

Abstract
Objective
To test the hypothesis that the trajectory of functional connections over time of the striatum and the cerebellum differs between presymptomatic patients with the Huntington disease (HD) gene expansion (GE) and patients with a family history of HD but without the GE (GNE), we evaluated functional MRI data from the Kids-HD study.
Methods
We utilized resting-state, functional MRI data from participants in the Kids-HD study between 6 and 18 years old. Participants were divided into GE (CAG 36–59) and GNE (CAG <36) groups. Seed-to-seed correlations were calculated among 4 regions that provide input signals to the anterior cerebellum: (1) dorsocaudal putamen, (2) globus pallidus externa, (3) subthalamic nucleus, and (4) pontine nuclei; and 2 regions that represented output from the cerebellum: the dentate nucleus to the (1) ventrolateral thalamus and (2) dorsocaudal putamen. Linear mixed effects regression models evaluated differences in developmental trajectories of these connections over time between groups.
Results
Four of the six striatal–cerebellum correlations showed significantly different trajectories between groups. All showed a pattern where in the early age ranges (6–12 years) there was hyperconnectivity in the GE compared to the GNE, with those trajectories showing linear decline in the latter half of the age range.
Conclusion
These results parallel previous findings showing striatal hypertrophy in children with GE as early as age 6. These findings support the notion of developmentally higher connectivity between the striatum and cerebellum early in the life of the child with HD GE, possibly setting the stage for cerebellar compensatory mechanisms.
Huntington disease (HD) is an inherited neurodegenerative disease that produces mutant huntingtin protein (mHTT).1 Traditionally, HD has been thought of as a toxic gain-of-function disease where the presence of mHTT results in neuronal death.2 However, loss of the normal function of wild-type huntingtin protein in HD significantly affects normal brain development.3–7 The neurodevelopmental hypothesis of neurodegeneration posits that early compensatory mechanisms allow for normal function in childhood despite abnormal neurodevelopment. Thus a developmentally aberrant circuit remains in “mutant steady state” due to compensation.2,8,9
Our group studies children ages 6–18 years at risk for HD. These children are genotyped into a gene-expanded (GE) group or a gene nonexpanded (GNE) group. We recently demonstrated that GE children who were decades away from their predicted motor onset showed striatal hypertrophy as early as age 6, and had a different developmental trajectory compared to GNE children.10 Because these participants are asymptomatic, compensation for the developmentally abnormal striatum may be occurring, but it is unclear which brain regions are responsible.
The indirect pathway (figure 1) is a primary site of initial decompensation in HD pathogenesis.11 The cerebellum has been shown to be integrated into the indirect pathway.12–14 It is possible that in development, the cerebellum may attempt to compensate for a developmentally aberrant striatum. This study was designed to evaluate the development of functional connectivity in striatal–cerebellar circuity as measured by resting-state fMRI (rs-fMRI) in children in the GE group compared to the GNE group.
Figure 1. Cerebellar integration with indirect pathway.
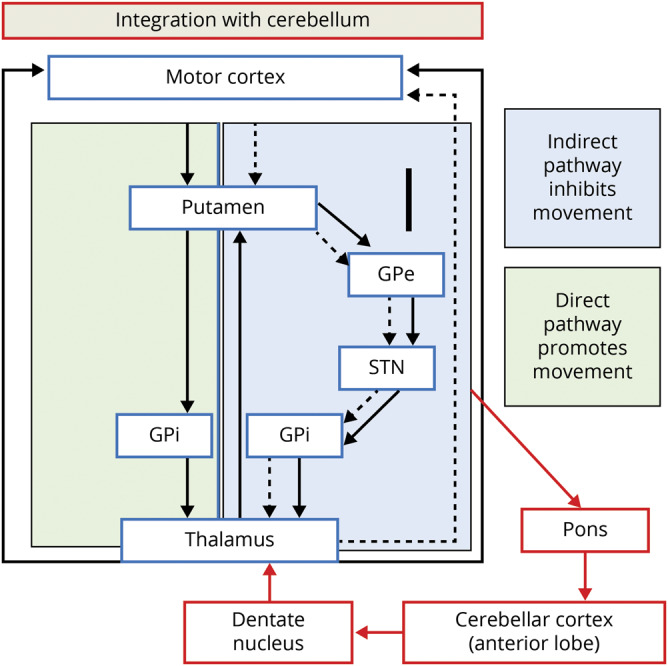
The portion of the figure in the green box represents the direct pathway (promotes movement) and the section in the blue box represents the indirect pathway (inhibits movement). In Huntington disease, it is the indirect pathway that degenerates first, leading to lack of inhibition and involuntary movements (chorea). The cerebellum is integrated into striatal circuity through the indirect pathway. Thus the cerebellum could compensate for a faulty indirect pathway, restoring balance and preventing the development of involuntary movements. Red arrows indicate where the cerebellum is integrated into the indirect pathway. GPe = globus pallidus externa; GPi = globus pallidus interna; STN = subthalamic nucleus. Figure adapted from references 13 and 14.
Methods
Description of data
This study utilized data obtained from the Kids-HD study, a novel observational study that collected clinical and neuroimaging data from participants between the ages of 6 and 18. The study recruited participants from around the United States to the University of Iowa who were at risk for developing HD based on their family history (i.e., all participants had a parent or grandparent with HD). Recruitment and assessments occurred between May 2009 and January 2018. All participants were considered by their parents to have no signs or symptoms of HD. These children underwent genetic testing for the HD gene mutation. These results were not disclosed to participants or their family members, nor were the results included in their medical record. Genetic results were obtained for research purposes only. Furthermore, the deidentified results of the genetic tests were only made available to a third party to ensure that all members of the research team remained blinded to the genetic results. Participants with a CAG repeat length of ≥36 were included in the GE group and participants with CAG <36 were included in the GNE group.
The Kids-HD study employed an accelerated longitudinal design (ALD). This method allows for more efficient study of brain development in pediatric patients by approximating growth curves by gathering data from participants over a large span of ages within a relatively short time frame.15 Using an ALD, participants can be seen once or multiple times.
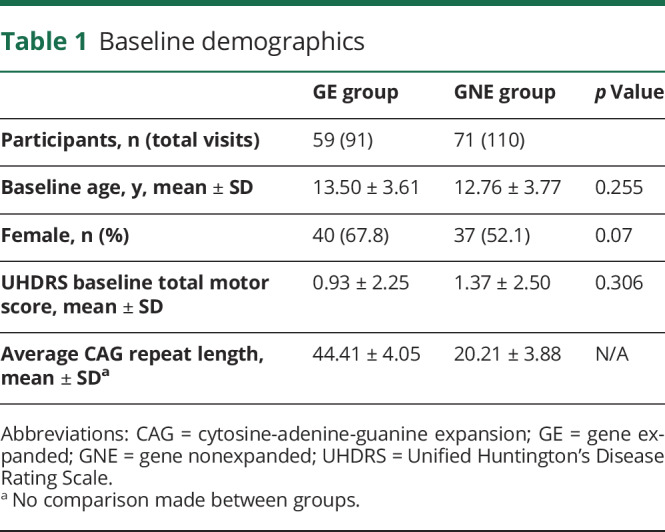
The current sample is a subsample of a recently published article on striatal growth and development and includes only those children with resting state data.10 The original sample included 75 participants in the GE group and 97 in the GNE group. In the current subsample, there were 59 unique participants in the GE group who accounted for 91 visits compared to 71 participants in the GNE group who accounted for 110 visits. Within the GE group, 34 participants (57.6%) had only 1 visit. Among the participants with multiple visits, 1 participant had 4 visits, 5 participants had 3 visits, and 19 participants had 2 visits. For the 25 participants with more than 1 visit, the mean time being followed in the study was 2.67 years (SD, 1.03; range, 0.91–4.98) and among all participants in the GE group, the mean years in the study was 0.88 (SD, 1.34; range, 0–4.98). Within the GNE group, 43 participants (60.5%) had only 1 visit. There were 11 participants who had 3 visits and 17 participants who had 2 visits. The average time in the study for those 28 participants with more than 1 visit was 3.02 years (SD, 1.28; range, 1.06–5.02). Among all patients in the GNE group, the mean years in the study was 1.19 (SD, 1.69; range, 0–5.02) (figure e-1, doi.org/10.5061/dryad.qbzkh18cw). There were no statistically significant differences between groups regarding sex distribution or baseline age. Because all participants were considered to be asymptomatic, there was no significant difference between groups regarding the baseline total motor score as measured by the Unified Huntington's Disease Rating Scale (table 1).16
Table 1.
Baseline demographics
Standard protocol approvals, registrations, and patient consents
The institutional review board at the University of Iowa approved this study. The participants in this study were younger than 18 years at enrollment. Therefore, parents or guardians provided written informed consent and children provided written assent.
Imaging acquisition
A total of 201 imaging studies were included in these analyses. Of those, 155 (GE, 66 scans; GNE, 89 scans) were acquired on a research-dedicated 3T Siemens TIM Trio scanner (Siemens Medical Solutions, Malvern, PA). The remaining 46 (GE, 25 scans; GNE, 21 scans) were acquired on a 3T GE Discovery scanner (GE Healthcare, Waukesha, WI). T2*-weighted echoplanar images were acquired (echo time, 30 ms; repetition time, 2,000 ms; matrix, 64 × 64; field of view, 220 × 220 mm) for 6 minutes. Participants were instructed to keep their eyes closed, stay awake, and not think of anything specific. Preprocessing of rs-fMRI data included slice timing correction, motion correction, and bandpass temporal filter of 0.008 < f < 0.08 Hz.17 Potential confounding sources of signal variance, such as white matter, CSF, motion parameter covariates, and global signal regression, were removed using linear regression and data deletion techniques.18 We performed motion scrubbing to censor time of repetitions with excess motion.18 The primary outcome measures were those of striatal–cerebellar circuitry. The mean time series blood oxygenation level–dependent signal from a primary seed region was extracted and correlated with the time series signal of the target region of interest (ROI). This measure indicates functional connectivity strength between the 2 seeds. Seeds were placed based on previously published reports of resting state connectivity and 4 primary seeds were selected to target measures of input into the anterior cerebellum: (1) dorsocaudal putamen (dPU),19 (2) globus pallidus externus (GPE),20 (3) subthalamic nucleus (STN),20 and (4) pontine nuclei (PN). For PN seeds, novel ROIs were created as 6 mm diameter spheres from peak group mean coactivation. Seeding was started in the motor cortex hand ROI to validate functional connections with the cerebellum.21 As both the cerebellum and motor cortex assist in movement control, additional analysis was carried out to examine connections between the striatum and the motor cortex. The target region of the anterior cerebellum was defined by a mask that included the entire region as determined by BRAINSTools. It was decided to use the global mask of this region rather than a specific seed that would have been restrained to certain motor areas (such as hand, mouth, foot). This is important given the potential for poor coverage of the inferior cerebellum when specific cerebellar regional maps are employed. Preliminary analysis utilizing individual seeds (mouth, hand, foot) produced similar but less robust findings than the global anterior cerebellar mask (data not shown). To represent cerebellar output to the striatum, a primary seed was placed in the dentate nucleus (DN)22 with target seeds of the ventrolateral nucleus of the thalamus (VL)23 and dPU.19
Statistical analysis
The primary outcome measure was the developmental trajectory of each seed-to-seed correlation compared across the 2 groups. Linear mixed effects regression models were constructed. The dependent variable was the seed-to-seed functional correlation (R2) between the ROIs. The models included random effects per participant to account for participants who had multiple visits, and per family to account for siblings within the sample. Sex and scanner (to account for potential scanner effects) were included as fixed variables in the model. Post hoc analyses were conducted on a subset of healthy controls and demonstrated that a significant scanner effect was not detectable for any of the dependent variables of interest. A sex by group interaction term was included in all applicable models. The age by group interaction term was the main effect of interest to quantify the age-based trajectory of the functional correlations between the GE and GNE groups.
The secondary outcome was to measure the effect of CAG repeat on developmental trajectory. This analysis was limited to those correlations that had a significant group by age interaction to limit the number of analyses done and minimize the potential for a type II error. Similar linear mixed effects regression models were used to predict strength of seed to seed correlation based on CAG repeat within the GE group only. An age by CAG interaction term was the effect of interest, evaluating whether developmental trajectories changed depending upon the length of the gene mutation. RStudio (version 3.4.4) was used for all analyses and a p value of <0.05 was considered significant for all analyses.
Data availability
Deidentified data will be shared by reasonable request from any qualified investigator by the corresponding author for purposed of replicating procedures and results.
Results
Table 2 shows the primary effects of age, sex, and group on connectivity as well as the significant interactions. For the primary outcome of interest (a group by age interaction), there were significant differences in developmental trajectory between the GE and GNE groups in the strength of 3 of the 4 inputs to the cerebellum seed-to-seed correlations: (1) GPE to anterior lobe of the cerebellum (aCB) (p = 0.003), (2) STN to aCB (p = 0.0001), and (3) PN to aCB (p = 0.033) (table 2 and figure 2, A–C). In all 3, the pattern was that the GE group began at the youngest age with significantly higher connectivity compared to the GNE group, and then had a declining strength of connection over time. In comparison, the GNE trajectories for these 3 input ROIs were slightly positive with a small increase in connectivity over time (GPE to aCB) or with no significant change in connectivity (STN and PN to aCB). The trajectories between the dPU and aCB were not significantly different between groups (p = 0.403).
Table 2.
Primary outcome measures

Figure 2. Striatal input to cerebellum.

(A–C) Predicted values from a linear mixed effects regression model of the functional connectivity (R2) between the striatal–cerebellar regions of interest (dependent variables) over time between groups (age × group interaction term). The model controlled for age, sex, and scanner, and included a sex × group interaction term and a random effect term per the participant's slope of age, and a random effect term per family to account for participants who were siblings. aCB = anterior lobe of the cerebellum; dPU = dorsocaudal putamen; GE = gene-expanded; GNE = gene nonexpanded; GPE = globus pallidus externus; PN = pontine nucleus; STN = subthalamic nucleus.
In regard to the cerebellar output ROIs, the functional connectivity between the DN and VL changed at significantly different rates between the groups (p = 0.045) (figure 3). Similar to the input seed-to-seed correlations, the connectivity in the GE group began at the youngest age remarkably higher than the GNE, but then had a significant decline in connectivity over time. In contrast, the GNE group showed a positive slope with a modest increase in connectivity in this age range. There was no group difference in trajectory for the DN to dPU seed-to-seed correlations (p = 0.444).
Figure 3. Cerebellar output to striatum.
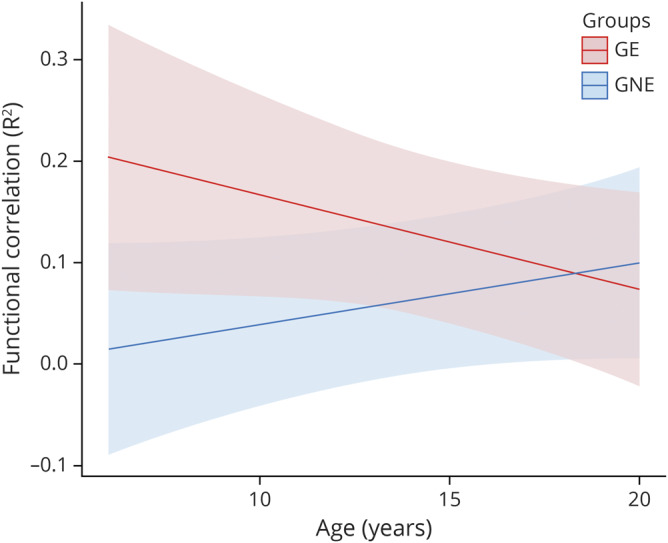
This figure represents the predicted values from a linear mixed effects regression model of the functional connectivity (R2) between the dentate nucleus and ventrolateral nucleus of the thalamus (dependent variable) over time between groups (age × group interaction term). The model controlled for age, sex, and scanner, and included a sex × group interaction term and a random effect term per participant's slope of age, and a random effect term per family to account for participants who were siblings. GE = gene expanded; GNE = gene nonexpanded.
In the models examining group by age interaction for the input regions GPE to aCB and STN to aCB, there were also significant sex by group interactions (table 2). Post hoc analysis showed that there was a significant difference in overall functional connectivity in these 2 seed-to-seed correlations between male participants in the GE group compared to the GNE group; however, there were no differences between the groups among female participants (figure e-2, doi.org/10.5061/dryad.qbzkh18cw). Given these sex differences, we aimed to determine if the trajectory over time was significantly different between male and female participants. Therefore, we reran the models that had a significant sex by group effect (GPE to aCB and STN to aCB) and included a triple interaction of group by age by sex. The triple interaction term was not significant for either input region (GPE to aCB, t = 0.925, p = 0.356; and STN to aCB, t = −0.445, p = 0.657) (figure e-3, doi.org/10.5061/dryad.qbzkh18cw), suggesting that although GE male participants had higher overall connectivity compared to GNE male participants while the female participants did not, there were no sex differences in the trajectories over time. Figure e-4 (doi.org/10.5061/dryad.qbzkh18cw) shows the trajectories for both GPE to aCB and STN to aCB broken down by sex. Although not statistically different, the slope of the trajectory for the GE female participants is steeper than the male participants and crossing over the GNE group at an early age, with a pattern leaving young GE female participants with hyperconnectivity in these regions but older GE female participants having hypoconnectivity (figure e-4, doi.org/10.5061/dryad.qbzkh18cw). In contrast, the GE male participants had a slower decline in connectivity over time, leaving their mean connectivity measures higher than the GNE male participants for most of the age range, resulting in a mean higher connectivity measure. These sex-specific differences in neurodevelopment may be attributable to the fact that the onset of puberty (and subsequent neurodevelopmental changes) is earlier in girls than in boys.24
After controlling for the higher level group by age and group by sex interactions, there remains a significant group effect for the GPE to aCB and STN to aCB seed-to-seed correlations indicating a global measure of hyperconnectivity in these regions for the GE group compared to the GNE group. Similarly, there remained a primary effect of sex on the output DN to VL correlation, with male participants having higher connectivity compared to female participants. Finally, there was a primary effect of age on the input GPE to aCB and STN to aCB correlations, suggesting these regions have an overall decrement in connectivity over this age range; however, this seems to be driven primarily by the GE group (table 2).
For the secondary outcomes, the CAG repeat number as a function of age did not significantly predict functional correlations between the GPE to aCB (p = 0.732), the PN to aCB (p = 0.075), or the DN to VL (p = 0.413). However, the CAG by age interaction term significantly predicted the functional connectivity between the STN and aCB (table 3). Specifically, the longer the CAG repeat length, the faster the rate of decline in functional connectivity between these brain regions (figure 4).
Table 3.
Secondary outcome measures
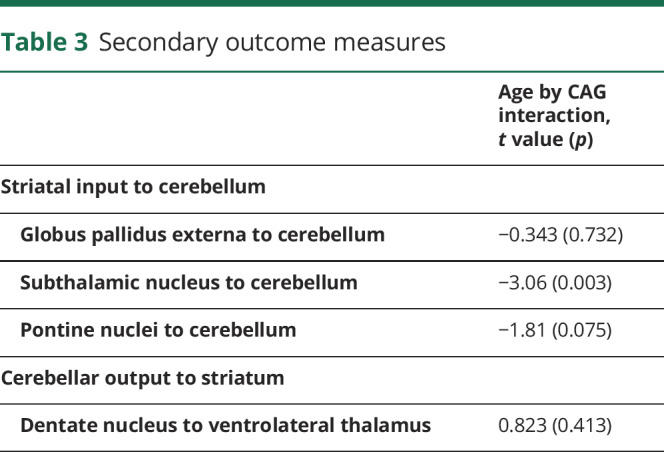
Figure 4. CAG effect over time in functional correlations between anterior lobe of the cerebellum and subthalamic nucleus.
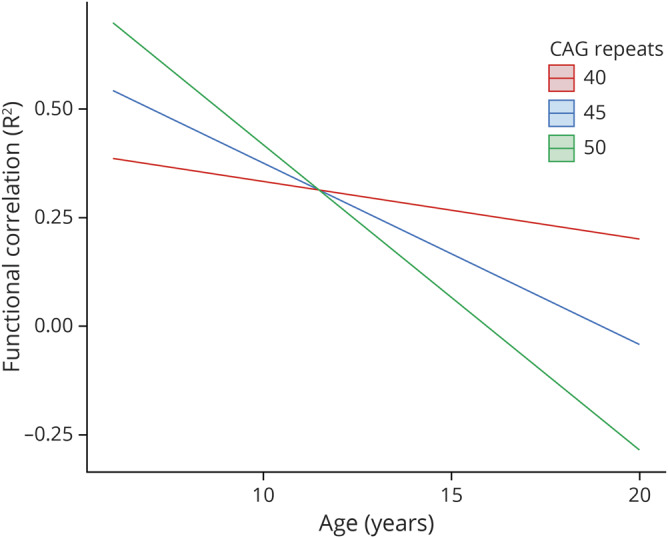
CAG = cytosine-adenine-guanine.
Discussion
In this study, we demonstrate for the first time that the presence of mHTT seems to significantly alter cerebellar–striatal circuitry decades prior to the anticipated onset of HD. Specifically, we observed hyperconnectivity of both the inputs from the striatum to the cerebellum, and the outputs of the cerebellum, early in life of GE children, and this connectivity decreases substantially over time. This is in contrast to the connectivity of the striatum and the cerebellum in the GNE children, which showed either no significant change or slight increase in connectivity over time. These findings parallel the structural imaging findings of the same group showing that the striatum is enlarged in GE children compared to GNE children around the age of 6, but then declines in volume over time, implicating that the striatal enlargement is associated with stronger cerebellum connectivity.10
These results provide compelling evidence for the neurodevelopmental theory of neurodegeneration that posits the primary pathology of HD is that of abnormal development of specific regions or circuits, and that despite abnormal development, functioning of the brain circuits remain normal during childhood due to developmental compensatory mechanisms. In the case of HD, it suggests that the striatum is preferentially affected by mHTT in development and that the functional connectivity to the cerebellum may play a compensatory role to maintain normal function during childhood. These compensatory mechanisms may also explain why significant striatal degeneration is appreciable via structural imaging studies in the absence of chorea.25,26 Enlarged volumes of brain structures typically signal abnormal development,10 and there is no functional evidence that the GE children are different from the GNE children in terms of motor function. Therefore, the early striatal enlargement could be considered to be abnormal and the hyperconnectivity to the cerebellum allows the abnormal striatum to function normally (i.e., prevent motor symptoms). Tracing studies have shown the bidirectional communication that occurs between the cerebellum and basal ganglia. Specifically, output from the cerebellum to the striatum occurs via the DN, and the cerebellum receives input from the striatum via output from the STN.27 The increased connectivity of the striatal input to the cerebellum may be expected given striatal enlargement, but the increase in the output from the cerebellum back to the striatum (via the thalamus) is the finding that most closely supports the concept of potential cerebellar compensation.
Also in parallel with the structural imaging findings, the current report found that some aspects of connectivity (specifically the STN to aCB) were modified by CAG repeat length. Similarly, the functional connectivity between the PN and aCB demonstrated a similar pattern of CAG modification, but the results did not reach the threshold for significance (p = 0.075). The shape of the GE trajectory showed higher functional connectivity earlier in life that declined linearly over time. This is true for both striatal volume in the previous article10 and the connectivity measures here. CAG repeat affects this pattern by altering the slope of the change, with higher repeats creating higher initial values and steeper slopes over time. One potential explanation for this finding is that the STN is presumed to be the only excitatory nucleus within the basal ganglia, making it an important player in regards to basal ganglia output.14 We know that the basal ganglia is the primary brain region affected in HD,26 so STN-mediated output may be greatly reduced. If the cerebellum is playing a compensatory role, it is reasonable to assume that the primary output region of the diseased region would be targeted. The STN also seems to target the pontine nuclei, which may explain why CAG modification of that cerebellar functional connection approaches significance.14
Given the loss of strength of connectivity over time in the GE participants, there seems to be some evidence to suggest that it is the loss of these connections over time that may underlie eventual striatal dysfunction and symptom onset. Previous PET studies have shown cerebellar and thalamic hypermetabolism in preHD individuals28,29 and only when these measures dropped below normal did the patients cross over into motor diagnosis.29 If the cerebellum is indeed acting as compensation for a faulty striatum, it may be the loss of compensation (rather than primary striatal dysfunction) that is responsible for motor onset of HD.
There are important limitations to this work. First, it is important to note that our use of rs-fMRI does not allow us to present physical connections between the cerebellum and striatum; rather, these results demonstrate functional connectivity between these brain regions. Another limitation is the potential for scanner effects to affect the results. As noted previously, post hoc analyses on healthy controls demonstrated that scanner effects did not significantly alter the results of the variables of interest. A covariate was included in all models to account for differences in scanner used. This variable did not significantly contribute to any of the models presented. Another limitation of this study is that the participants are, on average, decades from their anticipated age at motor onset. While this represents a unique opportunity to investigate the effect of mHTT on normal development, it is difficult to correlate these findings to clinical manifestations of HD. For example, it is unclear if the functional connections between the cerebellum and basal ganglia are strengthened in participants who are beginning to exhibit chorea. Furthermore, it is unclear if the strength of these functional connections affects cognitive or neuropsychological test results. As a result, it is difficult to hypothesize further about potential cause and effect relationships between these findings and the clinical phenotype of HD.
Strengths of this study include the relatively large sample size of a unique participant population. All the participants in this analysis have a family history of HD. Given that this study only included pediatric participants, we presume that all participants are living in a household with another person with HD. Environmental stressors that may arise secondary to this should be relatively balanced between groups. Volumetric and functional changes may also occur because of normal neurodevelopment. Our ability to have a comparator group to account for this is also a major strength of this study. Furthermore, the use of mixed modeling allows us to utilize all available data from qualifying individuals.
Overall, this study provides the first evidence that mHTT may affect cerebellar–striatal circuits early in life in children with the gene mutation for HD. It also may provide further evidence that the cerebellum may play a compensatory role in HD. Regardless, this study demonstrates the need to understand how the mHTT affects neurodevelopment. This is especially important given the emergence of new mHTT-lowering therapies. It is imperative that we understand the potential ramifications of lowering HTT levels on neurodevelopment to help guide the timing of treatment with these agents. This study also outlines the importance of placing more focus on the role of the cerebellum in HD, as it may have the potential to serve as a therapeutic target for HD in the future.
Glossary
- aCB
anterior lobe of the cerebellum
- ALD
accelerated longitudinal design
- DN
dentate nucleus
- dPU
dorsocaudal putamen
- GE
gene expanded
- GNE
gene nonexpanded
- GPE
globus pallidus externus
- HD
Huntington disease
- mHTT
mutant huntingtin protein
- PN
pontine nuclei
- ROI
region of interest
- rs-fMRI
resting-state fMRI
- STN
subthalamic nucleus
- VL
ventrolateral nucleus of the thalamus
Appendix. Authors
Study funding
R01 NS055903 (PI: Nopoulos).
Disclosure
The authors report no disclosures. Go to Neurology.org/N for full disclosures.
References
- 1.Norremolle A, Riess O, Epplen JT, Fenger K, Hasholt L, Sorensen SA. Trinucleotide repeat elongation in the Huntingtin gene in Huntington disease patients from 71 Danish families. Hum Mol Genet 1993;2:1475–1476. [DOI] [PubMed] [Google Scholar]
- 2.Nopoulos PC. Huntington disease: a single-gene degenerative disorder of the striatum. Dialogues Clin Neurosci 2016;18:91–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nasir J, Floresco SB, O'Kusky JR, et al. Targeted disruption of the Huntington's disease gene results in embryonic lethality and behavioral and morphological changes in heterozygotes. Cell 1995;81:811–823. [DOI] [PubMed] [Google Scholar]
- 4.Bhide PG, Day M, Sapp E, et al. Expression of normal and mutant huntingtin in the developing brain. J Neurosci 1996;16:5523–5535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zeitlin S, Liu JP, Chapman DL, Papaioannou VE, Efstratiadis A. Increased apoptosis and early embryonic lethality in mice nullizygous for the Huntington's disease gene homologue. Nat Genet 1995;11:155–163. [DOI] [PubMed] [Google Scholar]
- 6.Rigamonti D, Bauer JH, De-Fraja C, et al. Wild-type huntingtin protects from apoptosis upstream of caspase-3. J Neurosci 2000;20:3705–3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cattaneo E, Rigamonti D, Goffredo D, Zuccato C, Squitieri F, Sipione S. Loss of normal huntingtin function: new developments in Huntington's disease research. Trends Neurosci 2001;24:182–188. [DOI] [PubMed] [Google Scholar]
- 8.Arteaga-Bracho EE, Gulinello M, Winchester ML, et al. Postnatal and adult consequences of loss of huntingtin during development: implications for Huntington's disease. Neurobiol Dis 2016;96:144–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mehler MF, Gokhan S. Mechanisms underlying neural cell death in neurodegenerative diseases: alterations of a developmentally-mediated cellular rheostat. Trends Neurosci 2000;23:599–605. [DOI] [PubMed] [Google Scholar]
- 10.van der Plas ELD, Conrad AL, Tereshchenko A, Epping EA, Magnotta VA, Nopoulos PC. Abnormal brain development in child and adolescent carriers of mutant huntingtin. Neurology 2019;93:e1021–e1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Galvan L, Andre VM, Wang EA, Cepeda C, Levine MS. Functional differences between direct and indirect striatal output pathways in Huntington’s disease. J Huntingtons Dis 2012;1:17–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Milardi D, Arrigo A, Anastasi G, et al. Extensive direct subcortical cerebellum-basal ganglia connections in human brain as revealed by constrained spherical deconvolution tractography. Front Neuroanat 2016;10:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bostan AC, Dum RP, Strick PL. The basal ganglia communicate with the cerebellum. Proc Natl Acad Sci USA 2010;107:8452–8456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bostan AC, Strick PL. The basal ganglia and the cerebellum: nodes in an integrated network. Nat Rev Neurosci 2018;19:338–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vijayakumar N, Mills KL, Alexander-Bloch A, Tamnes CK, Whittle S. Structural brain development: a review of methodological approaches and best practices. Dev Cogn Neurosci 2018;33:129–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huntington Study Group. Unified Huntington's Disease Rating Scale: reliability and consistency. Mov Disord 1996;11:136–142. [DOI] [PubMed] [Google Scholar]
- 17.Fox MD, Snyder AZ, Zacks JM, Raichle ME. Coherent spontaneous activity accounts for trial-to-trial variability in human evoked brain responses. Nat Neurosci 2006;9:23–25. [DOI] [PubMed] [Google Scholar]
- 18.Power JD, Barnes KA, Snyder AZ, Schlaggar BL, Petersen SE. Spurious but systematic correlations in functional connectivity MRI networks arise from subject motion. Neuroimage 2012;59:2142–2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Di Martino A, Scheres A, Margulies DS, et al. Functional connectivity of human striatum: a resting state fMRI study. Cereb Cortex 2008;18:2735–2747. [DOI] [PubMed] [Google Scholar]
- 20.Keuken MC, Bazin PL, Crown L, et al. Quantifying inter-individual anatomical variability in the subcortex using 7 T structural MRI. Neuroimage 2014;94:40–46. [DOI] [PubMed] [Google Scholar]
- 21.Buckner RL, Krienen FM, Castellanos A, Diaz JC, Yeo BT. The organization of the human cerebellum estimated by intrinsic functional connectivity. J Neurophysiol 2011;106:2322–2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Diedrichsen J, Maderwald S, Kuper M, et al. Imaging the deep cerebellar nuclei: a probabilistic atlas and normalization procedure. Neuroimage 2011;54:1786–1794. [DOI] [PubMed] [Google Scholar]
- 23.Behrens TE, Johansen-Berg H, Woolrich MW, et al. Non-invasive mapping of connections between human thalamus and cortex using diffusion imaging. Nat Neurosci 2003;6:750–757. [DOI] [PubMed] [Google Scholar]
- 24.Tanner JM, Davies PS. Clinical longitudinal standards for height and height velocity for North American children. J Pediatr 1985;107:317–329. [DOI] [PubMed] [Google Scholar]
- 25.Kipps CM, Duggins AJ, Mahant N, Gomes L, Ashburner J, McCusker EA. Progression of structural neuropathology in preclinical Huntington's disease: a tensor based morphometry study. J Neurol Neurosurg Psychiatry 2005;76:650–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aylward EH, Sparks BF, Field KM, et al. Onset and rate of striatal atrophy in preclinical Huntington disease. Neurology 2004;63:66–72. [DOI] [PubMed] [Google Scholar]
- 27.Bostan AC, Dum RP, Strick PL. Cerebellar networks with the cerebral cortex and basal ganglia. Trends Cogn Sci 2013;17:241–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gaura V, Lavisse S, Payoux P, et al. Association between motor symptoms and brain metabolism in early Huntington disease. JAMA Neurol 2017;74:1088–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Feigin A, Leenders KL, Moeller JR, et al. Metabolic network abnormalities in early Huntington's disease: an [(18)F]FDG PET study. J Nucl Med 2001;42:1591–1595. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Deidentified data will be shared by reasonable request from any qualified investigator by the corresponding author for purposed of replicating procedures and results.