

Abstract
Many pathologic conditions lead to the development of tissue scarring and fibrosis, which are characterized by the accumulation of abnormal ECM and changes in tissue mechanical properties. Cells within fibrotic tissues are exposed to dynamic microenvironments that may promote or prolong fibrosis, which makes it difficult to treat. Biomaterials have proved indispensable to better understand how cells sense their extracellular environment and are now being employed to study fibrosis in many tissues. As mechanical testing of tissues becomes more routine and biomaterial tools become more advanced, we are beginning to understand the impact of biophysical factors in fibrosis. Here we review fibrosis from a materials perspective, including the role and mechanical properties of ECM components, the spatiotemporal mechanical changes that occur during fibrosis, current biomaterial systems to study fibrosis, and emerging biomaterial systems and tools that can further our understanding of fibrosis initiation and progression. We conclude this review by highlighting considerations in promoting wide-spread use of biomaterials for fibrosis investigations and by suggesting future in vivo studies that we hope will inspire the development of even more advanced biomaterial systems.
Keywords: Extracellular matrix, fibrosis, mechanics, hydrogels, mechanobiology
1. Introduction
Fibrosis is defined as the accumulation and abnormal distribution of ECM with associated tissue stiffening in response to injury. It affects almost all tissues in the body, including skin, liver, lung, kidney, and vasculature. Progressive fibrosis leads to a loss of tissue function and eventual tissue failure, which makes it a major indication for organ transplantation, the only effective treatment in most cases. The lack of success in developing therapies via the conventional path of identifying soluble factor signaling pathway inhibitors has led researchers to turn to alternative approaches, including the manipulation of mechanobiological pathways.[1] Fibrosis is a palpable disease, and just as physicians can feel the increased stiffness of fibrotic tissues, cells can sense this also. Mechanical properties of tissues change during fibrosis development due to increased matrix deposition, remodeling, chemical modification, and degradation, as well as the actions of cells (e.g., contraction) and changes in their phenotype (e.g., myofibroblast activation). It is becoming clear that ECM mechanical changes affect the function of all adherent cells within an organ, and it is imperative to understand the contribution of these outside-in signals in the context of both healthy and diseased tissues.
Initial in vitro studies on fibrosis were carried out using tissue culture polystyrene (TCPS) or glass, where the rigid environment activates cells towards a fibrotic phenotype without exogenous signals. Although much progress in fibrosis research has been made using these two-dimensional (2D) systems, it is difficult to separate the effects of soluble factors and mechanical inputs in cell phenotype, and cell behavior in 2D (seeded atop a surface) may differ from that in three-dimensions (3D) (embedded within a material). Engineered systems which decouple soluble and mechanical factors and permit 3D culture have now been developed to address many of these concerns. Here we will review the ECM and its composition along with the mechanical changes and key cellular players in fibrosis of tissues including the skin, heart, lung and liver. We will also describe major advances in our understanding of the mechanobiology of fibrosis that have been discovered using engineered systems and will identify emerging biomaterial technologies to study fibrosis in the future.
2. Fibrosis from a materials perspective
Fibrosis is a normal part of wound healing; however, with injury, particularly chronic injury, ECM homeostasis is disrupted, and aberrant healing occurs, leading to prolonged and often permanent changes in the composition and concentration of the ECM. These changes directly affect cells residing within tissues through both biochemical and biophysical signals. Materials based on the ECM are being developed to help understand how changes that occur during fibrosis signal to cells and, potentially, to serve a therapeutic role in reversing these changes. To provide a foundation for understanding the fibrotic microenvironment that engineered systems are trying to emulate, we will first cover the structure, function, and mechanical properties of the major components of the ECM.
Collagen is the major matrix component and source of structure and strength in both normal and fibrotic tissues, resisting tensile forces. The “collagen domain” of collagens has multiple repeats, potentially hundreds, of the sequence Gly-X-Y (where X is often Pro and Y often OH-Pro), which facilitates formation of a rigid, tightly packed hetero- or homomeric triple helix. While there are nearly thirty collagens, the fibrillar collagens (primarily I, II, III, and V) predominate. The collagens in this family are almost entirely triple helical and form rigid rods that self-assemble to form large fibrils and larger fibers. The bending modulus of isolated type I collagen fibrils, measured with atomic force microscopy (AFM), is 1.4 GPa and 3.8 GPa for native and glutaraldehyde-crosslinked collagen, respectively.[2] Collagen undergoes a variety of forms of physiological cross-linking, most notably intra- and inter-molecular covalent cross-linking mediated by lysyl hydroxylase and lysyl oxidase family enzymes. Collagen can also rapidly form weak dynamic bonds with other collagen molecules that might contribute to tissue remodeling; however, the nature of these bonds is unclear. [3,4]As a result of these bonds, fibrillar collagen forms a fibrous network (Box 1). Such networks are critical to the function of collagen, as they enable cell contractility-mediated collagen reorganization and alignment, that is often plastic (permanent).[3,5,6] Increases in collagen cross-linking have also been shown to be responsible for initial tissue stiffening in both lung and liver fibrosis, and to drive progressive fibrosis.[7,8]
Box 1. Mechanical terms in biomaterials and mechanobiology.
Stress
The force over a given area (Pa). 1 Pa = 1 pN/μm2. Cells exert traction forces on the ECM in the range of pN-nN over small areas (nm2−μm2), resulting in stresses of 0.02−50 kPa.
Strain
A change in shape (deformation) resulting from applied stress. Cells strain the ECM through traction stress.
Elastic modulus (E)
The slope of the stress-strain curve, and an intrinsic property of a material. Tissues have distinct E that influences cell function and differentiation in vitro. AFM measurement of tissue suggests ~E for (i) Dermis: 0.1−10 kPa,[34] (ii) Heart: 10 kPa, Fibrotic Heart: 20–100 kPa,[38] (iii) Lung 2 kPa, Fibrotic Lung: 17 kPa,[93] (iv) Liver: 0.150 kPa, Fibrotic Liver: 1–6 kPa.[47]
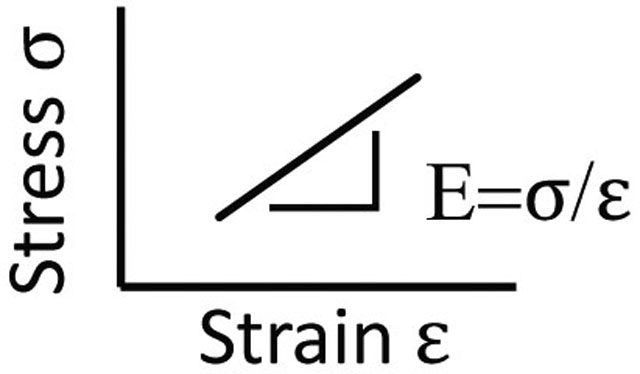
Stiffness
The degree to which an object resists deformation in response to stress. The stiffness of the ECM is unique to different tissues, and is determined by variables such as ECM density, tension and crosslinking. Stiffness differs from E in that it is dependent on the object’s geometry.
Linear elasticity
Materials that display a linear increase in stress with strain have a constant elastic modulus. Synthetic hydrogels typically have linearly elastic responses.
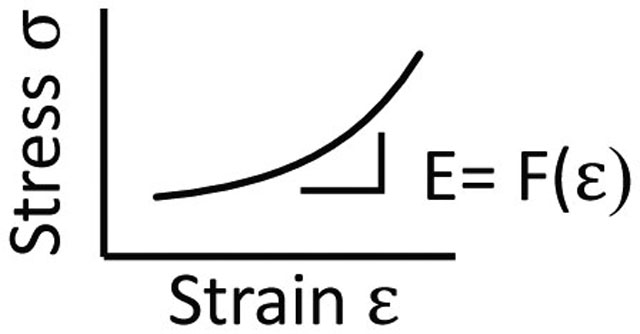
Non-linear elasticity
A dynamic hyperelastic property of fibrous ECM, where stiffness increases non-linearly in response to strain. Non-linearly elastic ECM stiffens when cells contract (strain) the matrix.
Viscoelasticity
Property of materials exhibiting solid- (elastic) and fluid-like (viscous) properties that are dependent on time. ECM entanglement and charge can influence interactions with surrounding ECM and interstitial fluid, which dissipate force and give viscoelastic behavior. The factors that determine both elasticity and viscosity in cells and tissues are not fully understood.
Plasticity
Permanent changes in shape in response to stress. Plastic remodeling of the ECM can result in persistent alignment or densification of ECM fibers through the breakage and formation of crosslinks.
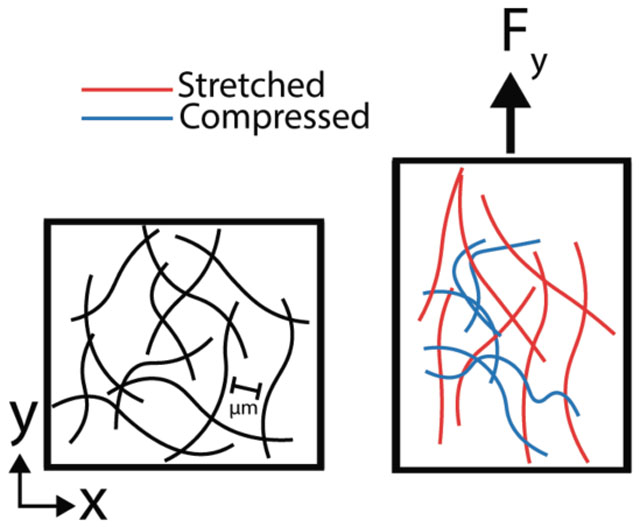
Fibrous network
A loosely connected web of ECM proteins that have a fibrillar structure. Fiber networks have large pores relative to fiber diameter and undergo architectural rearrangements in response to stress. Propagation of traction force can be more than 10 times the cell diameter in fibrous networks.
Collagen in fibrosis is typically dense and aligned, structure that has significant functional impact. For example, the deposition of fibrillar collagens in fibrotic lung prevents expansion and air exchange; in fibrotic liver, collagen septae disrupt normal blood flow. Collagen is non-linearly elastic, softening under compression and stiffening in tension.[9] Tissues show the opposite, compression stiffening and tension softening. Recent work suggests that the interaction between collagen fibrous networks and the cells which adhere to them is critical in defining tissue mechanics.[10] Another important mechanical feature of collagen is that it is piezoelectric. None of these properties of collagen have been easy to mimic in engineered systems. The majority of biomaterials are non-fibrillar and many, including polyacrylamide, are linearly elastic, in contrast to the fibrous network structure of collagen, and few materials are piezoelectric. Although biomaterials are often coated with collagen to enable cell binding, this thin layer of collagen lacks the mechanical properties of collagen fibrous networks. Biomaterials based on collagen, on the other hand, have the problem that, while they permit cell-mediated reorganization, they are difficult as a result to tune mechanically.
Non-fibrillar collagens including the basement membrane collagen IV and the filamentous collagen VI are also important to normal and, in many cases, fibrotic tissues. These collagens may have significant non-collagenous domains, which add flexibility and enable them to serve a variety of functions including as linkers or meshes. Collagen IV is a major component of Matrigel™, a matrix mixture often used to mimic the basement membrane in in vitro systems. However, Matrigel™ lacks the organized structure of the basement membrane and is isolated from tumor tissue, which might represent a pre-fibrotic niche. Additionally, these materials are notorious for batch-to-batch variation, which makes it difficult to probe the effects of ECM perturbations on cell outcomes.
Elastins make up the second category of major structural proteins. Rich in proline and glycine, elastins have extensible hydrophobic domains as well as α-helical domains that are heavily cross-linked by lysyl oxidases; they provide resilience to tissues. Mechanical testing of elastins highlight their contribution to tissue properties, showing a low stiffness, 1MPa, and high extensibility (~150% strain at failure) compared to collagen. Fibrin is another highly extensible ECM protein; it accounts for the mechanical properties of early blood clots. Fibrin can be strained to three times its original length before failing, and also has relatively low stiffness (1–10MPa). Fibrin is a common component of in vitro systems because it is easy to use for device fabrication and undergoes high levels of cell-mediated remodeling; however, it is still difficult to tune the properties of this protein. Interestingly, fibrin, similarly to collagen, has adaptive properties whereby fibrils and fibers rapidly form weak dynamic bonds with each other under mechanical force, which contributes to changes in mechanics and ECM structure without the activity of ECM cross-linking enzymes.[11,12]
Glycosaminoglycans (GAGs) and proteoglycans (GAG-modified proteins) are another key family of ECM components. This group includes the GAG hyaluronic acid (HA), the small leucine-rich proteoglycans lumican, fibromodulin, decorin, and biglycan, the basement membrane proteoglycan perlecan, and the large interstitial proteoglycans versican and aggrecan. These ECM components, which typically increase in fibrosis (although to different degrees), have a major impact on the mechanics of tissues. GAGs are highly-charged, space-filling components of the ECM, forming hydrated gels. GAGs and proteoglycans, by virtue of their water associations, enable tissues to resist compression; they are very stiff and provide significant turgor. They can both interact with and regulate the fibrillogenesis of collagen. The interaction between GAG networks and collagen fibrous networks increases the stiffness of collagen. While HA is the base of many engineered materials for fibrosis studies, it is often cross-linked and highly modified, preventing physiological binding to cells; in physiological form it is rarely used for in vitro studies.
Other matrix components include the multi-domain adhesive glycoproteins such as fibronectin and laminin, which have cell-binding, collagen-binding, and GAG-binding domains and serve as linkers in tissues, connecting cells and different parts of the ECM. Fibronectin increases in fibrosis and is often one of the first ECM molecules to do so; in particular, the cellular (as opposed to plasma) variant of fibronectin increases, is bound and organized by cells (via its RGD domain) and in its relaxed state plays a necessary role in the organization of collagen and other matrix proteins.[13] Fibronectin has unusual mechanical properties – it can be stretched up to 8-fold, becoming remarkably stiff in the process.[14] While it is frequently used to coat hydrogels, and is deposited atop engineered materials by cells, it is rarely used in materials to study fibrosis.
It is key to note that, in vivo, ECM components are organized in large heterogenous aggregates. These are important to the function of the ECM and are essential to the organization and mechanics of the fibrillar collagens, which is particularly relevant to fibrosis. It is clear that the complexity of the ECM is a challenge and not a surprise that it is rarely represented in engineered biomaterials.
3. Multi-scale mechanical properties of tissues
Healthy, injured and fibrotic tissues all experience forces including compressive (e.g. squeezing due to ECM-mediated confinement), tensile (e.g. stretching as the result of cell contractility), and shear (e.g. blood flow). These forces exert stress on tissues, which is measured as the force over a given area (Newtons/m2) and expressed in pascals (Pa) (Box1). Tissues resist forces with their stiffness, and these forces, along with soluble factors, regulate cell phenotype and tissue architecture in healthy and diseased tissues.[15,16] Understanding such mechanical properties and forces are important as biomaterial platforms are developed to model fibrosis.
The macroscale (bulk) multi-axial properties of tissues can be analyzed by taking whole tissues and straining them in tension (e.g. stretching) or compression (e.g. squeezing) while measuring the resultant stress with a mechanical testing machine (e.g. Instron). By calculating the slope of the stress-strain curve, one can obtain the elastic modulus (E), a commonly used measure of the properties of healthy and fibrotic tissues (Box1). The properties of tissues under shear as opposed to axial stress can be measured using a rheometer. The shear storage modulus (G’), which is analogous to E, and loss modulus (G”), which is a measure of viscosity, can be measured under different levels of compression or tension (Box1).[9,17]Non-invasive techniques such as elastography and pressure-based elasticity measurement have been developed for measuring these tissue mechanical properties in vivo. Depending on the degree of fibrosis, fibrotic tissues will generally have higher Young’s, storage, and loss moduli than healthy tissue.
The major limitation of bulk tissue mechanical testing for mechanobiological studies is scale. Cells sense their environments on a small scale (e.g., a single collagen fiber versus the entire tissue), and it is unlikely that the bulk properties of a tissue reflect what a cell experiences. For example, bulk measurements of type I collagen hydrogels using shear rheology show low storage moduli (G’, 100s of pascals), while individual collagen fibers, measured via three point bending with AFM probes, are rigid (gigapascal range). Thus, AFM has been widely employed to characterize the properties of tissues. Most AFMs can assess tissue mechanics with probe sizes relevant to cells (from <1μm up to 50μm), can measure cell scale forces (pN-nN) and strains (<1–15μm), and can be positioned above confocal microscopes to combine mechanical analysis with fluorescence guidance.[18] In liver fibrosis research, AFM has shown that tissue stiffness is heterogenous at the sub-cellular scale, significantly changing the way we think about the cell microenvironment in fibrosis.[19] It is important to note that sample preparation could dramatically influence measured outcomes when measuring mechanical properties at this scale due to anisotropic ECM alignment, which is common in fibrotic tissues and has been demonstrated in the meniscus.[20]
To begin to understand forces generated within tissues, in vitro self-assembled microtissues have been developed.[21] Lung microtissues composed of type I collagen, human lung small airway epithelial cells (SAECs) and lung fibroblasts produced contractile forces of 95 μN, while TGFß1 treatment increased forces up to 210 μN.[22] Similarly, microtissues assembled with 3T3 fibroblasts increased contractile forces from 20 μN to 75 μN with TGFß1 treatment.[23] Although microtissues are useful to begin to uncover the traction forces generated in multicellular fibrotic tissues, the nature of these forces (e.g., tensile, compressive, shear) and their magnitude is still unclear.
Other mechanical properties are now being studied in tissues across different scales (Box1). For example, hyperelasticity of tissues has been observed with AFM and compression testing and changes with fibrosis progression – fibrotic tissues often display linearly elastic responses to strain while pre-fibrotic tissues display non-linearly elastic responses.[10,24] The viscoelasticity of tissues can be measured from the macro to microscale using stress relaxation tests, where a constant strain is applied and the relaxation or decrease in stress is measured over time. As stress decays, a characteristic relaxation time is obtained and the time it takes for the stress to relax to 50% of its peak value (T1/2) can be reported. Purely elastic materials (without a viscous component to dissipate force) will not undergo stress relaxation. Lastly, tissue plasticity (or viscoplasticity) has recently emerged as a material property that is altered in cancer progression and can be measured using creep-recovery tests where a stress is applied and then removed to assess permanent changes in strain (Box1).[3,25]It is unclear if plasticity is altered in fibrosis. Creep-relaxation tests on cardiac, lung and liver tissues have shown viscoplastic behavior, and that plasticity decreases with age. [25] Highly-crosslinked fibrotic tissues likely have low plasticity, while pre-fibrotic tissues likely have relatively high levels of plasticity; however, this has not been well studied in fibrotic tissues.[26]
4. Mechanical changes and pathogenesis of fibrosis across tissues
Mechanisms for the development of fibrosis are conserved across tissues. Common etiologies of tissue damage include traumatic injury, metabolic disease, viral and bacterial infection, and inflammatory responses (Figure 1). Fibrotic tissues show disruption of tissue architecture, activation of local tissue fibroblasts into ECM-producing myofibroblasts and stiffening of the tissue ECM. Chronic injury as well as the properties of the fibrotic tissue itself serve as potentiating factors driving fibrosis progression. In addition to common mechanisms of fibrosis, tissues also have unique responses to injury that include differences in cellular mediators of myofibroblast activation, ECM composition, time course, and propensity for fibrosis regression. These appear to arise out of differences in the normal function of the tissue. This section will describe several tissue systems to illustrate critical differences between them that are important to consider in biomaterial platform design.
Figure 1. A simplified schematic of fibrosis pathogenesis.
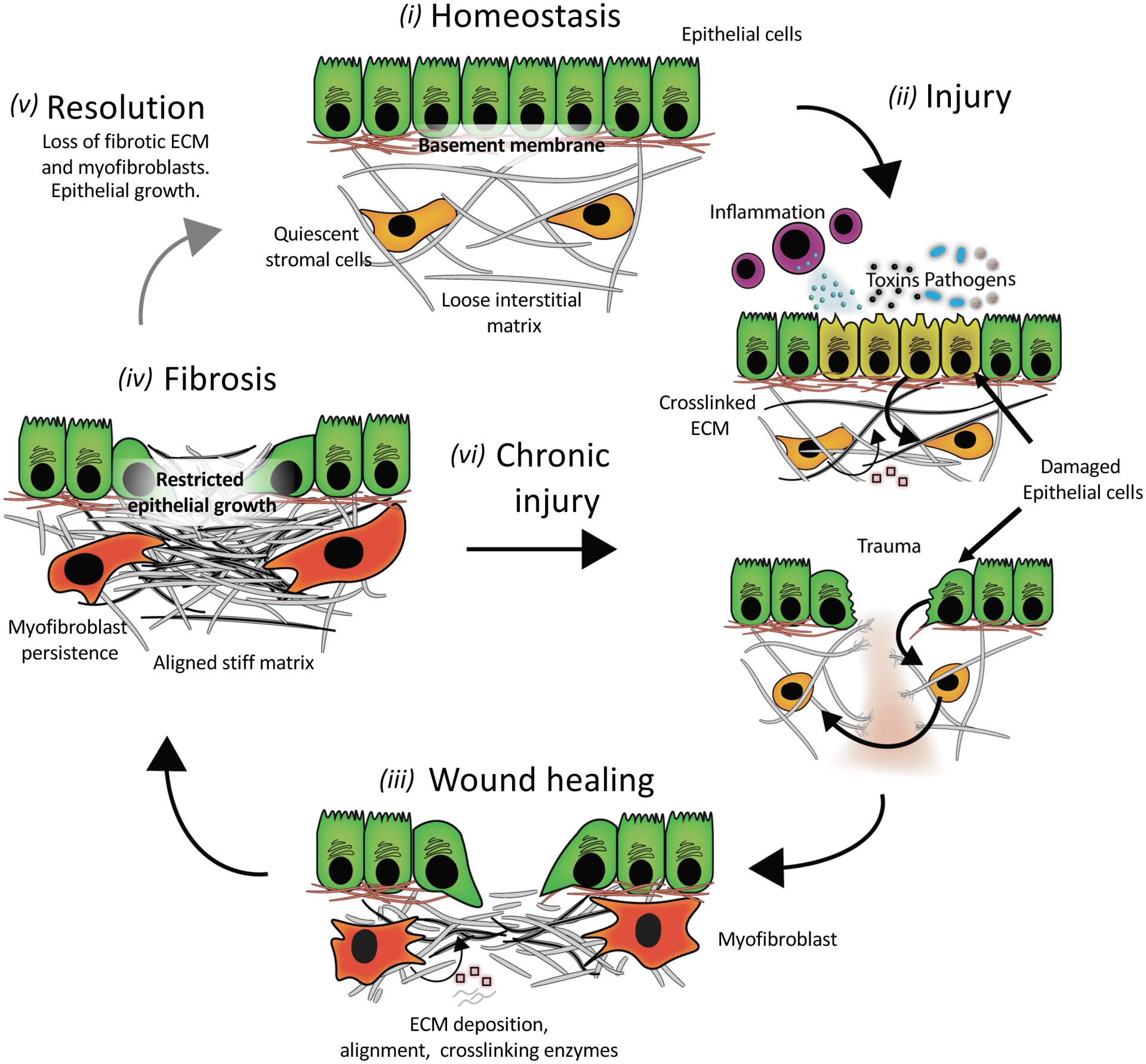
(i) An ECM network in a healthy tissue has a stiffness that supports epithelial cell function and stromal cells in a quiescent state. (ii) Common injuries to tissues include trauma, autoimmune reactions (inflammation), chemicals (toxins), and pathogens (bacteria and viral infections). Acute signaling between injured epithelial and stromal cells triggers ECM remodeling (degradation and/or stiffening (pink squares are ECM modifying proteins)). (iii) During the wound-healing response, myofibroblasts secrete ECM (grey lines) and ECM-modifying proteins while also contracting the matrix to re-establish tensional homeostasis. This provisional matrix can provide a substrate for re-epithelialization; however, excessive matrix deposition and stiffening can lead to (iv) scarring or fibrosis that alters epithelial growth and differentiation. Fibrosis is marked by high levels of stiff ECM such as crosslinked collagen, with resulting myofibroblast persistence. In some tissues, fibrosis resolves (v) if the underlying stimulus for injury is removed, but often (vi) chronic injury leads to a progressive cycle of further wound healing and fibrosis without removal of excess ECM.
4.1. Fibrosis in skin
The skin is a multilayered organ with an outer epidermal layer composed mostly of keratinocytes and an inner dermal layer composed of sweat glands, hair follicles, vasculature, fibroblasts, and ECM.[27] Skin fibrosis occurs in the dermal layer, and the major causes of skin injury that lead to fibrosis include trauma (e.g. abrasions and burns), while immune reactions are less common (Figure 1). [28,29] Fibrotic conditions of the skin include hypertrophic scars that last for years, keloids, hypertrophic and proliferative scars that spread beyond the initial injured area, and scleroderma, a widespread and idiopathic thickening and stiffening of the skin.[30,31]Wounds that do not heal within 3–4 weeks post injury will form scars that lack the properties of healthy skin tissue. [32,33] In vitro systems where mechanical properties can be dynamically changed during culture could be useful to study the influence of mechanics over time in wound healing responses.
Surface tension measurements of human skin show that hypertrophic scars have a 4-fold increase compared to healthy skin (3 N/mm for scars and 0.75 N/mm for healthy). Healthy human skin is heterogeneous in stiffness at the cell scale, and this is likely exacerbated in fibrosis.[34] The dermal ECM composition is also complex with various biochemical ligands (e.g, fibrillar (I, III and V) and non-fibrillar (VI, XII, XIV and XVI) collagens, proteoglycans, glycoproteins, laminins and elastins) that change dramatically during fibrosis (e.g., increases in collagen I, III, IV and V such that they predominate). [28,31,32] Thus, biomaterials with the ability to pattern ligands and mechanical properties with high spatial control will be useful to determine the role of heterogeneity in skin fibrosis initiation and progression.
During the course of wound healing, dermal fibroblasts and pericytes migrate into the injured tissue or blood clot and undergo myofibroblastic activation, contract the wound, and secrete ECM to form granulation tissue.[32] While this myofibroblast activation is necessary for proper wound healing, prolonged activation can lead to excessive ECM deposition and hypertrophic scarring. Myofibroblast persistence is likely caused by paracrine signaling with immune cells and keratinocytes, as well as mechanical abnormalities in the ECM. Platforms that enable tight control over cell-cell signaling and biophysical properties should open up new opportunities to probe the factors that drive myofibroblast persistence in skin fibrosis.
4.2. Fibrosis in cardiac tissue
Cardiac fibrosis has three typical histological presentations: perivascular, interstitial, and focal.[37] The major etiology of cardiac fibrosis is myocardial infarction (MI); however, other causes include cardiotoxins, metabolic disease and hypertension. Altered cardiac mechanical properties occur early in disease,[37] and AFM-based mechanical testing shows the Young’s modulus of the healthy heart to be around 10 kPa, while that of the fibrotic heart ranges between 20–100 kPa.[37,38] AFM stiffness mapping studies have found heterogeneity in heart stiffness, as seen in other tissues, and studies of fibrotic hearts show that damaged and scarred areas have higher moduli than healthy regions.[39]
Stiffening of the ECM can have adverse effects on the function of cardiomyocytes and can potentiate the activation of cardiac fibroblasts to further promote fibrosis. [37] Thus, it is generally accepted that cardiac fibrosis is not reversible without therapeutic intervention.[40]Developing material platforms that undergo large dynamic stiffness (~1 order of magnitude) changes with high spatiotemporal resolution should improve our understanding of how changes in mechanics influence the progression of cardiac fibrosis.
The major stimulus triggering myofibroblast activation is cardiomyocyte death; however, pressure overload has similar effects.[41] Since cardiomyocyte proliferation is limited, myofibroblasts maintain the integrity of the tissue by using scar tissue to fill in areas of cardiomyocyte death. Cardiac myofibroblasts secrete type I and III collagens and their crosslinkers the lysyl oxidases, which stiffen the ECM and increase its resistance to mechanical stress and degradation. Integrating biomaterial systems with engineered functional cardiac tissues (e.g., microtissues or organoids) will help elucidate how myofibroblast-mediated ECM modifications (e.g., stiffening and biochemical changes) locally influence cardiomyocyte function in fibrosis.
4.3. Fibrosis in lung tissue
Lung fibrosis has a variable presentation depending on its etiology. For instance, pulmonary artery hypertension leads to vascular fibrosis, while idiopathic pulmonary fibrosis (IPF) presents with patchy fibrotic regions that distort the normal lung architecture into a microscopic honeycomb pattern.[42] Although the etiology of lung fibrosis is often unknown (as in IPF), it can also be caused by viral infections, chronic exposure to aerosolized containments, chemotherapeutics, and radiotherapy.[43] Spontaneous lung fibrosis resolution has been reported in patients with acute respiratory distress syndrome; however, there is no evidence that IPF can resolve and lung failure typically occurs 2–5 years after diagnosis.[44] Due to the complex nature of lung fibrosis pathogenesis, biomaterial systems that are amendable to personalized cell culture systems (e.g., iPSC-derived and organoids) will be important to improve our understanding of the causes of lung fibrosis.
In IPF, chronic micro-injuries to the lung epithelium are thought to drive abnormal healing and the deposition of excessive matrix by lung fibroblasts.[45] Animal models of IPF have shown that collagen crosslinking (as opposed to increased collagen concentration) is a major reason for tissue stiffening and that inhibiting crosslinking limits stiffening and fibrosis-related dysfunction.[8] Topography scans of collagen deposits in diseased tissue found that collagen fibers had smaller diameters and increased stiffness and crosslinking when compared with normal tissue.
Recent work has also found that normal lung and regions of lung with active fibrogenesis (fibrotic foci) both display nonlinear elastic properties, whereas mature fibrotic areas of the lung have linearly elastic responses to strain. [24] To elucidate the consequences of changes in nanoscale structure, stiffness and non-linear ECM properties in lung myofibroblast activation, new biomaterial systems that accurately capture these properties are needed.
4.4. Fibrosis in liver tissue
Liver fibrosis is the result of chronic (not acute) injury, including from chronic viral infection, alcoholic and non-alcoholic fatty liver disease, and autoimmune syndromes. The histological presentation varies depending on the underlying etiology and may initially show a periportal, pericentral, or pericellular distribution. Stiffening of the liver, assessed via shear rheology, precedes ECM deposition and myofibroblast activation and is likely due to collagen crosslinking shortly after injury.[7] AFM based mechanical testing of the human liver shows that cirrhotic tissue ex vivo is ~360 Pa, while normal tissue is ~175 Pa,[46] and animal studies have confirmed that liver stiffness is heterogenous with fibrosis.[7][47] The liver has a high capacity for regeneration; however, chronic injury leads to aberrant healing and fibrosis, which limits the regenerative capacity of hepatocytes.[48]
The liver is different from most epithelial tissues in that epithelial and endothelial cell populations (hepatocytes and sinusoidal endothelial cells) lack a traditional basement membrane and are instead bounded by the space of Dissé, an interstitial space with low density, loosely-organized ECM (mainly composed of collagen IV and laminin) that transitions towards a stiff and viscous ECM (e.g., Col I, III, GAGs, and glycoproteins) during fibrosis.[48]
Liver rheology has shown that with fibrosis progression there can be an increase in the loss modulus or viscosity, which could be due to increases in GAGs.[7] Liver, like other soft tissues, undergoes compression stiffening, and this is dramatically increased with fibrosis.[10,17,24] To elucidate the role of ECM structure and organization and force dependent materials properties (e.g., compression stiffening) on hepatocyte function and myofibroblast activation, new dynamic material systems (tunable compression stiffening hydrogels) and material fabrication strategies (e.g., electrospinning) could be combined to accurately address these questions. Additionally, to understand how small spatiotemporal changes in liver stiffness influence hepatocyte function and myofibroblast activation, new biomaterial systems should be developed where small changes (100s of pascals) can be introduced in a spatiotemporal manner.
5. Biomaterial design to study fibrosis
Over the past decades, numerous biomaterials have been developed that have enabled researchers to gain a better understanding of how cells perceive their environment. The majority of these biomaterials have been hydrogels (water-swollen polymer networks), although other systems such as elastomers have also been investigated. Since mechanical changes clearly occur during fibrosis progression, mechanically-variable biomaterials have been designed to mimic aspects of healthy and diseased tissues. Beyond understanding how the microenvironment influences cell behavior, an overarching goal of mechanobiological studies is the identification of new therapeutic targets for fibrosis.[1] Surprisingly, the majority of in vitro fibrosis studies are carried out on tissue culture plastic dishes, which are rigid and cause spontaneous myofibroblast activation. In order to accurately predict efficacious anti-fibrotic therapies, culture systems are needed where the mechanics can be tailored to investigate questions related to myofibroblast activation and reversion to a quiescent state. Before we discuss in vitro fibrosis studies carried out using biomaterials, we will first describe various properties that can be introduced into biomaterial systems to probe mechanobiological questions (Figure 2) and review the general material systems available (Table 1).
Figure 2. Schematic of important structural and mechanical features to incorporate into biomaterial platforms to investigate fibrosis.
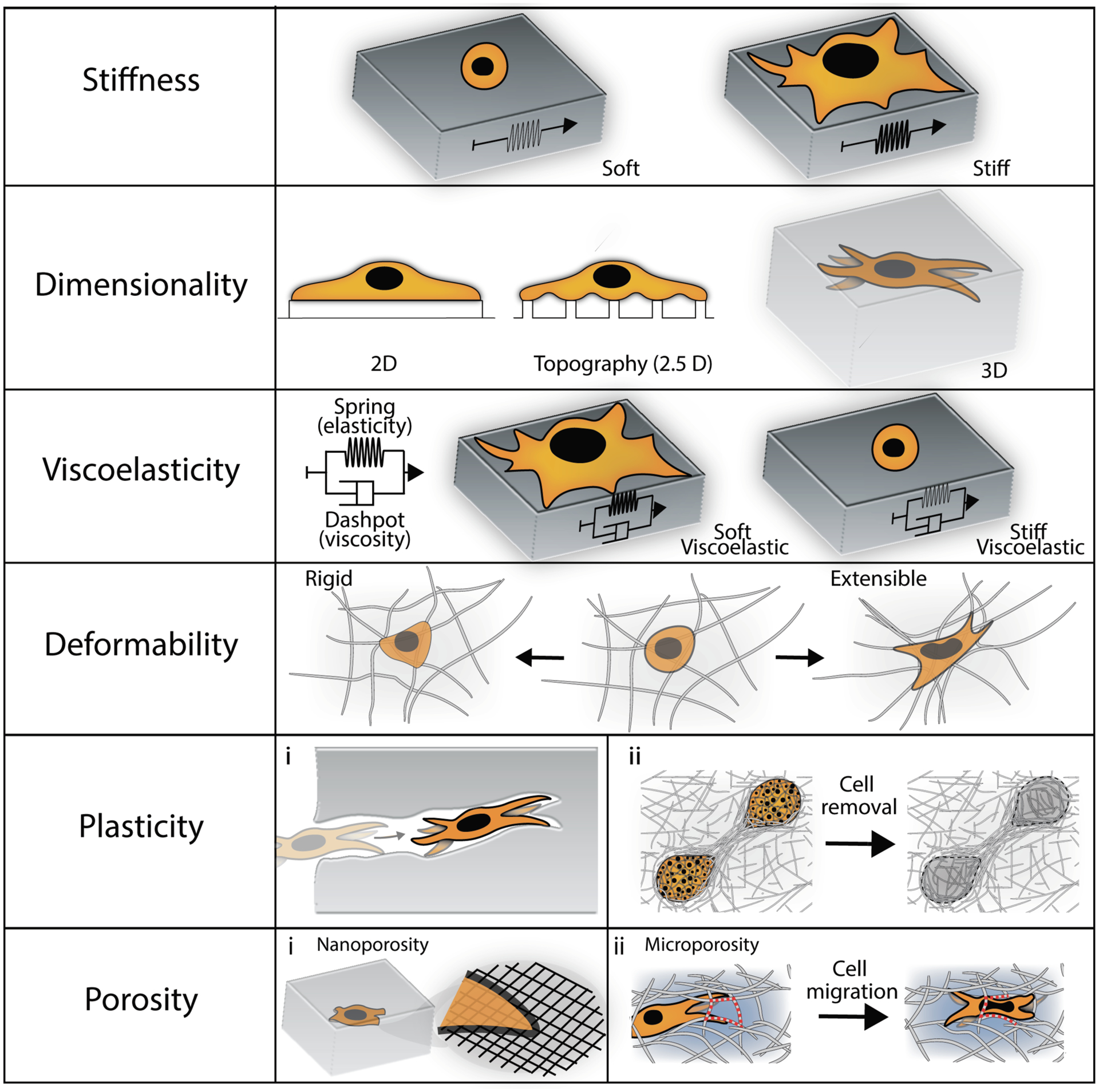
These include various features of the extracellular matrix, as well as outcomes of cellular behavior, such as spreading, morphology and migration.
Table 1.
List of important structural and mechanical features investigated in fibrosis platforms, commonly used biomaterials to model these features, and representative experimental outcomes.
| Material Features | Biomaterials Investigated | Representative Outcomes |
|---|---|---|
| Stiffness | 2D: Alginate, HA, PDMS, PEG, PAA | Stiff, relative to normal tissue stiffness, biomaterials:
|
| Dimensionality |
2D: Alginate, HA, PDMS, PEG, PAA 2.5D: Electrospun Dextran/HA/PEG Fibrin, Collagen 3D: Alginate, Dextran, HA, PEG, Polypeptides, Fibrin, Collagen |
2.5D biomaterials (e.g., electrospun and natural fibrillar proteins):
|
| Viscoelasticity |
2D: Alginate, HA, PAA, PDMS, PEG 3D: Alginate, HA, PEG |
Viscoelastic biomaterials: |
| Deformability |
2D: Alginate, PEG 2.5D: Electrospun Dextran/HA/PEG, Fibrin, Collagen 3D: Alginate, HA, PEG, Fibrin, Collagen |
Extensible (soft) fibrous materials:
|
| Plasticity |
2.5D: Electrospun Dextran Fibrin, Collagen 3D: Alginate, Fibrin, Collagen |
Plastically deformable hydrogels: |
| Porosity |
Nanoscale: Alginate, Dextran, HA, PEG Microscale: Alginate, HA Electrospun Dextran/HA/PEG Fibrin, Collagen |
Nanoscale porosity:
|
2D: two-dimensions, 3D: three-dimensions, HA: hyaluronic acid, PEG: polyethylene glycol, PDMS: polydimethylsiloxane, PAA: polyacrylamide
When designing biomaterials it is critical to have a system where biophysical and biochemical variables such as stiffness, polymer concentration, porosity, and of adhesion site concentration can be tuned and decoupled. One disadvantage of using natural polymer systems (e.g., collagen) is that such decoupling is difficult, making it a challenge to understand how one specific parameter influences cell behavior. Engineered material systems, however, have been developed where decoupling is possible. Synthetic systems allow for tight control over adhesion ligand density and porosity, while stiffness can be changed with crosslinker concentration. Other major features to consider in designing engineered systems are stability in culture and potential off-target effects. For example, cell surface CD44 receptors could bind to engineered hyaluronic acid hydrogels or cell-secreted factors (e.g. ECM) could block cell-material interactions.[49–51] Additionally, it is important to understand in engineered systems how changing a given material property will alter the material from the molecular to microscale. Keeping this in mind, we will next discuss some of the natural, synthetic, and hybrid materials that have been engineered and their limitations.
A common natural ECM hydrogel used for cell studies is type I collagen. Collagen is inherently adhesive and has a fibrillar architecture that can be remodeled by cells. Since collagen is degradable via cell-secreted proteases, it can be difficult to determine how mechanical properties influence cell behavior since these properties are coupled. Additionally, tuning the properties of natural materials may alter degradability, microstructure, or adhesion ligand density.[52] In addition to collagen, other commonly used natural materials include fibrin and Matrigel™. Since all of these materials are purified forms of natural ECM, they enable incorporation of many aspects of the ECM, including non-linear mechanics (e.g. strain stiffening, viscoelasticity), nano- to micro-structure, the potential for cell remodeling, and the capacity for plasticity, into engineered systems. [3,25,53]
The most common synthetic material used for studies of ECM stiffness is polyacrylamide (PAA). PAA hydrogels have been utilized for multiple applications (e.g. electrophoresis) due to their limited non-specific interactions with proteins and are useful for cell culture studies since cells can not interact with them through surface receptors or foul them with secreted proteins. PAA hydrogels are often modified using full-length ECM proteins (e.g. fibronectin) or peptides, which are chemically linked to the PAA, to facilitate cell adhesion.[54] PAA hydrogels allow for decoupled changes in stiffness and adhesion ligand density, which makes them a useful tool for understanding how these factors modulate cell phenotype.[55] Unfortunately, PAA hydrogels are restricted to 2D studies, since the catalysts used for PAA polymerization are cytotoxic and prevent cell encapsulation.
Poly(ethylene glycol) (PEG) hydrogels are often used as an alternative to PAA hydrogels since they are similarly inert and tunable, but can be polymerized into hydrogels using cytocompatible conditions. PAA and PEG polymers have been engineered with tunable viscoelasticity by changing polymer:crosslinker ratios or tuning the number of dynamic bonds that undergo stress relaxation and are increasingly used to study the cell response to viscoelasticity. [56,57] Additionally, PEG-based hydrogels have been developed with protease- and photo-degradable crosslinkers, which enable cell- or user-directed modification of hydrogel properties. [58,59] Another commonly-used synthetic polymer is polydimethylsiloxane (PDMS), which has been engineered to undergo photo-crosslinking-based stiffening and is widely used for fabricating microfluidic devices.[60] One major benefit to the use of PDMS is that the optical properties of this elastomer allow for the visualization of cell-mediated substrate contraction (wrinkling) with phase contrast imaging, which is not possible with other isotropic hydrogels.[61] PDMS is considered an elastomer and is not hydrated, so it is stable under a range of conditions.
Common chemically-modified natural polymers (hybrid materials) include alginate and hyaluronic acid (HA). Hydrogels made from these materials are highly tunable and can be crosslinked under cytocompatible conditions. These polymers have been engineered with functional groups that allow for spatiotemporal changes in stiffness with exposure to light and provide the ability to tune dynamic crosslinks that regulate time-dependent properties such as viscoelasticity and viscoplasticity.[50,62–65] HA has also been engineered with protease- and photo-degradable crosslinkers.[66,67] Other common hybrid materials used to make tunable hydrogels include dextran, chitosan, and gelatin.[68,69] Gelatin is typically modified with methacrylate groups (e.g. GelMA), which can be rapidly photo crosslinked into gels. Gelatin is widely used due to its natural adhesive ligands for cell attachment and ability to be degraded by cell-secreted matrix metalloproteinases. The latter feature of gelatin makes mechanobiological studies difficult to interpret due to the potential for material relaxation and softening with degradation. It is unclear whether cell-secreted proteins interfere with the signals presented by synthetic and hybrid materials in 2D; however, this does occur in 3D culture since nascent proteins are secreted at the cell-material interface and diffusion of the ECM out of the polymer network is limited.[50]
6. Applications of biomaterials to study fibrosis
6.1. Influence of stiffness on myofibroblast activation
Both mesenchymal stromal cells (MSCs) and fibroblasts are important myofibroblast precursors in fibrosis, and both have been studied extensively using model biomaterial systems.[70–72]. Initial studies with collagen hydrogels showed that stiff substrates promote stress fiber formation and α-smooth muscle actin (SMA) expression (typical of myofibroblasts) and that myofibroblasts only respond to the pro-fibrotic factor TGF-ß1 in stiff collagen hydrogels under tension.[73] Collagen hydrogels in early experiments were not chemically modified. Some were tethered to a substrate and were considered stiff because cell traction forces increased tensile stresses (tension) within the hydrogel and stiffened the matrix, while other hydrogels were free-floating (with minimal tension) and were considered soft.
To gain further insight into how mechanical properties affect myofibroblast activation, researchers began using materials that permitted more fine tuning of mechanical properties. PDMS was used to understand lung fibroblast activation, and the threshold for α-SMA incorporation into actin stress fibers was found to be ~16 kPa, which is similar to the stiffness measured for fibrotic skin.[74] Similarly, dermal fibroblasts undergo myofibroblast activation on stiff PDMS substrates (50 kPa). Initial studies with cardiac fibroblasts found that collagen-coated agar hydrogels prevented myofibroblast activation, while rigid tissue culture polystyrene (TCPS) led to myofibroblast activation, and recent work confirmed this observation with soft PDMS substrates (5 kPa).[75,76] Surprisingly, cardiac fibroblasts cultured on HA hydrogels with a range of stiffnesses in the range of healthy myocardium (8 kPa) developed α-SMA positive stress fibers; however, a pronounced myofibroblast phenotype was observed with stiffer 50 kPa hydrogels.[77] Activation of latent TGF-ß1 to the active form also requires a stiff microenvironment, highlighting the intersection between mechanical and soluble factor signaling and the overall importance of mechanics on myofibroblast behavior.[34][74]
Stiffness gradients created using PAA hydrogels were used to study the migration behaviors of lung fibroblasts, with the highest migration speeds observed on intermediate stiffness gels (~3 kPa); however, fibrosis-like stiffnesses (50 kPa) promoted high expression of COL1A1 and COL3A1 and low expression of MMP-1 relative to fibroblasts on soft gels.[19] Similarly, PAA hydrogels have been used to show that hepatic stellate cells and portal fibroblasts require a stiff environment (~12 kPa) for TGF-ß1-mediated myofibroblast activation.[78,79] Recent work has shown that isolated MSCs cultured on stiff PDMS (100 kPa) form a mechanical memory of this fibrosis-like environment and that this memory persists when MSCs are implanted into dermal wounds where they promote fibrosis.[72] Importantly, implanted MSCs cultured directly on soft PDMS (5 kPa) do not promote dermal wound fibrosis.
6.2. Dynamic mechanical properties
Fibrosis takes months to years to develop in humans, while most in vitro fibrosis studies take place over days. Additionally, in vitro studies place cells into environments that mimic the final properties of diseased tissue such that cells fail to experience the dynamic changes that occur within tissues. To recapitulate the temporal aspects of fibrosis, biomaterials capable of dynamic changes in mechanical properties have been developed.[67] Cardiac fibroblasts cultured on soft HA hydrogels (8 kPa) that were stiffened (to 50 kPa) underwent myofibroblast activation in response to stiffening (Figure 3).[77] Cardiac fibroblasts also displayed increased spreading on dynamically-stiffened PDMS.[60] Similarly, dynamically-stiffened HA hydrogels activated hepatic stellate cells, and nuclear translocation of YAP (an indicator of mechanotransduction in a stiff environment) occurred more rapidly when cells were precultured on soft hydrogels for multiple days rather than seeding directly on stiff hydrogels.[80] This work suggests that myofibroblast activation in vivo might occur rapidly, and that in vitro activation (~7 days) does not accurately recapitulate the timing of phenotypic changes (perhaps reflecting the need for a recovery period after cell isolation). Supporting this, α-SMA expression increases in the injured liver after just 3 days.[7]
Figure 3. Dynamic hydrogels mimic changes in mechanical properties observed in fibrosis.
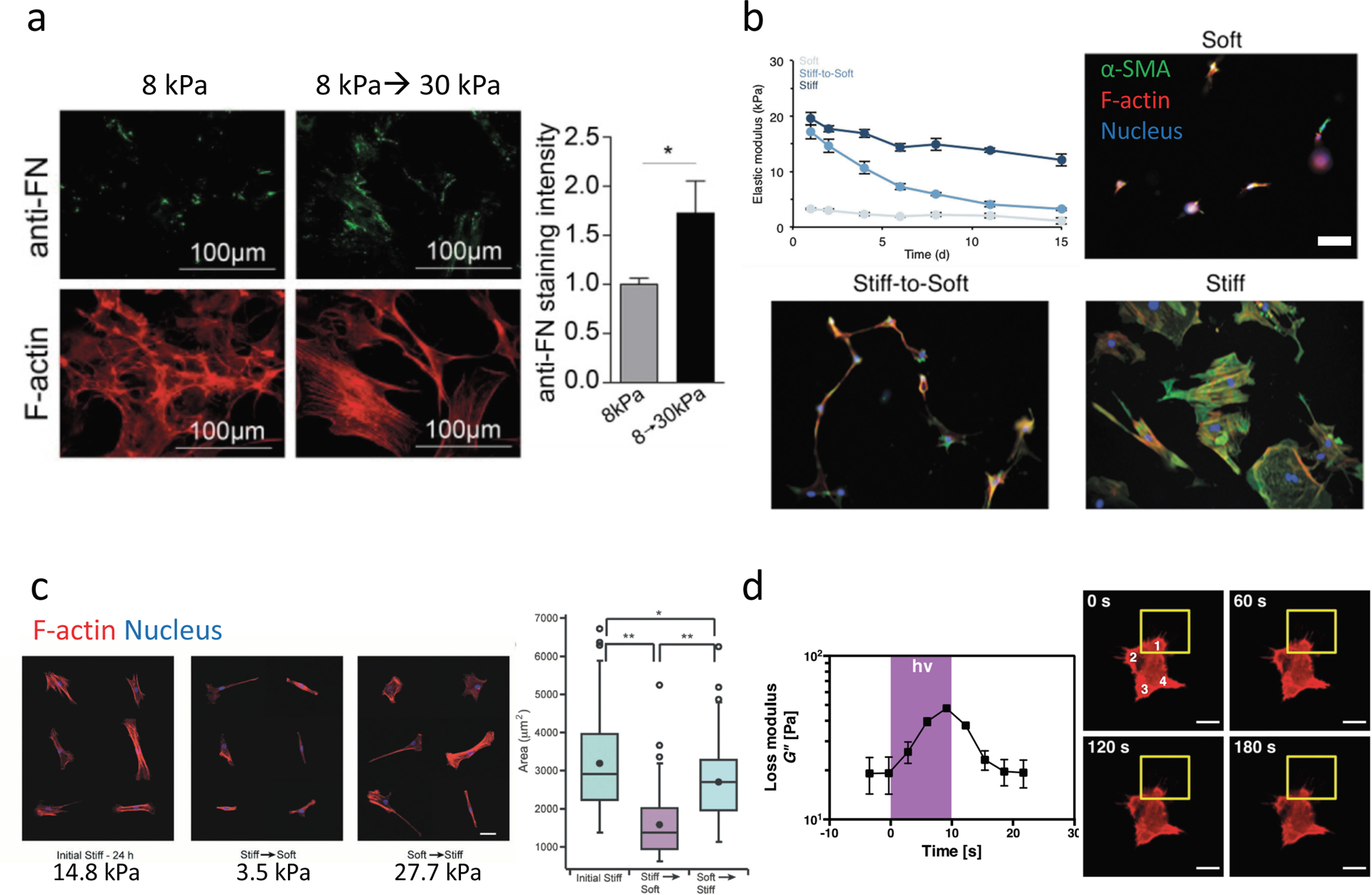
(a) Stiffening methacrylated-HA hydrogels can be used to mimic the changes in stiffness that occur during cardiac fibrosis though through photoinitiated (UV) crosslinking of methacrylate groups to stiffen gels from 8 kPa to 30 kPa in the presence of cells. Cardiac fibroblasts seeded on 8 kPa hydrogels and either stiffened to 30 kPa or maintained at 8 kPa showed increased fibrosis markers with increased fibronectin deposition (top and quantified on right) and large f-actin stress fibers (bottom). (b) Degradable HA hydrogels that gradually soften over time due to crosslinker hydrolysis can be used to mimic regression of liver fibrosis. Activated hepatic stellate cells (myofibroblasts) seeded on softening hydrogels (stiff-to-soft) develop an intermediate phenotype (with regards to α-SMA expression and cell spreading) between quiescent stellate cells continuously cultured on soft hydrogels (soft), and activated stellate cells continuously cultured on stiff hydrogels (stiff). Scale bar 100 μm. (c) Dynamic softening and subsequent stiffening hydrogels can be designed to mimic repeated changes in ECM mechanics using HA hydrogels synthesized with photodegradable o-nitrobenzyl and methacrylate crosslinking groups that undergo softening (14.8 kPa→3.5 kPa) through photodegradation of the o-nitrobenzyl crosslinker, and subsequent stiffening (3.5 kPa--> 27.7 kPa) through photoinitiated methacrylate crosslinking. MSCs seeded on hydrogels and then softened showed reduced spread area, while subsequent stiffening increased spread area, as assessed by staining cells for f-actin after each change in stiffness. (d) Photoinitiator (Irgacure 2959)-modified PEG hydrogels polymerized with crosslinkers that participate in additional fragmentation chain transfer reactions (e.g., allyl sulfide bis(azide)) undergo acute changes in loss modulus (G”) upon exposure to light through crosslinker rearrangement, which subsides after cessation of light (left). Live imaging of MSC f-actin shows responses to local changes in viscoelasticity (yellow square) over time by retracting cell projections and locally reducing cell area. Scale bar 20μm. *,**, represent p≤0.05, p≤0.01. Part (a) is adapted with permission from ref.[70] CC-BY-3.0 (https://creativecommons.org/licenses/by-nc-sa/3.0/) 2017 ASCB®, part (b) is adapted with permission from ref. [79] 2016 Oxford University Press, part (c) is adapted with permission from ref. [63] 2017 John Wiley and Sons, part (d) is adapted with permission from ref. [93] 2019 ] CC-BY-3.0 (https://creativecommons.org/licenses/by-nc-sa/3.0/) IOP Publishing.
The matrix softening that occurs during fibrosis regression can also be modeled with dynamic hydrogels, eliminating the need to study myofibroblast reversion by trypsinizing cells and plating them on new substrates. These systems allow for studies of differences between quiescent fibroblasts and previously-activated myofibroblasts. Studies using photodegradable PEG hydrogels that undergo softening during cell culture suggest that MSCs form a mechanical memory of past stiff environments that is dependent on the time spent in the stiff environment and that stiff environments promote persistent changes in chromatin architecture.[106] Dynamically-softened PEG gels have also been used to study the regression of valvular interstitial myofibroblasts and have shown that myofibroblasts transition towards a quiescent phenotype within 2 hours of gel softening.[107,108] Thus, basic studies with these novel hydrogels suggest the possibility that cardiac fibrosis might be reversible with ECM targeting therapies.
Gradually-softening HA hydrogels with crosslinks that slowly undergo hydrolysis (20 kPa to 3 kPa over ~2 weeks) were used to study how previously-activated hepatic stellate cells respond to subsequent stiffening (Figure 3),[85] and showed that cells underwent rapid activation in response to a second exposure to a stiff environment (as has been observed in vivo). Phototunable HA-based systems have also been engineered where repeated changes in stiffness are possible.[67] MSCs in this system reduce cell spreading with initial softening (14 kPa to 3.5 kPa), while subsequent stiffening (3.5 kPa to 28 kPa) leads to a return of spreading in these cells. Dynamic systems will be useful to investigate how mechanical changes in fibrosis affect multiple cells within tissues.[86] These experiments are difficult to plan due to differential growth of cells with varied mechanics, which can alter paracrine signaling, and require many experimental controls to account for the various chemical reactions and light exposures that cells experience. Additionally, ECM deposition may interfere with mechanical dosing experiments as cells are continuously cultured on the same substrate and previously deposited ECM is not removed. Nonetheless, this kind of multi-cell dynamic experiment offers the potential to better understand fibrosis development and regression at the tissue level.
6.3. Modeling ECM heterogeneity
Spatial heterogeneity of mechanics and ECM components is found in all fibrotic tissues; however, all of the previously discussed in vitro systems model the fibrotic environment as a homogenous substrate. To understand how heterogeneity affects cell phenotype in fibrosis, multiple material platforms have been used. Spatially patterning stiffness on HA hydrogels (~24 kPa stiff areas within a soft hydrogel of ~2 kPa) showed that critical areas of stiffness (areas large enough for cells to spread) are required for primary hepatic stellate cell myofibroblast activation (Figure 4).[87] Patterned areas of stiffness on PEG hydrogels have been used to study how cardiac fibroblasts are recruited to stiff regions and how fibroblasts respond to antifibrotic therapeutics differently based on the local mechanical environment.[88] To mimic the fractal heterogeneity observed in fibrotic tissues, a hybrid system was developed whereby stiff collagen fibers were incorporated into PAA hydrogels during polymerization, yielding a hydrogel with homogenous adhesion ligand density and heterogenous stiffness and topography.[89] MSCs in this system expressed α-SMA more slowly than cells on stiff hydrogels; however, they showed less cell-to-cell heterogeneity in activation than cultures on stiff PAA gels. Microscale mechanical heterogeneity similar to that observed in AFM studies of fibrotic tissue can be modeled using photodegradable PEG hydrogels; recent work has shown that valvular interstitial cells respond differently to regular as opposed to random patterns of softness (focal adhesion sized (4μm2) patches), with the cells showing a less activated phenotype on the random patterns (Figure 4).[90]
Figure 4. Mechanical heterogeneity in fibrosis and biomaterial systems to mimic this.
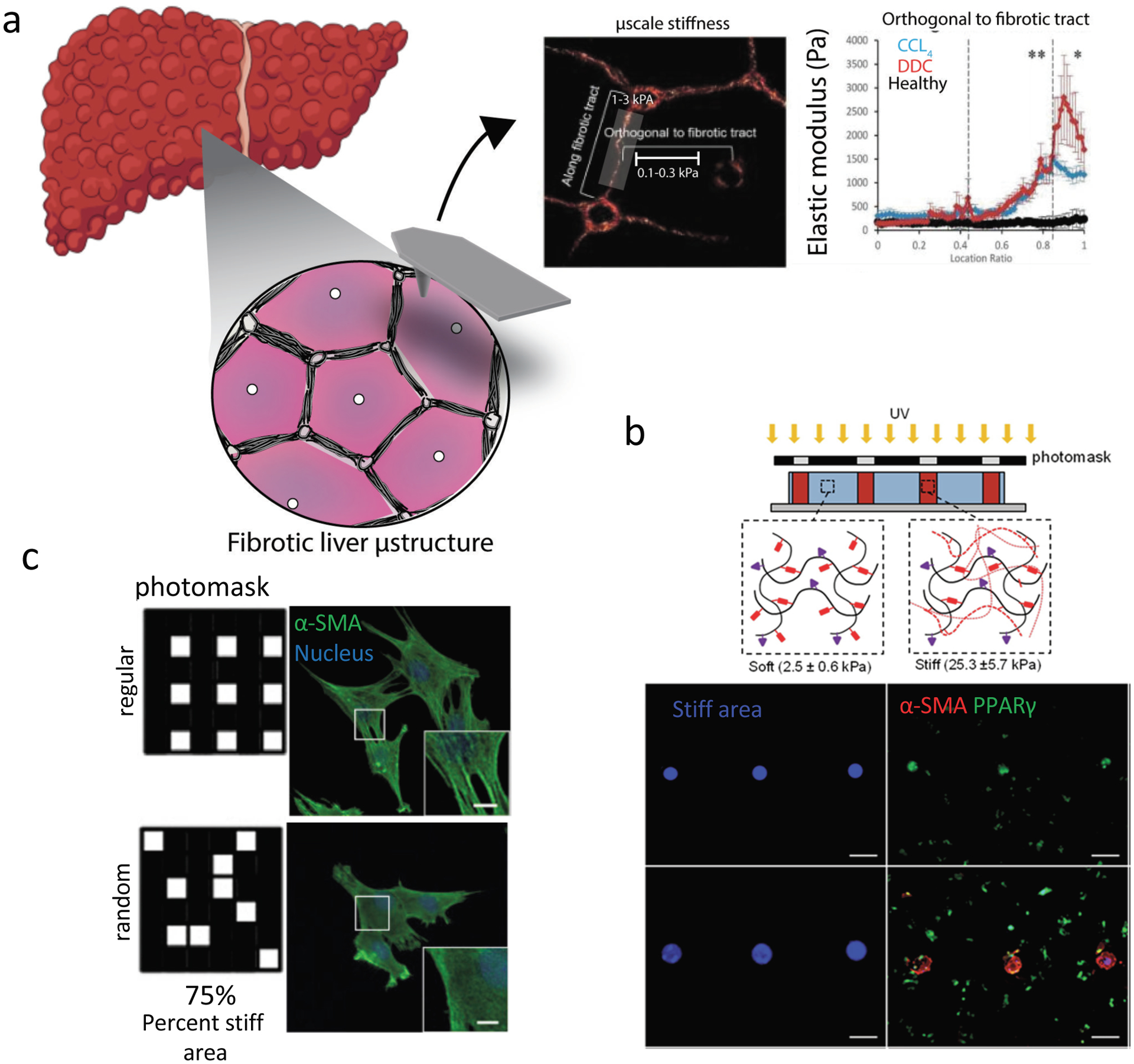
. (a) Fibrosis in the liver is characterized by fibrotic tracts which ultimately (in cirrhosis) form nodules. When characterized by atomic force microscopy (AFM), fibrotic tracts of the diseased liver have higher elastic moduli than non-fibrotic regions. (b) Patterned stiffness, similar to fibrotic regions in tissue, can be created within soft hydrogels to probe how stiff area influences myofibroblast activation. Stiff patterns can be created within methacrylated-HA hydrogels using a photomask and photoinitiated crosslinking (top, bottom left (blue)). Quiescent hepatic stellate cell activation, assessed by loss of PPARγ and increased α-SMA expression (bottom, right), is promoted by large (multicellular sized) stiff patterns, while cell-sized patterns to not lead to activation. Scale bar is 200 μm. (c) To mimic subcellular heterogeneity in ECM mechanical properties observed in tissues, high resolution photomasks can be used to create small 4 μm2 patterns of softened (photodegraded) gel within photodegradable PEG-o-nitrobenzyl hydrogels. Regular and random patterns of soft matrix can be patterned on stiff matrix, and the response of valvular interstitial cells (VICs) can be probed. VICs seeded on regular patterns have increased myofibroblast features (including α-SMA-containing stress fibers), when compared to VICs seeded on random patterns. Scale bar is 2 μm. Part (a) is adapted with permission from ref. [43] 2016 John Wiley and Sons, part (b) is adapted with permission from ref. [81] 2014 Elsevier, part (c) is adapted with permission from ref. [84] 2017 Elsevier.
Engineered systems have been developed to recapitulate the biochemical as well as the mechanical heterogeneity of the ECM. Polyelectrolyte multi-layers (PEMs) composed of proteoglycans and fibrillar collagens can be fabricated on top of PAA gels without altering substrate mechanics. Hepatic stellate cells in the same mechanical environment show different behaviors in response to the incorporation of lumican in PEMs.[91] A composite fiber-hydrogel system where fibers can be coated with fibronectin (RGD) or collagen (GFOGER) adhesion sequences was developed to permit separate control over fiber and bulk hydrogel properties.[92] Another strategy is to use decellularized matrices from healthy and fibrotic tissues, which capture many of the complex changes that occur during fibrosis.[93] However, decellularized matrices suffer from the same limitations in tunability that come with natural materials such as collagen.
6.4. The role of dimensionality on fibrotic outcomes
Most cells reside within a 3D environment in vivo; however, few studies have investigated the effects of mechanical properties in 3D. Studies with MSCs show that stiff 3D environments prevent spreading and nuclear localization of YAP, while soft 3D environments promote spreading, the opposite of the trends that are observed for cells seeded atop hydrogels. [94] Matrix degradability is also important for cell spreading in 3D, and MSCs spread and differentiate into osteoblasts more frequently in degradable hydrogels when compared to stiffness-matched non-degradable hydrogels.[66] Similarly, when valvular interstitial cells are initially cultured in 3D soft (0.24 kPa) protease-degradable PEG gels, they develop a spread myofibroblastic phenotype, while subsequent stiffening causes fibroblasts to revert to a quiescent state. [95] Thus, in contrast to what 2D studies and in vivo tissue characterization of tissues suggest, highly crosslinked and stiff 3D environments might limit rather than promote fibroblast activation in tissues. This is likely due to reduced cell spreading in restrictive 3D environments, which influences traction generation and signaling towards activation. Consistent with this, scar tissue within the cirrhotic liver is mostly acellular, which might be explained by cells creating a rigid 3D environment that is not permissive to reorganization and cell infiltration.[96] These findings emphasize the importance of modeling cell-biomaterial interactions as a way to understand cell behaviors in tissues.
Microarray studies of the transcriptional changes associated with valvular interstitial cell culture in 2D versus 3D show slightly higher levels of myofibroblast markers in 2D versus 3D degradable hydrogels engineered to have the same stiffness.[97] Future work is needed to develop culture systems that capture other aspects of the in vivo 3D environment (fibrous topography, viscoelasticity, microporosity) in order to more closely understand how dimensionality influences myofibroblast activation and persistence. It is likely that cells activate in stiff 3D environments, but that at stiffness extremes, fiber stiffness prevents cell-mediated fiber reorganization and prevents activation – suggesting that there is a biphasic stiffness-response curve in fibrous, 3D environments. Notably however, although 3D culture more closely represents the in vivo environment, it is difficult to decouple the effects of nutrient and oxygen diffusion within these systems from the mechanics.
6.5. Influence of time-dependent mechanical properties on cell behavior
Tissues and the ECM display complex mechanical properties that go beyond stiffness, such as the non-linear properties of stress relaxation and strain stiffening, and these properties change as fibrosis progresses. Material systems with tunable viscoelasticity and stress relaxation are now under development. MSCs and hepatic stellate cells on PAA substrates with constant storage moduli (~4.5kPa) and varied loss moduli (1–130 Pa) showed dramatic increases in cell spreading with even small increases in the loss modulus, which correlates with the increased viscosity observed with fibrosis progression in the liver.[56] Subsequent studies found that stress-relaxing materials promote the spreading of MSCs in both 2D[64] and 3D,[57,65] and that faster relaxation promotes a myofibroblast-like phenotype (with higher spreading, increased nuclear YAP, and osteogenic differentiation).
Methods have been developed to limit cell adhesion to the elastic components of multi-component viscoelastic gels (elastic crosslinked PAA networks mixed with viscous linear PAA), and these studies show that higher loss moduli reduce cell spreading and hepatic stellate cell activation when cells are attached to elastic components.[98] Similarly, PEG hydrogels with phototunable viscoelasticity showed that the introduction of local areas of viscoelasticity induces rapid retraction of MSC protrusions, suggesting that subcellular-sized areas of viscoelasticity may determine cell phenotype (Figure 3).[99] The effects of tissue viscoelasticity, unlike stiffness, are not straightforward and it is likely that the specific ECM factors that contribute to tissue viscoelasticity and ECM storage modulus determine cellular responses to viscoelasticity. [100] Additionally, further characterization of viscoelastic hydrogels need to be carried out and clearly described as some materials display viscoplastic behavior,[65] while others do not have plastic behavior within the cell force-relevant regime.[98]
6.6. Strain-stiffening ECM and fibrous biomaterials
Multiple collagen-based platforms have been developed to exploit the non-linear properties of collagen. As previously discussed, cells polymerized within collagen hydrogels contract and compact the matrix. The Chen group used this property of collagen to create contracted microtissues around microfabricated posts, such that the collective traction forces exerted by cells can be quantified by measuring the bending of the calibrated micro post boundaries.[101] In this system, introduction of a gap mimicking a wound in a contracted fibroblast microtissue led to the observation that cell-secreted fibronectin acts as a provisional matrix that permits wound closure.[102] Contracted micro- tissues composed of lung myofibroblasts have been fabricated using similar methods. A fibrosis-like response was induced in this model with TGF-ß1 treatment and used to screen for efficacious anti-fibrosis therapies (Figure 5).[22]
Figure 5. Fibrous biomaterials to investigate fibrosis.
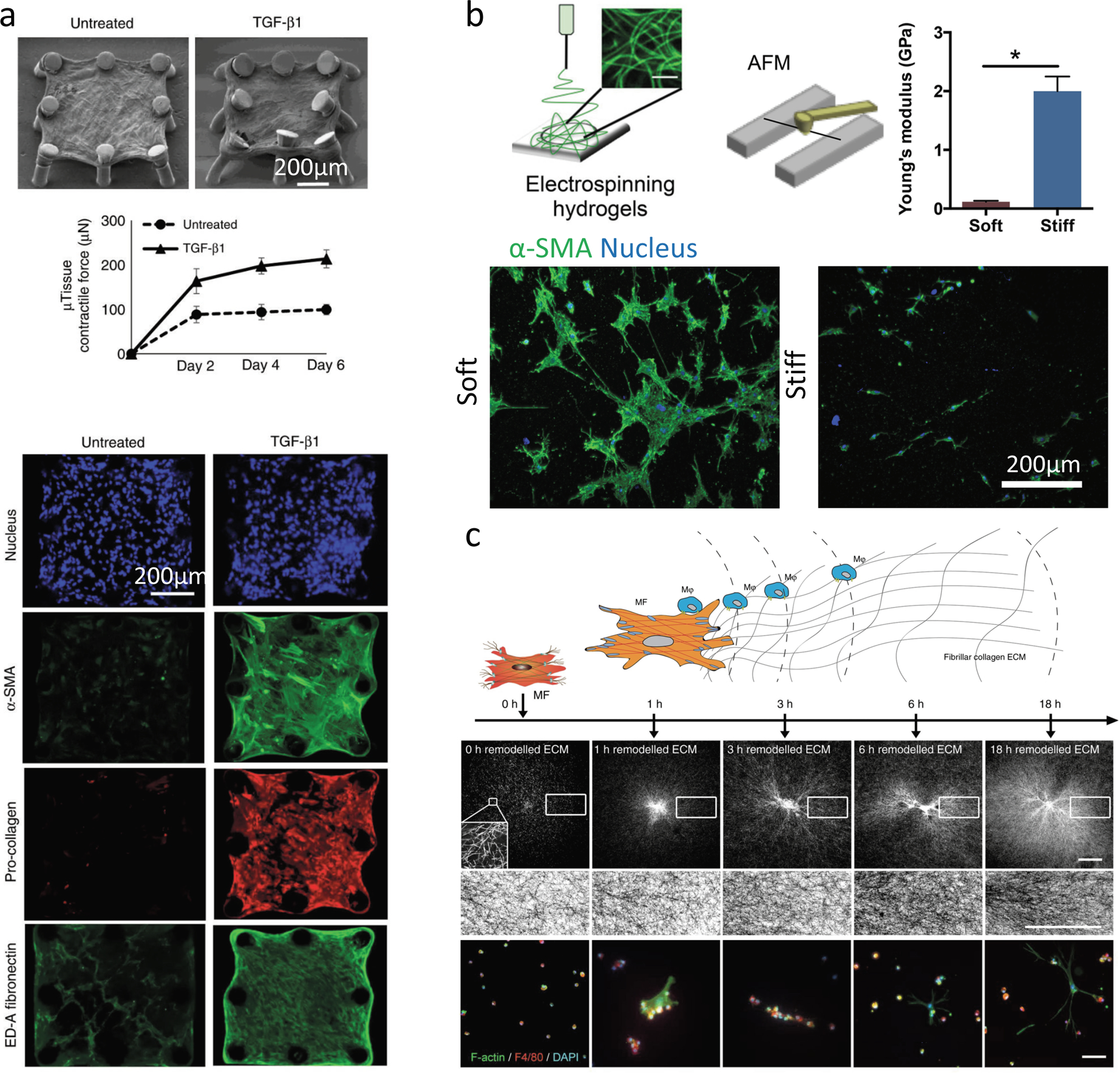
(a) Lung fibrosis models are fabricated with lung fibroblasts embedded in fibrillar type I collagen hydrogels. Lung fibroblasts contract collagen matrix around microfabricated elastomeric (PDMS) posts to create microtissues, which bend soft microposts in response to microtissue contractile forces (observed through SEM). Lung fibroblast microtissues treated with TGF-ß1 display fibrosis-relevant phenotypes with increased contraction of the matrix (higher levels of micro-post bending in SEM image, (top)), and fibrosis relevant markers (bottom) such as α-SMA labeling and collagen/ED-A fibronectin deposition. (b) To mimic fibrotic fibrous ECM, HA can be modified with different levels of methacrylate groups to give soft (low modification) and stiff (high modification) electrospun fibrous hydrogel networks (top). Fibrous networks are created atop microfabricated wells to isolate cell-fiber interactions. Quiescent hepatic stellate cells undergo myofibroblast activation and form large α-SMA positive multicellular clusters over 7 days on soft fibers (bottom, left), while stiff fibers prevent myofibroblast activation and highly spread cluster formation (bottom, right). (c) To investigate heterotypic cell-cell interactions in fibrous environments, macrophages and fibroblasts can be co-seeded on top of type I collagen gels and their interactions tracked over time with f-actin (myofibroblast) and F4–80 (macrophages) (bottom) labeling, while observing collagen remodeling (center panels). Macrophages are seeded at various times after myofibroblast seeding, and are attracted to myofibroblasts during initial ECM contraction due to dynamic tugging of the ECM which activates macrophage integrin signaling and directs migration (top). Myofibroblast-induced ECM alignment does not appear to direct macrophage migration. Scale bar 100 μm. Part (a) is adapted with permission from ref.[97] CC-BY-4.0 (https://creativecommons.org/licenses/by/4.0/) 2018 Springer Nature, Part (b) is adapted with permission from ref.[99] 2019 American Chemical Society, Part (c) is adapted with permission from ref.[102] CC-BY-4.0 (https://creativecommons.org/licenses/by/4.0/) 2019 Springer Nature.
Although these different platforms provide relevant methods to study fibrosis, multiple studies point to collagen crosslinking as an important factor in fibrosis initiation, and this is difficult to tune in reconstituted collagen. To overcome this limitation, synthetic fibrous hydrogels have been developed where single fiber mechanics can be tuned separately from adhesion ligand density, fiber network porosity, and fiber diameter. Initial studies with MSCs showed unexpected trends with cell proliferation, spreading, and focal adhesions being higher in soft fibers rather than stiff.[103] Similar trends were observed with hepatic stellate cells, which underwent or maintained myofibroblast activation on soft fibrous hydrogels while stiff fibrous hydrogels prevented myofibroblast activation.[104] To understand the impact of dimensionality in fibrous environments, composite fiber-hydrogel systems have shown that increased fiber density promotes fibroblast spreading and myofibroblast phenotype in 3D (Figure 6).[92]
Figure 6. Emerging technologies to study fibrosis.
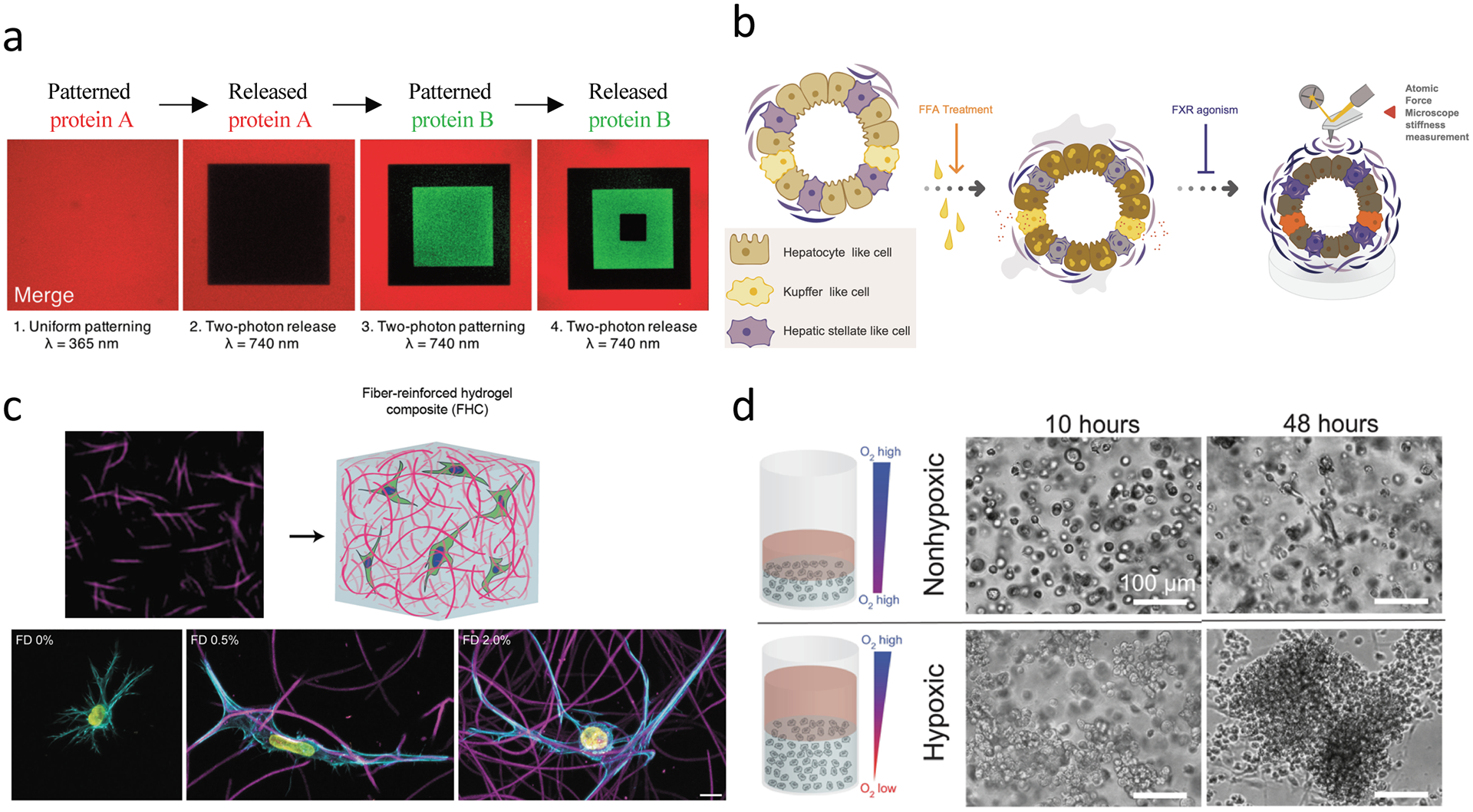
(a) Sequential tethering and release of signaling factors (e.g., ECM ligands or fibrogenic factors such as TGF-ß1) can be achieved using allyl sulfide-modified PEG hydrogels, and could be used to understand spatial heterogeneity in ECM factors/properties and their contribution to fibrosis development. (b) Multicellular organoids have been developed from iPSCs, which enable stromal cell-epithelial cell interactions using personalized cell cultures. These methods could be expanded to use in biomaterial systems where the influence of ECM properties on fibrosis outcomes could be studied. (c) Heterogeneity in ECM topography and mechanical properties can be recapitulated with composite hydrogel systems composed of Dextran-vinyl sulfone (VS) electrospun fibers embedded in tunable bulk photocrosslinked hydrogels (gelatin-methacrylate hydrogel shown here). Fibroblasts display increased spreading and protrusions in 3D in response to local fiber density. Scale bar is 10 μm. (d) Hypoxia-inducible hydrogels can be fabricated with ferulic acid-modified polymers, where oxygen is consumed in a laccase-mediated reaction that crosslinks ferulic acid groups and simultaneously creates a hypoxic environment. Hypoxia-inducible gels promote multi-cellular vascular network formation with endothelial-colony-forming cells (ECFCs, bottom), while non-hypoxic gels show low levels of single cell network formation (top). Scale bar is 100 μm. Hypoxia-inducible gels have tunable mechanical properties, and thus could be explored to probe the impact of hypoxia and mechanics on fibrosis outcomes. Part (a) is adapted with permission from ref.[117] https://pubs.acs.org/doi/full/10.1021/acscentsci.8b00325 2019 American Chemical Society, part (b) is adapted with permission from ref.[121] 2019 Elsevier, part (c) is adapted with permission from ref.[86] 2019 American Chemical Society, part (d) is adapted with permission from ref.[131] CC BY-NC 4.0 https://creativecommons.org/licenses/by-nc/4.0/legalcode 2019 AAAS.
Fibrous networks enable mechanical coupling and cell-cell interactions over long distances.[5,6] This is emerging as an important factor in fibrosis development, where high levels of cell-cell interactions are colocalized with active areas of ECM deposition (e.g. fibrotic foci) and disruption of normal tissue architecture. Initial work showed that myofibroblast interactions on collagen matrices occur through N- and OB-cadherin junctions, the latter of which reinforce cell contraction and α-SMA recruitment to stress fibers.[105] Recent work has demonstrated that the mechanical properties of electrospun fibrous networks regulate cell-cell interactions through the matrix, with softer fibers enabling the formation of vascular-like structures by endothelial cells and fibrotic foci-like clusters by hepatic stellate cells (Figure 5).[104,106]
Heterotypic interactions between myofibroblasts and surrounding cells also appear to be important for fibrosis outcomes. Myofibroblast contraction rapidly attracts surrounding macrophages, which can further promote myofibroblast persistence through cadherin-11 signaling (Figure 5).[107,108] Additionally, endothelial cell contraction of fibrillar ECM might promote myofibroblast activation of hepatic stellate cells through tugging forces that activate mechanosensitive pro-fibrotic pathways.[109] Since long-range force transmission in fibrous matrices gives rise to aligned patterns of fibers reminiscent of histological patterns of fibrotic ECM, it is likely that cell contraction of fibrous networks are responsible for ECM patterns such as bridging fibrosis.[110,111]
6.7. The response of non-fibroblast cells to mechanical changes
Although myofibroblasts are the major contributors to fibrosis and the most studied cells in regards to their response to mechanical stimuli, it is clear that other cells in tissues suffer functional consequences with changes in stiffness. Cardiomyocyte beating is optimal on compliant substrates that mimic the stiffness of the heart and facilitate coherent beating, suggesting that the beating of cardiomyocytes on the border zone of cardiac scars is impeded.[38,112] Additionally the propagation of electrical signals through cardiac scar tissue differs from that through healthy myocardium, which contributes to arrythmias and increased contraction of myofibroblasts within scars, which in turn can slow electrical conduction to border zone cardiomyocytes.[113]
Hepatocytes, the major epithelial cells of the liver and cells whose metabolic and detoxification functions are critical to the survival of the organism, are highly sensitive to the stiffness of their underlying substrate. Hepatocyte differentiation and function, assayed via HNF4-α expression and albumin production, were the highest on soft PAA substrates (0.14 kPa) and lost on substrates with the stiffness of the fibrotic liver (1–60 kPa).[47] Thus, hepatocytes adjacent to fibrotic septae or encased in fibrotic ECM likely dedifferentiate, contributing to liver failure in fibrosis. Small changes in stiffness can have large effects and it appears that epithelial and mesenchymal cells have different thresholds for responding to changes in stiffness, even within the same organ. The high sensitivity of epithelial cells might permit initial recognition of small changes in stiffness and lead to paracrine signaling to effector cells, such as fibroblasts, that modulate further mechanical changes in the ECM.
Lung epithelial AT2 cells cultured on stiff PAA substrates undergo a transition to myofibroblast-like cells, with increased fibrillar fibronectin and laminin deposition, suggesting that epithelial cells might initiate changes in lung stiffness.[114,115] Keratinocytes cultured on stiff PDMS substrates, when compared to those on compliant substrates, undergo significant proliferation, which might explain the abnormal growth observed in keloid scars.[116] Numerous materials systems have been developed to study myofibroblasts in fibrosis, while only few studies have characterized the response of other cells; thus, this is an area ripe for exploration.
7. Remaining questions and emerging materials to study fibrosis
Many questions remain regarding the nature of ECM changes during fibrosis and the consequence of these changes on cells within injured and fibrotic tissues. Biomaterials are indispensable for defining the effects of ECM properties on cellular outcomes; however, as with any models, biomaterials reflect only a subset of the properties of tissues that are found in vivo. To conclude this review, we will cover features of the environment in vivo that will help inform biomaterial design and the development of new materials and technologies to further expand our understanding of fibrosis development and progression. In particular, new materials may enable the identification of unifying mechanisms of fibrosis across organs which could be used to identify new targets for therapies.
A clear theme that has emerged is that as fibrosis progresses, changes in tissue mechanics are highly dynamic. Along with stiffness, other mechanical properties such as viscoelasticity and elasticity change at the tissue and subcellular level. Thus, it is critical to characterize the multiple mechanical properties of tissues at high spatiotemporal resolution in order to better inform materials development and in vitro systems. Label-free imaging techniques such as 2nd and 3rd harmonic generation imaging integrated with mechanical imaging techniques such as Brillouin microscopy should aid in these characterizations.[117] Defining the spatial organization of ECM heterogeneity and interactions between different ECM molecules and cells will similarly contribute to the development of new biomaterials. Additionally, the micro- to nanoscale changes that occur in the ECM during fibrosis progression are often overlooked. Moving forward, it will be important to characterize the spatiotemporal porosity and alignment of the ECM along with assessing the posttranslational modifications (most notably crosslinking), morphology, and distribution of specific proteins. Lastly, an improved understanding of ECM-ECM (e.g., GAG-collagen) interactions in health and disease will further our understanding of the ways ECM-protein interactions influence mechanical properties and how to properly model them in vitro.[118]
At the macroscale, large topographical changes occur during fibrosis. In the skin, hypertrophic scars remain within the confines of the regenerated tissue; however, keloid scars grow beyond the initial scar and require surgically removal.[30] Within the liver, there is the development of macroscale nodules of fibrotic ECM and epithelial cells. It is not clear what causes this characteristic pattern and how the pattern itself and the size of the nodules are influenced by ECM mechanics. During liver and lung fibrosis, abnormal ECM deposition begins in focal regions, but eventually bridges between foci. At the macroscopic scale, this ECM deposition occurs in fractal patterns, yet we still do not understand what dictates these patterns or the time-line of ECM deposition.[89] Models of the ECM and in vitro studies suggest that scars might interact over long distances through mechanical coupling, and that matrix properties are important for coupling.[111,119,120] Supporting this, recent studies with synthetic fibrous materials suggest that cells are mechanically coupled through the matrix and that soft, rather than stiff, matrix permits physical remodeling and long range cell-cell signaling to promote cell accumulation and formation of fibrotic foci.[104] Thus, long range cell-cell communication needs to be studied in vivo during the onset and progression of fibrosis to better inform in vitro models.
As we gain a deeper understanding of the spatiotemporal dynamics of fibrosis mechanics, matrix heterogeneity, and abnormal tissue architecture, we can start developing new materials to understand how these properties affect cell phenotype. It is still unclear exactly which changes initiate myofibroblast activation. Areas of active fibrogenesis are non-linearly elastic, at least in the lung, and recently developed stress-stiffening hydrogels could be employed to understand how changes in stress-stiffening properties of the ECM (for example, the critical strains where stiffening begins) influence fibrotic phenotypes.[121] MSCs within these materials display myofibroblast phenotypes (spreading, osteogenic differentiation) when the materials stiffen at a higher critical stress (19.3 Pa versus 9.4 Pa) with matched storage moduli (~250 Pa). Thus, it is likely that soft strain-stiffening ECM promotes myofibroblast activation, while soft elastic materials do not.
Since lung and liver tissues stiffen prior to ECM deposition, it would be informative to have tunable materials that mimic LOX-mediated crosslinking of collagen fibers, such that fiber diameter decreases and stiffness increases.[8] Methods to produce fibrous hydrogels on the scale of collagen fibers that could also be modified to undergo dynamic changes from wide extensible fibers to narrow rigid fibers using photochemistry would also be useful to develop.
Local soluble signals, either matrix-derived or paracrine, are important for initiating and prolonging fibrosis. Multiple biomaterial systems have been developed that could help understand the role of signaling factor sequestration and release from the ECM in fibrosis. Recently-developed PEG hydrogels allow for the repeated tethering and release of modified (i.e. thiolated) proteins with photochemical reactions (Figure 6).[122] Using this system, TGF-ß1 could be patterned and released on cue in the presence of embryonic fibroblasts, which respond to dynamic changes in ligand presentation with altered TGF-ß1 signaling. Using this system, other proteins could be tethered and released in different locations over time to introduce spatiotemporal ligand heterogeneity.
Similarly, engineered proteins have recently been developed that can be tethered to hydrogels and released with spatiotemporal control through photochemical reactions.[123] These materials could contribute to understanding how cell organization and paracrine signaling drive patterns of ECM deposition and migration in fibrosis. DNA-based materials (aptamers) have also been engineered that mimic the Large Latent Complex protein that sequesters TGFß for release under cell generated tractions.[124] These aptamers with bound growth factors (PDGF or VEGF) can be linked to hydrogels and activated with primary human dermal fibroblast-mediated traction forces to cause growth factor release. Integrating these technologies into existing hydrogel systems should provide further understanding of how sequestered factors within the matrix modulate fibrosis initiation and progression.
Another area for future biomaterial and fibrosis research will be to elucidate how cell-cell interactions are modulated by ECM mechanics. Since non-fibrotic cells appear to contribute to myofibroblast activation through mechanical coupling between cells,[107,109] it will be important to understand how mechanical changes to the matrix modulate cell-cell interactions. Stiff matrix limits the mechanical coupling of cells, and thus cell-cell interactions through the matrix are likely to be more important in the initiation of fibrosis when the matrix is still extensible. Emerging 3D composite fibrous material platforms could help to elucidate the importance of these cell-cell interactions (Figure 6). Recent advances in melt electrowriting (MEW),[125] 3D printing,[126] and the combination of these techniques makes it possible to precisely control fiber deposition within 3D hydrogels, which could offer new avenues to study cell-cell interactions through synthetic ECMs.
The electrical impedance of tissues can be measured non-invasively to assess tissue scarring. For instance, scarred myocardial tissue has lower impedance than healthy myocardium.[127] Importantly, impedance varies between tissues and can be used to identify different layers of tissues (e.g., muscle, adipose) or to distinguish between malignant and benign tumors.[128] To our knowledge, biomaterials with tunable electrical impedance have not been used in fibrosis or mechanobiology studies.[129] This is a future area for materials development that could have important implications in understanding the role of bioelectricity in fibrosis and wound healing.
Organoid and spheroid cultures are also emerging as important tools for drug development since they can be fabricated from patient-specific tissue samples, are composed of multiple cell types, and retain physiologically-relevant cell-cell interactions and function (Figure 6).[130,131] Integrating these cultures into dynamic materials will bridge the gap between knowledge of single cell responses to changes in ECM properties to tissue level responses. Specifically, it will be interesting to probe how local mechanical changes (such as softening and stiffening, ECM density, and porosity) influence fibrosis initiation within organoids composed of epithelial and mesenchymal cells. Organoid cultures could also shed light on how fibrosis of organ membranes, such as capsular fibrosis in the liver, affects parenchymal cell function and intra-organ fibrosis.[132]
Vascular signals and hypoxia are also important in fibrosis, and biomaterials can help elucidate how these factors influence fibrosis and synergize with mechanical properties. Angiocrine signals implicated in fibrosis[109,133] could be introduced into fibrosis models by creating vasculature in tunable biomaterial systems. Microscale molding and 3D printing technologies (e.g. inks and support materials) provide controlled methods for producing in vivo-like vasculature within tunable biomaterials and have been integrated into microfluidic chips where flow can be introduced.[134–136] Normal tissue oxygen tension is much lower (1–5% O2)[137] than that used in most fibrosis studies and excessive ECM deposition is known to increase local hypoxia within tissues. Importantly, hypoxia differentially regulates fibroblast activation depending on organs; [138][139] thus, available oxygen needs be tightly regulated in fibrosis studies. Microfluidic devices and tunable hypoxia-inducing materials could be used to create oxygen gradients (Figure 6) in order to understand how oxygen availability couples with mechanical properties, such as stiffness and viscoelasticity, to promote fibrotic phenotypes.[139–141]
8. Conclusions
Over the past decades, our understanding of the way cells respond to the properties of their microenvironment has grown substantially. This can be attributed to increased interest in the mechanobiology of disease, the accessibility of new tools for studying tissue, cell, and ECM mechanics, and the integration of new properties into biomaterial platforms. As the field continues to grow, collaborative efforts between biologists, clinicians, materials scientists and computational biophysicists should enable significant breakthroughs in our understanding of the ECM and its role in fibrosis. Emphasis should be placed on the clinical relevance of the questions at hand and on how discoveries will bring us closer to new therapies. Multiple therapies that target ECM stiffness are currently in clinical trials;[1] however, pirfenidone is one of the only currently approved drugs for IPF and it only prolongs the inevitable need for lung transplantation. The small number of efficacious fibrosis therapies is likely due to the lack of appropriate pre-clinical models of human disease – biomaterial platforms should help us to identify and understand targets for future therapies.
It is clear that relevance of biomaterial platforms designed to probe fibrosis is increasing, with the inclusion of more complex mechanical properties, more relevant structures, and increased heterogeneity. New organ-on-a-chip and microfluidic platforms, made possible by advances in techniques such as microfabrication and 3D-printing, are emerging to add further complexity to models of fibrosis. A potential limitation to current biomaterial platforms is the complexity and high level of expertise needed to fabricate such systems. As biomaterial systems progress, efforts should be made to increase the accessibility of these systems to their intended users and to explore the development of simple alternative methods of fabrication and synthesis. Fortunately, some tunable systems are now offered by commercial vendors,[49] which should help make the study of mechanobiological questions more routine.
Acknowledgements:
The authors would like to acknowledge Dr. Jessica Llewellyn, Dr. Claudia Loebel, and Dr. Katrina Wisdom for helpful discussions. This study was supported by the National Institutes of Health (F32 DK117568, R01 AR056624, R01 EB017753) and the National Science Foundation MRSEC (DMR-1720530) and Center for Engineering MechanoBiology (CMMI: 15-48571).
References:
- [1].Lampi MC, Reinhart-King CA, Sci. Transl. Med 2018, 10, 475. [DOI] [PubMed] [Google Scholar]
- [2].Yang L, Van Der Werf KO, Koopman BFJM, Subramaniam V, Bennink ML, Dijkstra PJ, Feijen J, J. Biomed. Mater. Res. - Part A 2007, 82, 160. [DOI] [PubMed] [Google Scholar]
- [3].Ban E, Franklin JM, Nam S, Smith LR, Wang H, Wells RG, Chaudhuri O, Liphardt JT, Shenoy VB, Biophys. J 2018, 114, 450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, Brown RA, Nat. Rev. Mol. Cell Biol 2002, 3, 349. [DOI] [PubMed] [Google Scholar]
- [5].Wang H, Abhilash AS, Chen CS, Wells RG, Shenoy VB, Biophys. J 2014, 107, 2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Abhilash AS, Baker BM, Trappmann B, Chen CS, Shenoy VB, Biophys. J 2014, 107, 1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Georges PC, Hui JJ, Gombos Z, McCormick ME, Wang AY, Uemura M, Mick R, Janmey PA, Furth EE, Wells RG, Am. J. Physiol. - Gastrointest. Liver Physiol 2007, 293, G1147. [DOI] [PubMed] [Google Scholar]
- [8].Jones MG, Andriotis OG, Roberts JJ, Lunn K, Tear VJ, Cao L, Ask K, Smart DE, Bonfanti A, Johnson P, et al. , Elife 2018, 7, DOI 10.7554/eLife.36354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Van Oosten ASG, Vahabi M, Licup AJ, Sharma A, Galie PA, MacKintosh FC, Janmey PA, Sci. Rep 2016, 6, 19270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].van Oosten ASGG, Chen X, Chin LK, Cruz K, Patteson AE, Pogoda K, Shenoy VB, Janmey PA, Nature 2019, 573, 96. [DOI] [PubMed] [Google Scholar]
- [11].Kurniawan NA, Vos BE, Biebricher A, Wuite GJL, Peterman EJG, Koenderink GH, Biophys. J 2016, 111, 1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Vos BE, Liebrand LC, Vahabi M, Biebricher A, Wuite GJL, Peterman EJG, Kurniawan NA, MacKintosh FC, Koenderink GH, Soft Matter 2017, 13, 8886. [DOI] [PubMed] [Google Scholar]
- [13].Kubow KE, Vukmirovic R, Zhe L, Klotzsch E, Smith ML, Gourdon D, Luna S, Vogel V, Nat. Commun 2015, 6, DOI 10.1038/ncomms9026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Klotzsch E, Smith ML, Kubow KE, Muntwyler S, Little WC, Beyeler F, Gourdon D, Nelson BJ, Vogel V, Proc. Natl. Acad. Sci. U. S. A 2009, 106, 18267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Wells RG, Biochim. Biophys. Acta - Mol. Basis Dis 2013, 1832, 884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Discher DE, Janmey P, Wang Y-L, Science (80-.) 2005, 310, 1139. [DOI] [PubMed] [Google Scholar]
- [17].Perepelyuk M, Chin L, Cao X, van Oosten A, Shenoy VB, Janmey PA, Wells RG, PLoS One 2016, 11, e0146588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Sen S, Kumar S, J. Biomech 2010, 43, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Liu F, Mih JD, Shea BS, Kho AT, Sharif AS, Tager AM, Tschumperlin DJ, J. Cell Biol 2010, 190, 693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Li Q, Qu F, Han B, Wang C, Li H, Mauck RL, Han L, Acta Biomater. 2017, 54, 356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Legant WR, Pathak A, Yang MT, Deshpande VS, McMeeking RM, Chen CS, Proc. Natl. Acad. Sci. U. S. A 2009, 106, 10097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Asmani M, Velumani S, Li Y, Wawrzyniak N, Hsia I, Chen Z, Hinz B, Zhao R, Nat. Commun 2018, 9, 2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Zhao R, Chen CS, Reich DH, Biomaterials 2014, 35, 5056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Fiore VF, Wong SS, Tran C, Tan C, Xu W, Sulchek T, White ES, Hagood JS, Barker TH, JCI Insight 2018, 3, DOI 10.1172/JCI.INSIGHT.97597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Nam S, Lee J, Brownfield DGG, Chaudhuri O, Biophys. J 2016, 111, 2296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Mohammadi H, Sahai E, Nat. Cell Biol 2018, 20, 766. [DOI] [PubMed] [Google Scholar]
- [27].Watt FM, Fujiwara H, Cold Spring Harb. Perspect. Biol 2011, 3, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Eming SA, Martin P, Tomic-Canic M, Sci. Transl. Med 2014, 6, 265sr6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Andrews JP, Marttala J, Macarak E, Rosenbloom J, Uitto J, Matrix Biol. 2016, 51, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Trace AP, Enos CW, Mantel A, Harvey VM, Am. J. Clin. Dermatol 2016, 17, 201. [DOI] [PubMed] [Google Scholar]
- [31].Uitto J, Jimenez S, Arch. Dermatol 1990, 126, 661. [DOI] [PubMed] [Google Scholar]
- [32].Xue M, Jackson CJ, Adv. Wound Care 2015, 4, 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Profyris C, Tziotzios C, Do Vale I, J. Am. Acad. Dermatol 2012, 66, 1. [DOI] [PubMed] [Google Scholar]
- [34].Achterberg VF, Buscemi L, Diekmann H, Smith-Clerc J, Schwengler H, Meister JJ, Wenck H, Gallinat S, Hinz B, J. Invest. Dermatol 2014, 134, 1862. [DOI] [PubMed] [Google Scholar]
- [35].Hsu CK, Lin HH, Harn HIC, Hughes MW, Tang MJ, Yang CC, J. Dermatol. Sci 2018, 90, 232. [DOI] [PubMed] [Google Scholar]
- [36].Klingberg F, Hinz B, White ES, J. Pathol 2013, 229, 298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Herum K, Lunde I, McCulloch A, Christensen G, J. Clin. Med 2017, 6, 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Engler AJ, Carag-Krieger C, Johnson CP, Raab M, Tang HY, Speicher DW, Sanger JW, Sanger JM, Discher DE, J. Cell Sci 2008, 121, 3794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Andreu I, Luque T, Sancho A, Pelacho B, Iglesias-García O, Melo E, Farré R, Prósper F, Elizalde MR, Navajas D, Acta Biomater. 2014, 10, 3235. [DOI] [PubMed] [Google Scholar]
- [40].Frangogiannis NG, J. Am. Coll. Cardiol 2019, 73, 2283. [DOI] [PubMed] [Google Scholar]
- [41].González A, Schelbert EB, Díez J, Butler J, J. Am. Coll. Cardiol 2018, 71, 1696. [DOI] [PubMed] [Google Scholar]
- [42].Noble PW, Barkauskas CE, Jiang D, J. Clin. Invest 2012, 122, 2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Sundarakrishnan A, Chen Y, Black LD, Aldridge BB, Kaplan DL, Adv. Drug Deliv. Rev 2018, 129, 78. [DOI] [PubMed] [Google Scholar]
- [44].Herridge MS, Crit. Care Clin 2011, 27, 685. [DOI] [PubMed] [Google Scholar]
- [45].Sgalla G, Iovene B, Calvello M, Ori M, Varone F, Richeldi L, Respir. Res 2018, 19, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Zhao G, Cui J, Qin Q, Zhang J, Liu L, Deng S, Wu C, Yang M, Li S, Wang C, J. Surg. Oncol 2010, 102, 482. [DOI] [PubMed] [Google Scholar]
- [47].Desai SS, Tung JC, Zhou VX, Grenert JP, Malato Y, Rezvani M, Español-Suñer R, Willenbring H, Weaver VM, Chang TT, Hepatology 2016, 64, 261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Lee Y, Friedman SL, in Prog. Mol. Biol. Transl. Sci, Academic Press, 2010, pp. 151–200. [DOI] [PubMed] [Google Scholar]
- [49].Caliari SR, Burdick JA, Nat. Publ. Gr 2016, 13, 405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Loebel C, Mauck RL, Burdick JA, Nat. Mater 2019, 18, 883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Dicker KT, Gurski LA, Pradhan-Bhatt S, Witt RL, Farach-Carson MC, Jia X, Acta Biomater. 2014, 10, 1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Doyle AD, Carvajal N, Jin A, Matsumoto K, Yamada KM, Nat. Commun 2015, 6, 8720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Nam S, Hu KH, Butte MJ, Chaudhuri O, Proc. Natl. Acad. Sci. U. S. A 2016, 113, 5492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Brandley BK, Schnaar RL, Anal. Biochem 1988, 172, 270. [DOI] [PubMed] [Google Scholar]
- [55].Tse JR, Engler AJ, Curr. Protoc. Cell Biol 2010, 47, 10.16.1. [DOI] [PubMed] [Google Scholar]
- [56].Cameron AR, Frith JE, Cooper-White JJ, Biomaterials 2011, 32, 5979. [DOI] [PubMed] [Google Scholar]
- [57].McKinnon DD, Domaille DW, Cha JN, Anseth KS, Adv. Mater 2014, 26, 865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Kloxin AM, Kasko AM, Salinas CN, Anseth KS, Science (80-.) 2009, 324, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].West JL, Hubbell JA, Macromolecules 1999, 32, 241. [Google Scholar]
- [60].Yeh YC, Corbin EA, Caliari SR, Ouyang L, Vega SL, Truitt R, Han L, Margulies KB, Burdick JA, Biomaterials 2017, 145, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Harris AK, Wild P, Stopak D, Science (80-.) 1980, 208, 177. [DOI] [PubMed] [Google Scholar]
- [62].Jeon O, Bouhadir KH, Mansour JM, Alsberg E, Biomaterials 2009, 30, 2724. [DOI] [PubMed] [Google Scholar]
- [63].Guvendiren M, Burdick JA, Wang N, Butler JP, Ingber DE, Vogel V, Sheetz M, Mitrossilis D, Allioux-Guerin M, Lo CM, et al. , Nat. Commun 2012, 3, 792. [DOI] [PubMed] [Google Scholar]
- [64].Chaudhuri O, Gu L, Darnell M, Klumpers D, Bencherif SA, Weaver JC, Huebsch N, Mooney DJ, Nat. Commun 2015, 6, 6365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Chaudhuri O, Gu L, Klumpers D, Darnell M, Bencherif SA, Weaver JC, Huebsch N, Lee HP, Lippens E, Duda GN, et al. , Nat. Mater 2016, 15, 326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Khetan S, Guvendiren M, Legant WR, Cohen DM, Chen CS, Burdick JA, Nat. Mater 2013, 12, 458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Rosales AM, Vega SLSL, DelRio FW, Burdick JA, Anseth KS, Angew. Chemie Int. Ed 2017, 56, 12132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Van Tomme SR, Hennink WE, Expert Rev. Med. Devices 2007, 4, 147. [DOI] [PubMed] [Google Scholar]
- [69].Berger J, Reist M, Mayer JM, Felt O, Peppas NA, Gurny R, Eur. J. Pharm. Biopharm 2004, 57, 19. [DOI] [PubMed] [Google Scholar]
- [70].Hinz B, Phan SH, Thannickal VJ, Prunotto M, Desmoulire A, Varga J, De Wever O, Mareel M, Gabbiani G, Am. J. Pathol 2012, 180, 1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Hinz B, Dugina V, Ballestrem C, Wehrle-Haller B, Chaponnier C, Mol. Biol. Cell 2003, 14, 2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Li CX, Talele NP, Boo S, Koehler A, Knee-Walden E, Balestrini JL, Speight P, Kapus A, Hinz B, Nat. Mater 2017, 16, 379. [DOI] [PubMed] [Google Scholar]
- [73].Arora PD, Narani N, McCulloch CAG, Am. J. Pathol 1999, 154, 871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Goffin JM, Pittet P, Csucs G, Lussi JW, Meister JJ, Hinz B, J. Cell Biol 2006, 172, 259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Wang J, Chen H, Seth A, Mcculloch CA, Onai Y, Suzuki J-I, Maejima Y, Haraguchi G, Muto S, Isobe M, et al. , Am J Physiol Hear. Circ Physiol Am J Physiol Hear. Circ Physiol J. Cell Sci. Am J Physiol Hear. Circ Physiol Am J Physiol Hear. Circ Physiol AJP-Heart Circ. Physiol. Febr 2003, 285, 1871. [Google Scholar]
- [76].Zimina E, Hinz B, in Card. Fibros. Hear. Fail. Cause or Eff, Springer International Publishing, Cham, 2015, pp. 71–92. [Google Scholar]
- [77].Herum KM, Choppe J, Kumar A, Engler AJ, McCulloch AD, Mol. Biol. Cell 2017, 28, 1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Olsen AL, Bloomer SA, Chan EP, Gaça MDA, Georges PC, Sackey B, Uemura M, Janmey PA, Wells RG, Am. J. Physiol. - Gastrointest. Liver Physiol 2011, 301, DOI 10.1152/ajpgi.00412.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Li Z, Dranoff JA, Chan EP, Uemura M, Sévigny J, Wells RG, Hepatology 2007, 46, 1246. [DOI] [PubMed] [Google Scholar]
- [80].Caliari SR, Perepelyuk M, Cosgrove BD, Tsai SJ, Lee GY, Mauck RL, Wells RG, Burdick JA, Sci. Rep 2016, 6, 21387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Yang C, Tibbitt MW, Basta L, Anseth KS, Nat. Mater 2014, 13, 645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Killaars AR, Grim JC, Walker CJ, Hushka EA, Brown TE, Anseth KS, in Trans. Annu. Meet. Soc. Biomater. Annu. Int. Biomater. Symp, John Wiley & Sons, Ltd, 2019, p. 670. [Google Scholar]
- [83].Wang H, Tibbitt MW, Langer SJ, Leinwand LA, Anseth KS, Proc. Natl. Acad. Sci. U. S. A 2013, 110, 19336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Wang H, Haeger SM, Kloxin AM, Leinwand LA, Anseth KS, PLoS One 2012, 7, e39969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Caliari SR, Perepelyuk M, Soulas EM, Lee GY, Wells RG, Burdick JA, Integr. Biol. (United Kingdom) 2016, 8, 720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Rosales AM, Anseth KS, Nat. Rev. Mater 2016, 1, 15012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Guvendiren M, Perepelyuk M, Wells RG, Burdick JA, J. Mech. Behav. Biomed. Mater 2014, 38, 198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Zhao H, Li X, Zhao S, Zeng Y, Zhao L, Ding H, Sun W, Du Y, Biofabrication 2014, 6, 045009. [DOI] [PubMed] [Google Scholar]
- [89].Dingal PCDP, Bradshaw AM, Cho S, Raab M, Buxboim A, Swift J, Discher DE, Nat. Mater 2015, 14, 951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Ma H, Killaars AR, DelRio FW, Yang C, Anseth KS, Biomaterials 2017, 131, 131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Saums MK, Wang W, Han B, Madhavan L, Han L, Lee D, Wells RG, Langmuir 2014, 30, 5481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Matera DL, Wang WY, Smith MR, Shikanov A, Baker BM, ACS Biomater. Sci. Eng 2019, 5, 2965. [DOI] [PubMed] [Google Scholar]
- [93].Booth AJ, Hadley R, Cornett AM, Dreffs AA, Matthes SA, Tsui JL, Weiss K, Horowitz JC, Fiore VF, Barker TH, et al. , Am. J. Respir. Crit. Care Med 2012, 186, 866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].S. R. Caliari, S. L. Vega, M. Kwon, E. M. Soulas, J. A. Burdick, Biomaterials 2016, 103, 314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Mabry KM, Lawrence RL, Anseth KS, Biomaterials 2015, 49, 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Lo RC, Kim H, Clin. Mol. Hepatol 2017, 23, 302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Mabry KM, Payne SZ, Anseth KS, Biomaterials 2016, 74, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Charrier EE, Pogoda K, Wells RG, Janmey PA, Nat. Commun 2018, 9, 449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Marozas IA, Cooper-White JJ, Anseth KS, New J. Phys 2019, 21, 045004. [Google Scholar]
- [100].Gong Z, Szczesny SE, Caliari SR, Charrier EE, Chaudhuri O, Cao X, Lin Y, Mauck RL, Janmey PA, Burdick JA, et al. , Proc. Natl. Acad. Sci. U. S. A 2018, 115, E2686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Boudou T, Legant WR, Mu A, Borochin MA, Thavandiran N, Radisic M, Zandstra PW, Epstein JA, Margulies KB, Chen CS, Tissue Eng. Part A 2012, 18, 910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Selman Sakar M, Eyckmans J, Pieters R, Eberli D, Nelson BJ, Chen CS, Nat. Commun 2016, 7, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Baker BM, Trappmann B, Wang WY, Sakar MS, Kim IL, Shenoy VB, Burdick JA, Chen CS, Nat. Mater 2015, 14, 1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Davidson MD, Song KH, Lee M-H, Llewellyn J, Du Y, Baker BM, Wells RG, Burdick JA, ACS Biomater. Sci. Eng 2019, 5, 3899. [DOI] [PubMed] [Google Scholar]
- [105].Hinz B, Pittet P, Smith-Clerc J, Chaponnier C, Meister J-JJ-J, Mol. Biol. Cell 2004, 15, 4310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Davidson CD, Wang WY, Zaimi I, Jayco DKP, Baker BM, Sci. Rep 2019, 9, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Pakshir P, Alizadehgiashi M, Wong B, Coelho NM, Chen X, Gong Z, Shenoy VB, McCulloch C, Hinz B, Nat. Commun 2019, 10, 1850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Lodyga M, Cambridge E, Karvonen HM, Pakshir P, Wu B, Boo S, Kiebalo M, Kaarteenaho R, Glogauer M, Kapoor M, et al. , Sci. Signal 2019, 12, eaao3469. [DOI] [PubMed] [Google Scholar]
- [109].Liu L, You Z, Yu H, Zhou L, Zhao H, Yan X, Li D, Wang B, Zhu L, Xu Y, et al. , Nat. Mater 2017, 16, 1252. [DOI] [PubMed] [Google Scholar]
- [110].Sanders EJ, Prasad S, J. Exp. Zool 1983, 226, 81. [DOI] [PubMed] [Google Scholar]
- [111].Monteiro AA, Borojevic R, Vitr. Cell. Dev. Biol 1987, 23, 10. [DOI] [PubMed] [Google Scholar]
- [112].Dasbiswas K, Majkut S, Discher DE, Safran SA, Nat. Commun 2015, 6, 6085. [DOI] [PubMed] [Google Scholar]
- [113].Thompson SA, Copeland CR, Reich DH, Tung L, Circulation 2011, 123, 2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Jones J, Res. Rep. Biol 2011, 2011, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Brown AC, Fiore VF, Sulchek TA, Barker TH, J. Pathol 2013, 229, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Kenny FN, Drymoussi Z, Delaine-Smith R, Kao AP, Laly AC, Knight MM, Philpott MP, Connelly JT, J. Cell Sci 2018, 131, jcs215780. [DOI] [PubMed] [Google Scholar]
- [117].Bevilacqua C, Sánchez-Iranzo H, Richter D, Diz-Muñoz A, Prevedel R, Biomed. Opt. Express 2019, 10, 1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Burla F, Tauber J, Dussi S, van der Gucht J, Koenderink GH, Nat. Phys 2019, 15, 549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Suki B, Majumdar A, Nugent MA, Bates JHT, Drug Discov. Today Dis. Model 2007, 4, 139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Bates JHT, Davis GS, Majumdar A, Butnor KJ, Suki B, Am. J. Respir. Crit. Care Med 2007, 176, 617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Das RK, Gocheva V, Hammink R, Zouani OF, Rowan AE, Nat. Mater 2015, 15, 318. [DOI] [PubMed] [Google Scholar]
- [122].Grim JC, Brown TE, Aguado BA, Chapnick DA, Viert AL, Liu X, Anseth KS, ACS Cent. Sci 2018, 4, 909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Shadish JA, Benuska GM, DeForest CA, Nat. Mater 2019, 18, 1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Stejskalová A, Oliva N, England FJ, Almquist BD, Adv. Mater 2019, 31, 1806380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Robinson TM, Hutmacher DW, Dalton PD, Adv. Funct. Mater 2019, 29, 1904664. [Google Scholar]
- [126].Moroni L, Burdick JA, Highley C, Lee SJ, Morimoto Y, Takeuchi S, Yoo JJ, Nat. Rev. Mater 2018, 3, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Schwartzman D, Chang I, Michele JJ, Mirotznik MS, Foster KR, J. Interv. Card. Electrophysiol 1999, 3, 213. [DOI] [PubMed] [Google Scholar]
- [128].Halter RJ, Schned A, Heaney J, Hartov A, Schutz S, Paulsen KD, J. Urol 2008, 179, 1580. [DOI] [PubMed] [Google Scholar]
- [129].Noshadi I, Walker BW, Portillo-Lara R, Sani ES, Gomes N, Aziziyan MR, Annabi N, Sci. Rep 2017, 7, DOI 10.1038/s41598-017-04280-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Fang Y, Eglen RM, SLAS Discov. 2017, 22, 456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Ouchi R, Togo S, Kimura M, Shinozawa T, Koido M, Koike H, Thompson W, Karns RA, Mayhew CN, McGrath PS, et al. , Cell Metab. 2019, 30, 374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Balog S, Li Y, Ogawa T, Miki T, Saito T, French SW, Asahina K, Hepatology 2019, hep. 30809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Sen Ding B, Cao Z, Lis R, Nolan DJ, Guo P, Simons M, Penfold ME, Shido K, Rabbany SY, Rafii S, Nature 2014, 505, 97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Trappmann B, Baker BM, Polacheck WJ, Choi CK, Burdick JA, Chen CS, Nat. Commun 2017, 8, 371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Polacheck WJ, Kutys ML, Yang J, Eyckmans J, Wu Y, Vasavada H, Hirschi KK, Chen CS, Nature 2017, 552, 258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Song KH, Highley CB, Rouff A, Burdick JA, Adv. Funct. Mater 2018, 28, 1801331. [Google Scholar]
- [137].Mendler N, Schuchhardt S, Sebening F, Res. Exp. Med. (Berl) 1973, 159, 231. [DOI] [PubMed] [Google Scholar]
- [138].van Putten S, Shafieyan Y, Hinz B, J. Mol. Cell. Cardiol 2016, 93, 133. [DOI] [PubMed] [Google Scholar]
- [139].Modarressi A, Pietramaggiori G, Godbout C, Vigato E, Pittet B, Hinz B, J. Invest. Dermatol 2010, 130, 2818. [DOI] [PubMed] [Google Scholar]
- [140].Park KM, Gerecht S, Nat. Commun 2014, 5, 4075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Blatchley MR, Hall F, Wang S, Pruitt HC, Gerecht S, in Trans. Annu. Meet. Soc. Biomater. Annu. Int. Biomater. Symp, 2019, p. 649. [Google Scholar]
- [142].Pelham RJ, Wang YL, Proc. Natl. Acad. Sci. U. S. A 1997, 94, 13661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Van Helvert S, Friedl P, ACS Appl. Mater. Interfaces 2016, 8, 21946. [DOI] [PubMed] [Google Scholar]
- [144].Wisdom KM, Adebowale K, Chang J, Lee JY, Nam S, Desai R, Rossen NS, Rafat M, West RB, Hodgson L, et al. , Nat. Commun 2018, 9, 4144. [DOI] [PMC free article] [PubMed] [Google Scholar]