Mazzucato M, De Marco L, Baralle FE, Wagner DD, Muro AF. Prothrombotic effects of fibronectin isoforms containing the eda domain. Arterioscler Thromb Vasc Biol. 2008;28:296–301 [DOI] [PubMed] [Google Scholar]
- 35.Prakash P, Kulkarni PP, Lentz SR, Chauhan AK. Cellular fibronectin containing extra domain a promotes arterial thrombosis in mice through platelet toll-like receptor 4. Blood. 2015;125:3164–3172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chamorro A, Blasco J, Lopez A, Amaro S, Roman LS, Llull L, Renu A, Rudilosso S, Laredo C, Obach V, Urra X, Planas AM, Leira EC, Macho J. Complete reperfusion is required for maximal benefits of mechanical thrombectomy in stroke patients. Sci Rep. 2017;7:11636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Goyal N, Tsivgoulis G, Frei D, Turk A, Baxter B, Froehler MT, Mocco J, Ishfaq MF, Malhotra K, Chang JJ, Hoit D, Elijovich L, Loy D, Turner RD, Mascitelli J, Espaillat K, Alexandrov AV, Arthur AS. Comparative safety and efficacy of modified tici 2b and tici 3 reperfusion in acute ischemic strokes treated with mechanical thrombectomy. Neurosurgery. 2018;83:593. [DOI] [PubMed] [Google Scholar]
- 38.Alexandrov AV, Hall CE, Labiche LA, Wojner AW, Grotta JC. Ischemic stunning of the brain: Early recanalization without immediate clinical improvement in acute ischemic stroke. Stroke. 2004;35:449–452 [DOI] [PubMed] [Google Scholar]
- 39.Enlimomab Acute Stroke Trial I. Use of anti-icam-1 therapy in ischemic stroke: Results of the enlimomab acute stroke trial. Neurology. 2001;57:1428–1434 [DOI] [PubMed] [Google Scholar]
- 40.Becker KJ. Anti-leukocyte antibodies: Leukarrest (hu23f2g) and enlimomab (r6.5) in acute stroke. Curr Med Res Opin. 2002;18 Suppl 2:s18–22 [DOI] [PubMed] [Google Scholar]
- 41.Krams M, Lees KR, Hacke W, Grieve AP, Orgogozo JM, Ford GA, Investigators AS. Acute stroke therapy by inhibition of neutrophils (astin): An adaptive dose-response study of uk-279,276 in acute ischemic stroke. Stroke. 2003;34:2543–2548 [DOI] [PubMed] [Google Scholar]
- 42.Akopov SE, Grigorian GS, Ovanessian GA. Deactivation of no by polymorphonuclear leukocytes in patients with ischemic cerebral infarction. Stroke. 1996;27:2337–2338 [PubMed] [Google Scholar]
- 43.Dhanesha N, Vazquez-Rosa E, Cintron-Perez CJ, Thedens D, Kort AJ, Chuong V, Rivera-Dompenciel AM, Chauhan AK, Leira EC, Pieper AA. Treatment with uric acid reduces infarct and improves neurologic function in female mice after transient cerebral ischemia. J Stroke Cerebrovasc Dis. 2018;27:1412–1416 [DOI] [PubMed] [Google Scholar]
- 44.Bederson JB, Pitts LH, Tsuji M, Nishimura MC, Davis RL, Bartkowski H. Rat middle cerebral artery occlusion: Evaluation of the model and development of a neurologic examination. Stroke. 1986;17:472–476 [DOI] [PubMed] [Google Scholar]
- 45.Garcia JH, Wagner S, Liu KF, Hu XJ. Neurological deficit and extent of neuronal necrosis attributable to middle cerebral artery occlusion in rats. Statistical validation. Stroke. 1995;26:627–634; discussion 635 [DOI] [PubMed] [Google Scholar]
- 46.Li X, Blizzard KK, Zeng Z, DeVries AC, Hurn PD, McCullough LD. Chronic behavioral testing after focal ischemia in the mouse: Functional recovery and the effects of gender. Exp Neurol. 2004;187:94–104 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.