

Abstract
Objectives
Alzheimer disease (AD) shows a broad array of clinical presentations, but the mechanisms underlying these phenotypic variants remain elusive. Aging-related astrogliopathy (ARTAG) is a relatively recent term encompassing a broad array of tau deposition in astroglia outside the range of traditional tauopathies. White matter thorn-shaped astrocyte (WM-TSA) clusters, a specific ARTAG subtype, has been associated with atypical language presentation of AD in a small study lacking replication. To interrogate the impact of WM-TSA in modifying clinical phenotype in AD, we investigated a clinicopathologic sample of 83 persons with pure cortical AD pathology and heterogeneous clinical presentations.
Methods
We mapped WM-TSA presence and density throughout cortical areas and interrogated whether WM-TSA correlated with atypical AD presentation or worse performance in neuropsychological testing.
Results
WM-TSA was present in nearly half of the cases and equally distributed in typical and atypical AD presentations. Worsening language and visuospatial functions were correlated with higher WM-TSA density in language-related and visuospatial-related regions, respectively. These findings were unrelated to regional neurofibrillary tangle burden. Next, unsupervised clustering divided the participants into 2 groups: a high–WM-TSA (n = 9) and low–WM-TSA (n = 74) pathology signature. The high–WM-TSA group scored significantly worse in language but not in other cognitive domains.
Conclusions
The negative impact of WM-TSA pathology to language and possibly visuospatial networks suggests that WM-TSA is not as benign as other ARTAG types and may be explored as a framework to understand the mechanisms and impact of astrocytic tau deposition in AD in humans.
Alzheimer disease (AD) features a stereotypical accumulation of neuritic plaques and neurofibrillary tangles (NFT)/neuropil threads. Despite this apparently homogeneous neuropathologic picture, AD manifests with different clinical presentations and rates of progression.1 Beyond the classic amnestic syndrome, AD can manifest with an atypical clinical presentation, including behavioral variant frontotemporal dementia, also known as frontal variant or behavioral/dysexecutive variant of AD,2 logopenic variant primary progressive aphasia (lvPPA),3 corticobasal syndrome4 and posterior cortical atrophy.5 Genetic and environmental factors have been implicated as underlying contributors to this clinical heterogeneity, but comorbid pathology may also play a role in the clinical phenotype. Co-occurrence of TAR DNA binding protein-43 (TDP-43) in the hippocampus with AD pathology, for instance, leads to worse memory impairment6 and faster rates of hippocampal atrophy.7
In 2007, Munoz et al.8 described a type of astroglial tau inclusion featuring eccentric, tau-positive perinuclear deposits, found predominantly in clusters at the gray-white matter junction, called argyrophilic thorn-shaped astrocyte clusters (ATACs). ATAC in many brain areas was the only identifiable difference between 8 cases of AD presenting clinically with a prominent aphasic syndrome (with or without an amnestic component) and 6 cases of AD manifesting as an amnestic syndrome. However, an independent study examining ATACs in 10 cases of AD presenting with a primary progressive aphasia (PPA) syndrome failed to confirm such an association.3 Thus, the question of whether ATAC is a modifier of AD clinical presentation or a cause of prominent language deficits remains unanswered.
Years after ATACs were described, an international group of neuropathologists published strategies to harmonize evaluation of so-called aging-related tau astrogliopathy (ARTAG), an umbrella term used to describe tau pathology in astrocytes that accumulates with aging.9 Because ATAC is equivalent to thorn-shaped astrocytes (TSA) in the white matter, in this study, we refer to ATAC as white matter TSA (WM-TSA).
To determine whether WM-TSA contributes to the clinical heterogeneity in AD, we investigated a large, well-characterized clinicopathologic series of individuals with autopsy-proven AD pathology and heterogeneous clinical presentations. We quantified WM-TSA burden in key language-related and other additional cortical regions and compared the results with the clinical phenotype and neuropsychological performance in language and other cognitive domains. First, we tested the hypothesis that cortical accumulation of WM-TSA, including in language-related areas, is associated with an atypical clinical diagnosis such as lvPPA. Next, we examined whether the cortical accumulation of WM-TSA correlates with worse neuropsychological scores on language and other cognitive domains. Finally, we assessed unbiased clustering of the presence and density of WM-TSA pathology in our cohort and compared how this grouping is related to cognitive function.
Methods
Participants
In this cross-sectional study, participants were identified from the clinicopathologic cohort of the Neurodegenerative Disease Brain Bank, which is sourced from the cohorts of the Memory and Aging Center (MAC) at the University of California, San Francisco (UCSF). Individuals in this study underwent an in-depth clinical assessment at least once. This assessment included neurologic history and examination and comprehensive neuropsychological and functional testing, including the Clinical Dementia Rating (CDR). Neuropathologic assessment included an extensive dementia-oriented investigation covering dementia-related regions of interest on the left hemisphere unless, on gross pathology, the right was noted to be more atrophic. Neuropathologic diagnosis followed currently accepted guidelines.10–12 Subtyping for frontotemporal lobar degeneration (FTLD)–TDP-43 and FTLD-tau followed the current harmonized nomenclature.13
From 2008 to 2016, 145 participants who underwent an autopsy at the Neurodegenerative Disease Brain Bank received a primary diagnosis of AD pathologic changes. From those, we excluded cases with comorbid FTLD (FUS, tau, or TDP-43), chronic traumatic encephalopathy, α-synuclein pathology staged Braak ≥3, hippocampal sclerosis, or contributing cerebrovascular lesions, analyzing a final number of 83 cases. The frequency of cerebrovascular changes was low in general in our cohort, similar to low rates of cerebrovascular changes in this age group found in other series.14
Standard protocol approvals, registrations, and patient consents
The study was approved by the UCSF Institutional Review Board, and all participants or their legal representatives signed a written informed consent that was obtained according to the Declaration of Helsinki and its further amendments.
Identifying and quantifying WM-TSA
A neuropathologist (A.L.N.) reviewed all cases, blinded to the clinical and neuropathologic diagnosis, to determine the presence and density of WM-TSA in 8-μm-thick histologic sections immunostained for phospho-tau (p-Ser202 tau, CP-13, 1:500, gift of Peter Davies, NY). Eight cortical regions were sampled: opercular inferior frontal gyrus, superior temporal gyrus at the level of the lateral geniculate body, inferior temporal gyrus at the level of the amygdala and angular gyrus (all associated with language), and middle frontal gyrus at the level of the genu of the corpus callosum, anterior cingulate cortex, anterior insula, and entorhinal cortex. Although we initially analyzed other forms of ARTAG, only WM-TSA was the focus of this study because we (data not shown) and others did not find clinicopathologic correlations with other ARTAG subtypes. WM-TSA was defined as the presence of TSA (prominent eccentric somatic tau deposition) located in the gray-white junction (figure 1). We annotated whether WM-TSA was present in each area of interest and, if so, the highest density in a ×20 microscopic field.
Figure 1. Argyrophilic thorn-shaped astrocytes clusters or WM-TSA typically found in the white-gray matter junction.

Representative images of white matter thorn-shaped astrocytes (WM-TSA) type of aging-related tau astrogliopathy in the middle frontal gyrus visualized with phosphorylated tau immunostaining (CP-13).
Quantifying NFT
To interrogate a correlation between WM-TSA and NFT burden, we quantified the NFT densities in the middle frontal, superior temporal, and angular gyri in histologic sections stained with thioflavin-S.15 Briefly, three 0.25-mm2 areas were sampled at random from each region, and quantitative NFT counts were averaged across these 3 areas to produce a density score. NFTs were quantified in all cases.
Clinical characterization
At MAC/UCSF, the clinical working diagnosis, including the diagnosis of lvPPA, is based on clinical, neuropsychological, and language test results that are reviewed by a multidisciplinary consensus of an expert neurologist, neuropsychologist, and speech and language pathologist. The clinical syndrome was determined by chart review of the first clinical encounter and based on published criteria for dementia of amnestic type,16 posterior cortical atrophy syndrome,17 lvPPA,18 corticobasal degeneration syndrome,19 behavioral variant frontotemporal dementia,20 Lewy body dementia,11 and mild cognitive impairment (MCI).21 The charts were reviewed by a behavioral neurologist (E.d.P.F.R. and/or Z.M.) who were blinded to the WM-TSA status or the severity of AD pathology. If there was a discrepancy in the chart, the final diagnosis was determined after a consensus. All participants were diagnosed with 1 of these syndromes, except for 1 individual who presented with rapid cognitive decline, parkinsonism, and ataxia and was classified as dementia without other specification.
For clarity, we use the term AD strictly to refer to the neuropathologic entity, whereas we use the term amnestic-type dementia to refer to the classic AD-associated clinical phenotype.
We also obtained information on the following variables from the MAC/UCSF Clinical Database: age at onset, age at death, disease duration, sex, and years of education. APOE allele genotyping was done with a TaqMan Allelic Discrimination Assay on an ABI7900HT Fast Real-Time PCR system (Applied Biosystems, Foster City, CA).
Determining neuropsychological scores
Composite z scores for the cognitive domains were obtained from the neuropsychological evaluation closest to death. The language z scores were calculated from normative data from a cohort of cognitively healthy older adults,22 and the final composite score was the sum of the z scores for the Boston Naming Test, Animal Fluency Test in 1 minute, and the Peabody Picture Vocabulary Test.23
We also calculated z scores for the following nonlanguage domains: a memory composite z score, defined as the sum of the z scores from the California Verbal Learning Test–Delayed Recall, sum of the learning trials, and recognition accounting for false positives, and the z score of the modified Rey Figure Delayed Recall; an executive composite z score, defined as the sum of the z scores for design fluency, letter fluency, Stroop test (correct naming), digital backwards, and Trail Making B (number of correct lines in 1 minute); and a visuospatial composite z score, defined as the sum of the z scores on the modified Rey Figure, Number Location for the Visual Object and Space Perception battery, and the block design of the Wechsler Adult Intelligence Scale–III.
Statistical analysis
First, we investigated whether WM-TSA presence correlated with an atypical AD presentation using χ2 tests. Then, to determine whether regional WM-TSA density correlates with impairment of associated cognitive domains, we compared the cognitive domain composite z scores closest to death (outcome) with regional WM-TSA densities (predictors). Because deficits in specific cognitive domain scores might also be a result of increased overall cognitive impairment rather than a target effect of WM-TSA, we also examined the relationship of WM-TSA densities (predictor) to the CDR Sum of Boxes (outcome), a measure of overall clinical severity,24 obtained at the last visit before death. Participants classified as having MCI/controls in this study maintained a CDR score of ≤0.5, even in a postmortem interview with a caregiver.
Multiple linear regression models were assessed to analyze the relationships between WM-TSA densities and cognitive domain composite z score and between WM-TSA densities and CDR Sum of Boxes score. All the models were adjusted for age at death and sex, as well as for Braak staging of NFT25 and the time lag between cognitive testing and death. The fitness of the linear regression models was assessed by analyzing the residuals, and we did not observe any clear pattern, although there was an imbalance in the x-axis because of the number of zero values.
To validate our previously described a priori approach in which we correlated regional WM-TSA with cognitive domain composite z scores, we additionally applied unbiased clustering analysis to classify the cases on the basis of the anatomic distribution and WM-TSA density throughout all brain regions examined using the K-means algorithm. We validated the robustness of the k-means clustering against other clustering methodologies based on 3 criteria: connectivity, silhouette width, and the Dunn index. The validation was performed in R (R Foundation for Statistical Computing, Vienna, Austria) using the Cluster Validation Package clValid.26 To select the optimal number of k-means clusters, we used the elbow method for a plot of within-groups sum of squares and the number of clusters. Similarity of k-means and the Ward method of hierarchical clustering was assessed to appraise the robustness of the partitioning. The 2 resulting k-means clusters were a high–WM-TSA (n = 9) cluster, corresponding to an overall high density of WM-TSA throughout brain regions, and a low–WM-TSA (n = 74) cluster, with an overall low density of WM-TSA in few brain regions. These 2 groups were contrasted in terms of demographics and neuropsychological composite scores using a Mann-Whitney U test. A χ2 test was used to compare the proportion of men and APOE ε4 allele carriers across the groups. In addition, we performed 3 sensitivity analyses to validate and test the robustness of the k-means clustering groups by examining the classification of the WM-TSA groups used in our cluster analysis.
Finally, we assessed whether there was an association between WM-TSA densities and burden of AD pathology represented by NFT density by calculating the Pearson coefficients of correlation in middle frontal, superior temporal, and angular gyri. Then, we retested the multiple linear regression models described above, including the NFT densities in the model to assess whether the correlation between the density of WM-TSA and language performance was independent of the NFT density.
All analyses were performed in R Statistical Software (version 3.3.3), and values of p < 0.01 were considered significant to account for multiple testing and to mitigate the risk of false positives.
Data availability
All the data are available on reasonable request.
Results
Of the 83 participants, 62.6% (n = 52) were male. Their mean educational attainment was 15.7 (3.4) years; the mean age at death was 72.4 (10.9) years; the mean age at symptom onset was 61.9 (10.8) years; and the mean disease duration was 9.8 (3.4) years. Overall, the mean lag time between the latest cognitive assessment and death was 3.8 (2.2) years. At least 1 APOE ε4 allele was present in 44.7% (n = 37) of cases. The median Braak stage for neurofibrillary pathology was 6 (table 1). Almost half of the participants (47%, n = 39) had WM-TSA in at least 1 of the brain regions examined. A majority of the participants (81.9%, n = 68) were right-handed, and in most cases, the left hemisphere was sampled.
Table 1.
Demographic and clinical characteristics per clinical syndrome

Approximately half of the participants (53.5%, n = 46) were diagnosed with a classic amnestic syndrome, while 26.3% (n = 27) met criteria for an atypical clinical presentation, including 7 participants (8.4% of total) with a clinical diagnosis of lvPPA (table 1). Furthermore, 7 individuals met diagnostic criteria for MCI, and 3 were cognitively normal at death (table 1). Noteworthy, the clinical diagnosis was established at the time of first clinical encounter, whereas table 1 shows results from the last neuropsychological evaluation before death, a time when most participants already had significant deficits in multiple domains.
WM-TSA was similarly prevalent in cases with a classic amnestic syndrome, an lvPPA, or any other atypical clinical presentation.
Considering only the 73 participants who had dementia at death (CDR score postmortem > 1), we failed to find significant differences in the presence of WM-TSA comparing participants with typical and atypical presentations. WM-TSA was present in at least 1 region in 56% (n = 26) of the 46 participants manifesting a classic amnestic AD and in 37% (n = 10) of the 26 participants manifesting an atypical dementia syndrome (p = 0.141). When the 7 atypical cases manifesting a lvPPA were isolated, only 43% (n = 3) had WM-TSA in at least 1 region examined.
Higher density of WM-TSA correlated with deficits in language function and visuospatial ability
Assessing multiple linear regression models, we observed a significant correlation between higher density of WM-TSA in language-related areas and deficits in language function (table 2). The density of WM-TSA in inferior frontal gyrus was significantly correlated with worse language scores (df = 68, β = −0.04, standard error [SE] = 0.01, p = 0.002), as well as the density of WM-TSA in superior temporal gyrus (df = 65, β = −0.03, SE = 0.00, p = 0.008) and angular gyrus (df = 68, β = −0.02, SE = 0.01, p = 0.008). Next, we tested whether there was a specific association between each language test that composed the language composite z score, including z scores for PPVT, the Boston Naming Test, and Animal Fluency Test and the density of WM-TSA in the language-related brain regions. We found a significant association only between the Boston Naming Test and the density of WM-TSA in the superior temporal gyrus (table 2).
Table 2.
Density of WM-TSA in language-related regions predicts worse language scores

In addition, we observed a significant correlation between higher density of WM-TSA in angular gyrus, a brain area associated with visuospatial function and worse visuospatial scores (df = 68, β = −0.04, SE = 0.01, p = 0.003) (table 3). Next, we tested whether there was a specific association between 3 visuospatial tests, including z scores for the modified Rey Figure and Number Location for the Visual Object and Space Perception battery, and the density of WM-TSA in angular gyrus and found a significant association with the modified Rey Figure test. Finally, we failed to identify any significant correlations between higher density of WM-TSA in any region and deficits in executive function or memory.
Table 3.
Density of WM-TSA in visuospatial-related regions predicts worse visuospatial scores

To examine whether the association between higher density of WM-TSA and language and visuospatial dysfunction was simply a reflection of a worsening global cognition in cases with higher burden of WM-TSA, we analyzed the associations between the density of WM-TSA in each of the relevant regions and the CDR Sum of Boxes scores and failed to find a significant relationship (tables 2 and 3), suggesting that the correlation between WM-TSA density and worse language and visuospatial scores was independent of the clinical severity. For all of these results, a sensitivity analysis was done in a subgroup of participants with dementia (i.e., excluding the ones with MCI and the cognitively healthy controls, n = 73), which disclosed similar results for the association between inferior frontal gyrus and language scores and angular gyrus for visuospatial scores. The relationships between superior temporal and angular gyri and language scores were very close to statistical significance (table 4).
Table 4.
Correlation between the density of WM-TSA and language and visuospatial scores in the subgroup with dementia (n = 73), excluding those with MCI and controls

Unbiased clustering of pathology confirms an association of increased density of WM-TSA and language dysfunction
Next, we sought to look at the data using an unbiased approach and to validate our a priori findings. The K-means clustering algorithm was used to classify our cases by density and anatomic distribution of WM-TSA pathology in all brain regions analyzed. Two robust groups were identified: a high–WM-TSA (n = 9) group with increased densities throughout the cortex and a low–WM-TSA (n = 74) group with low to no WM-TSA pathology throughout the cortex.
The high–WM-TSA group had a significantly older age at dementia onset (t = 8.6, p = 0.021) and age at death (t = 8.7, p = 0.012), as well as a higher proportion of men (t = 42, p = 0.037) (table 5). Similar to our correlation results, the high–WM-TSA group exhibited significantly reduced language function (t = −2.6, p = 0.018). We failed to find a similar significant correlation for other domains, including memory (t = −0.4, p = 0.946), executive (t = −0.0, p = 0.816) and visuospatial (t = −0.2, p = 0.898) domains (table 5 and figure 2). We ran a sensitivity analyses comparing cases lacking WM-TSA with the remaining cases of the low–WM-TSA group combined with the high–WM-TSA group, cases lacking WM-TSA with the remaining of the low–WM-TSA group, and cases of low–WM-TSA group having some WM-TSA with the high–WM-TSA group and using the sample of 73 individuals with dementia at death and found similar results (table 5).
Table 5.
Characteristics of the high– and low–WM-TSA groups

Figure 2. Performance on language, but not other cognitive tests, was worse in the high– WM-TSA group.
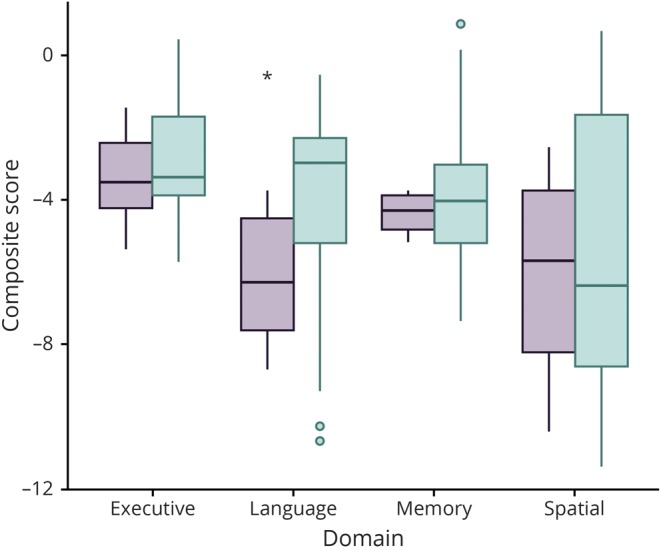
Comparison of the composite z score for each cognitive domain per high– and low–white matter thorn-shaped astrocytes (WM-TSA) groups. The only significant difference found was in the language domain (*p = 0.018). Green shows the low–WM-TSA group; purple shows the high–WM-TSA group.
WM-TSA contributed to worsening language and visuospatial functions independently of NFT burden
We also looked at whether there was an association between the density of WM-TSA and the burden of AD pathology represented by NFT density. The overlapping areas for NFT and the density of WM-TSA counts were middle frontal, superior temporal, and angular gyri. The Pearson coefficients of correlation between NFT and the density of WM-TSA were weak: 0.03 in the middle frontal gyrus, 0.11 in the superior temporal gyrus, and 0.12 in the angular gyrus. Similarly, there were no significant differences in NFT density between the participants who had WM-TSA in at least 1 brain region and the participants who did not have WM-TSA (data not shown). After NFT density was included in the linear regression models described above, the correlation between the density of WM-TSA and language composite z scores remained significant in the superior temporal and angular gyrus (df = 59, β = −0.02, SE = 0.01, p = 0.034 and df = 61, β = −0.02, SE = 0.00, p = 0.044, respectively). In addition, the correlation between the density of WM-TSA and visuospatial composite z scores remained significant in the angular gyrus (df = 60, β = −0.04, SE = 0.01, p = 0.009). These results suggest that, at least in the regions tested, WM-TSA contributes to worsening language and visuospatial function likely independently from regional NFT burden.
Discussion
Here, we investigated the clinical impact of TSA inclusion in the subcortical white matter in a well-characterized clinicopathologic series of 83 participants with AD pathology and heterogeneous clinical presentations. We found that higher WM-TSA densities in language-related and visuospatial-related brain regions, but not in other brain regions, correlate with worse language and visuospatial performance, respectively, measured by standardized neuropsychological testing, independently of global cognition deficits. Although we failed to find a direct association between the presence of WM-TSA and an atypical language presentation as reported by Munoz et al.,8 the strong correlation between WM-TSA densities in language-related brain regions and language supports their observations.
Clinicopathologic studies and, more recently, neuroimaging studies were critical in attributing brain regions to specific cognitive functions. Lesions in the classic Broca and Wernicke areas, corresponding to left inferior frontal gyrus, opercular area, and left temporoparietal junction, were associated with deficits in motor speech and language comprehension, respectively.27 Further studies using functional neuroimaging expanded the language network areas to include dorsolateral prefrontal cortex and inferior temporal gyrus.28 Investigation of neurodegenerative PPAs further clarified the role of specific brain regions in different aspects of language function.29 Language deficits in AD, for instance, correlate with atrophy and hypometabolism in the posterior parts of the superior and middle temporal gyrus and inferior parietal lobule and manifest primarily as impaired sentence repetition and impaired single word retrieval in spontaneous speech.29,30 Those deficits, when predominant in the early stages of a dementia syndrome and the absence of significant episodic memory impairment, characterize lvPPA, in which AD is the most common underlying pathology.18,31,32 Visuospatial dysfunction is an early event in AD.33 Visuospatial ability has been associated primarily with the right hemisphere with participation of the parietal, temporal, occipital regions, as well as the hippocampus. The right angular gyrus and, to a lesser extent, the left angular gyrus play a critical role in spatial cognition.34,35
Why some individuals with pure AD pathology manifest an atypical syndrome remains unanswered. Individuals presenting with PPA caused by AD pathology, for instance, tend to have a younger age at onset and to show an asymmetric distribution of NFT,36 a higher ratio of neocortical to entorhinal tangles,37 and a smaller influence of the APOE ε4 allele as a risk factor.38 Regarding anatomic distribution of pathology, recent clinicopathologic studies found that a higher semiquantitative burden of neuronal tau pathology in the superior and middle temporal gyrus and middle frontal gyrus in AD manifesting as a PPA syndrome,39,40 suggesting a distinct pattern of NFT distribution in atypical AD manifestation, may reflect a cortical proposed subtype of NFT deposition.41 Other autopsies studies unveiled comorbid conditions in cases of lvPPA with Lewy body pathology or FTLD.42 However, these cases are infrequent. To date, a common neuropathologic or genetic denominator unequivocally distinguishing AD presenting as PPA, including the logopenic variant, from an amnestic presentation remains elusive.
A converging body of evidence points to a role of astrocytic pathology in modifying the course of AD. Among astrocytic changes associated with aging, tau accumulation is common. However, this feature has been mostly neglected in neurodegenerative evaluations of the brain until recently when an international group of pathologists proposed a harmonizing strategy for classifying tau inclusions in astroglia collectively called ARTAG.9 A small but growing number of clinicopathologic studies have been exploring in-depth associations between ARTAG and cognitive deficits. In individuals ≥90 year of age, cortical ARTAG, but not limbic ARTAG, correlated with cognitive impairment.43 Another study found that ARTAG in the white matter correlated with worse verbal memory only in people in whom AD was the primary pathology.44 None of these recent studies considered WM-TSA as an independent variable because, in the ARTAG classification, all TSA were grouped regardless of their location. Thus, the very few studies investigating the role of WM-TSA as an independent driver of cognitive decline remain limited to the 1 study of Munoz et al.,8 2 others from the Mesulam3 and Bigio groups,45 and 1 from Lopez-Gonzalez et al.46 Munoz et al.8 suggested an association between WM-TSA and language presentations of AD because this pathology in language areas distinguished their language presentation from amnestic presentation cases. However, they also identified WM-TSA in other brain areas, mainly in frontotemporal regions, without a clear clinical impact. On the other hand, both studies from the Mesulam group failed to reproduce this association, although they found WM-TSA in cases of AD with severe language impairment.3,45 Lopez-Gonzalez and colleagues46 also failed to demonstrate an association in a pathologic series of 17 patients with WM-TSA in the medial temporal lobes that co-occurred with AD pathology. None of those cases were diagnosed with PPA in life, and 12 of them (Braak stage III–IV) died without any cognitive impairment. In our study with a larger sample size, we also failed to find a correlation between WM-TSA and an atypical AD presentation. We are unaware of studies investigating the role of WM-TSA in visuospatial function.
However, leveraging the in-depth clinical characterization of our cohort, we were able to identify a robust correlation between the accumulation of WM-TSA and language and possibly visuospatial dysfunction. Whether WM-TSA causes direct neurotoxicity or is a marker of an underlying process that increases neurotoxicity remains an unanswered question for future studies. In fact, the mechanisms underlying the possible neurotoxicity of WM-TSA are unknown, but experimental work suggests that accumulation of tau in astrocytes increases neuronal vulnerability to death and dysfunction.47 Astrocytes are essential in maintaining the integrity of the blood-brain barrier and synaptic function, among other functions. Increasing evidence points to a role of tau astrogliopathy in increasing neuronal vulnerability through inflammatory48 and blood-brain barrier dysfunction mechanisms.49 In Drosophila tauopathy models, the coexpression of tau in neurons, astrocytes, and other glial cells acts synergistically to cause cell death.50 We speculate that cell specificity may play a role because WM-TSA is thought to be specific to fibrous astrocytes located in the white matter and to interfere with brain connections important for language functioning.
Curiously, this association was not observed for other cognitive domains tested. Why the language and visuospatial networks are particularly vulnerable to WM-TSA is also unknown. We had a broader representation of language-related than other domain areas, and the neuropathologic examination was restricted mostly to the left hemisphere, which could confound the results. In fact, in our unbiased clustering analysis, despite an association with language functioning, we failed to find an association between higher accumulation of WM-TSA in the high–WM-TSA group and other domains, including visuospatial, suggesting that the impact of WM-TSA deposition is dose dependent and regional or possibly that language-related regions are particularly vulnerable to the effects of WM-TSA deposition. Further studies with better representation of the right hemisphere are needed to elucidate this question. Regardless, our results suggest that WM-TSA may represent a unique and less benign entity, distinct from other ARTAG subtypes, and isolating WM-TSA from other types of TSA deposits in future investigations may be necessary to pinpoint its role.
This study has strengths and limitations. Limitations of our study include a relatively low number of cases with an atypical presentation, including lvPPA containing WM-TSA. However, these diagnoses are rare, and our collection is relatively large for atypical AD cases, and the exclusion of cases with non-AD neuropathologic changes minimizes confounders. In addition, only 1 hemisphere (mostly left) was examined, preventing comparison of the densities of WM-TSA across the hemispheres and a better understanding of the impact of WM-TSA in cognitive function other than language. Finally, this is a series enriched for atypical AD presentations, which are rare; thus, the demographic features differ from community-based series on AD. Furthermore, the time gap between the last language evaluation and death was relatively long in some cases, mostly because it is impractical to measure neuropsychological function once the symptoms reach a certain level of severity. Our study design is not sensitive to inform at which stage WM-TSA started to develop and how it progressed. Thus, we cannot be certain whether WM-TSA was present at the time of the neuropsychological testing. Unfortunately, to date, methodologic limitations preclude the identification of any form of ARTAG in living patients.
Our findings corroborate the hypothesis that WM-TSA may contribute to clinical deficits in individuals with AD, especially regarding language dysfunction and possibly visuospatial function. WM-TSA should be added to the list of neuropathologic lesions routinely examined in postmortem brain assessments related to aging and dementia, in addition to the known limbic TDP-43 inclusions and comorbid α-synucleinopathy pathology. Our results justify future research exploring the mechanisms underlying WM-TSA accumulation and improving our understanding of the WM-TSA–specific contribution to cognitive impairment.
Acknowledgment
The authors thank the patients and their families for their invaluable contribution to brain aging and neurodegenerative disease research. Dr. Resende is an Atlantic Fellow for Equity in Brain Health and thanks the fellowship for supporting her work.
Glossary
- AD
Alzheimer disease
- ARTAG
aging-related tau astrogliopathy
- ATAC
argyrophilic thorn-shaped astrocyte clusters
- CDR
Clinical Dementia Rating
- FTLD
frontotemporal lobar degeneration
- lvPPA
logopenic variant primary progressive aphasia
- MAC
Memory and Aging Center
- MCI
mild cognitive impairment
- NFT
neurofibrillary tangles
- PPA
primary progressive aphasia
- SE
standard error
- TDP-43
TAR DNA binding protein-43
- TSA
thorn-shaped astrocytes
- UCSF
University of California, San Francisco
- WM-TSA
white matter thorn-shaped astrocytes
Appendix. Authors
Study funding
This study was supported by NIH grant K24AG053435 and institutional NIH grants P50AG023501, P01AG019724. M.L.G.T. is funded by NIH K24DC015544A and R01NS50915. E.P.F.R. was funded by the Global Brain Health Institute.
Disclosure
E. Resende’s fellowship was funded by the Global Brain Health Institute. A. Nolan and C. Petersen report no disclosures relevant to the manuscript. A. Ehrenberg has accepted financial compensation for scientific consulting services to Epiodyne, Inc. S. Spina is supported by NIH K08 AG052648. I. Allen, H. Rosen, and J. Kramer report no disclosures relevant to the manuscript. B. Miller serves as board member on the John Douglas French Alzheimer's Foundation and Larry L. Hillblom Foundation; serves as a consultant for TauRx, Ltd, Allon Therapeutics, Siemens, BMS, the Tau Consortium, and the Consortium for Frontotemporal Research; has received institutional support from Novartis; and is funded by NIH grants P50AG023501, P01AG019724, P50 AG1657303, and the State of California. W. Seeley is funded by NIH grants, Program Project Grant, Alzheimer Disease Research Center, the Bluefield Project to Cure FTD, and the Tau Consortium. M. Gorno-Tempini is supported by NIH K24DC015544A and R01NS50915. Z. Miller is funded by an NIH/National Institute on Aging K23 AG048291 grant and receives additional funds from the Hellman Foundation and the Arking Foundation for Frontotemporal Dementia. L. Grinberg is supported by NIH K24AG053435, R01AG060477, R56AG057528, U54 NS100717, and R01AG056573; the Rainwater Charity Foundation; and the Brightfocus Foundation. She also receives grants from AVID Radiotherapeutics and Eli Lilly. Go to Neurology.org/N for full disclosures.
References
- 1.Korczyn AD. Is Alzheimer's disease a homogeneous disease entity? J Neural Transm (Vienna) 2013;120:1475–1477. [DOI] [PubMed] [Google Scholar]
- 2.Ossenkoppele R, Pijnenburg YA, Perry DC, et al. The behavioural/dysexecutive variant of Alzheimer's disease: clinical, neuroimaging and pathological features. Brain 2015;138:2732–2749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mesulam M, Wicklund A, Johnson N, et al. Alzheimer and frontotemporal pathology in subsets of primary progressive aphasia. Ann Neurol 2008;63:709–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee SE, Rabinovici GD, Mayo MC, et al. Clinicopathological correlations in corticobasal degeneration. Ann Neurol 2011;70:327–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Crutch SJ, Lehmann M, Schott JM, Rabinovici GD, Rossor MN, Fox NC. Posterior cortical atrophy. Lancet Neurol 2012;11:170–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Josephs KA, Whitwell JL, Tosakulwong N, et al. TAR DNA-binding protein 43 and pathological subtype of Alzheimer's disease impact clinical features. Ann Neurol 2015;78:697–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Josephs KA, Dickson DW, Tosakulwong N, et al. Rates of hippocampal atrophy and presence of post-mortem TDP-43 in patients with Alzheimer's disease: a longitudinal retrospective study. Lancet Neurol 2017;16:917–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Munoz DG, Woulfe J, Kertesz A. Argyrophilic thorny astrocyte clusters in association with Alzheimer's disease pathology in possible primary progressive aphasia. Acta Neuropathol 2007;114:347–357. [DOI] [PubMed] [Google Scholar]
- 9.Kovacs GG, Ferrer I, Grinberg LT, et al. Aging-related tau astrogliopathy (ARTAG): harmonized evaluation strategy. Acta Neuropathol 2016;131:87–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cairns NJ, Bigio EH, Mackenzie IR, et al. Neuropathologic diagnostic and nosologic criteria for frontotemporal lobar degeneration: consensus of the Consortium for Frontotemporal Lobar Degeneration. Acta Neuropathol 2007;114:5–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McKeith IG, Dickson DW, Lowe J, et al. Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology 2005;65:1863–1872. [DOI] [PubMed] [Google Scholar]
- 12.Montine TJ, Phelps CH, Beach TG, et al. National Institute on Aging-Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease: a practical approach. Acta Neuropathol 2012;123:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mackenzie IR, Neumann M, Baborie A, et al. A harmonized classification system for FTLD-TDP pathology. Acta Neuropathol 2011;122:111–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Suemoto CK, Leite REP, Ferretti-Rebustini REL, et al. Neuropathological lesions in the very old: results from a large Brazilian autopsy study. Brain Pathol 2019;29:771–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Terry RD, Hansen LA, DeTeresa R, Davies P, Tobias H, Katzman R. Senile dementia of the Alzheimer type without neocortical neurofibrillary tangles. J Neuropathol Exp Neurol 1987;46:262–268. [DOI] [PubMed] [Google Scholar]
- 16.McKhann GM, Knopman DS, Chertkow H, et al. The diagnosis of dementia due to Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement 2011;7:263–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Crutch SJ, Schott JM, Rabinovici GD, et al. Consensus classification of posterior cortical atrophy. Alzheimers Dement 2017;13:870–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gorno-Tempini ML, Hillis AE, Weintraub S, et al. Classification of primary progressive aphasia and its variants. Neurology 2011;76:1006–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Armstrong MJ, Litvan I, Lang AE, et al. Criteria for the diagnosis of corticobasal degeneration. Neurology 2013;80:496–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rascovsky K, Hodges JR, Knopman D, et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain 2011;134:2456–2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Albert MS, Dekosky ST, Dickson D, et al. The diagnosis of mild cognitive impairment due to Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement 2011;7:270–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Staffaroni AM, Brown JA, Casaletto KB, et al. The longitudinal trajectory of default mode network connectivity in healthy older adults varies as a function of age and is associated with changes in episodic memory and processing speed. J Neurosci 2018;38:2809–2817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dunn LM, Dunn DM, Pearson A. PPVT-4 : Peabody Picture Vocabulary Test. Minneapolis, MN: Pearson Assessments; 2007. [Google Scholar]
- 24.O'Bryant SE, Waring SC, Cullum CM, et al. Staging dementia using Clinical Dementia Rating Scale Sum of Boxes scores: a Texas Alzheimer's Research Consortium study. Arch Neurol 2008;65:1091–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 1991;82:239–259. [DOI] [PubMed] [Google Scholar]
- 26.Brock G, Pihur V, Datta S, Datta S. clValid : an R package for cluster validation. J Stat Softw 2008;25:1–22. [Google Scholar]
- 27.Mesulam MM, Thompson CK, Weintraub S, Rogalski EJ. The Wernicke conundrum and the anatomy of language comprehension in primary progressive aphasia. Brain 2015:2423–2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fedorenko E, Thompson-Schill SL. Reworking the language network. Trends Cogn Sci 2014;18:120–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mesulam MM, Rogalski EJ, Wieneke C, et al. Primary progressive aphasia and the evolving neurology of the language network. Nat Rev Neurol 2014;10:554–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gorno-Tempini ML, Brambati SM, Ginex V, et al. The logopenic/phonological variant of primary progressive aphasia. Neurology 2008;71:1227–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Spinelli EG, Mandelli ML, Miller ZA, et al. Typical and atypical pathology in primary progressive aphasia variants. Ann Neurol 2017;81:430–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rohrer JD, Rossor MN, Warren JD. Alzheimer's pathology in primary progressive aphasia. Neurobiol Aging 2012;33:744–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Quental NB, Brucki SM, Bueno OF. Visuospatial function in early Alzheimer's disease: the use of the Visual Object and Space Perception (VOSP) battery. PLoS One 2013;8:e68398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Berryhill ME, Olson IR. The right parietal lobe is critical for visual working memory. Neuropsychologia 2008;46:1767–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tres ES, Brucki SMD. Visuospatial processing: a review from basic to current concepts. Dement Neuropsychol 2014;8:175–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mesulam MM, Weintraub S, Rogalski EJ, Wieneke C, Geula C, Bigio EH. Asymmetry and heterogeneity of Alzheimer's and frontotemporal pathology in primary progressive aphasia. Brain 2014;137:1176–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Josephs KA, Dickson DW, Murray ME, et al. Quantitative neurofibrillary tangle density and brain volumetric MRI analyses in Alzheimer's disease presenting as logopenic progressive aphasia. Brain Lang 2013;127:127–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gefen T, Gasho K, Rademaker A, et al. Clinically concordant variations of Alzheimer pathology in aphasic versus amnestic dementia. Brain 2012;135:1554–1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Giannini LAA, Irwin DJ, McMillan CT, et al. Clinical marker for Alzheimer disease pathology in logopenic primary progressive aphasia. Neurology 2017;88:2276–2284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Petersen C, Nolan AL, de Paula Franca Resende E, et al. Alzheimer's disease clinical variants show distinct regional patterns of neurofibrillary tangle accumulation. Acta Neuropathol 2019;138:597–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Murray ME, Graff-Radford NR, Ross OA, Petersen RC, Duara R, Dickson DW. Neuropathologically defined subtypes of Alzheimer's disease with distinct clinical characteristics: a retrospective study. Lancet Neurol 2011;10:785–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Teichmann M, Kas A, Boutet C, et al. Deciphering logopenic primary progressive aphasia: a clinical, imaging and biomarker investigation. Brain 2013;136:3474–3488. [DOI] [PubMed] [Google Scholar]
- 43.Robinson JL, Corrada MM, Kovacs GG, et al. Non-Alzheimer's contributions to dementia and cognitive resilience in the 90+ Study. Acta Neuropathol 2018;136:377–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kovacs GG, Robinson JL, Xie SX, et al. Evaluating the patterns of aging-related tau astrogliopathy unravels novel insights into brain aging and neurodegenerative diseases. J Neuropathol Exp Neurol 2017;76:270–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bigio EH, Mishra M, Hatanpaa KJ, et al. TDP-43 pathology in primary progressive aphasia and frontotemporal dementia with pathologic Alzheimer disease. Acta Neuropathol 2010;120:43–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lopez-Gonzalez I, Carmona M, Blanco R, et al. Characterization of thorn-shaped astrocytes in white matter of temporal lobe in Alzheimer's disease brains. Brain Pathol 2013;23:144–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kahlson MA, Colodner KJ. Glial tau pathology in tauopathies: functional consequences. J Exp Neurosci 2015;9:43–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chun H, Marriott I, Lee CJ, Cho H. Elucidating the interactive roles of glia in Alzheimer's disease using established and newly developed experimental models. Front Neurol 2018;9:797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Blair LJ, Frauen HD, Zhang B, et al. Tau depletion prevents progressive blood-brain barrier damage in a mouse model of tauopathy. Acta Neuropathol Commun 2015;3:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Colodner KJ, Feany MB. Glial fibrillary tangles and JAK/STAT-mediated glial and neuronal cell death in a Drosophila model of glial tauopathy. J Neurosci 2010;30:16102–16113. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All the data are available on reasonable request.