

Abstract
Background
In recent randomized, placebo-controlled, phase III trials, highly purified cannabidiol demonstrated efficacy with an acceptable safety profile in patients with Lennox–Gastaut syndrome or Dravet syndrome. It is anticipated that antiepileptic drugs such as stiripentol and valproate will be administered concomitantly with cannabidiol.
Objectives
This trial evaluated the effect of cannabidiol on steady-state pharmacokinetics of stiripentol or valproate in patients with epilepsy, and the safety and tolerability of cannabidiol.
Methods
This phase II, two-arm, parallel-group, double-blind, randomized, placebo-controlled trial recruited male and female patients with epilepsy aged 16–55 years. Patients receiving a stable dose of stiripentol or valproate were randomized 4:1 to receive concomitant double-blind cannabidiol or placebo. Patients received plant-derived, highly purified cannabidiol medicine (Epidiolex® in the USA; Epidyolex® in the EU; 100 mg/mL oral solution) at a dose of 20 mg/kg/day from day 12 to 26, following a 10-day dose-escalation period. Blood samples for pharmacokinetic evaluations were collected on days 1 and 26 before stiripentol/valproate dosing and up to 12 h postdose. Treatment-emergent adverse events (AEs) were recorded.
Results
In total, 35 patients were recruited to the stiripentol arm (n = 14) or the valproate arm (n = 21). Both the safety and the pharmacokinetic populations of the stiripentol arm comprised 14 patients (2 placebo; 12 cannabidiol). The safety population of the valproate arm comprised 20 patients (4 placebo; 16 cannabidiol; one withdrew before receiving treatment); the pharmacokinetic population comprised 15 patients (3 placebo; 12 cannabidiol). Concomitant cannabidiol led to a small increase in stiripentol exposure (17% increase in maximum observed plasma concentration [Cmax]; 30% increase in area under the concentration–time curve over the dosing interval [AUCtau]). Concomitant cannabidiol also had little effect on valproate exposure (13% decrease in Cmax; 17% decrease in AUCtau) or its metabolite, 2-propyl-4-pentenoic acid (4-ene-VPA) (23% decrease in Cmax; 30% decrease in AUCtau). All changes in exposure are expressed as the dose-normalized geometric mean (CV%) day 26 to day 1 ratio. The most common AE was diarrhea; most AEs were mild. Two patients discontinued cannabidiol because of serious AEs (rash [n = 1] in the stiripentol arm; hypertransaminasemia [n = 1] in the valproate arm). A separate in vitro study investigated the bidirectional effect of cannabidiol, or its metabolite 7-carboxy-cannabidiol, on valproate plasma protein binding; no change in plasma protein binding was observed for either compound.
Conclusions
The clinical relevance of the increase in stiripentol exposure is unknown; patients receiving cannabidiol and stiripentol concomitantly should be monitored for adverse reactions as individual patient responses may vary. Coadministration of cannabidiol did not affect the pharmacokinetics of valproate or its metabolite, 4-ene-VPA, in adult patients with epilepsy. Safety results were consistent with the known safety profile of cannabidiol at a dose of 20 mg/kg/day.
Clinicaltrials.gov: NCT02607891.
Electronic supplementary material
The online version of this article (10.1007/s40263-020-00726-4) contains supplementary material, which is available to authorized users.
Key Points
| A phase II trial was conducted to evaluate any pharmacokinetic drug–drug interactions between cannabidiol and stiripentol or valproate in patients with epilepsy. |
| The combination of cannabidiol and stiripentol led to a small increase in exposure to stiripentol. |
| The combination of cannabidiol and valproate did not cause clinically important changes in the pharmacokinetics of valproate or its metabolite, 2-propyl-4-pentenoic acid (4-ene-VPA). |
| The safety profile of cannabidiol in combination with stiripentol or valproate was consistent with that previously reported. |
Introduction
Highly purified cannabidiol is approved in the USA as Epidiolex® (Greenwich Biosciences, Inc., Carlsbad, CA, USA) for seizures associated with Lennox–Gastaut syndrome (LGS) or Dravet syndrome (DS) in patients aged ≥ 2 years [1] and in the EU as Epidyolex® (GW Pharmaceuticals [International] B.V., Amersfoort, The Netherlands) for seizures associated with LGS or DS, in conjunction with clobazam, in patients aged ≥ 2 years [2].
In recent randomized, placebo-controlled, phase III trials, cannabidiol demonstrated efficacy with an acceptable safety profile in patients with LGS or DS [3–6]. These findings are supported by published results from an open-label extension (OLE) trial (maximum of 3 years’ exposure to cannabidiol) that followed the above phase III trials [7, 8] and a cannabidiol open-label expanded access program in patients with treatment-resistant epilepsy, including DS and LGS [9].
DS and LGS are rare epileptic encephalopathies that are both associated with intractable seizures and a severe burden of illness [10, 11]. Onset of seizures typically occurs in the first year of life in DS [11] and from 1 to 8 years of age in LGS [12]. Intractable seizures are associated with a high rate of polypharmacy in patients with DS and LGS [13–15], leaving them vulnerable to a high risk of adverse reactions due to drug–drug interactions (DDIs).
It is important to investigate possible DDIs between cannabidiol and antiseizure drugs, commonly referred to as antiepileptic drugs (AEDs), because cannabidiol will likely be used concomitantly with other AEDs. Clobazam and valproate are the optimal first-line medications in DS, whereas stiripentol is a preferred second-line add-on treatment if seizure control is suboptimal [15, 16]. Valproate is the preferred first-line treatment for LGS [14]. Stiripentol may be used as an add-on therapy with clobazam or valproate; however, evidence to support the efficacy of stiripentol in LGS treatment is limited [14]. Previous trials have investigated potential DDIs between cannabidiol and clobazam, stiripentol, or valproate in healthy volunteers [17] and between cannabidiol and clobazam in patients with epilepsy [18].
Induction or inhibition of enzymes involved in drug metabolism are major mechanisms that underlie DDIs [19, 20]. Enzymes of the cytochrome P450 (CYP) and UDP-glucuronosyltransferase (UGT) families are integral in drug metabolism [19–21], including the metabolism of cannabidiol, stiripentol, and valproate [22–25]. In vitro studies suggest that cannabidiol can inhibit various CYP or UGT enzymes, increasing the risk of a DDI [26–29]. These preclinical studies suggest that cannabidiol is a potent inhibitor of both CYP2C19 and CYP3A4, a weak CYP2D6 inhibitor, and a potential inhibitor of UGT1A9 and UGT2B7 [26–28]. Trials performed in patients with epilepsy or healthy volunteers have reported conflicting results on the effect of cannabidiol on CYP3A4 activity. Cannabidiol has been shown to mildly increase exposure to clobazam, a substrate of CYP3A4 [30], in pediatric patients with refractory epilepsy [31, 32]. However, trials in healthy volunteers have separately reported that concomitant cannabidiol has little or no effect on the pharmacokinetics of clobazam [17], consistent with a lack of effect on CYP3A4 activity [33]. It should be noted that any observed increases in exposure to clobazam were relatively minor compared with significant increases in exposure to N-desmethylclobazam (N-CLB) when in combination with cannabidiol [17, 31]. In vitro studies have suggested that, in addition to effects on CYP enzymes, cannabidiol is an inhibitor of both UGT1A9 and UGT2B7 [29].
Stiripentol is extensively metabolized by demethylenation and glucuronidation. The primary CYP enzymes involved in the phase I metabolism of stiripentol are CYP1A2, CYP2C19, and CYP3A4 [25]. In healthy volunteers, coadministration of stiripentol and cannabidiol led to a small increase in exposure to stiripentol (55% increase in area under the concentration–time curve over the dosing interval [AUCtau]; 28% increase in maximum observed plasma concentration [Cmax]) [17]. Conversely, concomitant stiripentol did not alter exposure to cannabidiol, but it caused a small decrease in exposure to the cannabidiol metabolites 7-hydroxy-cannabidiol (28% decrease in AUCtau; 29% decrease in Cmax) and 7-carboxy-cannabidiol (7-COOH-CBD) (13% decrease in AUCtau; 13% decrease in Cmax) [17]. The clinical relevance of these minor changes is unknown.
The major routes of valproate metabolism are glucuronidation and β-oxidation, which account for 50% and 40% of biotransformation, respectively. A comparatively minor route of metabolism forms 2-propyl-4-pentenoic acid (4-ene-VPA) and other metabolites via CYP2C9, CYP2B6, and CYP2A6; this route accounts for approximately 10% of the dose [23]. Investigation of potential DDIs with valproate is important because both valproate and 4-ene-VPA are associated with hepatotoxicity [34–36], and increased hepatic transaminase levels have been observed in patients receiving concomitant cannabidiol and valproate [3–6, 37]. A trial investigating potential DDIs with cannabidiol in healthy volunteers, and a dose-ranging safety trial in pediatric patients with epilepsy, showed no effect of cannabidiol on pharmacokinetic parameters or systemic exposure to valproate [17, 38]. In addition, concomitant valproate did not alter exposure to cannabidiol in healthy volunteers [17]. Other than pharmacokinetic interactions, another potential source of DDIs is displacement of drugs from plasma protein binding (PPB) sites by other drugs [39], which is more relevant for drugs such as valproate that are highly bound to plasma proteins [40]. The overall clearance and elimination of valproate is influenced by the amount of circulating, pharmacologically active, unbound drug as well as metabolic flux [40]. Therefore, a need exists for further investigation of the potential for PPB interactions and alterations in free levels of either cannabidiol or valproate when these two drugs are combined.
The primary objective of this phase II trial in patients with epilepsy was to determine whether cannabidiol affects the pharmacokinetic profile of stiripentol or valproate. The secondary objectives were to assess the safety and tolerability of cannabidiol in the presence of stiripentol or valproate and to assess whether cannabidiol affects the pharmacokinetic profile of 4-ene-VPA. The findings from an in vitro study that aimed to explore PPB displacement of cannabidiol, 7-COOH-CBD, and valproate in human plasma are also reported (see the electronic supplementary material [ESM]).
Methods
Patients
This trial recruited male and female patients aged 16–55 years (inclusive) (18–55 years inclusive in Sweden). Patients must have been receiving stiripentol (for patients in the stiripentol arm) or valproate (for patients in the valproate arm) and no more than two other AEDs during the double-blind period. Patients in the valproate arm must not have been receiving stiripentol. AED doses, including stiripentol or valproate, were required to be stable for 4 weeks prior to screening and throughout the double-blind period. Patients must have experienced at least one countable seizure of any type (i.e., tonic-clonic, tonic, clonic, atonic, or focal onset; focal onset seizures with retained awareness and a motor component; focal onset seizures with impaired awareness; focal onset impaired awareness seizures evolving to bilateral tonic-clonic seizures) within 2 months prior to randomization. Patients were also required to have a documented magnetic resonance imaging or computerized tomography scan to rule out a progressive neurological condition. Key exclusion criteria were a clinically significant unstable medical condition or a clinically significant illness (other than epilepsy) in the 4 weeks prior to screening, current or recent use of medicinal or recreational cannabis, a history of alcohol or drug addiction, use of felbamate for < 12 months prior to screening, consumption of grapefruit or grapefruit juice and/or alcohol (including 7 days prior to screening), significantly impaired hepatic function, a history of orthostatic blood pressure changes, a prolonged QT interval corrected for heart rate with Bazett’s formula, or any history of suicidal behavior or any suicidal ideation in the month prior to or at screening.
Trial Design
This phase II, two-arm, parallel-group, double-blind, randomized, placebo-controlled, DDI trial was conducted between 9 November 2016 and 2 October 2018 at five trial sites: three in Spain and one each in the Netherlands and Sweden (ClinicalTrials.gov Identifier: NCT02607891). The trial consisted of two arms to investigate the effect of concomitant treatment with cannabidiol on the pharmacokinetics of stiripentol or valproate. Efficacy assessments were not measured as outcomes for this trial, as the patients may have had a low baseline seizure frequency.
Patients already receiving a stable dose of stiripentol or valproate were randomized in a 4:1 ratio to receive concomitant double-blind cannabidiol or placebo. Patients received a pharmaceutical formulation of highly purified cannabidiol derived from Cannabis sativa L. plant (100 mg/mL oral solution; Epidiolex® in the USA, Epidyolex® in the EU; GW Research Ltd, Cambridge, UK) or equivalent volume of placebo. All patients received their first dose of cannabidiol or placebo on day 2 immediately after stiripentol or valproate administration, which was administered in line with the physician’s preferred dosing regimen for each patient. A 10-day dose-escalation period was followed by a 14-day maintenance period (Fig. 1). The target dose of cannabidiol was 20 mg/kg/day administered as two equal doses in the morning and evening. Patients were instructed to be consistent in the timing of their food intake relative to dosing throughout the double-blind period; however, the exact timing of dosing in relation to meals was not recorded.
Fig. 1.

Trial design. CBD cannabidiol, OLE open-label extension, PK pharmacokinetics, STP stiripentol, VPA valproate
Following the end of blinded treatment (day 26), patients were invited to enter the OLE period (to be reported separately), which had a maximum duration of 1 year. If a patient chose not to enter the OLE period, the cannabidiol dose was tapered down over 10 days. Patients then returned to the clinic for an end-of-taper visit, followed by a safety follow-up visit 28 days later (Fig. 1).
Pharmacokinetic Assessments
Blood samples were taken either by direct venipuncture or via an indwelling cannula inserted into a forearm vein at the following time points on day 1 (AED alone) and day 26 (AED + cannabidiol or placebo): predose, 15 and 30 minutes, then 1, 1.5, 2, 4, 6, and 12 h postdose. The timing of each pharmacokinetic sample was relative to the morning dose of stiripentol or valproate.
Plasma concentrations of all analytes were determined using fully validated liquid chromatographic-tandem mass spectrometry bioanalytical methods (Covance Laboratories Ltd, Harrogate, UK; Pharmaron UK Ltd, Rushden, UK).
For stiripentol, valproate, and 4-ene-VPA, the following pharmacokinetic parameters were estimated by noncompartmental methods: Cmax, time to attain Cmax (tmax), and AUCtau.
Safety and Tolerability Assessments
Treatment-emergent adverse events (AEs) were recorded throughout the trial and at a post-trial safety follow-up. Safety assessments (laboratory parameters, vital signs, 12-lead electrocardiograms [ECGs], and physical examinations) were made at screening and on days 1, 12, and 26. The Columbia-Suicide Severity Rating Scale (C-SSRS) questionnaire [41] was completed by the investigator or their qualified designee at all trial visits. Drug abuse liability was monitored in response to triggering AEs of special interest, if cannabidiol was overused or went missing, and by patient interview. Patients (or caregivers) recorded seizure occurrence in a diary from screening until completion of dosing.
Statistical Analysis
There was no formal sample size calculation, and analyses were descriptive only. The planned sample size was 14 patients in the stiripentol arm and 20 patients in the valproate arm. Safety analyses were based on the safety population, which included all patients enrolled in the trial who received at least one dose of cannabidiol, placebo, or stiripentol/valproate during the double-blind period.
The pharmacokinetic population included all patients who received at least one dose of cannabidiol or placebo and who provided sufficient pharmacokinetic concentration data to derive pharmacokinetic parameters for day 1 or day 26.
Cmax and AUCtau for stiripentol, valproate, and 4-ene-VPA were dose normalized by dividing by the morning dose of stiripentol or valproate (expressed in mg/kg). Any patients who deviated from their regular dosing schedules were excluded from the statistical analysis for the treatment effect.
To assess whether the presence of cannabidiol altered the pharmacokinetic profile of stiripentol or valproate, a standard 90% confidence interval (CI) approach for the between-time-point ratios of geometric means of Cmax and AUCtau was carried out on logarithmic scale using a linear mixed-effect model with treatment (stiripentol or stiripentol + cannabidiol on a first model, valproate or valproate + cannabidiol on a second model, and 4-ene-VPA or 4-ene-VPA + cannabidiol on a third model) as a fixed effect and patient as a random effect. If the 90% CI for the ratio of the geometric means of a pharmacokinetic variable fell within the interval (0.5–2.0), a lack of meaningful effect was concluded.
Plasma Protein Binding (PPB) Displacement Study
Human plasma pooled from three healthy volunteers was used to assess the effect of cannabidiol and 7-COOH-CBD on the PPB of 14C-valproate and the effect of 14C-valproate on the PPB of cannabidiol and 7-COOH-CBD using an ultracentrifugation method (for detailed methods see the ESM) [42].
Results
Patient Disposition
Overall, 35 patients were screened and randomized to the stiripentol arm (n = 14) or the valproate arm (n = 21) (Fig. 2). Both the safety population and the pharmacokinetic population of the stiripentol arm comprised all 14 patients (2 placebo; 12 cannabidiol). One patient from the valproate arm withdrew before receiving treatment. The safety population of the valproate arm thus comprised 20 patients (4 placebo; 16 cannabidiol). Five patients were excluded from the pharmacokinetic population of the valproate arm due to insufficient or missed pharmacokinetic sampling (n = 4) or noncompliance (n = 1). In total, 30 patients completed the trial and entered the OLE period. Two patients, one from each treatment arm, discontinued from the double-blind period due to AEs. Two additional patients met protocol-specified withdrawal criteria: tetrahydrocannabinol was detected in the serum of one patient (stiripentol arm) and one patient withdrew consent to participate after experiencing moderate nausea and diarrhea (valproate arm).
Fig. 2.

Disposition of patients. AE treatment-emergent adverse event, CBD cannabidiol, STP stiripentol, VPA valproate. aPatient was excluded because of the presence of tetrahydrocannabinol in serum. bPatient did not want to participate because of diarrhea and nausea, which were recorded as AEs leading to withdrawal
Demographics and Baseline Characteristics
Overall, most patients enrolled in the trial were male (65%), and all patients were white (Table 1). The mean age of the patients was 29.5 years (range 17.4–54.5). The mean body mass index was 26.8 kg/m2. Most (97%) patients were taking an AED in addition to stiripentol or valproate. The most common baseline seizure types were focal onset with impaired awareness (50%), generalized onset tonic-clonic (36%), and myoclonic (36%) in the stiripentol arm; and focal onset with impaired awareness (55%), focal onset evolving to bilateral tonic-clonic seizures (40%), and generalized onset tonic-clonic (35%) in the valproate arm. The median daily doses of stiripentol and valproate received by patients in each arm were 625 mg (500–2000 mg) and 1115 mg (200–2500 mg), respectively.
Table 1.
Overall demographics and baseline characteristics (safety population)
| Placebo (n = 6) | CBD (n = 28) | Total (N = 34) | |
|---|---|---|---|
| Age (years) | 26.9 ± 7.0 | 30.1 ± 11.1 | 29.5 ± 10.5 |
| Sex | |||
| Male | 5 (83) | 17 (61) | 22 (65) |
| Female | 1 (17) | 11 (39) | 12 (35) |
| Race, white | 6 (100) | 28 (100) | 34 (100) |
| Height (cm) | 178.3 ± 6.9 | 174.3 ± 13.5 | 175.0 ± 12.6 |
| Weight (kg) | 86.0 ± 18.3 | 81.9 ± 22.2 | 82.7 ± 21.4 |
| Body mass index (kg/m2) | 27.0 ± 5.3 | 26.8 ± 5.4 | 26.8 ± 5.3 |
| STP arm | Placebo (n = 2) | CBD (n = 12) | Total (n = 14) |
|---|---|---|---|
| Daily STP dose (mg/day) | 1250 (500–2000) | 625 (500–2000) | 625 (500–2000) |
| No. receiving any AEDa | 2 (100) | 11 (92) | 13 (93) |
| Most common AEDs (≥ 10%) in any treatment group | |||
| Lacosamide | 2 (100) | 3 (25) | 5 (36) |
| Clobazam | 0 (0) | 4 (33) | 4 (29) |
| VPA | 0 (0) | 3 (25) | 3 (21) |
| Ethosuximide | 0 (0) | 2 (17) | 2 (14) |
| Lamotrigine | 0 (0) | 2 (17) | 2 (14) |
| Topiramate | 0 (0) | 2 (17) | 2 (14) |
| VPA sodium | 0 (0) | 2 (17) | 2 (14) |
| Levetiracetam | 1 (50) | 0 (0) | 1 (7) |
| Rufinamide | 1 (50) | 0 (0) | 1 (7) |
| VPA arm | Placebo (n = 4) | CBD (n = 16) | Total (n = 20) |
|---|---|---|---|
| Daily VPA dose (mg/day) | 1450 (900–2500) | 1015 (200–1950) | 1115 (200–2500) |
| No. receiving any AEDa | 4 (100) | 16 (100) | 20 (100) |
| Most common AEDs (≥ 10%) in any treatment group | |||
| Lacosamide | 0 (0) | 3 (19) | 3 (15) |
| Clobazam | 0 (0) | 6 (38) | 6 (30) |
| Lamotrigine | 2 (50) | 4 (25) | 6 (30) |
| Carbamazepine | 1 (25) | 1 (6) | 2 (10) |
| Levetiracetam | 0 (0) | 2 (13) | 2 (10) |
| Lorazepam | 1 (25) | 1 (6) | 2 (10) |
| Oxcarbazepine | 1 (25) | 1 (6) | 2 (10) |
| Rufinamide | 1 (25) | 2 (13) | 3 (15) |
| Clonazepam | 1 (25) | 2 (13) | 3 (15) |
| Zonisamide | 0 (0) | 2 (13) | 2 (10) |
Data are presented as mean ± standard deviation, n (%), or median (range) unless otherwise indicated
AED antiepileptic drug, CBD cannabidiol, STP stiripentol, VPA valproate
aOther than VPA or STP as required for each arm
Pharmacokinetics
Cannabidiol
The plasma concentration–time profiles of cannabidiol on day 26 were consistent with those previously derived following multiple twice-daily dosing [43], confirming that steady state was achieved during the maintenance period (see Supplementary Fig. 1 and Supplementary Fig. 2 in the ESM).
Stiripentol
Plasma concentration–time curves indicated that stiripentol plasma concentrations were higher in the presence of cannabidiol than with stiripentol alone; this effect was not observed in the two patients in the placebo group (Fig. 3a). However, coadministration of cannabidiol with steady-state stiripentol did not show a marked effect of cannabidiol on the pharmacokinetic parameters of stiripentol (Table 2). The treatment ratio (TR) point estimates for Cmax of 1.17 (90% CI 1.03–1.33) and for AUCtau of 1.30 (90% CI 1.09–1.55) represented a small increase in exposure to stiripentol following concomitant cannabidiol administration. Individual patient data revealed that increases in stiripentol exposure were not observed in all patients (Fig. 4a).
Fig. 3.
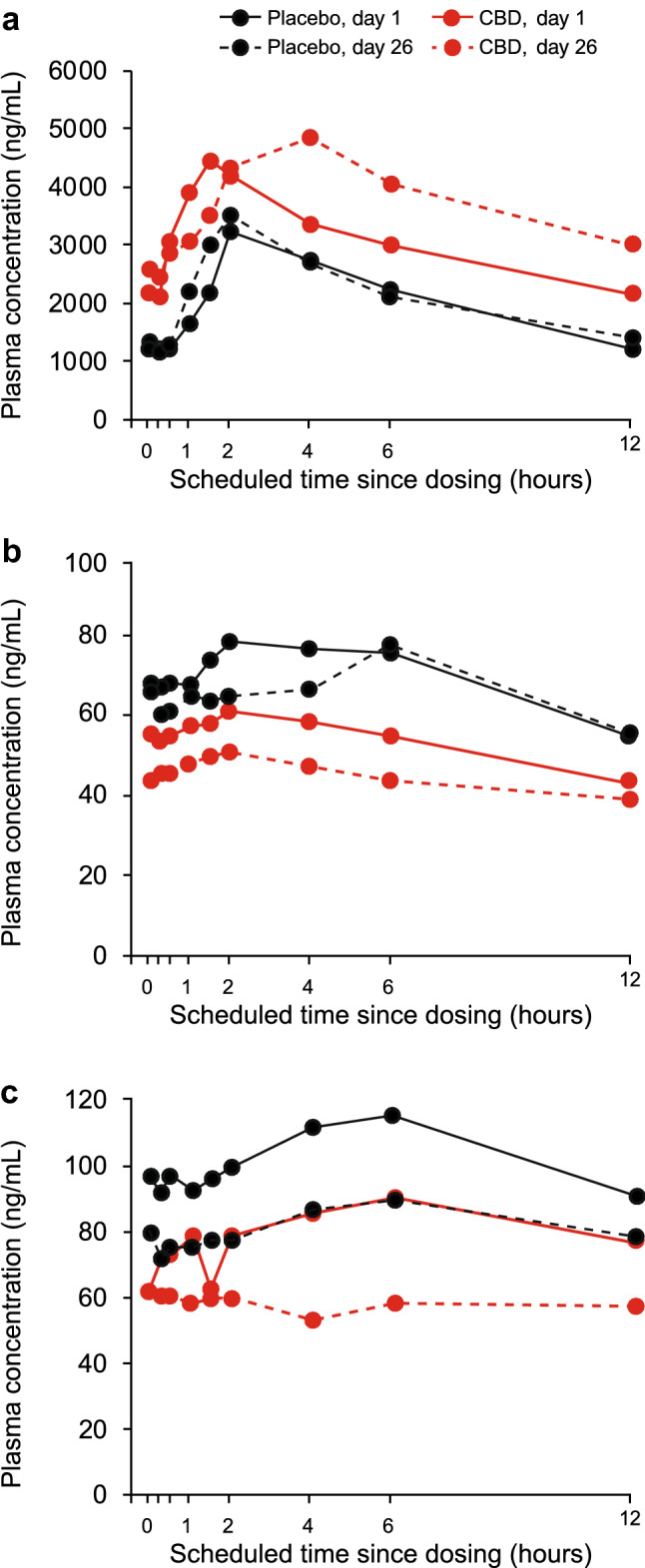
The effect of concomitant steady-state CBD on steady-state plasma concentrations of a STP, b VPA, c 4-ene-VPA (PK population). Patient numbers for an = 2 (placebo, day 1); n = 2 (placebo, day 26); n = 9 (CBD, day 1); n = 9 (CBD, day 26); bn = 3 (placebo, day 1); n = 3 (placebo, day 26); n = 10 (CBD, day 1); n = 10 (CBD, day 26); cn = 3 (placebo, day 1); n = 3 (placebo, day 26); n = 10 (CBD, day 1); n = 10 (CBD, day 26). 4-ene-VPA 2-propyl-4-pentenoic acid, CBD cannabidiol, STP stiripentol, VPA valproate
Table 2.
PK parameters on days 1 and 26 for STP and VPA and 4-ene-VPA (PK population)
| Parameter | Day 1 (AED alone) | Day 26 (AED + placebo) | Day 1 (AED alone) | Day 26 (AED + CBD) | Treatment ratiod (90% CI) |
|---|---|---|---|---|---|
| STP | Placebo (n = 2) | CBD (n = 12) | |||
| na | 2 | 2 | 11 | 9 | 9 |
| DN Cmax (ng/mL/mg)b | 7.7 (16.3) | 7.7 (18.4) | 7.6 (80.0) | 10.7 (59.4) | 1.17 (1.03–1.33) |
| DN AUCtau (ng.h/mL/mg)b | 49.7 (22.0) | 51.8 (41.3) | 52.3 (108.5) | 80.7 (66.8) | 1.30 (1.09–1.55) |
| tmax (h)c | 3.9 (2.0–5.9) | 1.7 (1.4–2.0) | 1.5 (0.3–6.1) | 2.1 (1.4–6.0) | |
| VPA | Placebo (n = 3) | CBD (n = 12) | |||
| na | 3 | 3 | 12 | 10 | 10 |
| DN Cmax (ng/mL/mg)b | 161 (54) | 168 (47) | 173 (55) | 143 (61) | 0.87 (0.79–0.95) |
| DN AUCtau (ng.h/mL/mg)b | 1620 (63) | 1540 (51) | 1710 (64) | 1350 (62) | 0.83 (0.75–0.92) |
| tmax (h)c | 3.9 (1.5–4.0) | 4.0 (0.0–6.0) | 3.0 (0.0–6.2) | 1.8 (0.0–12.0) | |
| 4-ene-VPA | Placebo (n = 3) | CBD (n = 12) | |||
| na | 3 | 3 | 12 | 10 | 10 |
| DN Cmax (ng/mL/mg)b | 0.23 (39) | 0.19 (45) | 0.25 (103) | 0.18 (90) | 0.77 (0.66–0.90) |
| DN AUCtau (ng.h/mL/mg)b | 2.35 (42) | 1.86 (42) | 2.54 (105) | 1.70 (103) | 0.70 (0.62–0.80) |
| tmax (h)c | 5.92 (4.00–6.00) | 3.85 (0.00–6.00) | 6.00 (0.25–12.48) | 1.73 (0.00–11.50) |
4-ene-VPA, 2-propyl-4-pentenoic acid, AED antiepileptic drug, AUCtau area under the plasma concentration-time curve over a dosing interval, where tau is the dosing interval, CBD cannabidiol, Cmax maximum observed plasma concentration, CI confidence interval, DN dose-normalized, PK pharmacokinetic, STP, stiripentol, tmax time to attain maximum observed plasma concentration, VPA valproate
aAll patients with PK data available
bGeometric mean (interpatient variability)
cMedian (range)
dGeometric least squares mean ratio for AED steady state + CBD steady state: AED steady state alone
Fig. 4.
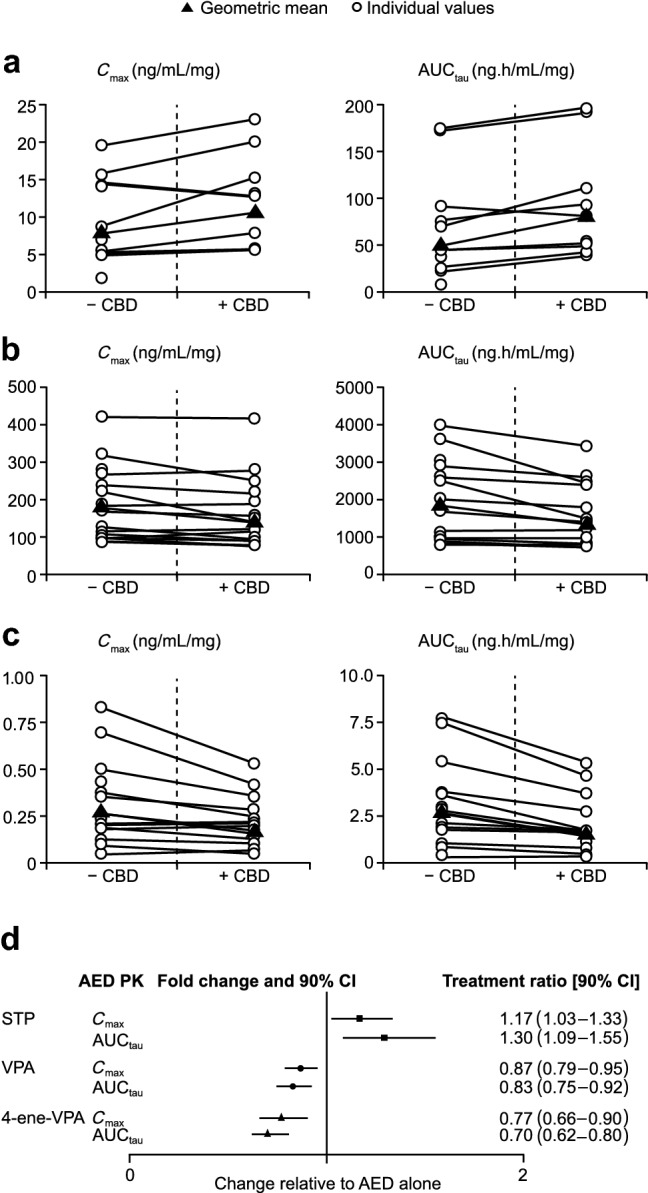
The effect of concomitant steady-state CBD on individual and geometric mean steady-state exposure (Cmax and AUCtau) of a STP, b VPA, and c 4-ene-VPA and d treatment ratios with 90% CIs for STP, VPA, and 4-ene-VPA (PK population). Patient numbers for dn = 9 (STP AUCtau); n = 9 (STP Cmax); n = 10 (VPA AUCtau); n = 10 (VPA Cmax); n = 10 (4-ene-VPA AUCtau); n = 10 (4-ene-VPA Cmax). 4-ene-VPA 2-propyl-4-pentenoic acid, AED antiepileptic drug, AUCtau area under the plasma concentration–time curve over a dosing interval, where tau is the dosing interval, CBD cannabidiol, CI confidence interval, Cmax maximum observed plasma concentration, PK pharmacokinetics, STP stiripentol, VPA valproate
Valproate
In the valproate arm, plasma concentrations of valproate were lower in the presence of cannabidiol than with valproate alone over the 12-h pharmacokinetic sampling period. By contrast, there was no clear difference between the valproate plasma concentration–time curves for patients receiving placebo on day 26 compared with day 1 (Fig. 3b). Plasma concentrations of 4-ene-VPA were lower in the presence of cannabidiol than with valproate alone; this effect was also observed in the placebo group (Fig. 3c).
Coadministration of cannabidiol with valproate did not show a marked effect (the 90% CIs of the TRs fell within the prespecified interval [0.5–2.0]) of cannabidiol on the pharmacokinetics of valproate or its metabolite, 4-ene-VPA (Table 2). For valproate, the point estimates for the geometric mean Cmax and AUCtau TRs were 0.87 (90% CI 0.79–0.95) and 0.83 (90% CI 0.75–0.92). For 4-ene-VPA, the point estimates for the geometric mean Cmax and AUCtau TRs were 0.77 (90% CI 0.66–0.90) and 0.70 (90% CI 0.62–0.80).
There was no obvious change in Cmax and AUCtau in any patients for valproate or 4-ene-VPA (Fig. 4b, c).
PPB Displacement Study
In an in vitro study, the combination of valproate and cannabidiol or 7-COOH-CBD at therapeutically relevant concentrations (0.1 × Cmax, Cmax, and 5 × Cmax) did not alter the PPB parameters of any of the test compounds (see the ESM for full results).
Safety and Tolerability
Eight (57%) patients in the stiripentol arm (all in the cannabidiol group) experienced an AE (Table 3). The most common AEs were diarrhea and fatigue; most AEs were mild. One patient in the stiripentol arm experienced a serious, severe rash that led to discontinuation of cannabidiol and withdrawal from the trial. The rash was generalized with no mucosal involvement and resolved after both cannabidiol and stiripentol were discontinued. Two patients receiving cannabidiol in the stiripentol arm experienced increased alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels. The ALT or AST values did not exceed three times the upper limit of normal (ULN) in either patient. These increases resolved during the trial, and both patients completed the trial.
Table 3.
Summary of AEs in STP arm (safety population)
| Number of patients, n (%) | Placebo (n = 2) | CBD (n = 12) |
|---|---|---|
| At least one AE | 0 (0) | 8 (67) |
| AEs leading to discontinuation | 0 (0) | 1 (8) |
| Serious AEs | 0 (0) | 1 (8) |
| Severe AEs | 0 (0) | 1 (8) |
| Death | 0 (0) | 0 (0) |
| AEs reported by ≥ 10% of patients | ||
| Diarrhea | 0 (0) | 5 (42) |
| Fatigue | 0 (0) | 3 (25) |
| Nausea | 0 (0) | 2 (17) |
| ALT increased | 0 (0) | 2 (17) |
| AST increased | 0 (0) | 2 (17) |
| Decreased appetite | 0 (0) | 2 (17) |
AE treatment-emergent adverse event, ALT alanine aminotransferase, AST aspartate aminotransferase, CBD cannabidiol, STP stiripentol
In the valproate arm, AEs were more common in the cannabidiol group (88%) than in the placebo group (25%) (Table 4). The most common AE was diarrhea; most AEs were mild. One patient in the valproate arm experienced a serious AE of moderate hypertransaminasemia (ALT 5.5× ULN), which led to discontinuation of cannabidiol and withdrawal from the trial. This AE resolved by day 40. A second patient receiving cannabidiol withdrew consent to participate in the trial after experiencing moderate diarrhea and moderate nausea. In addition, another patient receiving cannabidiol in the valproate arm had an ALT level of 365 U/L (12.2× ULN) and an AST level of 205 U/L (5.4× ULN) at the end of double-blind treatment (day 28). These findings in this patient were not reported as AEs in the double-blind period of the trial.
Table 4.
Summary of AEs in VPA arm (safety population)
| Number of patients, n (%) | Placebo (n = 4) | CBD (n = 16) |
|---|---|---|
| At least one AE | 1 (25) | 14 (88) |
| AEs leading to discontinuation | 0 (0) | 2 (13) |
| Serious AEs | 0 (0) | 1 (6) |
| Severe AEs | 0 (0) | 0 (0) |
| Death | 0 (0) | 0 (0) |
| AEs reported by ≥ 10% of patients | ||
| Diarrhea | 0 (0) | 11 (69) |
| Nausea | 0 (0) | 2 (13) |
| Nasopharyngitis | 0 (0) | 2 (13) |
AE treatment-emergent adverse event, CBD cannabidiol, VPA valproate
There were no findings of clinical significance in other laboratory parameters, vital signs, ECGs, or physical examination findings. No instances of laboratory findings met Hy’s law criteria for severe drug-induced liver injury, including the cases of increased ALT/AST.
There was no suicidal ideation or suicidal behavior as assessed by C-SSRS. No signals of potential substance abuse were exhibited by any patients in either treatment arm.
Evaluation of treatment efficacy by analysis of seizure frequency was not an objective of this trial. However, seizure frequency data were available for ten patients in the stiripentol arm and 16 patients in the valproate arm. In the cannabidiol group, a decrease in seizure frequency from baseline was observed in five patients in the stiripentol arm and five patients in the valproate arm. Data were not available for patients in the placebo group of the stiripentol arm, and two patients in the placebo group of the valproate arm experienced a decrease in seizure frequency from baseline (data not shown).
Discussion
The primary purpose of this trial was to assess the effect of cannabidiol on the pharmacokinetic profile of stiripentol or valproate in adult patients with epilepsy. The tested dose of cannabidiol in this trial was equivalent to the highest maintenance dose used in recent phase III trials in DS and LGS [3–6].
Coadministration of steady-state cannabidiol led to a small increase in exposure to steady-state stiripentol (30% increase in AUCtau; 17% increase in Cmax). These findings are consistent with those from a phase I pharmacokinetic trial that investigated possible DDIs between stiripentol and cannabidiol in healthy volunteers and reported a small increase in exposure to stiripentol (55% increase in AUCtau; 28% increase in Cmax) [17]. The slight discrepancy in pharmacokinetic results is likely due to differences in trial populations and designs. The mechanism for this increase in stiripentol exposure is not fully elucidated but may result from inhibition of CYP2C19 or some isoforms of UGT by cannabidiol. Cannabidiol inhibits both CYP2C19 and some UGT isoforms in human liver microsomes [28, 29] and led to a significant increase in exposure to N-CLB (238% increase in AUCtau; 239% increase in Cmax), a CYP2C19 substrate and the active metabolite of clobazam, in healthy volunteers [17]. There was a substantial increase in exposure to stiripentol in some patients receiving cannabidiol and stiripentol and a decrease in exposure to stiripentol in other patients, which suggests that responses of individual patients may vary. Patients receiving cannabidiol and stiripentol should therefore be monitored for adverse drug reactions.
Coadministration of cannabidiol did not cause clinically important changes in the pharmacokinetics of valproate or its metabolite, 4-ene-VPA, in adult patients with epilepsy. These findings are consistent with those from two previous trials, one performed in healthy volunteers and the other performed in pediatric patients with DS. Both trials reported no effect on valproate exposure following concomitant cannabidiol administration [17, 38]. In an in vitro study, combining valproate with cannabidiol or 7-COOH-CBD at therapeutically relevant concentrations did not alter PPB parameters, suggesting that valproate in combination with cannabidiol is not likely to increase the free drug fraction of either drug in human plasma. These findings collectively suggest that there is no pharmacokinetic or PPB interaction between cannabidiol and valproate. However, results from phase III trials in patients with DS or LGS indicate that dose-related elevations in hepatic transaminase levels are more common in patients receiving concomitant valproate and cannabidiol (10 or 20 mg/kg/day) than in those not receiving valproate. In these trials, the incidence of ALT elevations > 3× the ULN was 2% in patients receiving cannabidiol only, 3% in patients taking concomitant cannabidiol and clobazam, 17% in patients taking concomitant cannabidiol and valproate, and 23% in patients taking concomitant cannabidiol, valproate, and clobazam [2]. The mechanism behind the increased incidence of elevated hepatic transaminases with concomitant cannabidiol and valproate is unknown. However, results from this phase II trial and the in vitro PPB study appear to discount an increase in exposure to valproate or 4-ene-VPA due to concomitant cannabidiol exposure or any increase in unbound, active valproate as a likely cause for these transaminase increases. If clinically relevant elevations in hepatic transaminases occur in patients receiving concomitant valproate and cannabidiol, dose adjustment or discontinuation of valproate should be considered [2]. Dose adjustment of clobazam should be considered in this case if patients are also receiving concomitant clobazam [2]. Changes in exposure of stiripentol or valproate were described in accordance with the US FDA guidelines on clinical drug interaction studies. According to these guidelines, a weak inhibitor increases the AUC of a substrate by ≥ 1.25-fold to < 2-fold and a weak inducer decreases the AUC of a substrate by ≥ 20 to < 50% [44].
The safety results from this trial in patients with epilepsy are broadly consistent with the known safety profile of cannabidiol at a dose of 20 mg/kg/day in patients with DS or LGS [3–6]. Diarrhea was the most frequently reported AE in the cannabidiol group in both arms (stiripentol arm: 5/12 [42%]; valproate arm: 11/16 [69%]); no patients receiving placebo reported an AE of diarrhea. The frequency of diarrhea observed in patients receiving cannabidiol in this DDI trial (42–69%) was higher than that seen in previous phase III trials at 20 mg/kg/day (15–31%), although direct safety comparisons between these studies should be undertaken with caution because of the different patient age groups and trial durations [3–6].
There are a number of limitations to this trial. The effect of stiripentol or valproate on the pharmacokinetics of cannabidiol and its metabolites was not investigated, although such a trial has been conducted in healthy volunteers [17]. This trial also contained fewer patients in the stiripentol arm than in the valproate arm and low patient numbers in the placebo groups. However, the sample size in the cannabidiol groups was sufficient for robust pharmacokinetic conclusions. Most patients in this trial were receiving at least one AED other than valproate or stiripentol. This may have affected the results of this trial through interference with enzymes that metabolize cannabidiol, stiripentol, or valproate. The sample size was too low to analyze the effect of any other concomitant AED on the pharmacokinetics of stiripentol or valproate in combination with cannabidiol. While the PPB parameters of valproate in combination with cannabidiol were studied owing to the observed elevation in hepatic transaminases, investigation of such an interaction with stiripentol was not an objective of this trial. The combination of cannabidiol and stiripentol led to a small increase in stiripentol exposure and was generally well-tolerated in this trial, suggesting there is no important displacement effect. However, we cannot rule out cannabidiol displacing stiripentol PPB.
Conclusions
Coadministration of cannabidiol in patients with epilepsy led to a small increase in exposure to stiripentol. Patients receiving cannabidiol and stiripentol should be monitored for adverse drug reactions as responses of individual patients may vary. Coadministration of cannabidiol did not cause clinically important changes in the pharmacokinetics of valproate or its metabolite, 4-ene-VPA, in adult patients with epilepsy. Safety results were consistent with the known safety profile of cannabidiol at a dose of 20 mg/kg/day. This trial was conducted with Epidyolex®, and results do not apply to other cannabidiol-containing products.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
The authors thank the trial participants and the trial sites that participated in this trial, as well as Paula Stubbs (BSc Hons), GW Research Ltd, for her contributions as the clinical trial manager. Medical writing support was provided to the authors by Liam O’Reilly, PhD of Helios Medical Communications, Alderley Park, UK, and funded by Greenwich Biosciences, Inc., Carlsbad, CA, USA. Cannabidiol was provided by GW Research Ltd, Cambridge, UK.
Compliance with Ethical Standards
Data Availability
The sponsor adheres to current requirements of the USA and the EU so will not make individual de-identified participant data available; however, the protocol and statistical analysis plan will be made available upon request to the corresponding author.
Funding
Open access funding provided by Greenwich Biosciences, Inc. This trial was sponsored by GW Research Ltd, Cambridge, UK.
Conflicts of Interest
Elinor Ben-Menachem has received consulting honoraria from GW Pharmaceuticals companies, UCB, Sandoz, Eisai, and Bial and has been a principal investigator for GW Research Ltd. Boudewijn Gunning has received consultancy fees from GW Pharmaceuticals companies, Zogenix, and Ovid/Takeda; has been a principal investigator for GW Research Ltd, Zogenix, LivaNova, Marinus Pharmaceuticals, and UCB; and is a member of the International Speakers Bureau of LivaNova. Carmen María Arenas Cabrera has received consultancy fees from UCB, Eisai, Bial, and Esteve and has been a principal investigator for GW Research Ltd. Kevan VanLandingham is employed by Greenwich Biosciences, Inc. and owns shares in GW Pharmaceuticals plc. Bola Tayo and Gilmour Morrison are employed by GW Research Ltd and own shares in GW Pharmaceuticals plc. David Critchley, Louise Wray, and Julie Crockett are employed by GW Research Ltd and have share options in GW Pharmaceuticals plc. Manuel Toledo has received consulting honoraria and lecture fees from GW Pharmaceuticals companies, UCB Pharma, Eisai, Bial, Esteve, and Sanofi and has been a principal investigator for GW Research Ltd.
Ethical Approval
The protocol and informed consent forms were approved by local independent ethics committees. All procedures performed in studies involving human participants were in accordance with the International Council for Harmonisation Good Clinical Practice Consolidated Guideline, the applicable local laws and regulatory requirements of the countries in which the trial was conducted, and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed Consent
Written informed consent/assent was obtained from all individual patients included in the trial (or their parent/legal representative) before any trial-specific procedures were performed or any patient-related data were collected.
References
- 1.Epidiolex® (cannabidiol) oral solution, CV [prescribing information]. Carlsbad, CA: Greenwich Biosciences, Inc. 2018.
- 2.GW Pharmaceuticals, plc. Epidyolex 100 mg/ml oral solution: summary of product characteristics (SmPC). 2019.
- 3.Devinsky O, Cross JH, Laux L, Marsh E, Miller I, Nabbout R, et al. Trial of cannabidiol for drug-resistant seizures in the Dravet syndrome. N Engl J Med. 2017;376:2011–2020. doi: 10.1056/NEJMoa1611618. [DOI] [PubMed] [Google Scholar]
- 4.Devinsky O, Patel AD, Cross JH, Villanueva V, Wirrell EC, Privitera M, et al. Effect of cannabidiol on drop seizures in the Lennox-Gastaut syndrome. N Engl J Med. 2018;378:1888–1897. doi: 10.1056/NEJMoa1714631. [DOI] [PubMed] [Google Scholar]
- 5.Thiele EA, Marsh ED, French JA, Mazurkiewicz-Beldzinska M, Benbadis SR, Joshi C, et al. Cannabidiol in patients with seizures associated with Lennox-Gastaut syndrome (GWPCARE4): a randomised, double-blind, placebo-controlled Phase 3 trial. Lancet. 2018;391:1085–1096. doi: 10.1016/S0140-6736(18)30136-3. [DOI] [PubMed] [Google Scholar]
- 6.Miller I, Scheffer IE, Gunning B, Sanchez-Carpintero R, Gil-Nagel A, Perry MS, et al. Dose-ranging effect of adjunctive oral cannabidiol vs placebo on convulsive seizure frequency in Dravet syndrome: a randomized clinical trial. JAMA Neurol. 2020;10.1001/jamaneurol.2020.007, [Epub ahead of print]. [DOI] [PMC free article] [PubMed]
- 7.Devinsky O, Nabbout R, Miller I, Laux L, Zolnowska M, Wright S, et al. Long-term cannabidiol treatment in patients with Dravet syndrome: an open-label extension trial. Epilepsia. 2019;60:294–302. doi: 10.1111/epi.14628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thiele E, Marsh E, Mazurkiewicz-Beldzinska M, Halford JJ, Gunning B, Devinsky O, et al. Cannabidiol in patients with Lennox-Gastaut syndrome: interim analysis of an open-label extension study. Epilepsia. 2019;60:419–428. doi: 10.1111/epi.14670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Szaflarski JP, Bebin EM, Comi AM, Patel AD, Joshi C, Checketts D, et al. Long-term safety and treatment effects of cannabidiol in children and adults with treatment-resistant epilepsies: expanded access program results. Epilepsia. 2018;59:1540–1548. doi: 10.1111/epi.14477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Camfield PR. Definition and natural history of Lennox-Gastaut syndrome. Epilepsia. 2011;52:3–9. doi: 10.1111/j.1528-1167.2011.03177.x. [DOI] [PubMed] [Google Scholar]
- 11.Dravet C. The core Dravet syndrome phenotype. Epilepsia. 2011;52:3–9. doi: 10.1111/j.1528-1167.2011.02994.x. [DOI] [PubMed] [Google Scholar]
- 12.Crumrine PK. Management of seizures in Lennox-Gastaut syndrome: Paediatr Drugs. 2011;13:107–118. doi: 10.2165/11536940-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 13.Bourgeois BFD, Douglass LM, Sankar R. Lennox-Gastaut syndrome: a consensus approach to differential diagnosis. Epilepsia. 2014;55:4–9. doi: 10.1111/epi.12567. [DOI] [PubMed] [Google Scholar]
- 14.Cross JH, Auvin S, Falip M, Striano P, Arzimanoglou A. Expert opinion on the management of Lennox-Gastaut syndrome: treatment algorithms and practical considerations. Front Neurol. 2017;8:505. doi: 10.3389/fneur.2017.00505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ziobro J, Eschbach K, Sullivan JE, Knupp KG. Current treatment strategies and future treatment options for Dravet syndrome. Curr Treat Options Neurol. 2018;20:52. doi: 10.1007/s11940-018-0537-y. [DOI] [PubMed] [Google Scholar]
- 16.Wirrell EC, Laux L, Donner E, Jette N, Knupp K, Meskis MA, et al. Optimizing the diagnosis and management of Dravet Syndrome: recommendations from a North American consensus panel. Pediatr Neurol. 2017;68(18–34):e3. doi: 10.1016/j.pediatrneurol.2017.01.025. [DOI] [PubMed] [Google Scholar]
- 17.Morrison G, Crockett J, Blakey G, Sommerville K. A Phase 1, open-label, pharmacokinetic trial to investigate possible drug-drug interactions between clobazam, stiripentol, or valproate and cannabidiol in healthy subjects. Clin Pharmacol Drug Dev. 2019;8:1009–1031. doi: 10.1002/cpdd.665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Patsalos P, Gidal B, Szaflarski J, VanLandingham K, Critchley D, Morrison G. Drug-drug interaction (DDI) studies with coadministration of cannabidiol (CBD) and clobazam (CLB), valproate (VPA), stiripentol (STP) or midazolam (MDZ) in healthy volunteers (HVTS) and adults with epilepsy. Dev Med Child Neurol. 2019;61:5–14. [Google Scholar]
- 19.Kiang TKL, Ensom MHH, Chang TKH. UDP-glucuronosyltransferases and clinical drug-drug interactions. Pharmacol Ther. 2005;106:97–132. doi: 10.1016/j.pharmthera.2004.10.013. [DOI] [PubMed] [Google Scholar]
- 20.Manikandan P, Nagini S. Cytochrome P450 structure, function and clinical significance: a review. Curr Drug Targets. 2018;19:38–54. doi: 10.2174/1389450118666170125144557. [DOI] [PubMed] [Google Scholar]
- 21.Lynch T, Price A. The effect of cytochrome P450 metabolism on drug response, interactions, and adverse effects. Am Fam Physician. 2007;76:391–396. [PubMed] [Google Scholar]
- 22.Jiang R, Yamaori S, Takeda S, Yamamoto I, Watanabe K. Identification of cytochrome P450 enzymes responsible for metabolism of cannabidiol by human liver microsomes. Life Sci. 2011;89:165–170. doi: 10.1016/j.lfs.2011.05.018. [DOI] [PubMed] [Google Scholar]
- 23.Ghodke-Puranik Y, Thorn CF, Lamba JK, Leeder JS, Song W, Birnbaum AK, et al. Valproic acid pathway: pharmacokinetics and pharmacodynamics. Pharmacogenet Genomics. 2013;23:236–241. doi: 10.1097/FPC.0b013e32835ea0b2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ujváry I, Hanuš L. Human metabolites of cannabidiol: a review on their formation, biological activity, and relevance in therapy. Cannabis Cannabinoid Res. 2016;1:90–101. doi: 10.1089/can.2015.0012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Biocodex. Diacomit 250 mg hard capsules: summary of product characteristics (SmPC). 2018.
- 26.Yamaori S, Ebisawa J, Okushima Y, Yamamoto I, Watanabe K. Potent inhibition of human cytochrome P450 3A isoforms by cannabidiol: role of phenolic hydroxyl groups in the resorcinol moiety. Life Sci. 2011;88:730–736. doi: 10.1016/j.lfs.2011.02.017. [DOI] [PubMed] [Google Scholar]
- 27.Yamaori S, Okamoto Y, Yamamoto I, Watanabe K. Cannabidiol, a major phytocannabinoid, as a potent atypical inhibitor for CYP2D6. Drug Metab Dispos. 2011;39:2049–2056. doi: 10.1124/dmd.111.041384. [DOI] [PubMed] [Google Scholar]
- 28.Jiang R, Yamaori S, Okamoto Y, Yamamoto I, Watanabe K. Cannabidiol is a potent inhibitor of the catalytic activity of cytochrome P450 2C19. Drug Metab Pharmacokinet. 2013;28:332–338. doi: 10.2133/dmpk.DMPK-12-RG-129. [DOI] [PubMed] [Google Scholar]
- 29.Al Saabi A, Allorge D, Sauvage F-L, Tournel G, Gaulier J-M, Marquet P, et al. Involvement of UDP-glucuronosyltransferases UGT1A9 and UGT2B7 in ethanol glucuronidation, and interactions with common drugs of abuse. Drug Metab Dispos. 2013;41:568–574. doi: 10.1124/dmd.112.047878. [DOI] [PubMed] [Google Scholar]
- 30.Giraud C, Tran A, Rey E, Vincent J, Tréluyer J-M, Pons G. In vitro characterization of clobazam metabolism by recombinant cytochrome P450 enzymes: importance of CYP2C19. Drug Metab Dispos. 2004;32:1279–1286. doi: 10.1124/dmd.32.11.1279. [DOI] [PubMed] [Google Scholar]
- 31.Geffrey AL, Pollack SF, Bruno PL, Thiele EA. Drug-drug interaction between clobazam and cannabidiol in children with refractory epilepsy. Epilepsia. 2015;56:1246–1251. doi: 10.1111/epi.13060. [DOI] [PubMed] [Google Scholar]
- 32.Wheless JW, Dlugos D, Miller I, Oh DA, Parikh N, Phillips S, et al. Pharmacokinetics and tolerability of multiple doses of pharmaceutical-grade synthetic cannabidiol in pediatric patients with treatment-resistant epilepsy. CNS Drugs. 2019;33:593–604. doi: 10.1007/s40263-019-00624-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Morrison G, Taylor L, Crockett J, Critchley D, Tayo B. A phase 1 investigation into the potential effects of cannabidiol on CYP3A4-mediated drug-drug interactions in healthy volunteers. In: AES 2018 Annual Meeting Abstract Database. AESnet.org. 2018.
- 34.Rettie AE, Rettenmeier AW, Howald WN, Baillie TA. Cytochrome P-450 – catalyzed formation of delta 4-VPA, a toxic metabolite of valproic acid. Science. 1987;235:890–893. doi: 10.1126/science.3101178. [DOI] [PubMed] [Google Scholar]
- 35.Sadeque AJ, Fisher MB, Korzekwa KR, Gonzalez FJ, Rettie AE. Human CYP2C9 and CYP2A6 mediate formation of the hepatotoxin 4-ene-valproic acid. J Pharmacol Exp Ther. 1997;283:698–703. [PubMed] [Google Scholar]
- 36.Arroyo S, de la Morena A. Life-threatening adverse events of antiepileptic drugs. Epilepsy Res. 2001;47:155–174. doi: 10.1016/S0920-1211(01)00306-0. [DOI] [PubMed] [Google Scholar]
- 37.Gaston TE, Bebin EM, Cutter GR, Liu Y, Szaflarski JP, UAB CBD Program. Interactions between cannabidiol and commonly used antiepileptic drugs. Epilepsia. 2017;58:1586–92. [DOI] [PubMed]
- 38.Devinsky O, Patel AD, Thiele EA, Wong MH, Appleton R, Harden CL, et al. Randomized, dose-ranging safety trial of cannabidiol in Dravet syndrome. Neurology. 2018;90:e1204–e1211. doi: 10.1212/WNL.0000000000005254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Heuberger J, Schmidt S, Derendorf H. When is protein binding important? J Pharm Sci. 2013;102:3458–3467. doi: 10.1002/jps.23559. [DOI] [PubMed] [Google Scholar]
- 40.Perucca E, Grimaldi R, Gatti G, Pirracchio S, Crema F, Frigo GM. Pharmacokinetics of valproic acid in the elderly. Br J Clin Pharmacol. 1984;17:665–669. doi: 10.1111/j.1365-2125.1984.tb02401.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Posner K, Brown GK, Stanley B, Brent DA, Yershova KV, Oquendo MA, et al. The Columbia-Suicide Severity Rating Scale: initial validity and internal consistency findings from three multisite studies with adolescents and adults. Am J Psychiatry. 2011;168:1266–1277. doi: 10.1176/appi.ajp.2011.10111704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bowers WF, Fulton S, Thompson J. Ultrafiltration vs equilibrium dialysis for determination of free fraction. Clin Pharmacokinet. 1984;9(Suppl. 1):49–60. doi: 10.2165/00003088-198400091-00007. [DOI] [PubMed] [Google Scholar]
- 43.Taylor L, Gidal B, Blakey G, Tayo B, Morrison G. A phase I, randomized, double-blind, placebo-controlled, single ascending dose, multiple dose, and food effect trial of the safety, tolerability and pharmacokinetics of highly purified cannabidiol in healthy subjects. CNS Drugs. 2018;32:1053–1067. doi: 10.1007/s40263-018-0578-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.US Food & Drug Administration (FDA). Clinical drug interaction studies—cytochrome p450 enzyme- and transporter-mediated drug interactions guidance for industry [Internet]. 2020. https://www.fda.gov/media/134581/download. Accessed 26 Feb 2020.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The sponsor adheres to current requirements of the USA and the EU so will not make individual de-identified participant data available; however, the protocol and statistical analysis plan will be made available upon request to the corresponding author.