

Abstract
Objective
Dystonia is a complex movement disorder. Research progress has been difficult, particularly in developing widely effective therapies. This is a review of the current state of knowledge, research gaps, and proposed research priorities.
Methods
The NIH convened leaders in the field for a 2-day workshop. The participants addressed the natural history of the disease, the underlying etiology, the pathophysiology, relevant research technologies, research resources, and therapeutic approaches and attempted to prioritize dystonia research recommendations.
Results
The heterogeneity of dystonia poses challenges to research and therapy development. Much can be learned from specific genetic subtypes, and the disorder can be conceptualized along clinical, etiology, and pathophysiology axes. Advances in research technology and pooled resources can accelerate progress. Although etiologically based therapies would be optimal, a focus on circuit abnormalities can provide a convergent common target for symptomatic therapies across dystonia subtypes. The discussions have been integrated into a comprehensive review of all aspects of dystonia.
Conclusion
Overall research priorities include the generation and integration of high-quality phenotypic and genotypic data, reproducing key features in cellular and animal models, both of basic cellular mechanisms and phenotypes, leveraging new research technologies, and targeting circuit-level dysfunction with therapeutic interventions. Collaboration is necessary both for collection of large data sets and integration of different research methods.
Dystonia is a movement disorder characterized by sustained or intermittent muscle contractions causing abnormal, often repetitive, movements, postures, or both.1 Therapeutic options remain largely symptomatic, and progress in advancing new treatment options has been slow.
In October 2018, the National Institute of Neurological Disorders and Stroke (NINDS) conducted a workshop entitled “Defining Emergent Opportunities in Dystonia Research,” cosponsored by the Dystonia Medical Research Foundation, and including representatives from academic institutions, industry, patient advocacy groups, and funding and regulatory agencies. The participants were selected based on expertise in the field and ability to provide broad input in surveying the dystonia research landscape.
The overall goals were to (1) outline the current understanding of the natural history, pathology, and therapeutic space in dystonia; (2) identify gaps and limitations in treatment; and (3) generate research priorities for dystonia. Session organization promoted progressive discussion, addressing, in order, the natural history of the disease, defining the underlying etiology, elucidating the pathophysiology, developing research technologies, building and evaluating research resources, refining therapeutic approaches, and finally prioritizing dystonia research recommendations.
Natural history of dystonia
Dystonia is classified along 2 main axes: clinical characteristics and etiology.1 Understanding the temporal pattern of the symptoms and the timing of therapeutic response remains a critical need. The interaction between the age at onset, distribution, and site of onset is poorly understood.2 Regarding therapeutic response, for example, it is known that disease duration may affect the response to deep brain stimulation (DBS) in DYT1-TOR1A dystonia.3
Substantive phenotypic heterogeneity complicates the natural history of generalized dystonia. However, much can be learned from studying particular subtypes. The most frequent genetic form of early-onset generalized dystonia, due to mutation in TOR1A (DYT1-TOR1A dystonia),4 has provided a window for understanding brain circuitry through neuroimaging, yielded animal models, allowed throughput screening of FDA-approved compounds, and supported therapeutic efficacy for DBS and anticholinergic therapy. Another example is dopa-responsive dystonia, which is typically caused by mutations in GTP cyclohydrolase 1 (DYT/PARK‐GCH1),5 which demonstrates a dramatic response to l-DOPA therapy. This evidence suggests that even infrequent genetic etiologies can lead to breakthroughs and advances in therapies and may apply to other types of dystonia. Studies to determine additional genetic etiologies (including studies of families) are, therefore, needed, even when the specific genetic etiology is infrequent or of greater frequency in a particular group, as these will have ramifications for therapies in other etiologies of dystonia.
An even greater challenge surrounds focal dystonias, where smaller retrospective cohorts are typically available, and most cases are sporadic. The largest series are available for the most common focal dystonia, cervical dystonia (CD). The cross-sectional Dystonia Coalition study included 1,160 patients.6 This yielded important observations, including the 23% risk of spread to contiguous body regions.
Although the number of postmortem human brain samples from patients with dystonia is limited compared with most other common neurologic conditions, their collection is critical for the understanding of the relevant pathology, whereas cell-level studies can inform processes common to multiple dystonia forms or specific to certain genotypes or phenotypes.
Research priorities regarding natural history
The group identified the following as research priorities:
Larger sample sizes for natural history studies and a focus on multicenter studies enrolling diverse populations
Prospective data collection, with comprehensive inclusion criteria. For example, cohorts should include those with benefit from botulinum neurotoxin (BoNT) injections and those who discontinue therapy for various reasons
Consideration of all relevant genotypic and clinical characteristics when designing studies
Collection of DNA, other biospecimens, and brain, when available
Defining the underlying etiology of dystonia
One of the challenges in the field of dystonia is integrating the advances in knowledge accumulated across multiple levels of analysis. At a gene level, insights can be obtained from monogenic forms and other variants related to sporadic forms. At a biochemical level, abnormalities of dopaminergic, cholinergic, and GABAergic transmission are central. A network model of dystonia posits dysfunction along the pathways connecting the cortex, thalamus, basal ganglia, and cerebellum. The current challenge is to identify a framework linking genes, biochemistry, circuits, and clinical phenomenology (figure 1).
Figure 1. The etiology of dystonia can be thought of in a stepwise fashion.

One critical missing piece is the connection between these levels.
More than 20 genes have been implicated in dystonia. One of the clearest mechanisms derived from the identified genes is a role for dopamine. The dopamine synthesis pathway is implicated by mutations in GTP cyclohydrolase 1 (GCH1) and tyrosine hydroxylase (TH).7 In striatal medium spiny neurons, the targets of nigral dopamine cells, mutations in the guanine nucleotide-binding protein G(olf) subunit alpha (GNAL), adenylate cyclase 5 (ADCY5), and phosphodiesterase 10A (PDE10A) genes are all part of the signaling cascade affecting dopamine D1 receptors (D1Rs) in the direct pathway and the adenosine A2A receptors of the indirect pathway.8
About 90% of isolated dystonia cases are focal or segmental; however, very few of these cases have a known genetic cause, despite 5%–35% of cases having an affected relative. In addition, penetrance of the disease is estimated at <15%, suggesting that isolated focal/segmental dystonia is most likely multifactorial, with multiple genes and environmental factors interacting to trigger disease.9 Genome-wide association (GWA) studies in mainly sporadic patients and next-generation sequencing (NGS) studies in small multigeneration pedigrees have been attempted in this group with limited success.10–12 This is most likely due to the small numbers of cases studied in GWASs and the vast number of variants of unknown significance that arise through NGS in small pedigrees. Both types of studies have resulted in potential candidate genes that require follow-up in larger populations of focal/segmental dystonia patient samples.
Dopamine deficiency, supported by both genetic deficiencies in dopamine biosynthetic enzymes, such as GCH and TH,7 and clinical effects of dopamine receptor-blocking drugs, is a prominent example of genetic discoveries fueling the identification of molecular and cellular mechanisms for dystonia.
At least for some subsets of dystonias, there are examples in which multiple distinct genetic and clinical presentations of dystonia may share common cellular pathophysiologic mechanisms. Genetic and biochemical defects in a protein translation pathway regulated by activity of the eukaryotic initiation factor 2α (eIF2α) are found in multiple forms of inherited dystonia (DYT1, 6, and 11) and in sporadic CD.13,14 Activity of eIF2α is a central mechanism for responding to cellular stressors in the integrated stress response, whereas in the brain, it plays a critical role in inducing long-lasting synaptic plasticity.15
Discovery of shared cellular signaling mechanisms creates new opportunities to understand dystonia mechanisms and develop therapeutics. These efforts can be accelerated by the creation and sharing of dystonia cell and animal models and mine-able “omic” data sets. The ability to classify the various dystonias into cellular mechanism subgroups is a critical step to predict which populations may benefit as newer disease mechanism-targeting therapies emerge.
Animal models represent a critical tool to test mechanisms of dystonia pathogenesis.16 Such models can largely be divided into symptomatic and etiologic models. The latter have generally been related to specific genetic features, including gene-targeted (knock-in, knock-out, and conditional) and transgenic models. Examples of knock-in models include models for DOPA-responsive dystonia,17 THAP1 dystonia,18 and DYT1 dystonia. Knock-out models, transgenic models, and conditional models19 have also been generated for DYT1 dystonia. These numerous models have varying degrees of construct, face, and predictive validity for dystonia. Suitability of any given model will differ depending on the research question.
Research priorities regarding etiology
The group identified the following as research priorities:
Harnessing the multiple single gene insights and models to identify common relationships among dystonias, including hypothesis-driven studies in known pathways (like dopamine pathway or eIF2α)
Advancing parallel platforms that facilitate translation
Continued model development, as well as increased access to existing models
Integrating genetic information with data generated from animal and cellular models of dystonia, including various -omics in induced pluripotent stem cells (iPSCs) and brain specimens to identify candidate genes, as well as test for functionality of genes implicated by GWASs and NGS
Elucidating the pathophysiology of dystonia
Two axes of description are recognized for most neurologic conditions, including dystonia: clinical syndrome and etiology. A third axis can be considered, that of pathophysiology, which is the link between etiology and clinical syndrome. It provides the mechanism for how the etiology disrupts the normal function of the brain to produce signs and symptoms (figure 2).
Figure 2. Three axes approach to the pathophysiology of dystonia.
Photograph reprinted with permission from Tronnier V.M., Fogel W. Pallidal stimulation for generalized dystonia. J Neurosurg 2000;92:453–456.

Common pathophysiologic abnormalities in dystonia can be recognized in multiple sites in the nervous system20:
First is a loss of inhibition, which occurs both in the motor system, presumably predisposing to excess movement, and in the sensory system. These combined with abnormalities of sensorimotor integration give rise to subtle sensory abnormalities, such as elevated temporal discrimination thresholds.
Using noninvasive neurostimulation, such as transcranial magnetic stimulation, inhibition in the motor system can be evaluated in patients with dystonia.21 Overall, the findings have shown reduced inhibition (or excessive excitation) at the brainstem, spinal cord, and cortical levels.22 The loss of inhibition can be demonstrated by a prolonged cortical silent period (CSP). For instance, patients with spasmodic dysphonia have a prolonged CSP compared with healthy individuals and patients who have muscle tension dysphonia (a nondystonic vocal hyperfunction).23 This occurs in patients with other focal dystonias, such as CD, who have a prolonged CSP compared with healthy individuals.24 Loss of inhibition may demonstrate somatotopic organization, with some studies showing the greatest loss of inhibition in the cortical regions corresponding to the affected area. However, subclinical dysfunction may extend beyond these areas. Thus, loss of inhibition may represent a predisposition to develop dystonia rather than a causal mechanism for the abnormal movements.21
Sensorimotor abnormalities, including abnormal temporal and spatial discrimination, occur in isolated dystonia both before and after development of the abnormal motor behavior.20,25 This may reflect an endophenotype playing a role in the development of dystonia.26
Another key feature is the abnormality of brain plasticity (maladaptive plasticity), a failure of homeostatic plasticity, which is seen in several experimental paradigms.27 A recent study examined cortical excitability in patients with CD who were undergoing DBS. Pre-DBS cortical plasticity correlated with disease severity and a greater response to DBS.28
These pathophysiologic changes may lead to the development of predictive markers of response to therapeutic intervention, as seen in the DBS study above. Therapeutic target development may be guided by identification of pathophysiologically abnormal nodes in the sensorimotor system. Finally, the degree of inhibitory loss or the extent of aberrant cortical plasticity might be used in the development of predictive measures of therapeutic response.
Another hallmark of dystonia pathophysiology is its neural network dysfunction. The current state of knowledge conceptualized dystonias as disorders of large-scale functional networks29 (figure 3). When examining neural network connectivity dependent on dystonia genotype and phenotype, different patterns of altered connectivity emerge. Abnormally increased brain activity but decreased regional connectivity have been seen in patients with dystonia during both symptomatic and asymptomatic tasks, with abnormalities occurring in the basal ganglia, thalamus, cerebellum, and sensorimotor cortex. In addition, impairments have been seen in the parietal-premotor network,30 as well as in the frontoparietal and default mode networks.31,32 Neural abnormalities in dystonia extend to a large-scale network, forming the abnormal functional connectome. It has been shown across different forms of focal dystonia that an abnormal breakdown of a single neural community constituting the basal ganglia, thalamus, and cerebellum with subsequent formation of multiple communities exerts a network-wide effect on information transfer, especially in parietal and sensorimotor neural communities.33
Figure 3. Critical functional circuits in dystonia.
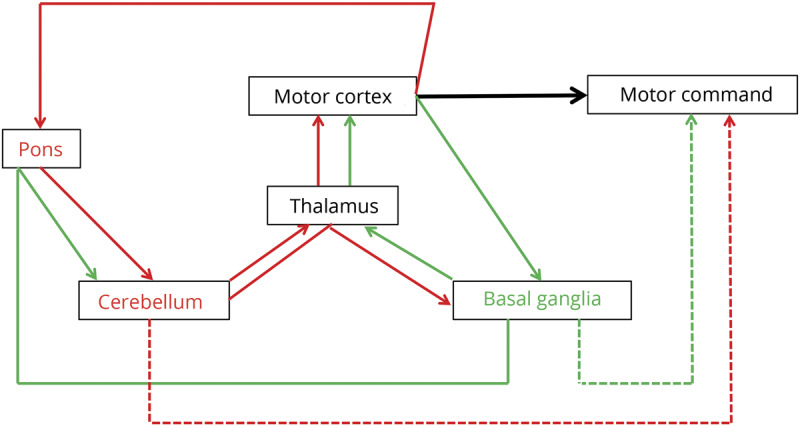
A specific pattern of functional disorganization involving influential regions of information transfer (hubs) has been shown to lead to distinct topology of the large-scale functional network in familial vs sporadic cases or in clinically different subtypes of dystonia (e.g., adductor vs abductor spasmodic dysphonia). The functional “network kernel” is a superlative network of the most influential hub regions. Aberrant diversity within the network kernel has been reported to shape the task-specificity of focal dystonia, for example in hand vs laryngeal dystonia or musician's vs nonmusician's dystonia.34
An important tool in the study of dystonia pathophysiology is the development of phenotypic models, focused on reproducing the essential disease features. Based on the key features that define human dystonia, namely the muscle overcontractions and characteristic twisting postures, a major goal—and hurdle—has been to reproduce these behaviors in animals. A rat model, the dt rat,35 which has a spontaneous mutation in the ATCAY gene, shows dystonic behaviors and neuronal phenotypes that motivated new views about brain circuits in dystonia.36 A remarkable and severe dystonia was reported for the mutant mouse tottering.37 Tottering mice have a spontaneous mutation in the CACNA1A gene, which encodes the P/Q-type voltage-gated calcium channel, Cav2.1. Engineered mutations in dyt1 have been necessary for revealing the molecular underpinnings of dystonia,38 especially given the intensive studies on human DYT1. Despite the lack of overt dystonia in dyt1 mutants, using these mice, the idea was reinforced that several motor regions are affected in dystonia.39 These studies inspired the development of pharmacologic 40 and conditional genetic41 approaches in mice that exhibit convincing behavioral defects resembling human dystonia. The robust behaviors in some rodent models have set the stage for resolving the in vivo electrophysiologic abnormalities of dystonia during development and in adults. At the same time, there is no single animal model that models the many forms of dystonia (nor should we expect one), although collectively the available experimental models and tools are well placed for uncovering the mechanisms of dystonia. Further studies using mammals, especially nonhuman primates, will provide additional insight into dystonia pathogenesis.
Research priorities regarding pathophysiology
The group identified the following as research priorities:
Reproducing specific pathophysiology features in animal models
Exploring the common underlying pathophysiology for the different phenotypic expressions in patients (e.g., disease manifestation in the neck vs the eyes) and identifying pathophysiology that is specific to one type or subtype of dystonia
Identifying which pathophysiologic abnormalities are predisposing and which correlate with the presence of dystonia
Identifying therapeutic targets and markers of therapy response
Studying the pathophysiology in the context of therapeutic interventions
Bridging the gap between cellular abnormality and network dysfunction
Research technology development
The NIH Brain Research through Advancing Innovative Neurotechnologies (BRAIN) Initiative is a prominent example of efforts to develop research tools and infrastructure for neuroscience research. The Initiative has among its goals the development of new technologies to map neural circuits and understand their function in health and disease, as well as enabling new discoveries to unlock the mysteries of the brain and treat its disorders. As of October 2018, the BRAIN Initiative issued 40 funding announcements and has 8 program areas managed by 6 project teams. The initiative includes a cell census consortium; tools for cells and circuits; recording and modulation technologies; human imaging and neuromodulation; understanding neural circuits; technology dissemination and training; data coordination and infrastructure; and neuroethics research.
Neuromodulation technology is a significant focus of the BRAIN Initiative, as well as a general opportunity for both refining and developing therapies and acquiring knowledge about normal and diseased brain function. DBS is a remarkably effective therapy for most isolated dystonias, yet the mechanism remains unclear. Any hypothesis of the mechanism of DBS in dystonia should take into account several clinical observations: there are many neurologic disorders in which the motor signs of parkinsonism and dystonia coexist, and the exact same DBS targets and stimulation parameters that are effective for Parkinson disease (PD) are also effective for isolated dystonias. Thus, a substantial overlap in the circuit mechanisms underlying these 2 conditions is hypothesized. A powerful technique for circuit analysis in movement disorders is multisite (cortical and subcortical) human brain recording, performed either acutely during surgery or chronically using implantable neural interfaces that have a sensing component. This approach has revealed common physiologic motifs in PD and isolated dystonia. In both conditions, there is strong beta (13–30 Hz) coherence between the globus pallidus and the motor cortex, at different peak frequencies.42 Rigid-bradykinetic PD off levodopa and generalized dystonias are associated with strong cortical synchronization in the beta range, manifested by coupling of high-frequency (broadband) activity to the phase of the motor beta rhythm.43 Isolated dystonias that have a hyperkinetic component and PD on levodopa with dyskinesias are both associated with cortical oscillatory activity in the 60–90 Hz (gamma range).44,45 It is hypothesized that DBS acts in both disorders by reducing these network abnormalities.
Research priorities regarding research technology development
The group identified the following as research priorities:
Leveraging existing resources, including the BRAIN Initiative, to continue to expand technologies
Studying network activities, and their response to therapy, including the utilization of the new-generation implantable devices with sensing capabilities
Performing anatomic and functional studies in models
Focus on translation research and therapeutic development studies
Building and leveraging research resources for dystonia
As mentioned previously, one important resource consists of the various genetic or phenotypic animal models. Their availability to the research community is critical to further progress in the field.
A list of additional resources to be considered also includes the following:
The NIH database of Phenotypes and Genotypes (dbGaP): the goal of this database is the broad and responsible sharing of genomic research data in a timely manner
The NINDS Human Genetics Resource Center: a public resource containing DNA and lymphoblastoid cell lines with corresponding deidentified clinical, demographic, and familial data. Since its establishment, specimens from over 41,000 individuals diagnosed with cerebrovascular diseases, parkinsonism, motor neuron diseases, epilepsy, Tourette syndrome, dystonia, and neurologically normal controls have been successfully banked and can be accessed through an online catalog at coriell.org/1/NINDS.
To date, the Dystonia Sub-Collection has received over 3,440 submissions, 2,887 of which are currently available for general research use. Focal dystonia is the primary diagnosis of the majority of the Dystonia Sub-Collection (N = 2035), followed by segmental dystonia (N = 452), generalized dystonia (N = 119), and multifocal dystonia (N = 110). Several less common diagnoses are also included in the Dystonia Sub-Collection. Over 5,800 DNA samples have been distributed back to clinical submitters, and over 600 DNA and LCL samples have been distributed to the research community at large.
The NINDS Human Cell and Data Repository (NHCDR): housed at Rutgers University, hosts an online catalog (bioq.nindsgenetics.org) that has 52 dystonia fibroblasts available for distribution. These lines consist of 26 control fibroblasts from unaffected family members, 6 cell lines with the TOR1A (DYT1) deletion mutation, 7 cell lines with the TAF1 SVA intronic insertion (DYT3) mutation, 1 line with an ATP1A3 (DYT12) mutation, and 1 line with the PRKRA (DYT16) mutation. Additional dystonia lines are in progress and expected to be released over the coming months. The dystonia fibroblast cell lines (and all other lines housed as part of the NHCDR at Rutgers University Cell and DNA Repository [RUCDR]) can be ordered through the online catalog at stemcells.nindsgenetics.org/. This catalog is searchable by disease, genetic disorder, etc., and cell lines are available to both nonprofit and for-profit customers. All of the dystonia fibroblasts have been tested for sterility, post-thaw viability, identity, and karyotype and are shipped with a certificate of analysis demonstrating the results of all these tests.
The NIH NeuroBioBank: the broad mission is to collect and distribute human postmortem tissue to the research community. It includes donors with neurologic, neurodevelopmental, and psychiatric disorders and unaffected controls. It includes 6 contracted brain and tissue repositories and centralized toxicology and IT services. There are 103 dystonia cases in the inventory with varying degrees of clinical characterization, encompassing multiple subtypes. Fifteen requests for tissue to support dystonia studies have been filled to date.
Patient registries: prospective natural history studies are important for designing clinical trials and studies of pathophysiology.6 They provide baseline data for metrics of disease progression, critical for power analyses to determine needed size and length of clinical trials for disease-modifying interventions. These data also permit better identification of homogenous groups for trial cohorts.
The Dystonia Coalition supported by the Office of Rare Disease Research in the National Center for Advancing Translational Sciences (NCATS) and the NINDS is a multicenter, international coalition of investigators (rarediseasesnetwork.org/cms/dystonia). The central focus has been clinical trial readiness, including appropriate diagnostic criteria and rating tools to measure outcomes. A cornerstone of this program is a natural history study that recruited more than 3,000 participants from 37 national and international sites with all data entered into a centralized web-based system and standardized videos stored on a centralized web-based video repository. More than 98% have DNA at the NINDS Repository at Coriell. A publicly available web site (clinportalquery.wustl.edu/pentaho/Home?userid=DYSTONIA_OPENI&password=DYSTONIA) permits anyone to determine what data have been collected. Similarly, the public can determine what videos are available at dystonia.wustl.edu. The videos and deidentified data can be accessed after permission granted by the Dystonia Coalition and proper IRB approval. Thus, the shared resources include cross-sectional phenotypic data, natural history with more than 850 follow-up visits, the video repository, and a patient registry with more than 5,450 patients that is managed by the patient advocacy groups (GlobalDystoniaRegistry.org).
Research priorities regarding research resources for dystonia
The group identified the following as research priorities:
Promoting additional collection and standardization of biosamples for genetics, epigenetics, proteomic, lipidomic, metabolomics and transcriptomics, and neuroimaging
Ensuring ongoing easy accessibility to repositories and shared resources and expanding existing repositories
Encouraging brain and tissue donation from patients and unaffected relatives, within the confines of ethical requirements
Refining therapeutic approaches for dystonia
Broadly speaking, dystonia therapy can be divided into etiologic and symptomatic; the latter is further divided, in the case of dystonia, into pharmacologic, chemodenervation, rehabilitation, and neuromodulation (both noninvasive and invasive).
There are currently no FDA-approved oral pharmacologic treatment options for isolated dystonia. The off-label symptomatic approaches to treat dystonia are generally empiric, with available evidence limited to anecdotal reports and small retrospective studies, except for some prospective trials in a small number of specific dystonia disorders. Initial oral treatment for isolated dystonia often involves trying a drug that targets one of the main neurotransmitter systems involved in dystonia. These include anticholinergic (e.g., trihexyphenidyl), GABAergic (e.g., baclofen and benzodiazepines), and dopaminergic (e.g., levodopa and VMAT-2 inhibitors) medications. Other pharmacologic treatments that are tried in dystonia include muscle relaxers, antiepileptics, and cannabinoids. Although current oral pharmacologic treatments can lessen symptoms of dystonia, they typically only lead to a modest reduction in dystonia at best and are frequently associated with intolerable adverse effects. More recently studied treatment options include sodium oxybate, a GABA agonist that has shown promise in relieving symptoms in alcohol-responsive laryngeal dystonia.46 Further research is needed to identify the most relevant pathophysiologic targets in dystonia and develop well-tolerated pharmacologic treatment options that can appreciably improve symptoms.
Chemodenervation of affected muscles with botulinum toxin injections is a first-line treatment for most forms of isolated dystonia. Presently, there are 4 brands in use in the United States with FDA-approved indications including blepharospasm and CD. Although the 4 formulations of botulinum toxin have important distinctions, their overall mechanisms of action are similar, and there is currently no evidence that one is superior to another. Treatment response, however, can vary depending on the injection technique used, and effectiveness may be limited due to adverse effects.47 Although chemodenervation can be effective, reinjection is needed approximately every 3 months.
Rehabilitation therapy for dystonia includes motor learning, sensory-motor training, mobilization, stretching and bracing, and biofeedback. These approaches can be used in isolation or in combination with other treatments, such as botulinum injections and neuromodulation.
Noninvasive neuromodulation is an active area of research,48 but additional clarity is needed regarding modality (magnetic or electric), settings, targets, and outcomes.
Invasive neuromodulation consists primarily of DBS, which has an established role and a track record of efficacy in patients with primary dystonia.49,50
For isolated generalized dystonia, DBS should be considered when there is significant disability despite optimal medical management. For isolated craniocervical or other isolated, relatively focal dystonias, DBS should be considered when there is significant disability despite optimal medical management and chemodenervation.
The most well-studied brain target for DBS in isolated dystonia is the globus pallidus. Although subthalamic nucleus DBS gives approximately equivalent benefit in isolated dystonias, in several case series, indications for selection of the STN remain undefined. Good results have, however, been reported with STN DBS.51
Given the functional organization of the basal ganglia, with distinct anatomic regions subserving motor, associative, and limbic function, it has been suggested that lead location within the internal segment of the globus pallidus (GPi) could be one critical factor in predicting outcome in patients with dystonia undergoing DBS.52 Given the somatotopic organization within the motor GPi,53 it has been suggested that placement of DBS leads within the sensorimotor region of the GPi may be dependent on each patient's phenotype.54
Early studies of lesion therapy reported significant improvement following thalamotomy yet this target is only rarely used.55 Bilateral lesions have a high incidence of side effects, currently limiting applications. Given the paucity of data examining the effect of thalamic DBS for dystonia, it remains unclear what site will provide the greatest benefit and if that would change based on other variables. The cerebellum has also been explored as a potential target and remains an area of research interest.56
Going forward these questions must be addressed with level 1 evidence examining the role of target sites, lead location, patients' individual phenotype, genotype, age, and other yet unidentified variables that may affect outcome. It is well known that DBS effect is highly dependent on genotype, and particular mutations and variants within the same gene can result in divergent effects.57 Better imaging technology would allow precisely defining the target and determining the location of each implanted lead, assessing the relative role of new DBS technology (directional leads, multiple current sources, sensing, and novel programming algorithms), and understanding the pathophysiologic basis underlying the development of dystonia, the network changes that occur, and the mechanism(s) underlying the improvement after DBS. Answers to these questions will be critically important to obtain the best outcomes for each patient with dystonia who undergoes DBS with the goal of developing “precision DBS for dystonia.”
Etiologic approaches for dystonia therapy have remained elusive, with some prominent exceptions. Dopa-responsive dystonia can be successfully treated, as discussed above. Gene therapy is primarily being studied in the context of specific gene defects, and cell therapy has had limited applications to date.
One particular interest is the potential use of iPSC technology. Somatic cells can be reprogrammed to iPSCs, and genome editing can be used as a tool for phenotype discovery. iPSC differentiation can recapitulate development in a dish,58 allowing disease modeling and functional characterization. This has been successful, for example, in X-linked dystonia-parkinsonism.59
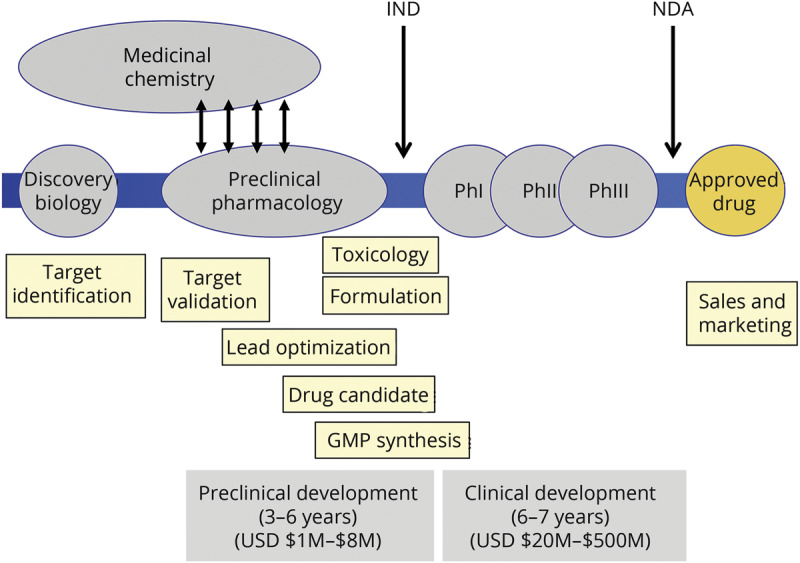
Predictive models can then be used to develop therapies, although therapeutic discovery and development is a lengthy and costly process (figure 4).
Figure 4. Therapeutic discovery and development.
Research priorities regarding therapeutic approaches for dystonia
The group identified the following as research priorities:
Better understanding of the cellular and circuit therapy targets, including continued development of tools like iPSC modeling
Improving the delivery and efficacy of existing therapies, including BoNT injections and DBS
Ongoing technology development, including new DBS paradigms and targets
Refining the standard approaches to therapy for individual patients, including the decisions on deploying invasive therapies
Ensuring effective infrastructures for clinical testing of proposed therapies, as well as the availability of relevant outcome measures, including updated clinical scales
Moving toward precision medicine, informed by patient genotype and phenotype
Discussion and overall research priorities
Overall, encompassing research priorities include the following:
Ongoing comprehensive data collection in clinical, biospecimen, genomic, and pathology domains, across genotypes and phenotypes, for continued target identification and clinical trial readiness
Continuing generation and integration of high-quality genetic data
Investigating molecular-cellular mechanisms and further understanding of which dystonias fit into mechanistic subgroups
Reproducing features of human pathophysiology in animal models and ensuring their wide availability
Bridging the gap between cellular and network dysfunction, and potentially translating the findings into therapies, using an iterative approach and designs ranging from observational studies to controlled trials
Advancing parallel research platforms to facilitate translation
Leveraging technologies like iPSCs to streamline therapeutic development
Developing novel technologies targeting circuit dysfunction, including new neuromodulation paradigms
Fostering collaborations and wide sharing and integration of resources
The field of dystonia research has evolved and seen significant progress over recent decades. However, treatments that are effective for a majority of patients remain elusive, and the development of disease-modifying therapies has been hindered by unanswered questions about the pathogenesis of the disease and optimal therapeutic targets.
Much of the challenge relates to the heterogeneity of the condition. As a consequence, the field is faced with difficult decisions regarding pursuit of specific therapies for well-defined pathogenic processes that may have only limited relevance to a minority of patients vs trying to elucidate the common pathways that could catalyze more globally relevant therapeutic targets.
A potential convergence point can be found in the circuit dysfunctions discussed in the section on pathophysiology. It is possible that targeting circuit abnormalities common across etiologies and phenotypes can ultimately yield the most benefit for most people.
However, the field can continue to learn a great deal about dystonia from specific mendelian forms or restricted syndromes. These can be more easily modeled, and using technologies like conditional models, iPSCs, and others, therapeutic targets can be discovered and validated, and therapies can be derived. Furthermore, the study of dystonia can yield a bounty of knowledge about the normal and abnormal function of the brain and can advance the field of neuroscience in general.
All outlined priorities for dystonia research are only achievable through close and continuing collaborations among all stakeholders, including across research teams. Isolated approaches are unlikely to yield the advances in knowledge and therapeutic approaches that the patient with dystonia and the advocacy community expect. Participants expressed enthusiasm for collaborative work.
Glossary
- BoNT
botulinum neurotoxin
- CD
cervical dystonia
- CSP
cortical silent period
- DBS
deep brain stimulation
- GWA
genome-wide association
- GPi
globus pallidus
- iPSC
induced pluripotent stem cell
- NGS
next-generation sequencing
- NINDS
National Institute of Neurological Disorders and Stroke
- PD
Parkinson disease
- NHCDR
NINDS Human Cell and Data Repository
- TH
tyrosine hydroxylase
Appendix 1. Authors
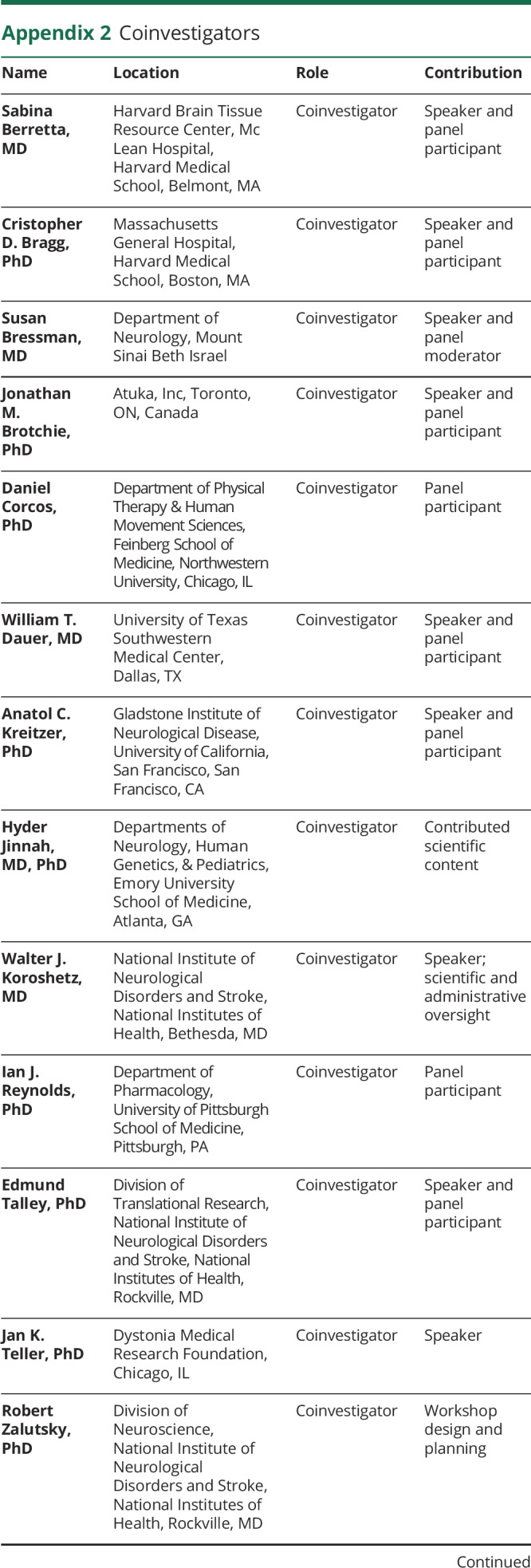
Appendix 2. Coinvestigators

Study funding
The workshop generating this article was funded by the National Institute of Neurological Disorders and Stroke and the Dystonia Medical Research Foundation.
Disclosure
C. Lungu reports no relevant disclosures. The work was conducted in the course of employment for the NIH, an agency of the US Government. L. Ozelius reports no relevant disclosures. D. Standaert is an investigator in studies funded by AbbVie, Inc., Avid Radiopharmaceuticals, the American Parkinson Disease Association, The Michael J. Fox Foundation for Parkinson Research, Alabama Department of Commerce, the Department of Defense, and NIH grants P01NS087997, P50NS108675, R25NS079188, P2CHD086851, P30NS047466, and T32NS095775. He has served as a consultant for or received honoraria from Serina Therapeutics, AbbVie Inc., Voyager Therapeutics, Blue Rock Therapeutics, Clintrex LLC, Revivo Therapeutics, Sanofi-Aventis Research and Development, Appello Pharmaceuticals, Avrobio, Inc., Extera Partners, Grey Matter Technologies, Theravance Inc., Alabama Academy of Neurology, McGraw Hill Publishers, and the University of Virginia. M. Hallett holds patents for an immunotoxin for the treatment of focal movement disorders and the H-coil for magnetic stimulation; in relation to the latter, he has received license fee payments from the NIH (from BrainsWay). He is on the medical advisory boards of CALA Health, BrainsWay, and Cadent. He receives royalties and/or honoraria from publishing from Cambridge University Press, Oxford University Press, Springer, and Elsevier. He has research grants from Allergan for studies of methods to inject botulinum toxins, Medtronic, Inc. for a study of DBS for dystonia, and CALA Health for studies of a device to suppress tremor. The work was conducted in the course of employment for the NIH, an agency of the US Government. B.-A. Sieber reports no relevant disclosures. The work was conducted in the course of employment for the NIH, an agency of the US Government. C. Swanson-Fisher reports no relevant disclosures. The work was conducted in the course of employment for the NIH, an agency of the US Government. B. D. Berman serves on the medical advisory board of the Benign Essential Blepharospasm Research Foundation and the National Spasmodic Torticollis Association. He reports grant support from the NIH (NIH/NCATS Colorado CTSI Grant Number KL2 TR001080), Dystonia Coalition (receives the majority of its support through NIH grant NS065701 from the Office of Rare Diseases Research in the National Center for Advancing Translational Science and National Institute of Neurological Disorders and Stroke), Dana Foundation, Benign Essential Blepharospasm Research Foundation, and from Mary Rossick Kern and Jerome H. Kern. He is a consultant for the National Football League Players Association. N. Calakos reports funding from Tyler's Hope for a Dystonia Cure Foundation, Dystonia Medical Research Foundation, and Cure Dystonia Now Foundation. J. C. Moore and J. S. Perlmutter report no relevant disclosures. S. E. Pirio Richardson has received research grant support from Dystonia Coalition Projects (NIH/NINDS/ORDR) and has received publishing royalties from Springer. S. E. Pirio Richardson serves on the scientific advisory board of the Benign Essential Blepharospasm Research Foundation. R. Saunders-Pullman reports funding from NINDS U01 NS107016, The Michael J. Fox Foundation, and the Bigglesworth Foundation. L. Scheinfeldt, N. Sharma, and R. Sillitoe report no relevant disclosures. K. Simonyan reports funding from the NIH (R01NS088160, R01DC011805, and R01DC012545), the Department of Defense (W911NF1810434), and Amazon Web Services and serves as a consultant to Jazz Pharmaceuticals and on the scientific advisory board of the Tourette Association of America. P. A. Starr reports research support from Boston Scientific Inc and Medtronic Inc. A. Taylor reports no relevant disclosures. The work was conducted in the course of employment for the NIH, an agency of the US Government. J. L. Vitek reports consulting work for LivaNova, Adamas, Insightec, SIS, Boston Scientific, Abbott/St. Jude Medical, and Medtronic. Go to Neurology.org/N for full disclosures.
References
- 1.Albanese A, Bhatia K, Bressman SB, et al. Phenomenology and classification of dystonia: a consensus update. Mov Disord 2013;28:863–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.O'Riordan S, Raymond D, Lynch T, et al. Age at onset as a factor in determining the phenotype of primary torsion dystonia. Neurology 2004;63:1423–1426. [DOI] [PubMed] [Google Scholar]
- 3.Markun LC, Starr PA, Air EL, Marks WJ Jr, Volz MM, Ostrem JL. Shorter disease duration correlates with improved long-term deep brain stimulation outcomes in young-onset DYT1 dystonia. Neurosurgery 2012;71:325–330. [DOI] [PubMed] [Google Scholar]
- 4.Ozelius LJ, Hewett JW, Page CE, et al. The early-onset torsion dystonia gene (DYT1) encodes an ATP-binding protein. Nat Genet 1997;17:40–48. [DOI] [PubMed] [Google Scholar]
- 5.Ichinose H, Ohye T, Takahashi E, et al. Hereditary progressive dystonia with marked diurnal fluctuation caused by mutations in the GTP cyclohydrolase I gene. Nat Genet 1994;8:236–242. [DOI] [PubMed] [Google Scholar]
- 6.Norris SA, Jinnah HA, Espay AJ, et al. Clinical and demographic characteristics related to onset site and spread of cervical dystonia. Mov Disord 2016;31:1874–1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wijemanne S, Jankovic J. Dopa-responsive dystonia-clinical and genetic heterogeneity. Nat Rev Neurol 2015;11:414–424. [DOI] [PubMed] [Google Scholar]
- 8.Carecchio M, Mencacci NE. Emerging monogenic complex hyperkinetic disorders. Curr Neurol Neurosci Rep 2017;17:97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Waddy HM, Fletcher NA, Harding AE, Marsden CD. A genetic study of idiopathic focal dystonias. Ann Neurol 1991;29:320–324. [DOI] [PubMed] [Google Scholar]
- 10.Tian J, Vemula SR, Xiao J, et al. Whole-exome sequencing for variant discovery in blepharospasm. Mol Genet Genomic Med 2018;6:601–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gomez-Garre P, Huertas-Fernandez I, Caceres-Redondo MT, et al. Lack of validation of variants associated with cervical dystonia risk: a GWAS replication study. Mov Disord 2014;29:1825–1828. [DOI] [PubMed] [Google Scholar]
- 12.Hammer M, Abravanel A, Peckham E, et al. Blepharospasm: a genetic screening study in 132 patients. Parkinsonism Relat Disord 2019;64:315–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rittiner JE, Caffall ZF, Hernandez-Martinez R, et al. Functional genomic analyses of mendelian and sporadic disease identify impaired eIF2alpha signaling as a generalizable mechanism for dystonia. Neuron 2016;92:1238–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zakirova Z, Fanutza T, Bonet J, et al. Mutations in THAP1/DYT6 reveal that diverse dystonia genes disrupt similar neuronal pathways and functions. PLoS Genet 2018;14:e1007169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Costa-Mattioli M, Sonenberg N. Translational control of long-term synaptic plasticity and memory storage by eIF2alpha. Crit Rev Neurobiol 2006;18:187–195. [DOI] [PubMed] [Google Scholar]
- 16.Richter F, Richter A. Genetic animal models of dystonia: common features and diversities. Prog Neurobiol 2014;121:91–113. [DOI] [PubMed] [Google Scholar]
- 17.Rose SJ, Yu XY, Heinzer AK, et al. A new knock-in mouse model of l-DOPA-responsive dystonia. Brain 2015;138:2987–3002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ruiz M, Perez-Garcia G, Ortiz-Virumbrales M, et al. Abnormalities of motor function, transcription and cerebellar structure in mouse models of THAP1 dystonia. Hum Mol Genet 2015;24:7159–7170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liang CC, Tanabe LM, Jou S, Chi F, Dauer WT. TorsinA hypofunction causes abnormal twisting movements and sensorimotor circuit neurodegeneration. J Clin Invest 2014;124:3080–3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Quartarone A, Hallett M. Emerging concepts in the physiological basis of dystonia. Mov Disord 2013;28:958–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Balint B, Mencacci NE, Valente EM, et al. Dystonia. Nat Rev Dis Primers 2018;4:25. [DOI] [PubMed] [Google Scholar]
- 22.Quartarone A, Ruge D. How many types of dystonia? Pathophysiological considerations. Front Neurol 2018;9:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Samargia S, Schmidt R, Kimberley TJ. Cortical silent period reveals differences between adductor spasmodic dysphonia and muscle tension dysphonia. Neurorehabil Neural Repair 2016;30:221–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pirio Richardson S. Enhanced dorsal premotor-motor inhibition in cervical dystonia. Clin Neurophysiol 2015;126:1387–1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Conte A, Rocchi L, Ferrazzano G, et al. Primary somatosensory cortical plasticity and tactile temporal discrimination in focal hand dystonia. Clin Neurophysiol 2014;125:537–543. [DOI] [PubMed] [Google Scholar]
- 26.Kimmich O, Bradley D, Whelan R, et al. Sporadic adult onset primary torsion dystonia is a genetic disorder by the temporal discrimination test. Brain 2011;134:2656–2663. [DOI] [PubMed] [Google Scholar]
- 27.Quartarone A, Bagnato S, Rizzo V, et al. Abnormal associative plasticity of the human motor cortex in writer's cramp. Brain 2003;126:2586–2596. [DOI] [PubMed] [Google Scholar]
- 28.Kroneberg D, Plettig P, Schneider GH, Kuhn AA. Motor cortical plasticity relates to symptom severity and clinical benefit from deep brain stimulation in cervical dystonia. Neuromodulation 2018;21:735–740. [DOI] [PubMed] [Google Scholar]
- 29.Battistella G, Termsarasab P, Ramdhani RA, Fuertinger S, Simonyan K. Isolated focal dystonia as a disorder of large-scale functional networks. Cereb Cortex 2017;27:1203–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gallea C, Horovitz SG, Najee-Ullah M, Hallett M. Impairment of a parieto-premotor network specialized for handwriting in writer's cramp. Hum Brain Mapp 2016;37:4363–4375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Delnooz CC, Pasman JW, Beckmann CF, van de Warrenburg BP. Task-free functional MRI in cervical dystonia reveals multi-network changes that partially normalize with botulinum toxin. PLoS One 2013;8:e62877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Battistella G, Fuertinger S, Fleysher L, Ozelius LJ, Simonyan K. Cortical sensorimotor alterations classify clinical phenotype and putative genotype of spasmodic dysphonia. Eur J Neurol 2016;23:1517–1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fuertinger S, Simonyan K. Connectome-wide phenotypical and genotypical associations in focal dystonia. J Neurosci 2017;37:7438–7449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fuertinger S, Simonyan K. Task-specificity in focal dystonia is shaped by aberrant diversity of a functional network kernel. Mov Disord 2018;33:1918–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lorden JF, McKeon TW, Baker HJ, Cox N, Walkley SU. Characterization of the rat mutant dystonic (dt): a new animal model of dystonia musculorum deformans. J Neurosci 1984;4:1925–1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.LeDoux MS, Lorden JF. Abnormal spontaneous and harmaline-stimulated Purkinje cell activity in the awake genetically dystonic rat. Exp Brain Res 2002;145:457–467. [DOI] [PubMed] [Google Scholar]
- 37.Campbell DB, Hess EJ. L-type calcium channels contribute to the tottering mouse dystonic episodes. Mol Pharmacol 1999;55:23–31. [DOI] [PubMed] [Google Scholar]
- 38.Tanabe LM, Martin C, Dauer WT. Genetic background modulates the phenotype of a mouse model of DYT1 dystonia. PLoS One 2012;7:e32245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ulug AM, Vo A, Argyelan M, et al. Cerebellothalamocortical pathway abnormalities in torsinA DYT1 knock-in mice. Proc Natl Acad Sci U S A 2011;108:6638–6643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Calderon DP, Fremont R, Kraenzlin F, Khodakhah K. The neural substrates of rapid-onset Dystonia-Parkinsonism. Nat Neurosci 2011;14:357–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.White JJ, Sillitoe RV. Genetic silencing of olivocerebellar synapses causes dystonia-like behaviour in mice. Nat Commun 2017;8:14912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang DD, de Hemptinne C, Miocinovic S, et al. Pallidal deep-brain stimulation disrupts pallidal beta oscillations and coherence with primary motor cortex in Parkinson's disease. J Neurosci 2018;38:4556–4568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Miocinovic S, de Hemptinne C, Qasim S, Ostrem JL, Starr PA. Patterns of cortical synchronization in isolated dystonia compared with Parkinson disease. JAMA Neurol 2015;72:1244–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Miocinovic S, Swann NC, de Hemptinne C, Miller A, Ostrem JL, Starr PA. Cortical gamma oscillations in isolated dystonia. Parkinsonism Relat Disord 2018;49:104–105. [DOI] [PubMed] [Google Scholar]
- 45.Swann NC, de Hemptinne C, Miocinovic S, et al. Gamma oscillations in the hyperkinetic state detected with chronic human brain recordings in Parkinson's disease. J Neurosci 2016;36:6445–6458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rumbach AF, Blitzer A, Frucht SJ, Simonyan K. An open-label study of sodium oxybate in Spasmodic dysphonia. Laryngoscope 2017;127:1402–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jinnah HA, Comella CL, Perlmutter J, Lungu C, Hallett M, Dystonia Coalition I. Longitudinal studies of botulinum toxin in cervical dystonia: why do patients discontinue therapy? Toxicon 2018;147:89–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Erro R, Tinazzi M, Morgante F, Bhatia KP. Non-invasive brain stimulation for dystonia: therapeutic implications. Eur J Neurol 2017;24:1228–e1264. [DOI] [PubMed] [Google Scholar]
- 49.Kupsch A, Benecke R, Muller J, et al. Pallidal deep-brain stimulation in primary generalized or segmental dystonia. N Engl J Med 2006;355:1978–1990. [DOI] [PubMed] [Google Scholar]
- 50.Volkmann J, Wolters A, Kupsch A, et al. Pallidal deep brain stimulation in patients with primary generalised or segmental dystonia: 5-year follow-up of a randomised trial. Lancet Neurol 2012;11:1029–1038. [DOI] [PubMed] [Google Scholar]
- 51.Ostrem JL, San Luciano M, Dodenhoff KA, et al. Subthalamic nucleus deep brain stimulation in isolated dystonia: a 3-year follow-up study. Neurology 2017;88:25–35. [DOI] [PubMed] [Google Scholar]
- 52.Tisch S, Zrinzo L, Limousin P, et al. Effect of electrode contact location on clinical efficacy of pallidal deep brain stimulation in primary generalised dystonia. J Neurol Neurosurg Psychiatry 2007;78:1314–1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Baker KB, Lee JY, Mavinkurve G, et al. Somatotopic organization in the internal segment of the globus pallidus in Parkinson's disease. Exp Neurol 2010;222:219–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Starr PA, Turner RS, Rau G, et al. Microelectrode-guided implantation of deep brain stimulators into the globus pallidus internus for dystonia: techniques, electrode locations, and outcomes. Neurosurg Focus 2004;17:E4. [DOI] [PubMed] [Google Scholar]
- 55.Andrew J, Fowler CJ, Harrison MJ. Stereotaxic thalamotomy in 55 cases of dystonia. Brain 1983;106:981–1000. [DOI] [PubMed] [Google Scholar]
- 56.Horisawa S, Arai T, Suzuki N, Kawamata T, Taira T. The striking effects of deep cerebellar stimulation on generalized fixed dystonia: case report. J Neurosurg Epub 2019 Mar 1. [DOI] [PubMed] [Google Scholar]
- 57.Park JE, Vanegas-Arroyave N, Hallett M, Lungu C. A woman with a novel mutation of THAP1 with a prominent response to deep brain stimulation of the globus pallidus internus. JAMA Neurol 2015;72:1369. [DOI] [PubMed] [Google Scholar]
- 58.Noakes Z, Fjodorova M, Li M. Deriving striatal projection neurons from human pluripotent stem cells with Activin A. Neural Regen Res 2015;10:1914–1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Aneichyk T, Hendriks WT, Yadav R, et al. Dissecting the causal mechanism of X-linked dystonia-parkinsonism by integrating genome and transcriptome assembly. Cell 2018;172:897–909 e821. [DOI] [PMC free article] [PubMed] [Google Scholar]