

Abstract
Rationale:
The heart undergoes dramatic developmental changes during the prenatal to postnatal transition, including maturation of cardiac myocyte energy metabolic and contractile machinery. Delineation of the mechanisms involved in cardiac postnatal development could provide new insight into the “fetal” shifts that occur in the diseased heart and unveil strategies for driving maturation of stem cell-derived cardiac myocytes.
Objective:
To delineate transcriptional drivers of cardiac maturation.
Methods and Results:
We hypothesized that estrogen-related receptors (ERRs) α and γ, known transcriptional regulators of postnatal mitochondrial biogenesis and function, serve a role in the broader cardiac maturation program. We devised a strategy to “knockdown” (KD) the expression of ERRα and γ in heart after birth in mice (pn-csERRα/γ KD mice). With high levels of KD, pn-csERRα/γ KD mice exhibited a cardiomyopathy with an arrest in mitochondrial maturation. RNA sequence (RNA-seq) analysis of pn-csERRα/γ KD hearts at five weeks of age combined with chromatin immunoprecipitation with deep sequencing (ChIP-seq) and functional characterization conducted in human induced pluripotent stem cell-derived cardiac myocytes (hiPSC-CM) demonstrated that ERRγ activates transcription of genes involved in virtually all aspects of postnatal developmental maturation including mitochondrial energy transduction, contractile function, and ion transport. In addition, ERRγ was found to suppress genes involved in fibroblast activation in hearts of pn-ERRα/γKD mice. Disruption of Esrra and Esrrg in mice during fetal development resulted in perinatal lethality associated with structural and genomic evidence of an arrest in cardiac maturation including persistent expression of early developmental and non-cardiac lineage gene markers including cardiac fibroblast signatures. Lastly, targeted deletion of ESRRA and ESRRG in hiPSC-CM de-repressed expression of early (TCF21) and mature (periostin, collagen type III) fibroblast gene signatures.
Conclusions:
ERRα and γ are critical regulators of cardiac myocyte maturation, serving as transcriptional activators of adult cardiac metabolic and structural genes, and suppressors of non-cardiac lineages including fibroblast determination.
Keywords: gene transcription, postnatal cardiac development, cardiomyocyte differentiation, mitochondria, fibroblast
Subject Terms: Developmental Biology, Fibrosis, Gene Expression and Regulation, Genetically Altered and Transgenic Models, Metabolism
Graphical Abstract
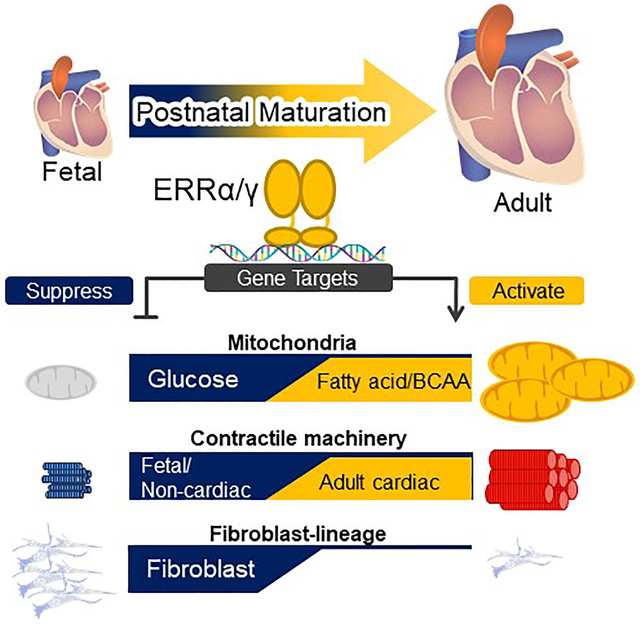
During the transition from the fetal to adult stages, the heart undergoes dramatic metabolic and structural maturation. Many adult cardiac processes revert to a fetal format during development to heart failure. Whereas many factors involved in cardiogenesis and early heart development have been defined, molecular mechanisms controlling postnatal cardiac development are largely unknown. We hypothesized that the transcriptional circuitry known to control mitochondrial biogenesis after birth is linked to a broader postnatal developmental program. Using conditional gene targeting in heart, we show that the estrogen-related receptors (ERRs) α and γ, known effectors of mitochondrial function, are necessary for normal postnatal and fetal cardiac maturation. Specifically, ERRα/γ deficiency resulted in a developmental maturation arrest of mitochondrial oxidative metabolism and gene regulatory programs involved in myocyte calcium handling, ion transport, and the adult sarcomeric machinery. In addition, ERR signaling was shown to be necessary for metabolic and electrophysiological maturation of hiPSC-CMs. Interestingly, ERRs also serve to repress early cardiac developmental pathways including the fibroblast lineage. Activation of ERR signaling could prove be an experimental tool to help drive hiPSC-CM maturation and as a candidate therapeutic target to antagonize “fetal” gene switches in the diseased heart.
INTRODUCTION
During the fetal to adult transition, the heart undergoes dramatic developmental maturation. Following birth, and during the postnatal period, many cardiac myocyte processes undergo transformation to adult programs including mitochondria, fuel utilization pathways, contractile machinery, and ion transport systems.1,2 Many of the perinatal and postnatal developmental events involve regulation at the level of gene transcription.3 Cardiac myocyte metabolic maturation includes a dramatic biogenic expansion of mitochondrial number at birth followed by gene regulatory events that equip mitochondria for high capacity fatty acid oxidation (FAO), the chief source of ATP in the adult heart. In parallel, the contractile machinery undergoes remodeling following birth driven by increased expression of adult sarcomeric gene isoforms encoding many contractile proteins including myosin heavy chains and troponins, concordant with downregulation of fetal isoforms. Dynamic cardiac chamber-specific gene expression shifts also occur during the perinatal period such as suppression of atrial isoform expression in ventricular chambers concordant with induction of ventricular-specific genes. Expression of specific ion transporters increase following birth allowing for enhanced capacity for uptake and release of ions such as calcium across the sarcolemmal and sarcoplasmic membranes via ATP-dependent processes. This finely orchestrated developmental maturation process results in the adult heart, an organ that maintains high capacity pumping function fueled by remarkably constant ATP production to meet the demands of postnatal life.
Considerable progress has been made in delineating the gene regulatory and signaling events involved in early cardiac development including specification of the cardiac myocyte lineage and the morphogenic events that lead to the development of a four chambered heart with proper alignment of the great vessels.3–5 In contrast, the upstream mechanisms involved in the developmental maturation of the cardiac myocyte during the fetal to adult transition are poorly understood. As the cardiac myocyte grows and matures in the largely post-proliferative postnatal period, fetal to adult re-programming of contractile and ion transport machinery must be tightly linked to augmentation of mitochondrial fuel oxidation and ATP synthesis. Key regulators of postnatal mitochondrial biogenesis in heart and other oxidative tissues have been defined over the past decade. Specifically, the transcriptional coregulators PPARgamma coactivator 1 (PGC-1) α and β are necessary and sufficient for the postnatal mitochondrial biogenesis in heart.6,7 PGC-1α was first discovered as a key regulator of mitochondrial biogenesis in brown fat.8 PGC-1α and β serve to coactivate downstream transcriptional events by interacting with specific transcription factors including the estrogen-related receptors (ERRs),9 peroxisome-proliferator-activated receptors (PPARs)10 and nuclear respiratory factor 1 (NRF-1).11 PGC-1α and β are induced in heart after birth to “boost” the activity of downstream effectors including the ERRs and PPARs triggering mitochondrial biogenesis.7,12 The mechanisms whereby this transcriptional cascade is coordinated with the gene regulatory circuits controlling structural and contractile maturation processes following birth are unknown.
Delineation of the mechanisms driving cardiac myocyte maturation is relevant to heart disease. During the development of pathological cardiac hypertrophy and in the failing heart, many metabolic and contractile processes shift to an immature or “fetal” state.13 For example, the high capacity for FAO and mitochondrial respiration is diminished in the failing heart. In addition, many of the adult contractile isoforms such as the myosin heavy chains revert to a fetal state. We know that the “fetal” shift of energy metabolism that occurs in the failing heart is due, at least in part, to deactivation of PGC-1/ERR, and PGC-1/PPAR signaling.14–16 However, whether these metabolic regulatory changes are linked to corresponding re-programming events in sarcomeric and ion transport genes is unknown. The answer to this question is important given reduced capacity for mitochondrial ATP-production contributes to the pathogenesis of heart failure.17,18 Delineation of the pathways involved in coordinating the fetal shift in the failing heart could enable experimental approaches to determine if this re-programming is adaptive or maladaptive. A second important rationale for defining the pathway involved in terminal cardiac maturation relates to the current challenge of generating adult cardiac myocytes from stem cells including human induced pluripotent stem cells (hiPSC). Full maturation of hiPSC-CM is necessary to re-capitulate heart disease using hiPSC-CM disease-in-a-dish approaches and, in the long-term, for effective regenerative therapeutic strategies.
In this report, we describe a series of studies designed to test the hypothesis that components of the PGC-1 transcriptional regulatory cascade are linked to the broad cardiac myocyte differentiation program. Using conditional gene disruption strategies in mice, we demonstrate that the central PGC-1 effectors, ERRα and γ, are necessary for proper control of cardiac myocyte differentiation gene switches, and for normal prenatal and postnatal cardiac maturation. Moreover, ERR signaling serves a developmental checkpoint function to suppress early developmental and non-cardiac lineage programs including cardiac fibroblast determination and activation.
METHODS
A Detailed Methods section is available in the Online Data Supplement. Please see the Major Resources Table in the Supplemental Materials. Animal studies were conducted in strict accordance with the National Institutes of Health guidelines for humane treatment of animals and approved by the Institutional Animal Care and Use Committee at the Sanford Burnham Prebys Medical Discovery Institute at Lake Nona and University of Pennsylvania. The sequencing datasets generated in this work have been deposited in NCBI’s Gene Expression Omnibus as a SuperSeries and are accessible through GEO Series accession number GSE113784.
RESULTS
ERRα/γ are necessary for normal postnatal cardiac developmental maturation.
If common upstream regulatory circuits orchestrate both energy metabolic and contractile maturation of the postnatal heart, perinatal patterns of expression of corresponding fetal and adult gene isoforms should be similar. We focused on the Acsl3/1 and Tnni1/3 fetal/adult isoform pairs which are known to be developmentally regulated.19,20 The Acsl isoform encodes long-chain acyl-CoA synthetase, a key mitochondrial enzyme that catalyzes the activation (thioesterification) of fatty acids prior to mitochondrial fatty acid oxidation (FAO), the main source of ATP in the adult heart. Troponin I, a component of the cardiac sarcomere, is encoded by fetal (Tnni1) and adult (Tnni3) gene isoforms. Cardiac levels of adult Acsl (Acsl1) and Tnni (Tnni3) gene isoforms were induced in a similar pattern during the first postnatal week in mice (Figure 1A, B). The mRNA and protein expression of Cox6a2, a nuclear gene that encodes a component of the electron transport chain was also coordinately induced during the postnatal period consistent with the known induction of mitochondrial biogenesis following birth (Figure 1A). Notably, the levels of the fetal transcripts (Acsl3 and Tnni1) fell dramatically beginning at P3 (Figure 1A, B).
Figure 1. Induction of Esrr gene expression during postnatal cardiac development.
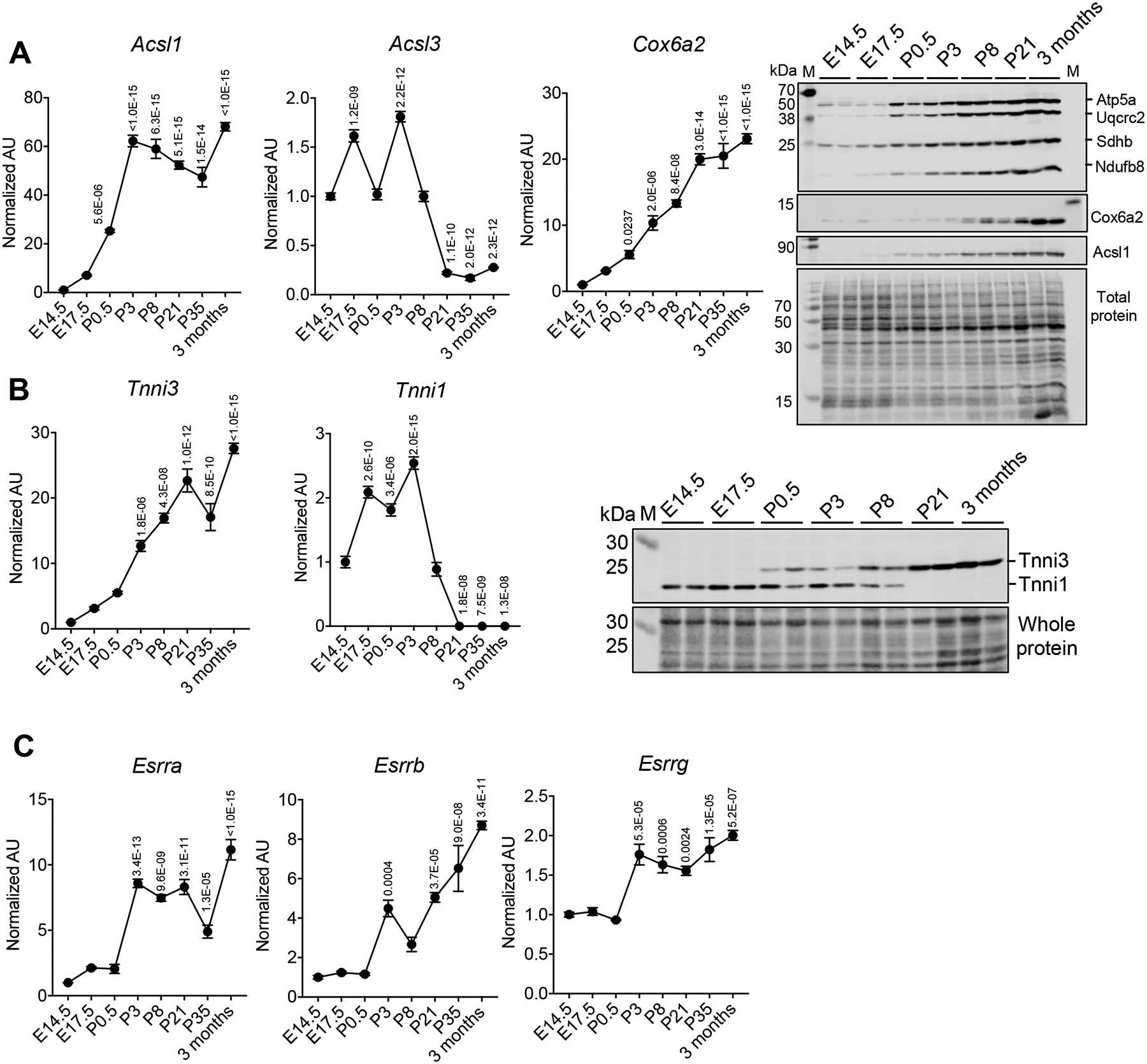
RT-qPCR-determined levels of the designated mRNAs during mouse postnatal cardiac development (from E14.5 to 3 months, n=3–5 per timepoint). Levels are shown as arbitrary units (AU) normalized to levels at E14.5 (A) Left: Levels of mRNA encoding Acsl1, Acsl3, and Cox6a2. Right: Representative immunoblot images of OXPHOS, Cox6a2, and Acsl1 proteins. Whole protein is shown as a control for loading. (B) Left: Levels of mRNA encoding Tnni3 and Tnni1. Right: Representative immunoblot image of Tnni3 and Tnni1 proteins. (C) Levels of mRNA encoding Esrra, Esrrb and Esrrg. p-values are indicated on the graphs, 1 way-ANOVA followed by Dunnett’s multiple comparisons test vs E14.5. All bars represent the mean ± SEM. Total protein staining at the corresponding molecular weight position was presented as a loading control for the representative immunoblot images. M indicates the protein marker lane. All immunoblot images were obtained with LI-COR Odyssey Fc. The qPCR raw data for Acsl1, Cox6a2, Tnni3, and Esrra are available in Online Table VIII.
Previously, we have shown that the transcriptional coactivators PGC-1α and β are necessary for postnatal cardiac mitochondrial biogenesis.6,7,21 PGC-1α serves as an inducible coactivator of several downstream nuclear receptor transcription factors including the estrogen-related receptors (ERRs) and peroxisome-proliferator activated receptors (PPARs).12 We found that the expression of the genes encoding ERRs, PPARα, and PPARδ were induced in parallel with Ascl1 and Tnni3 (Figure 1C and Online Figure IA, B).
Given the central role played by the ERRs in the PGC-1 cascade,12,22,23 we hypothesized that this nuclear receptor family serves a broader regulatory role in the postnatal heart beyond that of mitochondrial function. We developed a strategy to reduce ERR signaling specifically during the early postnatal developmental period. We focused on Esrra and Esrrg given that our quantitative PCR analyses demonstrated that expression of these ERRs is significantly higher than that of Esrrb in postnatal mouse ventricle (average qPCR Ct values for Esrra, Esrrb, and Esrrg transcripts were 24.023 (SD 0.3302), 26.412 (SD 0.5331), and 23.304 (SD 0.2374) in day P35 mouse heart, respectively). We sought to reduce activity of ERRα and ERRγ after the perinatal surge of mitochondrial biogenesis given that germline deletion of Esrrg24 or the ERR coactivators (Ppargc1a and b)7 results in early postnatal death related to disruption of perinatal mitochondrial biogenesis. Therefore, an inducible cardiac ERRα/γ knockdown (KD) mouse model system was employed using adeno-associated virus 9 (AAV9) expressing Cre recombinase under control of the cardiac troponin (cTnT) promoter (AAV-Cre) as outlined in the Online Detailed Methods section.25,26 Given that ERRα and ERRγ have overlapping cardiac target genes,23,27 both Esrra and Esrrg were targeted. The AAV-Cre was injected into ERRα/γflox/flox mice at P1 (Figure 2A) to generate postnatal ERRα/γ-deficient mice. Five weeks after the viral injection, the postnatal ERR KD showed markedly downregulated Esrra, Esrrg, Acsl1 and Cox6a2 expression and cardiac dysfunction as evidenced by increased Nppa and Nppb levels (Online Figure IIA), reduced left ventricular (LV) fractional shortening, and increased LV end-diastolic and end-systolic volume measurements compared to littermate controls injected with AAV9 expressing luciferase (AAV-Luc) (Online Figure IIB). The virus dose was reduced in order to assess the impact of postnatal ERRα/γ deficiency in the absence of cardiac dysfunction. The lower virus dose achieved significant downregulation of ERRα and ERRγ mRNA and protein levels (approximately 85 % reduction; Online Figure IIIA) in the absence of cardiac dysfunction as determined by the ratio of bi-ventricle to body weight (Online Figure IIIB) and echocardiography (Online Figures IIIC). Interestingly, ERRβ transcript levels were also reduced with ERRα/γ KD (Online Figure IIIA). ERRα and ERRγ protein levels were similarly reduced in myocytes isolated from the pn-csERRα/γ KD hearts, further confirming the efficacy of the AAV9-Cre vector (Online Figure IIIA). There were no differences between male and female in both degree of ERRα and ERRγ KD or cardiac function (Online Figure IIIA and C). This postnatal ERRα/ERRγ KD approach (pn-ERRα/γKD mice) was used for subsequent experiments to assess the role of ERR signaling in postnatal cardiac maturation.
Figure 2. Inducible cardiac ERRϖ/γ deficiency results in abnormalities in mitochondrial density and structure.
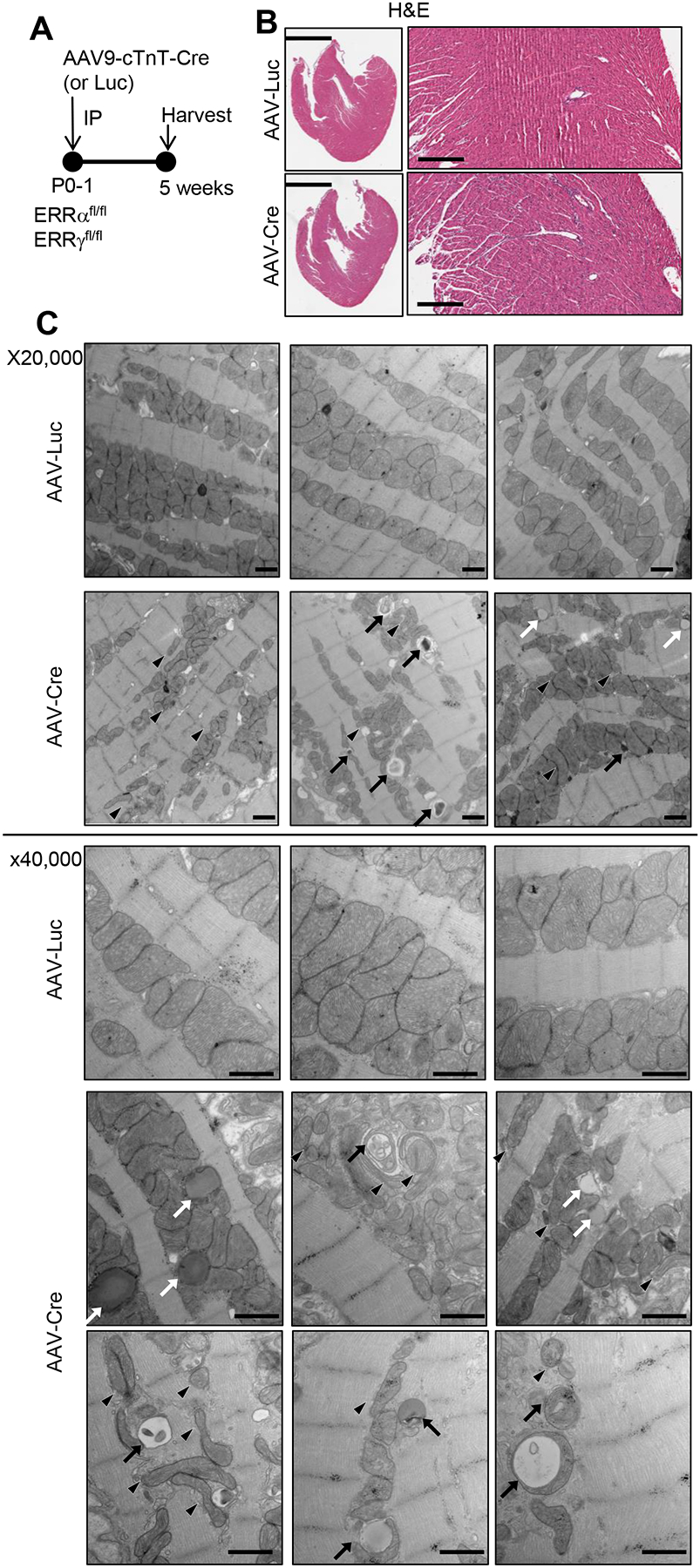
(A) Schematic showing the experimental timeline. AAV-Cre or AAV-Luc (control) was administered to P1 ERRα/γflox/flox pups at a dose that does not cause cardiac dysfunction as described in the text. (B) Representative H&E images of male AAV-Luc or AAV-Cre-injected 5 week-old LV. Scale bars represent 2 mm in lower magnification and 300 mm in higher magnification. (C) Representative electron microscope images of LV wall of the male or female postnatal ERR KD hearts (x20,000 and 40,000) magnification. Arrows indicate structurally abnormal mitochondria including engulfed organelles indicative of mitophagy (black arrows), and elongated or fragmented (black arrowheads). White arrows indicate droplet-like structures that may represent lipid. Scale bars represent 800 nm.
The cardiac phenotype of the pn-csERR KD mice was compared with littermate controls injected with AAV-Luc to further assess the impact of altered ERR signaling during postnatal cardiac development (Figure 2A). H&E staining of the left ventricle (LV) presented no obvious cellular damage in the postnatal ERRα/γ-deficient (pn-csERRα/γ KD) mice (Figure 2B). Ultrastructural imaging by electron microscopy at P35 revealed abnormal mitochondrial morphology in the pn-csERRα/γ KD hearts. Specifically, the mitochondria were smaller, often elongated, and the cristae were less dense and disorganized compared to controls (Figure 2C). In addition, we observed occasional globular structures within myocytes suggestive of lipid droplets and occasional double-membraned vesicles indicative of increased mitophagy in the pn-csERRα/γ KD hearts but not controls (Figure 2C). Consistent with these ultrastructural abnormalities, mitochondrial DNA content was significantly decreased in the hearts of pn-csERRα/γ KD mice along with reduced levels of Acsl1 and many OXPHOS proteins (Online Figure IIID). Metabolite profiling of pn-csERRα/γ KD ventricles was consistent with immature mitochondrial FAO as evidenced by reduced levels of several long-chain acylcarnitine species (e.g. C16-OH) (Online Figure IIIE). In addition, levels of short-chain acylcarnitine species including C3, C5–2-methylbutyryl, and C5 isovaleryl increased in the pn-csERRα/γ KD hearts (Online Figure IIIE) suggesting an alteration in branched-chain amino acid (BCAA) catabolism. Taken together, these data indicate that ERR signaling is critical for postnatal cardiac metabolic maturation.
We next performed whole genome RNA sequencing (RNA-seq) on cardiac ventricles of pn-csERRα/γ KD and control mice at 5 weeks of age. The RNA-seq dataset indicated a striking and widespread arrest in postnatal cardiac maturation in the pn-csERRα/γ KD hearts. Consistent with the observed mitochondrial defects, the expression of genes involved in multiple mitochondrial energy transduction pathways, including the TCA cycle, FAO, and OXPHOS, were all highly downregulated in the pn-csERRα/γ KD hearts and isolated cardiomyocytes (Figure 3A–C; Online Figure IVA). In addition, expression of genes related to BCAA catabolism (Bckdha, Bcat2, Bckdhb, Hibch, Acad8, Hibadh, and Auh) were significantly downregulated (Online Figure IVB). These gene expression changes were reflected in Gene Ontology (GO) and KEGG pathway results (Figure 3A and Online Table I). Consistent with reduced expression of genes involved in mitochondrial oxidative metabolism, a subset of glycolytic genes, including Eno2 and Pkm, were induced, and lactate dehydrogenase A (Ldha) expression was upregulated (data not shown) consistent with persistent glycolytic metabolism, characteristic of the fetal heart.
Figure 3. ERRα/γ is necessary for the postnatal cardiac maturation transcriptional program.
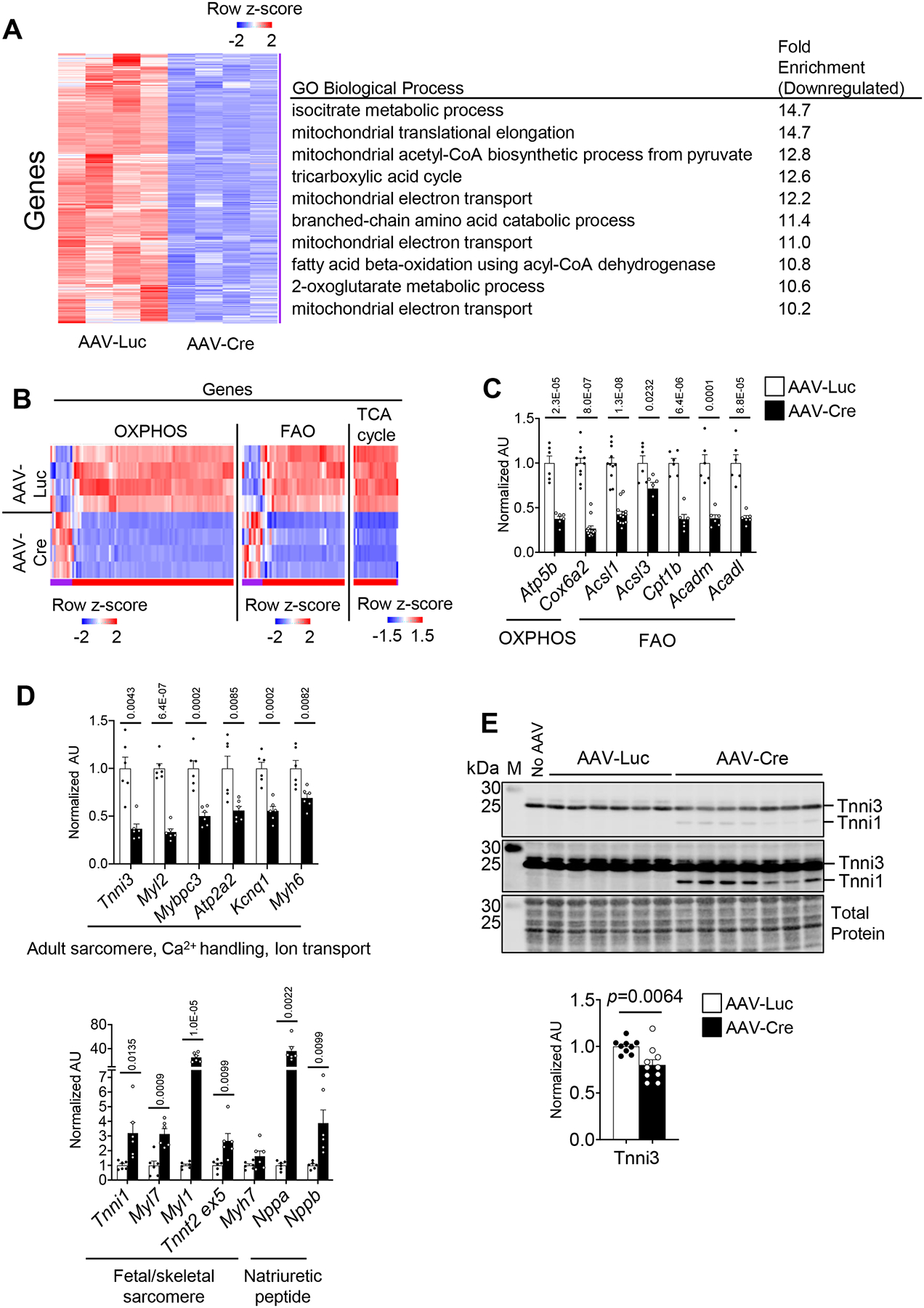
Whole genome RNA-seq was performed on ventricles from AAV-Luc or Cre-injected ERRα/γflox/flox. (A) Left: Heat map represents z-score of mRNA levels significantly downregulated from RNA-seq data performed on ventricles from AAV-Luc or Cre-injected ERRα/γflox/flox. 2032 genes downregulated in the Cre group are shown. Right: Significantly enriched GO Biological Process using transcripts downregulated in the AAV-Cre ventricles as compared to AAV-Luc controls. (B) Heat map represents z-score of mRNA levels of metabolic transcripts encoding OXPHOS, TCA cycle, and FAO proteins. (C) Levels of mRNA transcripts encoding energy metabolic genes, and (D) sarcomere proteins, ion channels, and natriuretic peptides in ventricles from male AAV-Luc (n=6–11) or Cre (n=6–13) -injected ERRα/γflox/flox mice as measured by RT-qPCR. p-values are indicated on the bar graphs comparing AAV-Luc vs AAV-Cre. Student’s t-test or Mann-Whitney U test was used (see Detailed Methods). Bars represent the mean ± SEM. (E) Upper: Representative immunoblot images of Tnni1/3 in 5-week old injected ERRα/γflox/flox ventricles subjected to AAV-Luc (n=6), AAV-Cre (n=7) or without AAV injection (No AAV) using an anti-pan Tnni antibody at two different exposures. Bottom: Bar graphs show the amount of Tnni3 protein expression in ERRα/γflox/flox ventricles injected with AAV-Luc (n=9) or AAV-Cre (n=10). Student’s t-test was used. M indicates the protein marker lane. Total protein staining was used as a loading control. All immunoblot images were obtained with LI-COR Odyssey Fc.
Interestingly, in addition to downregulation of mitochondrial and energy metabolic pathways, the pn-csERRα/γ KD RNA-seq dataset was significantly enriched (downregulated) for GO or KEGG terms related to cardiac structural pathways (Online Table I) including adult contractile protein gene isoforms. For example, expression of Tnni3 was downregulated, while the level of the fetal isoform, Tnni1, was upregulated in the pn-ERRα/γ KD hearts (Figure 3D; Online Figure IVC). This isoform switch was also reflected at the protein level based on immunoblotting, albeit more modestly (Figure 3E). These changes were also seen in isolated cardiac myocytes (Online Figure IVD). In addition, levels of the transcript encoding myosin regulatory light chain 2 (Myl2), a ventricular/cardiac muscle isoform, were downregulated in the pn-csERRα/γ KD hearts (Figure 3D; Online Figure IVC). Expression of other cardiac structural and ion channel/transporter genes including Myl3, Tnnc1, Mybpc3, Atp1a1, Atp1a2, Cacna2d3, and Kcnq1 were also reduced (Figure 3D; Online Figure IIIA; IVC). Moreover, levels of genes involved in Ca2+ handling functions such as Pln, Ryr2, and Atp2a2 were downregulated (Figure 3D; Online Figure IVC). The gene expression changes in the pn-csERRα/γ KD hearts were similar in males and females (Figure 3D; Online Figure IVE and F). Collectively, these data indicate that ERRα and ERRγ are not only necessary for energy metabolic maturation but also participate in the transcriptional control of a broad array of adult cardiac structural genes during postnatal developmental maturation.
ERRγ functions as a direct transcriptional activator of adult metabolic and structural cardiac genes.
ERRα and γ are known to exert direct transcriptional control upon genes involved in mitochondrial ATP production. However, we sought to determine if the postnatal regulation of structural ATP-consuming genes by ERR signaling described above occurs via direct transcriptional control or through indirect mechanisms. As an initial step to address this question, chromatin immunoprecipitation with high-throughput sequencing (ChIP-seq) was used to delineate the ERRγ cistrome in wild-type hiPSC-CMs (WT hiPSC-CMs) and ERRγ KO hiPSC-CMs generated with CRISPR/Cas9n (Online Figure V and in the Online Detailed Methods section). A total of 48,067 ERRγ peaks were identified in wild-type hiPSC-CM, of which 22,933 were deemed specific based on comparison with an ERRγ KO dataset. Peaks within 5kb of a known transcription start or within the first intron (Promoter-TSS) comprised 25.5% of the total peaks (Figure 4A). Many peaks were also distributed in intron (33.6 %) and intergenic (36.5%) regions, respectively (Figure 4A). Position-weight matrices (PWM)-based (de novo) motif analysis demonstrated that the canonical ERR response element was the most highly enriched motif in this ChIP-seq data (Figure 4B). Consensus binding motifs for GATA, MEF2 and TEAD, known cardiogenic transcription factors, were also significantly enriched within the ERRγ peaks (Figure 4B).
Figure 4. Analysis of the ERRγ cistrome indicates direct transcriptional control of adult cardiac metabolic and contractile protein genes.
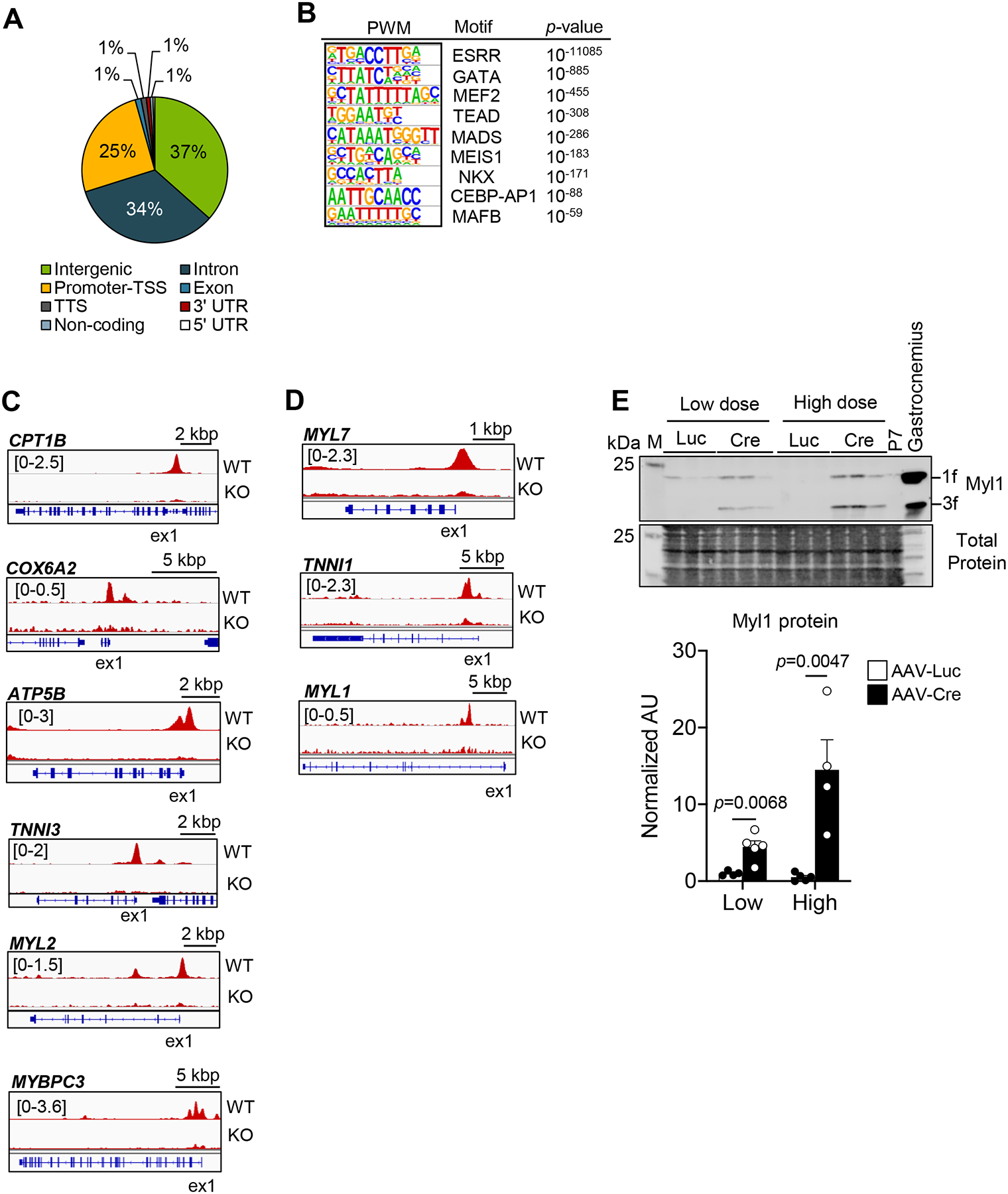
(A) Pie chart of ERRγ peak distribution determined from whole genome ChIP-seq data. Promoter transcription start site (TSS) was defined as −5 kbp or within the first intron. (B) Significantly enriched ERRγ binding motifs as defined by PWM motif analysis. Representative genome browser tracks of ERRγ peaks on the activated-targets (C) and the suppressed-targets (D) in WT and ERRγ KO hiPSC- CMs. Number in brackets indicates RPM (reads per million). (E) Left: Representative immunoblot images of Myl1 in 5-week old injected ERRα/γflox/flox ventricles subjected with low dose AAV-Luc or AAV-Cre. Right: Bar graphs depict quantification of Myl1 immunoblot analysis conducted on samples prepared from ERRα/γflox/flox ventricles subjected to low or high dose AAV-Luc (n=4) or AAV-Cre (n=4–5). Student’s t-test was used. Gastrocnemius muscle from aged-match wild type mouse was used as a positive control for Myl1. Total protein staining was used as a loading control. M indicates the protein marker lane. All immunoblot images were obtained with LI-COR Odyssey Fc.
We next conducted a cross-comparison of the ERRγ occupation peaks obtained by ChIP-seq of hiPSC-CMs with levels of transcripts defined in the RNA-seq dataset generated from the pn-csERRα/γ KD mouse hearts (approach outlined in Online Figure VIA and in the Online Detailed Methods section). The intersection of putative ERRγ target genes defined by the ChIP-seq data (defined by the gene closest to a given ERRγ peak) and regulation of its corresponding transcript in the pnERRα/γKD RNA-seq dataset were designated as direct ERRγ gene targets. As predicted, this intersection analysis identified direct ERRγ-activated gene targets across a broad spectrum of mitochondrial processes including biogenesis and energy transduction pathways as has been described in part previously23 including Nduf family members, OXPHOS (Atp5g3, Cox6a2, etc.), FAO (Acadm, Cpt1b, and Acsl1, etc.), BCAA catabolism (Bcat2, Bckdha, and Bckdhb), and TCA cycle (Mdh1, Idh3a, etc.) (Figure 4C; Online Figure VIB; Online Table II and III). Interestingly, in addition to canonical metabolic gene targets, the pathway analysis indicated that ERRγ directly activates many genes involved in cardiac contractile and ion channel function (Online Table II). The list of direct ERRγ-activated targets included adult sarcomeric gene isoforms such as Tnni3, Myl2, Mybpc3, Myl3, Tcap, and Tnnc1 (Figure 4C; Online Figure VIB; Online Table II and III). Ion transporter/channel ERR target genes included K+ transporters (Kcnj8, Kcnj11, Hcn2, and Scn4b), ATPase Na+/K+ transporter subunit (Atp1a1), potassium-dependent Na+/ Ca2+exchanger (Slc24a3), voltage-dependent Ca2+channel subunits (Cacna1h, Cacna2d1, and Cacna2d3) and Ca2+ handling proteins (Atp2a2 and Pln) (Online Table III).
Taken together, these results indicate that ERRγ directly regulates the transcription of genes involved in both ATP-producing and ATP-consuming processes in the cardiac myocyte during postnatal maturation.
ERRα/γ suppress a subset of fetal and non-cardiac myocyte genes.
The integrated ChIP-seq/RNA-seq intersection analysis also revealed a significant number of upregulated ERRγ target genes in the pn-csERRα/γ KD heart suggesting that they are suppressed by ERR signaling during postnatal development. Interestingly, the ERR-suppressed genes were identified in pathways related to cardiac fetal or atrial contractile programs, and the skeletal muscle sarcomere (Figure 4D; Online Figure VIB and Online Table II). Among fetal contractile protein targets, Tnni1 (Figure 3E; Figure 4D; Online Figure VIB) and the atrial/fetal ventricular isoforms Myl7 and Myl4 (Figure 4D and Online Table IV) were identified. An example of a non-cardiac lineage gene that was upregulated in pn-csERRα/γ KD ventricle is skeletal muscle contractile protein, Myl1 (Figure 4D–E; Online Table IV). These results indicate that in addition to activating genes involved in adult metabolic and contractile programs during postnatal development, ERRγ suppresses a subset of genes involved in early cardiac development and non-cardiac lineages.
Further analysis of enriched binding motifs determined from the ERRγ ChIP-seq comparing ERR-activated and -suppressed-targets revealed several interesting differences (Online Figure VIC). For the activated-targets, NFIC, NR5A1, TEAD, AC acceptor, COUPTFII, and MADS binding motifs were enriched (Online Figure VIC). Notably, TEAD1 has recently been identified as a core component of the cardiac transcriptional network and was shown to regulate mitochondrial metabolic genes in adult heart.28 Serum response factor (SRF) is the founding member of the MADS-box family of transcription factors29 and drives many aspects of cardiomyocyte maturation during mouse postnatal development.30 NR5A1 has been implicated in myocyte differentiation with MEF2A activation.31 For ERR-suppressed targets, Homeobox and NR2C2 binding motifs were enriched (Online Figure VIC). NR2C2 (also known as TR4) has been shown to work as a repressor for nuclear receptors including PPARα, retinoid receptors, and thyroid hormone receptors.32,33 These motif enrichment patterns suggest that distinct protein complexes could dictate ERR-mediated transcriptional activation or suppression.
ERRα/γ deficiency impacts functional maturation of hiPSC-CMs.
To further investigate the functional impact of ERR loss-of-function on cardiac myocyte maturation, hiPSC-CMs deficient for both ERRα and ERRγ were established using CRISPR/Cas9 (Online Figure VIIA). The loss of ERRα and ERRγ did not affect cardiac differentiation as evidenced by the number of cTnT-positive cells in the WT versus ERRα/γ KO hiPSC-CMs using flow cytometry (Online Figure VIIB). However, there were small populations expressing very low levels of cTnT in both ERR-mutant hiPSC-CM lines (Online Figure VIIB), suggesting that ERR-deficient hiPSC-CM culture contained a sub-population of immature cardiomyocytes.
We first assessed the metabolic maturation of the ERRα/γ KO hiPSC-CMs. ERR-deficient hiPSC-CMs exhibited a significantly decreased maximal oxygen consumption rate (OCR) which was largely related to diminished FAO (Etomoxir-sensitive OCR) as compared to control cells (Figure 5A). Consistent with the OCR studies, quantitative targeted metabolomics demonstrated reduction in many long-chain (C16–18) acylcarnitine species (Online Figure VIIIA). Electron microscope analysis revealed that loss of ERRα/γ caused smaller (or fragmented) mitochondria with less developed cristae structure (Online Figure VIIIB).
Figure 5. ERRα/γ drives hiPSC-CM metabolic and structural maturation.
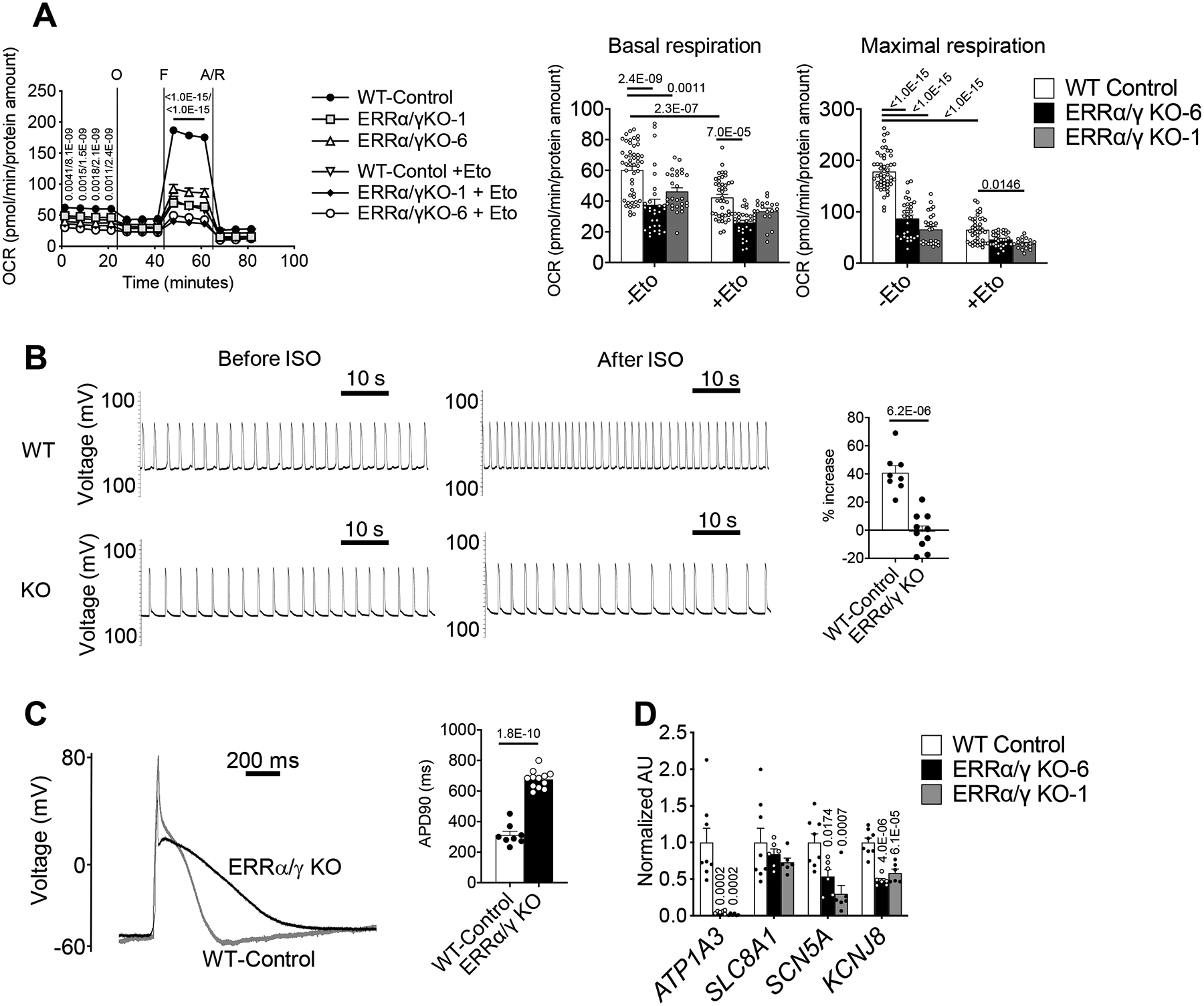
(A) Line graph represents oxygen consumption rates (OCR) in ERRα/γ KO hiPSC-CMs and controls (WT) with or without Etomoxir from 3–5 independent experiments, WT-Control (n=50), KO-6 (n=30), KO-1 (n=27), WT-Control + Eto (n=42), KO-1 + Eto (n=18), and KO-6 + Eto (n=30) were combined. Bar graphs show the mean basal (upper) and maximal (lower) respiration with ± SEM. p-values are indicated on each graph, 2-way ANOVA with Tukey’s multiple comparison test. p-values within the line graph indicate comparisons between WT-Control vs KO-1/ WT-Control vs KO-6. The OCR raw data is available in Online Table IX. (B) Representative spontaneous (left) and isoproterenol (Iso)-stimulated (right) action potential tracings of WT and ERRα/γ iPSC-CMs (line 1). Bar graph represents % rate increase to 1 μmol/L Iso in WT (n=8) and ERRα/γ iPSC-CMs line 1 (n=10). Error bars indicate SEM. Student’s t-test was used. (C) Representative tracing of the alteration in cell membrane potential during whole-cell patch clamp analysis. Quantification of the APD90 in WT (n=8) and ERRα/γ KO hiPSC-CMs line 1 (n=10) is presented in the bar graph. Student’s t-test was used. (D) Levels of mRNA transcripts encoding ion transport in ERRα/γ hiPSC-CMs as measured by RT-qPCR. p-values are indicated on each graph vs WT, 1-way ANOVA with Dunnet’s multiple comparisons test. Bars represent the mean ± SEM.
We next examined the electrophysiological properties of the ERR-deficient hiPSC-CMs. The spontaneous beating rate of the clusters of hiPSC-CMs was significantly lower in the ERR-deficient hiPSC-CMs (Figure 5B, left). Following exposure to isoproterenol, the ERR-deficient hiPSC-CMs did not increase rate in contrast to control cells (Figure 5B, right). Single cell whole-cell patch clamp studies demonstrated a significant increase in action potential duration indicative of prolonged repolarization in the ERR mutant CMs as compared to WT-control CMs (Figure 5C). Notably, expression levels of a subset of ion transporter genes (ATP1A3, SLC8A1, SCN5A, and KCNJ8) were downregulated in the KO CMs potentially accounting for the prolonged action potential (Figure 5D).
ERR signaling also contributes to late fetal maturation of the developing mouse heart.
ERRγ expression level has been shown to be dramatically upregulated from E7 to E11 in mouse embryo,34 and the cardiac expression has been confirmed at E8 with the LacZ knock-in reporter mouse.24 We sought to determine if ERR also impacts cardiac maturation during prenatal development. Mice in which both ERRα and ERRγ were targeted during fetal development (fetal-csERRα/γ−/− mice) were generated using an Nkx2–5-Cre recombinase driver35 (Online Figure IXA, B) and characterized. We confirmed absent ERRα and ERRγ protein expression at E17.5 (Online Figure IXB). Notably, transcript levels of ERRβ were increased in the fetal-csERRα/γ−/− hearts (Online Figure IXB), possibly as a compensatory regulatory response. The observed number of fetal-csERRα/γ−/− mice (Cre+ ERRαflox/flox ERRγflox/flox) was concordant with normal Mendelian ratios at day E17.5, but all died within 24 hours of birth (Figure 6A). The cardiac chambers of the fetal-csERRα/γ−/− mice developed normally but exhibited LV thinning (Figure 6B). High magnification images of fetal-csERRα/γ−/− LV free wall with H&E staining showed reduced cellular density resembling a ventricular non-compaction phenotype (Figure 6B). Electron microscopy demonstrated that mitochondria from the LV of fetal-csERRα/γ−/− mice were more sparse, smaller, and exhibited morphological abnormalities including poorly developed cristae structure compared to control littermates (Figure 6C), similar to that shown for pn-csERRα/γ KD hearts (Figure 2C) and ERRα/γ KO hiPSC-CMs (Online Figure VIIIB). In addition, sarcomeres were disorganized (Figure 6C). Metabolomic analyses of fetal-csERRα/γ−/− LV demonstrated reduced levels of long-chain acylcarnitine species indicative of reduced β−oxidation (Online Figure XA). Taken together these results indicate reduced capacity for mitochondrial FAO in prenatal ERR-deficient hearts.
Figure 6. ERRα/γ is necessary for the cardiac developmental maturation in vivo.
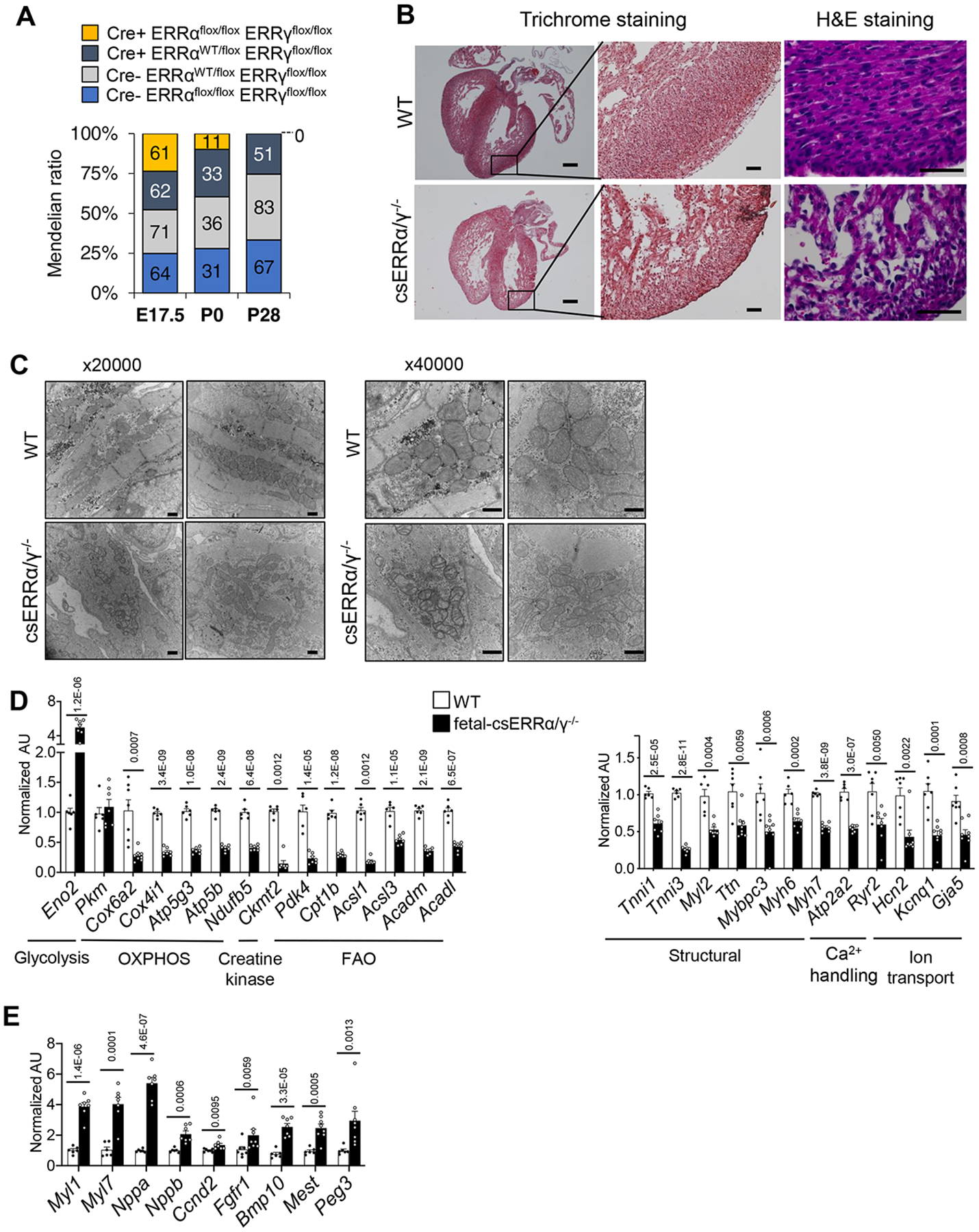
(A) Distribution of genotypes at indicated gestational ages in Cre- ERRαflox/flox ERRγflox/flox (WT), Cre- ERRαWT/flox ERRγflox/flox, Cre+ ERRαWT/flox ERRγflox/flox and Cre+ ERRαflox/flox ERRγflox/flox (fetal-csERRα/γ−/−) at E17.5 (n=258), P0 (n=111) and P28 (n=201). The numbers in each bar graph indicate the actual viable embryo or mouse number observed. Mendelian ratio (%) is shown on y-axis. At P0, chi squared =13.937 with 3 degrees of freedom. The two-tailed p value = 0.0030. At P28, chi squared = 48.467 with 3 degrees of freedom. The two-tailed p value < 0.0001. (B) Representative Masson Trichrome and H&E images of E17.5 hearts of littermate WT and fetal-csERRα/γ−/− mice. Scale bars represent 250 mm and 50 mm in the lower and higher magnification images respectively. (C) Representative electron microscope images of LV wall of E17.5 fetal-csERRα/γ−/− mice (x20,000 and 40,000) magnification. Scale bars represent 500 nm. Levels of mRNA transcripts encoding (D) energy metabolic, sarcomeric, ion transport, and Ca2+ handling protein genes as well as (E) the genes related to early cardiac development in E17.5 ventricles of control WT (n=6–7) and littermate fetal-csERRα/γ−/− mice (n=7–8) as measured by RT-qPCR. p-values are indicated on each graph vs WT. Student’s t-test or Mann-Whitney U test was used (see Detailed Methods). Bars represent the mean ± SEM.
RNA-seq of the fetal-csERRα/γ−/− ventricle at E17.5 compared to WT controls (Cre- ERRαflox/flox ERRγflox/flox) revealed a broad arrest in cardiac maturation and persistence of fetal gene markers (Online Table V). Similar to that observed in the pn-csERRα/γ KD hearts, expression of genes and proteins involved in mitochondrial energy transduction pathways (TCA cycle, FAO, and OXPHOS), cardiac contractile apparatus including Tnni3, ion channel, and Ca2+ handling proteins were reduced in the fetal-csERRα/γ−/− ventricles (Figure 6D and Online Figure XB, C, D; Online Table V). The fetal-csERRα/γ−/− ventricles also exhibited signatures of persistent activation of early fetal developmental markers including genes in fetal cardiac/skeletal sarcomere and the glycolysis, TGF-β, BMP, FGF, and Wnt signaling pathways (Figure 6E and Online Table V). Many of the upregulated genes were identified as ERR-direct targets in hiPSC-CMs (Online Table VI).
ERR signaling suppresses cardiac fibroblast and pro-inflammatory programs.
Analysis of the RNA-seq data from the pn-csERRα/γ KD ventricle revealed a significant number of cardiac ERRγ-suppressed genes involved in fibroblast activation, fibrosis, and pro-inflammatory pathways (Online Figure XIA). qPCR validation confirmed induction of genes involved in myofibroblast activation, including Tgfb2, Tgfb3, Postn, Col3a1, and Sox9, chemokine receptors (Ccl2 and Ccl7), and toll-like receptors (Tlr1, Tlr4, and Tlr8) in both male (Figure 7A) and female (data not shown) mice. Notably, TGFB2, TGFB3, POSTN, COL3A1, SOX9, and TLR4 were defined as ERRγ direct targets in hiPSC-CMs (Figure 7B; Online Figure XIB and Online Table IV). These results suggested that ERR signaling suppresses myofibroblast activation. To explore this possibility, Picro Sirius Red staining was performed on LV free wall sections prepared from male 5-week-old pn-csERRα/γ KD mice and littermate controls. Quantification of the fibrosis staining demonstrated modest but significantly increased collagen fiber deposition in the pn-csERRα/γ KD hearts (Figure 7C).
Figure 7. Loss of ERRα/γ induced fibroblast-lineage gene expression in cardiomyocytes.
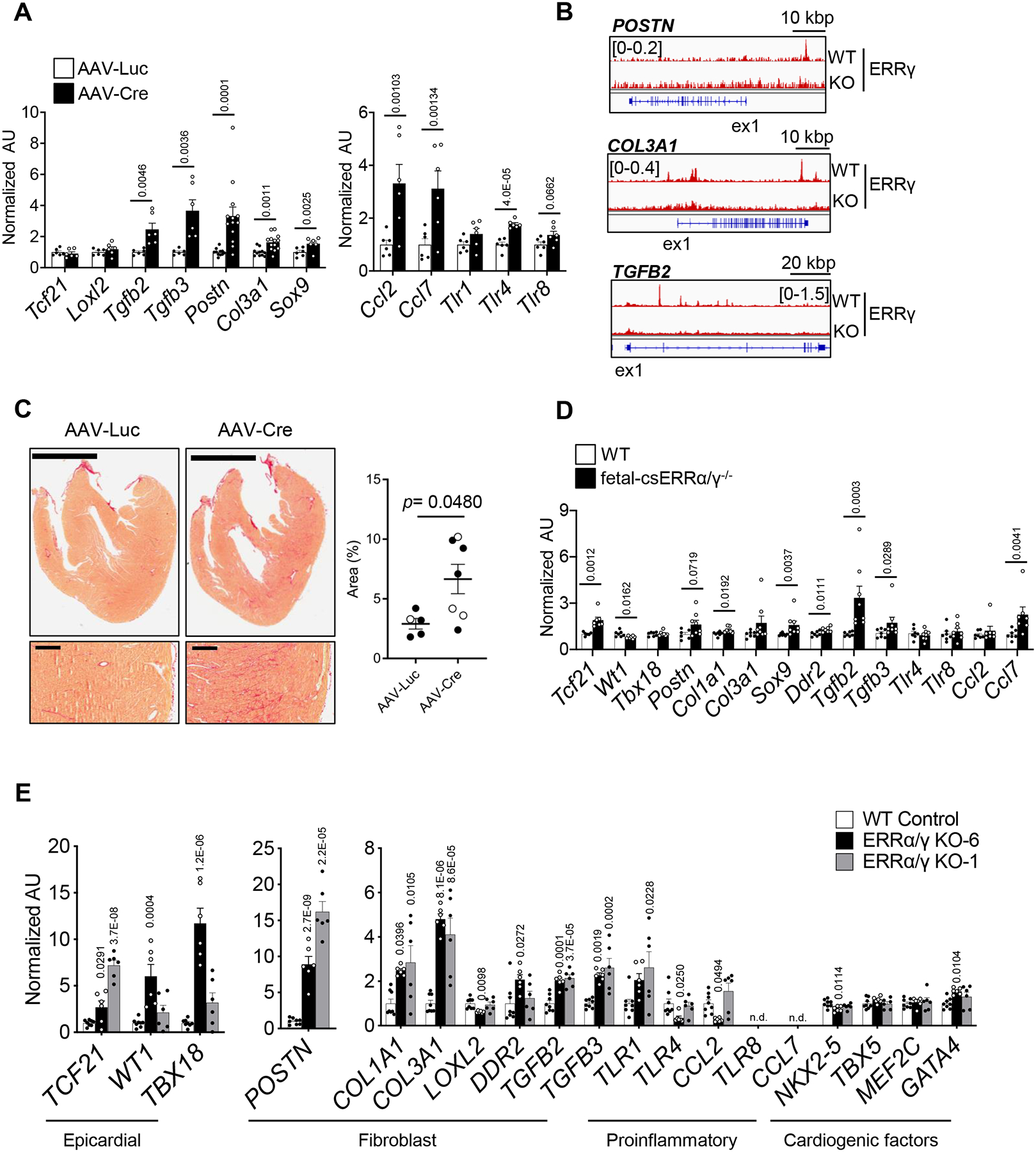
(A) Levels of mRNA transcripts encoding fibroblast and proinflammatory genes determined by RT-qPCR in ventricles from male pn-csERRα/γ KD mice and AAV-Luc-injected littermate controls (n=5–11) or Cre (n=7–13)-injected ERRα/γflox/flox mice as measured by RT-qPCR. p-values are indicated on the bar graphs vs AAV-Luc. Student’s t-test or Mann-Whitney U test was used (see Detailed Methods). Bars represent the mean ± SEM. (B) Representative genome browser tracks of ERRγ peaks on the suppressed-targets in WT and ERRγ KO hiPSC- CMs. Number in brackets indicates RPM (reads per million). (C) Left: Representative images of Picro Sirius Red staining of whole ventricles and LV free wall from AAV-Luc or Cre-injected ERRα/γflox/flox . Scale bars represent 2 mm (lower magnification) and 200 mm (higher magnification). Right: Bar graphs show the amount of % of Picro Sirius Red staining in ERRα/γflox/flox ventricles subjected with AAV-Luc or AAV-Cre. Open symbol indicates female, closed symbol indicates male. Mann-Whitney U test was used. (D) Levels of transcripts of ERR suppressed-targets involved in early development including genes involved in fetal contractile machinery, epicardial development, signaling and fibroblast processes in E17.5 ventricles of control WT (n=6–7) and littermate fetal-csERRα/γ−/− (n=7–8) mice as measured by RT-qPCR. p-values are indicated on the bar graphs vs WT. Student’s t-test or Mann-Whitney U test was used (see Detailed Methods). Bars represent the mean ± SEM. (E) Levels of mRNA transcripts encoding fibroblast, epicardial, and cardiac lineage markers in WT (n=8) or ERRα/γ KO hiPSC-CMs from two distinct ERRα/γ KO hiPSC lines (KO6, KO1; n=6) as measured by RT-qPCR. p-values are indicated on the bar graphs vs WT, 1-way ANOVA with Dunnet’s multiple comparisons test. Bars represent the mean ± SEM. n.d equals non-detectable.
Interestingly, there was also an increase in expression of a significant subset of fibroblast genes in the fetal-csERRα/γ−/− hearts. Specifically, transcript levels of the ERRγ target gene Tcf21, a known specification transcription factor for the cardiac fibroblast lineage expressed in the epicardial lineage,36 was upregulated in ERRα/γ depleted ventricles along with mature cardiac fibroblast specific markers (Postn, Ddr2, and Col1a1) and profibrotic cytokines (Tgfb2 and Tgfb3) (Figure 7D). In contrast to the pn-ERRα/γ KD hearts, fibrosis was not observed by histologic staining in the fetal-csERRα/γ−/− ventricles (Figure 6B), suggesting that myofibroblasts were not activated. In addition, expression of inflammatory markers, such as the Toll-like receptor genes, was not induced (Figure 7D).
The hiPSC-CM system was used to further explore the impact of cardiac myocyte ERR-deficiency on fibroblast programs, in a cell autonomous context.37 Expression of fibroblast lineage and activation marker genes was markedly induced in the two independent ERRα/γ KO hiPSC-CM lines including POSTN, COL3A1, TCF21, TGFB2, and TGFB3 (Figure 7E). The induction of POSTN, COL3A1 and TCF21 protein levels was confirmed (Online Figure XIC). Notably, transcript levels encoding cardiogenic factors (TBX5, MEF2C, NKX2–5, and GATA4) and the number of total cTnT-positive cells did not exhibit significant regulation, indicating that ERRα/γ are likely downstream of these cardiogenic factors (Figure 7E and Online Figure VIIB). In addition, most inflammatory markers observed in the pn-csERRα/γ KD heart were not induced in the ERRα/γ KO hiPSC-CMs including SOX9, toll-like receptors (except for TLR1), and chemokines (Figure 7E). The basis for the latter difference is unknown but could reflect the effects of myocardial immune response cells in vivo. Notably, the ERRα/γ KO hiPSC-CMs also expressed higher levels of several epicardial marker genes including WT1 and TBX18 along with TCF21 (Figure 7E).38 This latter finding is interesting given recent single cell RNA-seq evidence in hiPSC-CM demonstrating that the cardiac fibroblast lineage derives from epicardial origins.38
DISCUSSION
In this study, we sought to test the hypothesis that the PGC-1 transcriptional regulatory cascade, known to control mitochondrial biogenesis,6,7 is linked to other pathways involved in postnatal cardiac development. We focused on ERRα and ERRγ, two cardiac-enriched nuclear receptors downstream of PGC-1α. Cardiac-selective postnatal and prenatal Esrr gene targeting approaches, together with studies in hiPSC-CM demonstrated: 1) ERR signaling is not only necessary for postnatal mitochondrial maturation but also controls a broad array of adult gene cardiac myocyte maturation programs including those involved in contractile function and ion transport. As predicted, severe ERRα/γ deficiency during the postnatal period results in cardiac dysfunction; 2) ERRγ ChIP-seq experiments demonstrated that the coordinate regulation of both metabolic and structural genes by ERRs occurs, at least in part, through direct transcriptional control of genes in pathways across these various processes; 3) ERR signaling also serves as a cardiac myocyte differentiation factor during fetal development and in hiPSC-CM, and functions as a suppressor of many genes involved in early cardiac development; and 4) ERRs suppress non-cardiac genes during development including cardiac fibroblast lineage determination and myofibroblast activation factors. These findings expand the role of ERRs beyond regulators of mitochondrial biogenesis to the direct coordinate control of ATP consuming processes including genes involved in cardiac myocyte contractile function and ion homeostasis, and as suppressors of a subset of fetal and non-cardiac genes (Figure 8).
Figure 8. Activation of ERRα/γ is required for cardiomyocyte maturation.
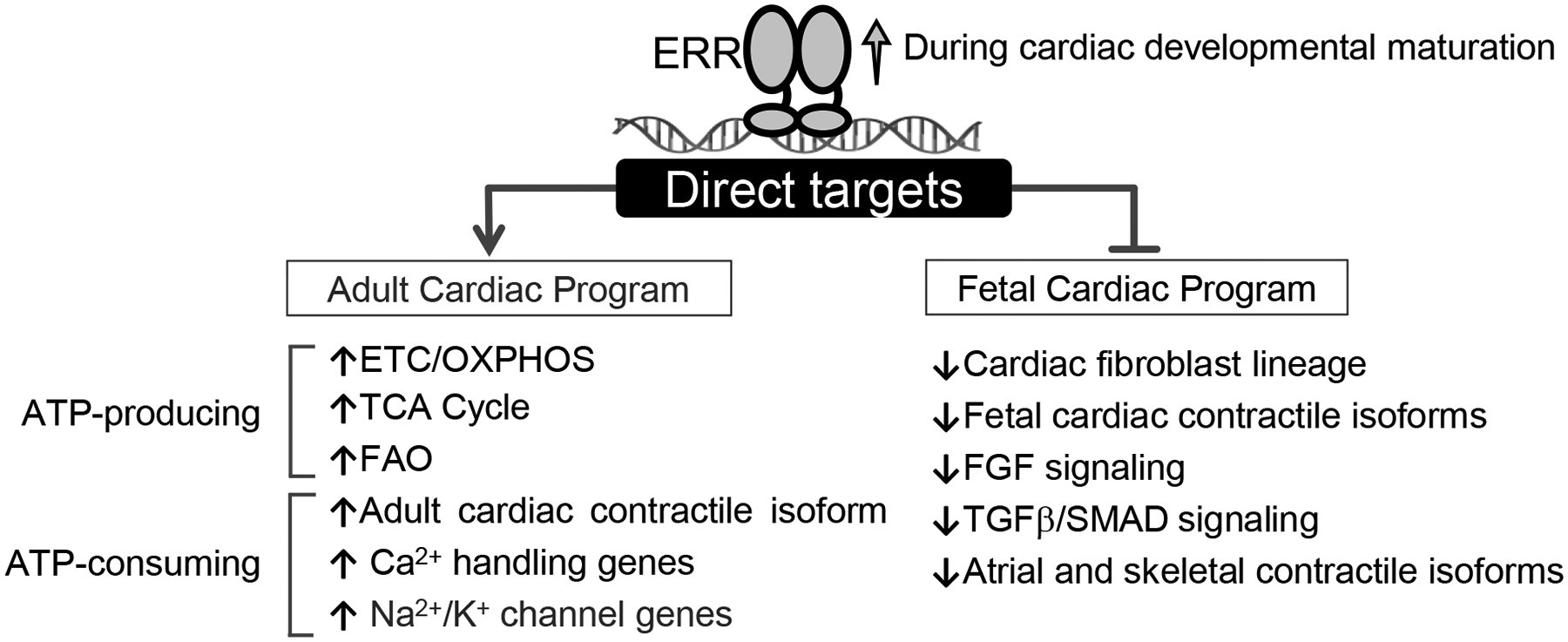
Schematic for proposed ERR functions as transcriptional activator and suppressor during cardiac developmental maturation.
It is well known that ERRγ and α regulate target genes involved in mitochondrial energy metabolism.22,23,39–41 Our results demonstrated the surprising finding that in addition to controlling transcription of genes involved in cardiac energy production, ERRγ serves as a direct transcriptional activator of a wide array of adult cardiac structural genes including those involved in contractility, excitation-contraction coupling, and ion transport. We speculate that this is a mechanism whereby ERR signaling coordinates the expression of genes involved in ATP generation with that of ATP-consuming processes. Consistent with this conclusion, ERRγ has been shown to coordinately regulate key metabolic and cell-specific functions in other tissues including mitochondrial respiratory function and angiogenesis in skeletal muscle,42–45 hippocampal metabolism and function in brain,46 functional maturation of pancreatic beta cells,47 and thermogenic machinery in brown adipose tissue.48,49 ERRs likely participate in a larger network that orchestrates postnatal cardiac myocyte differentiation. The transcriptional regulators and upstream signaling molecules that cooperate with and are upstream of PGC-1/ERR signaling involved in cardiac maturation remain unknown. It will be of interest to delineate the players involved in the complex transcriptional network that controls cardiac myocyte maturation. The ERRs provide a starting point for this goal.
Consistent with an arrest in cardiac maturation, mice with severe deficiency of ERRα and γ during the postnatal period developed cardiomyopathy. We and others have shown that single ERRα or ERRγ mouse gene knockout lines do not exhibit a baseline cardiac phenotype22,27 likely due to compensation given the overlap in target genes for these two nuclear receptors.23 One exception is a germline ERRγ knockout mouse line that exhibited lethality shortly after birth with evidence of persistent fetal fuel metabolism.24 This latter phenotype is quite similar to the perinatal lethal phenotype caused by PGC-1α/β deficiency due to an arrest in cardiac perinatal biogenesis.7 In this study, we sought to assess the function of ERR signaling in the postnatal and prenatal heart independent of the effects on perinatal mitochondrial biogenesis by employing cardiac-specific conditional gene targeting strategies. Our results indicate that the ERRs, likely in cooperation with the PGC-1 coactivators, play important roles in cardiac myocyte maturation during both the prenatal and postnatal periods in addition to the known function of regulating mitochondrial biogenesis and maturation. We speculate that the cardiomyopathy phenotypes caused by combined ERRα and ERRγ deficiency are due to a constellation of derangements caused by an arrest in cardiac myocyte maturation including mitochondrial dysfunction, contractile dysfunction and abnormal EC-coupling, features exhibited by ERRα/γ-deficient hiPSC-CM in this study.
Our results also identified interesting new roles for ERR signaling during fetal cardiac development. Whereas the hearts of fetal-csERRα/γ−/− mice did not exhibit gross morphologic abnormalities indicative of altered chamber formation or great vessel formation, disruption of ERR signaling during early stages of cardiac development imposed a substantial impact on the fetal cardiac myocyte differentiation process as reflected by mitochondrial abnormalities, myocardial wall thinning, and disorganized sarcomeres. Similar to the impact of ERRα/γ deficiency during the postnatal period, gene expression profiling of the fetal-csERRα/γ−/− heart demonstrated reduced expression of genes involved in mitochondrial metabolism and the adult contractile machinery. However, one of the most interesting findings was upregulation of many genes involved in early development including components of the Wnt,50,51 TGF-β,50,52 BMP,53 and FGF54 signaling pathways. Adult ventricular chamber-specific expression also exhibited persistent fetal patterns in the fetal-csERRα/γ−/− hearts as evidenced by increased ventricular expression of atrial myosin light chain gene isoforms at day E17.5. In addition, the fetal-csERRα/γ−/− heart exhibited inappropriate expression of non-cardiac lineage markers including fibroblast, skeletal muscle, smooth muscle and endothelial genes. We also found increased expression of a subset of cellular proliferation genes in the fetal-csERRα/γ−/−, including cell cycle activator (Ccnd2) recently implicated in myocyte proliferation and cardiac regeneration.55 Taken together, these results suggest that at early stages following cardiac myocyte determination, ERR signaling participates in placing a “brake” on early developmental and non-cardiac lineage programs while driving cardiac myocyte differentiation (Figure 8). This function appears to occur downstream of cardiogenic factors such as NKX2.5, GATA4, and MEF2C given that the expression of these key cardiac lineage determination genes was only minimally altered by ERR deficiency. The mechanism whereby ERRs can function as suppressors is unknown but the motif analysis from our ChiP-seq data (Online Figure VIC) suggests that distinct complexes may form with ERR on activated versus suppressed targets.
A surprising finding in the postnatal cardiac-specific ERR loss-of-function mouse lines was abnormal activation of a number of fibroblast and inflammatory genes. RNA-seq/ChIP-seq intersection data indicate that many cardiac fibroblast genes induced in pn-csERRα/γ KD hearts, including Postn and Col3a1, were directly suppressed by ERRγ. These findings confirm and extend the results of a recent study by the Liming Pei lab demonstrating increased expression of fibroblast markers in cardiac myocytes of P10 ERRα/γ-deficient hearts (germline targeted) using single nuclei RNA sequencing analysis.56 The finding of increased fibrosis in the LV of pn-csERRα/γ KD mice, indicative of myofibroblast activation, further corroborates the gene expression findings and suggests interesting cross-talk between the ERR-deficient myocytes and myofibroblasts. It is possible that the observed activation of fibroblast and inflammatory gene markers as well as the fibrotic response observed in the pn-csERRα/γ KD hearts, reflects a mild cardiomyopathic process caused by the energetic and contractile abnormalities related to ERRα/γ deficiency. However, several lines of evidence suggest the induction of the fibroblast lineage genes is a direct effect of the loss of ERR-mediated suppression in cardiac myocytes. First, fibroblast lineage markers (Tcf21, Postn, Ddr2, and Col1a1) were also upregulated in the fetal-csERRα/γ−/− hearts in the absence of inflammatory gene marker activation. Second, our ChIP-seq/RNA-seq studies demonstrated that many fibroblast genes in myocytes are direct ERR targets. Lastly, depletion of ERRα and ERRγ in hiPSC-CMs in culture resulted in robust induction of POSTN, COL3A1, and TCF21 indicating a cell autonomous effect. In addition and interestingly, several epicardial markers were also induced in the ERRα/γ KO hiPSC-CM including WT1, TBX18, along with TCF21 (Figure 7E). This latter finding is notable given the results of a recent study using hiPSC demonstrating that cardiac progenitors pass through the epicardial lineage en route to formation of cardiac fibroblasts.38 Accordingly, our results suggest that the suppressive effects of ERR signaling on cardiac fibroblast lineage determination occurs at or before the epicardial stage, perhaps serving a checkpoint function that directs cardiac progenitors to the cardiac myocyte differentiation pathway. In the adult heart, it is possible that myocyte ERR deficiency triggers a paracrine effect that cross-talks with cardiac fibroblast to elicit the fibrotic response. Interestingly, this type of cell-cell positive feedback loop has been described for cardiac myocyte TGFbeta signaling,57 which appears to be activated in ERR-deficient myocytes based on our gene expression and ChIP-seq data.38,56
Our work has several translational implications. During the development of heart failure, the heart is known to undergo a “fetal” shift of genes involved in many processes including mitochondrial fuel utilization and contractile proteins isoforms. The expression and activity of the PGC-1 transcriptional regulatory circuit, including ERR nuclear receptors, is downregulated during the development of heart failure.14,15,58 The results described here suggest that the fetal type of re-programming that occurs in the hypertrophied and failing heart may be related to reduced ERR signaling. Accordingly, it should be possible to manipulate ERR signaling in the failing heart to determine if the fetal re-programming is adaptive or maladaptive. In addition, the results shown here are relevant to the well-known lack of full differentiation of cardiac myocytes derived from stem cells when in culture conditions. This problem has hampered efforts to develop robust “disease-in-a-dish” experimental systems. Recent advances involving manipulation of hiPSC-CM culture conditions including addition of thyroid and glucocorticoid hormones59 and pacing protocols60 have improved the stage of differentiation of hiPSC-CM including the development of key maturation markers such as T-tubules. The effects of ERRγ shown here indicate that activation of ERR signaling should also be considered as a target pathway to enhance maturation of hiPSC-CM relevant to experimental systems.
Supplementary Material
NOVELTY AND SIGNIFICANCE.
What Is Known?
During postnatal development, cardiac mitochondria expand and mature into organelles that are capable of high capacity fuel combustion, with specialization for fatty acid oxidation.
The transcriptional coregulators PPARgamma coactivator 1 (PGC-1) α and β are necessary and sufficient to drive the postnatal mitochondrial biogenesis in heart.
Coincident with postnatal mitochondrial maturation, the heart undergoes growth and structural maturation that involves activation of adult contractile and ion transport gene regulatory programs.
What New Information Does This Article Contribute?
The orphan nuclear receptors estrogen-related receptors (ERRs) α and γ, known effectors of PGC-1α, are necessary for full maturation of both mitochondrial and structural processes (contractile function, Ca2+ handling, and ion transport) during postnatal cardiac development.
The coordinate regulation of metabolic and structural genes by ERRs occurs, in large part, through direct transcriptional control of genes in pathways across these various processes.
ERR signaling is also necessary for normal cardiac developmental maturation during fetal development including functioning as a suppressor of non-cardiac genes including the fibroblast lineage.
ERR signaling is necessary for normal metabolic and electrophysiological function in human induced pluripotent stem cell-derived cardiac myocytes (hiPSC-CMs).
ACKNOWLEDGMENTS
We thank Teresa C. Leone for insightful scientific discussions, critical review of the manuscript, and manuscript preparation. We are grateful to Dr. Johan Auwerx (École Polytechnique Fédérale de Lausanne) for generation and provision of the ERRα and ERRγ flox mice; Dr. Anastasia Kralli (Johns Hopkins Medical School) for our collaboration to generate the ERRγ antibody and helpful discussions regarding the ERR-targeted mouse lines and ERR biology. We would like to thank Dr. Laszlo Nagy at Johns Hopkins Medical School for providing assistance with the ChIP-sequencing protocol. We thank Peihong Ma for assistance with the animal studies and husbandry. We thank Kenneth C. Bedi for help with isolation of cardiomyocytes from mouse heart. We acknowledge the following Core Facilities at Sanford Burnham Prebys Medical Discovery Institute at Lake Nona (SBP-LN): Analytical Genomics Core in the Bioinformatics Core who did the RNA-seq analysis, Histology Core, and Metabolomics Core. We extend special thanks to Li Li in the Histology and Gene Expression Core and Dr. Swapnil Shewale in the Mouse Cardiovascular Phenotyping Core in the Cardiovascular Institute at the Perelman School of Medicine and Dr. Rachel Truitt in the iPSC Core Facility, University of Pennsylvania; the Electron Microscopy Resource Laboratory in the Department of Biochemistry and Biophysics at the University of Pennsylvania; Viral Vector Core (Diabetes Research Center (P30 DK019525) Y43), University of Pennsylvania; Pancreatic Islet Cell Biology Core, Institute for Diabetes, Obesity and Metabolism, University of Pennsylvania and the Metabolomics Core at the Translational Research Institute for Metabolism and Diabetes, Florida Hospital. The images of heart, fibroblast, and double stranded DNA in the Graphical Abstract and Figure 8 were obtained from © 2016 DBCLS TogoTV.
SOURCES OF FUNDING
This work was supported by NIH R01 HL058493 (D.P.K.), a postdoctoral fellowship from the American Heart Association #14POST20490309 (T.S.), and the NIH T32 HL00783 Training Grant (T.R.M.).
Nonstandard Abbreviations and Acronyms:
- BCAA
Branched-chain amino acid
- ChIP-seq
Chromatin immunoprecipitation with high throughput sequencing
- cs
Cardiac specific
- FAO
Fatty acid oxidation
- fetal-csERRγ−/−
Fetal ERRα/γ knockout mouse
- H&E
Hematoxylin and eosin
- hiPSC-CM
Human cardiac myocytes derived from induced pluripotent stem cells
- pn-csERRα/γ KD
Postnatal ERRα/γ-deficient mouse
- KO
Knockout
- KD
Knockdown
- LV
Left ventricular
- OXPHOS
Oxidative phosphorylation
- RNA-seq
RNA sequencing
Footnotes
REFERENCES
- 1.Galdos FX, Guo Y, Paige SL, VanDusen NJ, Wu SM and Pu WT. Cardiac Regeneration: Lessons From Development. Circ Res. 2017;120:941–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kannan S and Kwon C. Regulation of cardiomyocyte maturation during critical perinatal window. J Physiol. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Srivastava D and Olson EN. A genetic blueprint for cardiac development. Nature. 2000;407:221–6. [DOI] [PubMed] [Google Scholar]
- 4.Srivastava D Making or breaking the heart: from lineage determination to morphogenesis. Cell. 2006;126:1037–48. [DOI] [PubMed] [Google Scholar]
- 5.Sahara M, Santoro F and Chien KR. Programming and reprogramming a human heart cell. EMBO J. 2015;34:710–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lehman JJ, Barger PM, Kovacs A, Saffitz JE, Medeiros DM and Kelly DP. Peroxisome proliferator-activated receptor gamma coactivator-1 promotes cardiac mitochondrial biogenesis. J Clin Invest. 2000;106:847–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lai L, Leone TC, Zechner C, Schaeffer PJ, Kelly SM, Flanagan DP, Medeiros DM, Kovacs A and Kelly DP. Transcriptional coactivators PGC-1alpha and PGC-1beta control overlapping programs required for perinatal maturation of the heart. Genes Dev. 2008;22:1948–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Puigserver P, Wu Z, Park CW, Graves R, Wright M and Spiegelman BM. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell. 1998;92:829–39. [DOI] [PubMed] [Google Scholar]
- 9.Huss JM, Kopp RP and Kelly DP. Peroxisome proliferator-activated receptor coactivator-1alpha (PGC-1alpha) coactivates the cardiac-enriched nuclear receptors estrogen-related receptor-alpha and -gamma. Identification of novel leucine-rich interaction motif within PGC-1alpha. J Biol Chem. 2002;277:40265–74. [DOI] [PubMed] [Google Scholar]
- 10.Vega RB, Huss JM and Kelly DP. The coactivator PGC-1 cooperates with peroxisome proliferator-activated receptor alpha in transcriptional control of nuclear genes encoding mitochondrial fatty acid oxidation enzymes. Mol Cell Biol. 2000;20:1868–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wu Z, Puigserver P, Andersson U, Zhang C, Adelmant G, Mootha V, Troy A, Cinti S, Lowell B, Scarpulla RC and Spiegelman BM. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell. 1999;98:115–24. [DOI] [PubMed] [Google Scholar]
- 12.Scarpulla RC, Vega RB and Kelly DP. Transcriptional integration of mitochondrial biogenesis. Trends Endocrinol Metab. 2012;23:459–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Strauss AW and Kelly DP. The molecular basis of cardiomyopathies due to genetic deficiencies of mitochondrial proteins In: Haworth R. A. A. a. R. A., ed. Advances in Organ Biology Heart Metabolism in Failure: Haworth: JAI Press, Inc; 1998(4B): 323–340. [Google Scholar]
- 14.Finck BN, Lehman JJ, Barger PM and Kelly DP. Regulatory networks controlling mitochondrial energy production in the developing, hypertrophied, and diabetic heart. Cold Spring Harb Symp Quant Biol. 2002;67:371–82. [DOI] [PubMed] [Google Scholar]
- 15.Russell LK, Finck BN and Kelly DP. Mouse models of mitochondrial dysfunction and heart failure. J Mol Cell Cardiol. 2005;38:81–91. [DOI] [PubMed] [Google Scholar]
- 16.Huss JM and Kelly DP. Nuclear receptor signaling and cardiac energetics. Circ Res. 2004;95:568–78. [DOI] [PubMed] [Google Scholar]
- 17.Neubauer S The failing heart--an engine out of fuel. The New England journal of medicine. 2007;356:1140–51. [DOI] [PubMed] [Google Scholar]
- 18.Huss JM and Kelly DP. Mitochondrial energy metabolism in heart failure: a question of balance. J Clin Invest. 2005;115:547–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.de Jong H, Neal AC, Coleman RA and Lewin TM. Ontogeny of mRNA expression and activity of long-chain acyl-CoA synthetase (ACSL) isoforms in Mus musculus heart. Biochim Biophys Acta. 2007;1771:75–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bedada FB, Chan SS, Metzger SK, Zhang L, Zhang J, Garry DJ, Kamp TJ, Kyba M and Metzger JM. Acquisition of a quantitative, stoichiometrically conserved ratiometric marker of maturation status in stem cell-derived cardiac myocytes. Stem cell reports. 2014;3:594–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lehman JJ, Boudina S, Banke NH, Sambandam N, Han X, Young DM, Leone TC, Gross RW, Lewandowski ED, Abel ED and Kelly DP. The transcriptional coactivator PGC-1alpha is essential for maximal and efficient cardiac mitochondrial fatty acid oxidation and lipid homeostasis. American journal of physiology Heart and circulatory physiology. 2008;295:H185–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huss JM, Imahashi K, Dufour CR, Weinheimer CJ, Courtois M, Kovacs A, Giguere V, Murphy E and Kelly DP. The nuclear receptor ERRalpha is required for the bioenergetic and functional adaptation to cardiac pressure overload. Cell Metab. 2007;6:25–37. [DOI] [PubMed] [Google Scholar]
- 23.Dufour CR, Wilson BJ, Huss JM, Kelly DP, Alaynick WA, Downes M, Evans RM, Blanchette M and Giguere V. Genome-wide orchestration of cardiac functions by the orphan nuclear receptors ERRalpha and gamma. Cell Metab. 2007;5:345–56. [DOI] [PubMed] [Google Scholar]
- 24.Alaynick WA, Kondo RP, Xie W, He W, Dufour CR, Downes M, Jonker JW, Giles W, Naviaux RK, Giguere V and Evans RM. ERRgamma directs and maintains the transition to oxidative metabolism in the postnatal heart. Cell metabolism. 2007;6:13–24. [DOI] [PubMed] [Google Scholar]
- 25.Lin Z, von Gise A, Zhou P, Gu F, Ma Q, Jiang J, Yau AL, Buck JN, Gouin KA, van Gorp PR, Zhou B, Chen J, Seidman JG, Wang DZ and Pu WT. Cardiac-specific YAP activation improves cardiac function and survival in an experimental murine MI model. Circ Res. 2014;115:354–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Prendiville TW, Guo H, Lin Z, Zhou P, Stevens SM, He A, VanDusen N, Chen J, Zhong L, Wang DZ, Gao G and Pu WT. Novel Roles of GATA4/6 in the Postnatal Heart Identified through Temporally Controlled, Cardiomyocyte-Specific Gene Inactivation by Adeno-Associated Virus Delivery of Cre Recombinase. PLoS One. 2015;10:e0128105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang T, McDonald C, Petrenko NB, Leblanc M, Wang T, Giguere V, Evans RM, Patel VV and Pei L. Estrogen-related receptor alpha (ERRalpha) and ERRgamma are essential coordinators of cardiac metabolism and function. Mol Cell Biol. 2015;35:1281–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Akerberg BN, Gu F, VanDusen NJ, Zhang X, Dong R, Li K, Zhang B, Zhou B, Sethi I, Ma Q, Wasson L, Wen T, Liu J, Dong K, Conlon FL, Zhou J, Yuan GC, Zhou P and Pu WT. A reference map of murine cardiac transcription factor chromatin occupancy identifies dynamic and conserved enhancers. Nat Commun. 2019;10:4907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shore P and Sharrocks AD. The MADS-box family of transcription factors. Eur J Biochem. 1995;229:1–13. [DOI] [PubMed] [Google Scholar]
- 30.Guo Y, Jardin BD, Zhou P, Sethi I, Akerberg BN, Toepfer CN, Ai Y, Li Y, Ma Q, Guatimosim S, Hu Y, Varuzhanyan G, VanDusen NJ, Zhang D, Chan DC, Yuan GC, Seidman CE, Seidman JG and Pu WT. Hierarchical and stage-specific regulation of murine cardiomyocyte maturation by serum response factor. Nat Commun. 2018;9:3837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Estrella NL, Desjardins CA, Nocco SE, Clark AL, Maksimenko Y and Naya FJ. MEF2 transcription factors regulate distinct gene programs in mammalian skeletal muscle differentiation. J Biol Chem. 2015;290:1256–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hirose T, Apfel R, Pfahl M and Jetten AM. The orphan receptor TAK1 acts as a repressor of RAR-, RXR- and T3R-mediated signaling pathways. Biochem Biophys Res Commun. 1995;211:83–91. [DOI] [PubMed] [Google Scholar]
- 33.Yan ZH, Karam WG, Staudinger JL, Medvedev A, Ghanayem BI and Jetten AM. Regulation of peroxisome proliferator-activated receptor alpha-induced transactivation by the nuclear orphan receptor TAK1/TR4. J Biol Chem. 1998;273:10948–57. [DOI] [PubMed] [Google Scholar]
- 34.Hong H, Yang L and Stallcup MR. Hormone-independent transcriptional activation and coactivator binding by novel orphan nuclear receptor ERR3. J Biol Chem. 1999;274:22618–26. [DOI] [PubMed] [Google Scholar]
- 35.Stanley EG, Biben C, Elefanty A, Barnett L, Koentgen F, Robb L and Harvey RP. Efficient Cre-mediated deletion in cardiac progenitor cells conferred by a 3’UTR-ires-Cre allele of the homeobox gene Nkx2–5. Int J Dev Biol. 2002;46:431–9. [PubMed] [Google Scholar]
- 36.Acharya A, Baek ST, Huang G, Eskiocak B, Goetsch S, Sung CY, Banfi S, Sauer MF, Olsen GS, Duffield JS, Olson EN and Tallquist MD. The bHLH transcription factor Tcf21 is required for lineage-specific EMT of cardiac fibroblast progenitors. Development. 2012;139:2139–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Burridge PW, Matsa E, Shukla P, Lin ZC, Churko JM, Ebert AD, Lan F, Diecke S, Huber B, Mordwinkin NM, Plews JR, Abilez OJ, Cui B, Gold JD and Wu JC. Chemically defined generation of human cardiomyocytes. Nat Methods. 2014;11:855–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang H, Tian L, Shen M, Tu C, Wu H, Gu M, Paik DT and Wu JC. Generation of Quiescent Cardiac Fibroblasts From Human Induced Pluripotent Stem Cells for In Vitro Modeling of Cardiac Fibrosis. Circ Res. 2019;125:552–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huss JM, Torra IP, Staels B, Giguere V and Kelly DP. Estrogen-related receptor alpha directs peroxisome proliferator-activated receptor alpha signaling in the transcriptional control of energy metabolism in cardiac and skeletal muscle. Mol Cell Biol. 2004;24:9079–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schreiber SN, Emter R, Hock MB, Knutti D, Cardenas J, Podvinec M, Oakeley EJ and Kralli A. The estrogen-related receptor alpha (ERRalpha) functions in PPARgamma coactivator 1alpha (PGC-1alpha)-induced mitochondrial biogenesis. Proc Natl Acad Sci U S A. 2004;101:6472–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Giguere V Transcriptional control of energy homeostasis by the estrogen-related receptors. Endocr Rev. 2008;29:677–96. [DOI] [PubMed] [Google Scholar]
- 42.Arany Z, Foo SY, Ma Y, Ruas JL, Bommi-Reddy A, Girnun G, Cooper M, Laznik D, Chinsomboon J, Rangwala SM, Baek KH, Rosenzweig A and Spiegelman BM. HIF-independent regulation of VEGF and angiogenesis by the transcriptional coactivator PGC-1alpha. Nature. 2008;451:1008–12. [DOI] [PubMed] [Google Scholar]
- 43.Chinsomboon J, Ruas J, Gupta RK, Thom R, Shoag J, Rowe GC, Sawada N, Raghuram S and Arany Z. The transcriptional coactivator PGC-1alpha mediates exercise-induced angiogenesis in skeletal muscle. Proc Natl Acad Sci U S A. 2009;106:21401–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gan Z, Rumsey J, Hazen BC, Lai L, Leone TC, Vega RB, Xie H, Conley KE, Auwerx J, Smith SR, Olson EN, Kralli A and Kelly DP. Nuclear receptor/microRNA circuitry links muscle fiber type to energy metabolism. J Clin Invest. 2013;123:2564–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fan W, He N, Lin CS, Wei Z, Hah N, Waizenegger W, He MX, Liddle C, Yu RT, Atkins AR, Downes M and Evans RM. ERRgamma Promotes Angiogenesis, Mitochondrial Biogenesis, and Oxidative Remodeling in PGC1alpha/beta-Deficient Muscle. Cell Rep. 2018;22:2521–2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pei L, Mu Y, Leblanc M, Alaynick W, Barish GD, Pankratz M, Tseng TW, Kaufman S, Liddle C, Yu RT, Downes M, Pfaff SL, Auwerx J, Gage FH and Evans RM. Dependence of hippocampal function on ERRgamma-regulated mitochondrial metabolism. Cell Metab. 2015;21:628–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yoshihara E, Wei Z, Lin CS, Fang S, Ahmadian M, Kida Y, Tseng T, Dai Y, Yu RT, Liddle C, Atkins AR, Downes M and Evans RM. ERRgamma Is Required for the Metabolic Maturation of Therapeutically Functional Glucose-Responsive beta Cells. Cell Metab. 2016;23:622–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ahmadian M, Liu S, Reilly SM, Hah N, Fan W, Yoshihara E, Jha P, De Magalhaes Filho CD, Jacinto S, Gomez AV, Dai Y, Yu RT, Liddle C, Atkins AR, Auwerx J, Saltiel AR, Downes M and Evans RM. ERRgamma Preserves Brown Fat Innate Thermogenic Activity. Cell Rep. 2018;22:2849–2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Brown EL, Hazen BC, Eury E, Wattez JS, Gantner ML, Albert V, Chau S, Sanchez-Alavez M, Conti B and Kralli A. Estrogen-Related Receptors Mediate the Adaptive Response of Brown Adipose Tissue to Adrenergic Stimulation. iScience. 2018;2:221–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Olson EN and Schneider MD. Sizing up the heart: development redux in disease. Genes Dev. 2003;17:1937–56. [DOI] [PubMed] [Google Scholar]
- 51.Estaras C, Hsu HT, Huang L and Jones KA. YAP repression of the WNT3 gene controls hESC differentiation along the cardiac mesoderm lineage. Genes Dev. 2017;31:2250–2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Azhar M, Schultz Jel J, Grupp I, Dorn GW, Meneton P, Molin DG, Gittenberger-de Groot AC and Doetschman T. Transforming growth factor beta in cardiovascular development and function. Cytokine Growth Factor Rev. 2003;14:391–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen H, Shi S, Acosta L, Li W, Lu J, Bao S, Chen Z, Yang Z, Schneider MD, Chien KR, Conway SJ, Yoder MC, Haneline LS, Franco D and Shou W. BMP10 is essential for maintaining cardiac growth during murine cardiogenesis. Development. 2004;131:2219–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dell’Era P, Ronca R, Coco L, Nicoli S, Metra M and Presta M. Fibroblast growth factor receptor-1 is essential for in vitro cardiomyocyte development. Circ Res. 2003;93:414–20. [DOI] [PubMed] [Google Scholar]
- 55.Zhu W, Zhao M, Mattapally S, Chen S and Zhang J. CCND2 Overexpression Enhances the Regenerative Potency of Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes: Remuscularization of Injured Ventricle. Circ Res. 2018;122:88–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hu P, Liu J, Zhao J, Wilkins BJ, Lupino K, Wu H and Pei L. Single-nucleus transcriptomic survey of cell diversity and functional maturation in postnatal mammalian hearts. Genes Dev. 2018;32:1344–1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Koitabashi N, Danner T, Zaiman AL, Pinto YM, Rowell J, Mankowski J, Zhang D, Nakamura T, Takimoto E and Kass DA. Pivotal role of cardiomyocyte TGF-beta signaling in the murine pathological response to sustained pressure overload. J Clin Invest. 2011;121:2301–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Finck BN and Kelly DP. PGC-1 coactivators: inducible regulators of energy metabolism in health and disease. J Clin Invest. 2006;116:615–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Parikh SS, Blackwell DJ, Gomez-Hurtado N, Frisk M, Wang L, Kim K, Dahl CP, Fiane A, Tonnessen T, Kryshtal DO, Louch WE and Knollmann BC. Thyroid and Glucocorticoid Hormones Promote Functional T-Tubule Development in Human-Induced Pluripotent Stem Cell-Derived Cardiomyocytes. Circ Res. 2017;121:1323–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ronaldson-Bouchard K, Ma SP, Yeager K, Chen T, Song L, Sirabella D, Morikawa K, Teles D, Yazawa M and Vunjak-Novakovic G. Advanced maturation of human cardiac tissue grown from pluripotent stem cells. Nature. 2018;556:239–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Burridge PW, Holmstrom A and Wu JC. Chemically Defined Culture and Cardiomyocyte Differentiation of Human Pluripotent Stem Cells. Curr Protoc Hum Genet. 2015;87:21 31–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA and Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013;8:2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Miklas JW, Clark E, Levy S, Detraux D, Leonard A, Beussman K, Showalter MR, Smith AT, Hofsteen P, Yang X, Macadangdang J, Manninen T, Raftery D, Madan A, Suomalainen A, Kim DH, Murry CE, Fiehn O, Sniadecki NJ, Wang Y and Ruohola-Baker H. TFPa/HADHA is required for fatty acid beta-oxidation and cardiolipin re-modeling in human cardiomyocytes. Nat Commun. 2019;10:4671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Xiong Y, Bedi K, Berritt S, Attipoe BK, Brooks TG, Wang K, Margulies KB and Field J. Targeting MRTF/SRF in CAP2-dependent dilated cardiomyopathy delays disease onset. JCI Insight. 2019;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Judd J, Lovas J and Huang GN. Isolation, Culture and Transduction of Adult Mouse Cardiomyocytes. J Vis Exp. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mogen AB, Carroll RK, James KL, Lima G, Silva D, Culver JA, Petucci C, Shaw LN and Rice KC. Staphylococcus aureus nitric oxide synthase (saNOS) modulates aerobic respiratory metabolism and cell physiology. Mol Microbiol. 2017;105:139–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Horton JL, Martin OJ, Lai L, Riley NM, Richards AL, Vega RB, Leone TC, Pagliarini DJ, Muoio DM, Bedi KC Jr., Margulies KB, Coon JJ and Kelly DP. Mitochondrial protein hyperacetylation in the failing heart. JCI Insight. 2016;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M and Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Robinson MD, McCarthy DJ and Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Daniel B, Nagy G, Hah N, Horvath A, Czimmerer Z, Poliska S, Gyuris T, Keirsse J, Gysemans C, Van Ginderachter JA, Balint BL, Evans RM, Barta E and Nagy L. The active enhancer network operated by liganded RXR supports angiogenic activity in macrophages. Genes Dev. 2014;28:1562–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Langmead B and Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H and Glass CK. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell. 2010;38:576–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Thorvaldsdottir H, Robinson JT and Mesirov JP. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform. 2013;14:178–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Coordinators NR. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2016;44:D7–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.