

Abstract
The pathophysiology and treatment of depression has been the focus of intense research and while there is much that remains unknown, modern neurobiological approaches are making progress. This work demonstrates that stress and depression are associated with atrophy of neurons and reduced synaptic connectivity in brain regions such as the hippocampus and prefrontal cortex that contribute to depressive behaviors, and conversely that antidepressant treatment can reverse these deficits. The role of neurotrophic factors, particularly brain derived neurotrophic factor (BDNF) have been of particular interest as these factors play a key role in activity dependent regulation of synaptic plasticity. Here we review the literature demonstrating that exposure to stress and depression decreases BDNF expression in the hippocampus and PFC and conversely that antidepressant treatment can up-regulate BDNF in the adult brain and reverse the effects of stress. We then focus on rapid acting antidepressants, particularly the NMDA receptor antagonist ketamine, which produces rapid synaptic and antidepressant behavioral actions that are dependent on activity dependent release of BDNF. This rapid release of BDNF differs from typical monoaminergic agents that require chronic administration to produce a slow induction of BDNF expression, consistent with the time lag for the therapeutic action of these agents. We review evidence that other classes of rapid acting agents also require BDNF release, demonstrating that this is a common, convergent downstream mechanism. Finally, we discuss evidence that the actions of ketamine are also dependent on another growth factor, vascular endothelial growth factor (VEGF) and its complex interplay with BDNF.
Keywords: ketamine, synaptic plasticity, stress, neurotrophic factor
Graphical Abstract
Chronic stress decreases the expression of BDNF and causes atrophy of neurons in cortical and limbic brain regions.
Rapid acting antidepressants, notably the NMDA receptor antagonist ketamine, cause activity dependent release of BDNF that contributes to rapid increases in synapse number and function.
Typical monoaminergic antidepressants cause a delayed increase in BDNF expression but not activity dependent release.
Introduction
Despite the enormous personal suffering, risk of suicide, and economic consequences, our ability to diagnose and successfully treat depression has significant limitations (Flint & Kendler, 2014). Depression is a heterogeneous disorder with two-fold higher rates in women compared to men and a relatively low, 37 percent rate of heritability (Sullivan et al., 2018). These factors likely impact the difficulties in treating depression as current medications, comprised largely of monoaminergic agents, have a slow response rate of weeks to months and are only effective in approximately two thirds of patients (Trivedi et al., 2006). These limitations highlight the discovery that ketamine, an NMDA receptor (NMDAR) antagonist, produces rapid antidepressant actions after a single dose, even in patients considered treatment resistant (Berman et al., 2000; Zarate et al., 2006; Krystal et al., 2019). The discovery of an agent that produces rapid therapeutic actions by a mechanism completely different from currently available monoaminergic agents represents one of the biggest breakthroughs for the treatment of depression in over sixty years.
The (S)-enantiomer of ketamine referred to as esketamine was recently approved in the form of a nasal application for treatment resistant depression. Although esketamine as well as ketamine represent profound, significant alternatives for depressed patients who do not respond to typical antidepressants, these drugs have an undesirable side effect profile, including dissociative and psychotomimetic effects (Krystal et al., 2019). This has prompted efforts to identify the neurobiological mechanisms underlying the antidepressant actions of ketamine and thereby inform development of novel, ketamine-like agents with fewer side effects. Initial studies demonstrated that ketamine rapidly increases synaptic number and function in the medial prefrontal cortex (mPFC), and reverses the synaptic deficits caused by chronic stress exposure (Li et al., 2010; Li et al., 2011; Duman et al., 2016). These effects could contribute to reversal of the synaptic deficits and reduced connectivity associated with depression (Drevets et al., 2008; MacQueen & Frodl, 2011; Kang et al., 2012). These findings as well as evidence from studies of typical antidepressants have led to studies of neurotrophic factors and related signaling pathways that are critical for synaptic alterations.
The focus of the current review is to discuss evidence for a role of brain derived neurotrophic factor (BDNF) in the pathophysiology and treatment of depression. A brief description of BDNF is provided, and then a review of the preclinical and clinical literature demonstrating a role for BDNF in depression is presented. This includes evidence that stress and depression are associated with decreased expression of BDNF in limbic and cerebral cortical brain regions associated with depression, and conversely that antidepressant treatments increase the expression of BDNF. Particular attention is given to ketamine, and other classes of rapid acting antidepressants, and evidence that these agents cause activity dependent release of BDNF, which is thought to underlie their rapid synaptic and antidepressant behavioral responses. This review will also discuss recent evidence that the rapid actions of ketamine involve activity dependent release of vascular endothelial growth factor (VEGF) and its interactions with BDNF signaling.
Neurobiology of BDNF
BDNF is the most highly expressed member of the nerve growth factor (NGF) family that also includes NGF, neurotrophin-3, and neurotrophin-4 (Castren & Kojima, 2017). The neurotrophins were first identified as critical regulators of cell proliferation, migration, maturation, and survival during development, but are also expressed in the adult brain where they are involved in synaptic plasticity, neuronal function and survival (Martinowich et al., 2007). BDNF is multi-exon gene with multiple 5’ promoters that control constitutive and activity regulated expression of BDNF (Adachi et al., 2014; Castren & Kojima, 2017). BDNF is constitutively expressed and released but is also controlled by neuronal activity largely via Ca2+ signaling and activation of Ca2+/ and cAMP response elements (Bjorkholm & Monteggia, 2016). The initial transcript is translated to pre-proBDNF which then undergoes further processing and cleavage to proBDNF and then to mature BDNF, which bind to the p75 neurotrophin receptor (p75NTR) and the tyrosine kinase receptor TrkB, respectively, to activate different downstream signaling pathways (Castren & Kojima, 2017)(Figure 1). Both are packaged into vesicles and undergo trafficking to dendrite as well as axon terminals where mature BDNF undergoes activity dependent release while proBDNF undergoes low levels of constitutive release (Figure 1).
Figure 1. Processing of BDNF and receptor coupled signaling pathways.

BDNF gene expression is controlled by multiple signaling pathways included neuronal activity. BDNF transcripts can be translated in the soma or transported to other cellular compartments, notably dendrite as well as axon terminals. The transcripts are translated to pre-proBDNF which undergoes processing and cleavage to proBDNF, which is further processed by to mature BDNF. Mature BDNF is associated with synaptic plasticity and undergoes activity dependent release, while proBDNF undergoes low levels of constitutive released. The BDNF prodomain contains a common single nucleotide polymorphism, BDNF Val66Met. The Met allele impairs the trafficking and processing of proBDNF and therefore reduces activity dependent release of mature BDNF. Mature BDNF binds to the TrkB and activates downstream signaling pathways associated with synaptic plasticity, synapse formation, neuronal differentiation and survival. The proBDNF isoform binds to the p75 neurotrophin receptor (p75NTR) and activates a different set of downstream signaling pathways that are linked with disruption of synaptic plasticity, long-term depression, synaptic pruning, decreased growth and apoptosis.
The BDNF prodomain contains a common single nucleotide polymorphism, Val66Met expressed in approximately 30 percent of Caucasians, although higher levels, approximately 80 percent has been reported in a Chinese population (Dincheva et al., 2016; Su et al., 2017). The BDNF Val allele is required for processing of proBDNF to mature BDNF and is therefore required for activity dependent release of BDNF (Dincheva et al., 2016). The Met allele blocks the processing and activity dependent release of proBDNF and has been linked to cognitive deficits and decreased hippocampal volume, although a meta-analysis demonstrates that the effect size on hippocampal volume is smaller than originally reported (Molendijk et al., 2012; Edelmann et al., 2014; Mizui et al., 2016; Castren & Kojima, 2017). Activity dependent release of BDNF is required for long-term, protein synthesis dependent synaptic potentiation, and studies of the Met allele provide further evidence for BDNF release (Edelmann et al., 2014; Mizui et al., 2016). The Met allele is associated with reduced executive function and increased vulnerability to depression in patients exposed to early life stress (Kaufman et al., 2006; Kim et al., 2007; Gatt et al., 2009; Frodl & O’Keane, 2013). Met carriers also display increased anxiety symptoms, and PTSD patients who are Met carriers are less responsive to cognitive behavioral therapies (Dincheva et al., 2016). Together these studies indicate that carriers of the Met allele may have synaptic deficits in the hippocampus and PFC that contribute to decreased neuroplasticity and result in reduced coping mechanisms.
Role of BDNF in stress and depression
The impact of BDNF on synapse formation and synaptic plasticity, as well as neuronal survival and function led to studies of BDNF in the atrophy of neurons caused by chronic stress exposure. Chronic stress exposure causes atrophy of hippocampal CA3 pyramidal neurons as well as PFC layer 2/3 and layer 5 pyramidal neurons (Liu & Aghajanian, 2008; Popoli et al., 2011; McEwen et al., 2012; Morrison & Baxter, 2012). Early studies reported that exposure to different types of chronic stress, including unpredictable and social defeat also decreased the expression of BDNF in the hippocampus and PFC (Smith, 1995a; Duman & Monteggia, 2006; Krishnan & Nestler, 2008; 2010). In addition, BDNF expression is regulated by repeated administration of adrenal glucocorticoids (Adachi et al., 2014) and also undergoes regulation by estrogen and progesterone (Bath et al., 2013). In contrast, there is evidence that that contract stress increases BDNF expression and signaling in other brain regions, notably the nucleus accumbens and amygdala where increased function could contribute to synaptic plasticity that underlies depressive symptoms (Krishnan & Nestler, 2008; Roozendaal et al., 2009; Krishnan & Nestler, 2010). There has also been interest in epigenetic alterations that could underlie long-lasting changes in BDNF expression (Krishnan & Nestler, 2010; Sun et al., 2013).
Studies of BDNF deletion mutant mice, either constitutive or conditional forebrain deletion mutants, do not show clear depressive-like phenotypes, as would be expected if BDNF was required for normal emotional behaviors (Duman & Monteggia, 2006; Autry & Monteggia, 2012). In addition, mice with a knock-in of the BDNF Val66Met polymorphism, which decreases the transport of BDNF transcripts to dendrite compartments, do not display overt depressive or anxiety like behaviors (Chen et al., 2006). Interesting, these mice display a reduction of dendrite length and branching, and decreased hippocampus spine-synapse number and function in the hippocampus and/or PFC (Chen et al., 2006; Liu et al., 2012). The lack of effect of global or forebrain BDNF deletion or Met knockin could be due to opposing effects of BDNF in different brain regions (i.e., PFC and hippocampus vs. depressive effects in mesolimbic dopamine system). In support of this possibility, selective knockdown of BDNF in the hippocampus is reported to be sufficient to cause depressive behaviors (Taliaz et al., 2010). Another possibility is that current rodent models have limited validity for studies of depression and are insensitive to relevant behavioral phenotypes in rodents.
The impact and relevance of these preclinical studies is supported by postmortem studies demonstrating that levels of BDNF are decreased in the cerebral cortex of depressed, as well as suicide subjects (Duman & Monteggia, 2006; Dwivedi, 2009; Duman et al., 2016). Levels of TrkB and signaling pathways downstream of BDNF-TrkB (e.g., ERK and Akt) are also decreased in suicide subjects (Castren & Kojima, 2017). There is also evidence that levels of p75NTR are increased in the PFC of suicide subjects, which could contribute to neuronal atrophy (Castren & Kojima, 2017). In addition, levels of MAP kinase phosphatase 1, a negative regulator of TrkB-ERK signaling, are increased by stress and in depressed subjects, and viral mediated expression of MAP kinase phosphatase 1 in hippocampus is sufficient to cause depressive behaviors (Duric et al., 2010). These studies are consistent with the possibility that decreased BDNF-TrkB signaling contributes to reduced volume of PFC and hippocampus in depressed patients (Drevets et al., 2008; MacQueen & Frodl, 2011), as well as synaptic loss in humans (Kang et al., 2012).
Role of BDNF in response to typical antidepressants
As studies were being carried out to test the effects of stress and depression, others were beginning to examine the role of BDNF in the actions of antidepressant treatments. This research found that in contrast to stress, administration of antidepressant drugs increased the expression of BDNF in the hippocampus and PFC (Nibuya et al., 1995; Nibuya et al., 1996; Duman & Monteggia, 2006; Castren & Kojima, 2017). The up-regulation of BDNF was observed after chronic, but not acute antidepressant administration, consistent with the time course for the therapeutic actions of these agents (Duman & Monteggia, 2006; Bjorkholm & Monteggia, 2016; Castren & Kojima, 2017). Increased expression of BDNF was observed with different classes of antidepressants, including 5-HT and norepinephrine selective reuptake inhibitors, tricyclic antidepressants, and monoamine oxidase inhibitors, as well as electroconvulsive seizure (ECS). Only administration of ECS, which causes neuronal depolarization rapidly increased BDNF expression in the hippocampus and PFC (Nibuya et al., 1995). A role for BDNF in antidepressant induced synaptic plasticity is supported by studies demonstrating that chronic fluoxetine administration reinstates ocular dominance plasticity in the visual cortex and extinction learning in adult mice and that these effects require BDNF (Maya Vetencourt et al., 2008; Karpova et al., 2011).
The functional impact of increased BDNF expression was further examined using a number of different approaches. Infusion of BDNF into the hippocampus or PFC is sufficient to produce an antidepressant response, even after a single administration (Shirayama, 2002; Duman & Monteggia, 2006; Deyama et al., 2019a). However, as discussed above BDNF produces a depressive phenotype when infused into the mesolimbic dopamine system (Krishnan & Nestler, 2008). The role of BDNF in the actions of typical antidepressants has also been tested in BDNF mutant mice. These studies demonstrate that constitutive heterozygous deletion (homozygous deletion is lethal) or conditional forebrain deletion of BDNF blocks the antidepressant actions of typical antidepressant treatments (Duman & Monteggia, 2006; Bjorkholm & Monteggia, 2016; Castren & Kojima, 2017). In addition, mice with a knockin of the BDNF Met allele are nonresponsive to typical antidepressants such as fluoxetine (Chen et al., 2006; Dincheva et al., 2016).
Together the BDNF infusion and deletion studies demonstrate that BDNF is sufficient and necessary for the behavioral actions of typical antidepressant treatments. The delay in up-regulation of BDNF as well as the therapeutic actions of antidepressants also supports a role for BDNF in depression. The mechanisms underlying the delayed increase in BDNF are not clear but could be due to the delayed increase in intracellular signaling pathways that influence BDNF expression (Duman & Monteggia, 2006; Krishnan & Nestler, 2010; Castren & Kojima, 2017).
Role of BDNF in the response to rapid acting antidepressants
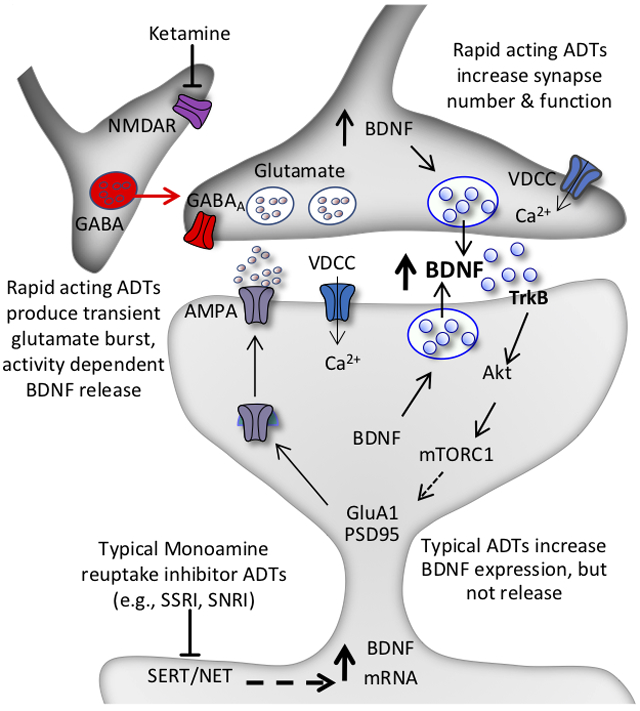
Studies of BDNF in the in the treatment of depression have been extended to rapid acting antidepressants. Initial studies reported that levels of BDNF were rapidly increased in the hippocampus after a single dose of ketamine, in contrast to the requirement for chronic administration of a typical monoaminergic antidepressant (Autry et al., 2011). Another study by this group replicated this effect and further reported that memantine, a structurally different NMDAR antagonist that does not produce rapid antidepressant therapeutic effects, does not increase BDNF levels (Gideons et al., 2014). Ketamine also increases the phosphorylated and activated form of TrkB (Kohtala et al., 2019), and racemate ketamine as well as both the (R)- and (S)-stereoisomers of ketamine reverse the deficit in BDNF and pTrkB in PFC and/or hippocampus of rodents exposed to social defeat stress or chronic unpredictable stress (Yang et al., 2015; Zhang et al., 2015; Liu et al., 2016; Sun et al., 2016). Moreover, the rapid antidepressant behavioral actions of ketamine are blocked in inducible BDNF deletion mutant mice (Autry et al., 2011). Further evidence for a role of BDNF in the actions of ketamine were provided by studies in mice with a knockin of the BDNF Val66Met polymorphism, the human variant that blocks the processing and activity dependent release of BDNF (Figure 1) (Dincheva et al., 2016). These studies demonstrate that the antidepressant behavioral actions of ketamine, as well as the increase in number and function of synapses in the mPFC, are blocked in BDNF Met mice (Liu et al., 2012). These results suggest that the rapid synaptic and behavioral actions of ketamine require activity dependent release of BDNF, not just increased expression (Duman et al., 2016; Duman et al., 2019) (Figure 2).
Figure 2. Model for the loss of spine synapses in stress and depression: rapid reversal by ketamine and comparison with typical monoamine reuptake inhibitor antidepressants.

Synaptic number and function in the PFC and hippocampus are maintained by homeostatic mechanisms, and proper control of synaptic connectivity in these regions is required for control of mood as well as other cortical functions. This includes activity dependent regulation of synaptic proteins, such as GluA1, PSD95, and synapsin 1. Chronic stress exposure (left) decreases the number and function of spine synapses in the PFC and hippocampus, in part via decreased BDNF expression, and decreased TrkB-mTORC1 signaling. Increased expression of a negative regulator of mTORC1 signaling, regulated in DNA damage and repair (REDD1) contributes to this effect. Antidepressant treatment (right) increases BDNF and reverses the effects of stress and depression. The NMDA receptor antagonist ketamine causes a rapid burst of glutamate that causes activity dependent release of BDNF resulting in rapid induction of synapse number and function via stimulation of TrkB-mTORC1 signaling and increased translation of synaptic proteins including GluA1. In contrast, typical antidepressants including the monoamine reuptake inhibitors (e.g., SSRI, SNRI) produce a slow induction of BDNF expression and constitutive release over several weeks of treatment, which contributes to slow adaptive changes that reverse the effects of stress and produce antidepressant responses. There is no evidence that typical antidepressants cause activity dependent release of BDNF, which is necessary for the rapid antidepressant actions of ketamine. Depression as well as relapse that occurs after 7 to 10 days in patients treated with ketamine could result from environmental factors such as sustained or uncontrollable stress or other susceptibility factors that decrease the stability of new synaptic connections.
To further test this hypothesis, studies were conducted to block the actions of extracellular BDNF by infusing a function blocking anti-BDNF antibody directly into the mPFC. Previous studies have demonstrated that the mPFC is necessary and sufficient for the actions of ketamine, providing the rationale for targeting this brain region (Fuchikami et al., 2015). The results demonstrate that infusion of an anti-BDNF antibody into the mPFC completely blocks the rapid antidepressant actions of a single dose of ketamine in several behavioral models (Lepack et al., 2014). Studies were also conducted to directly measure the release of BDNF, although this is problematic as in vivo microdialysis of large proteins such as BDNF are technically difficult. To address this issue we turned to primary cell cultures studies, and found that low concentrations of ketamine increase the release of BDNF into the culture media (Lepack et al., 2014; Lepack et al., 2016)(see below for further discussion of primary culture studies). Together these studies demonstrate that ketamine causes activity dependent release of BDNF and that extracellular BDNF is required for the rapid synaptic and antidepressant behavioral actions of ketamine. This differs from typical monoaminergic agents that slowly increase the expression of BDNF, but not the release of BDNF (Figure 2).
The role of BDNF release in the rapid therapeutic actions of ketamine in depressed patients is more difficult to address as it is currently not possible to measure BDNF in the brains of patients. There is a report that ketamine increases levels of BDNF in the blood of depressed patients and that levels are correlated with treatment response (Laje et al., 2012). Another approach is to examine patients carrying the BDNF Val66Met polymorphism as we would predict that Met carriers would have a decreased response to ketamine. An early study comprised primarily of Caucasian patients reported that BDNF Met carriers displayed a fifty percent reduction in their response to ketamine (Laje et al., 2012). Most of these patients were heterozygous Met carriers due to the relatively low prevalence of homozygous carriers in Caucasians. A more recent study in a Chinese population reported no effect of the BDNF Met allele on the response to ketamine (Su et al., 2017), although reanalysis of this data found that the BDNF Met allele is associated with a reduction in the sensitivity of the anti-suicidal actions of ketamine (i.e., 0.5 but not 0.2 mg/kg ketamine is effective) (Chen et al., 2019). These studies suggest a difference in the function of the BDNF Met allele depending on race, an/or that other polymorphisms interact in a race dependent manner to influence the ketamine response.
Role of BDNF in the actions of other rapid acting agents
Scopolamine: Acetylcholine Muscarinic Receptor Antagonist
Identification of other rapid acting antidepressants has been a major focus of the field to address the side effect profile of ketamine. Early studies of the acetylcholine muscarinic receptor antagonist scopolamine also demonstrate rapid antidepressant actions after a single dose (Furey & Drevets, 2006; Drevets & Furey, 2010), although a recent study reports that scopolamine is less effective for severe, treatment resistant depression (Park et al., 2019). Rodent studies demonstrate that scopolamine has effects that are convergent with ketamine, including increased mTORC1 signaling, and increased number and function of synapses in the mPFC (Voleti et al., 2013). In addition, the rapid antidepressant behavioral actions of scopolamine are blocked in BDNF Met knockin mice and by infusion of an anti-BDNF neutralizing antibody into the mPFC (Ghosal et al., 2018). These findings indicate that the actions of scopolamine, like ketamine, require activity dependent release of BDNF into the mPFC (Ghosal et al., 2018) (Figure 2).
Rapastinel: NMDA receptor positive allosteric modulator
Another rapid acting agent is rapastinel, an NMDAR positive allosteric modulator (Burgdorf et al., 2013; Burgdorf et al., 2015). Phase 2 clinical trials reported that a single dose of rapastinel produces rapid antidepressant actions (Preskorn et al., 2008), although this has not held up in larger phase 3 clinical trials possibly due to the dosing schedule. In rodent studies rapastinel produces rapid antidepressant behavioral actions, increases mTORC1 signaling, and increases synaptic number and function in the mPFC (Burgdorf et al., 2013; Burgdorf et al., 2015; Liu et al., 2016; Kato et al., 2018). Moreover, the rapid synaptic and antidepressant behavioral actions of rapastinel are blocked in BDNF Met mice and by infusion of an anti-BDNF neutralizing antibody into the mPFC (Kato et al., 2018). This study also shows that rapastinel or ketamine increase the phosphorylated and activated form of TrkB in the mPFC and that infusion of a TrkB antagonist into the mPFC blocks the rapid antidepressant actions of rapastinel (Kato et al., 2018). Finally, the results show that infusion of an inhibitor of the Rho GTPase Rac1, which contributes to BDNF mediated synaptic plasticity (Harward et al., 2016; Hedrick et al., 2016) blocks the antidepressant actions of rapastinel (Kato et al., 2018). Another study of social defeat stress has reported that rapastinel does not reverse the BDNF and pTrkB deficits in this model of stress (Yang et al., 2016).
(2R,6R)-hydroxynorketamine (HNK): ketamine metabolite with an unidentified binding site
Due in part to the long-lasting actions of ketamine there have also been studies of metabolites that could be responsible for its long-lasting, sustained effects. One of particular interest is (2R,6R)-HNK. Gould and colleagues have reported that a single dose of (2R,6R)-HNK produces rapid antidepressant behavioral actions and increases levels of BDNF in the hippocampus (Zanos et al., 2016). Importantly, (2R,6R)-HNK has very low affinity for the NMDAR channel and does not have the side effect profile of ketamine in rodent models (Zanos et al., 2016; Lumsden et al., 2019), although there is evidence that high concentrations of (2R,6R)-HNK produce effects similar to ketamine in hippocampal slices (Suzuki et al., 2017). Rather (2R,6R)-HNK is thought to act via an mGlu2 receptor-like presynaptic mechanism to increase glutamate release (Riggs et al., 2019; Zanos et al., 2019). Regardless of the initial target the induction of BDNF indicates that the actions of (2R,6R)-HNK converge with the downstream effects of ketamine. This possibility is supported by evidence that the actions of (2R,6R)-HNK are blocked in BDNF Met mice and by infusion of an anti-BDNF neutralizing antibody into the mPFC (Fukumoto et al., 2019). There have also been preliminary studies of another metabolite, (S)-norketamine reporting that it reverses the deficits of BDNF and phosphor-TrkB in a social defeat model, and that infusion of a TrkB antagonist (ANA-12) blocks the antidepressant actions of (S)-norketamine (Yang et al., 2018).
Metabotropic GluR2/3 receptor antagonists
Studies of glutamatergic agents have been extended to mGluR2/3 receptor antagonists, as blockade of mGluR2/3 autoreceptors represents another approach to increase glutamate transmission. Several studies have reported that mGluR2/3 receptor antagonists, including LY341495 and MGS0039 produce rapid antidepressant actions in rodent models (Koike et al., 2011b; a; Dwyer et al., 2012; Witkin et al., 2016; Chaki, 2017). Moreover, MGS0039 reverses the deficit in BDNF and pTrkB levels in the PFC of mice exposed to social defeat stress (Dong et al., 2017). The antidepressant actions of LY341495 are blocked by pretreatment with the nonselective Trk receptor antagonist K252a (Koike et al., 2013), and another study has reported that enhancement of the antidepressant actions of ketamine by LY341495 are blocked by a selective TrkB antagonist ANA-12 (Palucha-Poniewiera et al., 2019). Primary neuronal culture studies also demonstrate that LY341495 increases the release of BDNF (Lepack et al., 2016). Further studies are needed to determine if the antidepressant actions of mGluR2 antagonists require BDNF release in the mPFC but the evidence to date is consistent with this hypothesis.
Other rapid acting agents
There has also been interest in other classes of anesthetics, including isoflurane and nitrous oxide and these agents are reported to produce rapid antidepressant actions similar to ketamine phosphor-TrkB (Antila et al., 2017; Kohtala et al., 2019). Moreover, these effects are associated with increased levels of the phosphorylated and activated form of TrkB. Further studies are needed to test the role of BDNF release in the antidepressant actions of gaseous anesthetics.
Evidence that rapid acting antidepressants increase BDNF release
Primary neuronal culture systems have been used to directly test the effects of ketamine and other rapid acting agents on the release of BDNF. Primary cerebral cortical neurons from embryonic rat brains (E18) differentiate into neurons, both glutamate and GABA, as well as glia, and form an organized system comprised of mature soma, axons, dendrites and dendritic spines (Lepack et al., 2016; Ghosal et al., 2018). Because levels of BDNF release are low and/or because BDNF binds to both TrkB and p75NTR, an immunoprecipitation step is needed to enrich BDNF for detection by ELISA (Lepack et al., 2014; Lepack et al., 2016). We have found that incubation of primary cortical cultures with several different types of rapid-acting antidepressants, including ketamine, scopolamine, rapastinel, (2R,6R)-HNK and LY341495, but not classical monoaminergic drugs, rapidly (15 to 60 min) increase BDNF release (Lepack et al., 2014; Lepack et al., 2016; Ghosal et al., 2018; Kato et al., 2018; Fukumoto et al., 2019). These effects are observed at low, submicromolar or nanomolar concentrations and are completely abolished by pre-incubation with an AMPAR antagonist, or muscimol, a GABAA receptor agonist and neuronal silencing agent, indicating that the release of BDNF is activity dependent (Lepack et al., 2016; Ghosal et al., 2018). In addition, BDNF release in response to ketamine, (2R,6R)-HNK, rapastinel and scopolamine is blocked by verapamil, an L-type VDCCs blocker, providing further evidence for a requirement of activity-dependent AMPAR- and VDDCs-mediated neuronal depolarization (Lepack et al., 2014; Ghosal et al., 2018; Kato et al., 2018; Fukumoto et al., 2019)
Recently, d-methadone, the d-isomer of methadone that acts as an NMDA receptor antagonist, was also shown to activate the mTORC1 pathway and increase BDNF release in primary cortical cultures (Fogaca et al., 2019). Methadone is best known for treatment of opiate abuse disorder, based on the activity of l-methadone at opiate receptors (Gorman et al., 1997). However, d-methadone has relatively low affinity for opiate receptor subtypes compared to l-methadone (Callahan et al., 2004), and early studies show that d-methadone produces antidepressant behavioral actions in the FST (Hanania et al., 2019) as well as several other behavioral paradigms and in the chronic unpredictable stress model (Fogaca et al., 2019). Early clinical studies have supported the possibility that d-methadone could be used for the treatment of depression, with fewer side effects compared to ketamine (Bernstein et al., 2019).
Taken together, the in vitro and in vivo results demonstrate that rapid-acting antidepressants stimulate BDNF release through activation of AMPAR and VDCCs, resulting in activation of mTORC1 signaling, increased levels of synaptic proteins, and increased synaptic number and function (Lepack et al., 2014; Lepack et al., 2016; Ghosal et al., 2018). These findings indicate that fast, activity-dependent BDNF release is a critical factor that distinguishes rapid-acting antidepressants from monoaminergic drugs, which induce a delayed increase in BDNF expression, but for which there is no direct evidence for BDNF release (Martinez-Turrillas et al., 2005; Seo et al., 2014; Lepack et al., 2016; Harmer et al., 2017). Typical monoaminergic antidepressants are reported to rapidly increase the phosphorylation of TrkB, which has been used as a surogate marker for increased BDNF activity (Saarelainen et al., 2003). However, recent studies demonstrate that these monoaminergic antidepressants rapidly activate TrkB receptors via interactions with other regulatory sites on the receptor, including the endocytic adaptor complex AP-2 (Fred et al., 2019) and a cholesterol regulatory site (Casarotto et al., 2019) that are independent of BDNF release. Importantly, the rapid effects of monoaminergic antidepressants on TrkB do not correspond with the time lag for the therapeutic response to these agents, and there is no direct evidence that these agents produce activity dependent synaptic effects. Further studies are needed to examine this issue, notably the ability of typical antidepressants to produce rapid, activity dependent BDNF release and synaptic plasticity.
Role of VEGF in the actions of ketamine
There has also been interest in other growth factors in the pathophysiology and treatment of depression, and here we discuss the role of VEGF, a pleiotrophic growth factor expressed by neurons, astrocytes and perivascular macrophages, as well as endothelial cells, in the brain (Greene et al., 2009; Jais et al., 2016). Several splice variants of VEGF are generated by alternative splicing, and VEGF120 and VEGF164 are the most abundant (about 20% and 75%, respectively) in adult mouse brain (Ng et al., 2001). Both VEGF120 and VEGF164 bind to high-affinity tyrosine kinase receptors, Flt-1 (VEGFR1) and Flk-1 (VEGFR2), but only VEGF164 binds to the co-receptors, neuropilin-1 and −2 (Neufeld et al., 1999). VEGF has not only angiogenic but also potent neurotrophic and neuroprotective activity mainly through binding to Flk-1 (Rosenstein et al., 2003; Rosenstein et al., 2010).
Recent clinical studies demonstrate that VEGF levels are decreased in the cerebrospinal fluid of patients who have attempted suicide (Isung et al., 2012) and patients with at least one severe treatment-resistant depressive episode (Kranaster et al., 2019). Stress exposure is also reported to decrease VEGF in rodent models (Nowacka & Obuchowicz, 2013). In addition, viral-mediated non-cell type-specific VEGF knockdown in the hippocampal dentate gyrus is reported to produce depression-like behaviors in the forced swim test and novelty-suppressed feeding test, which were partially blocked by ketamine (Choi et al., 2016). A role for VEGF in the actions of antidepressant was first provided by evidence that chronic, but not acute, administration of a typical monoaminergic antidepressant increases VEGF expression in the hippocampus and PFC, and that the antidepressant effects of these drugs are blocked by infusion of a selective Flk-1 inhibitor (Warner-Schmidt & Duman, 2007; Greene et al., 2009).
Recently, we have demonstrated that neuronal VEGF-Flk-1 signaling plays a key role in the rapid antidepressant actions of ketamine (Deyama et al., 2019b). For these studies we generated mice with excitatory neuron-specific deletion of VEGF (CaMKIIα-Cre;VEGFflox/flox, hereafter, VEGFNEURON−/−) or Flk-1 (CaMKIIα-Cre;Flk-1flox/flox, hereafter, Flk-1NEURON−/−) in the forebrain. Similar to BDNF mutant mice (Duman et al., 2007; Advani et al., 2009; Yu et al., 2012), both VEGFNEURON−/− and Flk-1NEURON−/− mice did not show depression-like behaviors, indicating that loss of neuronal VEGF-Flk-1 signaling is not sufficient to produce a depression-like state under non-stressed conditions. However, the antidepressant behavioral effects of ketamine are blocked in VEGFNEURON−/− and Flk-1NEURON−/− mice. In addition, infusion of an anti-VEGF neutralizing antibody, which could bind and sequester extracellular VEGF, into the mPFC 30 min before ketamine administration also blocks the antidepressant actions of ketamine. In contrast, intra-mPFC infusion of the same antibody 2 hours after ketamine does not block the behavioral effects of ketamine. Further evidence for a role of VEGF in the mPFC is provided by studies demonstrating that infusion of recombinant VEGF164 into the mPFC produces ketamine-like rapid and sustained antidepressant effects via neuronal Flk-1 (Deyama et al., 2019b). Moreover, viral-mediated knockdown of Flk-1 selectively in mPFC excitatory neurons blocks the antidepressant behavioral actions of ketamine. Together, these findings indicate that ketamine rapidly and transiently increases VEGF release in the mPFC and that neuronal VEGF-Flk-1 signaling during the initial 2-hour period is necessary and sufficient for the rapid antidepressant actions of ketamine.
In addition to these behavioral actions, neuronal VEGF-Flk-1 signaling is necessary for the ketamine-induced increase in dendritic complexity of primary cortical neurons and the number of spine synapses in layer V mPFC pyramidal neurons, as these effects are blocked by an Flk-1 inhibitor and in VEGFNEURON−/− mice, respectively (Deyama et al., 2019b). Together, these findings indicate that neuronal VEGF-Flk-1 signaling, as well as BDNF signaling (Liu et al., 2012; Lepack et al., 2014), plays an essential role in the neurotrophic/synaptogenic effects of ketamine.
Since both BDNF and VEGF signaling are reported to mediate the neurotrophic and behavioral actions of ketamine, we have examined whether BDNF and VEGF act in series or parallel (Deyama et al., 2019a). Intra-mPFC infusion of recombinant BDNF produces ketamine-like antidepressant effects, and these effects are blocked by co-infusion of a VEGF neutralizing antibody or in VEGFNEURON−/− mice, indicating a requirement for excitatory neuron-derived extracellular VEGF. In primary cortical neurons, BDNF-TrkB signaling stimulates VEGF release (Deyama et al., 2019a), consistent with a previous study in a neuroblastoma cell line (Nakamura et al., 2006). The neurotrophic effects of BDNF on dendritic complexity also require VEGF-Flk-1 signaling in primary cortical neurons. The antidepressant actions of intra-mPFC VEGF164 infusion are also blocked by coinfusion of BDNF neutralizing antibody. Moreover, VEGF stimulates BDNF release and requires BDNF-TrkB signaling to produce neurotrophic effects on dendritic complexity in primary cortical neurons. Together, these findings highlight an important role for interplay between BDNF and VEGF in the rapid synaptic and antidepressant behavioral actions of ketamine. Future studies of co-localization and release of BDNF and VEGF could provide further evidence for the mechanisms underlying this interplay.
Future directions
Together, these findings highlight the complex role of BDNF, as well as VEGF in the actions of different classes of antidepressant treatments. This includes typical monoaminergic agents that increase BDNF expression, as well as fast acting antidepressants like ketamine that cause rapid, activity dependent release of BDNF and VEGF. Increased expression and release of these factors reverses the deficits caused by stress and depression and provides neurotrophic support that reverses the atrophy and synaptic loss that contribute to the pathophysiology of depression. These findings also underscore the possibility of targeting BDNF and VEGF for the treatment of depression. This could include behavioral therapies to enhance activity dependent increases in BDNF and VEGF expression in brain regions implicated in depression, as well as the well-known effects of exercise on BDNF expression (Intlekofer & Cotman, 2013; Marosi & Mattson, 2014). In addition, it may be possible to develop novel therapeutic drugs that increase BDNF and VEGF expression and/or release, but with fewer side effects than ketamine. Another approach would be to develop small molecular therapeutics that target the TrkB as well as Flk-1 receptors, although this approach has been difficult to date.
Acknowledgements:
This research was supported by NIMH Grants MH045481 and MH093897 to R.S.D and the State of CT.
Abbreviations
- BDNF
brain derived neurotrophic factor
- TrkB
tropomysin receptor kinase B
- p75NTR
p75 neurotrophin receptor
- VEGF
vascular endothelial growth factor
- Flt-1
VEGF receptor 1
- Flk-1
VEGF receptor 2
- NGF
nerve growth factor
- NMDA
N-methyl-D-aspartate
- NMDAR
NMDA receptor
- PFC
prefrontal cortex
- mPFC
medial prefrontal cortex
- PTSD
posttraumatic stress disorder
- ECS
electroconvulsive seizure
- ERK
extracellular signal regulated kinase
- Akt
serine threonine kinase
- MAP kinase
mitogen activated protein kinase
- mTORC1
mechanistic target of rapamycin complex 1
- (2R,6R)-HNK
(2R,6R)-hydroxynorketamine
- mGlu2/3
metabotropic glutamate 2/3 receptor
- MGS0039
mGlu2/3 receptor antagonist
- LY341495
mGlu2/3 receptor antagonist
- AMPAR
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor
- VDCC
voltage dependent calcium channel
Footnotes
Declaration of interest statement: Dr. Duman has received consulting fees from Janssen, Taisho, Naurex, and Aptinyx and has received research support from Lilly, Taisho, Allergan, Janssen, Naurex, Aptynix, Navitor, and Relmada. He is also listed as a co-inventor with Drs. Abdallah, Krystal, and Sanacora on Combination Therapy for Treating or Preventing Depression or Other Mood Diseases. U.S. Provisional Patent Application No. 047162-7177P1 (00754) filed on August 20, 2018, by Yale University Office of Cooperative Research OCR 7451 US01.
Literature Cited
- Adachi N, Numakawa T, Richards M, Nakajima S & Kunugi H (2014) New insight in expression, transport, and secretion of brain-derived neurotrophic factor: Implications in brain-related diseases. World J Biol Chem, 5, 409–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Advani T, Koek W & Hensler JG (2009) Gender differences in the enhanced vulnerability of BDNF+/− mice to mild stress. Int J Neuropsychopharmacol, 12, 583–588. [DOI] [PubMed] [Google Scholar]
- Antila H, Ryazantseva M, Popova D, Sipila P, Guirado R, Kohtala S, Yalcin I, Lindholm J, Vesa L, Sato V, Cordeira J, Autio H, Kislin M, Rios M, Joca S, Casarotto P, Khiroug L, Lauri S, Taira T, Castren E & Rantamaki T (2017) Isoflurane produces antidepressant effects and induces TrkB signaling in rodents. Sci Rep, 7, 7811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Autry AE, Adachi M, Nosyreva E, Na ES, Los MF, Cheng PF, Kavalali ET & Monteggia LM (2011) NMDA receptor blockade at rest triggers rapid behavioural anatidepressant responses. Nature, 475, 91–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Autry AE & Monteggia LM (2012) Brain-derived neurotrophic factor and neuropsychiatric disorders. Pharmacol Rev, 64, 238–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bath KG, Schilit A & Lee FS (2013) Stress effects on BDNF expression: effects of age, sex, and form of stress. Neuroscience, 239, 149–156. [DOI] [PubMed] [Google Scholar]
- Berman RM, Cappiello A, Anand A, Oren DA, Heninger GR, Charney DS & Krystal JH (2000) Antidepressant effects of ketamine in depressed patients. Biol Psychiatry, 47, 351–354. [DOI] [PubMed] [Google Scholar]
- Bernstein G, Davis K, Mills C, Wang L, McDonnell M, Oldenhof J, Inturrisi C, Manfredi PL & Vitolo OV (2019) Characterization of the Safety and Pharmacokinetic Profile of D-Methadone, a Novel N-Methyl-D-Aspartate Receptor Antagonist in Healthy, Opioid-Naive Subjects: Results of Two Phase 1 Studies. J Clin Psychopharmacol, 39, 226–237. [DOI] [PubMed] [Google Scholar]
- Bjorkholm C & Monteggia LM (2016) BDNF - a key transducer of antidepressant effects. Neuropharmacology, 102, 72–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgdorf J, Zhang XL, Nicholson KL, Balster RL, Leander JD, Stanton PK, Gross AL, Kroes RA & Moskal JR (2013) GLYX-13, a NMDA receptor glycine-site functional partial agonist, induces antidepressant-like effects without ketamine-like side effects. Neuropsychopharmacology, 38, 729–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgdorf J, Zhang XL, Weiss C, Gross A, Boikess SR, Kroes RA, Khan MA, Burch RM, Rex CS, Disterhoft JF, Stanton PK & Moskal JR (2015) The long-lasting antidepressant effects of rapastinel (GLYX-13) are associated with a metaplasticity process in the medial prefrontal cortex and hippocampus. Neuroscience, 308, 202–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callahan RJ, Au JD, Paul M, Liu C & Yost CS (2004) Functional inhibition by methadone of N-methyl-D-aspartate receptors expressed in Xenopus oocytes: stereospecific and subunit effects. Anesth Analg, 98, 653–659, table of contents. [DOI] [PubMed] [Google Scholar]
- Casarotto PC, Girych M, Fred SM, Moliner R, Enkavi G, Biojone C, Cannarozzo C, Brunello CA, Steinzeig A, Winkel F, Patil S, Vestring S, Serchov T, Laukkanen L, Cardon I, Antila H, Rog T, Bramham CR, Normann C, Lauri S, Vattulainen I & Castren E (2019) Antidepressants act by binding to the cholesterol-interaction site at TRKB neurotrophin receptor, bioRxiv. [Google Scholar]
- Castren E & Kojima M (2017) Brain-derived neurotrophic factor in mood disorders and antidepressant treatments. Neurobiology of disease, 97, 119–126. [DOI] [PubMed] [Google Scholar]
- Chaki S (2017) mGlu2/3 Receptor Antagonists as Novel Antidepressants. Trends Pharmacol Sci, 38, 569–580. [DOI] [PubMed] [Google Scholar]
- Chen MH, Lin WC, Wu HJ, Cheng CM, Li CT, Hong CJ, Tu PC, Bai YM, Tsai SJ & Su TP (2019) Antisuicidal effect, BDNF Val66Met polymorphism, and low-dose ketamine infusion: Reanalysis of adjunctive ketamine study of Taiwanese patients with treatment-resistant depression (AKSTP-TRD). J Affect Disord, 251, 162–169. [DOI] [PubMed] [Google Scholar]
- Chen ZY, Jing D, Bath KG, Ieraci A, Khan T, Siao CJ, Herrera DG, Toth M, Yang C, McEwen BS, Hempstead BL & Lee FS (2006) Genetic variant BDNF (Val66Met) polymorphism alters anxiety-related behavior. Science, 314, 140–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi M, Lee SH, Chang HL & Son H (2016) Hippocampal VEGF is necessary for antidepressant-like behaviors but not sufficient for antidepressant-like effects of ketamine in rats. Biochim Biophys Acta, 1862, 1247–1254. [DOI] [PubMed] [Google Scholar]
- Deyama S, Bang E, Kato T, Li XY & Duman RS (2019a) Neurotrophic and Antidepressant Actions of Brain-Derived Neurotrophic Factor Require Vascular Endothelial Growth Factor. Biol Psychiatry, 86, 143–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deyama S, Bang E, Wohleb ES, Li XY, Kato T, Gerhard DM, Dutheil S, Dwyer JM, Taylor SR, Picciotto MR & Duman RS (2019b) Role of Neuronal VEGF Signaling in the Prefrontal Cortex in the Rapid Antidepressant Effects of Ketamine. Am J Psychiatry, 176, 388–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dincheva I, Lynch NB & Lee FS (2016) The Role of BDNF in the Development of Fear Learning. Depress Anxiety, 33, 907–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong C, Zhang J. c., Yao W, Ren Q, Ma M, Yang C, Chaki S & Hashimoto K (2017) Rapid and sustained antidepressant action of the mGlu2/3 receptor antagonist MGS0039 in the social defeat stress model: comparison with ketamine. International Journal of Neuropsychopharmacology, 20, 228–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drevets WC & Furey ML (2010) Replication of scopolamine’s antidepressant efficacy in major depressive disorder: a randomized, placebo-controlled clinical trial. Biol Psychiatry, 67, 432–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drevets WC, Price JL & Furey ML (2008) Brain structural and functional abnormalities in mood disorders: implications for neurocircuitry models of depression. Brain Struct Funct, 213, 93–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duman CH, Schlesinger L, Kodama M, Russell DS & Duman RS (2007) A role for MAP kinase signaling in behavioral models of depression and antidepressant treatment. Biol Psychiatry, 61, 661–670. [DOI] [PubMed] [Google Scholar]
- Duman RS, Aghajanian GK, Sanacora G & Krystal JH (2016) Synaptic plasticity and depression: new insights from stress and rapid-acting antidepressants. Nat Med, 22, 238–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duman RS & Monteggia LM (2006) A neurotrophic model for stress-related mood disorders. Biol Psychiatry, 59, 1116–1127. [DOI] [PubMed] [Google Scholar]
- Duman RS, Sanacora G & Krystal JH (2019) Altered Connectivity in Depression: GABA and Glutamate Neurotransmitter Deficits and Reversal by Novel Treatments. Neuron, 102, 75–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duric V, Banasr M, Licznerski P, Schmidt H, Stockmeier C, Simen A, Newton S & Duman RS (2010) Negative regulator of MAP kinase is increased in depression and is necessary and sufficient for depressive behavior. Nat Med, 16, 1328–1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dwivedi Y (2009) Brain-derived neurotrophic factor: role in depression and suicide. Neuropsychiatr Dis Treat, 5, 433–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dwyer JM, Lepack AE & Duman RS (2012) mTOR activation is required for the antidepressant effects of mGluR(2)/(3) blockade. Int J Neuropsychopharmacol, 15, 429–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edelmann E, Lessmann V & Brigadski T (2014) Pre- and postsynaptic twists in BDNF secretion and action in synaptic plasticity. Neuropharmacology, 76 Pt C, 610–627. [DOI] [PubMed] [Google Scholar]
- Flint J & Kendler KS (2014) The Genetics of Major Depression. Neuron, 81, 1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fogaca MV, Fukumoto K, Franklin T, Liu RJ, Duman CH, Vitolo OV & Duman RS (2019) N-Methyl-D-aspartate receptor antagonist d-methadone produces rapid, mTORC1-dependent antidepressant effects. Neuropsychopharmacology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fred SM, Laukkanen L, Brunello CA, Vesa L, Goos H, Cardon I, Moliner R, Maritzen T, Varjosalo M, Casarotto PC & Castren E (2019) Pharmacologically diverse antidepressants facilitate TRKB receptor activation by disrupting its interaction with the endocytic adaptor complex AP-2. J Biol Chem. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frodl T & O’Keane V (2013) How does the brain deal with cumulative stress? A review with focus on developmental stress, HPA axis function and hippocampal structure in humans. Neurobiology of disease, 52, 24–37. [DOI] [PubMed] [Google Scholar]
- Fuchikami M, Thomas A, Liu R, Wohleb ES, Land BB, DiLeone RJ, Aghajanian GK & Duman RS (2015) Optogenetic stimulation of infralimbic PFC reproduces ketamine’s rapid and sustained antidepressant actions. Proc Natl Acad Sci U S A, 112, 8106–8111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukumoto K, Fogaca MV, Liu RJ, Duman C, Kato T, Li XY & Duman RS (2019) Activity-dependent brain-derived neurotrophic factor signaling is required for the antidepressant actions of (2R,6R)-hydroxynorketamine. Proc Natl Acad Sci U S A, 116, 297–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furey ML & Drevets WC (2006) Antidepressant efficacy of the antimuscarinic drug scopolamine: a randomized, placebo-controlled clinical trial. Arch Gen Psychiatry, 63, 1121–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatt JM, Nemeroff CB, Dobson-Stone C, Paul RH, Bryant RA, Schofield PR, Gordon E, Kemp AH & Williams LM (2009) Interactions between BDNF Val66Met polymorphism and early life stress predict brain and arousal pathways to syndromal depression and anxiety. Molecular Psychiatry, 14, 681–695. [DOI] [PubMed] [Google Scholar]
- Ghosal S, Bang E, Yue W, Hare BD, Lepack AE, Girgenti MJ & Duman RS (2018) Activity-Dependent Brain-Derived Neurotrophic Factor Release Is Required for the Rapid Antidepressant Actions of Scopolamine. Biol Psychiatry, 83, 29–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gideons ES, Kavalali ET & Monteggia LM (2014) Mechanisms underlying differential effectiveness of memantine and ketamine in rapid antidepressant responses. Proc Natl Acad Sci U S A, 111, 8649–8654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorman AL, Elliott KJ & Inturrisi CE (1997) The d- and l-isomers of methadone bind to the non-competitive site on the N-methyl-D-aspartate (NMDA) receptor in rat forebrain and spinal cord. Neurosci Lett, 223, 5–8. [DOI] [PubMed] [Google Scholar]
- Greene J, Banasr M, Lee B, Warner-Schmidt J & Duman RS (2009) Vascular endothelial growth factor signaling is required for the behavioral actions of antidepressant treatment: pharmacological and cellular characterization. Neuropsychopharmacology, 34, 2459–2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanania T, Manfredi P, Inturrisi C & Vitolo OV (2019) The N-methyl-D-aspartate receptor antagonist d-methadone acutely improves depressive-like behavior in the forced swim test performance of rats. Exp Clin Psychopharmacol. [DOI] [PubMed] [Google Scholar]
- Harmer CJ, Duman RS & Cowen PJ (2017) How do antidepressants work? New perspectives for refining future treatment approaches. The lancet. Psychiatry, 4, 409–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harward SC, Hedrick NG, Hall CE, Parra-Bueno P, Milner TA, Pan E, Laviv T, Hempstead BL, Yasuda R & McNamara JO (2016) Autocrine BDNF-TrkB signalling within a single dendritic spine. Nature, 538, 99–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedrick NG, Harward SC, Hall CE, Murakoshi H, McNamara JO & Yasuda R (2016) Rho GTPase complementation underlies BDNF-dependent homo- and heterosynaptic plasticity. Nature, 538, 104–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Intlekofer KA & Cotman CW (2013) Exercise counteracts declining hippocampal function in aging and Alzheimer’s disease. Neurobiology of disease, 57, 47–55. [DOI] [PubMed] [Google Scholar]
- Isung J, Aeinehband S, Mobarrez F, Martensson B, Nordstrom P, Asberg M, Piehl F & Jokinen J (2012) Low vascular endothelial growth factor and interleukin-8 in cerebrospinal fluid of suicide attempters. Transl Psychiatry, 2, e196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jais A, Solas M, Backes H, Chaurasia B, Kleinridders A, Theurich S, Mauer J, Steculorum SM, Hampel B, Goldau J, Alber J, Forster CY, Eming SA, Schwaninger M, Ferrara N, Karsenty G & Bruning JC (2016) Myeloid-Cell-Derived VEGF Maintains Brain Glucose Uptake and Limits Cognitive Impairment in Obesity. Cell, 165, 882–895. [DOI] [PubMed] [Google Scholar]
- Kang HJ, Voleti B, Hajszan T, Rajkowska G, Stockmeier CA, Licznerski P, Lepack A, Majik MS, Jeong LS, Banasr M, Son H & Duman RS (2012) Decreased expression of synapse-related genes and loss of synapses in major depressive disorder. Nat Med, 18, 1413–1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpova NN, Pickenhagen A, Lindholm J, Tiraboschi E, Kulesskaya N, Agustsdottir A, Antila H, Popova D, Akamine Y, Sullivan R, Hen R, Drew LJ & Castren E (2011) Fear Erasure in Mice Requires Synergy Between Antidepressant Drugs and Extinction Training. Science, 334, 1731–1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato T, Fogaca MV, Deyama S, Li XY, Fukumoto K & Duman RS (2018) BDNF release and signaling are required for the antidepressant actions of GLYX-13. Mol Psychiatry, 23, 2007–2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman J, Yang BZ, Douglas-Palumberi H, Grasso D, Lipschitz D, Houshyar S, Krystal JH & Gelernter J (2006) Brain-derived neurotrophic factor-5-HTTLPR gene interactions and environmental modifiers of depression in children. Biol Psychiatry, 59, 673–680. [DOI] [PubMed] [Google Scholar]
- Kim JM, Stewart R, Kim SW, Yang SJ, Shin IS, Kim YH & Yoon JS (2007) Interactions between life stressors and susceptibility genes (5-HTTLPR and BDNF) on depression in Korean elders. Biological Psychiatry, 62, 423–428. [DOI] [PubMed] [Google Scholar]
- Kohtala S, Theilmann W, Rosenholm M, Penna L, Karabulut G, Uusitalo S, Jarventausta K, Yli-Hankala A, Yalcin I, Matsui N, Wigren HK & Rantamaki T (2019) Cortical Excitability and Activation of TrkB Signaling During Rebound Slow Oscillations Are Critical for Rapid Antidepressant Responses. Mol Neurobiol, 56, 4163–4174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koike H, Fukumoto K, Iijima M & Chaki S (2013) Role of BDNF/TrkB signaling in antidepressant-like effects of a group II metabotropic glutamate receptor antagonist in animal models of depression. Behav Brain Res, 238, 48–52. [DOI] [PubMed] [Google Scholar]
- Koike H, Iijima M & Chaki S (2011a) Involvement of AMPA receptor in both the rapid and sustained antidepressant-like effects of ketamine in animal models of depression. Behav Brain Res, 224, 107–111. [DOI] [PubMed] [Google Scholar]
- Koike H, Iijima M & Chaki S (2011b) Involvement of the mammalian target of rapamycin signaling in the antidepressant-like effect of group II metabotropic glutamate receptor antagonists. Neuropharmacology, 61, 1419–1423. [DOI] [PubMed] [Google Scholar]
- Kranaster L, Blennow K, Zetterberg H & Sartorius A (2019) Reduced vascular endothelial growth factor levels in the cerebrospinal fluid in patients with treatment resistant major depression and the effects of electroconvulsive therapy-A pilot study. J Affect Disord, 253, 449–453. [DOI] [PubMed] [Google Scholar]
- Krishnan V & Nestler EJ (2008) The molecular neurobiology of depression. Nature, 455, 894–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan V & Nestler EJ (2010) Linking molecules to mood: new insight into the biology of depression. Am J Psychiatry, 167, 1305–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krystal JH, Abdallah CG, Sanacora G, Charney DS & Duman RS (2019) Ketamine: A Paradigm Shift for Depression Research and Treatment. Neuron, 101, 774–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laje G, Lally N, Mathews D, Brutsche N, Chemerinski A, Akula N, Kelmendi B, Simen A, McMahon FJ, Sanacora G & Zarate C (2012) Brain-Derived Neurotrophic Factor Val66Met Polymorphism and Antidepressant Efficacy of Ketamine in Depressed Patients. Biological psychiatry, 72, e27–e28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepack A, Fuchikami M, Dwyer J, Banasr M, Aghajanian G & Duman R (2014) BDNF release is required for the behavioral actions of ketamine. Int J Neuropsychopharmacol, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepack AE, Bang E, Lee B, Dwyer JM & Duman RS (2016) Fast-acting antidepressants rapidly stimulate ERK signaling and BDNF release in primary neuronal cultures. Neuropharmacology, 111, 242–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Lee B, Liu RJ, Banasr M, Dwyer JM, Iwata M, Li XY, Aghajanian G & Duman RS (2010) mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science, 329, 959–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Liu RJ, Dwyer JM, Banasr M, Lee B, Son H, Li XY, Aghajanian G & Duman RS (2011) Glutamate N-methyl-D-aspartate receptor antagonists rapidly reverse behavioral and synaptic deficits caused by chronic stress exposure. Biol Psychiatry, 69, 754–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu RJ & Aghajanian GK (2008) Stress blunts serotonin- and hypocretin-evoked EPSCs in prefrontal cortex: role of corticosterone-mediated apical dendritic atrophy. Proc Natl Acad Sci U S A, 105, 359–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu RJ, Lee FS, Li XY, Bambico F, Duman RS & Aghajanian GK (2012) Brain-derived neurotrophic factor Val66Met allele impairs basal and ketamine-stimulated synaptogenesis in prefrontal cortex. Biol Psychiatry, 71, 996–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu WX, Wang J, Xie ZM, Xu N, Zhang GF, Jia M, Zhou ZQ, Hashimoto K & Yang JJ (2016) Regulation of glutamate transporter 1 via BDNF-TrkB signaling plays a role in the anti-apoptotic and antidepressant effects of ketamine in chronic unpredictable stress model of depression. Psychopharmacology (Berl), 233, 405–415. [DOI] [PubMed] [Google Scholar]
- Lumsden EW, Troppoli TA, Myers SJ, Zanos P, Aracava Y, Kehr J, Lovett J, Kim S, Wang FH, Schmidt S, Jenne CE, Yuan P, Morris PJ, Thomas CJ, Zarate CA Jr., Moaddel R, Traynelis SF, Pereira EFR, Thompson SM, Albuquerque EX & Gould TD (2019) Antidepressant-relevant concentrations of the ketamine metabolite (2R,6R)-hydroxynorketamine do not block NMDA receptor function. Proc Natl Acad Sci U S A, 116, 5160–5169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacQueen G & Frodl T (2011) The hippocampus in major depression: evidence for the convergence of the bench and bedside in psychiatric research? Mol Psychiatry, 16, 252–264. [DOI] [PubMed] [Google Scholar]
- Marosi K & Mattson MP (2014) BDNF mediates adaptive brain and body responses 1600 to energetic challenges. 2014, 25, 89–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Turrillas R, Del Rio J & Frechilla D (2005) Sequential changes in BDNF mRNA expression and synaptic levels of AMPA receptor subunits in rat hippocampus after chronic antidepressant treatment. Neuropharmacology, 49, 1178–1188. [DOI] [PubMed] [Google Scholar]
- Martinowich K, Manji H & Lu B (2007) New insights into BDNF function in depression and anxiety. Nat Neurosci, 10, 1089–1093. [DOI] [PubMed] [Google Scholar]
- Maya Vetencourt JF, Sale A, Viegi A, Baroncelli L, De Pasquale R, O’Leary OF, Castren E & Maffei L (2008) The antidepressant fluoxetine restores plasticity in the adult visual cortex. Science, 320, 385–388. [DOI] [PubMed] [Google Scholar]
- McEwen BS, Eiland L, Hunter RG & Miller MM (2012) Stress and anxiety: Structural plasticity and epigenetic regulation as a consequence of stress. Neuropharmacology, 62, 3–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizui T, Ishikawa Y, Kumanogoh H & Kojima M (2016) Neurobiological actions by three distinct subtypes of brain-derived neurotrophic factor: Multi-ligand model of growth factor signaling. Pharmacol Res, 105, 93–98. [DOI] [PubMed] [Google Scholar]
- Molendijk ML, Bus BA, Spinhoven P, Kaimatzoglou A, Oude Voshaar RC, Penninx BW, van IMH & Elzinga BM (2012) A systematic review and meta-analysis on the association between BDNF val(66)met and hippocampal volume--a genuine effect or a winners curse? Am J Med Genet B Neuropsychiatr Genet, 159B, 731–740. [DOI] [PubMed] [Google Scholar]
- Morrison JH & Baxter MG (2012) The ageing cortical synapse: hallmarks and implications for cognitive decline. Nat Rev Neurosci, 13, 240–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura K, Martin KC, Jackson JK, Beppu K, Woo CW & Thiele CJ (2006) Brain-derived neurotrophic factor activation of TrkB induces vascular endothelial growth factor expression via hypoxia-inducible factor-1alpha in neuroblastoma cells. Cancer research, 66, 4249–4255. [DOI] [PubMed] [Google Scholar]
- Neufeld G, Cohen T, Gengrinovitch S & Poltorak Z (1999) Vascular endothelial growth factor (VEGF) and its receptors. FASEB J, 13, 9–22. [PubMed] [Google Scholar]
- Ng YS, Rohan R, Sunday ME, Demello DE & D’Amore PA (2001) Differential expression of VEGF isoforms in mouse during development and in the adult. Dev Dyn, 220, 112–121. [DOI] [PubMed] [Google Scholar]
- Nibuya M, Morinobu S & Duman RS (1995) Regulation of BDNF and trkB mRNA in rat brain by chronic electroconvulsive seizure and antidepressant drug treatments. J Neurosci, 15, 7539–7547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nibuya M, Nestler EJ & Duman RS (1996) Chronic antidepressant administration increases the expression of cAMP response element binding protein (CREB) in rat hippocampus. J Neurosci, 16, 2365–2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowacka M & Obuchowicz E (2013) BDNF and VEGF in the pathogenesis of stress-induced affective diseases: an insight from experimental studies. Pharmacol Rep, 65, 535–546. [DOI] [PubMed] [Google Scholar]
- Palucha-Poniewiera A, Podkowa K & Pilc A (2019) Role of AMPA receptor stimulation and TrkB signaling in the antidepressant-like effect of ketamine co-administered with a group II mGlu receptor antagonist, LY341495, in the forced swim test in rats. Behav Pharmacol, 30, 471–477. [DOI] [PubMed] [Google Scholar]
- Park L, Furey M, Nugent AC, Farmer C, Ellis J, Szczepanik J, Lener MS & Zarate CA Jr. (2019) Neurophysiological Changes Associated with Antidepressant Response to Ketamine Not Observed in a Negative Trial of Scopolamine in Major Depressive Disorder. Int J Neuropsychopharmacol, 22, 10–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popoli M, Yan Z, McEwen BS & Sanacora G (2011) The stressed synapse: the impact of stress and glucocorticoids on glutamate transmission. Nat Rev Neurosci, 13, 22–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preskorn SH, Baker B, Kolluri S, Menniti FS, Krams M & Landen JW (2008) An innovative design to establish proof of concept of the antidepressant effects of the NR2B subunit selective N-methyl-D-aspartate antagonist, CP-101,606, in patients with treatment-refractory major depressive disorder. J Clin Psychopharmacol, 28, 631–637. [DOI] [PubMed] [Google Scholar]
- Riggs LM, Aracava Y, Zanos P, Fischell J, Albuquerque EX, Pereira EFR, Thompson SM & Gould TD (2019) (2R,6R)-hydroxynorketamine rapidly potentiates hippocampal glutamatergic transmission through a synapse-specific presynaptic mechanism. Neuropsychopharmacology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roozendaal B, McEwen BS & Chattarji S (2009) Stress, memory and the amygdala. Nat Rev Neurosci, 10, 423–433. [DOI] [PubMed] [Google Scholar]
- Rosenstein JM, Krum JM & Ruhrberg C (2010) VEGF in the nervous system. Organogenesis, 6, 107–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenstein JM, Mani N, Khaibullina A & Krum JM (2003) Neurotrophic effects of vascular endothelial growth factor on organotypic cortical explants and primary cortical neurons. J Neurosci, 23, 11036–11044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saarelainen T, Hendolin P, Lucas G, Koponen E, Sairanen M, MacDonald E, Agerman K, Haapasalo A, Nawa H, Aloyz R, Ernfors P & Castren E (2003) Activation of the TrkB neurotrophin receptor is induced by antidepressant drugs and is required for antidepressant-induced behavioral effects. J Neurosci, 23, 349–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo MK, Lee CH, Cho HY, Lee JG, Lee BJ, Kim JE, Seol W, Kim YH & Park SW (2014) Effects of antidepressant drugs on synaptic protein levels and dendritic outgrowth in hippocampal neuronal cultures. Neuropharmacology, 79, 222–233. [DOI] [PubMed] [Google Scholar]
- Shirayama Y, Chen A C-H, Nakagawa S, Russell RS, Duman RS (2002) Brain derived neurotrophic factor produces antidepressant effects in behavioral models of depression. J Neurosci, 22, 3251–3261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MA, Makino S, Kvetnansky R, and Post RM (1995a) Stress alters the express of brain-derived neurotrophic factor and neurotrophin-3 mRNAs in the hippocampus. J Neurosci, 15, 1768–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su TP, Chen MH, Li CT, Lin WC, Hong CJ, Gueorguieva R, Tu PC, Bai YM, Cheng CM & Krystal JH (2017) Dose-Related Effects of Adjunctive Ketamine in Taiwanese Patients with Treatment-Resistant Depression. Neuropsychopharmacology, 42, 2482–2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan PF, Agrawal A, Bulik CM, Andreassen OA, Borglum AD, Breen G, Cichon S, Edenberg HJ, Faraone SV, Gelernter J, Mathews CA, Nievergelt CM, Smoller JW, O’Donovan MC & Psychiatric Genomics C (2018) Psychiatric Genomics: An Update and an Agenda. Am J Psychiatry, 175, 15–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun H, Kennedy PJ & Nestler EJ (2013) Epigenetics of the depressed brain: role of histone acetylation and methylation. Neuropsychopharmacology, 38, 124–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun HL, Zhou ZQ, Zhang GF, Yang C, Wang XM, Shen JC, Hashimoto K & Yang JJ (2016) Role of hippocampal p11 in the sustained antidepressant effect of ketamine in the chronic unpredictable mild stress model. Transl Psychiatry, 6, e741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki K, Nosyreva E, Hunt KW, Kavalali ET & Monteggia LM (2017) Effects of a ketamine metabolite on synaptic NMDAR function. Nature, 546, E1–E3. [DOI] [PubMed] [Google Scholar]
- Taliaz D, Stall N, Dar DE & Zangen A (2010) Knockdown of brain-derived neurotrophic factor in specific brain sites precipitates behaviors associated with depression and reduces neurogenesis. Mol Psychiatry, 15, 80–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trivedi MH, Rush AJ, Wisniewski SR, Nierenberg AA, Warden D, Ritz L, Norquist G, Howland RH, Lebowitz B, McGrath PJ, Shores-Wilson K, Biggs MM, Balasubramani GK, Fava M & Team SDS (2006) Evaluation of outcomes with citalopram for depression using measurement-based care in STAR*D: implications for clinical practice. Am J Psychiatry, 163, 28–40. [DOI] [PubMed] [Google Scholar]
- Voleti B, Navarria A, Liu RJ, Banasr M, Li N, Terwilliger R, Sanacora G, Eid T, Aghajanian G & Duman RS (2013) Scopolamine rapidly increases mammalian target of rapamycin complex 1 signaling, synaptogenesis, and antidepressant behavioral responses. Biol Psychiatry, 74, 742–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warner-Schmidt JL & Duman RS (2007) VEGF is an essential mediator of the neurogenic and behavioral actions of antidepressants. Proc Natl Acad Sci U S A, 104, 4647–4652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witkin JM, Monn JA, Schoepp DD, Li X, Overshiner C, Mitchell SN, Carter G, Johnson B, Rasmussen K & Rorick-Kehn LM (2016) The Rapidly Acting Antidepressant Ketamine and the mGlu2/3 Receptor Antagonist LY341495 Rapidly Engage Dopaminergic Mood Circuits. Journal of Pharmacology and Experimental Therapeutics, 358, 71–82. [DOI] [PubMed] [Google Scholar]
- Yang B, Zhang JC, Han M, Yao W, Yang C, Ren Q, Ma M, Chen QX & Hashimoto K (2016) Comparison of R-ketamine and rapastinel antidepressant effects in the social defeat stress model of depression. Psychopharmacology (Berl), 233, 3647–3657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C, Kobayashi S, Nakao K, Dong C, Han M, Qu Y, Ren Q, Zhang JC, Ma M, Toki H, Yamaguchi JI, Chaki S, Shirayama Y, Nakazawa K, Manabe T & Hashimoto K (2018) AMPA Receptor Activation-Independent Antidepressant Actions of Ketamine Metabolite (S)-Norketamine. Biol Psychiatry, 84, 591–600. [DOI] [PubMed] [Google Scholar]
- Yang C, Shirayama Y, Zhang JC, Ren Q, Yao W, Ma M, Dong C & Hashimoto K (2015) R-ketamine: a rapid-onset and sustained antidepressant without psychotomimetic side effects. Transl Psychiatry, 5, e632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H, Wang DD, Wang Y, Liu T, Lee FS & Chen ZY (2012) Variant brain-derived neurotrophic factor Val66Met polymorphism alters vulnerability to stress and response to antidepressants. J Neurosci, 32, 4092–4101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanos P, Highland JN, Stewart BW, Georgiou P, Jenne CE, Lovett J, Morris PJ, Thomas CJ, Moaddel R, Zarate CA Jr. & Gould TD (2019) (2R,6R)-hydroxynorketamine exerts mGlu2 receptor-dependent antidepressant actions. Proc Natl Acad Sci U S A, 116, 6441–6450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanos P, Moaddel R, Morris PJ, Georgiou P, Fischell J, Elmer GI, Alkondon M, Yuan P, Pribut HJ, Singh NS, Dossou KS, Fang Y, Huang XP, Mayo CL, Wainer IW, Albuquerque EX, Thompson SM, Thomas CJ, Zarate CA Jr. & Gould TD (2016) NMDAR inhibition-independent antidepressant actions of ketamine metabolites. Nature, 533, 481–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarate CA Jr., Singh JB, Carlson PJ, Brutsche NE, Ameli R, Luckenbaugh DA, Charney DS & Manji HK (2006) A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Arch Gen Psychiatry, 63, 856–864. [DOI] [PubMed] [Google Scholar]
- Zhang JC, Yao W, Dong C, Yang C, Ren Q, Ma M, Han M & Hashimoto K (2015) Comparison of ketamine, 7,8-dihydroxyflavone, and ANA-12 antidepressant effects in the social defeat stress model of depression. Psychopharmacology (Berl), 232, 4325–4335. [DOI] [PubMed] [Google Scholar]